

Abstract
Transition-metal-mediated cleavage of C−C single bonds can enable entirely new retrosynthetic disconnections in the total synthesis of natural products. Given that C−C bond cleavage inherently alters the carbon framework of a compound, and that, under transition-metal catalysis, the generated organometallic or radical intermediate is primed for further complexity-building reactivity, C−C bond-cleavage events have the potential to drastically and rapidly remodel skeletal frameworks. The recent acceleration of the use of transition-metal-mediated cleavage of C−C single bonds in total synthesis can be ascribed to a communal recognition of this fact. In this Review, we highlight ten selected total syntheses from 2014 to 2019 that illustrate how transition-metal-mediated cleavage of C−C single bonds at either the core or the periphery of synthetic intermediates can streamline synthetic efforts.
Keywords: C−C bond cleavage, cleavage reactions, natural products, total synthesis, transition metals
Graphical Abstract

1. Introduction
For over half a century, natural products have served as prime targets of chemical synthesis. The structural complexity of natural products continues to inspire the development of new strategies and methods for synthesis, and their often medicinally relevant biological activity motivates the design of efficient syntheses to access larger amounts of these compounds or their derivatives. Historically, a major focus in the development of synthetic strategies and methods has been the forging of C−C single bonds to construct the unique carbon frameworks associated with complex natural products; indeed, the pattern and prevalence of C−C single bonds is often the predominant distinction between these natural products and simple chemical building block precursors. As a result, some of the most practiced reactions in total synthesis—for example, the Diels-Alder reaction, the aldol reaction, and Pd-catalyzed cross-coupling reactions—feature the formation of C−C single bonds.
Although the progressive formation of C−C single bonds constitutes a logical approach for the efficient synthesis of natural products from commercially available starting materials, chemists have also long-recognized the potential of C−C single-bond cleavage to streamline syntheses.[1] Indeed, Woodward˛s landmark synthesis of quinine (4) in 1944 featured the cleavage of a C−C single bond of a cyclohexanone by treatment with ethyl nitrite (1→3; Scheme 1A).[2] In addition to classical organic reactions such as those employed in the Woodward synthesis, the development of transition-metal-mediated processes for the cleavage of C−C single bonds has enabled access to a wider array of chemical reactivity. Specifically, transition-metal-mediated cleavage of C−C single bonds often generates reactive organometallic intermediates, which offer the opportunity for further functionalization across the cleaved C−C bond by two-electron reactions. Trost˛s synthesis of (+)-frondosin A (10) showcases this mode of reactivity, where a key ruthenium-catalyzed vinylcyclopropane [5+2] cycloaddition proceeds via ruthena-cyclopentene intermediate 7; cleavage of the C−C single bond of the adjacent cyclopropane ring then generates reactive organoruthenium intermediate 8, thereby enabling subsequent formation of a C−C single bond by reductive elimination (Scheme 1B).[3] Overall, the transition-metal-mediated two-electron reactivity facilitated functionalization across a strained C−C single bond, with generation of an additional C−C σ-bond and a C−C π-bond.
Scheme 1.

Examples of previous syntheses: A) with non-transition-metal-mediated C−C bond cleavage, B) transition-metal-mediated cleavage of C−C single bonds resulting in a reactive organometallic intermediate (two-electron reactivity), and C) transition-metal-mediated cleavage of C−C single bonds resulting in a reactive radical intermediate (one-electron reactivity). Cp = cyclopentadienyl, DMPU = dimethylpropylene urea, Piv = pivaloyl, TBS = tert-butyldimethylsilyl, TIPS = triisopropylsilyl.
As an alternative mode of reactivity, transition metals can participate in single electron transfer events with a substrate; subsequent cleavage of a C−C bond would result in the formation of a reactive radical intermediate, which may recombine with a metal complex or engage in further radical processes.[4] Carreirąs synthesis of (+)-crotogoudin (16) exemplifies this reactivity, with the SmI2-mediated single-electron reduction of lactone 11 yielding ketyl radical 12 (Scheme 1C). Cleavage of a cyclopropane C−C bond then occurs to generate reactive radical intermediate 13, which undergoes a subsequent C−C bond formation through a 6-exo-trig radical cyclization to yield intermediate 14. Reduction, elimination of the pivalate group, and protonation then delivered compound 15, which was elaborated to the natural product. Overall, the installation of a C−C single bond in the core of crotogoudin was accomplished by cleavage/functionalization of a cyclopropane C−C bond through a one-electron transition-metal-mediated reaction.[5]
The accessibility of these multiple modes of reactivity upon transition-metal-mediated cleavage of a C−C single bond has increasingly inspired synthetic chemists to consider C−C single bonds as functional groups in retrosynthetic analysis. Recently, there has been a profusion of methods involving transition-metal-mediated cleavage/functionalization of C−C single bonds, which has resulted in the increased application of both novel and established tactics for C−C bond cleavage to develop new strategies for total synthesis. Although mechanistic aspects and methodological applications of C−C single bond cleavage processes[6–12] and transition-metal-catalyzed cleavage of C−C single bonds in total syntheses prior to 2015[13] have recently been reviewed by others, here we focus on selected total syntheses from 2014 to 2019 that utilize transition-metal-mediated cleavage of C−C single bonds. We will highlight how the cleavage of C−C single bonds in either the core or on the periphery of synthetic intermediates has provided access to both two-electron and one-electron transition-metal-mediated reactions, thereby enabling new retrosynthetic opportunities for each natural product discussed.
2. Cleavage of Core C−C single Bonds in Total Synthesis
For the purposes of this Review, we will define “core” C−C single bonds as those that occur within a central ring of a synthetic intermediate. Thus, the cleavage of such a core C−C single bond would transform a monocyclic system into a linear system, a bicyclic system into a monocyclic system, etc. When paired with ring-forming processes, the cleavage of core C−C single bonds can result in the rapid remodeling of synthetic intermediates to more closely resemble the molecular architecture of the target natural product. This may be the case irrespective of whether a C−C single-bond cleavage immediately precedes (Scheme 2A) or succeeds (Scheme 2B) a ring-forming event. As a result, synthetic tactics involving the cleavage of core C−C single bonds and ring-forming events occurring in rapid succession may enable novel, counterintuitive retrosynthetic disconnections that result in proposed synthetic intermediates that may not “map on” well to the target natural product. These tactics, therefore, expand the range of synthetic methods, extending the boundaries of retrosynthesis and inviting synthetic practitioners to develop new strategies. The following gives discussions on selected recent total syntheses employing the cleavage of a core C−C single bond as a strategic transformation.
Scheme 2.

Conceptual examples of paired cleavage and ring formation of core C−C single bonds, with cleavage of the core C−C single bonds occurring either: A) before or B) after formation of the ring-forming C−C bond.
2.1. Phomactin Terpenoids (Sarpong, 2018)
The phomactin natural products are a family of highly oxygenated diterpenoids first isolated in 1991 from fungi of the genus Phoma.[14] Early studies showed that (+)-phomactin A (17; Scheme 3) is a platelet activating factor receptor antagonist, with potential implications in cancer therapy.[15,16] Aside from this interesting bioactivity, the phomactins also contain a unique bicyclo[9.3.1]pentadecane core comprised of a densely functionalized cyclohexenyl fragment and a macrocyclic strap, thus presenting a formidable synthetic challenge. In 2018, Sarpong and co-workers successfully established the first unified approach to several congeners of the phomactin family.[17] Key to their strategy was the design of the versatile common intermediate 18 with functionality that could guide an array of downstream oxidation events to obtain various congeners (Scheme 3). Retrosynthetically, this intermediate could arise from two fragments—a highly functionalized cyclohexenyl ring 20 and known vinyl iodide 19[18]—which in the forward sense could undergo fragment coupling followed by macrocyclization. Cyclohexene 20 could be rapidly synthesized from (S)-carvone (21) through cleavage of a C−C bond.
Scheme 3.

Retrosynthesis of phomactins according to Sarpong and co-workers.
Although efficient access to the macrocyclic strap has been well-established in previous syntheses, accessing highly functionalized cyclohexenyl fragments such as 20 has previously been a particularly laborious endeavor, adding greatly to overall step counts.[18,19] To address the challenge of accessing fragments of this nature more efficiently, Sarpong and co-workers envisioned obtaining this cyclohexenyl fragment from the chiral pool terpene (S)-carvone by employing a C−C bond-cleavage reaction, which would result in a substantial remodeling of the carvone core framework and enable efficient access to 20. Previously, building on work pioneered by others on the C−C cleavage/functionalization of cyclobutanols,[20–22] Sarpong and co-workers had established a powerful strategy that entailed using enantioenriched cyclobutanols 22 derived from carvone in a transition-metal-mediated C−C bond-cleavage reaction to produce a versatile organometallic intermediate 23 primed for further functionalization, including C−C bond-forming processes[23] (Scheme 4). Overall, this strategy provides efficient access to a variety of stereochemically rich cyclohexenones that can be elaborated to diverse natural product-like scaffolds.
Scheme 4.
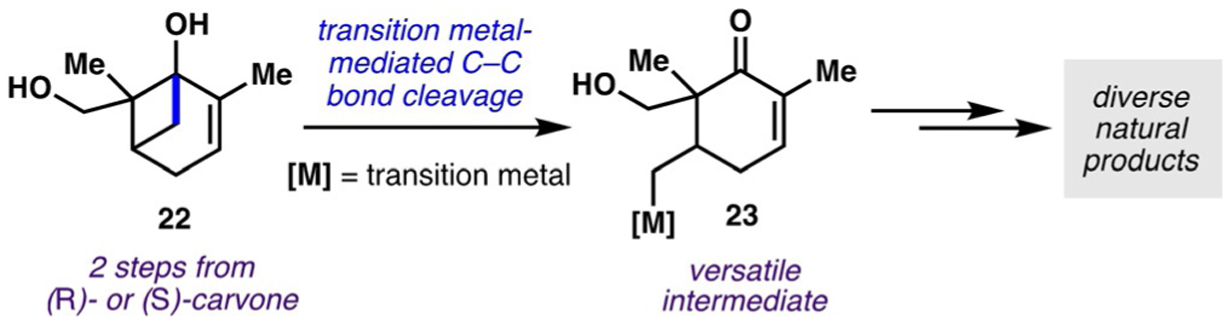
Strategy employed by Sarpong and co-workers for the cleavage of cyclobutanol C−C bonds.
To carry out this strategy in the context of the phomactins, Sarpong and co-workers began with the synthesis of cyclohexene 20 (Scheme 5). Following a known sequence by Bermejo and co-workers, epoxidation of the isopropenyl group of (S)-carvone followed by a TiIII-mediated reductive coupling of the epoxide and carbonyl afforded diastereomeric cyclobutanols 24 and 25.[24] A Mitsunobu-type displacement of the primary hydroxy group of the minor diastereomer furnished phenyl sulfide 26, which then set the stage for the key C−C bond cleavage. Treatment of 26 with [Rh(cod)OH]2 promoted a ligand exchange with the tertiary hydroxy group to provide Rh-alkoxide 27. β-Carbon elimination of the less-substituted C−C bond then yielded alkyl-Rh species 28, which readily afforded cyclohexenone 29 upon formal protodemetalation. This sequential installation of a strained cyclobutanol from carvone followed by strain-releasing C−C bond cleavage overall engendered a rearrangement of the carvone skeleton, thereby resulting in the integration of the isopropenyl group into the core framework (Scheme 5, dashed box, 21→29). This key sequence was followed by oxidation of the sulfide to the sulfone to afford 30; a subsequent addition of MeLi to the carbonyl moiety followed by a Burgess elimination of the resulting alcohol then furnished the exo-methylene moiety. Allylic oxidation of the vinylic methyl group delivered aldehyde 20. Lithiation of 19 promoted 1,2-addition to the aldehyde, and a subsequent protection of the resulting alcohol afforded 31. Cleavage of the TBS group followed by conversion of the hydroxy group into an allylic bromide provided 32, which underwent macrocyclization upon treatment with sodium bis(trimethylsilyl)amide (NaHMDS) to give 33. Reductive desulfonylation, cleavage of the 2-(trimethylsilyl)ethoxymethyl (SEM) group, and oxidation of the unveiled hydroxy group to a carbonyl moiety with a subsequent diastereoselective reduction furnished 18, which served as a common intermediate. From this intermediate, several congeners of the phomactin family, including phomactin A (17), were obtained efficiently through various oxidation tactics. In this work on the phomactins, Sarpong and co-workers effectively demonstrated the power of ring formation/C−C bond cleavage in remodeling the core framework of chiral pool molecules such as carvone, thus enabling efficient access to traditionally challenging motifs.
Scheme 5.

