

Abstract
Neuroinflammation leads to tissue injury causing many of the clinical symptoms of Multiple Sclerosis, an autoimmune disorder of the central nervous system (CNS). While T cells, specifically Th1 and Th17 cells, are the ultimate effectors of this disease, dendritic cells (DCs) mediate T-cell polarization, activation, etc. In our previous study, Apigenin, a natural flavonoid, has been shown to reduce EAE disease severity through amelioration of demyelination in the CNS as well as the sequestering of DCs and other myeloid cells in the periphery. Here, we show that Apigenin exerts its effects possibly through shifting DC modulated T-cell responses from Th1 and Th17 type towards Treg directed responses evident through the decrease in T-bet, IFN-γ (Th1), IL-17 (Th17) and increase in IL-10, TGF-β and FoxP3 (Treg) expression in cells from both normal human donors and EAE mice. RelB, an NF-κβ pathway protein is central to DC maturation, its antigen presentation capabilities and DC-mediated T-cell activation. Apigenin reduced mRNA and protein levels of RelB and also reduced its nuclear translocation. Additionally, siRNA-mediated silencing of RelB further potentiated the RelB-mediated effects of Apigenin thus confirming its role in Apigenin directed regulation of DC biology. These results provide key information about the molecular events controlled by Apigenin in its regulation of DC activity marking its potential as a therapy for neuroinflammatory disease.
Keywords: Flavonoid, Apigenin, Multiple Sclerosis, EAE, Immune Cells, Dendritic Cells, RelB, NF-κβ
Graphical Abstract
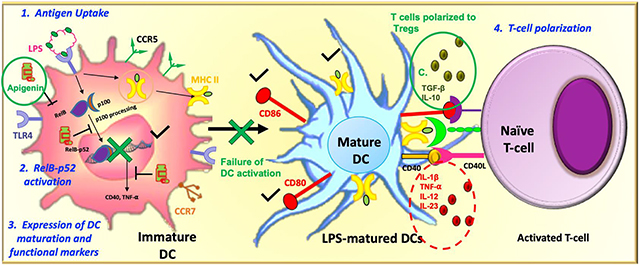
Background
Natural flavonoids belong to a class of polyphenolic compounds, ubiquitously present in several plants, fruits, vegetables, flowers, and its preparation products such as tea and wines. Because of their lower molecular weight and hydrophilic nature, these compounds can easily penetrate through the blood-brain barrier (BBB) and exert biological influences including antiviral, anti-carcinogenic, antioxidant and notably anti-inflammatory effects (Kumar and Pandey 2013). Of these compounds, Apigenin, which is abundantly present in celery, parsley, chamomile, and other fruits, vegetables, herbs and spices (Ginwala et al. 2016) has been seen to possess less toxicity even at higher doses (Venigalla et al. 2015). Also, as with other flavonoids, Apigenin demonstrates anti-inflammatory, and anti-oxidant properties in several cell and tissue systems (Millington et al. 2014; Wang et al. 2015). As a result, Apigenin has been used to treat many diseases for centuries such as Parkinson’s, neuralgia, shingles, and others (Reviewed in ref (Ginwala et al. 2016)).
In a wide variety of organ systems and their related disorders the underlying factor that maintains a crucial balance between steady-state and disease is the generation of inflammatory mediators. Since, Apigenin is a potent anti-inflammatory agent with low intrinsic toxicity, non-mutagenic, with slower decomposition rates in the liver and delayed plasma clearance it is an attractive target for development as a therapeutic anti-inflammatory agent. It is possible thus, that its anti-carcinogenic (Bruno et al. 2011; Shukla and Gupta 2010; Sung et al. 2016), anti-diabetic (Gentile et al. 2018; Jung et al. 2016), and more importantly to this study, its anti-neurodegenerative properties can be attributed to Apigenin’s ability to modulate immune responses. Apigenin mediated stimulation of immune cell responses, and modulation of multiple signaling pathways including PI3K/AKT, MAPK/ERK, JAK/STAT, NF-κB and Wnt/β-catenin may drive its anti-cancer activities (Shukla and Gupta 2010; Tong and Pelling 2013; Yan et al. 2017). Likewise, Apigenin has been shown to exert its anti-inflammatory activity by inactivating nuclear factor kappa B (NF-κB), and reducing the production of pro-inflammatory cytokines leading to decreased fat accumulation, improved insulin resistance, and decreased blood fat cholesterol in Apigenin supplemented obese mice (Gentile et al. 2018; Jung et al. 2016). Chronic inflammatory activities such as the kind associated with neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Multiple Sclerosis (MS), results in progressive and irreversible loss of neurons in the brain (Onasanwo et al. 2016). Prolonged activation of central nervous system (CNS)-resident macrophage-like immune cells, and overproduction of pro-inflammatory cytokines results in chronic CNS neuroinflammation (Ota and Ulrih 2017). Despite its well-studied role as an anti-inflammatory agent, Apigenin’s potential in modulating neuroinflammation and its subsequent effects on immune cell activity in the CNS and periphery in neurodegenerative diseases is surprisingly under-explored. Few studies show direct effects of Apigenin on CNS-resident microglial cells in in vitro studies showing anti-inflammatory activity on BV-2 cells and primary microglial cells through inhibition of p38 and JNK.
We looked to investigate the potential of Apigenin as a treatment for the management and cure of neuroinflammatory disease like MS because of its aforementioned traits and its reportedly high penetration and distribution in the CNS (Zhao et al. 2013). In our previous study using experimental autoimmune encephalomyelitis (EAE), the most widely used model of neuroinflammation, we found that Apigenin delayed EAE disease onset and ameliorated disease severity (Ginwala et al. 2016). The contributing cellular events leading to reduction in disease severity were decreased number of myeloid cells infiltrating the CNS in the Apigenin treated mice. Furthermore, Apigenin shifted the immune cell response from Th1/Th17 to a prominent T regulatory cell (Treg) lead response. While Apigenin-mediated regulation of inflammation has been attributed to several pathways, the exact molecular players in its control of dendritic cells (DC)s, key cells in immune regulation that are capable of inducing Tregs as well as other helper T-cell subsets, remain to be elucidated. Accumulating evidences have shown that DCs play a prominent role in maintaining central and peripheral tolerance, and the failure of which can lead to various autoimmune and neuroinflammatory diseases (Manuel et al. 2007; Wu and Laufer 2007). In the context of EAE, DCs are known to have important functions in T-cell polarization (Mohammad et al. 2012). They are known to secrete high levels of IL-10, while specifically inducting CD4+Foxp3+ Tregs and Type 1 Tregs. These Tregs have been seen to downregulate immune responses via IL-10 specifically in the CNS, while they can also secrete other anti-inflammatory cytokines such as TGF-β (Xie et al. 2015). Only a handful of studies have described the role of Apigenin in modulation of DC activity. In murine bone marrow derived DCs Apigenin was shown to reduce cell-surface expression of antigen presentation and co-stimulatory molecules in a NF-κB (p65)-dependent manner (Yoon et al. 2006). In the context of rheumatoid arthritis (RA), Apigenin has been seen to inhibit DC maturation, as well as reduce cytokine production, such as TNF-α, IL-12p70 and IL-10 (Li et al. 2016). However, the effect of Apigenin on DC phenotypical and functional characteristics under neuroinflammatory conditions and its associated molecular mechanism remains to be clearly outlined.
DC differentiation and activation is controlled by intracellular signaling pathways, most notably, the NF-κB pathway (Cejas et al. 2005; Li et al. 2007). The NF-κB family of proteins in mammals consists of c-Rel, RelA/p65, RelB, NF-κB1 (p50 and its precursor, p105) and NF-κB2 (p52 and its precursor, p100). While the canonical pathway involving the p65-p50 complex has been implicated in DC function, it is RelB that is more indispensable to DC differentiation and function through both canonical (RelB-p50) and non-canonical (RelB-p100) pathways (Cejas et al. 2005). The RelB-p100 complex, the non-canonical pathway, is activated by phosphorylation of the C-terminal region of p100. This phosphorylation leads to partial degradation of p100 to p52. Once active, RelB is able to translocate into the nucleus and affect NF-κB-related transcription (Li et al. 2007). RelB is critical for Th1 responses as well as DC maturation; this is in contrast to the canonical p65-p50 pathway, while inhibition of RelB has been seen to prevent maturation and reduce expression of costimulatory HLA-DR, CD80 and CD86 (Li et al. 2007). Crucially, RelB is a master regulator of DC differentiation, and has been suggested to be a possible target for manipulation of DC differentiation, especially in the context of autoimmune disease. No studies have so far described the effect of Apigenin on RelB expression, its nuclear translocation and subsequent control of DC functions. In this study we will show the effect of Apigenin on both the phenotypical and functional maturation of human peripheral blood DCs after confirming the optimum dose and kinetics of Apigenin in vitro treatments. Further, we will explore the effects of any Apigenin mediated alteration in DC function in the ultimate change in naïve T-cell differentiation. Any findings will be further investigated in splenocytes from EAE mice to confirm Apigenin role in neuroinflammatory disorders. Since, RelB is crucial to DC differentiation and maturation we will seek to analyze Apigenin effect on RelB expression and confirm RelB role in Apigenin mediated DC functional regulation by siRNA directed RelB silencing in DCs.
Methods
Blood dendritic cell isolation
Blood dendritic cells were isolated from human whole blood as described previously (Jain et al. 2007). Briefly, buffy coat isolated from whole blood from normal donors was purchased from Biological Specialty Corporation (Colmar, PA). Peripheral blood mononuclear cells (PBMCs) were then isolated from the buffy coat via density centrifugation using Ficoll-Paque Plus (GE, Pittsburg, PA). Isolated PBMCs were washed with hank’s balanced salt solution (HyClone, GE) followed by treatment with red blood cell (RBC) lysis buffer (Biolegend, San Diego, CA) for removal of any RBCs that were carried over from the buffy coat. DCs were subsequently enriched by negative selection using EasySep™ human pan-DC pre-enrichment kit (Stemcell Technologies, Vancouver Canada) as per the manufacturer’s protocol.
Apigenin dosing and kinetics
DCs were plated at a density of 0.6 × 106 per well in 12 well plates and cultured in RPMI supplemented with 10% fetal bovine serum, 5% penicillin-streptomycin, 1x non-essential amino acids, and 0.5mM sodium pyruvate (hereafter referred to as ‘complete medium’). DCs were treated with and without LPS (Sigma, St. Louis, MO) at 2μg/mL for 24h. Apigenin (Sigma, St. Louis, MO) dissolved in dimethyl sulfoxide (DMSO) (Fisher BioReagents, Fair Lawn, NJ) was administered at doses of 1, 3, 8 and 20μM for 5h at the end of LPS stimulation. To assess kinetics of Apigenin treatment effect on DC functionality Apigenin was administered at doses of 8 and 20μM for a period of 0.5, 1.5, 3, 6, 12 and 24h. All DC incubations were performed at 37°C. Culture supernatants were then extracted by centrifugation at 300g at room temperature for 10m. Supernatants were stored at −80°C for later analysis. Cell pellets were resuspended in 500μL of TRIzol reagent (Invitrogen, Carlsbad, CA) and stored at −20°C for later analysis.
Cytoplasmic/nuclear protein isolation and quantitative western analysis
DCs isolated from PBMCs from normal human donors were activated with LPS at 2μg/mL for 24h as described earlier. At the end of LPS treatment the DCs were further incubated with 20μM Apigenin for another 3h. DCs were then pelleted by centrifugation, lysed and cytoplasmic/nuclear fractions were extracted using the NucBuster Protein Extraction Kit (EMD Millipore, Billerica, MA) as per the manufacturer’s protocol. Crude protein concentration was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA). Protein detection and quantification by WES Simple Western size based capillary electrophoresis (ProteinSimple, San Jose, CA) was performed as described previously (Visavadiya et al. 2016), with samples loaded at a concentration of 0.03 mg/mL (Visavadiya et al. 2016). Primary antibodies used included: RelB (1:300, Cell Signaling Technology, Danvers, MA) and Vinculin (1:50 for cytoplasmic/1:25 for nuclear extracts, R&D Systems, Minneapolis, MN). The standard running conditions listed in the manufacturer’s protocol were used for size-based separation. Quantitation was performed using the Compass software (ProteinSimple) with peak representations that display the areas under the curve normalized to vinculin loading control.
DC: PBL co-culture
DCs from normal human blood donor were plated at a density of 2.5 × 106 per well in 12-well plates followed by treatment with LPS at 2μg/mL for 24h. At 24 hours, 20μM Apigenin was administered to the cells for a period of 3h. 1×106 DCs were then collected for phenotypic analysis for the expression of cell surface markers such as CD11c, HLA-ABC, HLA-DR, CD40, CD80, CD83, CD86, CCR5 and CCR7 by flow cytometry using the LSRII Fortessa flow cytometer (BD Biosciences, San Jose, CA) and subsequent data was analyzed using FlowJo software 10.4.2. (Treestar, Ashland, OR). Culture supernatants were also collected and stored at −80°C for later analysis. The remaining DCs were washed twice with PBS and then added to autologous peripheral blood lymphocytes (PBLs) (DC:PBL cell ratio: 1:5) at a density of 1 × 106. DCs and PBLs were co-cultured for 6 days in 12-well plates in complete medium supplemented with IL-2 (10ng/ml). On day 3 of the co-culture, fresh media supplemented with IL-2 was added to the culture. On day 6 of the co-culture, cells were collected for flow cytometric analyses for expression of T-cell surface markers such as CD3, CD4, CD25, CCR4, CCR5, CCR6 and intracellular markers such as T-bet, IFN- γ, IL-6, IL-17, FoxP3 and IL-10. Cell supernatants were collected and stored at −80°C for later analysis of cytokine secretion.
Naïve T-cell stimulation and polarization
Spleen from five naïve C57BL/6 mice were harvested and a single cell suspension was prepared followed by RBC lysis. Naïve T cells were negatively selected using EasySep™ Mouse T-cell Isolation Kit (Stemcell Technologies, Vancouver Canada) as per manufacturer’s instructions. Naïve T cells were then polarized in to different subsets as described previously (Noubade et al. 2011; Noubade et al. 2007). Briefly, 1×105 T cells were stimulated for 72h in complete RPMI media (Genesee, San Diego, CA) with plate bound anti-CD3 (5μg/ml) and soluble CD28 antibodies (2μg/ml) (Biolegend) for the Th0 condition. In addition, the following cytokines and antibodies were added to the Th0 conditions for polarization: Th1 (10μg/ml anti-IL-4 (BioLegend), 4ng/ml IL-12 (Peprotech)), Th2 (10μg/ml anti-IFN-γ (Biolegend), 30ng/ml IL-4 (Peprotech)), Th17 (30ng/ml IL-6, 1ng/ml TGF-β (Biolegend), 10μg/ml anti-IFN-γ, 10μg/ml anti-IL-4) and Treg (2ng/ml TGF-β). Cultures were harvested for RNA and supernatants.
