

Abstract
Cysteines existing in the deprotonated thiolate form or having a tendency to become deprotonated are important players in enzymatic and cellular redox functions and frequently exploited in covalent drug design; however, most computational studies assume cysteines as protonated. Thus, developing an efficient tool that can make accurate and reliable predictions of cysteine protonation states is timely needed. We recently implemented a generalized Born (GB) based continuous constant pH molecular dynamics (CpHMD) method in Amber for protein pKa calculations on CPUs and GPUs. Here we benchmark the performance of GB-CpHMD for predictions of cysteine pKa’s and reactivities using a data set of 24 proteins with both down- and upshifted cysteine pKa’s. We found that 10-ns single-pH or 4-ns replica-exchange CpHMD titrations gave root-mean-square errors of 1.2–1.3 and correlation coefficients of 0.8–0.9 with respect to experiment. The accuracy of predicting thiolates or reactive cysteines at physiological pH with single-pH titrations is 86 or 81% with a precision of 100 or 90%, respectively. This performance well surpasses the traditional structure-based methods, particularly, a widely used empirical pKa tool which gives an accuracy less than 50%. We discuss simulation convergence, dependence on starting structures, common determinants of the pKa downshifts and upshifts as well as the origin of the discrepancies from the structure-based calculations. Our work suggests that CpHMD titrations can be performed on a desktop computer equipped with a single GPU card to predict cysteine protonation states for a variety of applications, from understanding biological functions to covalent drug design.
INTRODUCTION
Cysteines are important players in cellular redox regulation.1 For example, oxidative protein folding involves thiol-disulfide exchange reactions,2 and under oxidative stress cysteine thiols in antioxidant enzymes and cytoplasmic glutathiones (a tripeptide Glu-Cys-Gly) are invoked to eliminate reactive oxygen species.3–6 All these processes are believed to involve a reactive cysteine that is in the negatively charged thiolate form (–S−)1,6,7 or has a high tendency to shift from the neutral thiol (–SH) to the thiolate form. The inverse relationship between cysteine reactivity and its pKa is well established through measurements of the kinetic rate constants of thiol containing compounds or peptides and the pKa’s of the thiol groups.7,8 Reactive cysteines are highly nucleophilic and can undergo thiol-Michael addition with electrophiles, making them suitable as covalent linkage sites for covalent inhibitors.9,10 Thus, knowledge of reactive cysteines would assist in covalent drug design for kinases and other targets that are not amenable to traditional reversible inhibition. Over the past decade, isoTOP-ABPP (isotopic tandem orthogonal proteolysisactivity-based protein profiling) techniques have been developed to scan for reactive cysteines in human proteins.11,12 The data from the isoTOP-ABPP experiments were used to develop structure-based bioinformatics13 and sequence-based machine learning tools14 to predict reactive cysteines. However, the reliability of these data-driven tools remains unclear, particularly because the protein profiling techniques can generate false positives.15 Thus, developing a physics-based in silico capability to accurately and reliably predict thiol reactivities is highly desirable.16
Recently, using a data set of 18 proteins, Rowley and coworker17 evaluated several computational methods for predicting cysteine pKa’s, including free energy simulations based on thermodynamic integration (TI), traditional structure-based pKa calculations based on solving the Poisson-Boltzmann (PB) equation,18–20 and the empirical pKa calculations using PROPKA.21 They found that the computationally costly TI method with the CHARMM C3622 and the modified Amber ff99SB-ILDNP23 force fields gave respective root-mean-square errors (RMSE’s) of 2.4 and 3.2 relative to experimental pKa’s, which are lower than the RMSE’s of 3.4–4.7 from the PB and empirical calculations.17 However, these errors are on par with the RMSE of 2.7 from the null model, which assumes that all cysteines have the model or solution pKa value.17 Thus, the work of Rowley and coworker demonstrated a need to improve the accuracies of computational methods for predicting cysteine pKa’s.
While TI methods are not routinely used for pKa calculations due to the extremely high computational cost, structure-based PB and empirical methods have been widely used. Over the past decade, constant pH molecular dynamics (MD) simulations have emerged as alternative powerful tools for pKa calculations.24 In constant pH MD,25–33 protonation states of titratable sites are determined on the fly based on the free energies of protonation relative to that in solution. Since constant pH MD describes the coupling between conformational changes and protonation/deprotonation events, pKa predictions can be more accurate than structure-based methods, particularly for buried residues34–36 and when protonation/deprotonation of two or more residues is highly coupled.37 The interested reader is referred to the reviews38,39 as well as the original articles on the discrete constant pH MD techniques,25,27,30–32 which combine MD with Monte-Carlo sampling of discrete protonation states, and the continuous constant pH MD (CpHMD) techniques,26,28,29,35,40,41 which make use of the extended Hamiltonian λ-dynamics approach42 to sample protonation states through an auxiliary set of continuous titration coordinates.
In CpHMD, the free energies of protonation are calculated for all titratable sites in a single MD trajectory; this is in contrast to TI simulations, wherein the free energy of titrating a single site is calculated while fixing the protonation states of all other sites. Consequently, in addition to having a substantially lower computational cost, CpHMD can be more accurate than TI due to its ability to account for the coupling between interacting titration sites. In an effort to develop an accurate and efficient pKa prediction tool, we recently implemented the CPU43 and GPU44 versions of the CpHMD method based on the Amber generalized Born (GB) model GBNeck245 in the Amber molecular dynamics package (Amber1846). In comparison to older GB models, such as GBSW47 employed in the GB-based CpHMD method in CHARMM,26,28 GBNeck245 more accurately reproduces the solvation free energies from PB calculations and the experimental structures and stabilities of model peptides and proteins.45,48
CpHMD simulations can be carried out in two modes. A single or fixed pH titration returns the protonation probabilities of specified titratable sites at a specified pH. Running the titration at several pH allows one to calculate the pKa’s by fitting the protonation probabilities vs. pH to the Henderson-Hasselbalch equation. CpHMD can also be performed with the pH replica-exchange protocol,40 whereby multiple independent titrations are run at a set of pH conditions and periodic swaps between adjacent pH conditions are accepted according to the Metropolis criterion. Compared to single pH titrations, the replica-exchange protocol can accelerate pKa convergence and better resolve the pKa’s of coupled residues due to enhanced sampling of both conformational and protonation states.30–32,40,44 However, single pH titrations are more convenient for routine use on a desktop computer equipped with one or two graphics processing units (GPU) cards. Encouragingly, our recent work44 showed that 2 ns single pH titrations with the GBNeck2-CpHMD method gave comparable overall accuracy as replica-exchange titrations for the pKa’s of Asp, Glu, and His residues in 10 benchmark proteins. While CpHMD simulations can also be used to predict Cys pKa’s,10 a systematic evaluation of the prediction accuracy and precision has not been conducted.