Total synthesis of phomactin natural products. cod = 1,5-cyclooctadiene, m-CPBA = meta-chloroperbenzoic acid, MsCl = methanesul-fonyl chloride, TBAF = tetrabutylammonium fluoride.
2.2. (−)-Xishacorene B (Sarpong, 2018)
In addition to their work on the phomactins, Sarpong and co-workers showcased the utility of C−C bond cleavage in 2018 with the first total synthesis of xishacorene B (34; Scheme 6A).[25] The xishacorene diterpenes, hydrocarbon natural products first isolated in 2017 from the soft coral Sinularia polydactyla off the coast of China,[26] exhibit immunopotentiating activity and contain a unique bicyclo-[3.3.1]nonane core. Retrosynthetically, xishacorene B could arise from triol 35, which in turn could be constructed from cyclohexenone 36 through a radical cyclization, with the reactive enone serving as a radical acceptor. This cyclohexenone was proposed to arise from cyclobutanol 37 and vinyl iodide 38 through a Pd-catalyzed C−C bond-cleavage/cross-coupling reaction. Cyclobutanol 37 was synthesized in two steps from (R)-carvone following Bermejo˛s procedure[24] (Scheme 5; 21→24).
Scheme 6.
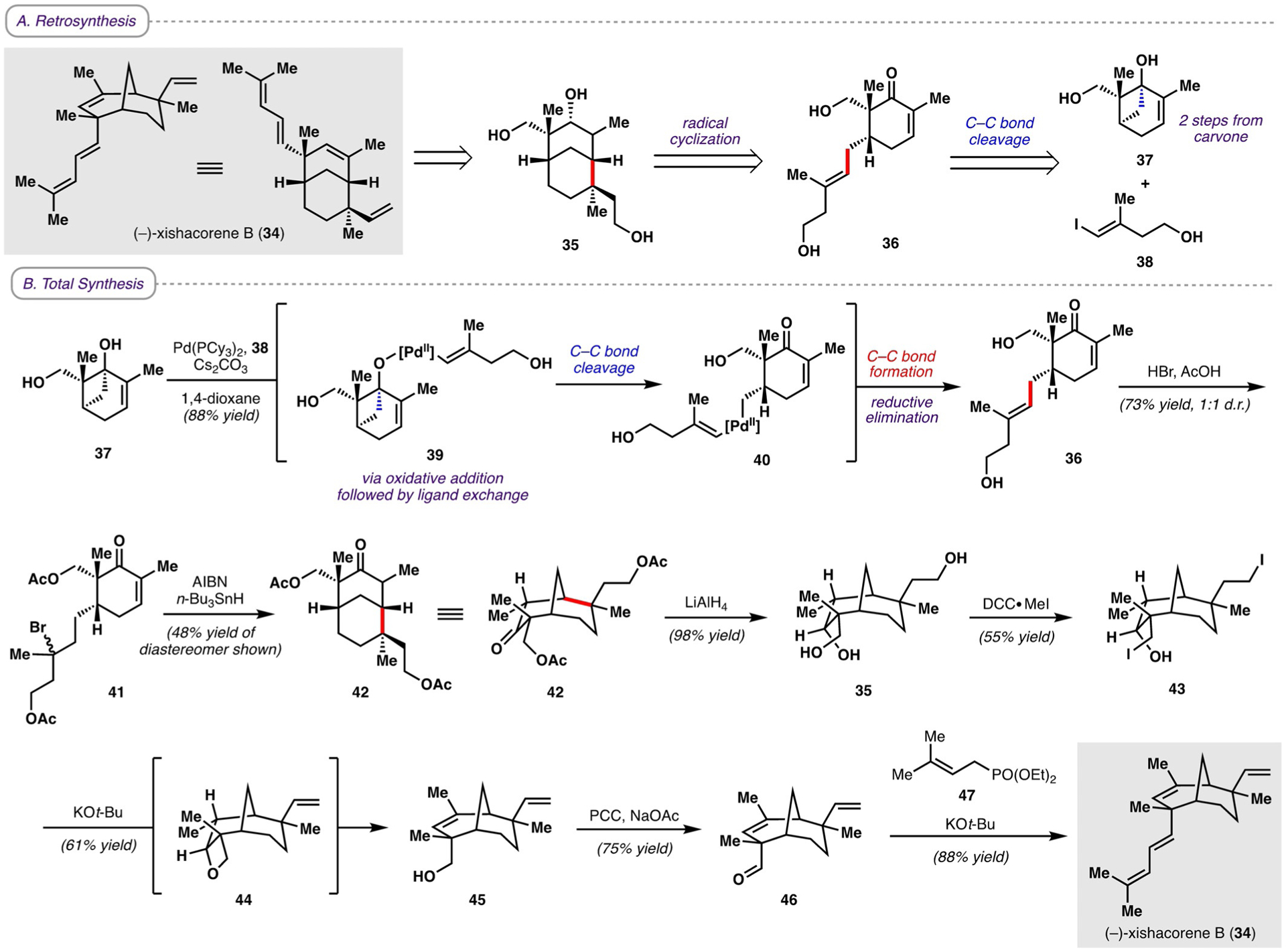
A) Retrosynthesis and B) total synthesis of (−)-xishacorene B. AIBN = azobis(isobutyronitrile), Cy = cyclohexyl, PCC = pyridinium chlorochromate.
The forward synthesis commenced with treatment of cyclobutanol 37 and vinyl iodide 38 with a Pd0 catalyst. This reaction formed Pd-alkoxide 39 after oxidative addition of the vinyl iodide and ligand exchange with the tertiary hydroxy group of 37 (Scheme 6B). The Pd-alkoxide then underwent β-carbon elimination to afford alkyl-Pd intermediate 40, which, after reductive elimination, yielded the desired cross-coupled product 36. This strategy, in which C−C bond cleavage of a carvone-derived cyclobutanol facilitated the synthesis of a highly functionalized cyclohexenone, was also employed by Sarpong and co-workers in their syntheses of phomactin terpenoids (Scheme 5); however, in contrast to the syntheses of the phomactins, in which an alkyl-Rh intermediate underwent formal protodemetalation, the alkyl-Pd intermediate 40 instead underwent reductive elimination, enabling the formation of an additional C−C bond. Bromination of the trisubstituted olefin group of 36 with concomitant acetylation of the hydroxy groups provided alkyl bromide 41. Treatment with AIBN and n-Bu3SnH then promoted the formation of a tertiary radical, which underwent a 6-exo-trig cyclization to the enone, thereby forging the [3.3.1] bicyclic core of xishacorene B. Notably, by employing various cross-coupling partners in place of 38 in the Pd-catalyzed C−C cleavage/cross-coupling sequence followed by an analogous radical cyclization, a variety of different [3.3.1] and [3.2.1] bicycles could be obtained (not shown).
Global reduction of 42 with LiAlH4 afforded triol 35, which was then iodinated with N,N’-dicyclohexylcarbodiimide·MeI (DCC·MeI) to yield bisiodinated alcohol 43. Treatment with KOt-Bu led, via oxetane 44, to alcohol 45, which upon oxidation with PCC afforded aldehyde 46. Finally, Horner-Wadsworth-Emmons olefination with phosphonate ester 47 provided (−)-xishacorene B in a total of 10 steps from (R)-carvone. Overall, Sarpong and co-workers were able to demonstrate a new strategy for preparing bicyclic compounds by performing a C−C bond-cleavage/cross-coupling reaction with subsequent radical cyclization, thereby showcasing the versatility of C−C bond cleavage for remodeling chiral pool molecules to provide unconventional access to complex frameworks.
2.3. (−)-Cycloclavine (Dong, 2018)
The ergot alkaloids are a large family of polycyclic indole alkaloids that exhibit a range of biological activities (Figure 1).[27] Cycloclavine (48), isolated from seeds of the flowering plant Ipomoea hildebrandtii in 1969,[28] exhibits insecticidal and antiparasitic activities.[29] Structurally, cycloclavine is unique among the ergot alkaloids as the only member that contains a cyclopropane ring. In addition, cycloclavine contains a pentacyclic core with three contiguous stereocenters, two of which are vicinal quaternary centers. These synthetic challenges, along with its biological activity, have made cycloclavine an intriguing target for synthetic chemists. Herein, we discuss the 2018 asymmetric total synthesis of (−)-cycloclavine by Dong and co-workers, which was accomplished through an enantioselective C−C activation/functionalization of a cyclobutanone.[30] For clarity, C−C activation in this Review will be defined as a specific form of C−C bond cleavage that is accomplished by an oxidative addition of a transition metal into a C−C bond.
Figure 1.
Representative ergot alkaloids with a benzo-fused tricyclic core (highlighted in purple).
Although transition-metal-catalyzed C−C activation of cyclobutanones is well-established,[7,9] there is an ongoing effort to utilize this powerful reactivity in new and interesting ways as a strategy for accessing natural product-like scaffolds. One prominent recent advancement in the C−C activation of cyclobutanone is the Rh-catalyzed carboacylation of olefins developed by Dong and co-workers, which provides efficient access to benzo-fused ring systems from benzocyclobutenones (Scheme 7). This specific C−C activation approach, first reported in 2012,[31] begins with the oxidative addition of a Rh complex into the C1−C2 bond of a strained cyclobutenone. Migratory insertion of the proximal olefin group and reductive elimination then forges two new bonds, giving rise to the desired benzo-fused ring system. It should be noted that although oxidative addition of the Rh complex into the less-substituted C1−C8 bond is kinetically favored (and usually observed in traditional methods), the tethered olefin group in this case serves as a directing group for the desired C1−C2 cleavage. This “cut-and-sew” approach therefore leads to the formation of novel benzo-fused tricycles, a key structural component in a number of biologically active natural products, including cycloclavine.
Scheme 7.

