

Abstract
Epigenetic changes are one underlying cause for cancer development and often due to dysregulation of enzymes modifying DNA or histones. Most Jumonji C domain-containing (JMJD) proteins are histone lysine demethylases (KDMs) and therefore epigenetic regulators. One JMJD subfamily consists of JMJD1A/KDM3A, JMJD1B/KDM3B and JMJD1C/KDM3C that are roughly 50% identical at the amino acid level. All three JMJD1 proteins are capable of removing di- and monomethyl marks from lysine 9 on histone H3 and might also demethylate histone H4 on arginine 3 and non-histone proteins. Analysis of knockout mice revealed critical roles for JMJD1 proteins in fertility, obesity, metabolic syndrome and heart disease. Importantly, a plethora of studies demonstrated that especially JMJD1A and JMJD1C are overexpressed in various tumors, stimulate cancer cell proliferation and invasion, and facilitate efficient tumor growth. However, JMJD1A may also inhibit the formation of germ cell tumors. Likewise, JMJD1B appears to be a tumor suppressor in acute myeloid leukemia, but a tumor promoter in other cancers. Notably, by reducing methylation levels on histone H3 lysine 9, JMJD1 proteins can profoundly alter the transcriptome and thereby affect tumorigenesis, including through upregulating oncogenes such as CCND1, JUN and MYC. This epigenetic activity of JMJD1 proteins is sensitive to heavy metals, oncometabolites, oxygen and reactive oxygen species, whose levels are frequently altered within cancer cells. In conclusion, inhibition of JMJD1 enzymatic activity through small molecules is predicted to be beneficial in many different cancers, but not in the few malignancies where JMJD1 proteins apparently exert tumor suppressive functions.
Introduction
The seminal role of gene mutations in cancer development has been recognized for a long time, but more recent studies have revealed a similarly important role of epigenetic changes. Histone (de)methylation is intimately involved in the underlying chromatin alterations, and the family of Jumonji C (JmjC) domain-containing (JMJD) proteins is primarily responsible for histone lysine demethylation. In humans, JMJD proteins constitute a large protein family with more than 30 members, most of which are reportedly endowed with histone lysine demethylase activity and thus often referred to as KDMs (lysine demethylases). This lysine demethylation activity is a reflection of two consecutive chemical reactions: hydroxylation of the methylated ε-amino group followed by spontaneous release of formaldehyde, only the former being a truly JMJD dependent process (Fig. 1A). Consistent with their inherent hydroxylase activity, JMJD proteins have been shown to also hydroxylate amino acid residues, including aspartate, asparagine, histidine, unmethylated lysine or arginine, as well as tRNA (1, 2). For all these activities, JMJD proteins require the cofactors oxygen and 2-oxoglutarate (also known as α-ketoglutarate) while carbon dioxide and succinate are byproducts (Fig. 1A), which makes JMJD activity sensitive to the metabolic state of a cell.
Fig. 1.
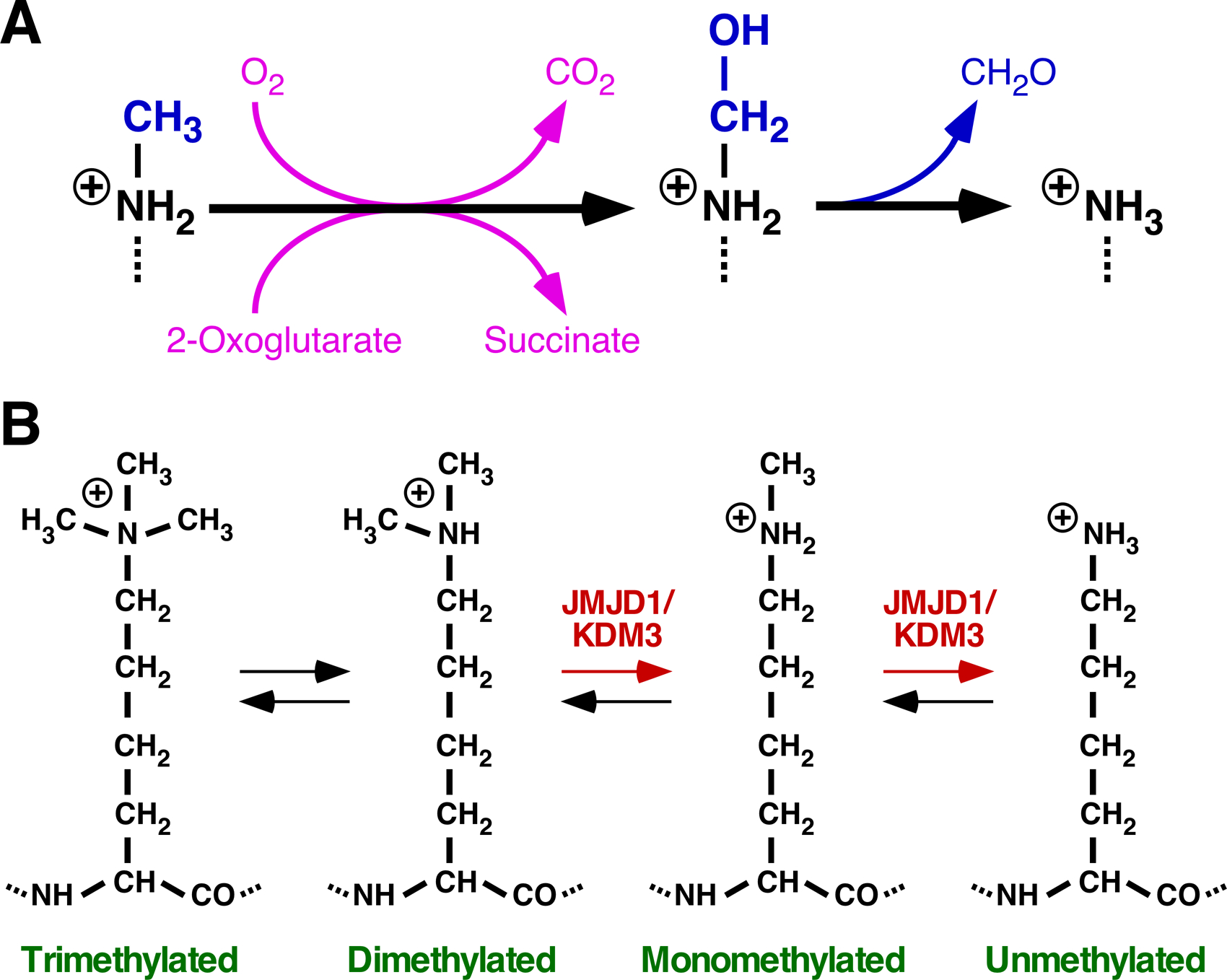
Lysine demethylation by JMJD1/KDM3 proteins. (A) First, hydroxylation of the methylated (in this case, monomethylated) ε-amino group occurs. The resultant hemiaminal is chemically labile, releasing formaldehyde (CH2O) and thereby leading to a demethylated ε-amino group. (B) Dynamic equilibrium between the unmethylated and methylated states of a lysine residue. JMJD1/KMD3 proteins catalyze the conversion of di- to monomethylated and of mono- to unmethylated lysine 9 on histone H3.
