

SUMMARY
Protein aggregates disrupt cellular homeostasis, causing toxicity linked to neurodegeneration. Selective autophagic elimination of aggregates is critical to protein quality control, but how aggregates are selectively targeted for degradation is unclear. We compared the requirements for autophagy receptor proteins: OPTN, NBR1, p62, NDP52, and TAX1BP1 in clearance of proteotoxic aggregates. Endogenous TAX1BP1 is recruited to and required for the clearance of stress-induced aggregates, whereas ectopic expression of TAX1BP1 increases clearance through autophagy, promoting viability of human induced pluripotent stem cell-derived neurons. In contrast, TAX1BP1 depletion sensitizes cells to several forms of aggregate-induced proteotoxicity. Furthermore, TAX1BP1 is more specifically expressed in the brain compared to other autophagy receptor proteins. In vivo, loss of TAX1BP1 results in accumulation of high molecular weight ubiquitin conjugates and premature lipofuscin accumulation in brains of young TAX1BP1 knockout mice. TAX1BP1 mediates clearance of a broad range of cytotoxic proteins indicating therapeutic potential in neurodegenerative diseases.
Graphical Abstract
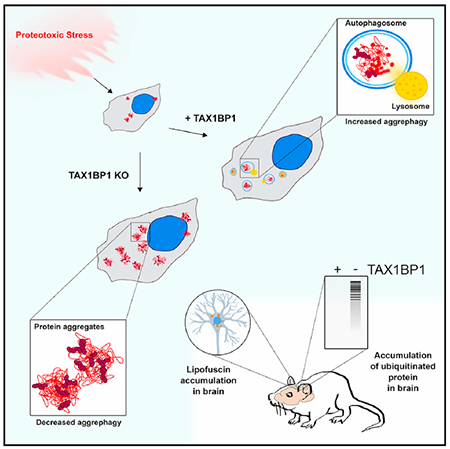
In Brief
Sarraf et al. discover a role for the autophagy receptor TAX1BP1 in clearance of protein aggregates induced by a broad range of proteotoxic stresses, including translational stress, proteasome inhibition, and expression of huntingtin protein. Mice lacking functional TAX1BP1 accumulate ubiquitinated protein conjugates in the brain.
INTRODUCTION
Maintenance of cellular and organismal health is intricately connected to protein quality control. A balance exists between protein translation, folding, and degradation that maintains the stoichiometry and function of cellular protein complexes and organelles. If this balance is perturbed, accumulation of misfolded proteins is toxic to the cell, causing disruption of cellular function, a pathological characteristic of numerous neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and Alzheimer’s disease (Balchin et al., 2016; Dikic, 2017). A number of protein quality control pathways exist within the cell to forestall dysfunction. Molecular chaperone systems function to monitor and refold proteins if possible, while misfolded or damaged proteins are targeted for elimination via autophagy or the ubiquitin proteasome system (UPS) (Mizushima and Komatsu, 2011; Rubinsztein, 2006). The UPS is generally responsible for routine turnover of short-lived proteins and targeted degradation of soluble or solubilized misfolded proteins, whereas macroautophagy, a catabolic process, culminates in the lysosomal degradation of long-lived proteins, damaged organelles, and portions of the cytoplasm (Feng et al., 2014; Hershko and Ciechanover, 1998; Sherman and Goldberg, 2001; Wong and Cuervo, 2010). Because the proteasome can only accommodate unfolded polypeptide chains, it is generally thought that autophagy is responsible for the removal of insoluble protein aggregates (Kopito, 2000; Lamark and Johansen, 2012). If either autophagy or the UPS is hindered, acute or chronic proteotoxic stress, such as that caused by expression of mutated or misfolded proteins in neurodegenerative disease, can result in accumulation of these aggregation-prone proteins (Bennett et al., 2005; Morimoto, 2008; Salomons et al., 2009). Inhibition of general autophagy in vivo results in intracellular protein aggregation contributing to neurodegeneration in mice (Hara et al., 2006; Komatsu et al., 2006).
In recent years, numerous studies have highlighted the ability of autophagy to selectively eliminate specific substrates, including endoplasmic reticulum, intracellular pathogens, mitochondria, and protein aggregates (Khaminets et al., 2015; Kirkin et al., 2009a; Lazarou et al., 2015). In these cases, the autophagic machinery employed in nonselective bulk degradation of cytosolic material is targeted to specific cargo. Stimulation of autophagy is a promising therapeutic strategy in the treatment of protein aggregation diseases and has been shown to enhance turnover of aggregated proteins, such as TDP-43, in neuronal ALS models and huntingtin protein in HD models (Barmada et al., 2014; Rubinsztein et al., 2015; Sarkar and Rubinsztein, 2008) . Key to this approach is an understanding of how autophagy is selectively targeted to aggregates. In addition to the specificity mediated by E3 ubiquitin (Ub) ligases that target their cognate substrates, there is selectivity in delivery to the proteasome via Ub receptors (Hjerpe et al., 2016) and in the recruitment of autophagic machinery via autophagy receptors, which generally associate with polyubiquitylated proteins, thus linking substrates to the degradation machinery (Kirkin et al., 2009a).
Numerous types of selective autophagy have been examined, and much progress has been made in determining the basis of cargo selectivity. The autophagy receptor proteins OPTN, NDP52, TAX1BP1 and p62 are known to be important in xenophagy (von Muhlinen et al., 2010; Tumbarello et al., 2015), whereas OPTN, NDP52, and to a lesser extent, TAX1BP1 are essential for PINK1/Parkin-mediated mitophagy (Heo et al., 2015; Lazarou et al., 2015; Wong and Holzbaur, 2014). A screen in yeast identified the ATG8 and Ub-binding protein, CUET, and its mammalian homolog, TOLLIP, as contributors to autophagy of expanded polyQ isoforms of huntingtin protein (Lu et al., 2014). NBR1 and p62 are linked to aggrephagy in mammalian cells (Lamark et al., 2009) ; however, p62 and NBR1 knockout (KO) mice exhibit only a mild increase in ubiquitylated aggregates, suggesting that additional receptors are involved (Komatsu et al., 2007). Here, we identify an essential role for TAX1BP1 in clearance of protein aggregates induced by various proteotoxic stressors. Furthermore, loss of endogenous TAX1BP1 in mice demonstrates that TAX1BP1 is essential for aggregate clearance in vivo.
RESULTS
TAX1BP1 Deletion Impairs Clearance of Protein Aggregates
To compare roles for five autophagy receptor proteins in aggrephagy, we examined production and clearance of puromycin-induced misfolded proteins in the cytosol (Eggers et al., 1997), termed “DRiPs” for defective ribosomal products (Yewdell et al., 1996) in individual KOs of the autophagy receptor proteins NBR1, p62, NDP52, OPTN, and TAX1BP1 (Figures 1A, 1B, 1C, and S1A). Consistent with published reports (Bjørkøy et al., 2005; Kirkin et al., 2009b), we found a drastic reduction in puromycin-induced foci formation in p62 and NBR1 KO cell lines, as well as in NDP52 KO cells compared to wild-type (WT) cells after 2 h of puromycin (Figures 1C, 1D, and 1E). Although aggregate formation was decreased in NBR1 and NDP52 KO cells, any foci that did form were effectively cleared by 3 h after washout of puromycin (Figures 1C, 1F, and 1G). In contrast, individual OPTN KO and TAX1BP1 KO cell lines showed robust Ub-positive foci formation, equal to or greater than WT cells after 2 h of puromycin (Figures 1C, 1D, and 1E) and a significant block in aggregate clearance after puromycin washout. More than twice as many cells with foci (~54%) persisted in OPTN KO cells compared to WT (~22%), while in TAX1BP1 KO cells, nearly 4 times as many cells retained foci (~85%) (Figures 1C, 1F, and 1G). In TAX1BP1 KO cells with foci, the number of foci per cell, although equivalent to the WT at 2 h of puromycin (Figures S1B and S1C), was substantially higher than WT cells upon puromycin washout (Figure S1D), indicating a block in clearance rather than increased aggregate formation. Our data reveal distinct roles for autophagy receptors in protein aggregate formation and clearance.
Figure 1. TAX1BP1 Depletion Impairs Clearance of Protein Aggregates.
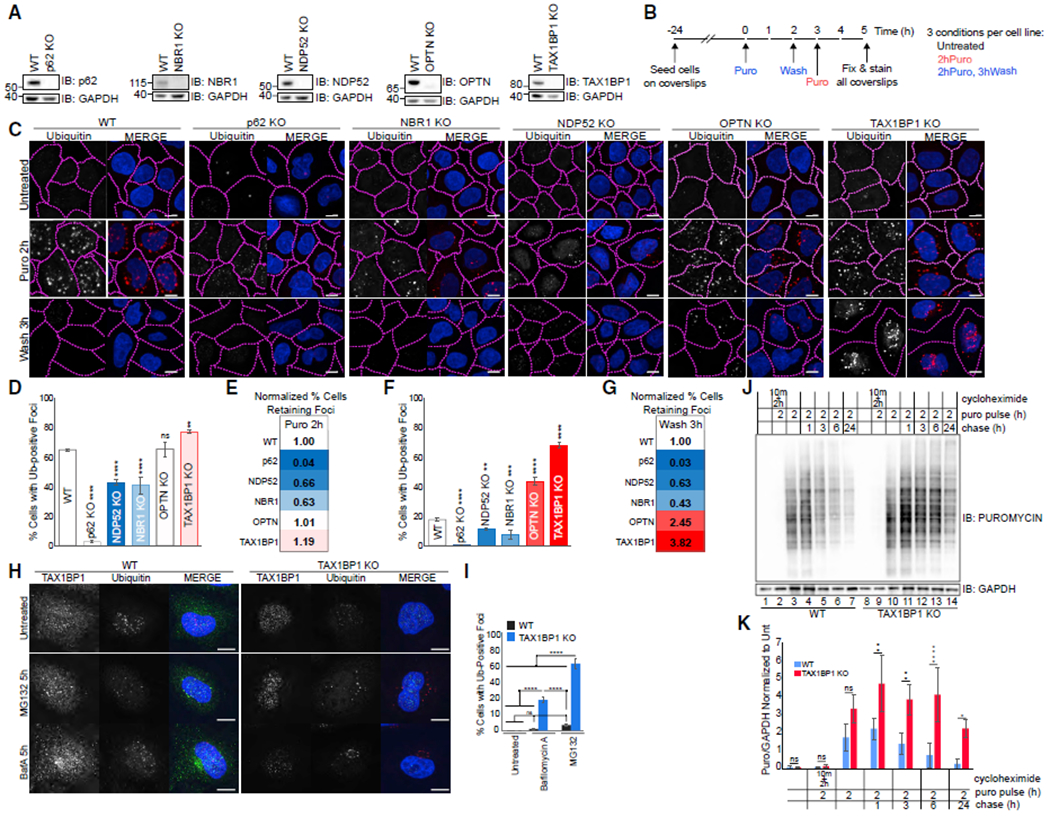
(A) Validation of knockout cell lines.
(B) Experimental outline for assessing aggregate formation and clearance.
(C) WT or individual knockouts for p62, NBR1, NDP52, OPTN, and TAX1BP1 cell lines exposed to 5 μg/mL puromycin for 2 h were either fixed for imaging or washed and followed for a further 3 h in full media as in (B); scale bar represents 10 μm.
(D and F) Quantification of (C): percent of cells containing Ub-positive foci was assessed in ~200 cells per condition at 2 h puromycin (D) or 2 h puromycin followed by 3 h washout (F). Quantification displayed as mean ± SD from three independent experiments using one-way ANOVA test (**p < 0.01, ***p < 0.001, ****p < 0.0001) comparing all to WT and Tukey’s post hoc test.
(E and G) WT-normalized comparisons of foci formation and clearance in autophagy receptor knockout cell lines.
(H) WT or TAX1BP1 KO cells were exposed to 100 nM Bafilomycin A or 1 μM MG132 for 5 h then fixed for imaging.
(I) Quantification of (H): percent of cells containing Ub-positive foci was assessed in ~200 cells per condition. Quantification displayed as mean ± SD from three independent experiments using one-way ANOVA test, (****p < 0.0001) comparing all to WT and Tukey’s post hoc test.
(J) WT or TAX1BP1 KO cells pulsed with 5 μg/mL puromycin and 15 μM cycloheximide as indicated, then chased/harvested at the indicated time points and immunoblotted for puromycin or GAPDH (loading control).
(K) Quantification of (J) determined by densitometry, normalized first to GAPDH and subsequently to untreated condition for each cell line. Quantification displayed as mean ± SD from three independent experiments using two-way ANOVA test (**p < 0.01, ***p < 0.001, ****p < 0.0001) and Tukey’s post hoc test. All blots and microscopy images are representative of at least three independent experiments.
We observed complementary results upon exposure of cells to low levels of proteasome inhibition, which induces juxtanuclear aggresome-like structures removed via autophagy (Kopito, 2000). Imaging revealed decreased Ub-labeled foci formation in p62, NBR1, and NDP52 KO cells (Figure S1E). Consistent with this, immunoblot of MG132-treated cellular lysates after fractionation revealed no accumulation of insoluble Ub-positive protein in the p62 KO and decreased levels in NBR1 and NDP52 KO cells relative to WT cells (Figures S1F, S1G, and S1H). However, in OPTN and TAX1BP1 KO cells, more Ub-foci accumulated than in WT cells (Figure S1E). Ub-conjugated protein was increased in the insoluble fraction of TAX1BP1 KO (~4× increase) and OPTN KO (~2x increase) compared to WT cells (Figures S1F and S1G), consistent with a block in aggrephagy.
We compared accumulation of Ub-positive punctae upon short term exposure to MG132 or Bafilomycin A, an autophagy inhibitor, to determine whether there is basal accumulation of misfolded or aggregated protein in TAX1BP1 KO cells. We saw little effect in WT cells (Figures 1H and 1I), but in TAX1BP1 KO cells, both inhibitors resulted in formation of Ub-positive foci (Figures 1H and 1I), although MG132 did so to a much greater extent (Figures 1H and 1I). This implies that the UPS is compensating for an increased basal proteotoxic load in TAX1BP1 KO cells, most likely due to a block in TAX1BP1-mediated selective aggrephagy. To further assess the effects of TAX1BP1 loss on protein turnover, we pulsed cells with puromycin for either 2 h (corresponding to the puromycin treatment in Figure 1C) to induce accumulation of misfolded protein or 10 min to briefly label newly synthesized proteins in the absence of global translation inhibition. We then assessed loss of puromycin-labeled peptides over 24 h via immunoblotting (Figures 1J, 1K, S2A, and S2B). In both cases, we observed prolonged retention of puromycin-labeled peptides in TAX1BP1 KO cells relative to WT cells, supporting a role for TAX1BP1 in clearance of misfolded proteins.