The main objective of the present work is to benchmark the performance and protocols of GBNeck2-CpHMD titrations in both single-pH and replica-exchange modes for predicting cysteine pKa values and reactive cysteines. We used a data set comprising the aforementioned targets compiled by the Rowley group which have mostly downshifted cysteine pKa’s17 and additional targets which have mostly upshifted pKa’s taken from a database developed by the Alexov group.49 In addition to assessing the accuracy of pKa calculations, we evaluated the accuracy and precision of predicting thiolates or reactive cysteines at physiological pH, defined as those with a pKa lower than 7.4 or 8.5, respectively.10 To determine the most accurate and yet efficient protocols for practical applications, we analyzed the convergence and accuracies of single-pH and replica-exchange titrations as well as the dependence of the pKa results on the starting structures. Finally, we examined the common determinants of cysteine pKa downshifts and upshifts and the origin of the discrepancies from the structure-based PB and empirical calculations. For 21 targets, the 10-ns single-pH CpHMD titrations predicted thiolates or reactive cysteines with an accuracy of 81 or 86% with a precision of 91 or 100%; which is in contrast to the PB-based calculations and the popular empirical method PROPKA21 which gives a prediction accuracy below 50%. However, our work also exposed a caveat of the current protocols or methodology which correctly predicted the protonation states at physiological pH, but were not able to yield a pKa value for the deeply buried cysteine involved in several hydrogen bonds in three phosphatases.
METHODS AND PROTOCOLS
Protein data set.
The first 15 proteins were taken from those evaluated by the Rowley group,17 which have an experimentally known Cys pKa. The protein names and PDB (Protein Data Bank) ID’s are as follows: α−1-antitrypsin (A1AT, PDB 1QLP50), acyl-coenzyme A binding protein wild type and M46C, S65C, and T17C mutants (ACBP, PDB 1NTI51), Salmonella alkyl hydroperoxide reductase C (AhpC, PDB 4MA952), human DJ-1 (DJ-1, PDB 1P5F53), human muscle creatine kinase and the S285A mutant (HMCK, PDB 1I0E54), sperm whale myoglobin G124C and A125C mutants (Mb, PDB 2MGE55), wild type and E115Q mutant of mouse methionine sulfoxide reductase A (MmsrA, PDB 2L9056), human O6-alkylguanine-DNA alkyltransferase (AGT, PDB 1EH657), papaya proteinase I (papain, PDB 1PPN58), and papaya protease omega (ppω, PDB 1PPO59).
Since only two of the above proteins have cysteine pKa’s upshifted relative to the model value, we added 6 proteins with upshifted experimental cysteine pKa’s. These proteins are: recombinant rat cathepsin B (Cathepsin B, PDB 1THE60), yeast ubiquitin-conjugating enzyme E2 (Ubc13, PDB 1JBB61), human ubiquitin-conjugating enzyme E2 wild type (Ubc2b, PDB 1JAS62) and E2 C114S mutant (UbcH10, PDB 1I7K,63 and acyl-coenzyme A binding protein V36C and E78C mutants (ACBP, PDB 1NTI51). We note, the Rowley data set also included human tyrosine phosphatase 1B (PTP1B, PDB 2HNP64) and Yersinia tyrosine phosphatase wild type and H402A mutant (YopH, PDB 1YPT65). These three proteins will be considered separately.
Structure preparation.
For wild type proteins, the initial structures were taken from the PDB files. Single mutations were performed with SWISS-MODEL.66 For AGT, missing residues (36–44) were added with SWISS-MODEL.66 For each structure, the CHARMM program (version c42a1)67 was used to add acetylated N terminus and amidated C terminus caps, disulfide bonds (if present), and hydrogen atoms. Initially, Asp/Glu were deprotonated, and His/Cys/Lys/Arg/Tyr were protonated. The system was then minimized with 50 steps of steepest descent method in the GBSW implicit solvent47 with a harmonic force constant of 50 kcal/mol/Å2 applied to heavy atoms. Dummy atoms were then added to Asp/Glu residues, and the structure was minimized for 10 steps of steepest descent and 10 steps of Newton-Raphson methods. Next, force field parameters and coordinate files were constructed from these structures with the Leap utility in AMBER.46 The structures were then energy minimized by 2000 steps of steepest descent followed by 8000 steps of conjugate-gradient methods in GB-Neck2 implicit solvent45 to obtain the initial structures for the CpHMD titration simulations.
Simulation protocol.
All GBNeck2-CpHMD simulations were performed using the pmemd engine of AMBER18.46 The replica-exchange titrations were performed on the CPUs43 and single pH titrations were performed on the GPUs.44 The proteins were represented by the ff14sb protein force field,68 and solvent represented by the GBNeck2 (igb=8) model with mbondi3 intrinsic Born radii and 0.15 M ionic strength.45 Modifications to the GB parameters and/or intrinsic Born radii were made for His and Cys residues, as discussed in our previous work.10,43,44 All simulations were run with an effectively infinite cutoff (999 Å) at a temperature of 300 K. SHAKE was used to allow a 2-fs time step. Asp, Glu, His, Lys, and free Cys residues were titrated, and the protonation states were recorded every 250 steps. The parameters for titrating model Asp/Glu/His/Lys/Cys were taken from our previous work.10,43,44 The model pKa is 8.55.69,70 The parameterization from thermodynamic integration and titration results of the model Cys peptide (AACAA) are given in Fig. S1. The pKa of the model Cys is 8.41±0.17 (Fig. S1). Replica-exchange titrations were performed for the first 15 proteins, whereby 12 independent replicas at pH conditions 0.5 unit apart were run for 4 ns each, and exchanges between adjacent pH conditions were attempted every 1000 MD steps unless otherwise noted. For the single pH titrations, each simulation was run for 50 ns for the first 15 proteins and 10 ns for the 6 proteins. Initially, the pH interval was 1 unit, but additional simulations at 0.5-unit interval were added if the titration curve appeared to be noisy. All other simulation settings were identical to our previous work.10,43,44 pKa’s from these simulations were obtained from fitting to the generalized Henderson-Hasselbalch (HH) equation. Error estimates were obtained from the estimated errors in the fit parameters.44
RESULTS AND DISCUSSION
Convergence and accuracy of the pKa calculations.