The JMJD protein family is quite heterogenous (Table 1). Its members can be subdivided by molecular weight into large (>100 kDa) and small (<100 kDa) proteins, by their specificity with regard to histone lysine demethylation, and by the presence of functional domains (1–3). One subfamily of JMJD proteins consists of three members, JMJD1A-C (or KDM3A-C). Two conserved domains have been identified in JMJD1A-C that are required for their catalytic activity (4–6): the JmjC catalytic center and a C6 zinc finger (Fig. 2), the latter one found in only one other JMJD protein, Hairless (HR). In addition, JMJD1 proteins possess a conserved LXXLL motif (Fig. 2), a protein-protein interaction motif originally found in coactivators mediating their binding to nuclear receptors, yet the role of this JMJD1 LXXLL motif has remained unresolved. JMJD1A/KDM3A is more closely related to JMJD1B/KDM3B (59.64% identity at the amino acid sequence level) than to JMJD1C/KDM3C (45.37% identity), and also JMJD1B shares less than half of its amino acids (47.88%) with JMJD1C, indicating that JMJD1C is evolutionarily most distant from the other two JMJD1 proteins. Although HR is very similar to JMJD1 proteins (Table 1), it is not regarded as an additional member of this subfamily, because HR is at most 33.47% identical to a JMJD1 protein and displays great variance even in the conserved functional domains as well as in the LXXLL motif (Supplementary Fig. S1).
Table 1.
Human JMJD proteins and their subfamilies.
| JMJD1A/KDM3A | >100 kDa | C6 Zn-finger | H3K9me2/1 |
| HR | >100 kDa | C6 Zn-finger | H3K9me2/1 |
| JMJD2A/KDM4A | >100 kDa | JmjN, PHD, Tudor | H3K9me3/2, H3K36me3/2 |
| JMJD2D/KDM4D | <100 kDa | JmjN | H3K9me3/2 |
| JARID1A/KDM5A | >100 kDa | JmjN, ARID, PHD, C5HC2 Zn-finger | H3K4me3/2 |
| JARID2/JMJ | >100 kDa | JmjN, ARID, C5HC2 Zn-finger | |
| KDM2A/FBXL11 | >100 kDa | CXXC Zn-finger, PHD, F-box, LRR | H3K36me2/1 |
| KDM2B/FBXL10 | H3K4me3, H3K36me2/1 | ||
| JMJD3/KDM6B | >100 kDa | H3K27me3/2 | |
| UTX/KDM6A | TPR | ||
| KDM7A/KIAA1718 | >100 kDa | PHD | H3K9me2, H3K27me2, H4K20me1 |
| KDM7B/PHF8 | H3K9me2/1, H3K27me2, H4K20me1 | ||
| KDM7C/PHF2 | H3K9me2 | ||
| HIF1AN/FIH | <100 kDa | ||
| HSPBAP1 | <100 kDa | ||
| JMJD4 | <100 kDa | ||
| JMJD5/KDM8 | <100 kDa | H3K36me2 | |
| JMJD6 | <100 kDa | ||
| JMJD7 | <100 kDa | ||
| JMJD8 | <100 kDa | ||
| NO66/JMJD9 | <100 kDa | H3K4me3/2/1, H3K36me3/2 | |
| MINA/MINA53/JMJD10 | <100 kDa | H3K9me3 | |
| TYW5 | <100 kDa |
ARID, AT-rich interaction domain; JmjN, Jumonji N domain; LRR, leucine-rich repeat; PHD, plant homeodomain; TPR, tetratricopeptide repeat domain.
Fig. 2.

Homology between the JMJD1/KMD3 proteins. (A) Scheme of the JMJD1/KMD3 proteins derived from the NCBI reference sequences for human JMJD1A (NP_060903.2), JMJD1B (NP_057688.3) and JMJD1C (NP_116165.1). However, there are many splice variants that can result into different length isoforms. (B) Alignment of the zinc fingers. Conserved cysteine residues are marked by asterisks, and amino acids conserved between all three proteins are highlighted in grey. (C) Alignment of the conserved LXXLL motifs spanning amino acids 885–889 in JMJD1A, 1293–1297 in JMJD1B and 2066–2070 in JMJD1C. The conserved leucine residues are marked by “#”. (D) Alignment of the JmjC domains (as defined by UniProt).
All three JMJD1 proteins have been established to utilize mono- and dimethylated lysine 9 on histone H3 (H3K9) as substrates in vitro and in vivo (Fig. 1B), whereas trimethylated H3K9 is not a substrate (4–6). But JMJD1 proteins can indirectly cause reduction of trimethylated H3K9 levels by depleting its precursor, dimethylated H3K9. No other methylated histone lysine residue bar H3K9 is targeted by JMJD1, and this limited specificity towards mono-/dimethylated H3K9 is another characteristic that sets JMJD1 enzymes apart from all other JMJD proteins except HR (Table 1). Given that tri- and dimethylated H3K9 are prominent marks of heterochromatin and associated with inactive genes (7), removal (indirectly and directly, respectively) of these histone marks by JMJD1 enzymes is expected to increase initiation of gene transcription. However, monomethylated H3K9 is frequently found at active genes (7), so its removal by JMJD1 proteins could potentially contribute to decreased transcription. As such, the net impact on gene transcription of JMJD1-mediated changes of the H3K9 methylation status cannot generally be predicted and may vary dependent on the gene promoter, cell type or physiological state of a cell.
JMJD1-mediated demethylation may not be restricted to H3K9. For instance, JMJD1A has been shown to demethylate in vitro peptides encompassing amino acids 1–15 of histone H3, in which lysine 9 was replaced by a monomethylated, asymmetrically dimethylated or symmetrically dimethylated arginine residue, suggesting that JMJD1A could also function as an arginine demethylase. However, multiple peptides with methylated arginine residues known to naturally exist in histone H3 or H4 were not targeted by JMJD1A, and no supporting in vivo evidence for an arginine demethylase activity has been supplied (8), raising doubts whether JMJD1A demethylates histone arginine residues under physiological conditions. But both in vitro and in vivo evidence strongly indicates that JMJD1B demethylates monomethylated and symmetrically dimethylated arginine 3 on histone H4 (9). Thus, JMJD1 enzymatic activity is likely to extend beyond the demethylation of H3K9.
The Normal: Upkeeping a Variety of Biological Functions
All three JMJD1 genes are widely expressed and their mRNA expression levels are mostly comparable (Fig. 3), although corresponding protein expression data are needed to better understand their tissue distribution. Regardless, this suggests JMJD1 proteins to be ubiquitously expressed and thus to potentially have pleiotropic functions in humans. However, no single JMJD1 protein appears to be essential, as first demonstrated with Jmjd1a knockout mice. But several abnormalities were observed: male Jmjd1a knockout mice were infertile, had smaller testes and defects in spermatogenesis, and often even displayed male-to-female sex reversal, which is probably due to the fact that JMJD1A-mediated removal of H3K9 methylation is needed for activation of the promoter of the Y-chromosomal Sry gene that encodes for the male sex determining factor (10, 11). In contrast, female Jmjd1a knockout mice were fertile. Male Jmjd1b−/− mice presented with greatly reduced fertility, which was caused by lower numbers of mature sperm, reduced sperm motility and impaired sexual behavior (12). Female Jmjd1b−/− mice had normally developed reproductive organs, yet they were essentially infertile due to multiple defects, including decreased ovulation capacity, fertilization rate, embryo implantation and survival. In addition, both male and female Jmjd1b knockout mice, although born at normal weight, were of smaller body size soon after birth and also frequently died before weaning (13); Jmjd1b−/− mice also presented with impaired hematopoiesis, including a reduction in red blood cell count and an increase in white blood cells and neutrophils (9). In contrast, Jmjd1c knockout mice displayed no change in body size and survival and females were reproductively normal. The same was true for young male Jmjd1c−/− mice, but fertility and testis size hugely decreased after three months of age, possibly due to increased apoptosis of germ cells (14). The common male infertility phenotype of Jmjd1 knockout mice may hint at overlapping functions, while the greatest severity of this phenotype observed upon Jmjd1a knockout may be a reflection of its expression being much higher in testes in comparison to Jmjd1b and Jmjd1c, as observed for human JMJD1 mRNA levels (Fig. 3). In support of JMJD1 redundancy, mice with knockout in any three, but not two, of the four Jmjd1a and Jmjd1b alleles died during embryogenesis (15).