Because we observed very different effects on cellular aggregate levels among the individual autophagy receptor KOs, we considered that, although this family of proteins is grouped as functionally similar, there are most likely significant biological distinctions in their roles. We compared DRiP clearance after puromycin treatment in WT cells or cells in which all five autophagy receptor proteins were knocked out (pentaKO) (Figure S2C). Because DRiP formation and clearance has been reported to require p62 (Pankiv et al., 2007), and on the basis of our observations in the single KO lines (Figure 1C), we were surprised to observe robust formation of Ub-positive foci upon 2 h of puromycin treatment in the pentaKO cell line (Figures S2D and S2E). We speculated that one explanation for this observation was that a strong block in aggregate clearance might overcome decreased aggregate formation, allowing us to observe aggregate accumulation in the absence of p62. Indeed, we observed a significant block in clearance of puromycin-induced aggregates in pentaKO compared to WT cells (Figures S2D and S2E), suggesting that loss of clearance-promoting autophagy receptors OPTN and TAX1BP1 was most likely allowing build-up of ubiquitinated material in the absence of p62. Proteasome inhibition similarly resulted in the appearance of Ub-positive foci in both WT and pentaKO cells, which accumulated to a greater extent in pentaKO cells (Figure S2F). While both WT and pentaKO cells accumulated Ub-positive material, foci were small, uniform, and generally perinuclear in WT cells, whereas in pentaKO cells, loosely associated masses of Ub-positive material persisted. We propose that the disordered appearance is due to an inability to promote clustering of ubiquitinated material in the absence of p62 and NBR1 (Kirkin et al., 2009b).
To further clarify distinctions between the roles of p62, NBR1, and TAX1BP1 in aggregate formation and clearance, we used CRISPR to make additional combinatorial KO lines: p62/NBR1 double KO (p62/NBR1 DKO), p62/TAX1BP1 double KO (p62/TAX1BP1 DKO), and p62/NBR1/TAX1BP1 triple KO (p62/NBR1/TAX1BP1 TKO) HeLa cells (Table S1). These cell lines, rather than the pentaKO cell line lacking additional autophagy receptor proteins, allow us to investigate roles of the individual proteins more specifically. Again, we observed little foci formation in p62 KO cells upon puromycin treatment and increased foci formation accompanied by a block in clearance in TAX1BP1 KO compared to WT cells (Figure S2G). The p62/NBR1 DKO behaved similarly to both the p62 and NBR1 single KOs, with little foci formation. Most informatively, we observed minimal foci formation in the p62/TAX1BP1 DKO and p62/NBR1/TAX1BP1 TKO (Figure S2G), allowing us to determine that foci formation, mediated by p62 and, to an extent, NBR1, is most likely upstream of clearance by TAX1BP1.
These results initially appear inconsistent with observations in the pentaKO cell line (Figures S2D and S2E) in which we observed foci formation and a mild block in aggregate clearance. Although it might be expected that no foci would form in the pentaKO because of the absence of p62 and NBR1, there are at least two possible reasons for this phenomenon. First, the additional KO of OPTN, which, like TAX1BP1, when singly knocked out caused a block in clearance (Figures 1C–1G), caused the level of misfolded protein to overwhelm the inability to form foci resulting in the appearance of foci in the pentaKO cell line (Figures S2D and S2E). As stated earlier, these foci appear less condensed and are perhaps less organized than foci that are formed in the presence of p62 (Figures S2D and S2F). To illustrate this better, we increased the duration of exposure time of pentaKO cells to puromycin and observed a striking difference in the appearance of the ubiquitinated material (Figure S2H). In WT cells exposed to puromycin for 18 h, we observed discrete ubiquitinated foci like those observed with shorter puromycin treatment. In contrast, in pentaKO cells, the ubiquitinated material was not present in discrete foci but diffuse or loosely clumped in the perinuclear region (Figure S2H). Second, we observed that the fitness of the pentaKO line and additional clones appears to be much lower than that of individual or combinatorial KOs of autophagy receptor proteins, exhibiting significantly decreased viability compared to WT HeLa cells (Figure S2I) as well as a mild but significant cell cycle defect affecting growth rate (Figure S2J). Therefore, the individual or double/ triple KO lines may be more informative in terms of examining the roles of the various autophagy receptor proteins. Thus, we place p62 and NBR1 in the roles of aggregate-forming autophagy receptor proteins, whereas TAX1BP1 and OPTN contribute to aggregate clearance, distinguishing functionally and temporally distinct roles for individual autophagy receptor proteins.
TAX1BP1 in Neurons and the Brain
We observed distinct roles in aggregate formation for p62, NBR1, and potentially NDP52, whereas TAX1BP1 and OPTN appear to contribute to aggregate clearance. To differentiate their roles in aggregate clearance, we further compared TAX1BP1, OPTN, and NDP52. TAX1BP1 and NDP52 are paralogous proteins of the CALCOCO gene family, containing a SKICH domain, a canonical LC3-interacting region (LIR), and coiled-coil domains while differing in primary sequence and in conservation of the C-terminal zinc finger (ZF) domains in various species (Figure S1A) (Tumbarello et al., 2015). On the basis of our initial observations, it seems likely that the specificity of TAX1BP1 and NDP52 may have diverged over time, allowing TAX1BP1 to assume a specific role in aggrephagy. Comparison of expression of TAX1BP1, OPTN, and NDP52 in a panel of human tissue lysates revealed that TAX1BP1 is highly and specifically expressed in brain, and to a lesser extent in kidney, while NDP52 was undetectable in brain and most likely does not function in neuronal tissues (Figure 2A). A database of transcriptome profiling of multiple CNS cell types purified from both mouse and human brains confirmed high mRNA expression of TAX1BP1 in the CNS compared to other autophagy receptors (Zhang et al., 2014, 2016). Consistent with a vital biological function, TAX1BP1, the most evolutionarily conserved member of the CALCOCO gene family, is present widely in vertebrates, unlike NDP52, which is sporadically lost or truncated (Tumbarello et al., 2015). Strikingly, OPTN was expressed broadly in all but one tissue type examined while TAX1BP1 was restricted (Figure 2A), suggesting that TAX1BP1 function may be especially important in the brain. OPTN is known to be an important autophagy receptor protein with roles in mitophagy, xenophagy, and aggrephagy, and although it has been extensively studied in selective autophagy pathways, TAX1BP1, perhaps because of its tissue specificity, is much less well understood.
Figure 2. TAX1BP1 Protein Responds to Proteotoxic Stress and Associates with Insoluble Protein in Neurons.
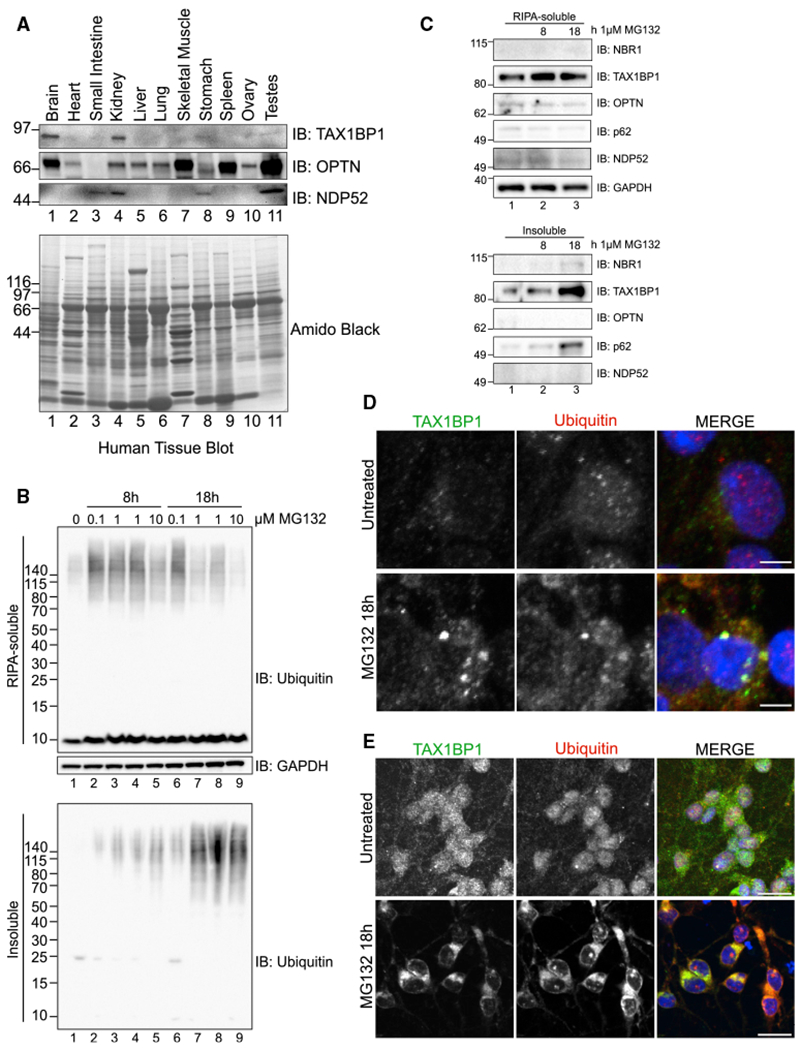
(A) Human tissue panel probed for TAX1BP1, OPTN, or NDP52.
(B) Primary rat cortical neurons were treated with the indicated amounts of MG132 for 8 or 18 h, fractionated into RIPA-soluble or -insoluble fractions, and immunoblotted for total Ub.
(C) Fractionated primary rat cortical neurons treated as in (B) were blotted for the indicated proteins.
(D) Primary rat cortical neurons were treated with 1 μM MG132 for18h, after which cells were fixed for imaging and stained with antibodies for TAX1BP1 and Ub; scale bar represents 5 μm. Larger fields of view in Figure S3D.
(E) Neurons derived from human induced pluripotent stem cells (iPSCs) treated with 1 μM MG132 for 18 h, fixed and stained with antibodies targeting TAX1BP1 and Ub; scale bar represents 20 μm. All blots and images are representative of at least three independent experiments. Larger fields of view in Figure S3E.
To investigate the role of TAX1BP1 in the brain, we examined primary rat cortical neurons exposed to proteotoxic stress. As in human cultured cells, insoluble Ub-conjugated protein accumulated upon MG132 exposure (Figure 2B). TAX1BP1 was expressed robustly in rat cortical neurons and accumulated to a greater extent in the insoluble fraction with increased proteotoxic stress compared to other autophagy receptor proteins (Figure 2C), and we observed a concomitant increase in TAX1BP1 protein levels (Figure 2C). A similar effect was observed in HeLa cells (Figures S3A–S3C), highlighting an important role forTAX1BP1 in aggrephagy.
To further examine the association between TAX1BP1 and stress-induced protein aggregates, we assessed TAX1BP1 localization in primary rat cortical neurons (Figures 2D and S3D) and in neurons derived from human iPSCs (Figures 2E and S3E). TAX1BP1 signal was diffuse in untreated cells but colocalized robustly in distinct punctae with Ub upon MG132 treatment (Figures 2D, 2E, S3D, and S3E). To our knowledge, this is the first study to identify a role for TAX1BP1 in aggrephagy and suggests that TAX1BP1 plays a specific role in neuronal aggrephagy.
TAX1BP1 Levels Modify Aggregate Clearance
To further validate a role for TAX1BP1 in aggrephagy, we reintroduced GFP-TAX1BP1 at near endogenous levels in the TAX1BP1 KO cell line (Figures 3A and S4A). In untreated cells, GFP-TAX1BP1 was diffuse or present in small punctae but colocalized with Ub-positive foci upon 2 h of puromycin treatment (Figure 3A). After puromycin washout, Ub foci were cleared to a similar extent in the TAX1BP1-rescued KO cells as in WT cells and GFP-TAX1BP1 returned to a diffuse appearance (Figures 3A and 3B).
Figure 3. TAX1BP1 Mediates Aggregate Clearance.
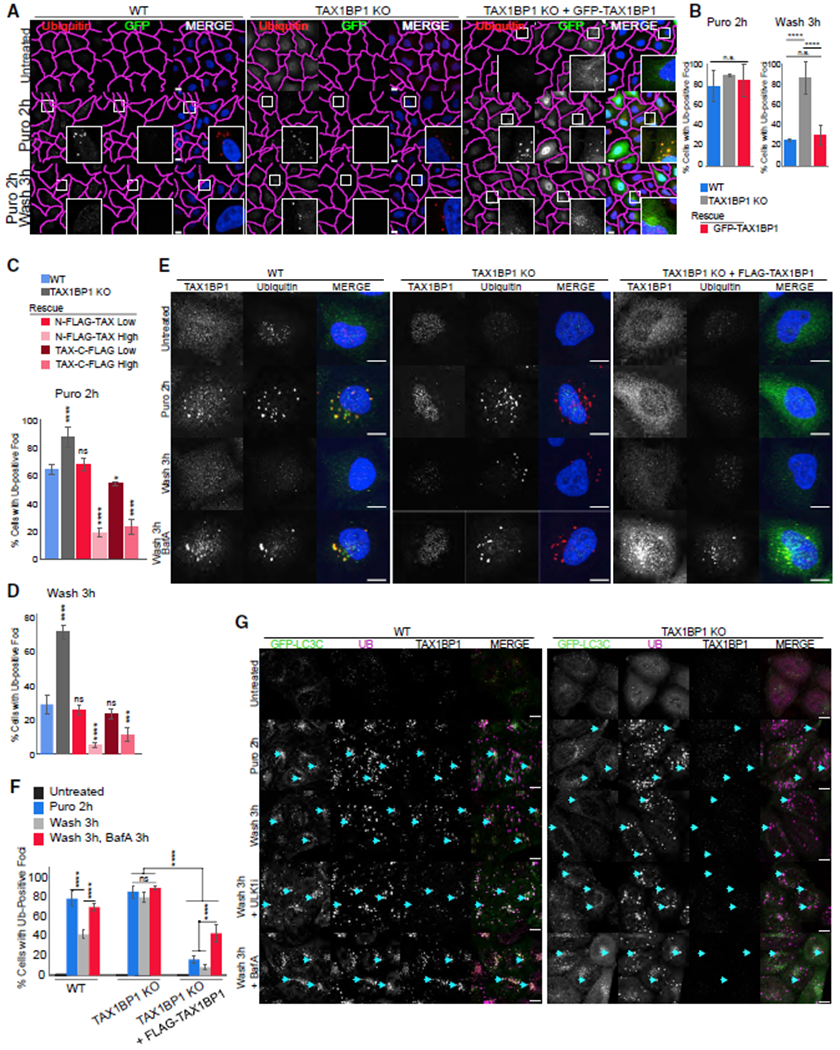
(A) WT, TAX1BP1 KO, and TAX1BP1 KO stably expressing GFP-TAX1BP1 were exposed to 5 μg/mL puromycin for 2 h, then fixed for imaging or washed and followed for a further 3 h in full media; scale bar represents 10 μm.
(B) Quantification of (A): percent of cells containing Ub-positive foci was assessed in ~200 cells per condition. Quantification displayed as mean ± SD from three independent experiments using one-way ANOVA test (****p < 0.0001) and Tukey’s post hoc test.
(C and D) Stably expressing TAX1BP1 rescue lines were created with N-FLAG or C-FLAG tag at high (H) or low (L) expression levels (see Figures S4A and S4B) and exposed to 5 μg/mL puromycin for 2 h, then either fixed for imaging (C) or washed and followed for 3 h in full media (D) and quantified as in (B). Quantification displayed as mean ± SD from three independent experiments using one-way ANOVA test (***p < 0.001, ****p < 0.0001) comparing all to WT and Tukey’s post hoc test.