EAE recall response
Three C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were induced for EAE with an immunization mixture containing 100μg MOG35–55 peptide, complete Freund’s adjuvant and 200μg Mycobacterium tuberculosis as previously described (Bearoff et al. 2016; Krementsov et al. 2014) and sacrificed at day 14, which is typically the earliest the mice start to show symptoms in this model. The mice were pre-symptomatic when sacrificed, hence, it is possible to find antigen-specific cells in the lymphoid tissue at this timepoint. Three experimental control mice were also sacrificed at day 14. Mice were housed at specific-pathogen-free Drexel University Laboratory Animal Resources (ULAR)’s animal facility and all animal procedures were performed in accordance with institutional guidelines. Spleen were harvested, a single cell suspension was prepared, and cells were stimulated for 72h with and without MOG35–55 (50μg/ml) for 3 days. Cells were then treated with Apigenin (20μM) for 3h and compared to the control (untreated cells and/or naïve mice). Cultures were harvested for RNA for qPCR and supernatants for ELISA to determine the mRNA and protein levels of transcription factors and cytokines related to DCs and T-cell function.
siRNA transfection
siRNAs for RelB (L-004767) and the negative control (D-001810–10) were purchased from Dharmacon Inc. (Lafayette, CO) while p65 siRNA (sc-29410) was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX) as a pool of 3–4 target-specific nucleotide sequences. 250,000 human peripheral blood DCs were plated in 24-well plates in 500μl complete RPMI medium without FBS and antibiotics. These cells were then treated with 2μg/mL LPS for 24h followed by treatment with 20μM Apigenin. After 24h transfection solution was prepared by mixing Genesilencer transfection reagent (Genlantis, San Diego, CA) and reduced serum medium (Opti-MEM, ThermoFisher, Scientific). RelB, p65 and negative control siRNAs were prepared by mixing siRNA with siRNA diluent (Genlantis) and Opti-MEM and incubated at room temperature for 5m. The transfection solution was then added to the individual siRNA solutions and the siRNA-transfection mix was incubated for a further 10m at room temperature. The mixture was then added to the DCs and incubated at 37°C. After 4h of transfection 500μl complete medium containing 20% FBS was added to the DCs and the cells were incubated for a total of 30–36h. Post-transfection DCs were analyzed for the expression of RelB and downstream targets by qPCR, ELISA and flow cytometry. Additionally, transfected DCs were plated with PBLs as described earlier in the mixed lymphocyte reaction. The effect of transfected DCs on T-cell polarization was assessed by flow cytometry.
RNA isolation and qPCR
RNA was extracted from culture pellets using TRIzol™ reagent (Invitrogen) following manufacturer’s protocol. Following RNA quantification, cDNA was prepared using high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) in a Mastercycler® pro S (Eppendorf, Hamburg, Germany). The cDNA was then used for qPCR according to the SYBR Select Master Mix protocol (Applied Biosystems) under standard conditions with a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). The following primers were used (human): TNF-α, 5’-AACGGAGCTGAACAATAGGC-3’ and 5’-CAGAGGCTCAGCAATGAGTG-3’; RelB, 5’-GAGCAAACGGCGGAAGAAAAA-3’ and 5’-CAGGGGACAGGGGCTTATG-3’; GAPDH, 5’-GAGTCAACGGATTTGGTCGT-3’ and 5’-TTGATTTTGGAGGGATCTCG-3’; FoxP3, 5’-TGCCTCCTCTTCTTCCTTGAAC-3’ and 5’-TCCTGGAGG AGT GCC TGT AAGT-3’; CD40, 5’-CGGTGAAAGCGAATTCCTAG-3’ and 5’-CAGCCTTCTTCACAGGTGC-3’. The following primers were used (murine): IL-17, 5’-TCTCCACCGCAATGAAGACC-3’ and 5’- CACACCCACCAGCATCTTCT-3’; TGF-β, 5’- CCTGCAAGACCATCGACATG-3’ and 5’-TGTTGTACAAAGCGAGCACC-3’; T-bet, 5’-GCCAGGGAACCGCTTATATG-3’ and 5’- GACGATCATCTGGGTCACATTGT-3’; IFN-γ, 5’-TGAACGCTACACACTGCATCTTGG-3’ and 5’-CGACTCCTTTTCCGCTTCCTGAG-3’; IL-4, 5’-AACGAGGTCACAGGAGAAGG-3’ and 5’-TCTGCAGCTCCATGAGAACA-3’; RelB, 5’-GCCGAATCAACAAGGAGAGCG-3’ and 5’-CATCAGCTTGAGAGAAGTCGGCA-3’; FoxP3, 5’-CCCAGGAAAGACAGCAACCTT-3’ and 5’-TTCTCACAACCAGGCCACTTG-3’; TNF-α, 5’-ATGAGCACAGAAAGCATGA-3’ and 5’-AGTAGACAGAAGAGCGTGGT-3’ The 2−ΔΔCT method was used to quantify fold changes in expression of all the genes relative to the control conditions and normalized to the housekeeping gene β-actin.
Enzyme-linked Immunosorbent Assay (ELISA) for cytokine detection in culture supernatants
Culture supernatants from both human and mouse cell samples were thawed and cytokines were assayed by sandwich ELISA. IFN-γ, IL-1β, IL-4, IL-6, IL-10, IL-12p70, IL-17, IL-23, TGF-β, and TNF-α, (Affymetrix, San Diego, CA and BioLegend, San Diego, CA) were assayed as per the manufacturer’s protocol. Absorbance was measured by a Tecan Sunrise 96-well plate reader (Research Triangle Park, NC) at 450 nm. A standard curve was generated and used to determine cytokine amounts in each sample.
Analysis of cell viability
The assessment of cell viability for the Apigenin dosing and kinetic experiments was performed by incubating cells with Resazurin (R&D systems, Minneapolis, MN) for a further 4h after end of each assay at 37°C. After incubation, absorbance was measured by a Tecan Sunrise 96-well plate reader (Research Triangle Park, NC) at 570 nm. For all other experiments, cell culture supernatants were treated with LDH assay buffer (Pierce LDH Cytotoxicity Assay Kit, ThermoFisher Scientific), for 30m at room temperature following manufacturer’s protocol. Extracellular LDH activity was then measured spectrophotometrically at 490nm and 680nm.
Statistical Analysis
Statistical analysis was performed using the Graphpad Prism software (version 5.00; GraphPad, San Diego, CA). All datasets were tested for normal distribution. Two-tailed, unpaired t-test were performed for comparison between sample groups where indicated. Statistical analysis was also conducted as indicated using 2-way ANOVA with post-hoc Sidak’s and Tukey’s tests of for comparison of multiple groups if required. An alpha level of <0.05 was considered significant for all analyses. Asterisks indicate significant difference (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Apigenin reduces soluble TNF-α release by dendritic cells in a dose- and time-dependent manner
To study the biological effects of Apigenin at the cellular and molecular level we first performed dose and kinetic studies of Apigenin activity. Myeloid cells including DCs and macrophages are key producers of TNF-α in response to toll-like receptor 4 (TLR4) stimulation critically influencing the nature of downstream inflammatory and immunological responses. To assess the appropriate dose of Apigenin-mediated DC functional modulation we treated normal human peripheral blood DCs with TLR4 ligand, lipopolysaccharide (LPS) to stimulate TNF-α secretion. We found that Apigenin reduces TNF-α secretion at all the concentrations tested (1–20μM) with the most potent reduction seen at 20μM where Apigenin was able to reduce both basal and LPS-stimulated TNF-α mRNA and protein levels (Fig. 1A). Several studies have noted anti-inflammatory responses to Apigenin at a dose of 20μM which we also observed in the modulation of TNF-α secretion by DCs.
Figure 1. Apigenin dose and kinetics tested in human peripheral blood DCs.
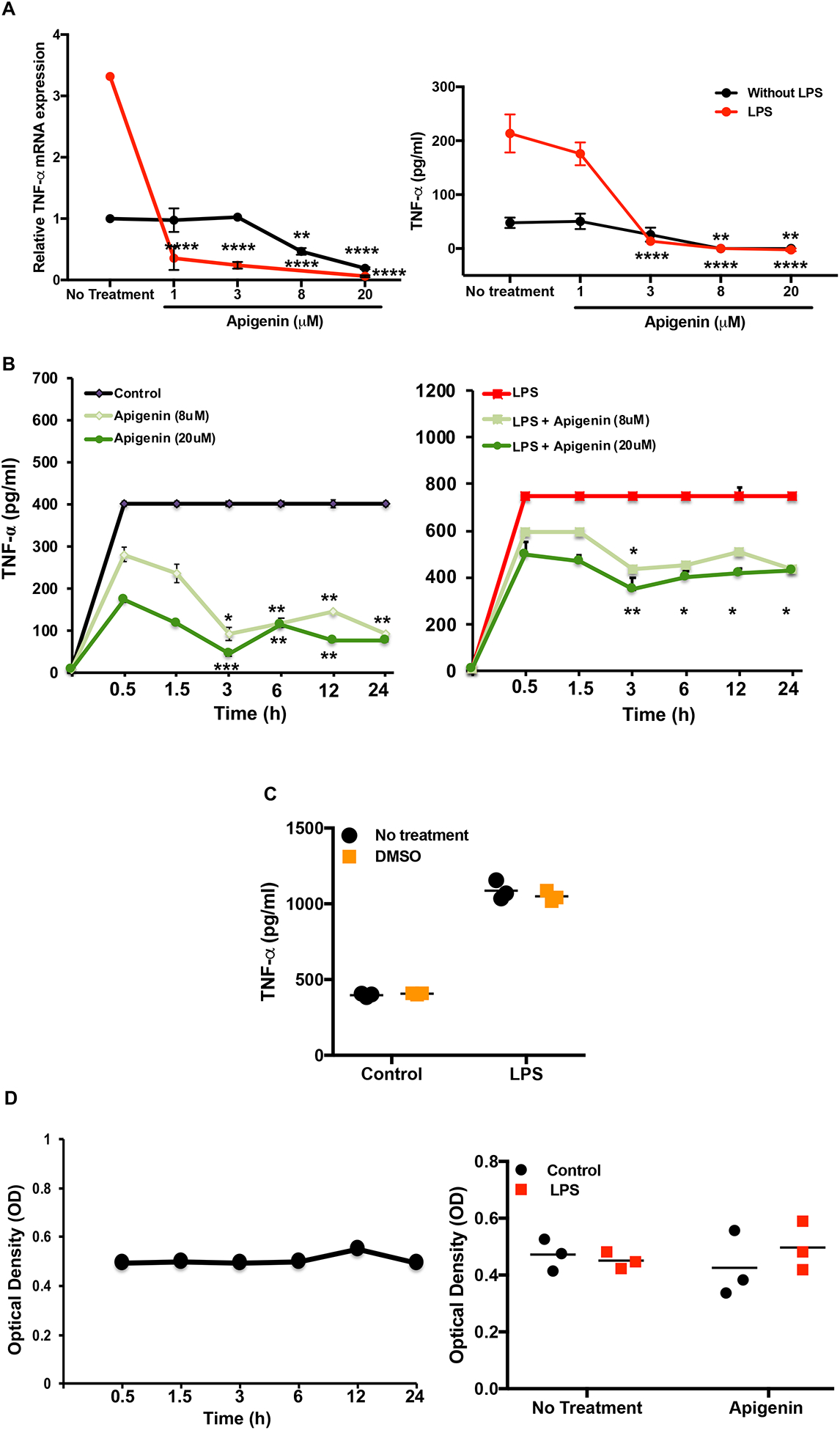
PBMCs were isolated from buffy coat via Ficoll-Pague density centrifugation. DCs were isolated from PBMCs using EasySepTM human pan-DC pre-enrichment kit and stimulated with LPS for 24h. DCs were subsequently treated with Apigenin at 1, 3, 8 and 20μM for 5h. RNA was extracted by TRIzol phenol-chloroform method and both RNA and culture supernatants were analyzed for TNF-α expression. A) TNF-α mRNA transcript levels were detected by qPCR (left). TNF-α cytokine levels were determined by ELISA (right). B) Similarly, DCs were stimulated with and without LPS for 24h and subsequently treated with Apigenin at 8 and 20μM for 0.5, 1.5, 3, 6, 12 and 24h. Culture supernatants were extracted and TNF-α level was assessed by ELISA. C) TNF-α secretion by DMSO-treated and untreated DCs. D) Cell viability as measured via resazurin fluorescence depicts DC viability over 24h under naïve (left), LPS stimulated and with or without 20μM Apigenin (right). Sample standard error (SE) is shown and statistical significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons for data shown in A-C. For D, statistical significance was determined by Student’s t-test. (*p<0.05, **p<0.01, ***p<0.001 ****p<0.0001). Data are representative of three independent experiments run in duplicate.
To determine the earliest time-point at which Apigenin could exert its effects, the levels of TNF-α upon LPS stimulation in DCs was again assessed by ELISA (Fig. 1B). LPS stimulation induced cytokine secretion in as little as 30m, which was paralleled by a decrease in cytokine production in the presence of Apigenin. Apigenin was observed to exert its effects most potently after 3h of incubation regardless of LPS stimulation. DMSO, the solvent used to dissolve Apigenin prior to use in cell culture, had no effect on TNF-α secretion (Fig. 1C).While DCs alone were viable for the 24h period, there was also no change in viability upon either activation with LPS or treatment with Apigenin (Fig. 1D).
Apigenin impairs DC phenotypic and functional maturation
DC precursors in the blood and those found at potential sites of pathogen entry reside as immature DCs. After antigen encounter these DCs partially mature with an upregulated expression of CCR7 enabling them to migrate to nearby lymphoid tissue for antigen presentation to naïve T cells. We assessed the ability of Apigenin to modulate DC phenotypic changes during maturation with LPS. As expected, LPS treatment brought about an increase in the number of activated DCs showing expression of HLA-DR, CD40, CD83 and CD86. After Apigenin treatment we saw a marked decrease in the level of antigen presentation molecules HLA-ABC and HLA-DR (Fig. 2A). Co-stimulatory molecules CD40, CD83 and CD86 increased upon LPS treatment and were reduced again close to the basal levels upon Apigenin treatment (Fig. 2B). CCR7 expression was also reduced suggesting loss of migratory potential upon Apigenin treatment (data not shown). Conversely, CCR5, a marker of immature DCs, expression reduced upon LPS induced maturation and increased slightly in the presence of Apigenin. Additionally, DCs produce numerous cytokines upon maturation (Leyva-Lopez et al. 2016), that help direct both innate and adaptive immune responses including those that polarize different T-cell subsets. Apigenin was able to reduce the production of IFN-γ, IL-1β, IL-6, IL-12, and IL-23 both in the presence and absence of LPS (Fig. 2C). Also, Apigenin was able to induce the production of anti-inflammatory TGF-β and IL-10 (p<0.0001) (Fig. 2D). It was interesting to note that for the release of some of the cytokines (IFN-γ, IL-1β, IL-6 and IL-10), the extent of Apigenin effect was dependent upon the activation state of DCs as evidenced by the significant interaction between these variables on performing a 2-way ANOVA. The preferential induction of anti-inflammatory cytokines with concurrent inhibition of Th1 and Th17 inducing cytokines may suggest that Apigenin is able to promote DCs towards a more tolerogenic phenotype.
Figure 2. Apigenin modulates both phenotypic and functional maturation in DCs.