“Cut and sew” C−C activation strategy according to Dong and co-workers.
In a retrosynthetic sense, it was proposed that cycloclavine (48) could arise from tetracycle 49 through an aza-Wittig/imine reduction/reductive amination sequence, which in the forward sense would forge the pyrrolidine moiety in the natural product (Scheme 8A). This tetracycle would arise from benzo-fused tricycle 50 through diazo formation α to the ketone followed by a diastereoselective Rh-catalyzed cyclopropanation. Tricycle 50 would then be constructed from benzocyclobutenone 51 through the key asymmetric Rh-catalyzed C−C activation reaction. Finally, benzocyclobutenone 51 could be constructed from triflate 52 by a tandem C−N coupling/allylic alkylation reaction.
Scheme 8.
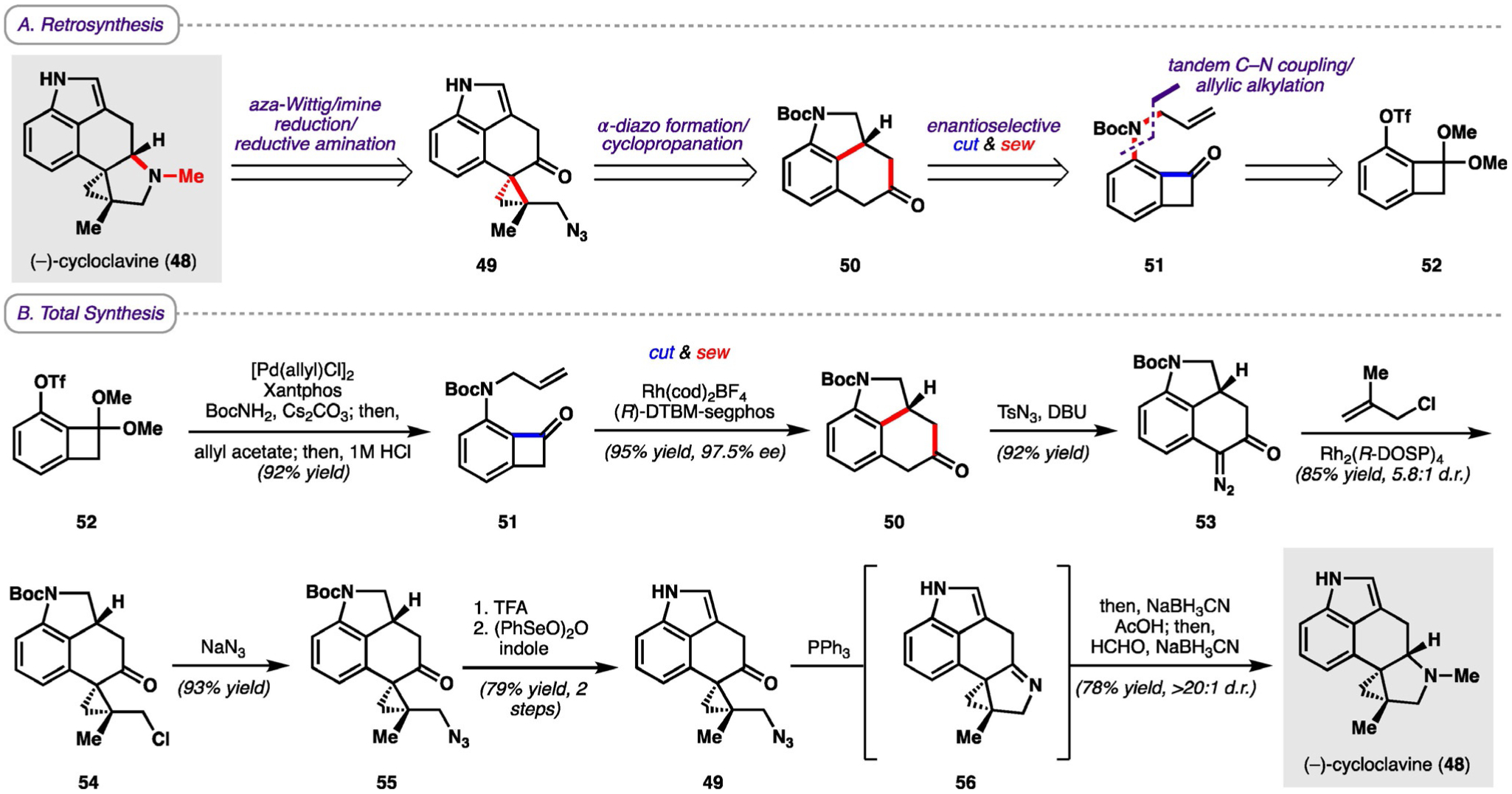
A) Retrosynthesis and B) total synthesis of (−)-cycloclavine. Boc = tert-butoxycarbonyl, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, DOSP = N-(dodecylbenzenesulfonyl)prolinate, (R)-DTBM-segphos = (R)-(−)-5,5’-bis[di(3,5-di-tert-butyl-4-methoxyphenyl)phosphino]4,4’-bi-1,3-ben-zodioxole, OTf = triflate, TFA = trifluoroacetic acid, Ts = tosyl.
The synthesis of cycloclavine (Scheme 8B) commenced with a known two-step sequence to obtain triflate 52 from 2-iodoresorcinol. Triflate 52 was first coupled to tert-butyl carbamate using a Pd-catalyzed C−N cross-coupling reaction, which was followed by an allylic alkylation catalyzed by the same Pd complex. Subsequent cleavage of the ketal upon acidic workup furnished benzocyclobutenone 51. Using conditions initially developed for the cut-and-sew of benzocyclobutenones with an allylic ether linkage[32] gave substandard results (i.e. low yields and enantioselectivities), which led to Dong and co-workers determining that the added bulkiness and rigidity of the amine linker required the use of a less bulky and more electron-deficient Rh complex. The use of the cationic RhI precatalyst Rh(cod)2BF4 proved to be effective in enhancing olefin coordination, and, when paired with (R)-DTBM-segphos as the ligand, provided the desired product with excellent yield and enantioselectivity (95% yield, 97.5% ee). With benzo-fused tricycle 50 in hand, Dong and co-workers turned their attention to constructing the [3.1.0] bicycle in the southern portion of the molecule. Treatment of 50 with TsN3 afforded the α-diazo ketone 53, which then underwent a Rh-catalyzed diastereoselective cyclopropanation with 2-methylallyl chloride to yield tetracycle 54. Azidation of the alkyl chloride moiety with NaN3 provided azide 55. Cleavage of the Boc protecting group followed by dehydrogenation afforded indole 49, which was converted into (−)-cycloclavine through the proposed aza-Wittig/imine reduction/reductive amination sequence. Although transition-metal-catalyzed C−C bond activation of strained ring systems has been an active field of research since the 1990s, there have been surprisingly few examples of its application in total synthesis. The synthesis of cycloclavine serves as a prime example of how C−C bond-activation strategies can be designed and implemented to build polycyclic ring systems efficiently and showcases the power that these strategies have in complex molecule synthesis.
2.4. (−)-Lingzhiol (Lan, Gong, and Yang, 2014)
Certain structural motifs occur frequently in a variety of diverse natural products; these conserved moieties often inspire synthetic chemists to develop strategies and methods for their preparation, which can ultimately facilitate entries into an array of natural products. One recent example is the work of Lan, Gong, Yang, and co-workers, who developed a highly versatile Rh-catalyzed formal (3+2) cycloaddition (facilitated by a C−C bond cleavage) to access [4.3.0] and [3.3.0] bicycles bearing vicinal quaternary centers,[33] structural motifs that appear in several diverse natural products (Figure 2). Herein, we discuss the application of this method to the total synthesis of (−)-lingzhiol (57), a meroterpenoid containing a [4.3.0] bicycle that was isolated in 2013 from the mushroom Ganoderma lucidum.[33,34]
Figure 2.
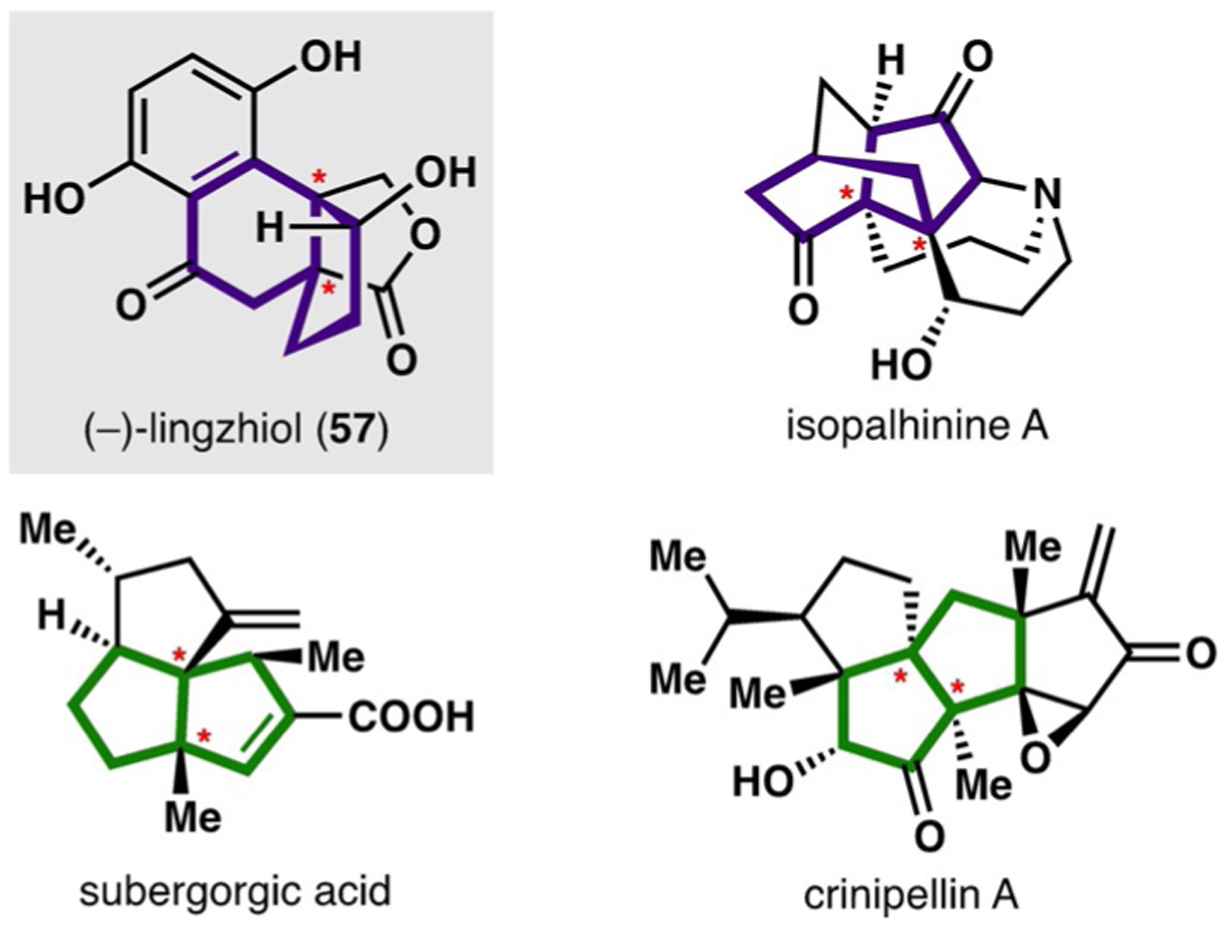
Representative natural products containing [4.3.0] (purple) and [3.3.0] bicycles (green) with vicinal quaternary stereocenters (red asterisk).
The proposed mechanism of the Rh-catalyzed formal (3+2) cycloaddition (Scheme 9) begins with formation of an alkyne-bound Rh-alkoxide 59 from cyclic enynol substrates 58. The Rh-alkoxide then undergoes a retro-propargylation, which results in C−C bond cleavage and formation of an allenyl-Rh intermediate 60. Conjugate addition of the allenyl-Rh species to the resulting enal forms the first C−C bond and leads to an oxygen-bound Rh-enolate 61. This enolate then likely undergoes a Conia-ene-type reaction with the allene to afford bicycle 62, with formation of the second C−C bond. Alcoholysis then leads to the desired bicyclic product 63, with concomitant regeneration of Rh-alkoxide 59. Notably, this process proved to be highly versatile, as the ring size in the starting material dictates the product outcome, with seven-membered rings yielding [4.3.0] bicycles and six-membered rings yielding [3.3.0] bicycles. Additionally, in contrast to examples discussed thus far, which leverage ring strain, this strategy can effect C−C bond cleavage in more thermodynamically stable six-membered rings.
Scheme 9.

Rh-catalyzed formal (3+2) cycloaddition.
Retrosynthetically, it was envisioned that lingzhiol could arise from lactone 64 through late-stage allylic and benzylic oxidations (Scheme 10). Lactone 64 could be synthesized from tricycle 65, which, in turn, could arise from 66 through the key Rh-catalyzed formal (3+2) sequence. Finally, 66 would arise from enone 67 through asymmetric reduction of the carbonyl group followed by a carboxylation/alkynylation sequence.
Scheme 10.
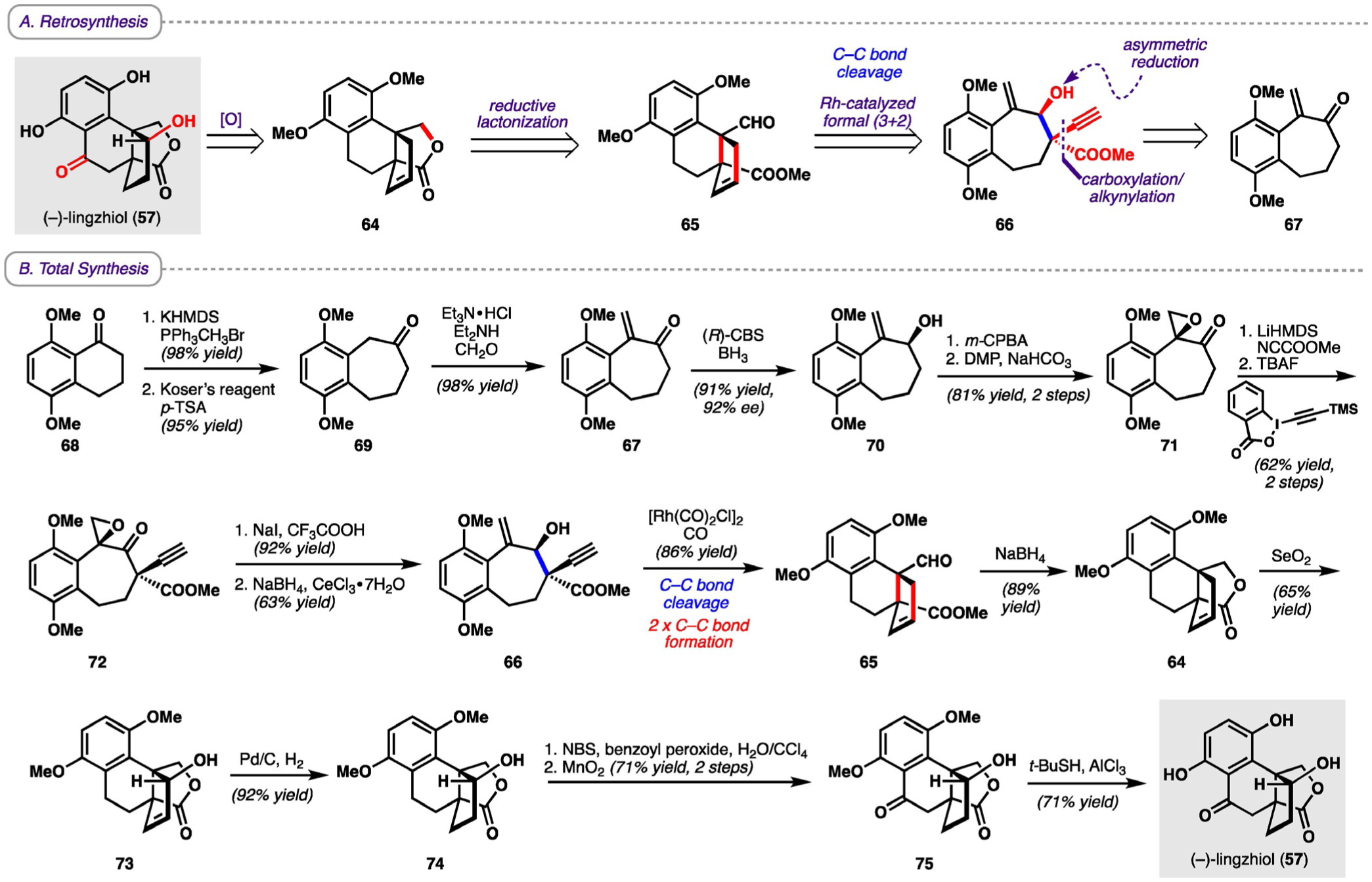
A) Retrosynthesis and B) total synthesis of (−)-lingzhiol. (R)-CBS = (R)-Corey-Bakshi-Shibata oxazaborolidine, HMDS = bis(trimethylsilyl)amide, NBS = N-bromosuccinimide, p-TSA = para-toluenesulfonic acid.
Beginning from 68, Wittig olefination followed by a ring expansion mediated by hydroxy(tosyloxy)iodobenzene (Koser˛s reagent) afforded 69, which was subsequently converted into enone 67 upon treatment with Eschenmoser˛s salt (generated in situ). A Corey-Bakshi-Shibata (CBS) reduction then afforded enantioenriched 70, which underwent epoxidation of the exo-methylene group with m-CPBA followed by oxidation of the hydroxy group to a carbonyl moiety with Dess-Martin periodinane (DMP) to deliver β-epoxy ketone 71. Carboxylation followed by propargylation afforded 72, which was then converted into allylic alcohol 66. Treatment with [Rh(CO)2Cl]2 converted 66 into tricyclic compound 65 through the key formal (3+2) cycloaddition with formation of two new C−C bonds. Reduction of the aldehyde with NaBH4 followed by lactonization of the resulting hydroxy group furnished lactone 64, which subsequently underwent allylic oxidation with SeO2 to provide allylic alcohol 73. Hydrogenation with Pd/C and H2 furnished 74, which was followed by a two-step sequence to effect a benzylic oxidation and yield the fully oxidized core 75. Finally, deprotection of the bisphenol moiety by cleavage of the methoxy groups completed the synthesis of (−)-lingzhiol (57). The total synthesis of (−)-lingzhiol represents a unique example of C−C bond cleavage as it is the only example in this Review in which core cleavage is not promoted by strain. This specific type of C−C bond cleavage expedites the overall process of remodeling a core framework by not requiring initial formation of a strained ring, but benefits instead from the formation of a π-allenyl intermediate.
2.5. (±)-Rhodomolleins XX and XXII (Ding, 2019)
Rhodomollein XX (76) and rhodomollein XXII (77) are grayanane diterpenoids isolated from Rhododendron molle from the Ericaceae family of flowering plants (Figure 3).[35] The grayanoid natural products exhibit a wide range of biological activities, including insecticidal, antifeedant, and analgesic effects, and contain a characteristic 5/7/6/5 tetracyclic carbon skeleton.[36] Many of these natural products are also densely functionalized with numerous stereogenic centers and various oxygen-containing substituents. Biosynthetically, the grayanoid natural products are thought to be related to the ent-kauranoid family of natural products because of the presence of the ent-kauranoid-like bicyclo[3.2.1]octane moiety (C/D rings). As an extension to their previous work on the synthesis of ent-kauranoid natural products,[37] Ding and co-workers sought to prepare structurally similar grayanoid natural products by focusing on establishing new strategies to access the bicyclo[3.2.1]octane moiety, which culminated in the first total syntheses of rhodomolleins XX and XXII.[38]
Figure 3.
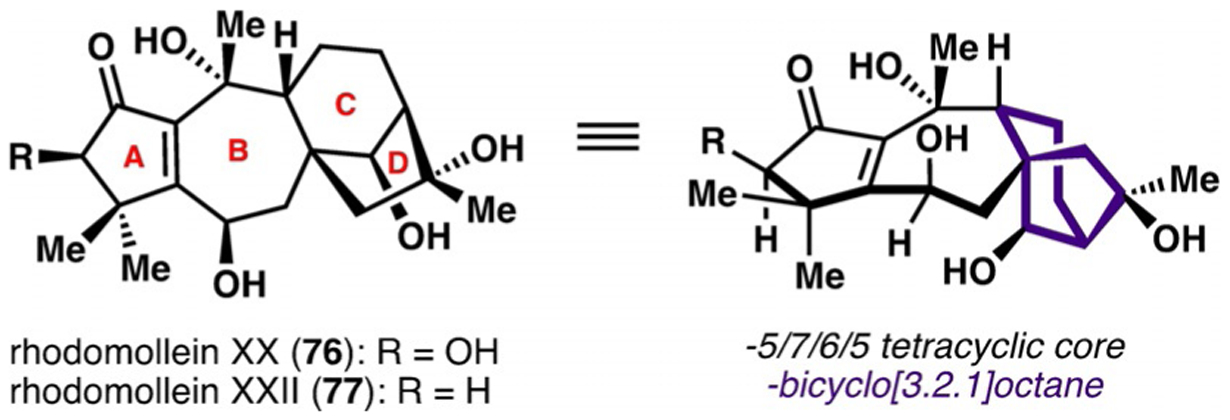
Rhodomollein XX and rhodomollein XXII.
Ding and co-workers envisioned obtaining rhodomollein XX and XXII in a divergent fashion from intermediate 78 through late-stage manipulations of the core framework (Scheme 11). The [3.2.1] bicycle containing 78 was anticipated to arise from epoxide 81 through a key TiIII-mediated Beckwith-Dowd rearrangement that would forge the [3.2.1] bicycle. In the forward sense, this process would begin with a TiIII-mediated epoxide ring opening to generate radical intermediate 80. Radical cyclization to the proximal carbonyl group to yield alkoxy radical 79 followed by C−C bond cleavage would then forge the [3.2.1] bicycle. Overall, this process would effect a rearrangement of the [2.2.2] bicycle to a [3.2.1] bicycle, which represents a substantial reorganization of the rhodomollein core framework. Finally, epoxide 81 would be obtained from phenol 82, which in the forward sense would undergo a tandem oxidative dearomatization/Diels-Alder sequence to build the [2.2.2] bicycle of 81.
Scheme 11.