Fig. 3.

JMJD1 mRNA expression in human tissues. Shown are the consensus datasets derived from the Human Protein Atlas (www.proteinatlas.org).
Other consequences of Jmjd1a knockout were the development of adult obesity and metabolic syndrome and a defective ability to respond to cold temperature. This involved defects in gene expression in both skeletal muscle and brown adipose tissue. In particular, loss of JMJD1A caused inhibition of expression of PPARα and UCP1, two important regulators of energy balance (16, 17). In addition, JMJD1A can form a complex with PPARγ in beige adipose cells, thereby reducing dimethylation of H3K9 and leading to the transcription of genes that mediate the response against chronic cold exposure (18). Similarly, JMJD1C may affect metabolism. For instance, phosphorylation of JMJD1C by mTOR strengthened its interaction with USF-1, a seminal transcription factor in lipogenesis. This was shown to reduce H3K9 methylation and increase promoter activity of many genes that become upregulated upon feeding, hinting at an important role for JMJD1C in normal lipogenesis (19). Likewise, JMJD1C depletion in murine 3T3-L1 cells increased dimethylation of H3K9 at the promoters of C/ebpα, C/ebpβ and Pparγ that should reduce the expression of these key adipogenic transcription factors. Consequently, JMJD1C depletion reduced accumulation of triglycerides and the size of lipid droplets upon adipogenesis induction in 3T3-L1 cells (20). Notably, Jmjd1c ablation in mouse livers also resulted into reduced lipogenesis and triglyceride levels and suppressed glucose intolerance and insulin resistance upon diet-induced obesity, suggesting that inhibition of JMJD1C may be beneficial for treating obesity-related maladies (19). This contrasts the predicted harmful inhibition of JMJD1A, as its catalytic inactivity should phenocopy the above described deleterious effects of its knockout on the metabolic state.
Analysis of the myocardium of patients with a hypertrophic heart revealed an upregulation of JMJD1A, suggesting a possible involvement of JMJD1A in the development of this disease. To prove so, both myocyte-selective transgenic and whole-body knockout Jmjd1a mice were analyzed and they displayed exacerbated or reduced, respectively, left ventricular hypertrophy and fibrosis of the heart in response to pressure overload. JMJD1A overexpression especially reactivated fetal genes that have been implicated in hypertrophic growth. Furthermore, JMJD1A bound to the promoter of the Timp1 gene, which encodes for an extracellular matrix protein and metallopeptidase inhibitor known to be involved in fibrosis, decreased dimethylation of H3K9 and thus induced Timp1 transcription. Importantly, knockdown of Timp1 in cardiomyocytes suppressed to a great extent the fibrotic effects of JMJD1A overexpression, indicating a seminal role of TIMP1 as a downstream effector of JMJD1A (21). This suggests that inhibition of JMJD1A may be helpful in the treatment of heart hypertrophy and fibrosis, and the same may apply for JMJD1C (22). In contrast, JMJD1A may be beneficial in the recovery from myocardial infarction. This was shown with a Jmjd1a−/− rat model, in which an abnormal macrophage polarization may explain the observed impaired cardiac repair process after infarction (23). Consistently, adenovirus-mediated delivery of JMJD1A to the heart mitigated the injuries associated with myocardial infarction (24).
JMJD1 proteins may also be important for normal intellectual development and behavior. For instance, truncating and missense mutations in JMJD1B were associated with intellectual disability and short stature (25), the latter reminiscent of the growth defects found in Jmjd1b−/− mice (13). In addition, JMJD1C mutations have been found in patients with intellectual disability and Rett syndrome that is phenotypically similar to autism (26). Also, a premature stop codon was identified in the JMJD1A gene and associated with developmental delay, autism and status epilepticus, but currently only one such patient has been identified (27). This indicates the need to screen for JMJD1 mutations in larger population studies. Experimental validation in cell culture and animal models is needed to substantiate that these JMJD1 mutations are underlying causes for the observed abnormalities.
OCT4, which is a crucial stem cell transcription factor, is capable of inducing Jmjd1a transcription in mouse embryonic stem cells. And JMJD1A downregulation induced differentiation of these cells, the first time showing the functional relevance of a JMJD1 protein in upkeeping a stem cell phenotype (28). However, another report showed that only combined loss of JMJD1A and JMJD1B compromised maintenance of embryonic stem cells (15). JMJD1A may additionally promote reactivation of OCT4 during reprogramming to pluripotency, but the underlying mechanism is not yet known (29). OCT4 is also capable of recruiting JMJD1C to oppose dimethylation of H3K9, and JMJD1C induces expression of both OCT4 and KLF4, another stem cell factor. Accordingly, JMJD1C was required for efficient somatic cell reprogramming and embryonic stem cell self-renewal (30, 31). This ability of JMJD1 proteins to promote stemness may also be relevant in cancer stem-like cells (32–34).
The Good: Keeping Tumors in Check
Aside from their roles in normal homeostasis and development, JMJD1 proteins appear to be sometimes involved in preventing the genesis of malignancies. Fittingly, analysis of human germ cell tumors (e.g., seminomas, embryonal carcinomas) revealed downregulation of JMJD1A at the mRNA and protein level. Notably, subcutaneous injection of Jmjd1a−/− compared to wild-type embryonic stem cells into nude mice led to larger tumors, but there was no growth difference between wild-type and Jmjd1a−/− embryonic stem cells in vitro. Rather, Jmjd1a knockout led to more microvessels in tumors. Jmjd1a knockout consistently induced changes in the expression of angiogenesis-related genes, especially downregulation of anti-angiogenic factors (35). This suggests that JMJD1A can repress angiogenesis and could thereby suppress germ cell tumorigenesis. Interestingly, germline missense mutations in JMJD1C were found to be enriched in intracranial germ cell tumors, but if these mutations inactivate JMJD1C function and thereby drive germ cell tumorigenesis is unknown (36).
JMJD1B was originally cloned as a candidate tumor suppressor gene located in the 5q31 chromosomal region that is often deleted in myelodysplasia and acute myeloid leukemia (AML). Accordingly, JMJD1B overexpression reduced clonogenic efficiency of a myelodysplasia or AML cell line with 5q deletion (37, 38). Analysis of AML patients revealed that JMJD1B is underexpressed and low mRNA levels correlate with an unfavorable prognosis (38, 39). In addition, JMJD1B downregulation promoted proliferation of NB4 cells, which express the PML-RARA fusion oncoprotein characteristic of the acute promyelocytic subtype of AML. Mechanistically, JMJD1B induced alterations of H3K9 methylation that were suggested to restrict chromatin accessibility for PML-RARA, thereby blunting the oncogenic potential of this transcription factor (39). However, this may seem counterintuitive, since JMJD1B activity is in general supposed to remove heterochromatin marks and thus make chromatin more accessible for transcription factors. JMJD1B also promoted PML-RARA degradation upon all-trans retinoic acid treatment of NB4 cells, thereby facilitating their differentiation into mature granulocytes (39). But one other study reported the conflicting observation of JMJD1B inhibiting the all-trans retinoic acid induced differentiation of the AML cell line, HL-60 (5). Altogether, a preponderance of data currently suggests that JMJD1B may be a tumor suppressor in AML.