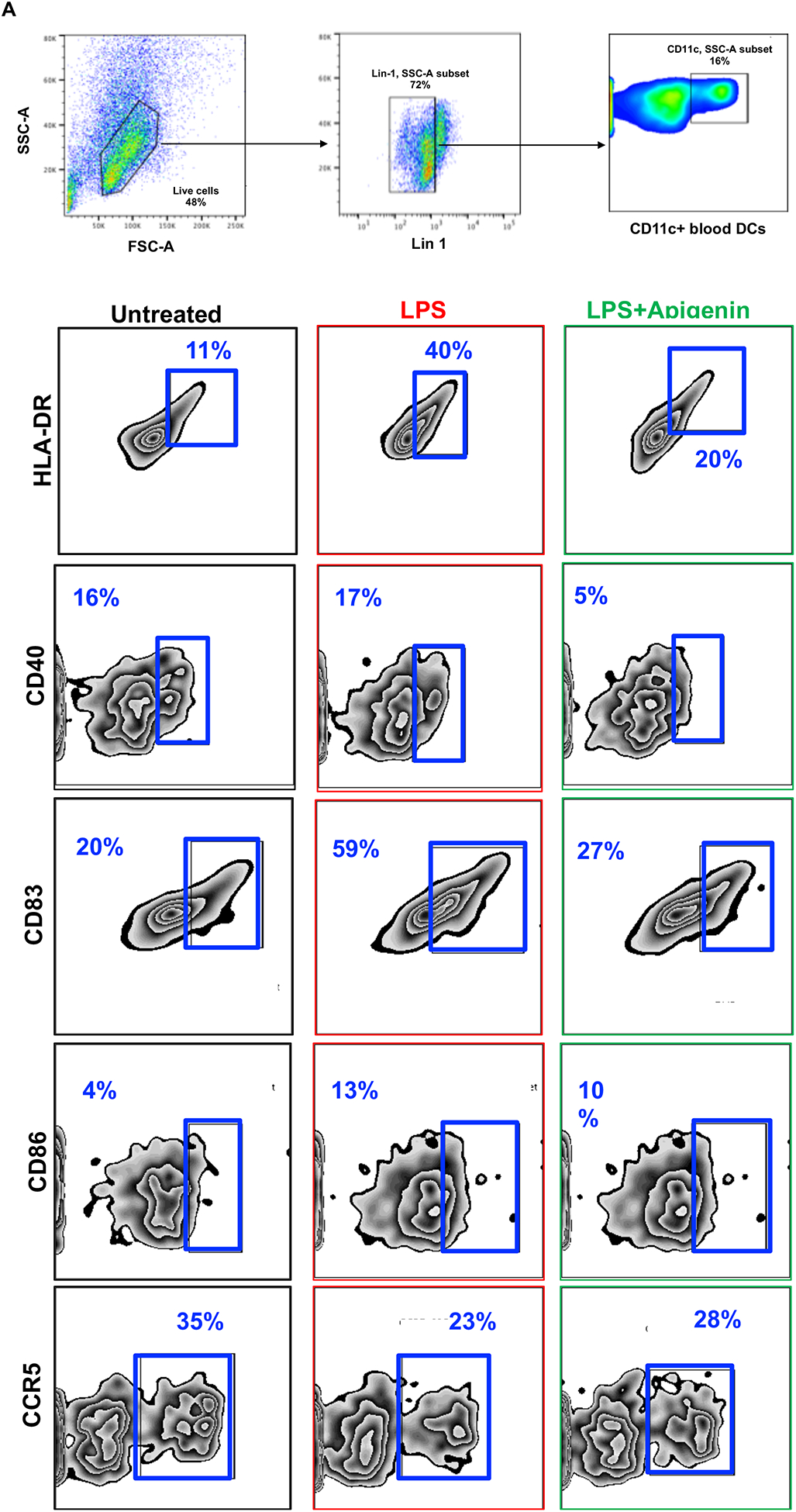
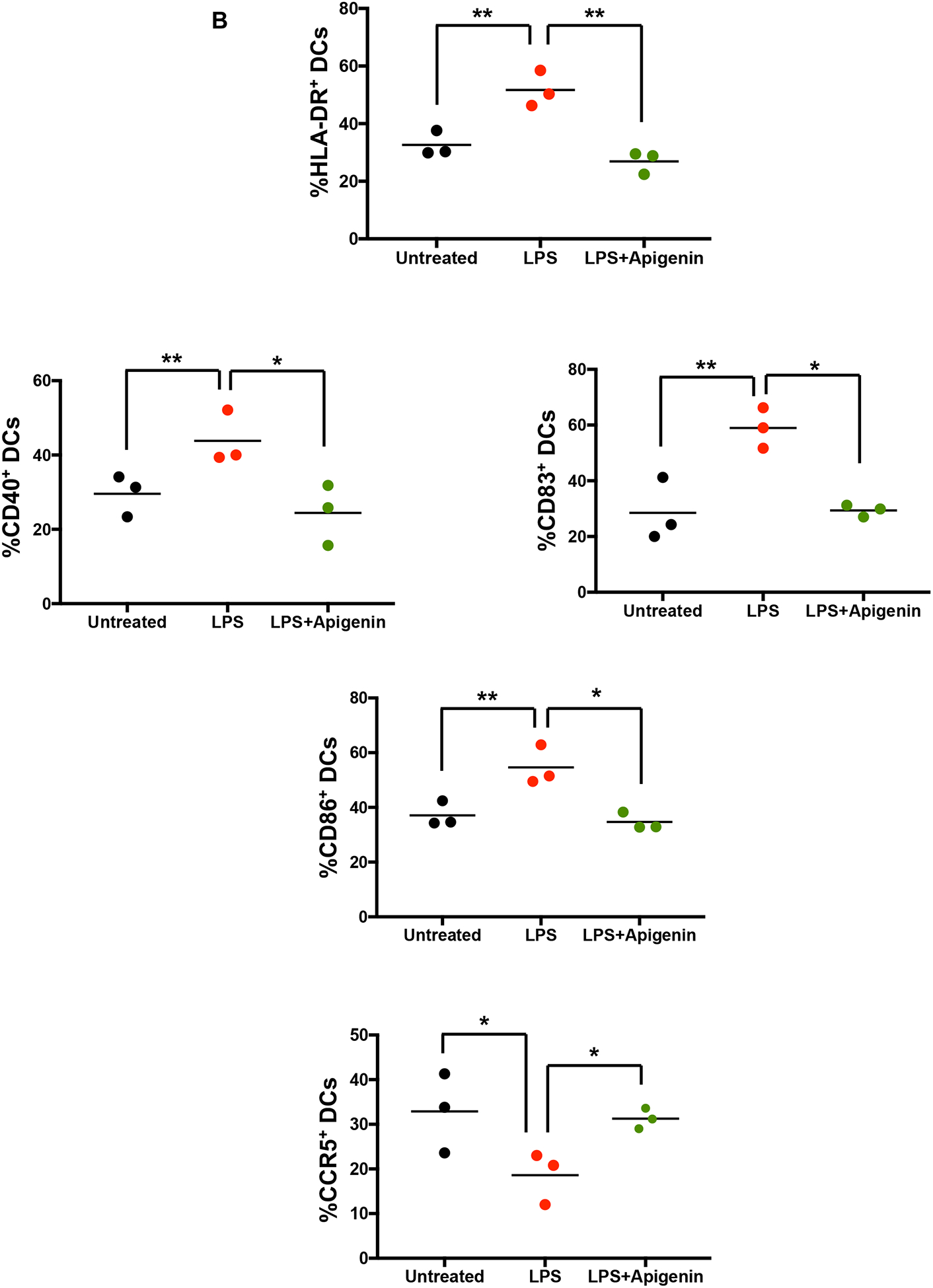
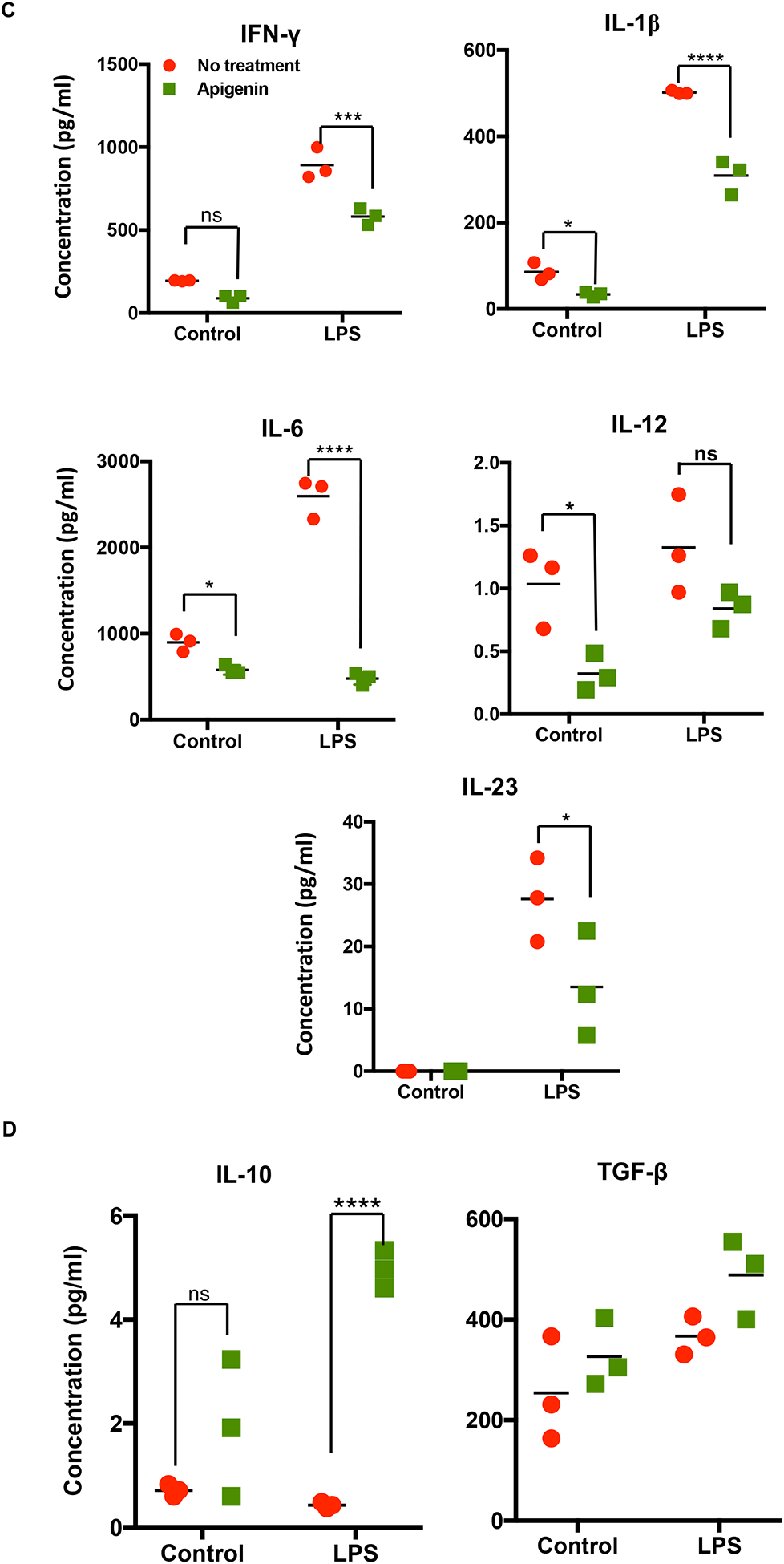
DCs were isolated as described in Figure 1. DCs were stimulated with and without LPS for 24h. DCs were then treated with Apigenin (20μM) for 3h. A) Antibodies against cell surface markers were used to stain human blood DCs and phenotypic expression was determined by flow cytometry. Each FACS plot is representative of three individual donors. B) Quantitative representation of FACS plots shown in A (N=3). Culture supernatants were extracted and C) pro-inflammatory cytokine IFN-γ, IL-1β, IL-6, IL-12, and IL-23 and D) anti-inflammatory cytokine IL-10 and TGF-β levels were detected by ELISA. FACS plots are representative of results from three individual donors. Statistical significance was determined by Student’s t-test for data shown in A and B. Statistical significance was derived by 2-way ANOVA using Sidak’s test for multiple comparisons for data shown in C and D (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Apigenin treated DCs lead to suppression of pro-inflammatory cytokine production and induction of a regulatory T-cell response
After analyzing the DC cytokine profile in response to Apigenin treatment, we analyzed whether this DC modulation had any effect on subsequent naïve T-cell differentiation. DCs were matured in the presence of 2μg/ml LPS for 24h. Apigenin was then administered for 3h after the end of LPS treatment and the DCs were then plated with PBLs. Th1 polarizing transcription factor T-bet was reduced in PBLs co-cultured with Apigenin treated mature DCs (Fig. 3A–B). Similar decrease was seen in the number of IL-17 producing cells. Conversely, the number of IL-10 (Treg) expressing CD4+ T cells were increased on incubation with Apigenin treated mature DCs while the number of CD4+ CCR4+ Th2 cells remain unchanged. Thus, Apigenin modulation of DC phenotype and function influences the subsequent polarization of naïve T cells thus directing the adaptive immune response to be more anti-inflammatory.
Figure 3. Modulation of T-cell responses by Apigenin treated DC.