Retrosynthesis of rhodomollein XX and XXII.
The total synthesis (Scheme 12) began with treatment of 83 with n-butyl vinyl ether and Hg(OAc)2, which promoted formation of a vinyl ether intermediate that underwent Claisen rearrangement through a Roskamp reaction to form β-ketoester 84. Diazo formation with a subsequent Cu-catalyzed cyclopropanation furnished cyclopropane 85, which then underwent Noyori ketalization[39] with concomitant ring opening of the cyclopropane to give ketal 86. Epoxidation of the benzylic olefin with NBS/H2O followed by a hydrogenative epoxide opening and Horner-Wadsworth-Emmons olefination with an in situ generated phosphonate ester provided enone 87. Cleavage of the triisopropylsilyl (TIPS) group unveiled the phenolic hydroxy group that, upon treatment with phenyliodine(III) diacetate (PIDA), underwent an oxidative dearomatization/Diels-Alder cascade to give the [2.2.2] bicycle as a mixture of diastereomers (88 + 89), with the desired bicycle 89 as the major diastereomer. Demethoxylation with SmI2 followed by epoxidation with dimethyldioxirane (DMDO) afforded the precursor, epoxide 81, for the key step. Subjecting this epoxide to Cp2TiCl2 and Mn proved effective for the key Ti-mediated cascade reaction, with the reaction affording a mixture of 90 and the desired [3.2.1] bicycle product 78, which was subsequently acetylated to give 91. Olefination with subsequent deacetylation provided triol 92, which was converted into 93 through an α-selenide elimination followed by a Mukaiyama hydration of the exo-methylene group. Protection of the ketone on the left-hand side as a silyl enol ether, methyl Grignard addition to the central ketone, and hydroxylation of the C1-position with MeReO3 afforded 95, which was elaborated to rhodomollein XX (76) by cleavage of the silyl ether. Likewise, rhodomollein XXII (77) was obtained from 94 after cleavage of the silyl enol ether and silyl ether with 2n HCl. Through their total synthesis of the rhodomollein natural products, Ding and co-workers effectively demonstrated the power of pairing robust ring-forming processes, such as the tandem oxidative dearomatization/Diels-Alder, with strategic C−C bond-cleavage events, such as the TiIII-mediated radical cascade, which can provide new opportunities for topological re-organization and enable efficient transition between complex bicyclic scaffolds. Notably, although the key C−C bond cleavage step in the total syntheses terminated with reduction of the C12-position, Ding and co-workers demonstrated through further studies that treatment of cyclopropanol 96 with VO(acac)3 could afford the C−C bond-cleavage product and generate 97 after a subsequent C12 hydroxylation, thus potentially providing access to the C12 oxygenated congeners of the grayanoid family of natural products (Scheme 13).
Scheme 12.

Total synthesis of rhodomollein XX and XXII. acac = acetylacetonato, DMAP = 4-(dimethylamino)pyridine, LDA = lithium diisopropyl-amide, PPTS = pyridinium p-toluenesulfonate, pyr = pyridine, TMS = trimethylsilyl, TsOH = para-toluenesulfonic acid.
Scheme 13.

C−C bond cleavage with subsequent C12 functionalization.
2.6. (±)-GB22 (Shenvi, 2019)
The bark of Galbulimima belgraveana and G. baccata exhibits hallucinogenic effects when chewed, and has been used in traditional medicine in Papua New Guinea, Malaysia, and Northern Australia, where these tropical flowering plants are found.[40] However, the constituent natural products present in the bark—collectively named the GB alkaloids—have not been extensively tested for their bioactivity, in part because their complex structures have made their chemical syntheses challenging, limiting the supply of material for biological study. For example, the most concise syntheses of GB alkaloid GB13 (98) required 18/19 steps;[41] the related compound GB22 (99) had not been synthesized prior to 2019 (Scheme 14). Here, we discuss the synthesis of GB22 by Shenvi and co-workers, who utilized a key Ir/Ni dual-catalytic siloxycyclopropane C−C bond cleavage/arylation that enabled access to GB22 in six steps in the longest linear sequence.[42]
Scheme 14.

GB alkaloids GB13 and GB22 as well as the original retrosynthesis of GB22 by Shenvi and co-workers.
Shenvi and co-workers originally envisioned that GB22 could arise through an intramolecular Friedel-Crafts addition to the phenol and ketone groups of 100 followed by diastereoselective hydrogenation of the pyridine ring; 100 in turn could be constructed by a Friedel-Crafts addition between phenol 101 and quinoline 102. It was hypothesized that quinoline 102 might yield an α,β-unsaturated oxocarbenium ion under acidic conditions to provide a reactive site for conjugate addition. However, all attempts to effect this Friedel-Crafts addition under Brønsted or Lewis acidic conditions failed, presumably because of the preferential Lewis acid coordination/protonation, and hence deactivation, of 101 over 102. As a result, Shenvi and co-workers turned to an alternative method for forging this first C−C bond by exploiting C−C bond cleavage.
To prepare a precursor substrate for C−C bond cleavage, commercially available ketone 103 was first converted into the silyl enol ether; a Simmons-Smith cyclopropanation then delivered siloxycyclopropane 104 (Scheme 15). It was discovered that single-electron oxidation of siloxycyclopropane 104 under iridium photocatalysis yielded radical cation 106, which prompted C−C bond cleavage to deliver radical intermediate 107. Nickel-catalyzed cross-coupling with bromoarene 105 (available in two steps from 1-naphthol) then afforded 108. Notably, this endo-selective siloxycyclopropane ring opening can be contrasted with the exo-selective cyclopropanol ring opening typically observed under two-electron Pd catalysis, thereby highlighting the complementarity of transition-metal reactivity in two-electron and one-electron reactions. In addition, these siloxycyclopropane arylation conditions could be successfully implemented with a variety of haloarene and haloalkene coupling partners.
Scheme 15.
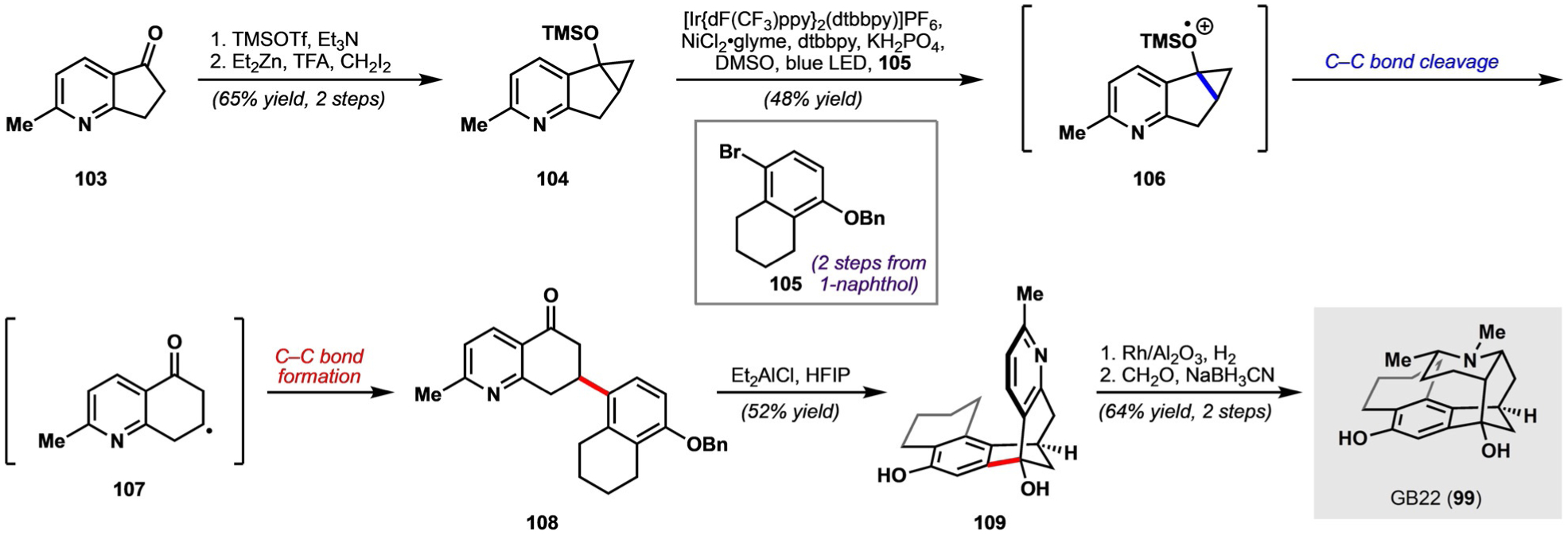
Synthesis of GB22 according to Shenvi and co-workers. dF(CF3)ppy = 3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl]phenyl, dtbbpy = 4,4’-di-tert-butyl-2,2’-dipyridyl.
To forge the second C−C bond (i.e. 108→109), several Lewis and Brønsted acids were screened but only resulted in retro-Friedel-Crafts additions. Ultimately, it was discovered that the combination of Et2AlCl and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was uniquely capable of effecting the desired Friedel-Crafts addition. It was hypothesized that a complex such as Al[OCH(CF3)2]2Cl may be formed in situ, which could serve as a hydrogen-bonding catalyst or a strong Lewis acid. After diastereoselective hydrogenation of the pyridine ring from the convex face and reductive amination of formaldehyde, the total synthesis of (±)-GB22 was completed in six steps. The brevity of the synthesis was enabled by a transition-metal-mediated C−C bond cleavage/C−C bond formation, where the formed C−C bond was otherwise difficult to install using more traditional methods.
3. Cleavage of Peripheral C−C single Bonds in Total Synthesis
In contrast to the cleavage of core C−C single bonds, the cleavage of “peripheral” C−C single bonds, as defined here, takes place outside the central ring(s) of a synthetic intermediate, with subsequent functionalizations potentially leading to an array of diverse products (Scheme 16). As a result, the cleavage of peripheral C−C single bonds does not inherently transform the core architecture of an intermediate and, therefore, does not tend to be employed in counterintuitive retrosynthetic strategies. In fact, the cleavage and functionalization of peripheral C−C bonds tends to replicate processes that are achievable with traditional reactive functional groups (e.g. a C−X bond, where X is a halogen). Nevertheless, the cleavage of peripheral C−C single bonds can present new retrosynthetic opportunities by 1) expanding the pool of starting materials to include building blocks with extra carbon atoms and 2) enabling the use of synthetic strategies and tactics that may not have been feasible with more traditional reactive functional groups, either as a result of synthetic inaccessibility or the instability of intermediates containing these moieties. The following gives discussions of recent total syntheses that highlight the utility of cleaving peripheral C−C single bonds as a strategic transformation.
Scheme 16.