(E) WT, TAX1BP1 KO, or TAX1BP1 KO + FLAG-TAX1BP1 (H) lines were exposed to 5 μg/mL puromycin in the presence or absence of 100 nM Bafilomycin A, then fixed for imaging or washed and followed for 3 h in full media or in media containing Bafilomycin A. Larger fields of view in Figure S4C.
(F) Quantification of (E): percent of cells containing Ub-positive foci was assessed in ~200 cells per condition in three independent experiments. Quantification displayed as mean ± SD from three independent experiments using two-way ANOVA test, (****p < 0.0001). All images are representative of at least three independent experiments.
(G) WT and TAX1BP1 KO cell lines stably expressing EGFP-LC3C were exposed to 5 μg/mL puromycin for 2 h in the presence or absence of 1 mM ULK1 inhibitor or 100 nM Bafilomycin A, after which cells were either fixed for imaging or washed and followed for 3 h in full media in the presence or absence of ULK1 inhibitor or Bafilomycin A; scale bar represents 10 μm.
To confirm the effect of TAX1BP1 on aggregate clearance, we titered expression of N- or C-terminally tagged TAX1BP1 constructs in the TAX1BP1 KO to create stable high- or low-expressing rescue lines (Figures S4A and S4B). Aggregate formation and clearance were similar to WT in the low-expressing TAX1BP1 rescue lines (Figures 3C, 3D, and S4B). However, upon puromycin treatment, Ub-positive foci were present in only ~20% of the high TAX1BP1-expressing rescue cells compared to ~65% of WT and ~90% of TAX1BP1 KO cells (Figure 3C). Furthermore, upon puromycin washout, foci were cleared to a greater extent in TAX1BP1 high-expressing rescue lines—only ~5%–10% of cells retained aggregates compared to 30% in WT cells and >70% in the parental TAX1BP1 KO (Figure 3D), suggesting an active role for TAX1BP1 in promoting aggregate clearance.
Because we did not observe robust foci formation in the high-expressing TAX1BP1 rescue lines (Figures 3C and S4B), we considered the possibilities that TAX1BP1 overexpression could either result in formation of fewer foci or increased clearance. Therefore, we inhibited autophagy during puromycin treatment and washout by addition of Bafilomycin A. In WT cells, autophagy inhibition resulted in increased aggregate retention after washout (Figures 3E and 3F, full field of view in Figure S4C), while in TAX1BP1 KO cells, Bafilomycin A barely exacerbated the defect in clearance, suggesting that aggrephagy is nonfunctional in the absence of TAX1BP1 (Figures 3E and 3F). Bafilomycin A was sufficient to restore Ub-positive foci in TAX1BP1 rescue cell lines (Figures 3E, 3F, and S4C), which colocalized with FLAG-TAX1BP1 and loosely accumulated in the perinuclear region, similar to foci observed in WT cells (Figures 3E and S4D).
To further confirm that TAX1BP1 contributes to aggrephagy in WT and TAX1BP1 KO cell lines, we examined colocalization of GFP-tagged versions of each member of the mammalian ATG8 protein family (LC3A, LC3B, LC3C, GABARAPL, GABARAPL1, and GABARAPL2) and Ub-positive protein aggregates. Puromycin induced colocalization between LC3A, LC3B, LC3C, GABARAPL1, and Ub foci in WT cells, with increased levels of colocalization in the presence of an ULK1 inhibitor or Bafilomycin A (Figures 3G, S5A, and S6A). The block in aggregate clearance in WT cells after washout of puromycin in the presence of ULK1 inhibitor suggests that clearance is occurring via autophagy. In contrast, in TAX1BP1 KO cells treated with ULK1 inhibitor, the existing block in clearance was not exacerbated, suggesting that TAX1BP1 is indeed directing aggregate clearance via autophagy (Figure 3G, S5B, and S6B). Furthermore, recruitment of all LC3 proteins was diminished in TAX1BP1 KO cells (Figures 3G and S5B). Strikingly, LC3C colocalized to the greatest extent with DRiPs and TAX1BP1 in WT cells (Figure 3G), consistent with previous reports for an interaction between LC3C and TAX1BP1 in other forms of selective autophagy (Tumbarello et al., 2015). We observed little colocalization between GABARAP or GABARAPL2 and DRiPs in our analysis in either WT or TAX1BP1 KO cells (Figure S6). Our data suggest that TAX1BP1 contributes to ATG8 protein recruitment. Recent reports suggest that NDP52 has the potential to initiate isolation membrane formation directly on ubiquitinated cargo (Ravenhill et al., 2019; Vargas et al., 2019) and TAX1BP1 may be capable of similar upstream regulation of autophagy as its paralog. Our data demonstrate that TAX1BP1-mediated aggregate clearance is dependent upon expression level and capable of accelerating degradation of protein aggregates by promoting selective flux through the autophagy pathway, suggesting that pharmacologic targeting to increase TAX1BP1 levels may reduce proteotoxicity.
TAX1BP1 Ub-Binding and SKICH Domains Are Important for Aggrephagy
TAX1BP1 contains several protein-interacting domains: an N-terminal SKICH domain, three coiled-coil regions, a canonical LIR motif, putative noncanonical CLIR motif, and two C-terminal ZF Ub-binding domains (Lu et al., 2014; Tumbarello et al., 2015; von Muhlinen et al., 2010; Wong and Holzbaur, 2014). We stably expressed a variety of truncation and point mutants in the TAX1BP1 KO to test the requirements for these domains in TAX1BP1-mediated aggrephagy (Figure 4A). Aggregate formation occurred similarly in WT and all mutant rescue lines as well as in a GFP-only control (Figures 4B and 4C, larger fields in Figure S7A). However, substantial differences were observed among the mutants in aggregate clearance.
Figure 4. Requirements for TAX1BP1 Domains in Aggrephagy.
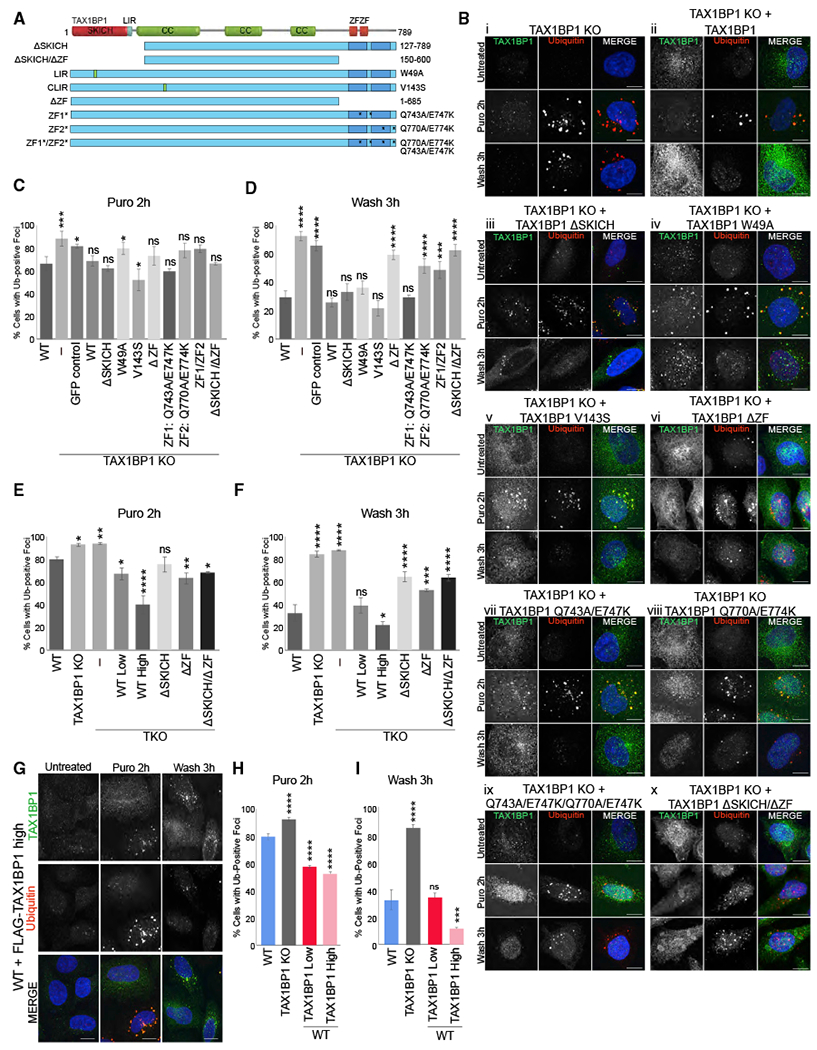
(A) TAX1BP1 truncation or point mutations used in this study.
(B) i–x, TAX1BP1 KO and TAX1BP1 KO with stable expression of TAX1BP1 mutants exposed to 5 μg/mL for 2 h were either fixed for imaging or washed and followed for a further 3 h in full media; scale bar represents 10 μm. Larger fields of view in Figure S7A.
(C and D) Quantification of Ub-foci formation (C) or clearance (D) observed in (B).
(E and F) Quantification of Ub-foci formation (E) or clearance (F) observed in TKO (OPTN/NDP52/TAX1BP1) ceils treated as in (B). See Figure S7B for images.
(G) WT cells stably expressing high levels of FLAG-TAX1BP1 exposed to 5 μg/mL puromycin for 2 h were either fixed for imaging or washed and followed for 3 h in full media; scale bar represents 10 μm.
(H and I) Quantification of Ub-foci formation (H) or clearance (I) observed in (G) and in Figure S7C. All images are representative of at least three independent experiments in which percent of cells containing Ub-positive foci was assessed in ~200 cells per condition. All quantification displayed as mean ± SD from three independent experiments using one-way ANOVA test comparing to WT (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and Tukey’s post hoc test.
N-terminal SKICH deletion (ΔSKICH) as well as both the LIR (W49A) and CLIR (V143S) point mutants localized to Ub-positive foci and rescued clearance similarly to WT TAX1BP1 (Figures 4Bi, 4Bii, 4Biii, 4Biv, 4Bv, 4D, and S7A). The structure of the TAX1BP1 SKICH domain shows that the canonical LIR motif (W49VGI) is mostly buried within the hydrophobic interior of the folded domain and, therefore, is most likely not functional (Yang et al., 2015). In contrast, a truncation mutant lacking both C-terminal ZF domains did not localize to puromycin-induced foci and was unable to rescue aggregate clearance (Figures 4Bvi and 4D). To determine whether one or both ZF domains was essential, we created point mutants targeting both ZF domains (ZF1/ZF2: Q743A/E747K/Q770A/E774K) or each individually (ZF1:Q743A/E747K, ZF2:Q770A/E774K). The double mutant, ZF1/ZF2, as well as the single ZF2 mutant, failed to localize to Ub foci after puromycin treatment and both were unable to rescue aggregate clearance (Figures 4Bviii, 4Bix, and 4D). In contrast, point mutation of ZF1 had little effect on TAX1BP1 localization and rescued aggregate clearance similarly to WT TAX1BP1 (Figures 4Bvii and 4D), consistent with a prior report that only ZF2 of TAX1BP1 is capable of binding Ub (Tumbarello et al., 2015). An N- and C-terminally truncated mutant (ΔSKICH/ΔZF) did not localize to aggregates but instead formed aberrant clusters in the cytosol and was inferior to the ZF mutants in rescuing clearance (Figures 4Bx, 4D, and S7A).
Because of concerns that potential redundancy might mask requirements for TAX1BP1 domains, we expressed a subset of TAX1BP1 mutants in a triple KO cell line lacking also the TAX1BP1 paralog, NDP52, and OPTN, which has been shown to contribute to aggregate clearance (Korac et al., 2013) (TKO: TAX1BP1, OPTN, NDP52 KO) (Figures S2C and S7B). Expression of WT TAX1BP1 at endogenous levels rescued aggregate clearance in TKO cells to levels in WT cells, indicating that TAX1BP1 alone is sufficient to promote aggrephagy (Figures 4E, 4F, and S7B). And, even in the TKO background, higher expression of TAX1BP1 increased aggregate clearance compared to WT cells (Figures 4E, 4F, and S7B). Consistent with observations in the TAX1BP1 KO background, neither the ΔZF nor ΔSKICH/AZF mutants were able to fully rescue aggregate clearance (Figures 4F and S7B). In contrast, the DSKICH mutant, which was able to rescue clearance in the TAX1BP1 KO background, was unable to fully rescue aggregate clearance in the TKO, demonstrating the need for this protein-interaction domain in the absence of NDP52 and OPTN (Figures 4F and S7B).
Increased TAX1BP1 Expression Promotes Aggrephagy
Comparison of expression levels of TAX1BP1 in rescue experiments in TAX1BP1 KO cells suggested that increased TAX1BP1 expression could promote aggregate clearance (Figures 3C and 3D). We therefore examined whether TAX1BP1 overexpression in WT cells above endogenous TAX1BP1 levels could promote aggregate clearance, thus exploring the therapeutic potential of TAX1BP1. TAX1BP1 protein expressed in WT cells behaved similarly to the endogenous protein, remaining diffuse in untreated cells and colocalizing with Ub-stained foci upon puromycin treatment (Figures 4G and S7C). Aggregate formation upon 2 h of puromycin treatment was decreased in both low (~20% decrease)- and high (~30% decrease)-overexpressing TAX1BP1 cells (Figure 4H). Clearance of aggregates in WT cells with low overexpression of TAX1BP1 was similar to that in WT cells expressing only endogenous TAX1BP1 (Figures 4I and S7C), whereas higher levels of TAX1BP1 expression increased clearance upon puromycin washout–only ~10% of cells retained Ub-positive foci in contrast to more than 30% of WT cells (Figures 4G and 4I).
TAX1BP1 Mediates Aggrephagy of Cytotoxic Aggregation-Prone Neurodegenerative Disease-Associated Proteins
Overexpression of polyQ huntingtin fragments and WTTDP-43 is cytotoxic in in vitro and in vivo model systems (Barmada et al., 2010, 2014; Iwata et al., 2005; Johnson et al., 2008; Krobitsch and Lindquist, 2000; Liachko et al., 2010; Ravikumar et al., 2002; Wang et al., 2011; Wils et al., 2010). Stimulation of autophagy has been shown to increase clearance of these aggregation-prone proteins and attenuate cytotoxicity (Barmada et al., 2014; Iwata et al., 2005; Menzies et al., 2011; Rose et al., 2010; Sarkar et al., 2007; Yamamoto and Simonsen, 2011). To determine whether TAX1BP1 may play a role in selective autophagy of proteins implicated in neurodegenerative disease, WT, TAX1BP1 KO, and high-expressing TAX1BP1 rescue cells were exposed to proteotoxic stress caused by expression of aggregation-prone proteins, including model substrates carrying expanded glutamine tracts (polyQ) expressed from exon 1 of the huntingtin-encoded gene (HttQ74-EGFP and HttQ104-EGFP) and the ALS-associated potein, EGFP-TDP43 (Figures S8A and S8B). Because of the low number of glutamine repeats, HttQ23-EGFP does not form inclusions and is a useful control for comparing to the aggregate-forming HttQ74-EGFP and HttQ103-EGFP proteins. Cells were infected or transfected on day 1 and assessed daily for 6 days. In order to study longer-term effects on cell survival and the ability of TAX1BP1 to modify it, we aimed for low to moderate expression of these aggregate-prone proteins, which is sufficient to promote aggregate formation without causing acute toxicity. A similar concept using late-onset disease models, which are not immediately toxic, underlies studies in flies, which have sought to examine longer-term effects of expression of polyQ expansions (Sang et al., 2005). Loss of TAX1BP1 decreased cell viability compared to WT cells upon exposure to both aggregate-forming Htt proteins and overexpressed TDP-43, both of which accumulated to a greater extent in TAX1BP1 KO cells (Figures 5A and S8C–S8G). Rescue with TAX1BP1 in the KO line was able to partially restore viability and GFP-TDP43 clearance (Figures S8C–S8E).