We first assess the convergence of the pKa calculations for the 15 proteins from the Rowley data set. Single pH titrations were performed for 50 ns at each pH. The pKa’s were calculated every 5 ns based on the cumulatively calculated unprotonated fractions at all pH (Fig. S2). Most pKa’s converged at around 25 ns; however, the pKa’s of HMCKS285A, and ACBPT17C converged more slowly and drifted towards slightly smaller values until 50 ns, while the pKa of A1AT kept decreasing past 50 ns. For this data set, the overall pKa accuracy, represented by the correlation coefficient R and root-mean-square error (RMSE) with respect to the experimental pKa’s, appeared to be best at 10 ns and slightly worsened as simulations were extended (Fig. 1a and b). The RMSE and R values are respectively 1.3 and 0.82 at 10 ns; 1.4 and 0.78 at 25 ns, and 1.4 and 0.78 at 50 ns. Compared to the single pH titrations of Asp/Glu/His in our previous work,44 the convergence rates for the present data set are much slower. This could be due to the selection biases in the two data sets. In our previous work, CpHMD titrations were performed on proteins where all titratable residues had measured pKa values, many of which do not have large shifts relative to the model values. In contrast, free cysteine residues are uncommon in nature, and the ones with experimental pKa’s are typically functional cysteines with large pKa shifts. We therefore expect the pKa calculations in the present data set to require longer sampling time to converge than those in our previous work.
Figure 1: Convergence of the simulation accuracy.
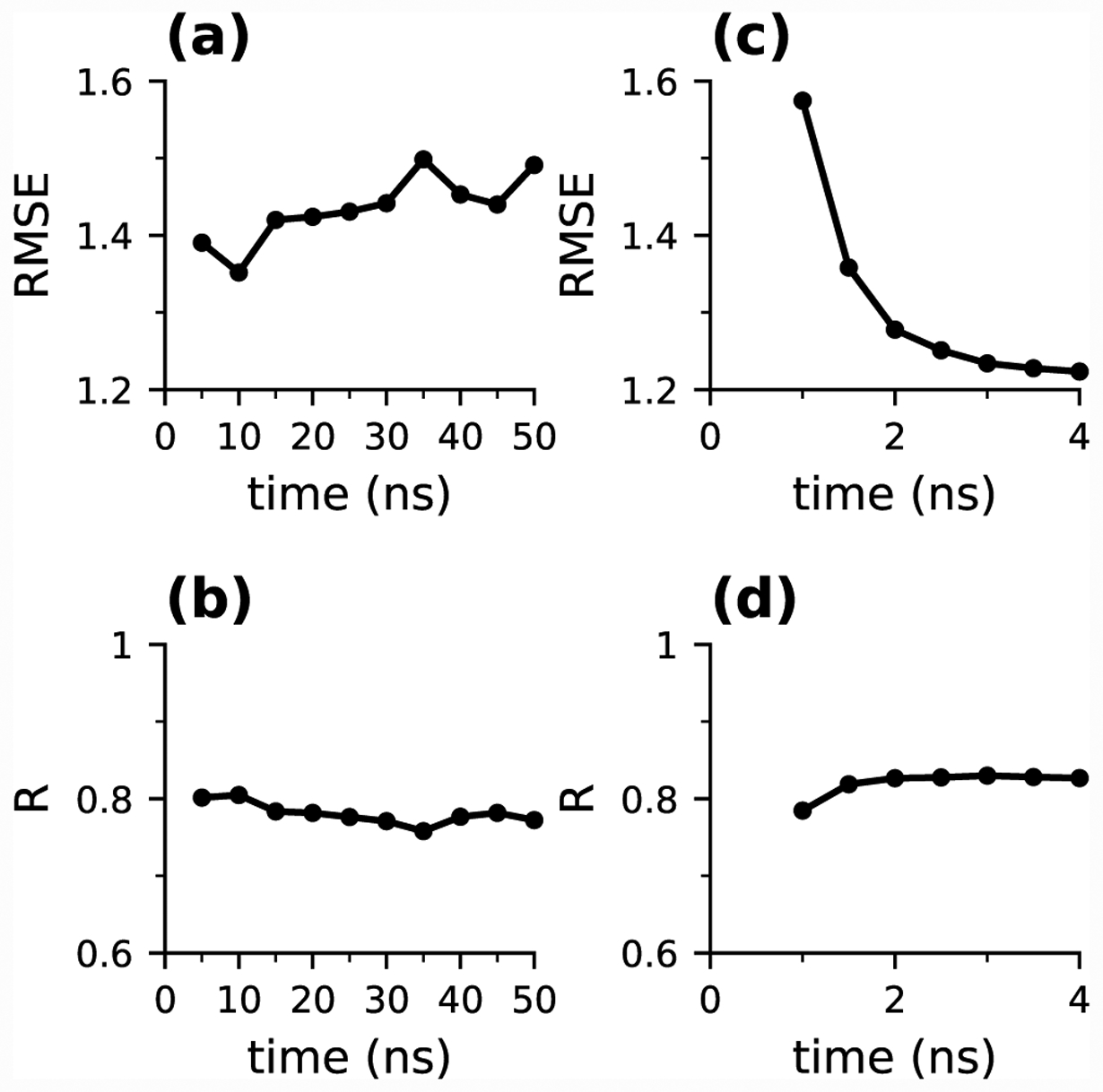
(a) and (b) Time series of root-mean-square error (RMSE) and correlation coefficient (R) with respect to the experimental data in the single-pH titrations of the 15 proteins. (c) and (d) Time series of RMSE and R in the replica-exchange titrations of the 15 proteins.