Conceptual examples of the cleavage and functionalization of peripheral C−C bonds.
3.1. (−)-Aspewentins A, B, and C (Grubbs and Stoltz, 2015)
Norditerpenoid natural products (+)-aspewentins A (110), B (111), and C (112) were first isolated from marine-derived fungus Aspergillus wentii in 2014 and exhibit potent growth inhibition activities against marine plankton species (Scheme 17A).[43] Aspewentin B, in particular, possesses a quaternary stereocenter bearing a vinyl group α to a carbonyl moiety, a structural motif that has been challenging for synthetic chemists to access enantioselectively. Indeed, even simple cyclohexanone 113 was unknown in the literature as a single enantiomer prior to 2015. To address this gap in synthetic methods, Stoltz and co-workers developed a strategy for accessing α-vinylcarbonyl compounds bearing an α-quaternary stereocenter by Pd-catalyzed decarbonylative dehydration.[44,45] Specifically, carboxylic acids such as 117 could be obtained as a single enantiomer by the enantioselective Michael addition of α,α-disubstituted enolates derived from carbonyl compounds 114 to acrylate acceptors 115; they could alternatively be accessed by the enantioselective Pd-catalyzed allylic alkylation of β-ketoesters 116 (Scheme 17B). A Pd-catalyzed decarbonylative dehydration of carboxylic acids 117 then afforded enantiopure α-vinylcarbonyl compounds 121 with an α-quaternary center; this process presumably occurs through the in situ formation of mixed anhydrides 118, followed by oxidative addition (→119), decarbonylation (→120), and β-hydride elimination. Stoltz and co-workers were able to implement this strategy on a variety of simple substrates to obtain the corresponding α-vinylcarbonyl products with an α-quaternary center.
Scheme 17.

A) (+)-Aspewentins A–C and cyclohexanone 113, and B) the strategy for accessing enantiopure α-vinylcarbonyl compounds with an α-quaternary center by Pd-catalyzed decarboxylative dehydration.
To further highlight the utility of this strategy, Stoltz and co-workers turned to the total synthesis of (−)-aspewentin B.[44] The cuprate derived from aryl bromide 122 was alkylated with 4-iodobutyrate; subsequent α-methylation afforded 123 (Scheme 18). Saponification and acid-mediated cyclization then delivered tricyclic ketone 124. Formation of the lithium enolate of 124 was followed by trapping with allyl chloroformate to yield enol carbonate 125. At this point, the aforementioned strategy for accessing α-vinylcarbonyl compounds was implemented. First, the Pd-catalyzed decarboxylative allylic alkylation of 125 with chiral ligand (S)-t-Bu-PHOX afforded α-quaternary carbonyl compound 126 with 94% ee. Hydroboration/oxidation then transformed the terminal alkene to a primary hydroxy group, which was further oxidized to deliver carboxylic acid 127. Finally, the key Pd-catalyzed decarbonylative dehydration yielded an α-vinyl-carbonyl compound through cleavage of a peripheral C−C bond, which was followed by cleavage of the phenyl methyl ether group to complete the synthesis of (−)-aspewentin B (128). Reduction of the benzylic carbonyl group of (−)-aspewentin B provided (−)-aspewentin A (129), which, in turn, could be transformed to (−)-aspewentin C (130) by oxidative dearomatization. The syntheses of (−)-aspewentins A–C highlight the ability of the cleavage/functionalization of peripheral C−C bonds to enable processes—namely, the enantiocontrolled formation of a quaternary center bearing a vinyl group α to a carbonyl moiety—that had otherwise been synthetically challenging.
Scheme 18.

Syntheses of (−)-aspewentins A–C according to Stoltz and co-workers. PHOX = phosphinooxazolines, Bz2O = benzoic anhydride, cap = caprolactamate, nbd = norbornadiene, DIBALH = diisobutylaluminum hydride, TBHP = tert-butyl hydroperoxide, TEMPO = 2,2,6,6-tetra-methylpiperidinyloxyl.
3.2. (−)-Chromodorolide B (Overman, 2018)
The chromodorolides are diterpenoid natural products originally isolated from nudibranchs in the genus Chromodoris and are hypothesized to originate from the sponges consumed by these nudibranchs.[46] Their structural analogy to other spongian diterpenes with potent effects on the Golgi apparatus suggests that the chromodorolides may also possess Golgi-modifying activity.[47] In particular, the structure of (−)-chromodorolide B (131; Scheme 19) is characterized by a highly oxygenated 5/5/5 all-cis-fused tricyclic fragment linked to a second trans-hydrindane fragment; this complex pentacyclic skeleton and its associated ten contiguous stereocenters establish (−)-chromodorolide B as a highly challenging synthetic target. In this section, we discuss the synthesis of (−)-chromodorolide B by Overman and co-workers, who used a transition-metal-photocatalyzed decarboxylative radical addition/cyclization/fragmentation cascade for the highly effective construction of the cis-oxabicyclo[3.3.0]octane ring system of the chromodorolides.[48]
Scheme 19.

Retrosynthesis of (−)-chromodorolide B according to Overman and co-workers. Bn = benzyl, Phth = phthaloyl.
Overman and co-workers hypothesized that (−)-chromodorolide B could arise from lactonization and other functional group manipulations of hydrindene 132. Hydrindene 132 was, in turn, envisioned to arise from a decarboxylative radical addition/cyclization/fragmentation cascade, ultimately beginning from N-acyloxyphthalimide fragment 137 and known butenolide fragment 136. In the forward sense, this process was envisioned to proceed with formation of an α-oxy radical 135 followed by conjugate addition to 136 to yield intermediate 133; a final radical cyclization would complete the oxabicyclo[3.3.0] ring system. Notably, four stereocenters (at carbon atoms 8, 12, 13, and 14) would have to be set with high levels of selectivity for this retrosynthetic disconnection to be reasonable. In particular, bond rotation of intermediate 133 to rotamer 134 and subsequent cyclization could afford an undesired C8-epimer of 132. It should be mentioned that the proposal of such a cascade sequence was enabled by the existence of decarboxylative methods for generating an α-oxy radical, as alternative radical precursors (e.g. C12-halides) would likely be unstable. To complete the retrosynthesis, N-acyloxyphthalimide 137 could ultimately be constructed from the coupling of two fragments derived from commercially available enedione 138 and the tartaric acid derived diester 139, both available in enantioenriched form.
Overman and co-workers began with the preparation of trans-hydrindenyl iodide fragment 142 from (S)-enedione 138 in nine steps (Scheme 20). Protection of the cyclopentanone carbonyl group as an ethylene glycol acetal, 1,2-reduction of the cyclohexenone, and acylation of the resulting secondary hydroxy group yielded allylic carbonate 140. A Pd-catalyzed reductive transposition provided a trans-hydrindene; subsequent cyclopropanation of the trisubstituted alkene, hydrolysis of the acetal, hydrogenolysis of the cyclopropane, and oxidation of the resulting secondary alcohol afforded trans-hydrindanone 141. Finally, 141 was converted into vinyl iodide 142 in two steps by Barton iodination.[49] Notably, although vinyl iodide 142 had been reported before, the previous routes were relatively inefficient and did not easily scale.
Scheme 20.
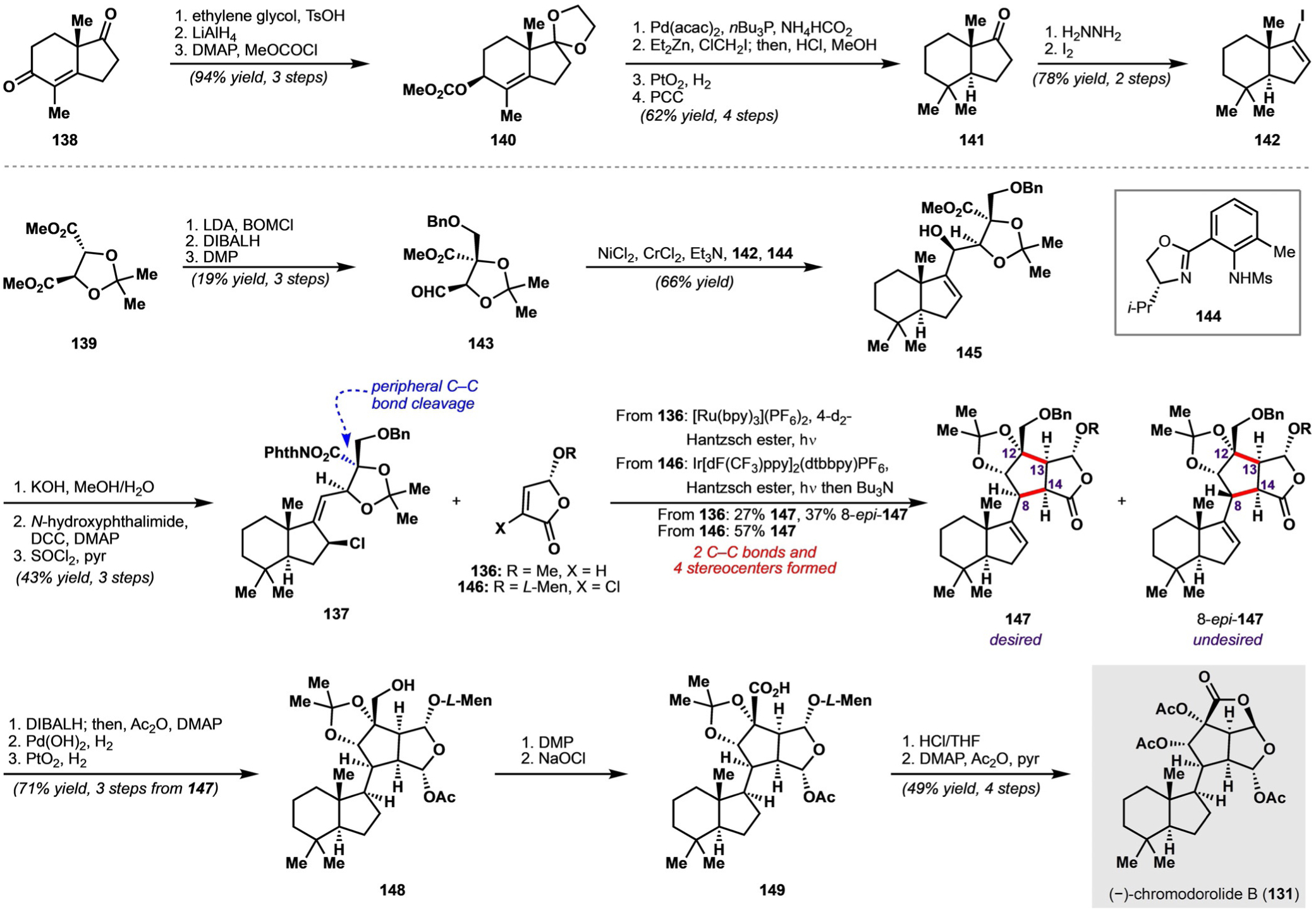
Synthesis of (−)-chromodorolide B according to Overman and co-workers. bpy = 2,2’-bipyridine, BOMCl = benzyloxymethyl chloride, L-Men = L-menthyl.
A second fragment, 143, was synthesized in three steps from tartrate-derived diester 139. Coupling of the two fragments (143 and 142) was then attempted. After some experimentation, it was discovered that Nozaki-Hiyama-Kishi coupling conditions with added oxazoline ligand 144 were optimal for the synthesis of allylic alcohol 145 as a single diastereomer; in contrast, similar reaction conditions without added ligand 144 or coupling conditions in which a vinyl-lithiate was generated resulted only in low to modest diastereoselectivity. Saponification of 145 and esterification of the resulting acid with N-hydroxyphthalimide provided an N-acyloxyphthalimide compound; allylic OH→Cl transposition then yielded allylic chloride 137.
Initial attempts at executing the key decarboxylative radical addition/cyclization/fragmentation sequence with N-acyloxyphthalimide 137 and butenolide 136 yielded a maximum of 27% of the desired product 147 along with 37% of its C8-epimer, with the latter arising from cyclization of conformer 134 (Scheme 20). A deuterated Hantzsch ester was used to disfavor premature quenching of the reactive α-acyl radical before the subsequent C−C bond formation; however, small quantities of a product resulting from this premature quenching were still isolated under the optimized conditions. Although the yield of the desired adduct (i.e. 147) was low, the favorable diastereoselectivity of 5.5:1 with respect to the stereocenters generated at C12, C13, and C14 suggested that the coupling could be rendered efficient if the process resulting in 8-epi-147 could be disfavored. Computational analysis of the transition states leading to 147 and 8-epi-147 led to a hypothesis that the addition of a chlorine substituent to the α position of the butenolide would favor the formation of 147 by enhancing steric interactions between this chlorine substituent and the allylic chlorine of fragment 137 in the transition state leading to 8-epi-147. This hypothesis was borne out experimentally, as the treatment of fragment 137 and chlorine-containing butenolide 146 under optimized photoredox-catalyzed conditions afforded desired product 147 exclusively in 57% yield, with dechlorination occurring in the same pot; notably, 8-epi-147 was not detected in the crude reaction mixture. Additionally, under these reaction conditions, replacing the Hantzsch ester with the deuterated Hantzsch ester was found to be unnecessary. Overall, the key addition/cyclization/fragmentation sequence, initiated by cleavage of a peripheral C−C bond through an iridium-photocatalyzed decarboxylation, resulted in the formation of two new C−C bonds and formed four stereocenters with high selectivity.
For the completion of the natural product, the lactone carbonyl group of 147 was reduced diastereoselectively to the aluminum hemiacetal, which was acetylated in situ; hydrogenolysis of the benzyl ether followed by diastereoselective reduction of the alkene delivered primary alcohol 148. Oxidation to the carboxylic acid over two steps afforded 149, after which treatment with HCl triggered formation of the lactone and cleavage of the acetonide. Finally, global acetylation completed the synthesis of (−)-chromodorolide B (131). The rapid generation of target-relevant complexity associated with the key addition/cyclization/fragmentation sequence in this synthesis exemplifies the power of radical reactivity, which may be facilitated by—or, in some situations, may even require—transition-metal-mediated C−C bond cleavage.
3.3. Pyrone Diterpenes (±)-Subglutinol A, (±)-Subglutinol B, (±)-Sesquicillin A, and (±)-Higginsianin A (Baran, 2018)
Pyrone diterpenes are a class of natural products that have become the subject of increasing study in recent years, as additional members of the subclass possessing interesting bioactivity continue to be discovered. Their bioactivity includes immunomodulatory, anti-infective, and cytotoxic effects.[50] In particular, subglutinol A (150), subglutinol B (151), and sesquicillin A (152) have been the targets of several previous synthetic efforts, with syntheses completed in 21–27 steps (Figure 4).[51] The related compound higginsianin A (153), on the other hand, had not been the subject of total synthetic efforts prior to 2018. Herein, we discuss the syntheses of these four pyrone diterpenes by Baran and co-workers, which were completed in 15–17 steps by relying on transition-metal-mediated cleavage of peripheral C−C bonds.[52]
Figure 4.