The Bad: Contribution to Tumorigenesis
In contrast to the putative tumor suppressor role of JMJD1B, JMJD1C has been identified as a promoter of AML by stimulating cell survival and stem cell self-renewal. Different mechanisms may apply, including by serving as a coactivator of the RUNX1-RUNX1T1 oncogenic fusion transcription factor to reduce dimethylated H3K9 at respective target genes or by cooperating with the HOXA9 transcription factor (40–43). Hitherto, a role for JMJD1A in AML is unknown, but a tumor promoting function has been described for many solid tumors. This was for the first time in colorectal cancer, where JMJD1A is overexpressed at the mRNA and protein level and high expression correlated with reduced survival and increased metastasis. Downregulation of JMJD1A in human colorectal cancer cell lines was reported to reduce cell growth in vitro and tumor size in xenograft models, impair in vitro cell invasion and suppress metastasis in vivo. Moreover, Jmjd1a−/− mice were protected from chemically induced colon carcinogenesis. Mechanistically, JMJD1A was shown to bind to the β-catenin oncoprotein and coactivate β-catenin targets such as the MYC and CCND1 oncogenes through reducing dimethylation of H3K9 (34, 44–47). Other potential ways how JMJD1A may facilitate colorectal tumorigenesis include coactivating the STAT3 transcription factor, which can be activated by interleukin-6 and −11 and thereby contribute to an inflammatory response furthering cancer formation, and stimulating the expression of the Hippo pathway effector, YAP1 (48, 49). Although less data is available for JMJD1B and JMJD1C, this also points to a tumor promoting role of these homologs in the colon and rectum and they may perform overlapping functions with JMJD1A, indicating potential redundancy (34, 50). Bioinformatics revealed that the expression of all three JMJD1 genes is significantly correlated in colorectal adenocarcinomas and many other tumors (Supplementary Table S1). Although the reason for this is unknown, it indicates that high JMJD1A expression is likely concurrent with high JMJD1B/JMJD1C levels in colorectal adenocarcinomas. This implies that simultaneous inhibition of all three JMJD1 proteins is desirable to combat this malignancy.
Both JMJD1A and JMJD1C are capable of interacting with the androgen receptor and thereby promoting androgen dependent gene transcription (4, 51). JMJD1A mRNA and protein levels are upregulated in human prostate tumors, while downregulation of JMJD1A led to reduced prostate cancer cell proliferation, survival and tumorigenesis in xenografts. This could be due to impaired recruitment of the androgen receptor to and activation of the MYC oncogene or due to reduced binding of JMJD1A to the promoter of SNAIL that encodes an important epithelial-to-mesenchymal transition factor. Further, JMJD1A-mediated activation of MYC and SNAIL transcription was associated with H3K9 demethylation (52, 53). Another mechanism by which JMJD1A could promote prostate tumorigenesis involves its interaction with the HNRNPF splicing factor, which resulted into alternative splicing leading to the generation of the V7 variant of androgen receptor that is one underlying cause of castration resistance of prostate tumors (54). JMJD1B can also stimulate growth of castration resistant prostate cancer cells, but it apparently does not affect androgen signaling (55). Altogether, JMJD1 proteins have convincingly been shown to be promoters of prostate cancer.
In breast cancer, JMJD1A is overexpressed and was necessary for estrogen receptor function, although it remains unresolved whether these two proteins bind each other. JMJD1A downregulation in breast cancer cells impaired their ability for invasion as well as growth in vitro and in xenografts. Thus, JMJD1A is predicted to have oncogenic functions in the breast, and this may entail upregulation of the MYC, JUN and CCND1 oncogenes through reducing H3K9 dimethylation as well as repression of the TP53 tumor suppressor protein. The latter may be due to JMJD1A-mediated reduction of lysine 372 methylation on TP53, a methylation mark known to increase TP53 stability and nuclear localization. However, compelling experiments are lacking to evaluate if JMJD1A brings this about through direct demethylation of TP53 (32, 56–58).
Apart from the above mentioned carcinomas, a variety of in vitro data with cancer cell lines, analysis of mouse models and/or overexpression in human tumor samples strongly implicated enzymatically active JMJD1A as a tumor promoter in many other cancers, including bladder cancer (59, 60), hepatocellular carcinoma (61, 62), non-small cell lung carcinoma (63), ovarian cancer (33), cervical cancer (64) and pancreatic ductal adenocarcinoma (65). JMJD1A may additionally function to drive Ewing sarcoma development (66) and is required for multiple myeloma cell survival (67). Also, JMJD1C is overexpressed in esophageal cancer and needed for efficient cell proliferation (68), and the same applies for JMJD1B and hepatocellular carcinoma (69). As such, there is a wealth of evidence to suggest that especially JMJD1A performs oncogenic functions in many different cancers through modulation of H3K9 methylation. It is noteworthy that this epigenetic function of JMJD1A and JMJD1C can be regulated through phosphorylation (19, 48, 70), indicating that altered signaling and kinase activities in tumor cells might profoundly affect the oncogenic potential of JMJD1 proteins.
Gain-of-function mutations are often a hallmark of an oncoprotein. Analysis of JMJD1 gene alterations revealed a low mutation and amplification frequency in most tumor types (Supplementary Fig. S2) and no clustering of mutations was observable in the catalytic center or elsewhere throughout the JMJD1 proteins (Supplementary Fig. S3). This argues that the tumor-driving function of JMJD1 proteins will rarely be linked to mutational events, but more likely associated with upregulation of JMJD1 transcription or stabilization/activation of JMJD1 proteins. However, this needs further investigation, including the experimental analysis whether any of the observed JMJD1 mutations lead to a gain of function (or a loss of function in cases where JMJD1 proteins act as tumor suppressors).
Hypoxia and JMJD1 Activity
One difference between a solid tumor and corresponding normal tissue is that the effective oxygen concentration is mostly lower in tumors. A key oxygen sensing system that lets tumors cope with hypoxia comprises the hypoxia-inducible factor (HIF), a transcriptional regulator consisting of an α and β subunit. The α subunit is exquisitely sensitive to oxygen as it is stabilized under hypoxia, which leads to the expression of a hypoxia-induced gene expression program (71). Notably, JMJD1A is a hypoxia-inducible gene, and both HIF-1α and HIF-2α were shown to bind to and activate the JMJD1A gene promoter (44, 72). Moreover, JMJD1A bound to HIF-1α and functioned as a coactivator to facilitate upregulation of many hypoxia sensitive genes (44, 60), highlighting how JMJD1A could promote tumorigenesis by furthering the adaptation of tumors to hypoxia. The interaction of JMJD1A with HIF-1α also establishes a feed-forward loop whereby JMJD1A autoregulates its own expression under hypoxia. Accordingly, hypoxia may (partially) account for the upregulation of JMJD1A expression observed in many human tumors. No corresponding reports have been published to assess if JMJD1B and JMJD1C transcription are also regulated by HIF.
Hypoxia may also temper JMJD1A activity. This is due to the fact that oxygen is an essential cofactor for its catalytic activity (see Fig. 1A) and the corresponding Michaelis-Menten constant is quite high with 7.6% O2 (73). Given that the oxygen concentration is approximately between 0.5% and 2% in tumors versus 4–10% in normal tissue, this can lead to a substantial reduction of JMJD1A catalytic activity within the hypoxic environment of a tumor (Supplementary Fig. S4). More investigations are needed to define the actual impact of tumor hypoxia on the in vivo catalytic activity of JMJD1A or its homologs and how this influences tumor initiation and progression.
JMJD1 Modulation by (Onco)Metabolites
Another essential cofactor of JMJD1 proteins is 2-oxoglutarate, which is an integral part of the tricarboxylic acid cycle (Fig. 4). Therefore, its intracellular concentration will vary depending on the supply of nutrients and along which metabolic pathways a cell proceeds, which can also be influenced by oxygen availability. Cancer cells present with a myriad of changes in metabolism, one of which is aerobic glycolysis that can potentially reduce 2-oxoglutarate levels by blocking the utilization of pyruvate to generate acetyl-CoA, which is required to replenish the tricarboxylic acid cycle by producing citrate. But this could be counterbalanced by increased glutaminolysis, which can produce 2-oxoglutarate and is often enhanced in cancer cells. Interestingly, there is some evidence that 2-oxoglutarate could function as an anti-cancer agent (74). Not only JMJD1 proteins but many more enzymes, including other JMJD proteins or the epigenetic TET proteins involved in DNA demethylation, are dependent on 2-oxoglutarate. Hence, altering 2-oxoglutarate levels would not be a specific means to modulate JMJD1 enzymatic activity, and increased JMJD1 catalytic activity may counteract a potential anti-cancer effect of 2-oxoglutarate in many carcinomas where JMJD1 proteins exert pro-tumorigenic functions.