We next examine the convergence of the replica-exchange titrations. The pKa’s were calculated every 0.5 ns per replica (Fig. S3). All pKa’s converged after about 2 ns per replica, including the pKa’s of the three proteins, for which single pH titrations were not able to converge until or after 50 ns. Consistently, the RMSE and R values also plateaued after about 2 ns, with the respective values of 1.3 and 0.83 at 2 ns per replica and 1.2 and 0.83 at 4 ns per replica (Fig. 1c and d). Thus, the overall accuracy of the replica-exchange titrations of 2–4 ns per replica is similar to that of the single pH titrations of 10–50 ns per pH. The significant acceleration in convergence can be attributed to the ability of the replica-exchange protocol to overcome local barriers of hydrogen bonding which is a major contributor to the pKa shifts of cysteines (see later discussion). This is consistent with the effect of replica exchange on the pKa’s of residues involved in salt-bridge interactions.40,71 In what follows, we will focus on 10-ns single-pH and 4-ns replica-exchange simulations (Table 1).
Table 1:
Comparison of calculated pKa’s from replica-exchange and single-pH GBNeck2-CpHMD titrations with experiment, Poisson-Boltzmann and empirical predictionsa
| Protein | PDB | Residue | Expt | Replica | Single pH | H++ | MCCE | DelPhiPKa | PROPKA |
|---|---|---|---|---|---|---|---|---|---|
| Crystal structures | |||||||||
| AGT | 1EH6 | C145 | 5.373 | 6.7 | 6.4±0.01 | 9.5 | 8.3 | 5.3 | 10.6 |
| HMCK | 1I0E | C283 | 5.674 | 5.6 | 6.3±0.13 | 9.1 | 6.8 | 6.0 | 10.4 |
| DJ-1 | 1P5F | C106 | 5.475 | 3.7 | 2.4±0.40 | 11.3 | 12.6 | 6.0 | 12.3 |
| papain | 1PPN | C25 | 3.376 | 4.0 | 3.7±0.05 | 9.3 | 8.8 | 5.6 | 10.5 |
| ppΩ | 1PPO | C25 | 2.976 | 3.7 | 3.1 ±0.12 | 9.4 | 7.6 | 4.9 | 7.5 |
| A1AT | 1QLP | C232 | 6.977 | 7.3 | 6.5±0.08 | 7.3 | 8.3 | 5.5 | 9.1 |
| MmsrA | 2L90 | C72 | 7.278 | 7.9 | 8.2±0.91 | >12.0 | 16.3 | 6.6 | 13.1 |
| AhpC | 4MA9 | C46 | 5.979 | 5.2 | 5.3±0.52 | 9.4 | 9.1 | 5.8 | 9.1 |
| Cathepsin B | 1THE | C29 | 3.660 | - | 3.4±0.02 | 11.2 | - | 6.4 | 11.2 |
| Ubc2 | 1JAS | C88 | 10.280 | - | 9.4±0.55 | 9.8 | - | 6.2 | 9.1 |
| Ubc13 | 1JBB | C87 | 11.180 | - | 11.2±0.07 | 9.3 | - | 6.5 | 9.9 |
| UbcH10C114S | 1I7K | C102 | 10.980 | - | 9.0±0.15 | >12.0 | - | 6.4 | 12.7 |
| RMSE | 0.95 | 1.3(1.1) | |||||||
| R | 0.81 | 0.74(0.92) | |||||||
| Computationally mutated structures | |||||||||
| HMCKS285A | 1I0E | C283 | 6.774 | 9.8 | 9.3±0.12 | 9.3 | 6.6 | 5.9 | 11.2 |
| ACBPM46C | 1NTI | C46 | 8.251 | 7.3 | 7.6±0.15 | 8.8 | 8.8 | 6.6 | 9.0 |
| ACBPS65C | 1NTI | C65 | 9.051 | 7.8 | 8.0±0.05 | 8.8 | 9.4 | 6.7 | 9.6 |
| ACBPT17C | 1NTI | C17 | 9.851 | 10.1 | 10.5±0.03 | 8.4 | 8.8 | 6.4 | 8.9 |
| ACBPV36C | 1NTI | C36 | 9.551 | - | 8.9±0.11 | 9.0 | - | 6.2 | 8.9 |
| ACBPE78C | 1NTI | C78 | 9.651 | - | 11.5±0.18 | 8.7 | - | 6.0 | 9.1 |
| MmsrAE115Q | 2L90 | C72 | 8.278 | 8.7 | 8.0±1.21 | >12.0 | 15.4 | 6.6 | 11.4 |
| MbA125C | 2MGE | C125 | 8.481 | 8.7 | 8.7±0.21 | 8.3 | 8.8 | 6.6 | 9.2 |
| MbG124C | 2MGE | C124 | 6.581 | 8.3 | 8.7±0.06 | 8.1 | 8.5 | 6.0 | 8.4 |
| RMSE | 1.5 | 1.4 (1.3) | |||||||
| R | 0.08 | 0.2 (0.5) | |||||||
| Overall RMSE | 1.2 | 1.3 (1.2) | 3.62 | 4.22 | 2.60 | 4.02 | |||
| Overall R | 0.83 | 0.82(0.89) | −0.48 | 0.23 | 0.61 | −0.10 | |||
The pKa’s from the 4 ns (per replica) replica-exchange and 10 ns (per pH) single pH titrations are listed. For single pH titrations, the RMSE and R values in parentheses include the additional 6 proteins (see main text). For the PB-based H++,18 MCCE,19 and DelPhiPka,20 as well as the empirical PROPKA21 methods, data were taken from the previous publications if available,17,82 or computed with the H++,18 DelPhiPka20 and PDB2PQR83 (for PROPKA21) online servers.
The RMSE and R of the pKa calculations by single-pH titrations are respectively 1.3 and 0.82, while those by replica-exchange titrations are respectively 1.2 and 0.83 (Table 1). This indicates that the accuracy is similar and replica-exchange titrations are perhaps slightly more accurate (see later discussion about the calculations with crystal structures). As expected and consistent with our previous work,43,44,71 the titration data (unprotonated fractions vs. pH) from the replica-exchange simulations display excellent fits to the HH equation (except for MmsrA-E115Q), while the titration data for several proteins from the single-pH simulations are very noisy (Fig. S4 and S5). The latter results in large uncertainty in the calculated pKa, e.g., for MmsrA and the the E115Q mutant the errors are 0.9 and 1.2 respectively (Table 1).