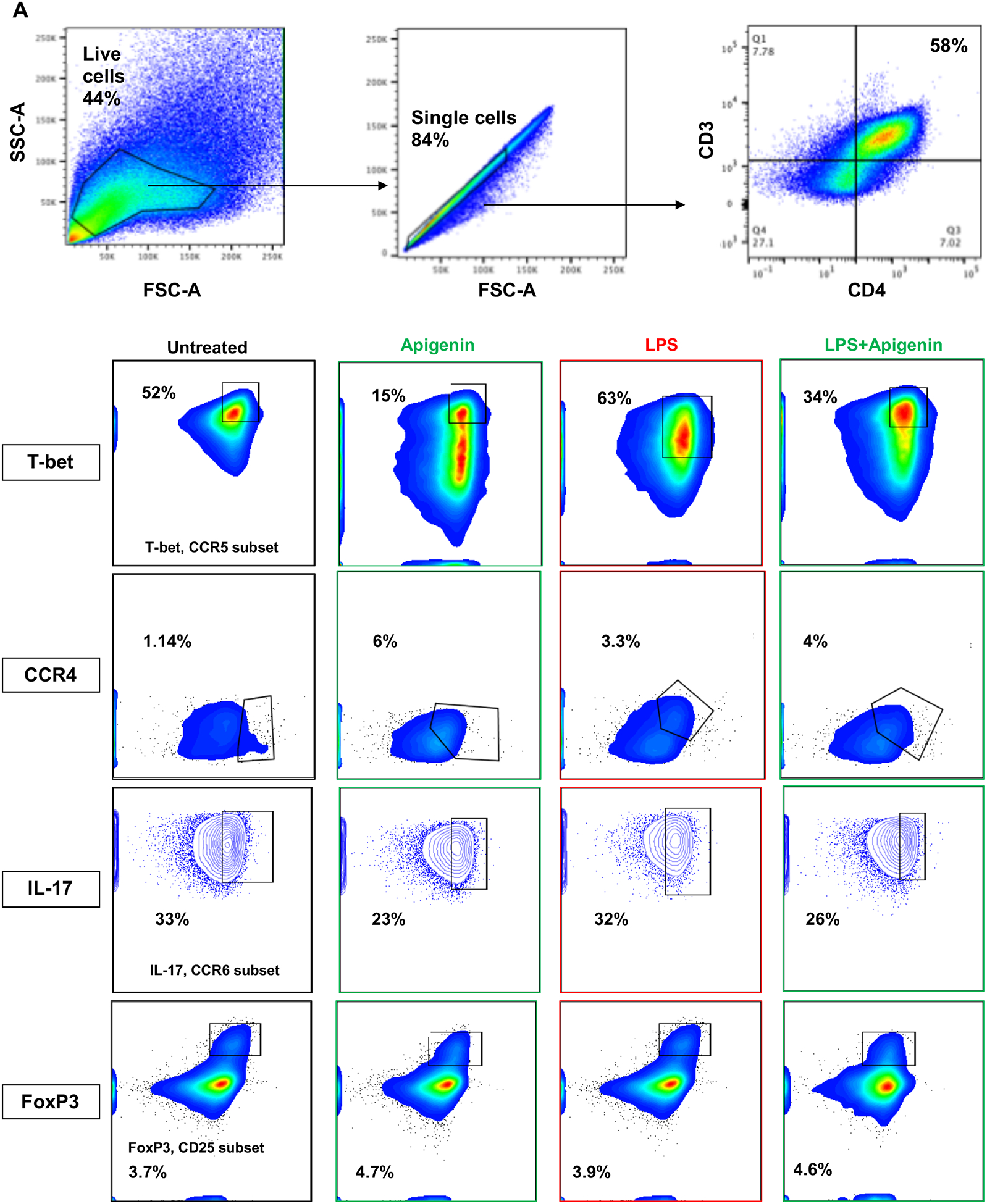
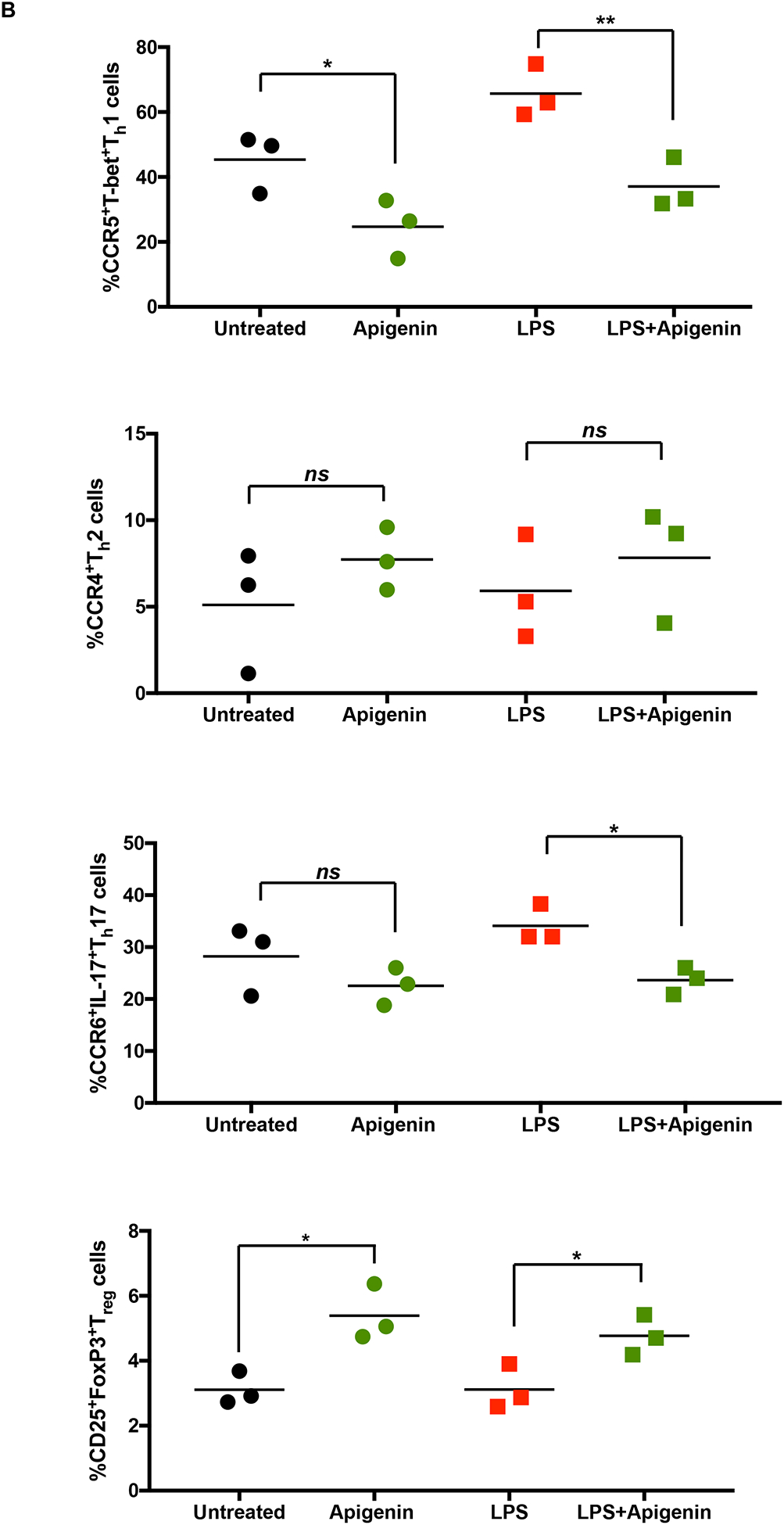
DCs from three donors were isolated as in Figure 1. DCs were cultured with and without LPS for 24h followed by 3h of 20μM Apigenin treatment. DCs were then co-cultured with autologous PBLs for 6 days with Brefeldin A and Monensin treatment in the last 16h of the co-culture. T-cell phenotype was elucidated by staining for various cell surface markers and transcription factors and the cells were analyzed by flow cytometry. A) FACS plots of T-cell subset-specific expression of transcription factors or cytokines. B) Quantitative representation of FACS plots shown in A (N=3). Statistical significance was determined by Student’s t-test (*p<0.05, **p<0.01, ***p<0.001).
Apigenin impairs both antigen-dependent and independent inflammatory responses in murine splenocytes
So far Apigenin exhibited a modulating effect on the pro-inflammatory functions of both DCs and T cells in response to non-specific LPS stimulation. We now aimed to study the effect of Apigenin on antigen-specific T cells isolated from mice with chronic EAE (closest available model mimicking progressive MS). EAE was induced in C57BL/6 mice using MOG35–55 peptide of the myelin protein. Splenocytes isolated from these mice were re-stimulated with MOG35–55 for 3d in the presence or absence of 20μM Apigenin to evaluate whether Apigenin is able to modulate an antigen-specific immune response in these cells. mRNA levels of Th1 polarizing transcription factor T-bet was markedly reduced upon treatment with Apigenin (Fig. 4). mRNA levels of IL-17, the key cytokine in maintaining inflammation within the CNS in MS, also demonstrated a downward trend upon Apigenin treatment with significant down-modulation in response to Apigenin in the control animals. At the same time, TGF-β mRNA expression was increased in the splenocytes of the EAE mice upon Apigenin treatment. Interestingly, levels of IL-4 mRNA, a Th2 cytokine, were not altered after Apigenin administration. The shift in balance from Th1 to Th2 has previously been attributed to suppress autoimmune phenotype and improve clinical course of MS. The anti-inflammatory activity of Apigenin could thus be due to its ability to modulate the production of Th1 and Th17 cytokines, increase levels of Treg cytokines while maintaining the protective levels of Th2 cytokines.
Figure 4. Antigen dependent assay with splenocytes from EAE mice.
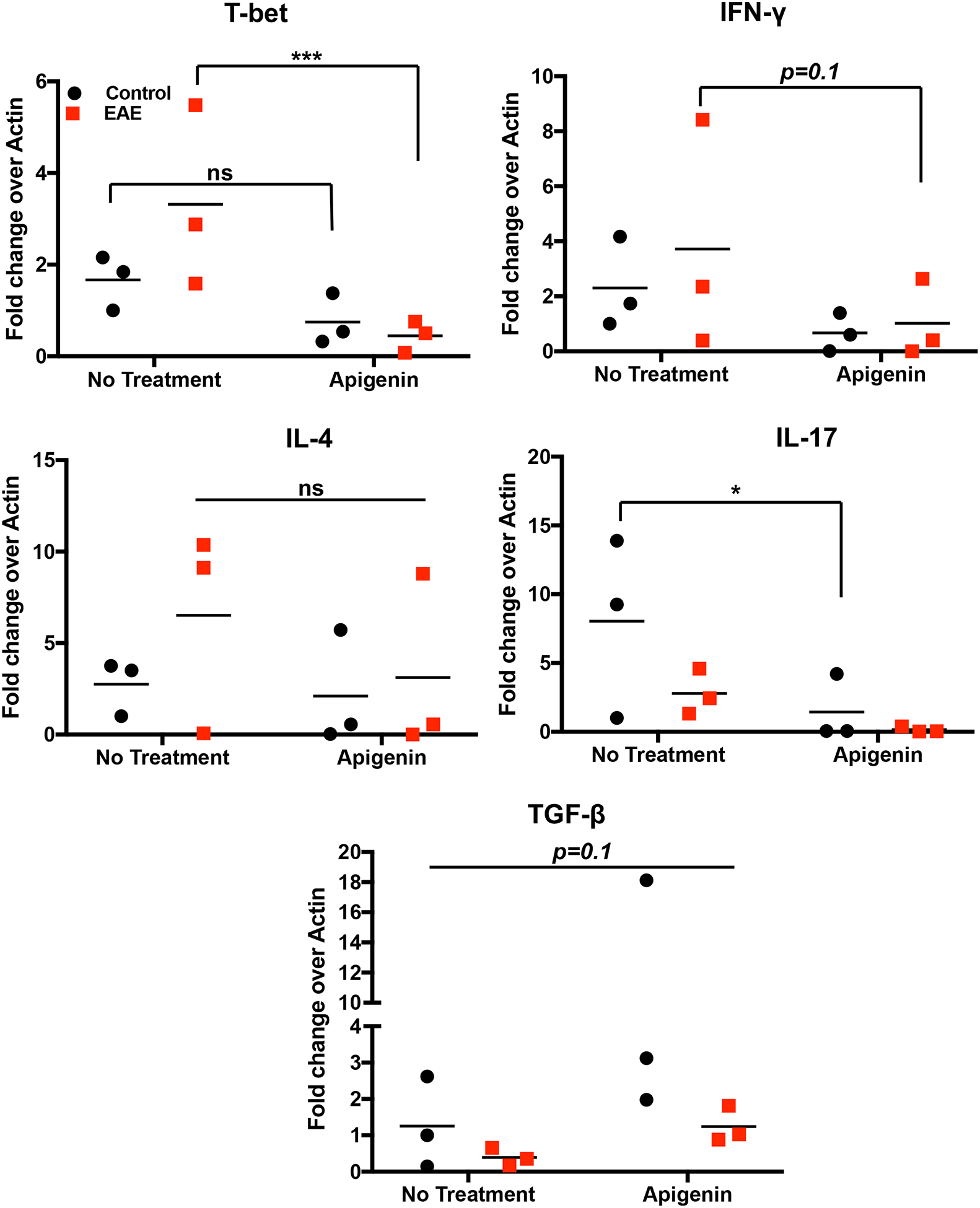
Single cell suspension was prepared from mouse spleen of EAE and naïve mice (n=3). Cells were stimulated with and without MOG35–55 peptide for 3 days in the presence of absence of 20μM Apigenin. Gene expression was compared to β-Actin and normalized to cells that were not treated with MOG35–55 peptide. T-bet, IFN-γ, IL-4, IL-17, and TGF-β mRNA expression for three EAE and three naïve mice were detected by qPCR. Statistical significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons (*p<0.05, ***p<0.001).
Further, we wanted to determine if Apigenin was able to control the differentiation of naïve T cells in the absence of specific antigen or antigen presenting cells by directly modulating T-cell cytokine secretion. We cultured naïve murine T cells in the presence of various T-cell subset polarizing factors as described in Materials and Methods in the presence or absence of 20μM Apigenin. mRNA quantification showed reduced levels of T-bet in the Th1 subset that were treated with Apigenin as compared to the untreated controls (Supplementary Fig 1, left). Similarly, IL-17 mRNA expression was also prominently reduced in the Th17 subsets. The mRNA levels of IL-4 were either maintained or increased in the presence of Apigenin. Release of pro-inflammatory Th1 cytokine TNF-α and Th17 cytokine, IL-17, was also inhibited by Apigenin whereas the release of anti-inflammatory TGF-β was boosted (Supplementary Fig. 1, right). These results are, however, preliminary and require further investigation to achieve accurate reproducible results establishing a direct effect of Apigenin on T-cell fate.
Apigenin downregulates RelB expression in dendritic cells and suppresses its nuclear translocation
The NF-κB protein RelB is central to DC maturation, its antigen presentation capabilities and DC-mediated T-cell activation (Shih et al. 2012; Zanetti et al. 2003). DC-specific RelB depletion has resulted in impaired T-cell responses affecting the balance of activated vs. regulatory T-cell (Hofmann et al. 2011; Li et al. 2007). To determine if Apigenin acts in a RelB dependent mechanism, we first measured RelB mRNA expression which was downregulated in the presence of Apigenin both in immature DCs and DCs stimulated with LPS (Fig. 5A). Further, we examined RelB protein expression in cytoplasmic and nuclear fractions of dendritic cells matured with LPS and treated with Apigenin. As RelB translocates to the nucleus upon DC maturation and differentiation, RelB was expected to translocate in response to LPS stimulation. Using Simple Western capillary based separation, RelB was seen to be present in higher amounts in the nuclear fraction after LPS stimulation (Fig. 5B) compared to the cytoplasmic fraction. Apigenin reduced RelB protein expression in both the cytoplasm and nucleus of immature and LPS-matured DCs. Further, there was less accumulation of RelB in the nuclei of LPS-matured DCs suggesting that Apigenin prevents nuclear translocation of RelB in response to inflammatory stimuli. RelB mRNA expression was then investigated in the splenocytes from EAE mice. Apigenin treatment in MOG35–55 specific cells reduced RelB mRNA expression 5-fold (Supplementary Fig. 2A). RelB expression was also reduced by Apigenin in Th1, Th2 and Th17 CD4+ T-cell subsets that were polarized in the absence of antigen and DCs (Supplementary Fig. 2B).
Figure 5. Apigenin downregulates RelB transcription.