Fig. 4.

Inhibition of JMJD1 proteins by oncometabolites. Shown are the tricarboxylic acid cycle and the production of the oncometabolites succinate, fumarate and 2-hydroxyglutarate that may inhibit catalytic activity of JMJD1 proteins. Loss-of-function mutations in succinate dehydrogenase and fumarate hydratase lead to the accumulation of fumarate and succinate, while neomorphic gain-of-function mutations in isocitrate dehydrogenase cause the reduction of 2-oxoglutarate into 2-hydroxyglutarate.
Succinate is a byproduct of JMJD1 catalytic activity (see Fig. 1A) and it functions as a competitive end product inhibitor. Likewise, fumarate competes with 2-oxoglutarate in the catalytic center of many 2-oxoglutarate dependent enzymes and thereby inhibits their activity. Thus, loss-of-function mutations in either succinate dehydrogenase or fumarate hydratase will result into accumulation of succinate or fumarate (Fig. 4), and such mutations have been observed in a wide variety of tumors, including paraganglioma, pheochromocytoma, neuroblastoma, renal cell carcinoma, thyroid cancer and gastrointestinal stromal tumors. Succinate and fumarate have been labeled as oncometabolites, since their abnormally high levels drive tumorigenesis (75). Neomorphic mutations in isocitrate dehydrogenase have been observed in low-grade glioma, glioblastoma, cholangiocarcinoma, chondrosarcoma and AML and lead to the production of another oncometabolite, 2-hydroxyglutarate (Fig. 4), which can also compete with 2-oxoglutarate in the catalytic center of JMJD proteins (75). Altogether, this indicates that changes in succinate, fumarate and 2-hydroxyglutarate levels likely alter JMJD1 catalytic activity, but if this is relevant in any tumor type needs to be explored.
Inactivation through Losing the Grip on Iron
JMJD proteins complex Fe2+ within their JmjC domain and this Fe2+ ion is required for their catalytic activity (1). Iron levels are generally elevated within cancer cells (76), suggesting that this may indiscriminately increase JMJD enzymatic activity. However, cancer cells also often present with elevated levels of reactive oxygen species (ROS), which may lead to oxidation of Fe2+ to Fe3+ (77), thereby potentially decreasing JMJD activity (Fig. 5). Furthermore, ascorbate (vitamin C) has been shown to promote the catalytic activity of many JMJD proteins, including JMJD1A (4), probably by preventing the oxidation of Fe2+ through scavenging ROS. Ascorbate has also been suggested to have anti-cancer activity, and this may in part be by counteracting harmful epigenetic changes induced by dysregulation of JMJD and TET proteins (78). In this regard, ascorbate stimulated JMJD1A/B activity in mouse embryonic fibroblasts and thereby triggered the demethylation of H3K9 (79), establishing that JMJD1 activity can potentially be regulated by ascorbate. However, more data is needed to discern how iron, ROS and ascorbate levels modulate JMJD1 proteins and whether this impacts tumorigenesis.
Fig. 5.
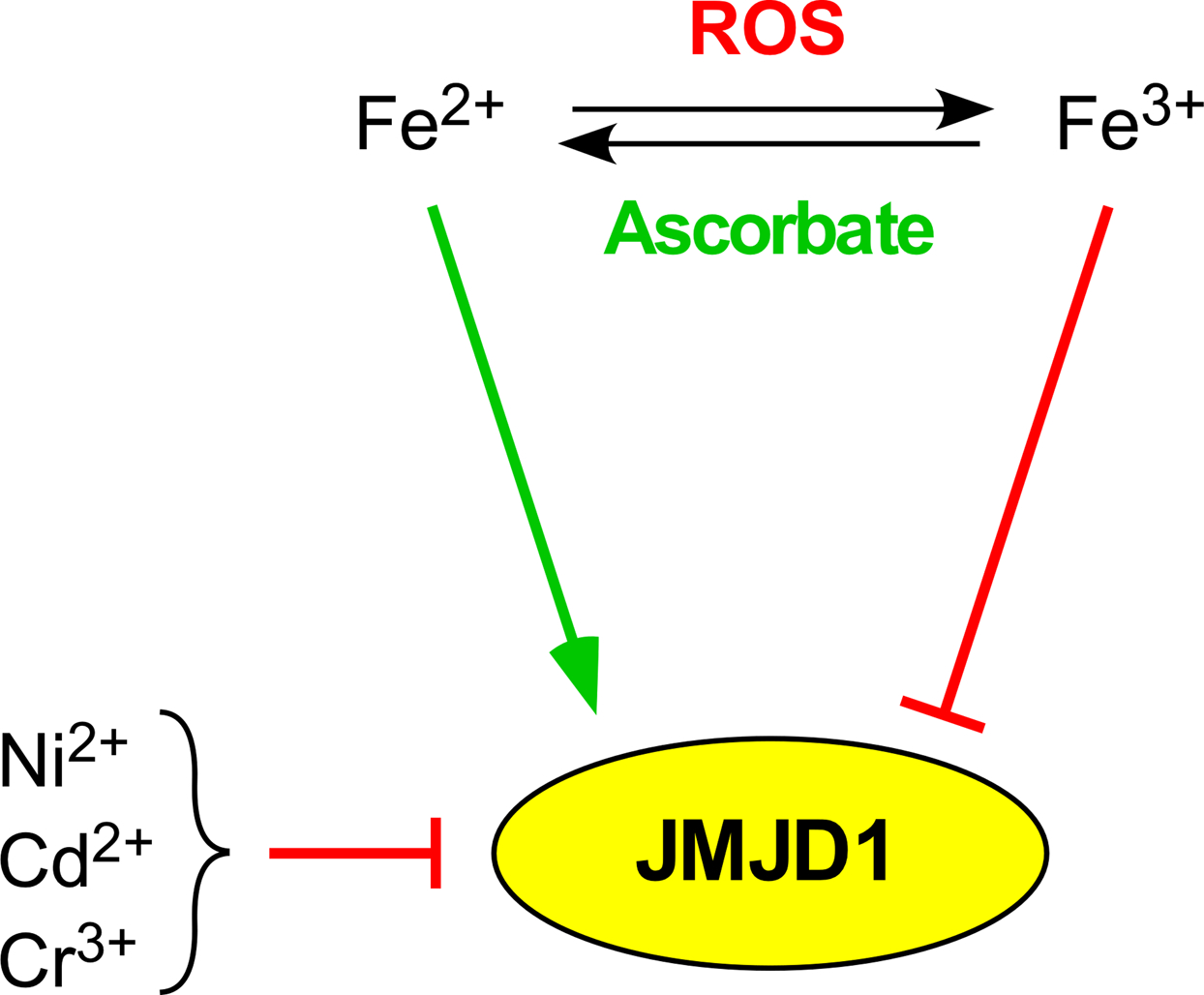
Essential role of Fe2+ for JMJD1 catalytic activity. ROS, ascorbate and heavy metals potentially modulate JMJD1 proteins by influencing their association with Fe2+.
Fe2+ in JMJD1A can be displaced by Ni2+ and this inhibited demethylase activity (80). This suggests that other heavy metals such as cadmium and chromium may also inactivate JMJD1 proteins through displacement of Fe2+ (Fig. 5). As such, the inactivation of JMJD1 proteins may play a role in heavy metal-induced carcinogenesis and development of other maladies, including hypertension, renal dysfunction, osteoporosis and mental retardation (81).