We note, for the three remaining proteins in the Rowley data set, human and Yersinia tyrosine phosphatase proteins, PTP1B and WT/mutant YopH, the relevant cysteines remained deprotonated in the entire pH range of the single pH and replica-exchange titrations due to multiple persistent hydrogen bonds. Although the experimental pKa downshifts were correctly predicted, no specific pKa values could be assigned from these simulations. We speculate that in order to reproduce the experimental pKa’s (4–7), these persistent hydrogen bonds would need to break, which would require much longer simulation times. It is also possible that sampling in explicit solvent is required to generate the conformational changes for a protonation state switch, as demonstrated in our previous work based on the hybrid-solvent CpHMD titrations in CHARMM72 as well as work from others.35 We will further examine these systems in future work.
Dependence of the pKa calculations on the starting structures.
Although compared to structure-based PB or empirical methods, MD-based pKa calculations are less sensitive to starting structures,24,34 it is worthwhile examining the dependence of the pKa results on the starting structures, particularly given the use of implicit solvent and limited conformational sampling. To do so, we divided the 15 targets into two groups: 8 Cys pKa’s were calculated using the crystal structures (all but one are of the wild type proteins) and 7 Cys pKa’s were calculated using the computationally mutated crystal structures.
For the pKa calculations with crystal structures, the RMSE and R from single-pH titrations are respectively 1.3 and 0.74, while those from replica-exchange titrations are respectively 0.95 and 0.81. Thus, the replica-exchange simulations appear to give somewhat more accurate pKa’s. The largest pKa error and also the largest difference between the single-pH and replica-exchange calculated pKa’s is for Cys106 of DJ-1, where the experimental pKa downshift of Cys106 is overestimated by 1.7 units by replica-exchange and 3.0 units by single-pH titrations. This difference is due to the persistence of hydrogen bonding in the single-pH simulations (see later discussion).
For the pKa calculations with the mutated crystal structures, the RMSE and R from single-pH titrations are respectively 1.4 and 0.2, while those from replica-exchange titrations are respectively 1.5 and 0.08. Thus, both methods gave larger errors compared to the calculations with crystal structures, and the decrease in performance is much worse for replica-exchange titrations. These observations indicate that for mutated proteins we may need to run longer simulations so that the conformation relaxes more closely to the natural state. We can see some evidence for this from the time evolution of the RMSE in the single-pH simulations. For single pH titrations with crystal structures, the RMSE is the lowest (1.2–1.25) at 5–10 ns and increases to about 1.5 in the next 40 ns. In contrast, using the computationally mutated structures the RMSE reached a minimum of 1.25–1.3 at 20–25 ns. Thus, prolonging the single pH titrations from 10 to 20 ns somewhat improved the pKa calculations with mutated structures but has a negligible effect on the calculations with crystal structures. We did not see any noticeable improvement in the RMSE with the mutant structures in the replica-exchange titrations between 2 and 4 ns, but perhaps we would observe some improvement with significantly longer simulations.
Common determinants of the cysteine pKa downshifts.
The Rowley data set is dominated by the downshifted pKa’s of cysteine; we first examine Cys145 of AGT. Both single pH and replica-exchange titrations revealed that the buried Cys145 accepts hydrogen bonds from the hydroxyl group of Tyr158 and the amino group of Asn137 when it becomes deprotonated. Fig. 2 demonstrates that the occupancies of these hydrogen bonds increase with pH and the pH-dependence is perfectly correlated with the pH-dependent deprotonation of Cys145, suggesting that Cys145 thiolate is stabilized by the hydrogen bond interactions. Consequently, the pKa of Cys145 is downshifted relative to the model value despite the lack of solvent exposure which would raise the pKa. Interestingly, none of the hydrogen bonds is present in the initial crystal structure. This may explain why the empirical method and PB calculations (except for DelPhiPKa which uniformly predicts pKa downshifts82) overestimate the pKa of Cys145 (Table 1), as they only consider the crystal structure. This observation is consistent with our previous finding that by capturing the pH-dependent hydrogen bond formation, CpHMD titrations can make more accurate pKa predictions than structure-based methods.37