Figure 5. TAX1BP1 Mediates Aggrephagy of Cytotoxic Aggregation-Prone Proteins.
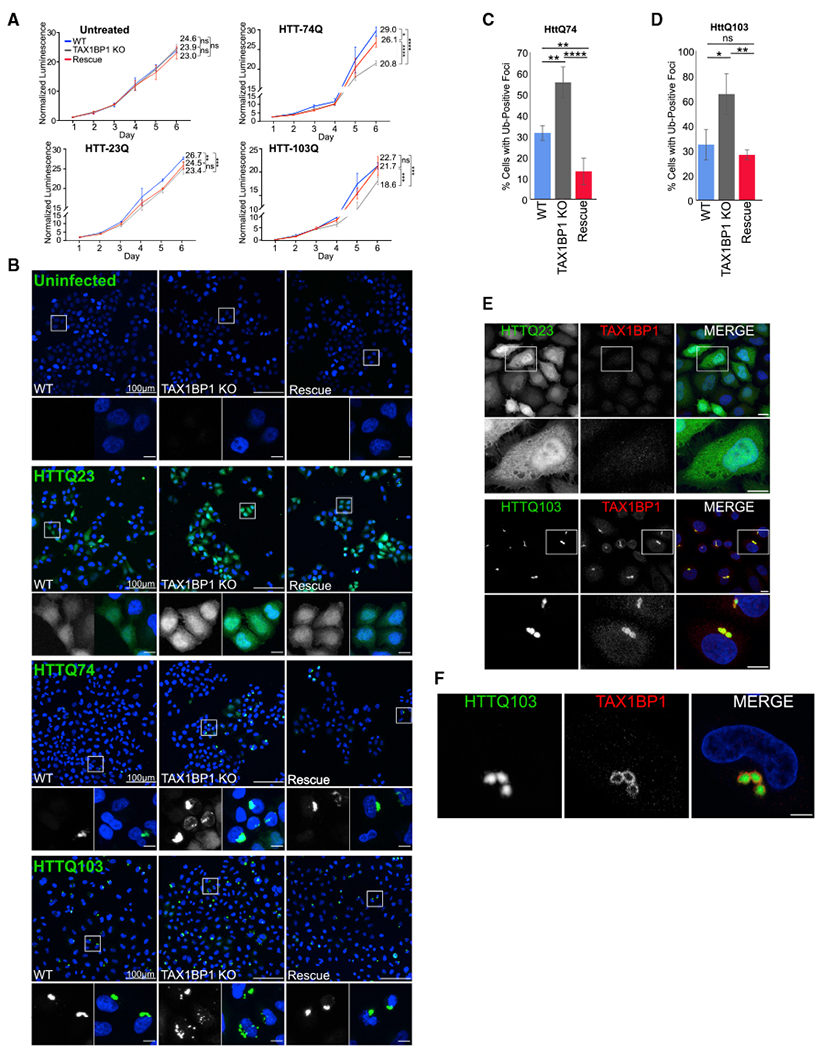
(A) WT, TAX1BP1 KO, or TAX1BP1 KO cells with stably expressed TAX1BP1 rescue were exposed to proteotoxic stressors as indicated on day 1 and followed for 6 days during which viability was measured by quantification of ATP production. Relative viability represents normalized luminescence displayed as mean ± SD from three independent experiments; significance was assessed using two-way ANOVA test (****p < 0.0001, ***p < 0.001, **p<0.01, *p< 0.05) with Tukey’s post hoc test. p values and normalized viability measurements shown on graphs are for day 6 comparisons.
(B) WT, TAX1BP1 KO, or rescue cells uninfected or infected with viruses expressing HttQ23-EGFP, HttQ74-EGFP, or HttQ103-EGFP were assessed for aggregate count 4 days post-infection.
(C) Quantification of HttQ74-EGFP aggregates observed in (B). Quantification is displayed as mean ± SD from three independent experiments using one-way ANOVA test (*p < 0.05, **p < 0.01, ****p < 0.0001) and Tukey’s post hoc test.
(D) Quantification of HttQ103-EGFP aggregates observed in (B), percent of cells containing GFP-positive foci was assessed in ~200 cells per condition in three independent experiments. Quantified as in (C).
(E) Immunofluorescence labeling of endogenous TAX1BP1 in cells infected with HttQ23-EGFP or HttQ103-EGFP. Maximum intensity projections shown.
(F) A single 1-μm slice of immunofluorescence labeling of endogenous TAX1BP1 in cells infected with HttQ103-EGFP as in (E). Scale bars represent 10 μm. All images are representative of at least three independent experiments.
To determine whether TAX1BP1 can aid in clearance of Htt-polyQ proteins, we expressed HttQ23-EGFP, HttQ74-EGFP, and HttQ103-EGFP in WT, TAX1BP1 KO, and TAX1BP1 rescue cells and assessed aggregate levels 4 days post-infection. HttQ23-EGFP expression was robust and, as expected, remained largely diffuse and cytosolic in all cell lines (Figure 5B). In contrast, TAX1BP1-deficient cells exhibited increased focal aggregates of both Htt74Q-EGFPand Htt103Q-EGFP compared to WT cells (Figures 5B–5D). Additionally, insoluble GFP-HttQ103 accumulated to a greater extent in TAX1BP1 KO cells than WT cells when assessed via filter-trap assay (Figures S8F and S8G). Rescue of the TAX1BP1 KO line with expression of FLAG-TAX1BP1 restored clearance of HttQ74-EGFP and Htt103Q-EGFP to levels observed in WT cells, indicating that TAX1BP1 is highly effective in directing clearance of huntingtin polyQ proteins (Figures 5B–5D). Endogenous TAX1BP1 colocalized with HttQ103-EGFP aggregates in WT cells (Figure 5E), appearing to enclose aggregates in many instances (Figures 5F and S8H).
Because TAX1BP1 overexpression can promote clearance of DRiPs beyond that in WT cells and because we found TAX1BP1 highly and specifically expressed in the brain (Figures 2A and 2C), we examined whether TAX1BP1 could provide a protective effect in iPSC-derived neurons exposed to huntingtin proteins (Figure 6A). iPSC-derived neurons expressing nuclear localized BFP (NLS-BFP) with/without stable overexpression of TAX1BP1 (Figure 6B) were infected with viruses expressing either the non-aggregating control, HttQ23-EGFP, or the aggregate-forming HttQ103-EGFP. iPSCs were then induced to differentiate and imaged daily for 2 weeks to assess viability via nuclei count (Figure 6A). As in HeLa cells, HttQ23-EGFP remained diffuse, while HttQ103-EGFP expression resulted in formation of aggregates throughout the cell body and neurites (Figure 6C). The rates of cell death, assessed by comparing slopes obtained from lines fitted to cell number over time, did not differ between WT and TAX1BP1-overexpressing neurons infected with the non-aggregate-forming HttQ23-EGFP (Figure 6D). However, TAX1BP1 overexpression significantly improved survival in neurons exposed to HttQ103-GFP protein (Figure 6E), consistent with our observations in HeLa cells.
Figure 6. TAX1BP1 Overexpression Preserves Viability in iPSC-Derived Neurons Exposed to Cytotoxic Aggregation-Prone Proteins.
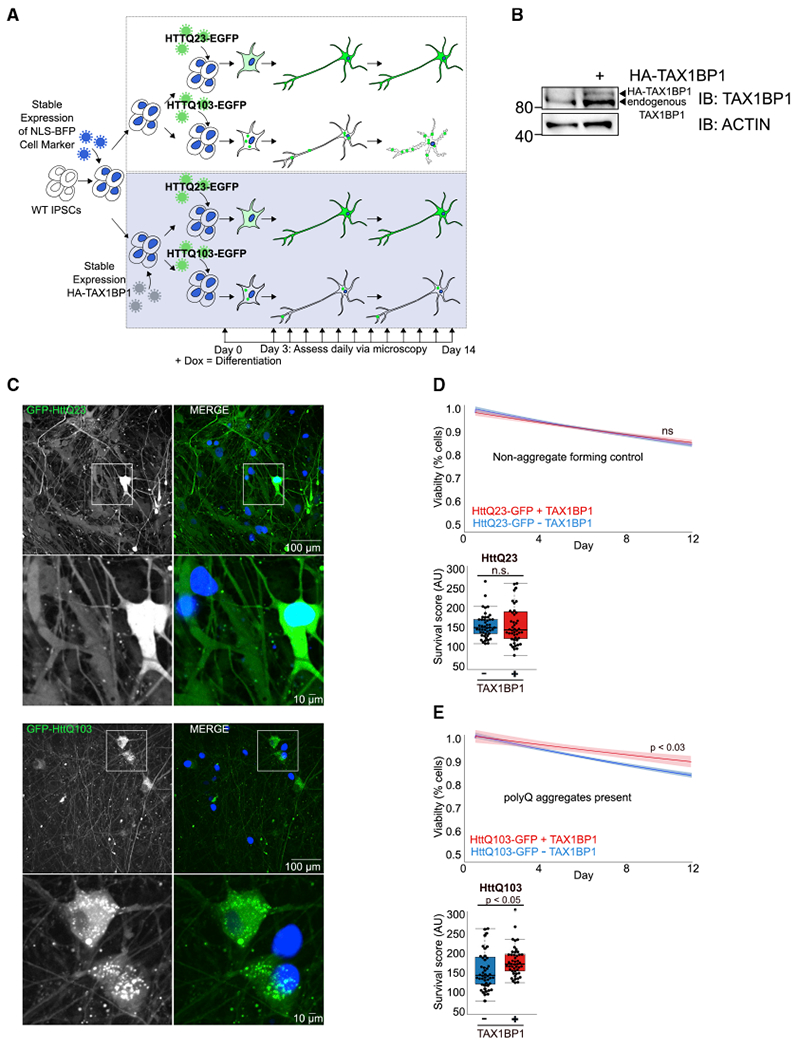
(A) iPSCs were transduced with viruses encoding NLS-BFP, sorted, and transduced with viruses encoding HA-TAX1BP1, followed by puromycin selection. WTiPSCs expressing NLS-BFP with/without HA-TAX1BP1 were transduced with viruses encoding HttQ23-EGFP or HttQ103-EGFP for 24 h, allowed to recover for 24 h, then treated with doxycycline to induce differentiation. Viability was assessed daily for 14 days via imaging of NLS-BFP and quantification of total cell number per well.
(B) Immunoblot confirming TAX1BP1 stable overexpression in iPSCs.
(C) NLS-BFP-expressing WT or TAX1BP1-overexpressing iPSC-derived neurons infected with viruses expressing either HttQ23-EGFP or HttQ103-EGFP.
(D and E) Graphs show line fitted to the number of BFP-positive nuclei counted daily for iPSC-derived neurons with/without stable TAX1BP1 overexpression infected with HttQ23-EGFP (D) or HttQ103-EGFP (E). Ribbon represents 95% confidence interval around the fitted line. Beeswarm boxplots compare survival scores determined by performing permutation analysis using the means of all slopes(center line = median, box limits = first to third quartile, whiskers = minimum and maximum).
TAX1BP1 Δ ZF Leads to Ub Accumulation and Lipofuscin Pathology In Vivo
Two TAX1BP1 KO mouse models produced via different approaches have resulted in distinct findings regarding the essentiality of TAX1BP1. Shembade et al. (2007) used a gene-trapping strategy resulting in expression of an exon 1–5-β-galactosidase fusion transcript and an alternatively spliced transcript consisting of full-length TAX1BP1 with an insertion between exons 5 and 6, most likely rendering it completely nonfunctional (Shembade et al., 2007), resulting in embryonic lethality at day 13.5. In contrast, Iha et al. (2008) used homologous recombination to disrupt the last exon of TAX1BP1, replacing the Ub-binding ZFs with a neomycin cassette (Figure S9A). This line is viable, resulting in significantly smaller animals with decreased lifespan and sensitivity to inflammatory stimuli (Iha et al., 2008; Nakano et al., 2013). Genotyping of animals and sequencing of genomic DNA show that the TAX1BP1 KO mouse does indeed contain a neomycin insertion replacing the final exon of TAX1BP1 (Figure S9B). Immunoblotting revealed a truncated TAX1BP1-positive protein product in the KO animals (Figure S9C). To further examine this, we immunoblotted whole brain lysate from WT, TAX1BP1 heterozygous, and TAX1BP1 KO animals with two antibodies targeting a central region of TAX1BP1, resulting in detection of the full-length protein in the WT and heterozygous animal and a truncated protein product in both the heterozygote and KO animal (Figures S9D and S9E). An additional antibody raised against the C terminus of the protein, precisely the region targeted by Iha et al. (2008), identified only the full-length protein in both the WT and heterozygote animals but gave no signal in the KO animal (Figures S9D and S9E). We confirmed the presence of TAX1BP1 peptides in the KO animal by using mass spectrometry (Figure S9F) and further determined the associated mRNA produced in the KO animal by using 3’ RACE PCR. We identified an mRNA transcript produced in the KO animal that contained intact TAX1BP1 exon 17 fused to 11 additional amino acids that are present because of partial read-through of intron 17–18 followed by a premature stop codon, confirming that exon 18, containing the ZF domains, is missing (Figures S10A and S10B). On the basis of these data, we have determined that the KO line produced by Iha et al. (2008) expresses a truncated and presumably partially functional TAX1BP1 protein product, allowing homozygous KO animals to survive but preventing Ub-dependent TAX1BP1 functions. Therefore, hereafter, we will refer to the TAX1BP1 KO mouse made by Iha et al. (2008) and used in this study as the TAX1BP1 ΔZF mouse.
To better understand the requirements for TAX1BP1 and its relevance in vivo, we compared multiple brain regions from 3538-week-old WT or TAX1BP1 ΔZF mice (Iha et al., 2008). Strikingly, we found that TAX1BP1 ΔZF animals had significantly increased levels of high molecular weight ubiquitinated protein conjugates in the striatum, cortex, cerebellum, and hippocampus (Figures 7A and 7B). Thus, it appears that TAX1BP1 is required for the steady-state removal or prevention of accumulation of ubiquitinated protein in vivo, consistent with phenotypes observed in mice lacking the essential autophagy gene, ATG7, in the CNS (Komatsu et al., 2006).