Pyrone diterpenes.
Previous syntheses of subglutinol A, subglutinol B, and sesquicillin A relied mainly on two-electron chemistry, requiring multiple protecting group, redox, and functional group manipulations to avoid functional group incompatibilities. In an attempt to avoid the high step counts associated with carrying out these manipulations, Baran and co-workers investigated one-electron reactions to assemble the pyrone diterpene skeletons. Specifically, they envisioned that the carbon skeletons of these natural products (conceptually represented as 154; Scheme 21) could arise from consecutive decarboxylative radical cross-coupling reactions between carboxylic acids (conceptually represented as 156) and simple coupling partners 155 and 157. The decalin system of compounds such as 156 could, in turn, arise from a radical polyene cyclization of linear β-ketoester 158.
Scheme 21.
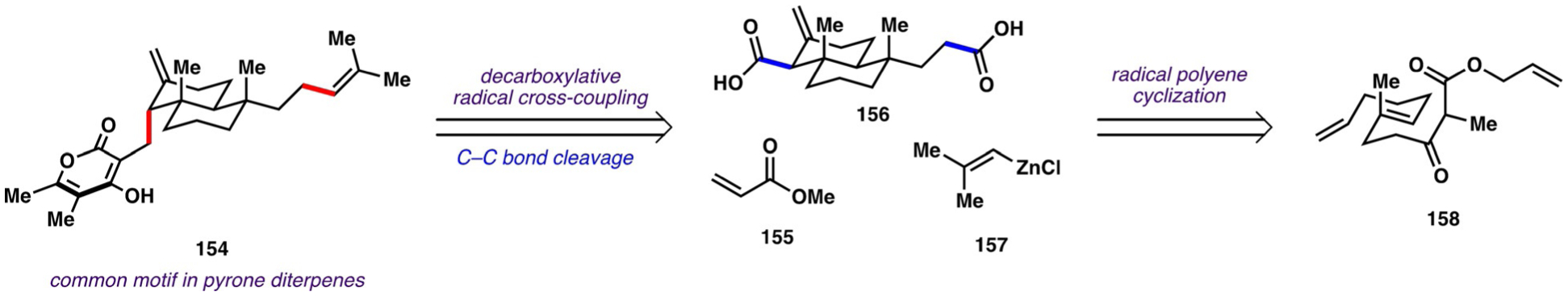
Strategy used by Baran and co-workers for the synthesis of pyrone diterpenes.
Baran and co-workers began with precursor 158 for the polyene cyclization, which was available in two steps from simple building blocks 159–161 (Scheme 22). Extensive optimization efforts culminated in an electrochemically assisted Mn-mediated radical polycyclization to furnish decalin system 162. Tsuji allylation constituted the first cleavage/formation of a peripheral C−C bond, which yielded intermediate 163; at this point, the synthetic route diverged toward the various pyrone diterpenes. For the syntheses of subglutinols A and B, reduction of the carbonyl group from the α-face was desired; this could be accomplished by treatment of intermediate 163 with LiAlH4. A vanadium-catalyzed hydroxy-directed epoxidation of the terminal alkene of 164 triggered closure of the THF ring to afford primary alcohol 165. Allylic oxidation and formation of a silyl ether provided 166, the alkene of which was converted into a carboxylic acid to deliver 167. A decarboxylative Giese reaction of 167 with methyl acrylate then yielded 169. Finally, cleavage of the silyl ether with pyridinium p-toluenesulfonate (PPTS), oxidation of the primary and secondary hydroxy groups by one oxidation state, and Takai-Lombardo olefination of both resulting carbonyl groups[53] afforded compound 170, thereby completing a formal synthesis of subglutinol A (5 steps from 170). Alternatively, from 169, cleavage of the silyl ether, oxidation of the primary hydroxy group to the carboxylic acid and the secondary hydroxy group to the ketone, and decarboxylative alkenylation with alkenylzinc species 168 resulted in diastereoselective installation of an isopropenyl group; Takai-Lombardo olefination then provided 171, thus completing the formal synthesis of subglutinol B (4 steps from 171).
Scheme 22.
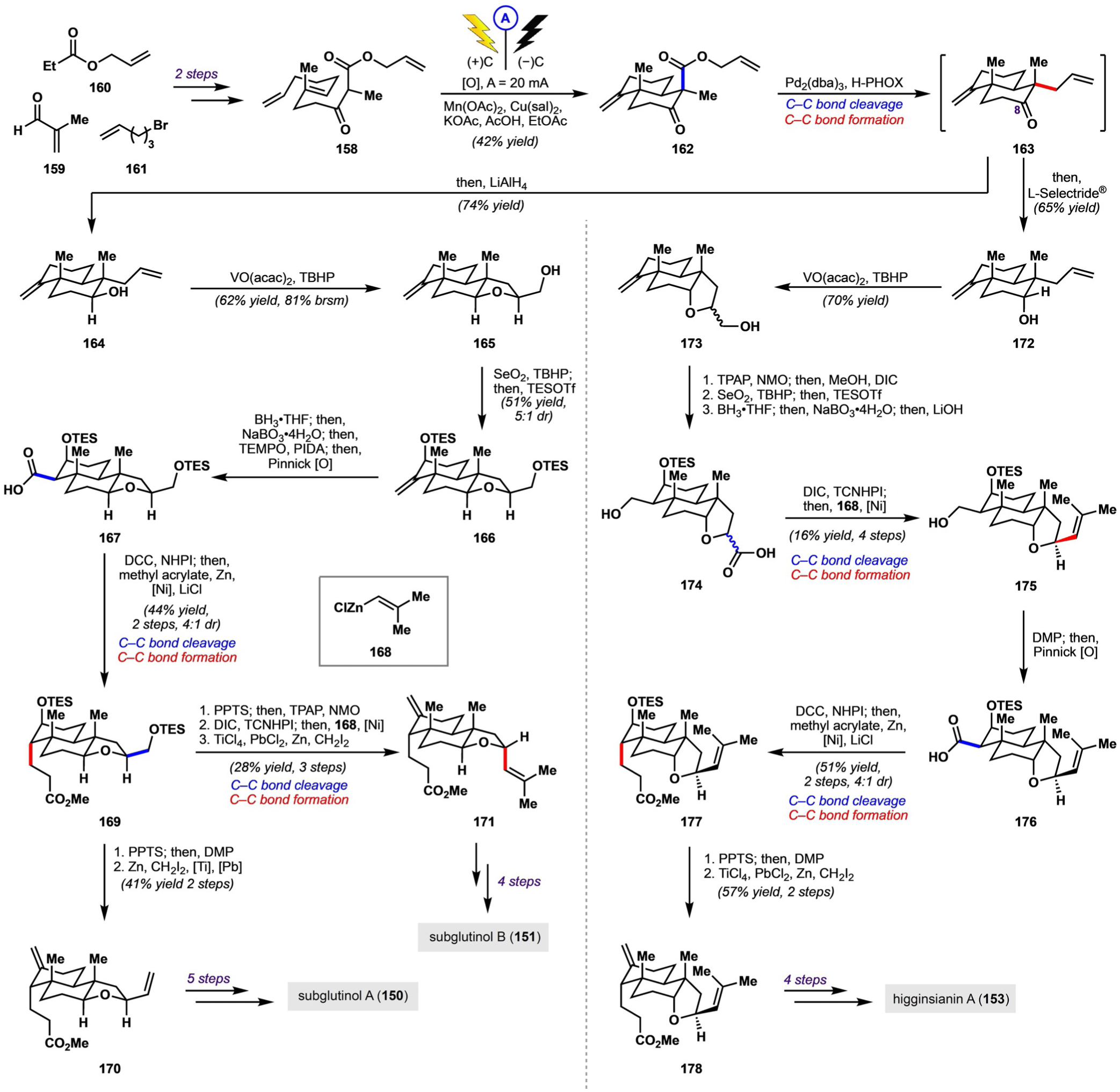
Synthesis of subglutinol A (150), subglutinol B (151), and higginsianin A (153) according to Baran and co-workers. dba = dibenzyl-ideneacetone, DIC = N,N’-diisopropylcarbodiimide, NHPI = N-hydroxyphthalimide, NMO = 4-methylmorpholine N-oxide, Cu(sal)2 = copper(II) 3,5-diisopropylsalicylate, TCNHPI = N-hydroxytetrachlorophthalimide, TES = triethylsilyl, TPAP = tetrapropylammonium perruthenate.
Compared to subglutinols A and B, higginsianin A is epimeric at C8; the desired stereoconfiguration for the synthesis of higginsianin A was established by reduction of intermediate 163 with L-Selectride® to give 172. A vanadium-catalyzed epoxidation and closure of the THF ring afforded primary alcohol 173 as a mixture of diastereomers; subsequent oxidation of the primary hydroxy group to the carboxylic acid was followed by methyl esterification. Allylic oxidation, triethylsilyl ether formation, hydroboration/oxidation, and saponification of the methyl ester then delivered carboxylic acid 174 as an inconsequential mixture of diastereomers. Diastereoselective installation of an isopropenyl group again could be accomplished by decarboxylative alkenylation with alkenylzinc species 168, which yielded primary alcohol 175. After oxidation to carboxylic acid 176, a decarboxylative Giese reaction with methyl acrylate afforded 177 as the major diastereomer. Cleavage of the silyl ether, oxidation of the resulting secondary hydroxy group to a ketone, and a Takai-Lombardo olefination delivered 178; in a four-step sequence analogous to that required for the syntheses of subglutinols A and B, 178 was transformed to higginsianin A (153).
Finally, sesquicillin A, which lacks the THF ring, was synthesized by using a similar strategy (Scheme 23). Decarboxylative Tsuji allylation of 162, reduction of the carbonyl group, and protection of the resulting hydroxy group as a TBS ether afforded compound 179. Hydroboration/oxidation, allylic oxidation, and formation of a triethylsilyl ether then delivered protected triol 181. Conversion of the alkene into a carboxylic acid provided 182, which underwent a decarboxylative Giese reaction to yield 183 as the major diastereomer. Cleavage of the silyl ether and global oxidation then afforded carboxylic acid 184, which underwent decarboxylative alkenylation with alkenylzinc species 168 to yield isopropenyl-containing compound 185. Finally, a Takai-Lombardo olefination provided 186, thereby completing the formal synthesis of sesquicillin A (6 steps from 186).
Scheme 23.