Figure 2: Cys145 thiolate in AGT is stabilized by hydrogen bonding.
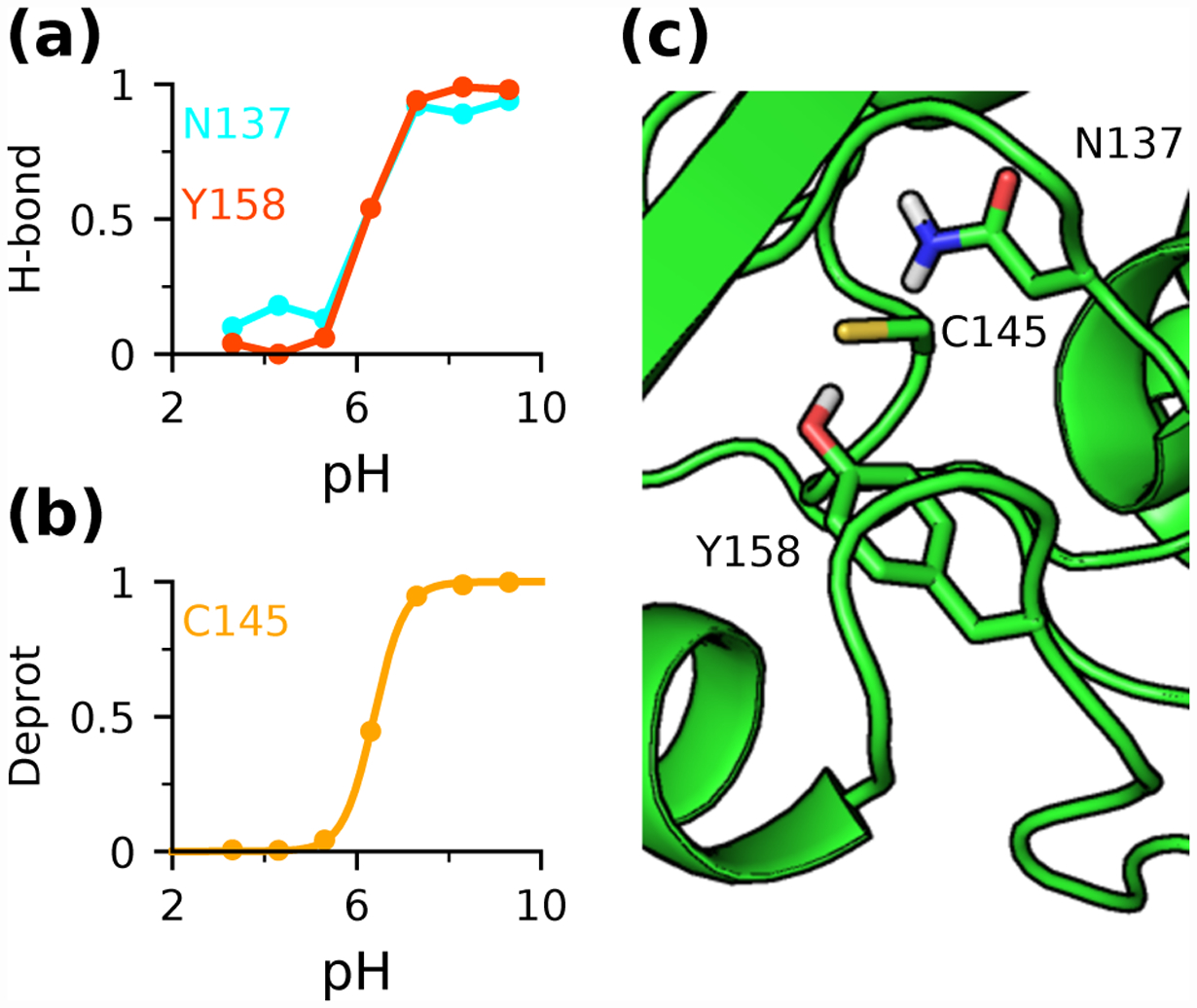
(a) Occupancies of the hydrogen bond between Cys145 and Asn137 (cyan) or Tyr158 (red) at different pH. (b) Deprotonated fractions of Cys145 (gold) at different pH. (c) Zoomed-in view of the structural environment of Cys145 in AGT (PDB ID: 1EH657). Data from (a) and (b) were obtained from 10-ns single pH titrations.
Cys283 in HMCK and HMCKS285A provides another interesting example. Consistent with our previous finding,10 both replica-exchange and single-pH simulations agree that, although Cys283 in HMCK is buried, its pKa is downshifted due to the formation of hydrogen bonds with the side chains and backbones of Ser285 and Asn286. Interestingly, when Ser285 is mutated to Ala in HMCKS285A, both sets of simulations predicted a significant increase in pKa to above the model value due to the loss of the hydrogen bond with Ser285 and destabilization of the conformation where it could form the other three hydrogen bonds. However, the experimental pKa of Cys283 in HMCKS285A is downshifted to 6.7, compared to 9.8 and 9.3 from the respective replica-exchange and single-pH titrations. One possible explanation for this discrepancy is that when modeling HMCKS285A we started with the wild-type crystal structure. It is possible that the mutant conformation differs in some way that leads to a large change in the pKa. Indeed, as the single-pH titrations were extended beyond 10 ns, a conformational change occurred such that Cys283 forms new hydrogen bonds with the backbone atoms of Thr71, Val72, and Gly73, stabilizing the thiolate form and lowering the pKa by 1.5 units to 7.8 at 50 ns, in closer agreement with the experimental value of 6.7 (Fig. S2). Notably, Cys283 became increasingly deprotonated in the simulations at pH 7.5 and above, it remained fully protonated at pH 7. Thus, we expect that with further prolonged sampling the pKa of Cys283 may stabilize just above pH 7, in better agreement with experiment. Nonetheless, it is also possible that the formation of new hydrogen bonds is an artifact of GB simulations which tend to overstabilize hydrogen bonds.40,45 A detailed investigation will be deferred to a future study using the GPU implementation of the fully explicit-solvent CpHMD as well as the asynchronous replica exchange scheme which allows replica exchange simulations to be performed on a single GPU card (Harris and Shen, work in progress).
Common determinants of the cysteine pKa upshifts.
Since the Rowley data set is dominated by downshifted Cys pKa’s, we added 5 targets with upshifted Cys pKa’s. We also included a protein with an extremely low experimental pKa to further test the accuracy and reliability for predicting downshifted Cys pKa’s (Table 1). Due to our limited computational resources, titrations were only performed in the single-pH mode for 10 ns at each pH (the sampling time found to give the smallest overall RMSE for the 15 aforementioned proteins). Including the additional 6 targets, the RMSE and R value of the single-pH titrations are 1.2 and 0.89, respectively (Table 1).
These additional data allowed us to examine the molecular determinants of the upshifted pKa’s of cysteine. It is well established that solvent exclusion favors the protonated thiol form, thus increasing the pKa; however, the pKa increase is often compensated by the ability of a buried thiolate to accept hydrogen bonds, which lowers the pKa of Cys, as demonstrated in our discussion of the downshifted pKa’s of Cys145 in AGT and Cys283 in HMCK as well as HMCKS285A. Thus, it is not surprising that the cysteines with upshifted pKa’s are (at least) partially buried but do not form frequent hydrogen bonds. Another mechanism for cysteine to have a pKa increase is to have acidic residues in the vicinity which can potentially form repulsive electrostatic interactions. For example, Cys87 in Ubc13 has the highest experimental pKa of 11.1 and the single pH titrations gave a pKa of 11.2. The simulations revealed that Cys87 is partially buried and its thiolate form is destabilized by the electrostatic repulsion with the negatively charged Asp81 that is about 4–6 Å away. Thus, both solvent exclusion and electrostatic interactions are major contributors to the pKa upshift of Cys87.
Overestimation of the pKa downshift of Cys106 in DJ-1.