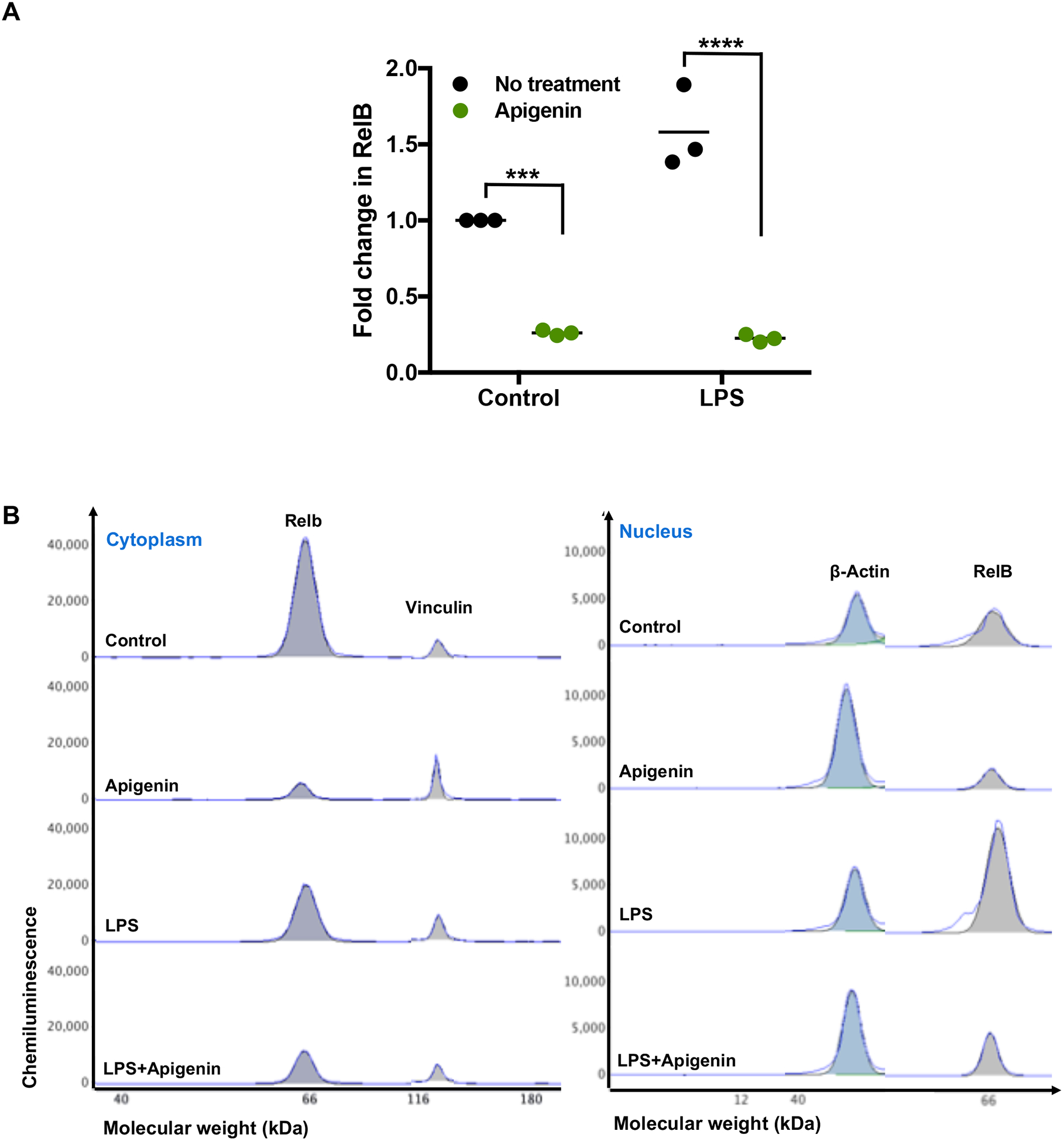
DCs were fractionated by centrifugation/lysis and extracted into cytoplasmic and nuclear fractions. After quantification via Bradford assay, protein determination was performed by WES size based capillary electrophoresis. A) RelB mRNA expression (N=3) was determined by qPCR. B) Graphical representation of RelB protein in both cytoplasm and nucleus as generated by Compass, a ProteinSimple software. Protein data is representative of two individual experiments. SE is shown, and significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons (***p<0.001, ****p<0.0001).
Confirmation of RelB mediated control of DC phenotype and function by Apigenin through RelB silencing using siRNA
To confirm that RelB is involved in Apigenin-mediated regulation of DC phenotype and function upon inflammatory insult, we measured key DC responses after siRNA-mediated silencing of RelB in the DCs. Human peripheral blood DCs were treated with different concentrations of RelB siRNA (10–50nM) for achieving optimal transfection efficiency. Figures 6 (A–C) indicate that RelB and its downstream transcriptional target TNF-α were effectively suppressed using 25nM RelB siRNA. We then analyzed the effect of LPS maturation and subsequent Apigenin treatment in DCs transfected with 25nM RelB siRNA. Apigenin treatment in transfected DCs further reduces RelB mRNA expression (Fig. 6D). Additionally, siRelB appears to potentiate Apigenin-mediated reduction in mRNA levels of TNF-α, downstream RelB target (Fig. 6E). Changes in DC phenotype upon siRelB silencing and Apigenin treatment were analyzed on Lin-1−CD11c+HLA-DR+ DCs (Fig. 7A). siRelB treatment induces semi-mature phenotype in DCs with reduction in cell surface expression of CD40, CD83 and CD86 in DCs pre-treated with LPS (Fig. 7B). This reduction is further enhanced upon Apigenin treatment indicating that the residual RelB is also down-modulated resulting in a further suppression of downstream targets. As comparison DCs were also transfected with siRNA targeted to p65, the nuclear translocation of which has been shown to be impaired by Apigenin in murine bone marrow derived DCs (Yoon et al. 2006). RelB silencing produces similar effects on DC maturation as seen with p65 knockdown with Apigenin potentiating both p65 and RelB mediated effects. Though RelB silencing in immature DCs reduces the cell surface expression of CD40, CD83 and CD86, this effect is not potentiated by Apigenin suggesting that Apigenin does not affect basal levels of these molecules maintaining steady-state DC phenotype in the absence of inflammatory stimulus (Supplementary Fig. 3). After confirming that Apigenin regulates DC maturation in a RelB-dependent manner we wished to see if DC functional traits were also regulated by Apigenin via a RelB-mediated mechanism. For this, transfected DCs treated with LPS and Apigenin were co-cultured with autologous PBLs for 6 days. At the end PBLs were collected and differences in the expression of different T-cell markers governing the various T-cell subtypes were analyzed. Figure 8 shows that RelB silencing in DCs promotes an anti-inflammatory phenotype in CD3+CD4+ T cells. CCR5+T-bet+ Th1 cells are reduced in cultures containing RelB-silenced DCs. Additionally, RelB-silenced DCs also cause decreased production of Th17 cells. Conversely, number of CD25+FoxP3+ Tregs increase in the presence of RelB-silenced DCs. Moreover, all these effects were further enhanced in the presence of Apigenin. Interestingly, knocking out RelB activity has a similar effect on DC phenotype and T cell polarization as does the addition of Apigenin, as can be seen from both figures 7 and 8. This further indicates the association of RelB with Apigenin-mediated control of DC activity. As previously p65 knockdown was used to compare Apigenin-mediated DC regulation via RelB. From figure 8 it appears that RelB is more critical to Apigenin-mediated DC directed polarization of CD4+T cells. Similar to the response of immature DCs to Apigenin, numbers of Th1 and Th17 cells did not further reduce upon RelB silencing. Steady-state numbers of Th2 and Tregs were higher when cultured with immature DCs both in presence and absence of Apigenin, however RelB knockdown marginally boosted the effect of Apigenin on Tregs causing an increase in their numbers (Supplementary Fig. 5).
Figure 6. siRNA directed silencing of RelB in human peripheral blood DCs.
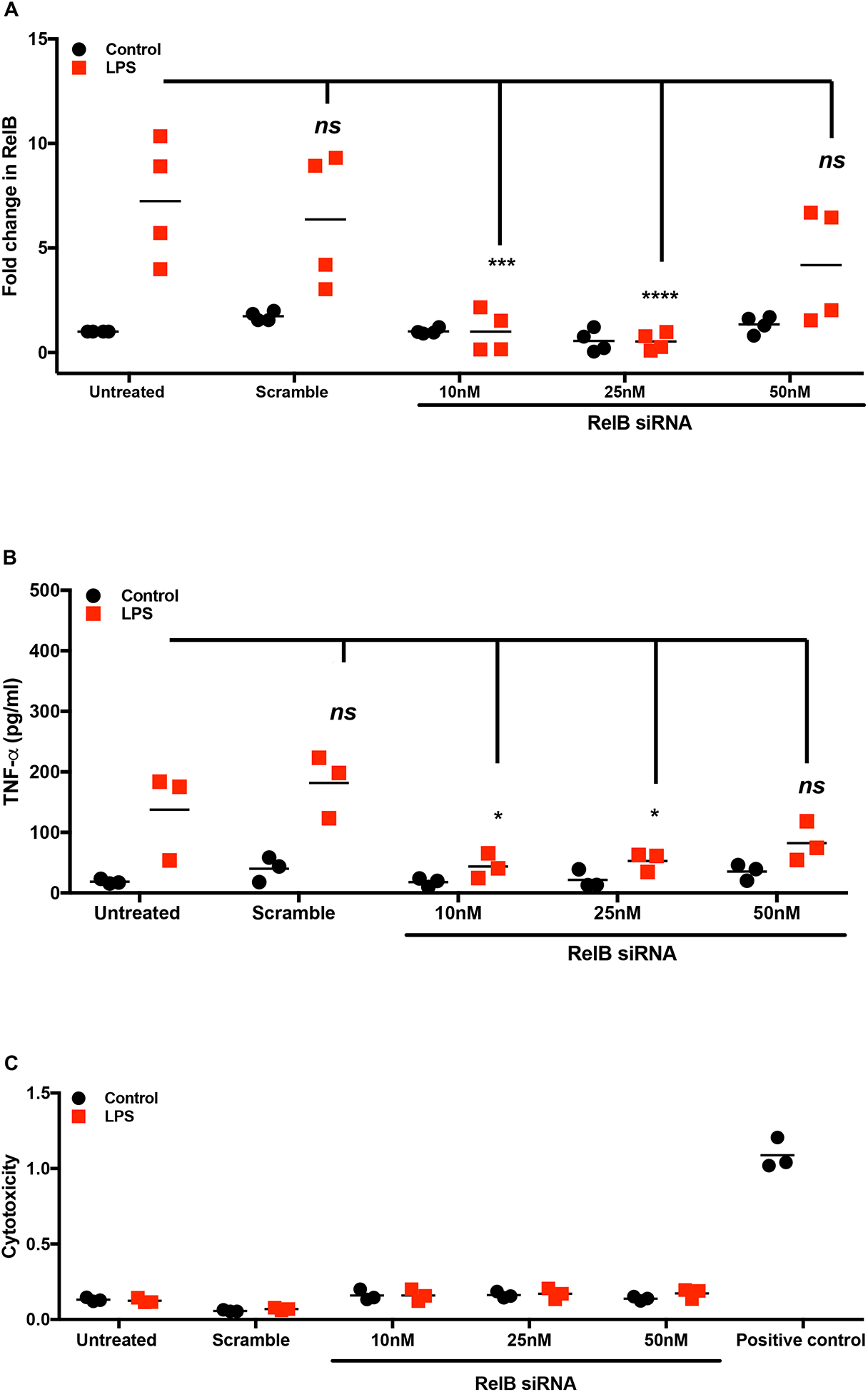
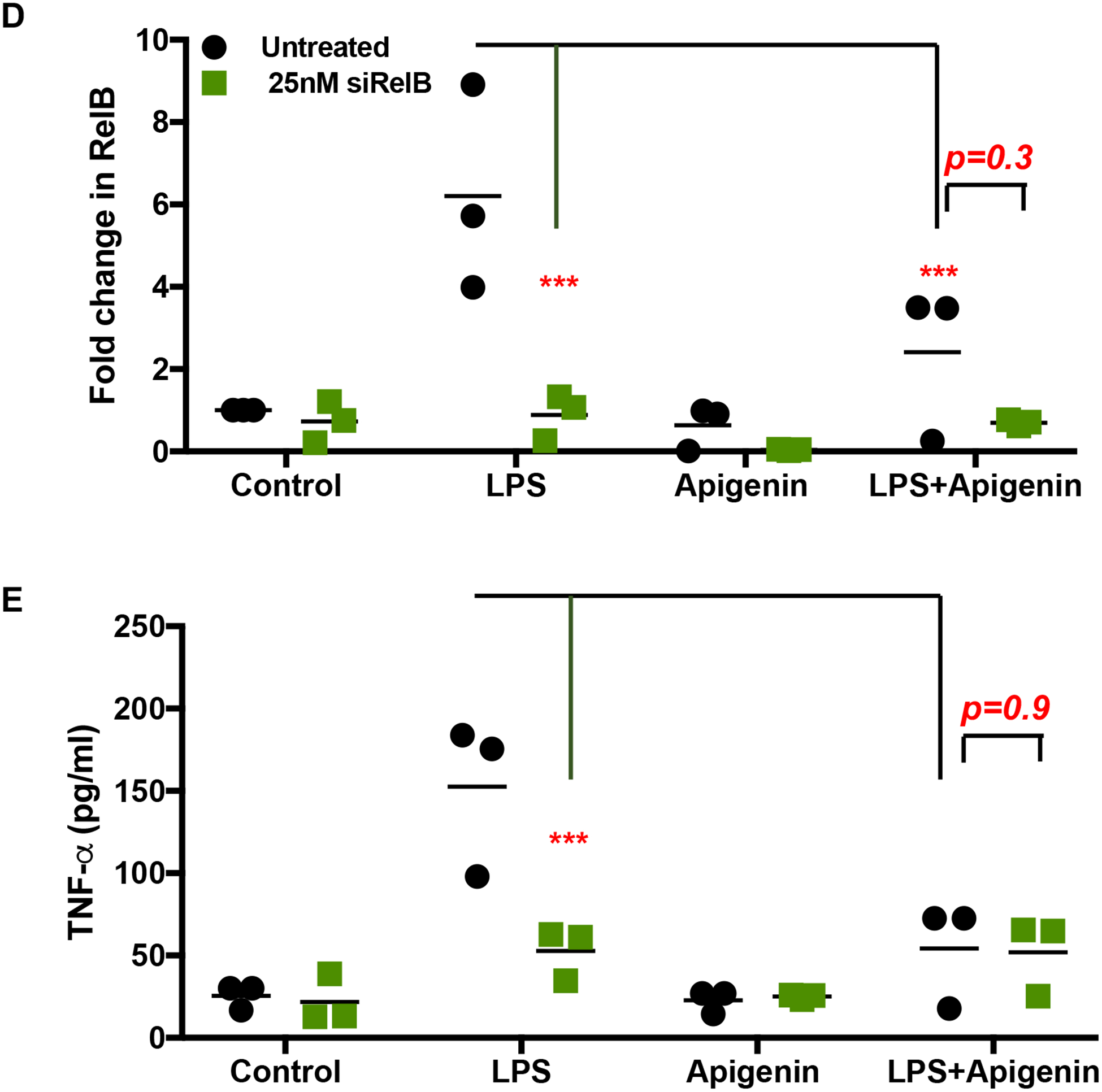
DCs from three donors were isolated as before. DCs were cultured with and without LPS for 24h. siRNA directed against RelB was transfected in to the DCs along with a negative control at different concentrations (10–50nM) using GeneSilencer transfection reagent for 36h. Cells and culture supernatants were collected for mRNA analysis and ELISA. Figures show dose-dependent changes in A) RelB mRNA expression B) TNF-α secretion and C) cellular cytotoxicity as determined by LDH assay. Following dosing experiments 25nM siRNA directed against RelB was transfected in to DCs matured with LPS and treated with 20μM Apigenin. Figures show changes in D) RelB mRNA expression, and E) TNF-α secretion. Statistical significance was determined by 2-way ANOVA using Tukey’s test (A-C) or Sidak’s test (D and E) for multiple comparisons (*p<0.05, ***p<0.001, ****p<0.0001).