Figure 7. TAX1BP1 KO Mice Accumulate Ubiquitin and Lipofuscin in Multiple Brain Regions.
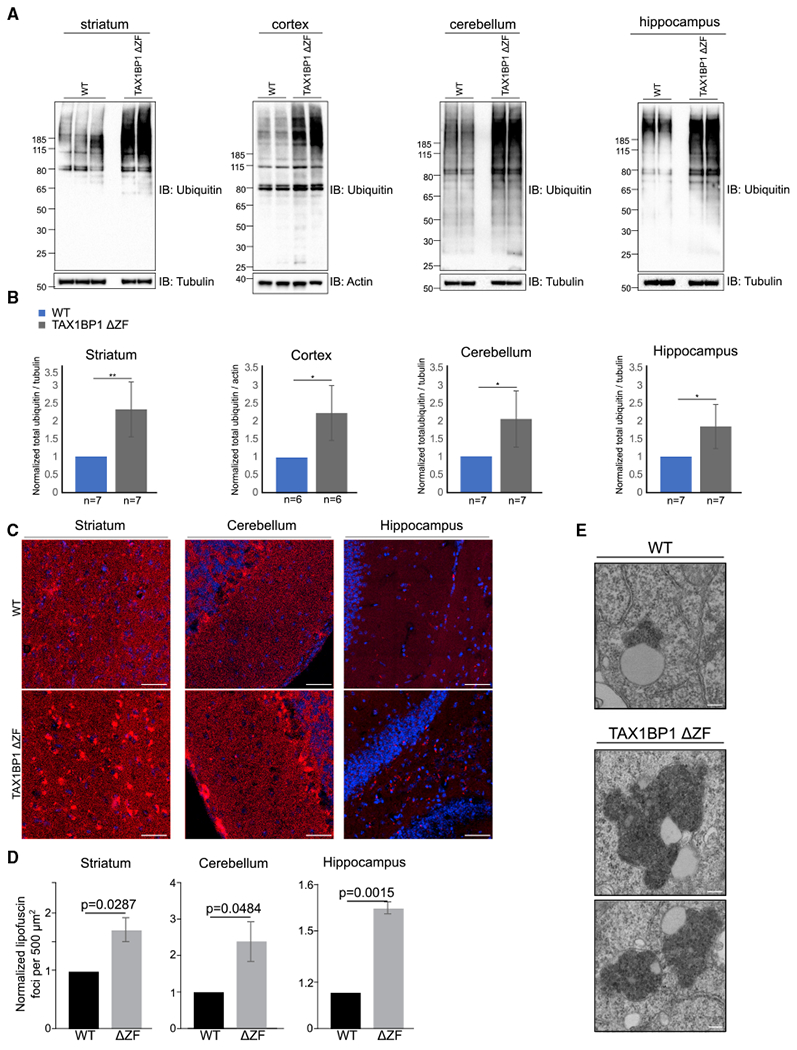
(A) Representative immunoblots of total Ub and loading controls in lysate from striatum, cerebellum, cortex, or hippocampus of 35–38-week-old WT and TAX1BP1 ΔZF mice.
(B) Quantification of total Ub signal normalized to loading control displayed as mean ± SEM.; significance was assessed using two-tailed Welch’s t test (**p < 0.01, *p < 0.05).
(C) Representative images of lipofuscin deposits (red puncta) in WT and TAX1BP1 ΔZF mouse striatum, cerebellum, or hippocampus sections imaged used 555 nm autofluorescence. Nuclei stained with DAPI. Scale bars represent 10 μm.
(D) Quantification of normalized counts of lipofuscin punctae per 500 μm2 displayed as mean ± SEM; significance was assessed using two-tailed Welch’s t test (*p < 0.05).
(E) Representative image of electron-dense storage material in postmortem cortical tissue from TAX1BP1 ΔZF mouse brain. Scale bars represent 200 nm.
Accumulation of lipofuscin, or “age-pigment,” occurs during normal aging as cellular proteostasis pathways become less effective. It is comprised of a highly variable mixture of oxidized cross-linked proteins, lipids, and sugars that cannot be degraded and thus accumulates in lysosomes of post-mitotic cells. In neurodegenerative disorders, lipofuscin aggregates in neurons appear earlier, accumulate to a greater extent, and may contribute to neurodegeneration (Moreno-Garcia et al., 2018). Strikingly, TAX1BP1 ΔZF brains revealed increased autofluorescent lipofuscin deposits in the cerebellum, hippocampus, and striatum compared to age-matched WT animals (Figures 7C, 7D, and S10C). Electron microscopy revealed abnormally large deposits of electron-dense storage material in a TAX1BP1 ΔZF mouse (Figure 7E), consistent with lipofuscin deposition and indicative of a role for TAX1BP1 in prevention of aggregate accumulation.
DISCUSSION
Here, we report a broad role for TAX1BP1 in protein homeostasis both in vitro and in vivo. Our results demonstrate that loss of TAX1BP1 leads to decreased ability to target insoluble protein for degradation. Overexpression of TAX1BP1 further promotes aggregate clearance and rescues cell viability upon exposure to varied proteotoxic insults, including translational stress, proteasome inhibition, and exposure to aggregate-prone proteins (Figure S8I). The fact that TAX1BP1 promotes clearance of such a wide range of ubiquitinated aggregates without apparent specificity for amino acid sequence suggests relevance to a wide range of diseases defined by aggregate pathologies.
Aggrephagy has the potential to mitigate neurodegenerative proteinopathies. Focus on the proteins that provide specificity in targeting aggregates, such as TAX1BP1, is valuable as these are most likely deciding factors in directing the autophagic response. To better delineate the roles of each autophagy receptor protein, we investigated the aggrephagy response to proteotoxic stress in single or combinatorial KOs of OPTN, NDP52, TAX1BP1, NBR1, and p62. At first glance, our data appear inconsistent with the established role of p62 in aggrephagy because we observed aggregate accumulation in pentaKO cells, leading us to ask how we observed accumulation of ubiquitinated material in cells lacking p62. Because p62 is generally required for aggregate formation, our data suggest that p62 most likely acts upstream of autophagic clearance, similar to its proposed role in mitophagy (Narendra et al., 2010; Okatsu et al., 2010). Accumulation of ubiquitinated protein suggests that the loss of TAX1BP1 acting downstream of p62 results in considerable protein accumulation, rendering superfluous the clustering activity of p62 in aggregate formation.
Although they may function en masse to maintain protein homeostasis, individual autophagy receptor proteins exhibit functional and spatial distinctions at the subcellular and tissue level. For example, autophagy receptor proteins are recruited independently to distinct microdomains surrounding bacteria and mitochondria where they perform nonredundant roles during xenophagy and mitophagy, respectively (Cemma et al., 2011; Heo et al., 2015). Furthermore, TAX1BP1 is robustly expressed in the human brain as well as in primary rat cortical neurons, distinguishing it from NDP52, its closest relative. Further detailed analysis of expression levels of each autophagy receptor in different cell types of the CNS may reveal functional distinctions beyond those observed at the tissue level. TAX1BP1 is also associated with the insoluble protein fraction in primary rat cortical neurons exposed to proteotoxic stress. Among the other autophagy receptor proteins, only p62 showed similar association with the insoluble fraction, although our data purport that p62 functions earlier in the aggregate-forming process. TAX1BP1 and its paralog, NDP52, share similar domain structures; however, recent studies suggest structural differences in the organization of the SKICH domains as well as distinct ATG8 binding affinities (Tumbarello et al., 2015; Yang et al., 2015). TAX1BP1 is also more broadly conserved than NDP52 among mammals (Tumbarello et al., 2015), suggesting that TAX1BP1 fills an essential nonredundant role. As of yet, there is no publicly available data identifying TAX1BP1 mutation in human disease. Per the Genome Aggregation Database (gnomAD), the probability of loss-of-function (LOF) intolerance (pLI) score for TAX1BP1 is 0, which suggests that TAX1BP1 haploinsufficiency is tolerable (https://gnomad.broadinstitute.org/gene/ENSG00000106052?dataset=gnomad_r2_1). However, there are a few interesting points to note: pLI does not identify genes that are haploinsufficient for adult-onset conditions, meaning diseases such as neurodegeneration, which generally become evident later in life, will not be identified. Furthermore, pLI does not identify genes involved in recessive conditions. Finally, perhaps most interestingly, there are no occurrences within gnomAD in which any individuals carry homozygous mutations for TAX1BP1 loss-of-function alleles—an unusual circumstance that suggests there are no living TAX1BP1-null individuals sampled in gnomAD, which contains data from more than 100,000 individuals, signifying that disease-causing mutations in TAX1BP1 may yet be identified. Studies examining TAX1BP1 expression levels during aging and in animal models of neurodegenerative disease are warranted.
TAX1BP1 recruitment to protein aggregates requires the C-terminal Ub-binding domain; additionally, its function in promoting aggrephagy utilizes the N-terminal SKICH domain. However, none of our TAX1BP1 mutants were completely dead in terms of rescue effect, suggesting that other cellular functions of TAX1BP1 may be involved. One such function is to act as a Myosin VI cargo adaptor protein for mediating autophagosome maturation, which could contribute to aggregate clearance dependent upon other upstream factors, thus explaining partial rescue (Tumbarello et al., 2015). Additionally, TAX1BP1 is best studied for its role in negatively regulating nuclear factor-kappa B (NF-κB) and interferon regulatory factor (IRF)-3 via an interaction with the deubiquitinase A20, thus restricting pro-inflammatory signaling and immune responses (Shembade et al., 2007, 2011). Although links between TAX1BP1 ’s role as an autophagy adaptor and the immune response are yet to be understood, it is well known that protein aggregation, inflammation, and autophagy are intertwined (Choi and Ryter, 2011). Studies have shown that exposure to disease-associated aggregates can elicit innate immune response in glial cells and that LPS-induced inflammation results in enhanced aggregate formation in disease models, suggesting a synergistic relationship between proinflammatory response, proteostasis, and neurodegeneration (Czirr and Wyss-Coray, 2012; Lim and Yue, 2015). Autophagy may function to downregulate inflammatory signaling (Prabakaran et al., 2018), and TAX1BP1 may be an important link between detection and monitoring of cellular protein aggregates and the inflammatory response.
We observed increased levels of ubiquitinated protein in the brains of TAX1BP1 ΔZF animals compared to WT controls. TAX1BP1 ΔZF animals appear normal, although they are smaller than WT animals upon gross evaluation, and they do die prematurely (Iha et al., 2008). More analysis is needed to understand the contributions to these phenotypes of both aggrephagy and the cellular functions described above. It has been observed that many mouse models of neurodegeneration with overexpression of neurodegenerative-associated aggregates do not show marked neurodegeneration (Ashe and Zahs, 2010; Irizarry et al., 1997). Because we are not exogenously triggering proteotoxic stress in TAX1BP1 ΔZF mice, the appearance of ubiquitinated material suggests that aggrephagy is an important process throughout aging and appears to be selectivity targeting deleterious material that nonselective autophagy is either not detecting or unable to address.
Notably, ectopic expression of TAX1BP1 in KO cells was able to rescue aggregate clearance to WT levels and overexpression of TAX1BP1 further reduced aggregate levels below those observed in WT cells. Furthermore, TAX1BP1 overexpression in iPSC-derived neurons was protective against huntingtin aggregate-induced toxicity. TAX1BP1 appears to play a specific role in promoting aggrephagy while responding broadly to varied proteotoxic stresses. Future studies aimed at increasing TAX1BP1 expression or stability in vivo present promising therapeutic potential in addressing proteinopathies. An increased understanding of targeting specificity of selective autophagy receptor proteins may make autophagy-stimulating approaches more specific and effective in treatment of protein-misfolding diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard Youle (youler@ninds.nih.gov).
Materials Availability
All materials generated in this study are available upon request.