Beyond Epigenetics
Consistent with an epigenetic function through demethylating H3K9, JMJD1 proteins are predominantly localized within the nucleus of many cell lines (4, 37, 82). But a significant cytoplasmic residence, even sometimes chiefly, has also been observed (10, 26, 46). JMJD1 intracellular localization may also be variable, as shown for mouse embryonic fibroblasts or MDA-MB-231 breast cancer cells where JMJD1A became more cytosolic upon growth arrest due to serum starvation or upon reduction of extracellular matrix stiffness, respectively. Moreover, cytoplasmic JMJD1A has been proposed to facilitate rearrangements of the cytoskeleton during spermatogenesis, and this activity may be conducted in cooperation with its interaction partner, the heat shock protein 90 (83, 84). Binding of JMJD1A to the cytoskeleton possibly impacts on actin dynamics and ciliogenesis (85). At the moment, the molecular details how JMJD1 proteins perform non-epigenetic functions within the cytosol are unclear, but this may plausibly involve the demethylation, or hydroxylation, of non-histone proteins.
Even nuclear JMJD1 proteins may perform functions unrelated to epigenetics. This was demonstrated for JMJD1C, which supported the RAP80-BRCA1 dependent pathway of DNA double-strand break repair via homologous recombination. The mechanism proposed is that JMJD1C induces the demethylation of dimethylated lysine 45 in the DNA repair factor MDC1, thereby promoting binding of MDC1 to the RNF8 ubiquitin ligase that leads to MDC1 polyubiquitylation and subsequent recruitment of RAP80-BRCA1 (86). This has the caveat that in vitro proof demonstrating that MDC1 is indeed a JMJD1C substrate has not yet been provided. In contrast, both in vitro and in vivo evidence for demethylation of monomethylated lysine 224 in the coactivator PGC-1α by JMJD1A has been furnished (73). Thus, JMJD1A is likely to demethylate and thereby regulate other non-histone proteins, many of which may perform functions unrelated to epigenetics.
Conclusion
A multitude of publications have implicated JMJD1A as a driver of tumorigenesis in a variety of different cancers. Less evidence hints at JMJD1B and JMJD1C as potential oncogenic proteins. And there is persuasive data to suggest that JMJD1B is a tumor suppressor, especially in AML, and JMJD1A and JMJD1C may also be tumor suppressive in germ cell tumors. Hence, there seems to be a context-dependent ability of JMJD1 proteins to be pro- or anti-tumorigenic.
Where JMJD1 proteins are thought to be oncogenic, it opens up obvious avenues of therapeutic intervention, particularly since the activity of JMJD1 proteins is most often dependent on their catalytic activity. In these cases, small molecule JMJD1 inhibitors should be developed and such inhibitors were already shown to be effective in leukemia cells (87). Because JMJD1 proteins seem to be often co-overexpressed in tumors, a pan-JMJD1 inhibitor might be most effective. However, this may not be when JMJD1 proteins exert opposite activities, such as in AML, where JMJD1B acts like a tumor suppressor while JMJD1C exerts pro-oncogenic activities. Here, a JMJD1C-specific inhibitor that does not affect JMJD1B would be needed.
It might be more difficult to therapeutically target JMJD1 proteins in those tumors where they function as tumor suppressors. One way would be through compounds that induce allosteric activation of JMJD1 activity and another way could be through forced overexpression, which is currently more hypothetical than practical. However, there might be an alternative: inhibit the opposing histone methyltransferase(s) with small molecule drugs to normalize the methylation status of H3K9. This strategy has been shown to be feasible: genetic or pharmacological inhibition of the G9a/EHMT2 methyltransferase reduced H3K9 methylation levels back to normal in Jmjd1a−/− germ cell tumors or male gonads. Importantly, this suppressed tumorigenesis or male-to-female sex reversal, respectively, induced by the absence of JMJD1A activity (35, 88).
While catalytic activity of JMJD1 proteins is required for many aspects of their function, there are instances when this is not essential. For instance, JMJD1A interacts with the SWI/SNF chromatin remodeling complex and this scaffolding function is not dependent on demethylase activity, yet facilitates induction of gene transcription (70). A similar demethylase independent scaffolding function has been proposed for the JMJD1A-mediated stabilization of the GLI1 transcription factor (89). Also independent of catalytic activity, JMJD1A may bind to and thereby inhibit the HUWE1 ubiquitin ligase, which could lead to stabilization of one of its clients, the MYC oncoprotein (52). For these reasons, inhibition of JMJD1 catalytic activity will not always disable JMJD1 function, and therefore different strategies to constrain JMJD1 activity (e.g., antisense oligonucleotides, shRNA-loaded nanoparticles) may be needed in some tumors for efficacious therapy.
Three decades have gone by since the first discovery of a JMJD1 protein (90). However, only the realization 15 years ago that these proteins are histone demethylases and epigenetic regulators has sparked a larger interest into these fascinating enzymes. Still, we do not know many mechanisms of action, including the range of enzymatic activities JMJD1 proteins are capable of, nor comprehensively their biological functions in normal and diseased tissues. But sufficient knowledge has been amassed to spur the development of therapies targeting JMJD1 proteins in many malignancies, including major ones such as colorectal, breast and prostate cancer. Hopefully, such JMJD1 focused therapies will find their way into the clinic in the not too distant future.
Supplementary Material
Acknowledgements
Research on JMJD proteins has been funded by grants from the National Institutes of Health/National Cancer Institute (R01 CA154745 and R03 CA223615; to R. Janknecht). In addition, R. Janknecht has been supported in part by the Oklahoma Tobacco Settlement Endowment Trust through an award made to the University of Oklahoma/Stephenson Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the granting agencies.
Footnotes
Conflict of Interest Disclosure
The authors declare that there is no conflict of interest.