Figure 7. RelB silencing may potentiate Apigenin effects on phenotype of mature DCs.
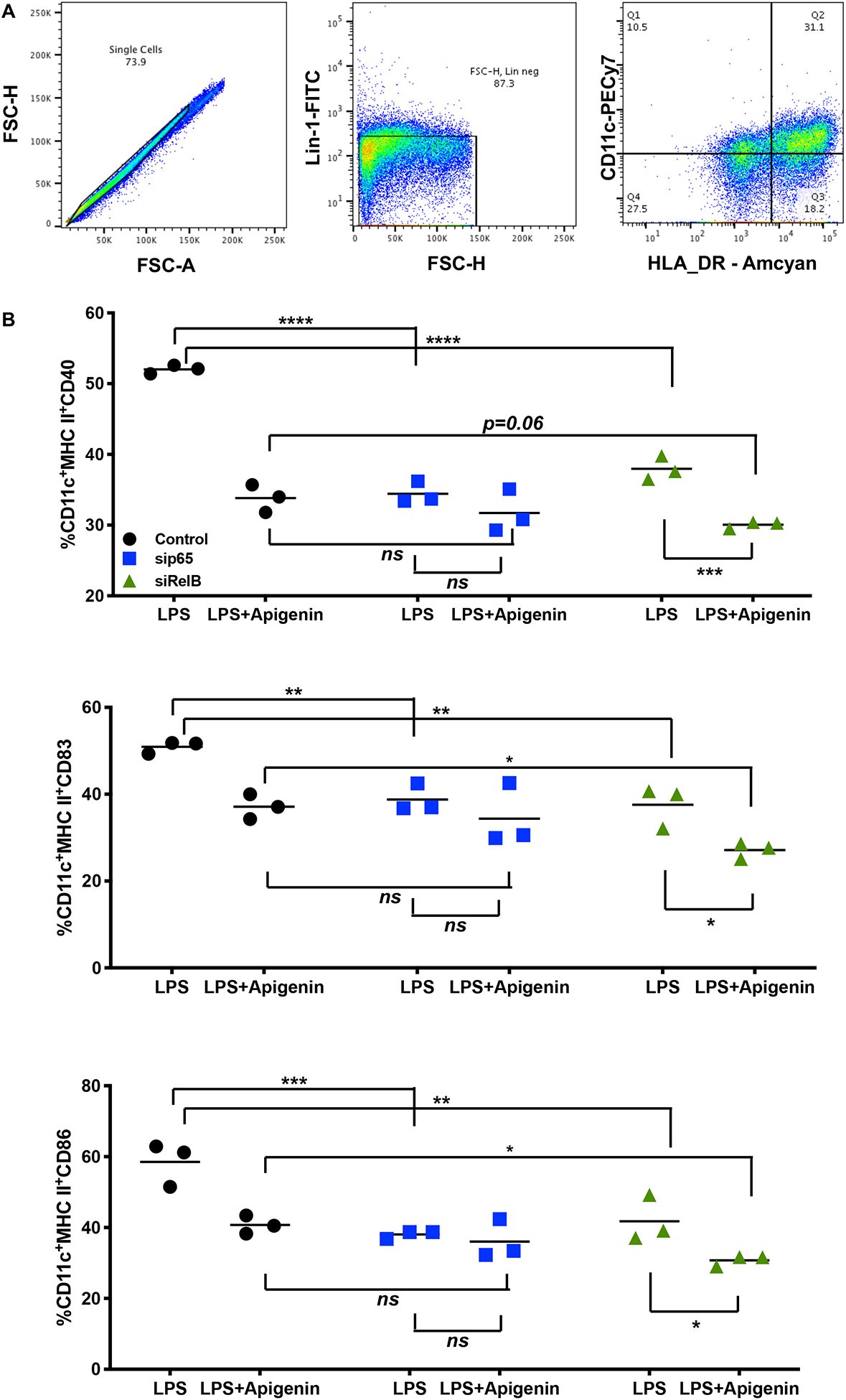
DCs from three donors were isolated as in Figure 1. DCs were cultured with and without LPS for 24h followed by treatment with 20μM Apigenin. 25nM siRNA directed against RelB was transfected in to DCs. sip65 was used as positive control. A) Live single cell populations were gated for Lin-1−CD11c+HLA-DR+ DCs. B) Changes in DC phenotype were determined by flow cytometry. Statistical significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Figure 8. RelB silencing boosts DC-mediated regulation of T-cell differentiation in presence of Apigenin.
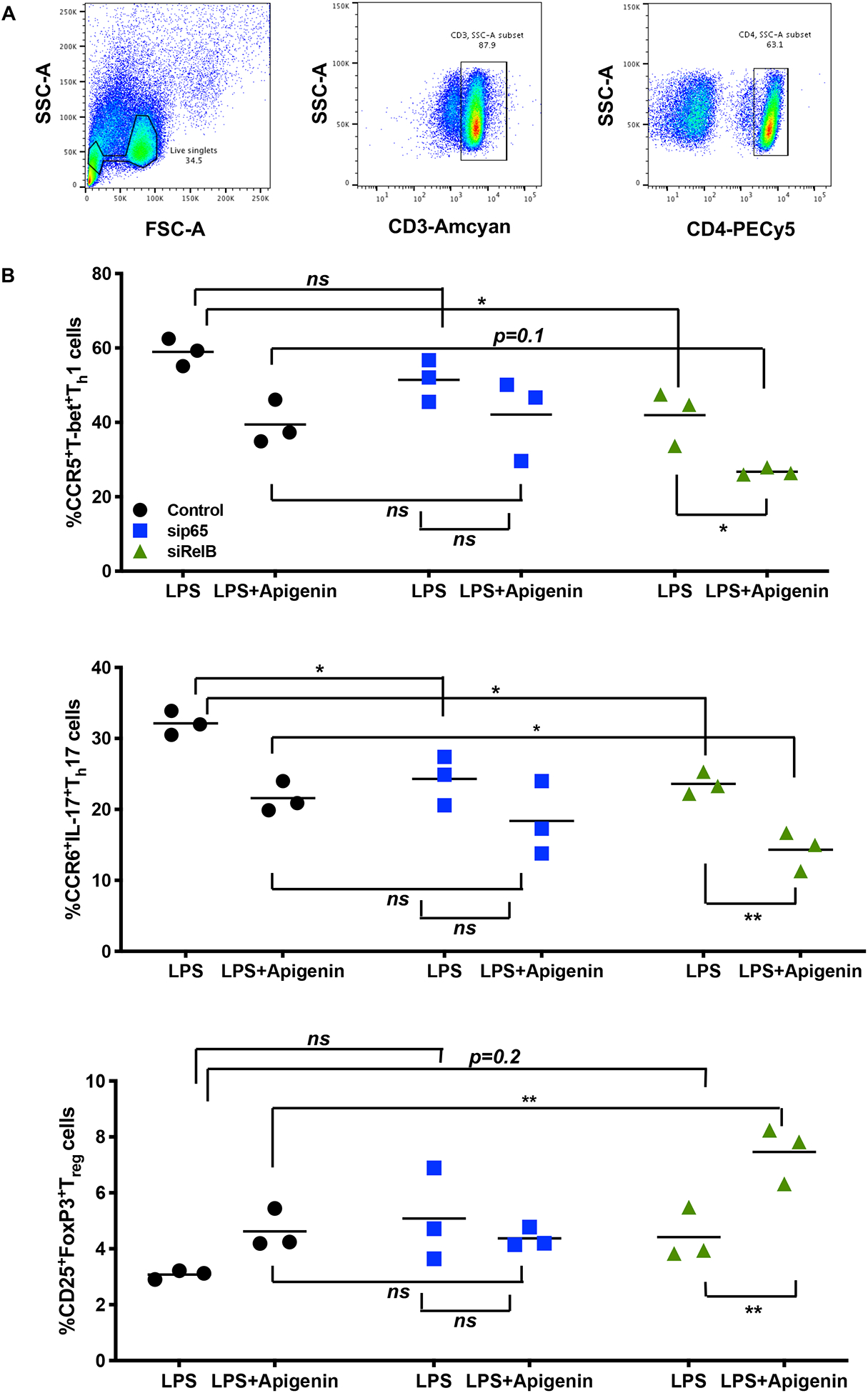
DCs from 3 donors were isolated as before and LPS-matured followed by treatment with 20μM Apigenin. 25nM siRelB was transfected in to DCs. sip65 at 25nM was used as positive control. 36h post transfection DCs were co-cultured with autologous PBLs for 6d. Cytokine secretion was blocked in the final 16h of culture by the addition of Brefeldin A and Monensin. Flow cytometry analysis of cell surface markers and intracellular transcription factors and cytokines was then performed. A) Live single cell populations were gated for CD3+CD4+ T cells. B) Enumeration of the various T-cell subsets upon both RelB knockdown and Apigenin treatment. Statistical significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons (*p<0.05, **p<0.01).
Discussion
In order to assess the therapeutic potential of Apigenin in neuroinflammatory disorders we recently decided to study its effects in mouse models of MS (Ginwala et al. 2016), a chronic autoimmune neuroinflammatory disease of the CNS characterized by episodes of reversible neurological deficits with no curative FDA approved therapy. Apigenin is of our specific interest as an immunomodulator since it exhibits low intrinsic toxicity and is not mutagenic as compared to other structurally related flavonoids (Chuang et al. 2009; Czeczot et al. 1990; Shukla and Gupta 2010). Several studies have proven the anti-inflammatory properties of flavones in general and Apigenin in particular (Byun et al. 2013; Middleton et al. 2000; Ross and Kasum 2002; Sae-Wong et al. 2011; Serafini et al. 2010). Despite numerous in vitro studies addressing the anti-inflammatory effects of Apigenin (Funakoshi-Tago et al. 2011; Jeong et al. 2009; Karamese et al. 2016; Kumar et al. 2018; Lee et al. 2007; Patil et al. 2016; Sadraei et al. 2017; Wang et al. 2014), its effect on key molecular targets controlling dendritic cell phenotypical and functional characteristics remain to be elucidated. DCs are potent antigen presenting cells that have been shown to efficiently interact and migrate across the inflamed microvasculature and accumulate in increased numbers in the CNS under inflammatory conditions (Colton 2013; Jain et al. 2010; Manuel et al. 2007; Sagar et al. 2012). Additionally, depending on their local environment, DCs are responsible for either priming or tolerizing naïve T cells, the key effectors in most neuroinflammatory conditions (Ludewig et al. 2016; Steinman 2003). Regulation of DC function is therefore critical in the management of neuroinflammatory diseases which is why we present here the molecular events that are regulated by Apigenin contributing to DC biology. For the efficient analyses of Apigenin effects on DC phenotype and function we initiated our studies with appropriate dosing and kinetic study of Apigenin activity. Myeloid cells including DCs and macrophages are key producers of TNF-α in response to TLR4 stimulation, critically influencing the nature of the inflammatory and immunological responses and its dysregulation contributes to the pathogenesis of several autoimmune diseases (Blanco et al. 2008; Krausgruber et al. 2010). Additionally, TNF-α has been shown previously to be successfully inhibited by apigenin in several cell types including macrophages and DCs (Kumazawa et al. 2006; Seo et al. 2014; Zhang et al. 2014). TNF-α is initially produced in a transmembrane form (tmTNF-α) and later released in the form of soluble protein (sTNF-α). At low levels, TNF-α exerts its protective effects of pathogen control and tissue regeneration through the interaction of tmTNF-α with TNFR2. At higher concentrations under inflammatory insult, sTNF-α induces the pro-inflammatory actions of TNF-α through TNFR1 signaling (Kemanetzoglou and Andreadou 2017; Olmos and Llado 2014). Since, selective inhibition of sTNF-α has been found beneficial in EAE, we chose to measure its secretion by DCs in response to LPS stimulation under the influence of different doses of Apigenin. We found that Apigenin is able to exert its anti-inflammatory activity in DCs at a dose of 20μM wherein it inhibits TNF-α mRNA and protein levels (Fig. 1A). This dose is in line with a number of studies showing similar results of Apigenin treatment in DCs and other cell types (Wang et al. 2018; Yoon et al. 2006). Apigenin showed maximum inhibition of TNF-α secretion by both immature and LPS-matured DCs when the cells where treated with Apigenin for 3h (Fig. 1B). Additionally, no cytotoxicity was seen over the time course of Apigenin treatment (Fig. 1E) reinstating the non-toxic nature of this flavonoid. These pharmacokinetics will be important for subsequent in vivo experiments aimed at projecting Apigenin as a therapeutic agent and suggest that Apigenin is well-tolerated and potent despite the traditionally difficult bioavailability associated with natural products.
Neuroinflammation, specifically in the context of MS, remains a serious concern for many patients. DCs have been seen to traffic into the CNS in MS and are responsible for subsequent T-cell trafficking into the CNS and their subsequent reactivation (Lüssi et al. 2016). DCs can be seen to promote the pro-inflammatory environment in the CNS and their presence in CNS lesions under inflammatory conditions has been seen in multiple studies (Colton 2013; Manuel et al. 2007; Sagar et al. 2012). In this study we measured the changes in expression of cell-surface molecules on DCs that are responsible for their maturation, migration and subsequent role in antigen presentation to T cells. Apigenin treatment reduced the cell surface expression of antigen presentation molecules HLA-ABC and HLA-DR, co-stimulatory molecules CD40, CD83 and CD86 (Fig. 2A) and CCR7 (data not shown) that aids in DC migration to lymphoid tissues thus affecting their ability to effectively prime T cells. DCs secrete TNF-α, IL-12, IL-23 and other pro-inflammatory cytokines/chemokines in many autoimmune disorders even beyond MS (Jensen and Gad 2010). IL-1β is also necessary for terminal Th17 differentiation (Lüssi et al. 2016) while in vivo differentiated Th17 cells known to expand in vitro in response to IL-23 have been found to be highly encephalitogenic (Yang et al. 2009). We found that Apigenin inhibits the production of multiple pro-inflammatory cytokines by DCs, notably Th17 polarizing cytokines IL-12, IL-23 and IL-1β (Fig. 2B). However, DCs also have roles in peripheral tolerance, which can lead to the induction of Tregs that can downregulate neuroinflammation via TGF-β and IL-10 secretion (Lüssi et al. 2016) (Colton 2013). Also critical in the polarization to Tregs is the lack of pro-inflammatory cytokines, namely IL-6 and IL-12, which can induce polarization to a Th1 response (Maldonado and von Andrian 2010). IL-6 is specifically known to inhibit the TGF-β induced expression of FoxP3, the canonical transcription factor of Tregs, and can shift to Th17 differentiation in the presence of TGF-β (Blanco et al. 2008). Apigenin regulation of pro-inflammatory cytokines like IL-6 was seen with a concurrent increase in Treg inducing TGF-β and IL-10 (Fig. 2C). This dual modulation of DC cytokine secretion suggests that Apigenin is able to confer neuroprotective effects by a decrease in pro-inflammatory cytokine secretion while also potentially inducing tolerogenic DCs and subsequent Treg polarization, as has been seen with DCs in EAE previously (Lüssi et al. 2016).