Data and Code Availability
All datasets generated are available from the corresponding author upon request. All macros created for use with ImageJ/Fiji are available on request. CellProfiler pipelines and detailed settings to reproduce the image analysis procedures are available upon request. Original data have been deposited to Mendeley Data: https://doi.org/10.17632/jv437rbcjk.1. Code generated for neuron viability analysis is publicly available at: https://github.com/gkanfer/Tax1bp_analysis.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Culture and reagents
HeLa and HEK293T were cultured in DMEM (Life Technologies) supplemented with 10% (v/v) FBS (Gemini Bio Products), 10 mM HEPES (Life Technologies), 1 mM sodium pyruvate (Life Technologies), 1mM non-essential amino acids (Life Technologies) and 2 mM glutamine (Life Technologies). HeLa cells were acquired from the ATCC and authenticated at the Johns Hopkins GRCF Fragment Analysis Facility using STR profiling. Testing for mycoplasma contamination was performed bimonthly using the PlasmoTest kit (InvivoGen). Plasmids were transfected using either X-tremeGENE 9 (Roche), polyethylenimine (Polysciences) or Avalanche-OMNI (EZ Bio-systems). The full list of antibodies and reagents are found in the Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin | CST (Cell Signaling Technology) | 4967, RRID:AB_330288 |
| GAPDH | Sigma-Aldrich | G9545; RRID:AB_796208 |
| GFP | Thermofisher | A10262, RRID:AB_2534023 |
| GFP | Sigma-Aldrich | 11814460001, RRID:AB_390913 |
| HA | CST | 3724S;RRID:AB_1549585 |
| NBR1 | Abnova | H00004077-M01, RRID:AB_1016849 |
| NDP52 | CST (Cell Signaling Technology) | 60732, RRID:AB_2732810 |
| OPTN | Proteintech | 10837-1-AP, RRID:AB_2156665 |
| p62 | Abnova | H00008878-M01; RRID:AB_437085 |
| TAX1BP1 | Bethyl | A303-792A-M, RRID:AB_2781469 |
| TAX1BP1 | Novus | NBP1-86662, RRID:AB_11043368 |
| TAX1BP1 | Sigma | HPA024432, RRID:AB_1857783 |
| Ubiquitin | Millipore | MAB1510, RRID:AB_2180556 |
| Ubiquitin | Santa Cruz | sc-8017 AC, RRID:AB_2762364 |
| Ubiquitin | Biomol International | BML-PW8810-0500, RRID:AB_2051891 |
| Alexa Fluor 488 goat anti-rabbit IgG (H+L) | Thermofisher | A-11070, RRID:AB_2534114 |
| Alexa Fluor 565 goat anti-mouse IgG (H+L) | Thermofisher | A-21425, RRID:AB_2535846 |
| Alexa Fluor 568 goat anti-mouse IgG (H+L) | Thermofisher | A-11004, RRID:AB_2534072 |
| Alexa Fluor 594 goat anti-mouse IgG (H+L) | Thermofisher | A-11032, RRID:AB_2534091 |
| Alexa Fluor 647 goat anti-mouse IgG (H+L) | Thermofisher | A-21236, RRID:AB_2535805 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accutase | StemCell Technologies | 07920 |
| B-27 Supplement | Thermofisher | 17504001 |
| Bafilomycin A | Sigma-Aldrich | B1793 |
| BamBanker | Wako | 302-14681 |
| Blasticidin | Invivogen | ant-bl-1 |
| BDNF | PeproTech | 450-02 |
| BrainPhys neuronal medium | StemCell Technologies | 05790 |
| Gateway BP Clonase II Enzyme Mix | Thermofisher | 11789100 |
| DAPI | Thermofisher | 62248 |
| DMEM | Thermofisher | 31053028 |
| DMEM/F12 | Thermofisher | 11320033 |
| doxycycline | Sigma-Aldrich | D9891 |
| Essential 8 Flex Medium | Thermofisher | A28583-01 |
| Essential 8 Flex Supplement | Thermofisher | Aa28584-01 |
| GlutaMax | Thermofisher | 35050061 |
| Goat serum | VWR Scientific Products | 76230-088 |
| HEPES | Thermofisher | 15630080 |
| Laminin | Thermofisher | 23017015 |
| LDS sample buffer (4x) | Thermofisher | NP0007 |
| Gateway LR Recombinase II Enzyme Mix | Thermofisher | 11791020 |
| Matrigel | Corning | 354277 |
| MG132 | Sigma-Aldrich | 133407-82-6 |
| MRT68921, ULK1 inhibitor | Selleckchem | S7949 |
| N-Ethylmaleimide (NEM) | Sigma-Aldrich | 04260-5G-F |
| N2 Suppplement | Thermofisher | 17502048 |
| Neurobasal Medium | Thermofisher | 21103049 |
| Nonessential amino acids | Thermofisher | 11140050 |
| Omni-Avalance | EZ Biosystem | EZT-OMNI-1 |
| Opti-MEM | Thermofisher | 31985062 |
| NT-3 | PeproTech | 450-03 |
| paraformaldehyde 16% solution, EM grade | Electron Microscopy Sciences | 15710 |
| Polybrene | Sigma-Aldrich | H9268 |
| Polyethylenimine (PEI 25K) | PolySciences | 23966-1 |
| poly-D-lysine | Sigma-Aldrich | P2780 |
| poly-L-ornithine | Sigma-Aldrich | P3655 |
| Protease inhibitor cocktail | Roche | 11873580001 |
| puromycin | Invivogen | ant-pr-1 |
| RIPA buffer | Thermofisher | 89900 |
| ROCK inhibitor Y-27632 | Tocris | 1254 |
| Sodium pyruvate | Thermofisher | 11360070 |
| TCEP | Sigma-Aldrich | C4706-2G |
| Triton X-100 | Sigma-Aldrich | T9284 |
| 0.05% Trypsin-EDTA | Thermofisher | 25300054 |
| TRIzol | Thermofisher | 15596026 |
| Trypsin | Promega | V5280 |
| Tween-20 | Sigma-Aldrich | P7949 |
| MRT68921, ULK1 inhibitor | Selleckchem | S7949 |
| Waters Oasis HLB μElution plate | Waters | 186001828BA |
| X-tremeGENE 9 | Sigma-Aldich | 6365787001 |
| Critical Commercial Assays | ||
| Amersham ECL Prime WB detection reagent | GE Healthcare Life Sciences | RPN2232 |
| Cell Titer-Glo | Promega | G7570 |
| DC Protein Assay | Biorad | 5000111 |
| NEBuilderHiFi DNA assembly master mix | New England BioLabs | M5520A |
| Phusion Hot Start II DNA polymerase | Thermofisher | F-549L |
| Q5 Hot Start High-Fidelity DNA Polymerase | New England BioLabs | M0493L |
| SuperSignal West Femto Luminol/Enhancer solution | Thermofisher | 1859022 |
| SuperSignal West Femto stable peroxide buffer | Thermofisher | 1859023 |
| SMARTer® RACE 5’/3’ Kit | Takara | 634858 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL-3216 |
| Lenti-X 293T | Takara | 632180 |
| HeLa | ATCC | CCL-2.2 |
| WTC11 hiPSC lines with doxycycline-inducible mNGN2 transgene at the AAVS1 locus | (Fernandopulle et al., 2018) | N/A |
| OPTN KO HeLa | (Lazarou et al., 2015) | N/A |
| NDP52 KO HeLa | (Lazarou et al., 2015) | N/A |
| NBR1 KO HeLa | This study | N/A |
| p62 KO HeLa | This study | N/A |
| TAX1BP1 KO HeLa | (Lazarou et al., 2015) | N/A |
| OPTN/NDP52/Taxbp1 TKO HeLa | (Lazarou et al., 2015) | N/A |
| OPTN/NDP52/Taxbp1/p62/NBR1 5KO HeLa | (Lazarou et al., 2015) | N/A |
| p62/NBR1 DKO HeLa | This study | N/A |
| p62/TAX1BP1 DKO HeLa | This study | N/A |
| p62/NBR1/TAX1BP1 TKO HeLa | This study | N/A |
| Deposited Data | ||
| All raw data | Mendeley Dataset | http://dx.doi.org/10.17632/gfy5tbthgc.1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: B6;Taxbp1tm1Ktj | (Iha et al., 2008) | N/A |
| Sprague Dawley outbred rat | Envigo | Cat#:002 |
| Oligonucleotides | ||
| TAX1BP1 exon 3 CRISPR FW | TATCCTTGAGAAATTGGATAGCA | N/A |
| TAX1BP1 exon 3 CRISPR Rev | TAGTACCTAAAAAGAAACCCACTCTTC | N/A |
| p62 exon 3 CRISPR FW | ACAGTGACGACAGAGGGGGA | N/A |
| p62 exon 3 CRISPR Rev | AATGCGAGCTTGGTGTGCC | N/A |
| NBR1 exon 5 CRISPR FW | GCCAGAGGATCCTGCAGTGC | N/A |
| NBR1 exon 5 CRISPR FW | AGAAACCTGTTCAGCTTTATTTA | N/A |
| BP-F for NBR1 | G GGG ACA ACT TTG TAC AAA AAA GTT GGC atggaaccacaggttact | N/A |
| BP-R for NBR1 | GGG GAC AAC TTT GTA CAA GAA AGT TGG TCA atagcgttggctgta | N/A |
| BP-F for NDP52 | G GGG ACA ACT TTG TAC AAA AAA GTT GGC atggaggagaccatcaaa | N/A |
| BP-R for NDP52 | GGG GAC AAC TTT GTA CAA GAA AGT TGG TCA gagagagtggcagaa | N/A |
| BP-F for p62 | G GGG ACA ACT TTG TAC AAA AAA GTT GGC atggcgtcgctcaccgtg | N/A |
| BP-R for p62 | GGG GAC AAC TTT GTA CAA GAA AGT TGG TCA caacggcgggggatg | N/A |
| BP-F for TAX1BP1 | G GGG ACA ACT TTG TAC AAA AAA GTT GGC atgacatcctttcaagaa | N/A |
| BP-R for TAX1BP1 | GGG GAC AAC TTT GTA CAA GAA AGT TGG GTA gtcaaaatttagaac | N/A |
| BP-F for OPTN | G GGG ACA ACT TTG TAC AAA AAA GTT GGC atgtcccatcaacctctc | N/A |
| BP-R for OPTN | GGG GAC AAC TTT GTA CAA GAA AGT TGG TTA aatgatgcaatccat | N/A |
| BP-F for TDP-43 | G GGG ACA ACT TTG TAC AAA AAA GTT GGC ATG TCTGAATATATTCGG | N/A |
| BP-R for TDP-43 | GGG GAC AAC TTT GTA CAA GAA AGT TGG CTACATTCCCCAGCCAGA | N/A |
| forward seq primer for TAX1BP1 | AAC TTG AAT GTC AGT TGA | N/A |
| forward seq primer for NBR1 | ctacatcccatctgtgga | N/A |
| SDM-F to make Q743A/E747K in TAX1BP1 | gactttcaacatgttctttaaatttgctcgcatcatagttaggaggaaacattaactcacagagg | N/A |
| SDM-R to make Q743A/E747K in TAX1BP1 | cctctgtgagttaatgtttcctcctaactatgatgcgagcaaatttaaagaacatgttgaaagtc | N/A |
| SDM-F to make Q770A/E774K in TAX1BP1 | gtctgcacatgccttttaaacacctgcgcgtcatagtcaggaggg | N/A |
| SDM-R to make Q770A/E774K in TAX1BP1 | ccctcctgactatgacgcgcaggtgtttaaaaggcatgtgcagac | N/A |
| SDM-F to mutate canonical LIR in TAX1BP1 at W49A | aaccttgaatataccaaccgcatcttttggatgtggatgaatatatggag | N/A |
| SDM-R to mutate canonical LIR in TAX1BP1 at W49A | ctccatatattcatccacatccaaaagatgcggttggtatattcaaggtt | N/A |
| SDM-F to mutate CLIR in TAX1BP1 at V143S | gaaggcctgcttttgtggtcgacactaacatgtcagaatttc | N/A |
| SDM-R to mutate CLIR in TAX1BP1 at V143S | gaaattctgacatgttagtgtcgaccacaaaagcaggccttc | N/A |
| BP-F for GFP | G GGG ACA ACT TTG TAC AAA AAA GTT GGC ATGgtgagcaagggcgaggag | N/A |
| BP-R for GFP | GGG GAC AAC TTT GTA CAA GAA AGT TGG ttacttgtacagctcgtccat | N/A |
| BP-R for TAX1BP1 delta ZFs | GGG GAC AAC TTT GTA CAA GAA AGT TGG CTA aacatcaaagctggaatcaaag | N/A |
| BP-F for TAX1BP1 CC domain only | G GGG ACA ACT TTG TAC AAA AAA GTT GGC ATG cttgagttgaaaattgagaaaa | N/A |
| BP-R for TAX1BP1 CC domain only | GGG GAC AAC TTT GTA CAA GAA AGT TGG CTA cctttctgctggattttctaga | N/A |
| BP-F for TAX1BP1 ZF domain only | G GGG ACA ACT TTG TAC AAA AAA GTT GGC ATG cacaagaagtgtcccctctg | N/A |
| BP-R for TAX1BP1 SKICH domain only | GGG GAC AAC TTT GTA CAA GAA AGT TGG CTA aactggagaagaagctcgaa | N/A |
| BP-R for TAX1BP1 STOP | GGG GAC AAC TTT GTA CAA GAA AGT TGG ctagtcaaaatttagaacat | N/A |
| genotyping primer for TAX1BP1 KO mouse | CGCCTTCTATCGCCTTCTTGACGAGTTCTT | N/A |
| genotyping primer for TAX1BP1 KO mouse | GGGAACATTAATTCACAGAGGGGACATTTC | N/A |
| genotyping primer for TAX1BP1 KO mouse | ATTAATTAATTCAACTTGTACACTGACAGG | N/A |
| R SMARTer RACE gene-specific primer (sits down in TAX mouse cDNA at 2057-2078 = Takara sequence for cloning) | GATTACGCCAAGCTTGGCCACCTGTGCGAGTCCCATC | N/A |
| Gibson primer to move long EF1a promoter into PLEX-PGK-N-EGFP-TDP43, pLEX-PGK-HTTQ25-C-mEMERALD | cgtgctgcaggtccgaggttctagaGTGCCCGTCAGTGGGCAG | N/A |
| Gibson primer to move long EF1a promoter into PLEX-PGK-N-EGFP-TDP43 | cccttgctcaccatgctagcggccgcAATGAACCAAGATCCAAACTCAAAAAGGGC | N/A |
| Gibson primer to move long EF1a promoter into PLEX-PGK-HTTQ25-C-mEMERALD | ctgagtccggacatgctagcggccgcAATGAACCAAGATCCAAACTCAAAAAGGGC | N/A |
| Gibson primer to move long EF1a promoter into PLEX-PGK-HTTQ103-C-mEMERALD | tccagggtcgccatgctagcggccgcAATGAACCAAGATCCAAACTCAAAAAGGGC | N/A |
| Gibson primer to move FLAG-HA-TAX1BP1 into pLEX-PGK | aggggatccgcggccgctagcATGGACTACAAGGATGAC | N/A |
| Gibson primer to move FLAG-HA-TAX1BP1 into pLEX-PGK | aatccagaggttgattgttacgcgtCTAGTCAAAATTTAGAACATTCTG | N/A |
| Gibson primer to move FLAG-HttQ103 into pLEX-PGK | aggggatccgcggccgctagcATGGACTACAAGGACGACGATGACAAGC | N/A |
| Gibson primer to move long EF1a promoter into PLEX-PGK-N-FLAG-HA-TAX1BP1 | gttgcgccttttccaaggcagccctggGTGCCCGTCAGTGGGCAG | N/A |
| Gibson primer to move long EF1a promoter into PLEX-PGK-N-FLAG-HA-TAX1BP1 | agtccatgctagcggccgcggatccAATGAACCAAGATCCAAACTCAAAAAGGGC | N/A |
| Gibson primer to move GFP-TDP-43 into pLEX-PGK | aggggatccgcggccgctagcATGGTGAGCAAGGGCGAG | N/A |
| Gibson primer to move GFP-TDP-43 into pLEX-PGK | aatccagaggttgattgttacgcgtCTACATTCCCCAGCCAGAAG | N/A |
| Gibson primer to move HttQ23 into pLEX-PGK | aggggatccgcggccgctagcatgTCCGGACTCAGATCTATGAAG | N/A |
| Gibson primer to move HttQ23 into pLEX-PGK | cttgctcaccatggtggcgaccggttTAGATCCGGTGGATCCCG | N/A |
| Gibson primer to move HttQ103 into pLEX-PGK | aggggatccgcggccgctagcaTGGCGACCCTGGAAAAGCTGATG | N/A |
| Gibson primer to move HttQ103 into pLEX-PGK | cttgctcaccatggtggcgaccggtAGGGATCCCCCGGGCTGC | N/A |
| Gibson primer to move TAX1BP1 into pLEX-PGK | aggggatccgcggccgctagcATGACATCCTTTCAAGAAG | N/A |
| Gibson primer to move TAX1BP1 into pLEX-PGK | aatccagaggttgattgttacgcgtctaTAGAACATTCTGATCAAAATGG | N/A |
| Recombinant DNA | ||
| pDONR223-OPTN | This study | N/A |
| pDONR223-p62 | This study | N/A |
| pDONR223-NBR1 | This study | N/A |
| pDONR223-NDP52 | This study | N/A |
| pDONR223-TAX1BP1 | This study | N/A |
| pHAGE-CMV-N-EGFP-TAX1BP1 | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 ΔSKICH | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 W49A | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 V143S | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 ΔZF | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 Q743A/E747K | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 Q770A/E774K | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 Q743A/E747K/Q770A/E774K | This study | N/A |
| pHAGE-CMV-N-FLAG-HA-TAX1BP1 ΔSKICH/ΔZF | This study | N/A |
| pDONR223-TDP-43 WT YFP | Aaron Gitler, (Johnson et al., 2008) | Addgene plasmid #27470 |
| pHAGE-N-EGFP-TDP43 | This study | N/A |
| pEGFP-Q23 | David Rubensztein, (Narain et al., 1999) | Addgene plasmid #40261 |
| EGFP-HttQ74 | Harm Kampinga, (Carra et al., 2009) | N/A |
| pYES2/103Q | Michael Sherman, (Meriin et al., 2002) | Addgene plasmid #1385 |
| pLEX-EF1α-Q23-EGFP | This study | N/A |
| pLEX-EF1α-Q74-EGFP | This study | N/A |
| pLEX-EF1α-Q103-EGFP | This study | N/A |
| pLEX-EF1α-FLAG-HA-TAX1BP1 | This study | N/A |
| pBMN-mEGFP-C-LC3A | (Lazarou et al., 2015) | N/A |
| pCHAC-mEGFP-C-LC3B | (Lazarou et al., 2015) | N/A |
| pBMN-mEGFP-C-LC3C | (Lazarou et al., 2015) | N/A |
| pBMN-mEGFP-C-GABARAP | (Lazarou et al., 2015) | N/A |
| pBMN-mEGFP-C-GABARAPL1 | (Lazarou et al., 2015) | N/A |
| pBMN-mEGFP-C-GABARAPL2 | (Lazarou et al., 2015) | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | (Ran et al., 2013) | Addgene plasmid #62988 |
| Software and Algorithms | ||
| Fiji | (Schindelin et al., 2012)(Schindelin et al., 2012) | https://fiji.sc/ |
| Cell Profiler | (Carpenter et al., 2006) | https://cellprofiler.org/ |
| Prism V7 | https://www.graphpad.com/scientific-software/prism/ | |
| R | (RCoreTeam, 2018) | https://www.R-project.org/ |
| Image Lab | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z | |
| Mascot 2.6 | (Perkins et al., 1999) | http://www.matrixscience.com/help/november2016.html |
| Neuron Viability Analysis | This study | https://github.com/gkanfer/Tax1bp_analysis |
Dissociated neuronal culture
The use of rats in this study followed the guidelines of the NIH Animal Care and Use Committee (Protocol number: 1171) approved by the National Institutes of Health NINDS Institutional Animal Care and Use Committee. Primary cultured neurons were isolated from both male and female embryonic day 18 Sprague-Dawley rats. Cortical neurons were plated in 6 well dishes coated with poly-D-lysine (Sigma) in Neurobasal media containing 2 mM Glutamax (100X) (GIBCO) and B27 supplement (50X) (GIBCO). Primary neurons were treated with MG132 on day-in-vitro 10.