The largest pKa error in this data set is for Cys106 in DJ-1. Although the direction of the pKa shift was correctly predicted by both replica-exchange and single pH titrations, the downshift was overestimated by 1.7 and 3 units, respectively. Considering the crystal structure, the downshift may be surprising, as Cys106 is largely buried, which would raise the pKa, and its proximity to Glu16 would also be thought to increase the pKa, as Glu residues are generally negatively charged at physiological pH. However, the CpHMD simulations revealed that while Cys106 becomes increasingly deprotonated as pH increases from 1.5 to 3, it accepts hydrogen bonds from the protonated carboxyl group of Glu16 and the backbone amide group of Gly75 (Fig. 3a and b). Additionally, the thiolate form is stabilized by the salt-bridge interaction with the protonated His126 (Fig. 3a and b). The latter two interactions are not present in the crystal structure; however, it is consistent with our previous finding that new hydrogen bond interactions may form to facilitate deprotonation events.37 This may explain why the structure-based PB and empirical methods failed to predict a downshifted pKa for Cys106 (Table 1), except for DelPhiPka which gave downshifted pKa’s for all targets.82 We suggest that the overestimation of the pKa downshift may be attributed to the overstabilization of hydrogen bonding, a well-known limitation of GB simulations.40,45 This problem is less severe in the replica-exchange titrations, as hydrogen bonds were able to break and reform, illustrating the benefit of the enhanced sampling provided by replica exchange.
Figure 3: Cys106 thiolate in DJ-1 is stabilized by a protonated Glu and a doubly protonated His.
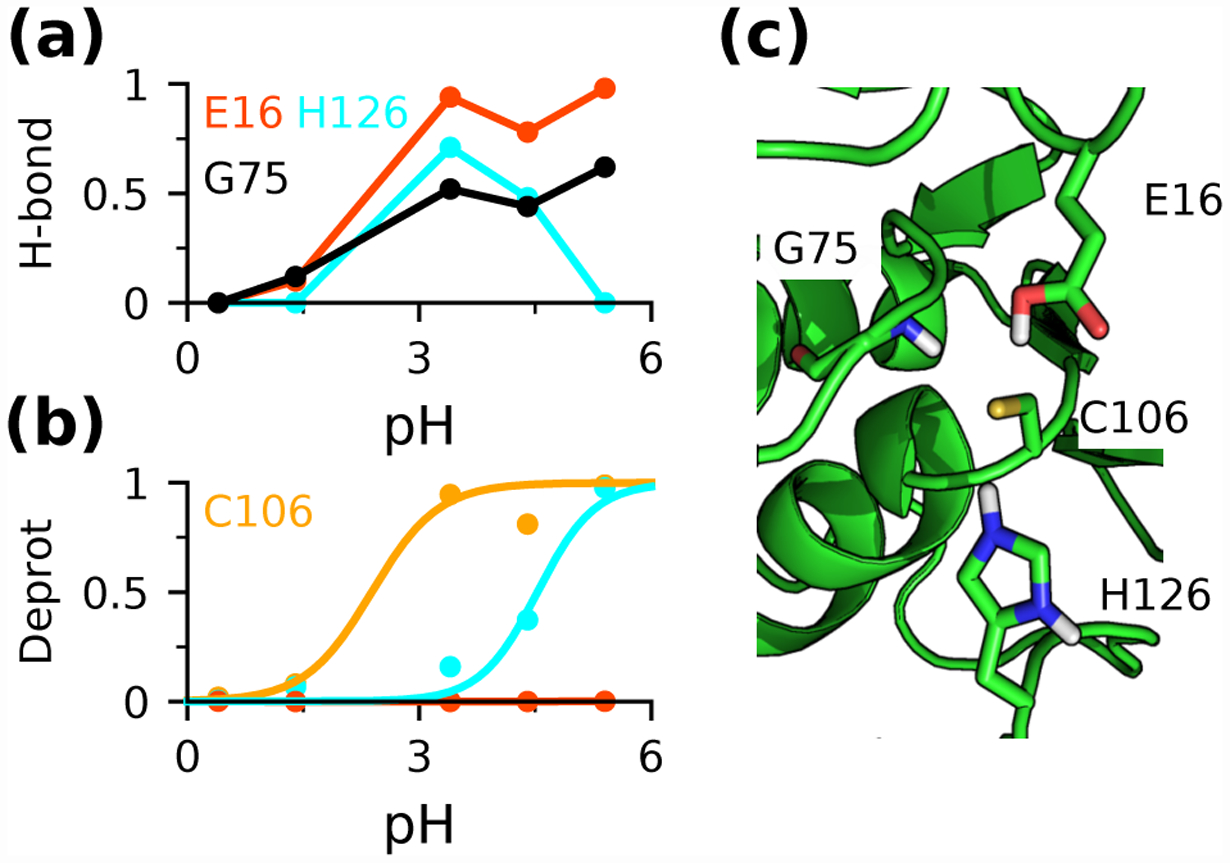
(a) Occupancies of the hydrogen bonds between Cys106 and Glu16 (red), His126 (cyan), or Gly75 (black) at different pH. (b) Deprotonated fractions of Cys106 (yellow) and His126 (cyan) at different pH. (c) Zoomed-in view of the structural environment of Cys106 in DJ-1 (PDB ID: 1P5F53).
Performance of predicting thiolates and reactive cysteines.
Another objective of the present work is to evaluate whether CpHMD can reliably predict thiolates at pH 7.4 (physiological condition) or pH 8.5. The latter can be used to identify reactive cysteines, which are defined as those existing in the thiolate form for at least 10% at physiological pH.10 To do the evaluations, we grouped the data of predicted vs. experimental pKa’ into four quadrants around the dividing pH of 7.4 for protonation state prediction and 8.5 for cysteine reactivity prediction. Data in the lower left quadrant are the true positives (TP), i.e., both predicted and experimental pKa’s are lower than 7.4 or 8.5. Data in the upper left quadrant are the false positives (FP), i.e., the predicted pKa is lower and the experimental pKa is higher than 7.4 or 8.5. Data in the lower right quadrant are the false negatives (FN), i.e., the predicted pKa is higher and the experimental pKa is lower than 7.4 or 8.5. Data in the upper right quadrant are the true negatives (TN), i.e., both predicted and experimental pKa’s are higher than 7.4 or 8.5.
The percentages of TP, FP, TN, and TP are entered in a confusion matrix for evaluating the accuracy and reliability of single pH CpHMD predictions based on the 21 targets (Table 1). The accuracy for predicting thiolates or reactive cysteines at physiological pH, defined as (TN+TP)/(TN+TP+FN+FP), is 86% or 81%, respectively (Fig. 4). The precision (also known as the positive predictive value) of the predictions, defined as TP/(TP+FP), is 100% or 91%, respectively. The miss rate (also known as the false negative rate) of the predictions, defined as FN/(TP+FN), is 27% or 21%, respectively. A similar analysis for the replica-exchange titrations gave an accuracy of 74% with a precision of 87% or 90% for predicting thiolates or reactive cysteines at physiological pH (Fig. S6). The accuracy is slightly lower due to the smaller data set (the additional targets with upshifted pKa’s were not included).