T cells that are able to penetrate the blood brain barrier and infiltrate into the CNS classically secrete pro-inflammatory cytokines that cause neuroinflammation, namely IFN-γ, TNF-α, IL-17, etc. (Becher et al. 2017). The discovery that Th17 responses play an important role in MS manifestations has been a more recent development but is now considered to be more critical than the role of Th1 cells in this inflammatory disease (Segura et al. 2013). Notably, EAE mice lacking IL-17A have a notable delay in the kinetics of EAE development (Becher et al. 2017). In this study we did see that the altered phenotype conferred by Apigenin led the DCs to induce a more tolerogenic effect on naïve T cells. Th1/Th17 cells were reduced in number while Th2 cells and Tregs were boosted by Apigenin treated DCs (Fig. 3). The switch from Th1/Th17 immunity toTh2 immunity via increased secretion of IL-4 and IL-10 produces an anti-inflammatory response and aids in tissue repair following an inflammatory insult (Allen and Wynn 2011; Wu et al. 2017). Consistent with surface marker expression we showed decrease in IFN-γ and IL-17 secretion by T cells differentiated under the influence of Apigenin-treated DCs. Both phenotypical and functional shift from Th1/Th17 to Th2 may contribute towards the protective role of Apigenin in inflammatory conditions.
CD11c+ DCs were shown to be sufficient to present myelin antigen to naive T cells leading to the development of EAE in mice (Greter et al. 2005). Furthermore, their interaction with naive CD4+ T cells driving Th17 differentiation, a T-cell subset involved in chronic inflammatory disease (Bailey et al. 2007; Isaksson et al. 2009) is a hallmark of EAE. We thus investigated the effects of Apigenin on DC-mediated polarization of MOG35–55 specific T cells from the periphery of EAE animals. Ex vivo antigen re-stimulation led to increases in Th1/Th17 related T-bet, IFN-γ, and IL-17 levels (Fig. 4A) which were then reduced upon Apigenin treatment. We also verified these effects in the absence of any antigen or antigen presenting cells, wherein naïve T cells were differentiated with the help of polarizing antibodies and cytokines. Even in this case we see for the first time that Apigenin is able to drive T-cell responses to a more regulatory phenotype demonstrating direct effects on T cells (Fig. Supplementary Fig. 1).
As a member of the NF-κβ transcription factor family, RelB is considered a master regulator of DC maturation (Platzer et al. 2004). RelB is involved in the non-canonical NF-κβ pathway of DC activation and maturation, where heterodimeric p100/RelB is cleaved to p52/RelB and translocated into the nucleus for downstream transcription (Platzer et al. 2004). Additionally, RelB activation through the RelB-p50 canonical pathway has also been implicated in DC maturation (Shih et al. 2012). Notably, both RelB pathways are activated rapidly in response to LPS stimuli in DCs leading to the production of several pro-inflammatory cytokines (Shih et al. 2012) (Gasparini et al. 2013). As was discussed previously, Apigenin has been linked with inhibition of NF-κB pathways and subsequent downregulation of inflammation; however, most studies have focused on the canonical pathways of NF-κβ mediated by p65 translocation (Cardenas et al. 2016). In particular, Apigenin has been seen to downregulate NF-κβ expression and subsequent IL-6, IL-1β and TNF-α secretion (Wang et al. 2014). Focusing on the RelB pathway, we show that RelB is translocated to the nucleus in response to LPS stimuli (Fig.5B). Apigenin is able to reduce both transcriptional levels and protein levels of RelB. Surprisingly, Apigenin treatment also seems to cause comparatively lower amounts of RelB to translocate into the nucleus. This is reflective of RelB’s ability to bind multiple partners prior to nuclear translocation. RelB is also known to associate with other proteins such as aryl hydrocarbon receptor (AhR), which may be responsible for its switching of the DC phenotype to a more regulatory pathway (Thomas 2013). RelB-AhR dimers have been previously seen to regulate DC tolerance responses during inflammation (Thomas 2013). In mature DCs RelB translocates to the nucleus for CD28-mediated co-stimulation of FoxP3+ Tregs. Other co-stimulatory cell surface markers, such as PDL1, are also involved in this induction (Thomas 2013). Thus, the anti-inflammatory activity of Apigenin may be reflective of both decreased canonical NF-κB activity as well as increased RelB-AhR translocation, however, further studies are required to confirm this partnership. Role of RelB in Apigenin-mediated regulation of DC phenotypical and functional characteristics was further confirmed through RelB silencing via siRNA. This showed an obvious potentiation of Apigenin effects on DC phenotypical traits as described earlier in Fig. 2 as well as functional activities in T-cell polarization (Fig. 6D–G, Fig. 7, and Fig. 8). Overall, this study suggests that Apigenin is able to regulate DC cytokine secretion by suppressing Th1/Th17 inducing inflammatory cytokines with a concomitant potentiation of anti-inflammatory Th2 and Treg cytokines in a RelB-dependent mechanism.
Conclusions
DCs amongst other myeloid cells are critical to the initiation and maintenance of inflammatory events that lead to the pathological outcomes in MS. So far there is no therapy targeting DC activity and its subsequent role in T-cell activation and reactivation in the CNS. Our efforts in defining the effects of plant phenolic compound Apigenin on DCs have shown an induction of a regulatory phenotype in both DCs and DC-mediated polarized T cells through downmodulation of the NF-κB protein RelB post Apigenin treatment. Apigenin can thus serve as a pharmaceutical lead compound for development as a potential therapy against MS and other neuroinflammatory conditions.
Supplementary Material
Supplementary Figure 1. Antigen independent naïve T-cell polarization. Splenocytes were pooled from naïve mice and total T cells were isolated as per the EasySep mouse T-cell isolation protocol. T cells were stimulated for 72h with different mixtures of cytokines and antibodies to induce T-cell polarization. The T cells were cultured in the presence or absence of 20μM Apigenin. T-bet, IL-4, IL-17, and FoxP3 mRNA expression was detected by qPCR and compared to appropriate control (left). Culture supernatants were extracted and cytokines TNF-α, IL-17, IL-4, and TGF-β levels were detected by ELISA (right). Each data point is representative of 2 individual experiments with two replicates.
Supplementary Figure 2. Reduced RelB expression in EAE and naïve mouse splenocytes. Splenocytes from EAE and naïve mice were stimulated with and without MOG35–55 peptide for 3 days in the presence or absence of 20μM Apigenin. Cells were pelleted, and RNA was extracted using the TRIzol method followed by cDNA preparation and qPCR. A) RelB mRNA expression compared to β-Actin and normalized to cells that were not treated with MOG35–55 peptide. Naïve T cells that were polarized as described in Figure 5 were analyzed for RelB mRNA expression in the Th1, Th2 and Th17 subsets. B) RelB mRNA expression compared to β-Actin and normalized to the Th0 subset. Sample standard error (SE) is shown and statistical significance was determined by Student’s t-test. (**p<0.01)
Supplementary Figure 3. RelB silencing does not potentiate Apigenin effect in immature DCs. DCs were isolated as in Figure 1 followed by treatment with 20μM Apigenin. 25nM siRNA directed against RelB was transfected in to DCs. sip65 was used as positive control. Changes in DC phenotype were determined by flow cytometry. Figures show FACS plot of cell surface markers expressed on DCs.
Supplementary Figure 4. RelB silencing does not significantly modulate differentiation of Th2 cells. DCs and autologous PBLs from 3 donors were treated as described in Figure 8. Enumeration of the Th2 cell subset upon both RelB knockdown and Apigenin treatment is shown. Statistical significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons.
Supplementary Figure 5. RelB silencing increases numbers of Tregs cultured in the presence of Apigenin treated immature DCs. DCs were isolated as described. DCs were treated with 20μM Apigenin or left untreated. 25nM siRelB was transfected in to DCs. sip65 was used as positive control. Following transfection DCs were co-cultured with autologous PBLs for 6d as described. FACS plots describe T-cell subset polarization in presence of immature DCs treated/untreated with Apigenin.
Funding
These studies have been funded in part with NIH/NINDS R01 NS097147 to PJ. Authors also wish to acknowledge the National MS Society grant RG 4471A6/2 that supported FB.
List of Abbreviations
- BBB
Blood-brain barrier
- TNF-α
Tumor necrosis factor
- IFN-γ
Interferon-γ
- NF-κB
Nuclear factor kappa B
- MS
Multiple Sclerosis
- CNS
Central nervous system
- EAE
Experimental autoimmune encephalomyelitis
- Treg
T regulatory cell
- DC
Dendritic cells
- PBMC
Peripheral blood mononuclear cell
- PBL
Peripheral blood lymphocyte
- LPS
Lipopolysaccharide
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Competing interests
The authors declare that they have no competing interests.
Ethics approval
The C57BL/6 mice were treated in accordance to NIH guidelines under protocols approved by the Institutional Animal Care and Use Committee of Drexel University.
Availability of supporting data
Please contact the corresponding author for requests for obtaining the datasets generated during this study.
References:
- Allen JE, Wynn TA (2011) Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens PLoS Pathog 7:e1002003 doi: 10.1371/journal.ppat.1002003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD (2007) CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE Nature immunology 8:172–180 doi: 10.1038/ni1430 [DOI] [PubMed] [Google Scholar]
- Bearoff F et al. (2016) Natural genetic variation profoundly regulates gene expression in immune cells and dictates susceptibility to CNS autoimmunity Genes Immun 17:386–395 doi: 10.1038/gene.2016.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Spath S, Goverman J (2017) Cytokine networks in neuroinflammation Nat Rev Immunol 17:49–59 doi: 10.1038/nri.2016.123 [DOI] [PubMed] [Google Scholar]
- Blanco P, Palucka AK, Pascual V, Banchereau J (2008) Dendritic cells and cytokines in human inflammatory and autoimmune diseases Cytokine Growth Factor Rev 19:41–52 doi: 10.1016/j.cytogfr.2007.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno A et al. (2011) Apigenin affects leptin/leptin receptor pathway and induces cell apoptosis in lung adenocarcinoma cell line Eur J Cancer 47:2042–2051 doi: 10.1016/j.ejca.2011.03.034 [DOI] [PubMed] [Google Scholar]
- Byun S et al. (2013) Src kinase is a direct target of apigenin against UVB-induced skin inflammation Carcinogenesis 34:397–405 doi: 10.1093/carcin/bgs358 [DOI] [PubMed] [Google Scholar]
- Cardenas H et al. (2016) Dietary Apigenin Exerts Immune-Regulatory Activity in Vivo by Reducing NF-kappaB Activity, Halting Leukocyte Infiltration and Restoring Normal Metabolic Function Int J Mol Sci 17:323 doi: 10.3390/ijms17030323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejas PJ et al. (2005) Regulation of RelB expression during the initiation of dendritic cell differentiation Mol Cell Biol 25:7900–7916 doi: 10.1128/MCB.25.17.7900-7916.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CM, Monie A, Wu A, Hung CF (2009) Combination of apigenin treatment with therapeutic HPV DNA vaccination generates enhanced therapeutic antitumor effects Journal of biomedical science 16:49 doi: 10.1186/1423-0127-16-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA (2013) Immune heterogeneity in neuroinflammation: dendritic cells in the brain J Neuroimmune Pharmacol 8:145–162 doi: 10.1007/s11481-012-9414-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeczot H et al. (1990) Isolation and studies of the mutagenic activity in the Ames test of flavonoids naturally occurring in medical herbs Mutation research 240:209–216 [DOI] [PubMed] [Google Scholar]
- Funakoshi-Tago M, Nakamura K, Tago K, Mashino T, Kasahara T (2011) Anti-inflammatory activity of structurally related flavonoids, Apigenin, Luteolin and Fisetin Int Immunopharmacol 11:1150–1159 doi: 10.1016/j.intimp.2011.03.012 [DOI] [PubMed] [Google Scholar]
- Gasparini C, Foxwell BM, Feldmann M (2013) RelB/p50 regulates TNF production in LPS-stimulated dendritic cells and macrophages Cytokine 61:736–740 doi: 10.1016/j.cyto.2012.12.029 [DOI] [PubMed] [Google Scholar]
- Gentile D et al. (2018) The flavonoid compound apigenin prevents colonic inflammation and motor dysfunctions associated with high fat diet-induced obesity PLoS One 13:e0195502 doi: 10.1371/journal.pone.0195502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginwala R et al. (2016) Apigenin, a Natural Flavonoid, Attenuates EAE Severity Through the Modulation of Dendritic Cell and Other Immune Cell Functions J Neuroimmune Pharmacol 11:36–47 doi: 10.1007/s11481-015-9617-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M et al. (2005) Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis Nature medicine 11:328–334 doi: 10.1038/nm1197 [DOI] [PubMed] [Google Scholar]
- Hofmann J, Mair F, Greter M, Schmidt-Supprian M, Becher B (2011) NIK signaling in dendritic cells but not in T cells is required for the development of effector T cells and cell-mediated immune responses J Exp Med 208:1917–1929 doi: 10.1084/jem.20110128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson M, Ardesjo B, Ronnblom L, Kampe O, Lassmann H, Eloranta ML, Lobell A (2009) Plasmacytoid DC promote priming of autoimmune Th17 cells and EAE Eur J Immunol 39:2925–2935 doi: 10.1002/eji.200839179 [DOI] [PubMed] [Google Scholar]
- Jain P, Ahuja J, Khan ZK, Shimizu S, Meucci O, Jennings SR, Wigdahl B (2007) Modulation of dendritic cell maturation and function by the Tax protein of human T cell leukemia virus type 1 J Leukoc Biol 82:44–56 doi: 10.1189/jlb.1006641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Coisne C, Enzmann G, Rottapel R, Engelhardt B (2010) Alpha4beta1 integrin mediates the recruitment of immature dendritic cells across the blood-brain barrier during experimental autoimmune encephalomyelitis J Immunol 184:7196–7206 doi: 10.4049/jimmunol.0901404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen SS, Gad M (2010) Differential induction of inflammatory cytokines by dendritic cells treated with novel TLR-agonist and cytokine based cocktails: targeting dendritic cells in autoimmunity J Inflamm (Lond) 7:37 doi: 10.1186/1476-9255-7-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong GS, Lee SH, Jeong SN, Kim YC, Kim EC (2009) Anti-inflammatory effects of apigenin on nicotine- and lipopolysaccharide-stimulated human periodontal ligament cells via heme oxygenase-1 Int Immunopharmacol 9:1374–1380 doi: 10.1016/j.intimp.2009.08.015 [DOI] [PubMed] [Google Scholar]