Synthesis of sesquicillin A (152) by Baran and co-workers. PHOX = phosphinooxazolines, Sia2BH = disiamylborane. (−)-effusin. HG-II = Hoveyda-Grubbs
In the four syntheses of these pyrone diterpenes by Baran and co-workers, eleven peripheral C−C bonds were cleaved/formed. In particular, the decarboxylative alkenylation events for the installation of isopropenyl groups deserve further comment, as they highlight the advantages of transition-metal-mediated C−C bond cleavage/functionalization: 1) many of the precursor carboxylic acids are located α to an ethereal oxygen atom, which renders alternative radical precursors (e.g. alkyl halides) potentially unstable; and 2) in comparison to decarboxylative Giese reactions, decarboxylative alkenylations take further advantage of the reactivity of transition metals, as the transition metals guide the C−C bond formation after re-engaging with the initially formed radical species. Overall, the four syntheses of pyrone diterpenes discussed here illustrate the power of retrosynthetic disconnections involving the cleavage/functionalization of peripheral C−C bonds.
3.4. (+)-Longirabdiol, (−)-Longirabdolactone, and (−)-Effusin (Li, 2019
The ent-kauranoid natural products, a family of diterpenoids isolated from the Isodon genus of flowering plants, exhibit a wide range of bioactivities, including antibacterial, antitumor, anti-inflammatory, and antifeedant effects.[54] Their biological activities, in addition to their intricate carbon skeletons, have made them the subject of intense synthetic interest over the years. However, syntheses of spirolactone ent-kauranoids (+)-longirabdiol (187; Scheme 24A),[55] (−)-longirabdolactone (188),[56] and (−)-effusin (189),[57] as well as their specific bioactivities, had not been reported prior to 2019. Here, we discuss the first syntheses of these natural products by Li and co-workers, who made use of decarboxylative tactics for the formation of key C−C bonds.[58] Specifically, Li and co-workers envisioned that the bicyclo[3.2.1]octane of the three natural products could be furnished through a vinyl radical cyclization from vinyl bromide 190; 190 could in turn arise from a decarboxylative Giese reaction between acid 191 and a suitable α,β-unsaturated ester radical acceptor 192. The carboxylic acid of 191 would be obtained from oxidative cleavage of alkene 193, with two carbon-carbon bonds forged in a novel tandem decarboxylative cyclization/alkenylation sequence from acid 195 via radical intermediate 194.
Scheme 24.
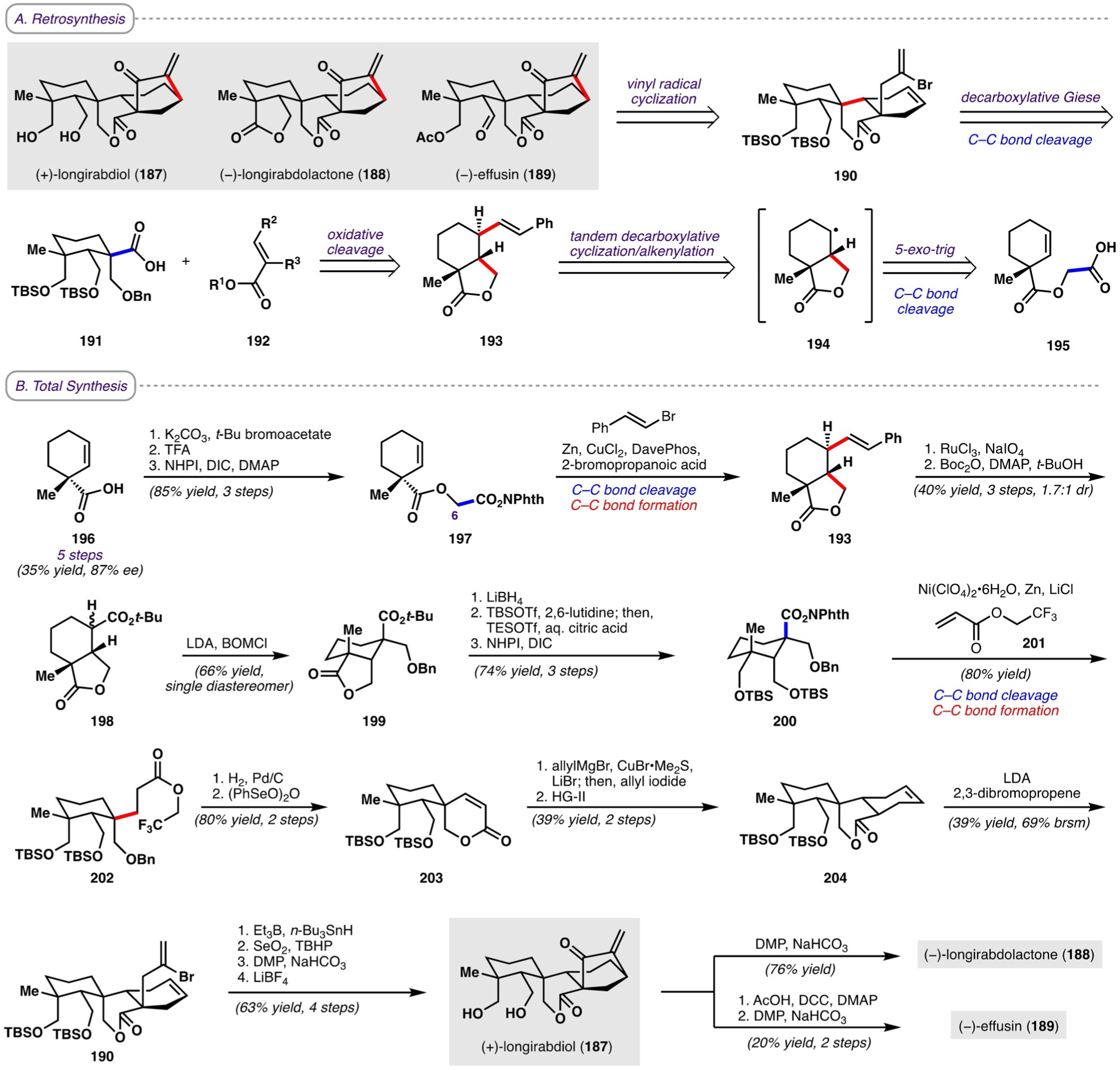
A) Retrosynthesis and B) total syntheses of (+)-longirabdiol, (−)-longirabdolactone, and generation). catalyst (2nd generation).
Li and co-workers began their synthesis with a known five-step sequence, in which commercially available ethyl 2-cyclohexanonecarboxylate was converted enantioselectively into carboxylic acid 196; in an additional three steps, N-acyloxyphthalimide 197 was synthesized, which serves as a precursor for the key tandem decarboxylative cyclization/alkenylation sequence (Scheme 24B). After extensive optimization, it was found that reductive cross-coupling conditions with CuCl2 as catalyst, DavePhos as ligand, and 2-bromopropanoic acid as additive were optimal for the decarboxylative cyclization/alkenylation with β-bromostyrene, delivering lactone 193 in 60% yield as a mixture of diastereomers (1H NMR analysis). These conditions were also found to be adequate for the syntheses of several other γ-lactones. Mechanistic studies indicated that both a radical addition/fragmentation pathway as well as a copper-catalyzed radical cross-coupling pathway may be operative. It should be noted that the construction of γ-lactones by Cγ–Cβ radical cyclization had not been previously achieved because of the relative rarity of alternative radical precursors (e.g. α-oxy halides); carboxylic acid derivatives are thus unique reagents in this regard. Oxidative cleavage of the alkene in 193 followed by esterification afforded 198 in 40% yield over three steps.
α-Alkylation of ester 198 with benzyloxymethyl chloride (BOMCl) provided benzyl ether 199 as a single diastereomer, which suggests that electrophile addition occurred exclusively from the α-face to avoid 1,3-diaxial interactions with the axial methyl group. As this diastereoselectivity would be undesired for the subsequent decarboxylative Giese reaction, efforts were taken to hinder the α-face. Thus, the lactone of 199 was reduced, and the resulting hydroxy groups were protected as bulky TBS ethers; esterification then yielded N-acyloxyphthalimide 200. Although efforts to install a tethered radical acceptor followed by effecting an intramolecular decarboxylative Giese reaction were unfruitful, an intermolecular decarboxylative Giese reaction with 201 successfully provided product 202 as a single diastereomer, thus validating the hypothesis that the bulky TBS ethers would drive radical addition from the β-face. Hydrogenolysis of the benzyl ether triggered spontaneous lactonization, after which oxidation with benzeneseleninic anhydride afforded unsaturated lactone 203.
Initial attempts to introduce an additional six-membered ring by a Diels-Alder cycloaddition with 1,3-butadiene were unsuccessful; therefore, a two-step alternative was performed instead, whereby conjugate addition of allyl cuprate followed by treatment with allyl iodide yielded a diolefin, and subsequent ring-closing metathesis delivered 204. α-Allylation with 2,3-dibromopropene provided vinyl bromide 190, which cyclized upon treatment with Et3B and n-Bu3SnH to furnish a bicyclo[3.2.1]octane. Sequential treatment with SeO2 and DMP afforded an allylic ketone intermediate, which after cleavage of the silyl ether yielded (+)-longirabdiol. (−)-Longirabdolactone and (−)-effusin could be accessed from (+)-longirabdiol in one and two steps, respectively. The completion of the first total syntheses of these natural products, enabled by peripheral decarboxylative C−C bond cleavage/C−C bond formation tactics, provided material for biological study. Each of the three natural products was found to exhibit low micromolar activity in cell viability assays against ten different cancer cell lines.
4. Summary and Outlook
The syntheses described above highlight the retrosynthetic disconnections that are uniquely enabled by the transition-metal-mediated cleavage of C−C single bonds. In several cases, synthetic strategies based on C−C bond cleavage resulted in shorter synthetic routes than had been accomplished thus far with more traditional strategies, often as a result of facilitating the formation of organometallic or radical intermediates that would otherwise be difficult to access. Despite the potential advantages of incorporating C−C bond-cleavage tactics into synthetic strategies, it remains a relatively rare phenomenon in total synthesis, in part because of the paucity of methods for transition-metal-mediated C−C bond cleavage/functionalization relative to those that are purely for bond construction.
In recent years, however, there has been a profusion of C−C bond-cleavage methods that enable bond constructions beyond those employed in the syntheses covered in this Review and offer new opportunities for innovative total syntheses. One area ripe for innovation is the cleavage of C−C bonds of unstrained rings, for which a number of effective methods have been published recently[59] but whose use in total synthesis has so far been limited (the synthesis of (−)-lingzhiol described above being the sole exception highlighted in this Review). In contrast to using approaches to cleave C−C bonds of strained rings, we anticipate that the cleavage of C−C bonds of unstrained rings in total synthesis would both enable myriad retrosynthetic opportunities—as unstrained rings are far more common than strained rings in simple chemical building blocks—as well as help further curtail the lengths of syntheses, given that synthetic steps often must be expended to construct strained rings before subsequent C−C bond cleavage. In addition, opportunities remain for advancing the use of peripheral C−C bond cleavage in total synthesis, moving beyond decarboxylative strategies; for example, the use of newly published methods for C−C bond cleavage on substrates at a lower oxidation state than the carboxylic acid (e.g. at the alcohol or aldehyde oxidation state)[60] promises to streamline syntheses that often involve multiple steps for functional group interconversions.
With the increasing abundance of methods for C−C bond cleavage, there is much reason to believe that the use of transition-metal-mediated C−C bond cleavage in total synthesis will only become more prominent in the future. The complexity of natural products generally makes them ideal “proving grounds” for synthetic strategies and tactics; therefore, the increasing utility of transition-metal-mediated cleavage of C−C single bonds in natural product synthesis may be a harbinger of its more widespread application in other synthetic fields. We expect this development, if it comes to pass, will have a welcome impact on drug discovery, materials synthesis, and chemical biology.
Acknowledgements
B.W. and M.A.P. thank the NSF for graduate research fellowships (DGE 1106400 and 1752814).
Biographies

Brian Wang received his B.A. in Chemistry from Cornell University in 2013. During his time as an undergraduate, he carried out research with Professor Hening Lin. He is currently a Ph.D. student in Chemistry under the supervision of Professor Richmond Sarpong at the University of California, Berkeley. His research interests include the total synthesis of natural products and other bioactive molecules.

Richmond Sarpong received his B.A. from Macalester College in 1995. During that time, he carried out research with Professor Rebecca Hoye. He completed his Ph.D. research with Professor Martin F. Semmel-hack at Princeton University in 2000. After three and a half years as a postdoctoral fellow with Professor Brian Stoltz at Caltech, he began his independent career at the University of California, Berkeley where he is at present Professor of Chemistry. His research interests include the development of new strategies for the synthesis of complex natural products including the diterpenoid alkaloids.