Neuronal differentiation from iPSCs
Induced pluripotent stem cells (iPSCs) were maintained in Essential 8 Flex medium (GIBCO) supplemented with Essential 8 Flex Supplement (GIBCO). iPSCs were grown on Matrigel-coated plates (Corning) and cell dissociation was performed using Accutase (StemCell Technologies). Stable expression of NLS-BFP (used for nuclei counting) was obtained by infecting cells with lentivirus expressing U6-NLS-BFP followed by sorting for BFP-positive cells. Stable expression of TAX1BP1 was obtained by infecting NLS-BFP-expressing cells with lentivirus expressing EF1α-FLAG-TAX1BP1 followed by puromycin selection. Neuronal differentiation from iPSCs followed a published protocol (Wang et al., 2017). Briefly, iPSCs were seeded on a Matrigel-coated 6-well plate in neural induction media: DMEM/F12 with HEPES (GIBCO) containing N2 Supplement (100X) (GIBCO), non-essential amino acids (100x) (GIBCO), glutamax (100X) (GIBCO), ROCK inhibitor Y-27632 (10mM) (Tocris), and doxycycline (2mg/mL) (Sigma). Induction medium was changed daily for 2 additional days. On day 3 post-induction, cells were dissociated using Accutase, counted, and plated at 50,000 cells/well in poly-L-ornithine-coated (PLO) (Sigma, P3655) 96-well plates (Perkin Elmer, 6055302) in BrainPhys neuronal medium (StemCell Technologies, 05790) containing B27 supplement (50X) (GIBCO, 17504044), BDNF (10μg/mL) (PeproTech, 450-02), NT-3 (10μg/mL) (PeproTech, 450-03), and laminin (1μg/mL) (GIBCO, 23017015). Half of the well volume was removed and replaced with fresh supplemented BrainPhys media every 2 days for the duration of any assay.
Mice
Male and female C57BL/6 Taxbp1tm1KtjTAX1BP1 mice provided by Drs. Hidekatsu Ihaand Jennifer Philips were described previously (Iha et al., 2008). WT C57BL/6L mice were obtained from Jackson Laboratories (Stock No: 000664). Mice were housed in a virus-antigen-free facility at the NIH Division of Veterinary Resources in a 12 h light/dark cycle at RT and fed ad libitum with a standard rodent diet. All mouse procedures were performed according to a protocol (#1306) approved by the National Institutes of Health NINDS Institutional Animal Care and Use Committee. Age and sex of mice used in the study is described in the associated figure legends.
METHOD DETAILS
TALEN and CRISPR gene knockout cell lines
Construction of OPTN, NDP52, TAX1BP1, TKO and pentaKO knockout HeLa lines were reported previously (Lazarou et al., 2015). The NBR1, p62, and TAX1BP1 single or combinatorial knockout lines were created as described in Lazarou et al., 2015. Briefly, CRISPR gRNAs were generated to target common exons shared by all splice variants and cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene plasmid #62988). HeLa cells were transfected with the gRNA plasmids and treated with 1 ug/mL puromycin for 2 days to enrich transfected cells, which were then diluted and placed into 96-well plates to produce single colonies. Primer sets used for PCR screening and sequencing are listed in the Key Resources Table. Cell lines were additionally validated via immunoblot.
Cloning and generation of stably infected cell lines
The Gateway Cloning (Life Technologies) system was used to generate pHAGE-GFP-TAX1BP1, pHAGE-N-FLAG-HA-TAX1BP1, and pHAGE-GFP-TDP43. Briefly, genes of interest were cloned into pDONR223 then recombined into destination vectors pHAGE-N-FLAG-HA or pHAGE-N-GFP using L Recombinase (Life Technologies) as per the manufacturer’s protocol. Mutations in cDNA sequences were introduced using PCR site-directed mutagenesis in the pDONR223 vector. All constructs generated in this study were verified by sequencing. Original plasmids containing EGFP-HttQ43 and EGFP-HttQ74 were kindly provided by Dr. Harm Kampinga (Groningen, Netherlands). pYES2/103Q was a gift from Michael Sherman (Addgene plasmid #1385) and pEGFP-Q23 was a gift from David Rubinzstein (Addgene plasmid #40261). All huntingtin protein constructs were cloned into pLEX-EF1α-EGFP for lentiviral expression. pDONR-TDP-43 WT YFP was a gift from Aaron Gitler (Addgene plasmid #27470).
For generation of stable cell lines, lentiviruses (pHAGE vectors) were packaged in HEK293T or Lenti-X 293T cells. HeLa cells were transduced with virus for 24 h with 8 μg/mL polybrene then selected for protein expression by drug-resistance (puromycin or blasticidin) or fluorescence activated cell sorting. Generation of low- and high-expressing cells was done via infection with various dilutions of virus and expression assessed via comparison to endogenous protein levels on immunoblot.
Aggregate formation and clearance studies
For acute treatments, HeLa cells were grown on poly-D-lysine-coated (Sigma-Aldrich) coverslips and treated with either 5 μg/mL of puromycin (Invivogen) for 2 h or 1 μM MG132 (Sigma-Aldrich) for 8 h at 37°C. Cells were either fixed for imaging or assessed for clearance. For clearance, cells were washed three times in DMEM with 10% FBS and released into drug-free medium for 3 h at 37°C. For long treatments, HeLa cells were grown on poly-D-lysine-coated coverslips and treated with 1 μg/mL of puromycin or 1 μM MG132 for 18 h at 37°C. Coverslips were fixed and processed for microscopy, as outlined above. For Bafilomycin A (Sigma-Aldrich) treatments, cells were treated with the indicated concentration of drug in full media and either harvested as described or washed three times in full media and returned to full media for the indicated durations.
For huntingtin poly-Q-protein clearance assays, HeLa cells were seeded on poly-D-lysine-coated coverslips and transduced the following day with lentivirus expressing either HttQ23-EGFP, HttQ74-EGFP or HttQ103-EGFP for 24 h with 8 μg/mL polybrene. Cells were fixed for imaging and assessed 4 days post-infection.
Subcellular fractionation
Cells were washed once and scraped in cold PBS, pelleted, and lysed in RIPA buffer (Thermofisher) supplemented with Complete Protease Inhibitor Cocktail (Roche), 1mM EGTA, 1mM EDTA, 100mM chloroacetamide (Sigma-Aldrich) and 100mM DTT for 15 min at 4°C with end over end rotation. Five to ten percent of the total volume was collected, mixed with 4X LDS (Life Technologies), boiled, and reserved for the input fraction. RIPA-soluble fraction was obtained after centrifugation at 20,000 rpm for 15 min at 4°C, 4X LDS was added, and samples were heated to 99°C with shaking for 10 min. The insoluble fraction consisting of the remaining pellet was washed once with lysis buffer, spun at 20,000 rpm for 10 min at 4°C, then lysed in 1X LDS, 100mM DTT in PBS and heated to 99°C with shaking for 15 min. Protein concentration was determined via DC Protein Assay (BioRad). Soluble and insoluble fractions were normalized to the protein concentration of the input fraction. For analysis, 10%–15% of RIPA-soluble fraction and 2.5%–5% of insoluble fraction were loaded on 4%–12% Bis-Trisgels and run using MOPS buffer (Life Technologies) and visualized by western blotting on PVDF. Western blotting was performed by wet transfer method in either NuPage transfer buffer or Tris-glycine transfer buffer (Life Technologies). Proteins were detected using horseradish peroxidase-coupled secondary antibodies (GE Healthcare Life Sciences) and ECL Prime (GE Healthcare Life Sciences) or Supersignal West Femto solution (Thermofisher) western blotting detection reagents. Images were acquired using an MPgel documentation system (Bio-Rad Laboratories). Quantification of immunoprecipitation bands was performed using the volume tools in Image Lab software (Bio-Rad Laboratories).
Immunoblotting
After the indicated treatments, cells were harvested by scraping in cold PBS and either fractionated as described above or lysed in 2% SDS/50mM Tris-HCl, pH 6.8/150mM NaCl/100mM DTT (Sigma-Aldrich)/1X PBS, heated to 99°C with shaking for 10 min, and spun at 20,000 rpm for 15 min at RT. Protein concentration was determined using DC Protein Assay (BioRad) and 20-50 ug of protein per sample was separated on 4%–12% Bis-Tris gels using MOPS or MES running buffer (Life Technologies). Western blotting was performed by wet transfer method using polyvinyl difluoride membranes in either NuPage transfer buffer or Tris-glycine transfer buffer (Life Technologies). Proteins were detected using horseradish peroxidase-coupled secondary antibodies (GE Healthcare Life Sciences, Piscataway, NJ) and ECL Prime or Supersignal West Femto solution (Thermofisher) western blotting detection reagents (GE Healthcare Life Sciences). Images were acquired using an MP gel documentation system (Bio-Rad Laboratories). Quantification of immunoprecipitation bands was performed using the volume tools in Image Lab software (Bio-Rad Laboratories). Human tissue panel blot was purchased (NOVUS Biologicals).
Immunofluorescence Microscopy
HeLa cells were seeded on poly-D-lysine-coated (Sigma-Aldrich, P7280) coverslips and treated as indicated. Following treatment, cells were fixed at room temperature in 4% paraformaldehyde for 15 min, permeabilized and blocked with filtered IF buffer (0.1% Triton X-100, 3% goat serum, 1X PBS) for 1 h at RT. For immunostaining, cells were incubated with indicated antibodies diluted in IF buffer overnight at 4°C, washed 3 times with IF buffer and incubated with Alexa Fluor-conjugated secondary antibodies (Life Technologies) 1:1000 in IF buffer for 1 h at RT. Cells were then incubated with DAPI (Sigma) diluted in IF buffer at 1:10,000 for 10 min at RT. Cells were washed 2 times with IF buffer and 1 time with 1X PBS. Coverslips were mounted on slides using Prolong Gold Antifade (Life Technologies). Representative images were collected with an inverted laser scanning LSM 880 microscope (Carl Zeiss) using a 63X/1.4 objective Plan-Apochromat (Carl Zeiss). Images were collected as z stacks captured at optimal thickness. Representative images shown are maximum intensity projections unless otherwise noted.
Cell viability
For HeLa cell viability assays, cells were seeded in quadruplicate in white clear-bottom 96-well plates (Corning). The next day, cells were treated as indicated in figure legends. Cell viability was measured by using the Cell Titer-Glo system (Promega) according to the manufacturer’s instructions. Values were normalized to the value for untreated samples for each cell line. Means and standard deviations were calculated, and statistical significance was calculated using one-way analysis of variance (ANOVA). One plate was measured each day using a Synergy H1Hybrid Multi-Mode Reader (Biotek).
Lipofuscin Capture
Anesthetized mice were perfused with ice-cold 1XPBS. Isolated brain hemispheres were submerged in 4% paraformaldehyde at 4°C overnight, cryoprotected in 30% sucrose 24 to 48 h at 4°C, embedded in Tissue Tek O.C.T. compound (EMS; Hatfield, PA), then frozen by submersion into 2-methylbutane cooled on dry ice and stored at −80pC. Cryosections (14μ) were cut on a Leica 3050S cryostat (Leica Biosystems; Nussloch, Germany) and mounted onto Superfrost Plus slides (Fisher Scientific; Pittsburgh, PA). Slides were incubated with DAPI and mounted with Prolong Diamond Antifade Mounting Media (Thermofisher). To capture lipofuscin autofluorescence, large regions of interest (ROIs) were captured in tile-scan mode with a centered Z stack at 555nm on a Zeiss LSM 800 confocal microscope equipped with a Plan-Apochromat 20X, 0.8NA lens (Zeiss; Thornwood, NY).
Brain Tissue Lysates
Anesthetized mice were perfused with ice-cold 1X PBS and brains were dissected regionally into cortex, striatum, hippocampus, and cerebellum. Brain regions were snap-frozen in LN2 and stored at −80°C prior to preparation. Tissue was homogenized on ice with RIPA buffer plus Complete Protease Inhibitor Cocktail (Roche) and deubiquitylase inhibitors (10μM PR-619, Selleck Chemicals, Cat#S7130). Homogenates were incubated on ice for 20 min and turned end over end at 4°C for 20 min. Protein concentration and immunoblotting were performed as described above.
Neuronal Viability Assay
iPSCs grown in 6-well plates were infected with lentivirus expressing EF1α-HttQ23-EGFP or EF1α-HttQ103-EGFP; the following day, media was changed, and cells were allowed to recover for 1 day. Two days post-infection, cells were plated in induction medium. On day 3 post-induction, cells were dissociated using Accutase, counted, and plated at 50,000 cells/well in poly-L-ornithine-coated (PLO) (Sigma, P3655) 96-well plates (Perkin Elmer, 6055302) in supplemented BrainPhys neuronal medium (StemCell Technologies, 05790). Half of the well volume was removed and replaced with fresh supplemented BrainPhys media every 3 days for the duration of the assay. iPSC-derived neurons were imaged daily for 20 days. Sixteen fields of view were imaged per each well of three independent biological replicates. Cells were imaged using a 20X air objective (NA 0.75) with 1.5X optical zoom on a Nikon Ti-2 CSU-W1 spinning disk system with a photometrics 95B camera operated by Nikon Elements software equipped with temperature regulation and CO2 control.