- Jung UJ, Cho YY, Choi MS (2016) Apigenin Ameliorates Dyslipidemia, Hepatic Steatosis and Insulin Resistance by Modulating Metabolic and Transcriptional Profiles in the Liver of High-Fat Diet-Induced Obese Mice Nutrients 8 doi: 10.3390/nu8050305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamese M, Erol HS, Albayrak M, Findik Guvendi G, Aydin E, Aksak Karamese S (2016) Anti-oxidant and anti-inflammatory effects of apigenin in a rat model of sepsis: an immunological, biochemical, and histopathological study Immunopharmacol Immunotoxicol 38:228–237 doi: 10.3109/08923973.2016.1173058 [DOI] [PubMed] [Google Scholar]
- Kemanetzoglou E, Andreadou E (2017) CNS Demyelination with TNF-alpha Blockers Curr Neurol Neurosci Rep 17:36 doi: 10.1007/s11910-017-0742-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausgruber T, Saliba D, Ryzhakov G, Lanfrancotti A, Blazek K, Udalova IA (2010) IRF5 is required for late-phase TNF secretion by human dendritic cells Blood 115:4421–4430 doi: 10.1182/blood-2010-01-263020 [DOI] [PubMed] [Google Scholar]
- Krementsov DN, Noubade R, Dragon JA, Otsu K, Rincon M, Teuscher C (2014) Sex-specific control of central nervous system autoimmunity by p38 mitogen-activated protein kinase signaling in myeloid cells Ann Neurol 75:50–66 doi: 10.1002/ana.24020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KS, Sabu V, Sindhu G, Rauf AA, Helen A (2018) Isolation, identification and characterization of apigenin from Justicia gendarussa and its anti-inflammatory activity Int Immunopharmacol 59:157–167 doi: 10.1016/j.intimp.2018.04.004 [DOI] [PubMed] [Google Scholar]
- Kumar S, Pandey AK (2013) Chemistry and biological activities of flavonoids: an overview ScientificWorldJournal 2013:162750 doi: 10.1155/2013/162750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumazawa Y, Kawaguchi K, Takimoto H (2006) Immunomodulating effects of flavonoids on acute and chronic inflammatory responses caused by tumor necrosis factor alpha Curr Pharm Des 12:4271–4279 [DOI] [PubMed] [Google Scholar]
- Lee JH, Zhou HY, Cho SY, Kim YS, Lee YS, Jeong CS (2007) Anti-inflammatory mechanisms of apigenin: inhibition of cyclooxygenase-2 expression, adhesion of monocytes to human umbilical vein endothelial cells, and expression of cellular adhesion molecules Arch Pharm Res 30:1318–1327 [DOI] [PubMed] [Google Scholar]
- Leyva-Lopez N, Gutierrez-Grijalva EP, Ambriz-Perez DL, Heredia JB (2016) Flavonoids as Cytokine Modulators: A Possible Therapy for Inflammation-Related Diseases Int J Mol Sci 17 doi: 10.3390/ijms17060921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M et al. (2007) Immune modulation and tolerance induction by RelB-silenced dendritic cells through RNA interference J Immunol 178:5480–5487 [DOI] [PubMed] [Google Scholar]
- Li X et al. (2016) Apigenin, a potent suppressor of dendritic cell maturation and migration, protects against collagen-induced arthritis J Cell Mol Med 20:170–180 doi: 10.1111/jcmm.12717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig P et al. (2016) Dendritic cells in brain diseases Biochim Biophys Acta 1862:352–367 doi: 10.1016/j.bbadis.2015.11.003 [DOI] [PubMed] [Google Scholar]
- Lüssi F, Zipp F, Witsch E (2016) Dendritic cells as therapeutic targets in neuroinflammation Cell Mol Life Sci 73:2425–2450 doi: 10.1007/s00018-016-2170-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado RA, von Andrian UH (2010) How tolerogenic dendritic cells induce regulatory T cells Adv Immunol 108:111–165 doi: 10.1016/B978-0-12-380995-7.00004-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel SL, Rahman S, Wigdahl B, Khan ZK, Jain P (2007) Dendritic cells in autoimmune diseases and neuroinflammatory disorders Front Biosci 12:4315–4335 [DOI] [PubMed] [Google Scholar]
- Middleton E Jr., Kandaswami C, Theoharides TC (2000) The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer Pharmacological reviews 52:673–751 [PubMed] [Google Scholar]
- Millington C et al. (2014) Chronic neuroinflammation in Alzheimer’s disease: new perspectives on animal models and promising candidate drugs Biomed Res Int 2014:309129 doi: 10.1155/2014/309129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad MG et al. (2012) Dendritic cells and multiple sclerosis: disease, tolerance and therapy Int J Mol Sci 14:547–562 doi: 10.3390/ijms14010547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubade R et al. (2011) Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis Blood 118:3290–3300 doi: 10.1182/blood-2011-02-336552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubade R, Milligan G, Zachary JF, Blankenhorn EP, del Rio R, Rincon M, Teuscher C (2007) Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice J Clin Invest 117:3507–3518 doi: 10.1172/JCI32792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos G, Llado J (2014) Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity Mediators Inflamm 2014:861231 doi: 10.1155/2014/861231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onasanwo SA, Velagapudi R, El-Bakoush A, Olajide OA (2016) Inhibition of neuroinflammation in BV2 microglia by the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant protective mechanism Mol Cell Biochem 414:23–36 doi: 10.1007/s11010-016-2655-8 [DOI] [PubMed] [Google Scholar]
- Ota A, Ulrih NP (2017) An Overview of Herbal Products and Secondary Metabolites Used for Management of Type Two Diabetes Front Pharmacol 8:436 doi: 10.3389/fphar.2017.00436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil RH et al. (2016) Anti-Inflammatory Effect of Apigenin on LPS-Induced Pro-Inflammatory Mediators and AP-1 Factors in Human Lung Epithelial Cells Inflammation 39:138–147 doi: 10.1007/s10753-015-0232-z [DOI] [PubMed] [Google Scholar]
- Platzer B, Jorgl A, Taschner S, Hocher B, Strobl H (2004) RelB regulates human dendritic cell subset development by promoting monocyte intermediates Blood 104:3655–3663 doi: 10.1182/blood-2004-02-0412 [DOI] [PubMed] [Google Scholar]
- Ross JA, Kasum CM (2002) Dietary flavonoids: bioavailability, metabolic effects, and safety Annual review of nutrition 22:19–34 doi: 10.1146/annurev.nutr.22.111401.144957 [DOI] [PubMed] [Google Scholar]
- Sadraei H, Asghari G, Khanabadi M, Minaiyan M (2017) Anti-inflammatory effect of apigenin and hydroalcoholic extract of Dracocephalum kotschyi on acetic acid-induced colitis in rats Res Pharm Sci 12:322–329 doi: 10.4103/1735-5362.212050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sae-Wong C, Matsuda H, Tewtrakul S, Tansakul P, Nakamura S, Nomura Y, Yoshikawa M (2011) Suppressive effects of methoxyflavonoids isolated from Kaempferia parviflora on inducible nitric oxide synthase (iNOS) expression in RAW 264.7 cells Journal of ethnopharmacology 136:488–495 doi: 10.1016/j.jep.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Sagar D, Foss C, El Baz R, Pomper MG, Khan ZK, Jain P (2012) Mechanisms of dendritic cell trafficking across the blood-brain barrier J Neuroimmune Pharmacol 7:74–94 doi: 10.1007/s11481-011-9302-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura E et al. (2013) Human inflammatory dendritic cells induce Th17 cell differentiation Immunity 38:336–348 doi: 10.1016/j.immuni.2012.10.018 [DOI] [PubMed] [Google Scholar]
- Seo HS, Sikder MA, Lee HJ, Ryu J, Lee CJ (2014) Apigenin Inhibits Tumor Necrosis Factor-alpha-Induced Production and Gene Expression of Mucin through Regulating Nuclear Factor-Kappa B Signaling Pathway in Airway Epithelial Cells Biomol Ther (Seoul) 22:525–531 doi: 10.4062/biomolther.2014.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini M, Peluso I, Raguzzini A (2010) Flavonoids as anti-inflammatory agents The Proceedings of the Nutrition Society 69:273–278 doi: 10.1017/S002966511000162X [DOI] [PubMed] [Google Scholar]
- Shih VF et al. (2012) Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways Nat Immunol 13:1162–1170 doi: 10.1038/ni.2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Gupta S (2010) Apigenin: a promising molecule for cancer prevention Pharm Res 27:962–978 doi: 10.1007/s11095-010-0089-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM (2003) The control of immunity and tolerance by dendritic cell Pathol Biol (Paris) 51:59–60 [DOI] [PubMed] [Google Scholar]
- Sung B, Chung HY, Kim ND (2016) Role of Apigenin in Cancer Prevention via the Induction of Apoptosis and Autophagy J Cancer Prev 21:216–226 doi: 10.15430/JCP.2016.21.4.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R (2013) RelB and the aryl hydrocarbon receptor: dendritic cell tolerance at the epithelial interface Immunol Cell Biol 91:543–544 doi: 10.1038/icb.2013.51 [DOI] [PubMed] [Google Scholar]
- Tong X, Pelling JC (2013) Targeting the PI3K/Akt/mTOR axis by apigenin for cancer prevention Anticancer Agents Med Chem 13:971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venigalla M, Gyengesi E, Munch G (2015) Curcumin and Apigenin - novel and promising therapeutics against chronic neuroinflammation in Alzheimer’s disease Neural Regen Res 10:1181–1185 doi: 10.4103/1673-5374.162686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visavadiya NP et al. (2016) Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3 Cell Commun Signal 14:32 doi: 10.1186/s12964-016-0157-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu YT, Xiao L, Zhu L, Wang Q, Yan T (2014) Anti-inflammatory effects of apigenin in lipopolysaccharide-induced inflammatory in acute lung injury by suppressing COX-2 and NF-kB pathway Inflammation 37:2085–2090 doi: 10.1007/s10753-014-9942-x [DOI] [PubMed] [Google Scholar]
- Wang Q, Cui W, Liu M, Zhang J, Liao R-q, Liao X-l, Yang J (2015) An improved synthesis of apigenin Journal of Chemical Research 39:67+ [Google Scholar]
- Wang X, Wang W, Wang JZ, Yang C, Liang CZ (2018) Effect of apigenin on apoptosis induced by renal ischemia/reperfusion injury in vivo and in vitro Ren Fail 40:498–505 doi: 10.1080/0886022X.2018.1497517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GF, Laufer TM (2007) The role of dendritic cells in multiple sclerosis Curr Neurol Neurosci Rep 7:245–252 [DOI] [PubMed] [Google Scholar]
- Wu Q, Wang Q, Mao G, Dowling CA, Lundy SK, Mao-Draayer Y (2017) Dimethyl Fumarate Selectively Reduces Memory T Cells and Shifts the Balance between Th1/Th17 and Th2 in Multiple Sclerosis Patients J Immunol 198:3069–3080 doi: 10.4049/jimmunol.1601532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZX, Zhang HL, Wu XJ, Zhu J, Ma DH, Jin T (2015) Role of the immunogenic and tolerogenic subsets of dendritic cells in multiple sclerosis Mediators Inflamm 2015:513295 doi: 10.1155/2015/513295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Qi M, Li P, Zhan Y, Shao H (2017) Apigenin in cancer therapy: anti-cancer effects and mechanisms of action Cell Biosci 7:50 doi: 10.1186/s13578-017-0179-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y et al. (2009) T-bet is essential for encephalitogenicity of both Th1 and Th17 cells J Exp Med 206:1549–1564 doi: 10.1084/jem.20082584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS et al. (2006) Apigenin inhibits immunostimulatory function of dendritic cells: Implication of immunotherapeutic adjuvant Mol Pharmacol 70:1033–1044 doi: 10.1124/mol.106.024547 [DOI] [PubMed] [Google Scholar]
- Zanetti M, Castiglioni P, Schoenberger S, Gerloni M (2003) The role of relB in regulating the adaptive immune response Ann N Y Acad Sci 987:249–257 [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang G, Gurley EC, Zhou H (2014) Flavonoid apigenin inhibits lipopolysaccharide-induced inflammatory response through multiple mechanisms in macrophages PLoS One 9:e107072 doi: 10.1371/journal.pone.0107072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang JL, Wang YR, Fa XZ (2013) Apigenin attenuates copper-mediated beta-amyloid neurotoxicity through antioxidation, mitochondrion protection and MAPK signal inactivation in an AD cell model Brain Res 1492:33–45 doi: 10.1016/j.brainres.2012.11.019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Antigen independent naïve T-cell polarization. Splenocytes were pooled from naïve mice and total T cells were isolated as per the EasySep mouse T-cell isolation protocol. T cells were stimulated for 72h with different mixtures of cytokines and antibodies to induce T-cell polarization. The T cells were cultured in the presence or absence of 20μM Apigenin. T-bet, IL-4, IL-17, and FoxP3 mRNA expression was detected by qPCR and compared to appropriate control (left). Culture supernatants were extracted and cytokines TNF-α, IL-17, IL-4, and TGF-β levels were detected by ELISA (right). Each data point is representative of 2 individual experiments with two replicates.
Supplementary Figure 2. Reduced RelB expression in EAE and naïve mouse splenocytes. Splenocytes from EAE and naïve mice were stimulated with and without MOG35–55 peptide for 3 days in the presence or absence of 20μM Apigenin. Cells were pelleted, and RNA was extracted using the TRIzol method followed by cDNA preparation and qPCR. A) RelB mRNA expression compared to β-Actin and normalized to cells that were not treated with MOG35–55 peptide. Naïve T cells that were polarized as described in Figure 5 were analyzed for RelB mRNA expression in the Th1, Th2 and Th17 subsets. B) RelB mRNA expression compared to β-Actin and normalized to the Th0 subset. Sample standard error (SE) is shown and statistical significance was determined by Student’s t-test. (**p<0.01)
Supplementary Figure 3. RelB silencing does not potentiate Apigenin effect in immature DCs. DCs were isolated as in Figure 1 followed by treatment with 20μM Apigenin. 25nM siRNA directed against RelB was transfected in to DCs. sip65 was used as positive control. Changes in DC phenotype were determined by flow cytometry. Figures show FACS plot of cell surface markers expressed on DCs.
Supplementary Figure 4. RelB silencing does not significantly modulate differentiation of Th2 cells. DCs and autologous PBLs from 3 donors were treated as described in Figure 8. Enumeration of the Th2 cell subset upon both RelB knockdown and Apigenin treatment is shown. Statistical significance was determined by 2-way ANOVA using Sidak’s test for multiple comparisons.
Supplementary Figure 5. RelB silencing increases numbers of Tregs cultured in the presence of Apigenin treated immature DCs. DCs were isolated as described. DCs were treated with 20μM Apigenin or left untreated. 25nM siRelB was transfected in to DCs. sip65 was used as positive control. Following transfection DCs were co-cultured with autologous PBLs for 6d as described. FACS plots describe T-cell subset polarization in presence of immature DCs treated/untreated with Apigenin.