Melecio Perea received his B.S. in Chemistry from New Mexico Highlands University in 2016. During his time as an undergraduate, he conducted research with Professor Brooks Maki. He is currently a Ph.D. student in Chemistry at the University of California, Berkeley in Professor Richmond Sarpong’s group. His current research focuses on the development of strategies for the total synthesis of taxoid natural products.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Drahl MA, Manpadi M, Williams LJ, Angew. Chem. Int. Ed 2013, 52, 11222–11251; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 11430–11461. [Google Scholar]
- [2] a).Woodward RB, Doering WE, J. Am. Chem. Soc 1944, 66, 849; [Google Scholar]; b) Woodward RB, Doering WE, J. Am. Chem. Soc 1945, 67, 860–874. [Google Scholar]
- [3].Trost BM, Hu Y, Horne DB, J. Am. Chem. Soc 2007, 129, 11781–11790. [DOI] [PubMed] [Google Scholar]
- [4]. C−C bond cleavage followed by further radical reactivity can, of course, also be initiated without a transition metal. For examples of recent total syntheses employing C−C bond cleavage followed by radical reactivity without a transition metal, see.; a) Imamura Y, Yoshioka S, Nagatomo M, Inoue M, Angew. Chem. Int. Ed 2019, 58, 12159–12163; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 12287–12291; [Google Scholar]; b) Fujino H, Nagatomo M, Paudel A, Panthee S, Hamamoto H, Sekimizu K, Inoue M, Angew. Chem. Int. Ed 2017, 56, 11865–11869; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 12027–12031; [Google Scholar]; c) Crossley SWM, Tong G, Lambrecht MJ, Burdge HE, Shenvi R, J. Am. Chem. Soc 2019, 142, 11376–11381; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Renata H, Zhou Q, Dünstl G, Felding J, Merchant RR, Yeh C-H, Baran PS, J. Am. Chem. Soc 2015, 137, 1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Breitler S, Carreira EM, Angew. Chem. Int. Ed 2013, 52, 11168–11171; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 11375–11379. [Google Scholar]
- [6].Fumagalli G, Stanton S, Bower JF, Chem. Rev 2017, 117, 9404–9432. [DOI] [PubMed] [Google Scholar]
- [7].Chen P, Billett BA, Tsukamoto T, Dong G, ACS Catal. 2017, 7, 1340–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Marek I, Masarwa A, Delaye P-O, Leibeling M, Angew. Chem. Int. Ed 2015, 54, 414–429; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 424–439. [Google Scholar]
- [9].Souillart L, Cramer N, Chem. Rev 2015, 115, 9410–9464. [DOI] [PubMed] [Google Scholar]
- [10].Sivaguru P, Wang Z, Zanoni G, Bi X, Chem. Soc. Rev 2019, 48, 2615–2656. [DOI] [PubMed] [Google Scholar]
- [11].Nairoukh Z, Cormier M, Marek I, Nat. Rev. Chem 2017, 1, 1–17. [Google Scholar]
- [12].Morcillo SP, Angew. Chem. Int. Ed 2019, 58, 14044–14054; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 14182–14192. [Google Scholar]
- [13].Murakami M, Ishida N, Cleavage of Carbon-Carbon Single Bonds by Transition Metals, Wiley-VCH, Weinheim, 2015, pp. 253–272. [Google Scholar]
- [14].Sugano M, Sato A, Iijima Y, Oshima T, Furuya K, Kuwano H, Hata T, Hanzawa H, J. Am. Chem. Soc 1991, 113, 5463–5464. [Google Scholar]
- [15].Onuchic AC, Machado CML, Saito RF, Rios FJ, Jancar S, Chammas R, Mediat. Inflamm 2012, 175408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sahu RP, Harrison KA, Weyerbacher J, Murphy RC, Konger RL, Garrett JE, Chin-Sinex HJ, Johnston ME, Dynlacht JR, Mendonca M, McMullen K, Li G, Spandau DF, Travers JB, Oncotarget 2016, 7, 20788–20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kuroda Y, Nicacio KJ, da Silva-Jr IA, Leger PR, Chang S, Gubiani JR, Deflon VM, Nagashima N, Rode A, Blackford K, Ferreira AG, Sette LD, Williams DE, Andersen RJ, Jancar S, Berlinck RGS, Sarpong R, Nat. Chem 2018, 10, 938–945. [DOI] [PubMed] [Google Scholar]
- [18].Miyaoka H, Saka Y, Miura S, Yamada Y, Tetrahedron Lett. 1996, 37, 7107–7110. [Google Scholar]
- [19].Mohr PJ, Halcomb RL, J. Am. Chem. Soc 2003, 125, 1712–1713. [DOI] [PubMed] [Google Scholar]
- [20] a).Matsumura S, Maeda Y, Nishimura T, Uemura S, J. Am. Chem. Soc 2003, 125, 8862–8869; [DOI] [PubMed] [Google Scholar]; b) Nishimura T, Uemura S, J. Am. Chem. Soc 1999, 121, 11010–11011; [Google Scholar]; c) Nishimura T, Ohe K, Uemura S, J. Am. Chem. Soc 1999, 121, 2645–2646. [Google Scholar]
- [21] a).Seiser T, Cramer N, J. Am. Chem. Soc 2010, 132, 5340–5341; [DOI] [PubMed] [Google Scholar]; b) Seiser T, Cramer N, Angew. Chem. Int. Ed 2008, 47, 9294–9297; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 9435–9438. [Google Scholar]
- [22].Ishida N, Sawano S, Masuda Y, Murakami M, J. Am. Chem. Soc 2012, 134, 17502–17504. [DOI] [PubMed] [Google Scholar]
- [23] a).Masarwa A, Weber M, Sarpong R, J. Am. Chem. Soc 2015, 137, 6327–6334; [DOI] [PubMed] [Google Scholar]; b) Weber M, Owens K, Masarwa A, Sarpong R, Org. Lett 2015, 17, 5432–5435. [DOI] [PubMed] [Google Scholar]
- [24] a).Bermejo FA, Fernández Mateos A, Marcos Escribano A, Martín Lago R, Mateos Burón L, Rodríguez López M, Rubio González R, Tetrahedron 2006, 62, 8933–8942; [Google Scholar]; b) Martín-Rodríguez M, Galán-Fernández R, Marcos-Escribano A, Bermejo FA, J. Org. Chem 2009, 74, 1798–1801. [DOI] [PubMed] [Google Scholar]
- [25].Kerschgens I, Rovira AR, Sarpong R, J. Am. Chem. Soc 2018, 140, 9810–9813. [DOI] [PubMed] [Google Scholar]
- [26].Ye F, Zhu Z-D, Chen J-S, Li J, Gu Y-C, Zhu W-L, Li X-W, Guo Y-W, Org. Lett 2017, 19, 4183–4186. [DOI] [PubMed] [Google Scholar]
- [27] a).MacLeod RM, Lehmeyer JE, Cancer Res. 1973, 33, 849–855; [PubMed] [Google Scholar]; b) de Groot ANJA, van Dongen PWJ, Vree TB, Hekster YA, van Roosmalen J, Drugs 1998, 56, 523–535; [DOI] [PubMed] [Google Scholar]; c) Berde B, Stürmer E, in Ergot Alkaloids and Related Compounds (Eds.: Berde B, Schild HO), Springer, Berlin, Heidelberg, 1978, pp. 1–28; [Google Scholar]; d) Schardl CL, Panaccione DG, Tudzynski P, in The Alkaloids: Chemistry and Biology (Ed.: Cordell GA), Academic Press, San Diego, 2006, pp. 45–86; [DOI] [PubMed] [Google Scholar]; e) Schiff PL, Am. J. Pharm. Educ 2006, 70, 98; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) McCabe SR, Wipf P, Org. Biomol. Chem 2016, 14, 5894–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stauffacher D, Niklaus P, Tscherter H, Weber HP, Hofmann A, Tetrahedron 1969, 25, 5879–5887. [DOI] [PubMed] [Google Scholar]
- [29].Kçrber K, Song D, Rheinheimer J, Kaiser F, Dickhaut J, Narine A, Culbertson DL, Thompson S, Rieder J, Cycloclavine and Derivatives Thereof for Controlling Invertebrate Pests, WO2014096238A1, 2014. [Google Scholar]
- [30].Deng L, Chen M, Dong G, J. Am. Chem. Soc 2018, 140, 9652–9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu T, Dong G, Angew. Chem. Int. Ed 2012, 51, 7567–7571; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 7685–7689. [Google Scholar]
- [32].Xu T, Dong G, Angew. Chem. Int. Ed 2014, 53, 10733–10736; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 10909–10912. [Google Scholar]
- [33].Long R, Huang J, Shao W, Liu S, Lan Y, Gong J, Yang Z, Nat. Commun 2014, 5, 1–10. [DOI] [PubMed] [Google Scholar]
- [34].Yan Y-M, Ai J, Zhou L, Chung ACK, Li R, Nie J, Fang P, Wang X-L, Luo J, Hu Q, Hu F-F, Cheng Y-X, Org. Lett 2013, 15, 5488–5491. [DOI] [PubMed] [Google Scholar]
- [35] a).Li C-J, Liu H, Wang L-Q, Jin M-W, Chen S-N, Bao G-H, Qin G-W, Acta Chim. Sinica 2003, 61, 1153–1156; [Google Scholar]; b) Zhou S-Z, Yao S, Tang C, Ke C, Li L, Lin G, Ye Y, J. Nat. Prod 2014, 77, 1185–1192. [DOI] [PubMed] [Google Scholar]
- [36] a).Li Y, Liu Y-B, Yu S-S, Phytochem. Rev 2013, 12, 305–325; [Google Scholar]; b) Chen X-Q, Gao L-H, Li Y-P, Li H-M, Liu D, Liao X-L, Li R-T, J. Agric. Food Chem 2017, 65, 4456–4463. [DOI] [PubMed] [Google Scholar]
- [37].He C, Hu J, Wu Y, Ding H, J. Am. Chem. Soc 2017, 139, 6098–6101. [DOI] [PubMed] [Google Scholar]
- [38].Yu K, Yang Z-N, Liu C-H, Wu S-Q, Hong X, Zhao X-L, Ding H, Angew. Chem. Int. Ed 2019, 58, 8556–8560; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 8644–8648. [Google Scholar]
- [39].Noyori R, Murata S, Suzuki M, Tetrahedron 1981, 37, 3899–3910. [Google Scholar]
- [40].Rinner U, Lentsch C, Aichinger C, Synthesis 2010, 3763–3784. [Google Scholar]
- [41] a).Movassaghi M, Hunt DK, Tjandra M, J. Am. Chem. Soc 2006, 128, 8126–8127; [DOI] [PubMed] [Google Scholar]; b) Zi W, Yu S, Ma D, Angew. Chem. Int. Ed 2010, 49, 5887–5890; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2010, 122, 6023–6026; [Google Scholar]; c) Larson KK, Sarpong R, J. Am. Chem. Soc 2009, 131, 13244–13245. [DOI] [PubMed] [Google Scholar]
- [42].Burdge HE, Oguma T, Kawajiri T, Shenvi R, 2019, 10.26434/chemrxiv.8263415.v1. [DOI]
- [43].Miao F-P, Liang X-R, Liu X-H, Ji N-Y, J. Nat. Prod 2014, 77, 429–432. [DOI] [PubMed] [Google Scholar]
- [44].Liu Y, Virgil SC, Grubbs RH, Stoltz BM, Angew. Chem. Int. Ed 2015, 54, 11800–11803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 11966–11969. [Google Scholar]
- [45].Liu Y, Kim KE, Herbert MB, Fedorov A, Grubbs RH, Stoltz BM, Adv. Synth. Catal 2014, 356, 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46] a).Dumdei EJ, De Silva ED, Andersen RJ, Choudhary MI, Clardy J, J. Am. Chem. Soc 1989, 111, 2712–2713; [Google Scholar]; b) Morris SA, de Silva ED, Andersen RJ, Can. J. Chem 1991, 69, 768–771. [Google Scholar]
- [47].Schnermann MJ, Beaudry CM, Egorova AV, Polish-chuk RS, Sütterlin C, Overman LE, Proc. Natl. Acad. Sci. USA 2010, 107, 6158–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48] a).Tao DJ, Slutskyy Y, Muuronen M, Le A, Kohler P, Overman LE, J. Am. Chem. Soc 2018, 140, 3091–3102; [DOI] [PubMed] [Google Scholar]; b) Tao DJ, Slutskyy Y, Overman LE, J. Am. Chem. Soc 2016, 138, 2186–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Barton DHR, Bashiardes G, Fourrey J-L, Tetrahedron Lett. 1983, 24, 1605–1608. [Google Scholar]
- [50].Al-Khdhairawi AAQ, Cordell GA, Thomas NF, Nagojappa NBS, Weber J-FF, Org. Biomol. Chem 2019, 17, 8943–8957. [DOI] [PubMed] [Google Scholar]
- [51] a).Zhang F, Danishefsky SJ, Angew. Chem. Int. Ed 2002, 41, 1434–1437; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2002, 114, 1492–1495; [Google Scholar]; b) Katoh T, Oguchi T, Watanabe K, Abe H, Heterocycles 2010, 80, 229; [Google Scholar]; c) Kim H, Baker JB, Lee S-U, Park Y, Bolduc KL, Park H-B, Dickens MG, Lee D-S, Kim Y, Kim SH, Hong J, J. Am. Chem. Soc 2009, 131, 3192–3194; [DOI] [PubMed] [Google Scholar]; d) Kikuchi T, Mineta M, Ohtaka J, Matsumoto N, Katoh T, Eur. J. Org. Chem 2011, 5020–5030; [Google Scholar]; e) Kim H, Baker JB, Park Y, Park H-B, DeArmond PD, Kim SH, Fitzgerald MC, Lee D-S, Hong J, Chem. Asian J 2010, 5, 1902–1910. [DOI] [PubMed] [Google Scholar]
- [52].Merchant RR, Oberg KM, Lin Y, Novak AJE, Felding J, Baran PS, J. Am. Chem. Soc 2018, 140, 7462–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Takai K, Kakiuchi T, Kataoka Y, Utimoto K, J. Org. Chem 1994, 59, 2668–2670. [Google Scholar]
- [54].Sun H-D, Huang S-X, Han Q-B, Nat. Prod. Rep 2006, 23, 673–698. [DOI] [PubMed] [Google Scholar]
- [55].Kido M, Ichihara T, Otsuka H, Takeda Y, Chem. Pharm. Bull 1992, 40, 3324–3326. [Google Scholar]
- [56].Takeda Y, Ichihara T, Otsuka H, Kido M, Phytochemistry 1993, 33, 643–646. [Google Scholar]
- [57].Kubo I, Kamikawa T, Isobe T, Kubota T, J. Chem. Soc. Chem. Commun 1980, 1206–1207. [Google Scholar]
- [58].Zhang J, Li Z, Zhuo J, Cui Y, Han T, Li C, J. Am. Chem. Soc 2019, 141, 8372–8380. [DOI] [PubMed] [Google Scholar]
- [59] a).Roque JB, Kuroda Y, Gçttemann LT, Sarpong R, Science 2018, 361, 171–174; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roque JB, Kuroda Y, Gçttemann LT, Sarpong R, Nature 2018, 564, 244–248; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xia Y, Lu G, Liu P, Dong G, Nature 2016, 539, 546–550; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhao K, Yamashita K, Carpenter JE, Sherwood TC, Ewing WR, Cheng PTW, Knowles RR, J. Am. Chem. Soc 2019, 141, 8752–8757; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hu A, Chen Y, Guo J-J, Yu N, An Q, Zuo Z, J. Am. Chem. Soc 2018, 140, 13580–13585; [DOI] [PubMed] [Google Scholar]; f) Guo J-J, Hu A, Chen Y, Sun J, Tang H, Zuo Z, Angew. Chem. Int. Ed 2016, 55, 15319–15322; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 15545–15548. [Google Scholar]
- [60] a).Xu Y, Qi X, Zheng P, Berti CC, Liu P, Dong G, Nature 2019, 567, 373–378; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Murphy SK, Park J-W, Cruz FA, Dong VM, Science 2015, 347, 56–60; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wu X, Cruz FA, Lu A, Dong VM, J. Am. Chem. Soc 2018, 140, 10126–10130; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang K, Chang L, An Q, Wang X, Zuo Z, J. Am. Chem. Soc 2019, 141, 10556–10564. [DOI] [PubMed] [Google Scholar]