References
- 1.Markolovic S, Leissing TM, Chowdhury R, Wilkins SE, Lu X, Schofield CJ. Structure-function relationships of human JmjC oxygenases-demethylases versus hydroxylases. Curr Opin Struct Biol 2016;41:62–72. [DOI] [PubMed] [Google Scholar]
- 2.Oh S, Shin S, Janknecht R. The small members of the JMJD protein family: Enzymatic jewels or jinxes? Biochim Biophys Acta Rev Cancer 2019;1871:406–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 2012;13:297–311. [DOI] [PubMed] [Google Scholar]
- 4.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006;125:483–95. [DOI] [PubMed] [Google Scholar]
- 5.Kim JY, Kim KB, Eom GH, Choe N, Kee HJ, Son HJ, et al. KDM3B is the H3K9 demethylase involved in transcriptional activation of lmo2 in leukemia. Mol Cell Biol 2012;32:2917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SM, Kim JY, Choe NW, Cho IH, Kim JR, Kim DW, et al. Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res 2010;38:6389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol 2015;16:499–513. [DOI] [PubMed] [Google Scholar]
- 8.Walport LJ, Hopkinson RJ, Chowdhury R, Schiller R, Ge W, Kawamura A, et al. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat Commun 2016;7:11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Ali S, Duan X, Liu S, Du J, Liu C, et al. JMJD1B demethylates H4R3me2s and H3K9me2 to facilitate gene expression for development of hematopoietic stem and progenitor cells. Cell Rep 2018;23:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 2007;450:119–23. [DOI] [PubMed] [Google Scholar]
- 11.Kuroki S, Matoba S, Akiyoshi M, Matsumura Y, Miyachi H, Mise N, et al. Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science 2013;341:1106–9. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Oyola MG, Zhou S, Chen X, Liao L, Tien JC, et al. Knockout of the histone demethylase Kdm3b decreases spermatogenesis and impairs male sexual behaviors. Int J Biol Sci 2015;11:1447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Chen X, Zhou S, Liao L, Jiang R, Xu J. The histone H3K9 demethylase Kdm3b is required for somatic growth and female reproductive function. Int J Biol Sci 2015;11:494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuroki S, Akiyoshi M, Tokura M, Miyachi H, Nakai Y, Kimura H, et al. JMJD1C, a JmjC domain-containing protein, is required for long-term maintenance of male germ cells in mice. Biol Reprod 2013;89:93. [DOI] [PubMed] [Google Scholar]
- 15.Kuroki S, Nakai Y, Maeda R, Okashita N, Akiyoshi M, Yamaguchi Y, et al. Combined loss of JMJD1A and JMJD1B reveals critical roles for H3K9 demethylation in the maintenance of embryonic stem cells and early embryogenesis. Stem Cell Reports 2018;10:1340–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009;458:757–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inagaki T, Tachibana M, Magoori K, Kudo H, Tanaka T, Okamura M, et al. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009;14:991–1001. [DOI] [PubMed] [Google Scholar]
- 18.Abe Y, Fujiwara Y, Takahashi H, Matsumura Y, Sawada T, Jiang S, et al. Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nat Commun 2018;9:1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viscarra JA, Wang Y, Nguyen HP, Choi YG, Sul HS. Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat Commun 2020;11:796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buerger F, Muller S, Ney N, Weiner J, Heiker JT, Kallendrusch S, et al. Depletion of Jmjd1c impairs adipogenesis in murine 3T3-L1 cells. Biochim Biophys Acta Mol Basis Dis 2017;1863:1709–17. [DOI] [PubMed] [Google Scholar]
- 21.Zhang QJ, Tran TAT, Wang M, Ranek MJ, Kokkonen-Simon KM, Gao J, et al. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat Commun 2018;9:5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang S, Lu Y, Jiang C. Inhibition of histone demethylase JMJD1C attenuates cardiac hypertrophy and fibrosis induced by angiotensin II. J Recept Signal Transduct Res 2020:1–9. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Chen J, Zhang B, Liu G, Zhao H, Hu Q. KDM3A inhibition modulates macrophage polarization to aggravate post-MI injuries and accelerates adverse ventricular remodeling via an IRF4 signaling pathway. Cell Signal 2019;64:109415. [DOI] [PubMed] [Google Scholar]
- 24.Zhang BF, Jiang H, Chen J, Hu Q, Yang S, Liu XP, et al. LncRNA H19 ameliorates myocardial infarction-induced myocardial injury and maladaptive cardiac remodelling by regulating KDM3A. J Cell Mol Med 2020;24:1099–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diets IJ, van der Donk R, Baltrunaite K, Waanders E, Reijnders MRF, Dingemans AJM, et al. De novo and inherited pathogenic variants in KDM3B cause intellectual disability, short stature, and facial dysmorphism. Am J Hum Genet 2019;104:758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saez MA, Fernandez-Rodriguez J, Moutinho C, Sanchez-Mut JV, Gomez A, Vidal E, et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet Med 2016;18:378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zahir FR, Tucker T, Mayo S, Brown CJ, Lim EL, Taylor J, et al. Intragenic CNVs for epigenetic regulatory genes in intellectual disability: Survey identifies pathogenic and benign single exon changes. Am J Med Genet A 2016;170:2916–26. [DOI] [PubMed] [Google Scholar]
- 28.Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev 2007;21:2545–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma DK, Chiang CH, Ponnusamy K, Ming GL, Song H. G9a and Jhdm2a regulate embryonic stem cell fusion-induced reprogramming of adult neural stem cells. Stem cells 2008;26:2131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shakya A, Callister C, Goren A, Yosef N, Garg N, Khoddami V, et al. Pluripotency transcription factor Oct4 mediates stepwise nucleosome demethylation and depletion. Mol Cell Biol 2015;35:1014–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao F, Liao B, Hu J, Li S, Zhao H, Sun M, et al. JMJD1C ensures mouse embryonic stem cell self-renewal and somatic cell reprogramming through controlling microRNA expression. Stem Cell Reports 2017;9:927–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramadoss S, Guo G, Wang CY. Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene 2017;36:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramadoss S, Sen S, Ramachandran I, Roy S, Chaudhuri G, Farias-Eisner R. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 2017;36:1537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork K, et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat Commun 2017;8:15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueda J, Ho JC, Lee KL, Kitajima S, Yang H, Sun W, et al. The hypoxia-inducible epigenetic regulators Jmjd1a and G9a provide a mechanistic link between angiogenesis and tumor growth. Mol Cell Biol 2014;34:3702–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature 2014;511:241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Z, Gomes I, Horrigan SK, Kravarusic J, Mar B, Arbieva Z, et al. A novel nuclear protein, 5qNCA (LOC51780) is a candidate for the myeloid leukemia tumor suppressor gene on chromosome 5 band q31. Oncogene 2001;20:6946–54. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, Nagel S, Quentmeier H, Wang Z, Pommerenke C, Dirks WG, et al. KDM3B shows tumor-suppressive activity and transcriptionally regulates HOXA1 through retinoic acid response elements in acute myeloid leukemia. Leuk Lymphoma 2018;59:204–13. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Fan H, Xu C, Jiang G, Wang H, Zhang J. KDM3B suppresses APL progression by restricting chromatin accessibility and facilitating the ATRA-mediated degradation of PML/RARalpha. Cancer Cell Int 2019;19:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sroczynska P, Cruickshank VA, Bukowski JP, Miyagi S, Bagger FO, Walfridsson J, et al. shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood 2014;123:1870–82. [DOI] [PubMed] [Google Scholar]
- 41.Chen M, Zhu N, Liu X, Laurent B, Tang Z, Eng R, et al. JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors. Genes Dev 2015;29:2123–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu N, Chen M, Eng R, DeJong J, Sinha AU, Rahnamay NF, et al. MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Invest 2016;126:997–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynch JR, Salik B, Connerty P, Vick B, Leung H, Pijning A, et al. JMJD1C-mediated metabolic dysregulation contributes to HOXA9-dependent leukemogenesis. Leukemia 2019;33:1400–10. [DOI] [PubMed] [Google Scholar]