Puromycin Pulse Chase
Cells were seeded on day 0 to achieve a density of 60% on day 1. Control samples were pretreated with 15 μM cycloheximide for 10 min and all samples were then pulsed with 5 μg/mL of puromycin (Invivogen) for 2 h or 10 m. After either 2 h or 10 m, cells were washed three times in complete media and released into drug-free medium for the time periods specified. After the indicated treatments, cells were harvested by scraping in cold PBS and lysed in 1X LDS, 100mM DTT in PBS and heated to 99°C with shaking for 15 min. Protein concentration was determined via DC Protein Assay (BioRad) and equal protein amounts were loaded per sample. Immunoblotting and quantitation was performed as described above.
Filter-trap Assay
Cells were lysed via scraping in 2% SDS in FTA buffer (pH 8.0: 10 mM Tris–Cl and 150 mM NaCl), sonicated, and protein concentration determined by DCA assay. One-, five-, or 25-fold dilutions of protein were prepared and slotted onto a washed (FTA buffer + 0.1% SDS) 0.22 μM cellulose acetate membrane (GE Healthcare Life Sciences) using a Hybri.Slot 24 slot blotting apparatus as per (van Waarde-Verhagen and Kampinga, 2018). Gentle suction was applied to trap the detergent and urea-insoluble protein followed by three washes. Membranes were then removed and washed once in PBS containing 1% Tween-20 then processed in the same manner as for immunoblotting described above.
Mass Spectrometry
SDS-PAGE gel regions with molecular weight between 65 and 115 KDa were excised followed by in-gel reduction with TCEP (Thermo Fisher Scientific), alkylation with NEM, and digestion with trypsin. After extraction, digests were desalted with Waters Oasis HLB μElution plate. An UltiMate 3000 RSLC-nano system (Thermo Fisher Scientific) was used for chromatography separation. Peptides were separated on a nano-ES802 column over a 145-min gradient from 5% to 24% acetonitrile at a flow rate of 300 nL/min. LC-MS/MS experiments were performed on an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) in data-dependent acquisition (DDA) mode. Other parameters include: MS resolution: 120K at m/z 400; MS scan range: 375-1500 m/z; Automated Gain Control (AGC) target: 2 × 10e5; quadrupole isolation window: 1.6 m/z; precursors with charge states 2–6 and intensity higher than 1x10e4 within a 3 s cycle between MS1 scans were selected for MS/MS acquisition in the linear ion trap.
Mascot 2.6 was used for database searches. Other parameters include: trypsin digestion with 1 missed cleavage allowed; N-ethylmaleimide on cysteines as fixed modification; oxidation (M) as variable modification; mass tolerance: 5 ppm for precursor ions and 0.6 Da for fragment ions; protein false discovery rate (FDR): 0.01.
Rapid amplification of cDNA ends (3’-RACE)
Total RNA from a TAX1BP1 knockout mouse cortex was isolated using TRIzol reagent (Thermofisher) per manufacturer’s instructions. Rapid amplification of cDNA ends (3’ RACE), 3’ RACE PCR purification, and cloning of 3’ RACE PCR products was conducted as per manufacturer’s instruction using the SMARTer® RACE 5’/3’ Kit (Takara). Total RNA (500 ng) was used for cDNA synthesis followed by 3’ RACE PCR using gene specific primers (included in Key Resources Table) targeting mouse TAX1BP1 exon 16 and a Universal Primer Mix (UPM) included in the SMARTer RACE 5’/3’ Kit.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for aggregate formation and clearance assays
For quantification, cells with puromycin-, MG132-, or HTT-induced foci were either counted manually in a blinded fashion or using the particle analysis plug-in in ImageJ after blinding. Any macros created are available on request. At least 200 cells per sample were analyzed in three biological replicates. Means and standard deviations were calculated, and statistical significance was assessed. For comparisons between three or more groups, a one or two way-ANOVA with Tukey’s post hoc analysis was performed using Prism V7 (GraphPad Software) as noted in the figure legend. Error bars represent standard deviation (SD): *p < 0.5, **p < 0.01, ***p < 0.001, ****p < 0.0001. To determine number of foci per individual cell, CellProfiler (Carpenter et al., 2006), version 2.2.0 was used. All CellProfiler pipelines and detailed settings to reproduce the image analysis procedures are available upon request. Specific details for each experiment (statistical test used, n, dispersion, and precision measures) can be found in the figure legends.
Quantitation and statistical analysis of lipofuscin punctae
Images were blinded and autofluorescent foci counted using the particle analysis plug-in in ImageJ. Means and standard error of measurement were calculated, and statistical significance was assessed via unpaired t test with Welch’s correction using Prism V7 (GraphPad Software) as noted in the figure legend. Error bars represent standard error of measurement (SEM): *p < 0.5, **p < 0.01, ***p < .001. The number of observations used in each experimental series was included in the figure legend.
Quantification and analysis of immunoblots
Immunoblots were analyzed using the volume tools in Image Lab software (Bio-Rad Laboratories). Values were normalized to the loading control and in some cases to WT or untreated samples. Specific details for each experiment (statistical test used, n, dispersion, and precision measures) can be found in the figure legends.
Statistical Analysis for Neuron Viability Assay
To determine iPCS-derived neuron survival, NLS-BFP-expressing nuclei were counted automatically using the R EBImage package (Pau et al., 2010). Three biological replicates per condition were pooled together and the data were fitted to an exponential decay model using the R DRC package (Ritz et al., 2015) and the formula: (f(x) = c + (d-c)(exp(−x/e))) where (e) is the slope and (d) and (c) represent the upper and the lower limits around the slope. Outliers were detected and removed using interquartile range using the R Outliers package (He et al., 2005). To model and correct for the basal reduction in cell number over time in untreated cells, an averaged correction factor was calculated like so: samples were median normalized at each time point and the corresponding experimental samples were corrected by multiplying each time point by the averaged correction factor. The survival score was calculated from the fitted model, in which the slope (e) represents the survival score of each population of cells counted at every time point for the designated treatment. For statistical comparison, a permutation test (a.k.a randomization test) was used. In brief, the delta mean score of the groups was compared to random delta mean scores of shuffled groups iterated 10,000 times. P value was determined by calculating the number of times the delta score was higher in the shuffled group than in the ground true group (Ritz et al., 2015). Code is provided in Supplementary Information. R based analysis code and data are at https://github.com/gkanfer/Tax1bp_analysis.
Supplementary Material
Highlights.
Loss of TAX1BP1 blocks clearance of protein aggregates in cells
Increased expression of TAX1BP1 promotes aggregate clearance via autophagy
Aggregate clearance is ubiquitin dependent
Mice lacking functional TAX1BP1 accumulate ubiquitin conjugates in the brain
ACKNOWLEDGMENTS
We thank Drs. Malavika Raman, Achim Werner, David Beck, and Richa Lomash for critical reading of the manuscript. We thank the NINDS Light Imaging Facility, NHLBI Flow Cytometry Core Facility, NINDS Protein/Peptide Sequencing Facility, and NINDS EM facility for technical assistance. We thank Dr. Chunxin Wang for the p62 KO cell line, Dr. Harm Kampinga for sharing the EGFP-HttQ43 and EGFP-HTTQ74 plasmids, and John Badgerand Dr. Katherine Roche for providing primary rat cortical neurons. We thank Drs. Jennifer Philips, Guozhe Yang, and Hidekatsu Iha for providing TAX1BP1 ΔZF mice. This work was supported in part by the NIH grants NINDS intramural program (R.J.Y., S.A.S., G.K., H.V.S, and M.E.W.), NICHD (NIH)-Microscopy Imaging Core (L.A.H.), and NIGMS Postdoctoral Research Associate (PRAT) Fellowship (S.A.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.10.041.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ashe KH, and Zahs KR (2010). Probing the biology of Alzheimer’s disease in mice. Neuron 66, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, and Finkbeiner S (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci 30, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol 10, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EJ, Bence NF, Jayakumar R, and Kopito RR (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17,351–365. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, and Johansen T (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra S, Brunsting JF, Lambert H, Landry J, and Kampinga HH (2009). HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2α phosphorylation. J. Biol. Chem 284, 5523–5532. [DOI] [PubMed] [Google Scholar]
- Cemma M, Kim PK, and Brumell JH (2011). The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 7, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AJS, and Ryter SW (2011). Autophagy in inflammatory diseases. Int. J. Cell Biol 2011, 732798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirr E, and Wyss-Coray T (2012). The immunology of neurodegeneration. J. Clin. Invest 122, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I (2017). Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem 86, 193–224. [DOI] [PubMed] [Google Scholar]
- Eggers DK, Welch WJ, and Hansen WJ (1997). Complexes between nascent polypeptides and their molecular chaperones in the cytosol of mammalian cells. Mol. Biol. Cell 8, 1559–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, and Klionsky DJ (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, and Ward ME (2018). Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr. Protoc. Cell Biol 79, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- He Z, Xu X, Huang JZ, and Deng S (2005). FP-Outlier: Frequent Pattern Based Outlier Detection. Comput. Sci. Inf. Syst 2, 103–118. [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, and Harper JW (2015). The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 60, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, and Ciechanover A (1998). The ubiquitin system. Annu. Rev. Biochem 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, Varghese J, Wood N, Wightman M, et al. (2016). UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 166, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iha H, Peloponese J-M, Verstrepen L, Zapart G, Ikeda F, Smith CD, Starost MF, Yedavalli V, Heyninck K, Dikic I, et al. (2008). Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 27, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, and Hyman BT (1997). APPSw transgenic mice develop age-related A β deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol 56, 965–973. [DOI] [PubMed] [Google Scholar]
- Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, and Kopito RR (2005). Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 102, 13135–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, and Gitler AD (2008). A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 105, 6439–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522,354–358. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, and Dikic I (2009a). A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269. [DOI] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Johansen T, and Dikic I (2009b). NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5, 732–733. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, and Tanaka K (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441,880–884. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou Y-S, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163. [DOI] [PubMed] [Google Scholar]
- Kopito RR (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. [DOI] [PubMed] [Google Scholar]
- Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, and Dikic I (2013). Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci 126, 580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krobitsch S, and Lindquist S (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA 97, 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, and Johansen T (2012). Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol 2012, 736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Kirkin V, Dikic I, and Johansen T (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8, 1986–1990. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, Guthrie CR, and Kraemer BC (2010). Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci 30, 16208–16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, and Yue Z (2015). Neuronal aggregates: formation, clearance, and spreading. Dev. Cell 32, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Psakhye I, and Jentsch S (2014). Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 158, 549–563. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Moreau K, and Rubinsztein DC (2011). Protein misfolding disorders and macroautophagy. Curr. Opin. Cell Biol 23, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, and Sherman MY (2002). Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol 157, 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, and Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Moreno-García A, Kun A, Calero O, Medina M, and Calero M (2018). An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci 12, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI (2008). Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Ikebe E, Tsukamoto Y, Wang Y, Matsumoto T, Mitsui T, Yahiro T, Inoue K, Kawazato H, Yasuda A, et al. (2013). Commensal microbiota contributes to chronic endocarditis in TAX1BP1 deficient mice. PLoS ONE 8, e73205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narain Y, Wyttenbach A, Rankin J, Furlong RA, and Rubinsztein DC (1999). A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet 36, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, and Youle RJ (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. (2010). p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-AA, Outzen H, Øvervatn A, Bjørkøy G, and Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- Pau G, Fuchs F, Skylar O, Boutros M, and Huber W (2010). EBImage–an R package for image processing with applications to cellular phenotypes. Bioinformatics 26, 979–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- Prabakaran T, Bodda C, Krapp C, Zhang BC, Christensen MH, Sun C, Reinert L, Cai Y, Jensen SB, Skouboe MK, et al. (2018). Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 87, e97858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J,Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, Foeglein A, Williams R, and Randow F (2019). The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 74, 320–329.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, and Rubinsztein DC (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet 11, 1107–1117. [DOI] [PubMed] [Google Scholar]
- RCoreTeam (2018). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D (2015). Dose-Response Analysis Using R. PLoS ONE 10, e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, Brown SD, and Rubinsztein DC (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet 19, 2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Bento CF, and Deretic V (2015). Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med 212, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomons FA, Menéndez-Benito V, Böttcher C, McCray BA, Taylor JP, and Dantuma NP (2009). Selective accumulation of aggregation-prone proteasome substrates in response to proteotoxic stress. Mol. Cell. Biol 29, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang TK, Li C, Liu W, Rodriguez A, Abrams JM, Zipursky SL, and Jackson GR (2005). Inactivation of Drosophila Apaf-1 related killer suppresses formation of polyglutamine aggregates and blocks polyglutamine pathogenesis. Hum. Mol. Genet 14, 357–372. [DOI] [PubMed] [Google Scholar]
- Sarkar S, and Rubinsztein DC (2008). Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 275, 4263–4270. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, and Rubinsztein DC (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem 282, 5641–5652. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Harhaj NS, Liebl DJ, and Harhaj EW (2007). Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 26, 3910–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Pujari R, Harhaj NS, Abbott DW, and Harhaj EW (2011). The kinase IKKα inhibits activation of the transcription factor NF-κB by phosphorylating the regulatory molecule TAX1BP1. Nat. Immunol 12, 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MY, and Goldberg AL (2001). Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32. [DOI] [PubMed] [Google Scholar]
- Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, and Buss F (2015). The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 11, e1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Waarde-Verhagen MAWH, and Kampinga HH (2018). Measurement of chaperone-mediated effects on polyglutamine protein aggregation by the filter trap assay. Methods Mol. Biol 1709, 59–74. [DOI] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, and Youle RJ (2019). Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 74, 347–362.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, and Randow F (2010). NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 6, 288–289. [DOI] [PubMed] [Google Scholar]
- Wang DB, Gitcho MA, Kraemer BC, and Klein RL (2011). Genetic strategies to study TDP-43 in rodents and to develop preclinical therapeutics for amyotrophic lateral sclerosis. Eur. J. Neurosci 34, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, et al. (2017). Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports 9, 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, and Kumar-Singh S (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 107, 3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, and Cuervo AM (2010). Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb. Perspect. Biol 2, a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, and Holzbaur ELF. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 111, E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, and Simonsen A (2011). The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol. Dis 43, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang G, Huang X, and Du Z (2015). Crystallographic and modelling studies suggest that the SKICH domains from different protein families share a common Ig-like fold but harbour substantial structural variations. J. Biomol. Struct. Dyn 33, 1385–1398. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Antón LC, and Bennink JR (1996). Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol 157, 1823–1826. [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated are available from the corresponding author upon request. All macros created for use with ImageJ/Fiji are available on request. CellProfiler pipelines and detailed settings to reproduce the image analysis procedures are available upon request. Original data have been deposited to Mendeley Data: https://doi.org/10.17632/jv437rbcjk.1. Code generated for neuron viability analysis is publicly available at: https://github.com/gkanfer/Tax1bp_analysis.