- 44.Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 2010;30:344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T, et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin Cancer Res 2010;16:4636–46. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Oh S, Song H, Shin S, Zhang B, Freeman WM, et al. A potential common role of the Jumonji C domain-containing 1A histone demethylase and chromatin remodeler ATRX in promoting colon cancer. Oncol Lett 2018;16:6652–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng K, Su G, Ji J, Yang X, Miao M, Mo P, et al. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/beta-catenin signaling. J Biol Chem 2018;293:10606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim H, Kim D, Choi SA, Kim CR, Oh SK, Pyo KE, et al. KDM3A histone demethylase functions as an essential factor for activation of JAK2-STAT3 signaling pathway. Proc Natl Acad Sci USA 2018;115:11766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang HY, Long QY, Tang SB, Xiao Q, Gao C, Zhao QY, et al. Histone demethylase KDM3A is required for enhancer activation of hippo target genes in colorectal cancer. Nucleic Acids Res 2019;47:2349–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C, Aihemaiti M, Zhang X, Qu H, Sun QL, He QS, et al. Downregulation of histone demethylase JMJD1C inhibits colorectal cancer metastasis through targeting ATF2. Am J Cancer Res 2018;8:852–65. [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf SS, Patchev VK, Obendorf M. A novel variant of the putative demethylase gene, s-JMJD1C, is a coactivator of the AR. Arch Biochem Biophys 2007;460:56–66. [DOI] [PubMed] [Google Scholar]
- 52.Fan L, Peng G, Sahgal N, Fazli L, Gleave M, Zhang Y, et al. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2016;35:2441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang DE, Dai Y, Fan LL, Geng XY, Fu DX, Jiang HW, et al. Histone demethylase JMJD1A promotes tumor progression via activating Snail in prostate cancer. Mol Cancer Res 2020. [DOI] [PubMed] [Google Scholar]
- 54.Fan L, Zhang F, Xu S, Cui X, Hussain A, Fazli L, et al. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci USA 2018;115:E4584–E93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarac H, Morova T, Pires E, McCullagh J, Kaplan A, Cingoz A, et al. Systematic characterization of chromatin modifying enzymes identifies KDM3B as a critical regulator in castration resistant prostate cancer. Oncogene 2020;39:2187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wade MA, Jones D, Wilson L, Stockley J, Coffey K, Robson CN, et al. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res 2015;43:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao QY, Lei PJ, Zhang X, Zheng JY, Wang HY, Zhao J, et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin Epigenetics 2016;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qin L, Xu Y, Yu X, Toneff MJ, Li D, Liao L, et al. The histone demethylase Kdm3a is required for normal epithelial proliferation, ductal elongation and tumor growth in the mouse mammary gland. Oncotarget 2017;8:84761–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho HS, Toyokawa G, Daigo Y, Hayami S, Masuda K, Ikawa N, et al. The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int J Cancer 2012;131:E179–E89. [DOI] [PubMed] [Google Scholar]
- 60.Wan W, Peng K, Li M, Qin L, Tong Z, Yan J, et al. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1alpha. Oncogene 2017;36:3868–77. [DOI] [PubMed] [Google Scholar]
- 61.Park SJ, Kim JG, Son TG, Yi JM, Kim ND, Yang K, et al. The histone demethylase JMJD1A regulates adrenomedullin-mediated cell proliferation in hepatocellular carcinoma under hypoxia. Biochem Biophys Res Commun 2013;434:722–7. [DOI] [PubMed] [Google Scholar]
- 62.Nakatsuka T, Tateishi K, Kudo Y, Yamamoto K, Nakagawa H, Fujiwara H, et al. Impact of histone demethylase KDM3A-dependent AP-1 transactivity on hepatotumorigenesis induced by PI3K activation. Oncogene 2017;36:6262–71. [DOI] [PubMed] [Google Scholar]
- 63.Zhan M, Wen F, Liu L, Chen Z, Wei H, Zhou H. JMJD1A promotes tumorigenesis and forms a feedback loop with EZH2/let-7c in NSCLC cells. Tumour Biol 2016;37:11237–47. [DOI] [PubMed] [Google Scholar]
- 64.Liu J, Zhu M, Xia X, Huang Y, Zhang Q, Wang X. Jumonji domain-containing protein 1A promotes cell growth and progression via transactivation of c-Myc expression and predicts a poor prognosis in cervical cancer. Oncotarget 2016;7:85151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dandawate P, Ghosh C, Palaniyandi K, Paul S, Rawal S, Pradhan R, et al. The histone demethylase KDM3A, increased in human pancreatic tumors, regulates expression of DCLK1 and promotes tumorigenesis in mice. Gastroenterology 2019;157:1646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parrish JK, Sechler M, Winn RA, Jedlicka P. The histone demethylase KDM3A is a microRNA-22-regulated tumor promoter in Ewing Sarcoma. Oncogene 2015;34:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohguchi H, Hideshima T, Bhasin MK, Gorgun GT, Santo L, Cea M, et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat Commun 2016;7:10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cai Y, Fu X, Deng Y. Histone demethylase JMJD1C regulates esophageal cancer proliferation Via YAP1 signaling. Am J Cancer Res 2017;7:115–24. [PMC free article] [PubMed] [Google Scholar]
- 69.An MJ, Kim DH, Kim CH, Kim M, Rhee S, Seo SB, et al. Histone demethylase KDM3B regulates the transcriptional network of cell-cycle genes in hepatocarcinoma HepG2 cells. Biochem Biophys Res Commun 2019;508:576–82. [DOI] [PubMed] [Google Scholar]
- 70.Abe Y, Rozqie R, Matsumura Y, Kawamura T, Nakaki R, Tsurutani Y, et al. JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nat Commun 2015;6:7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ivan M, Kaelin WG, Jr. The EGLN-HIF O2-sensing system: multiple inputs and feedbacks. Mol Cell 2017;66:772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, et al. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun 2008;372:892–7. [DOI] [PubMed] [Google Scholar]
- 73.Qian X, Li X, Shi Z, Bai X, Xia Y, Zheng Y, et al. KDM3A senses oxygen availability to regulate PGC-1alpha-mediated mitochondrial biogenesis. Mol Cell 2019;76:885–95. [DOI] [PubMed] [Google Scholar]
- 74.Abla H, Sollazzo M, Gasparre G, Iommarini L, Porcelli AM. The multifaceted contribution of alpha-ketoglutarate to tumor progression: An opportunity to exploit? Semin Cell Dev Biol 2020;98:26–33. [DOI] [PubMed] [Google Scholar]
- 75.Collins RRJ, Patel K, Putnam WC, Kapur P, Rakheja D. Oncometabolites: a new paradigm for oncology, metabolism, and the clinical laboratory. Clin Chem 2017;63:1812–20. [DOI] [PubMed] [Google Scholar]
- 76.Bystrom LM, Guzman ML, Rivella S. Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid Redox Signal 2014;20:1917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol 2018;80:50–64. [DOI] [PubMed] [Google Scholar]
- 78.Cimmino L, Neel BG, Aifantis I. Vitamin C in stem cell reprogramming and cancer. Trends Cell Biol 2018;28:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ebata KT, Mesh K, Liu S, Bilenky M, Fekete A, Acker MG, et al. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenetics Chromatin 2017;10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen H, Giri NC, Zhang R, Yamane K, Zhang Y, Maroney M, et al. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J Biol Chem 2010;285:7374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim HS, Kim YJ, Seo YR. An overview of carcinogenic heavy metal: molecular toxicity mechanism and prevention. J Cancer Prev 2015;20:232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Knebel J, De Haro L, Janknecht R. Repression of transcription by TSGA/Jmjd1a, a novel interaction partner of the ETS protein ER71. J Cell Biochem 2006;99:319–29. [DOI] [PubMed] [Google Scholar]
- 83.Kasioulis I, Syred HM, Tate P, Finch A, Shaw J, Seawright A, et al. Kdm3a lysine demethylase is an Hsp90 client required for cytoskeletal rearrangements during spermatogenesis. Mol Biol Cell 2014;25:1216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaukonen R, Mai A, Georgiadou M, Saari M, De Franceschi N, Betz T, et al. Normal stroma suppresses cancer cell proliferation via mechanosensitive regulation of JMJD1a-mediated transcription. Nat Commun 2016;7:12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yeyati PL, Schiller R, Mali G, Kasioulis I, Kawamura A, Adams IR, et al. KDM3A coordinates actin dynamics with intraflagellar transport to regulate cilia stability. J Cell Biol 2017;216:999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watanabe S, Watanabe K, Akimov V, Bartkova J, Blagoev B, Lukas J, et al. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol 2013;20:1425–33. [DOI] [PubMed] [Google Scholar]
- 87.Xu X, Wang L, Hu L, Dirks WG, Zhao Y, Wei Z, et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int J Cancer 2020;146:400–12. [DOI] [PubMed] [Google Scholar]
- 88.Kuroki S, Okashita N, Baba S, Maeda R, Miyawaki S, Yano M, et al. Rescuing the aberrant sex development of H3K9 demethylase Jmjd1a-deficient mice by modulating H3K9 methylation balance. PLoS Genet 2017;13:e1007034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schneider P, Bayo-Fina JM, Singh R, Kumar Dhanyamraju P, Holz P, Baier A, et al. Identification of a novel actin-dependent signal transducing module allows for the targeted degradation of GLI1. Nat Commun 2015;6:8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Höög C, Schalling M, Grunder-Brundell E, Daneholt B. Analysis of a murine male germ cell-specific transcript that encodes a putative zinc finger protein. Mol Reprod Dev 1991;30:173–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.