Figure 4: Performance of single-pH titrations for predicting cysteine pKa’s and identify thiolates at pH 7.5 or 8.5.
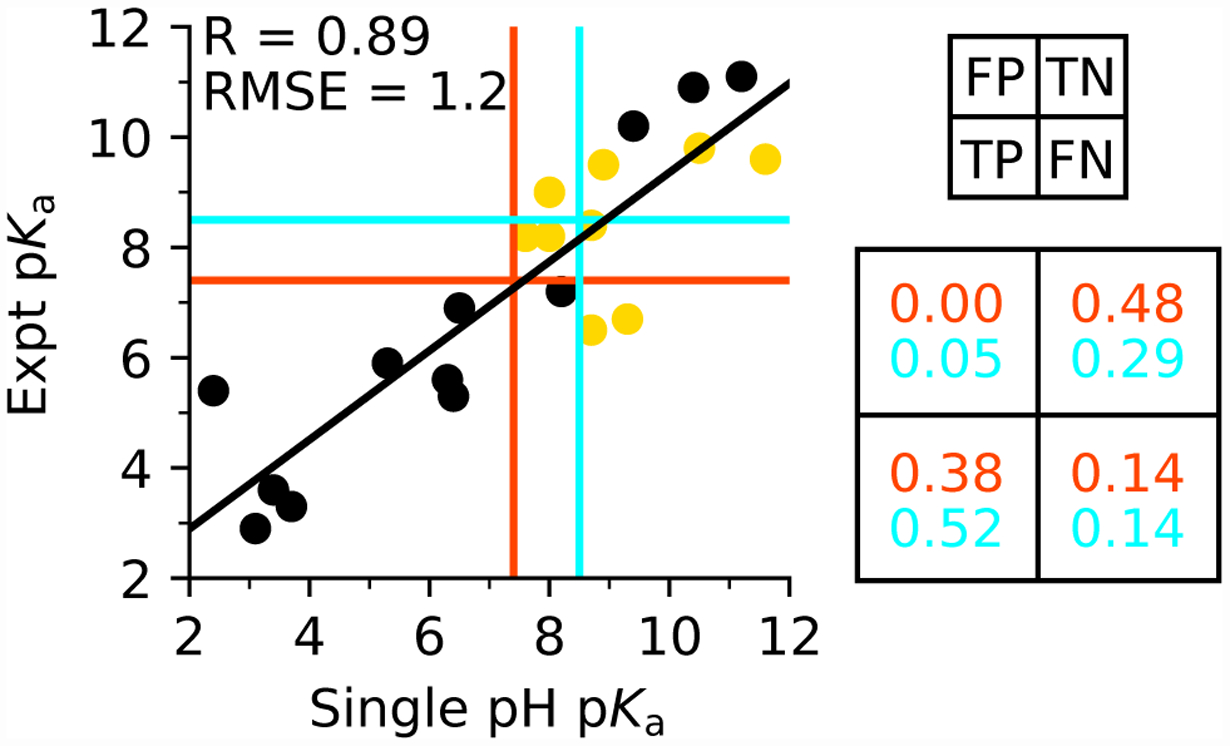
Comparison between experimental pKa’s and those obtained from 10-ns single-pH titrations of the entire data set of 21 proteins. Linear regression line is shown in black, and the correlation coefficient R and RMSE are given. Calculations based on crystal structures are shown in black, and those based on the computationally mutated structures in gold. The data are grouped into four quadrants around the dividing pH 7.4 (red) or pH 8.5 (cyan). A confusion matrix is shown on the right, with the rates for false positives (FP), true positives (TP), true negatives (TN), and false negatives (FN) given for thiolate predictions for pH 7.4 (red) and pH 8.5 (cyan). See main text for more explanation.
Comparison to the structure-based calculations.
To compare with the popular structure-based pKa calculation methods, Table 1 also lists the Cys pKa’s predicted by the PB solvers H++,18 MCCE,19 and DelphiPKa20 as well as the empirical method PROPKA.21 The RMSE’s range from 2.6 to 4.0, and the R values range from −0.48 to 0.61. The smallest error and best correlation is given by DelPhiPKa;20 however, it predicts that all Cys pKa’s are downshifted. With regards to prediction of thiolates or reactive cysteines at physiological pH, the accuracy of these methods is about 50%, with the exception of DelphiPKa which gave 67% for predicting thiolates although the precision also 67% (Table S1). We suggest that the better performance of CpHMD relative to structure-based methods is the ability to capture pH-dependent formation of hydrogen bonds or salt bridges that are not present in the starting structure. As discussed above, Cys145 in AGT and Cys106 in DJ-1 have downshifted experimental pKa’s, and these downshifts were correctly predicted by CpHMD; however, the structure-based PB and empirical methods (except for DelPhiPka) predict either an upshift or no significant shift. Analysis suggests that the reason for this discrepancy is that CpHMD sampled the hydrogen bonds that are not present in the initial crystal structure.
CONCLUDING DISCUSSION
Based on a test set of 21 protein targets, we benchmarked the performance of single-pH and replica-exchange GBNeck2-CpHMD titrations for cysteine pKa calculations and predictions of thiolates or reactive cysteines at physiological pH. We found that 10-ns single pH and 4-ns replica-exchange titrations gave similar RMSE (1.2–1.3) and R (0.8–0.9) for pKa calculations. The accuracy of predicting thiolates or reactive cysteines at physiological pH with single-pH titrations is 86 or 81% with a precision of 100 or 90%, respectively. The accuracy and precision with the 4-ns replica-exchange protocol are similar.
Given crystal structures, the calculated pKa’s from both protocols are in significantly better correlation with experiment. However, while the RMSE from the replica-exchange titrations using crystal structures is significantly decreased (0.95) relative to that (1.5) using the computationally mutated structures, the RMSE from the single-pH titrations based on the crystal structures is only slightly smaller (1.3 vs. 1.4). This difference can be attributed to the noise in the single-pH titrations, which also manifests itself in the poor fitting of the protonation fractions to the HH equation for several proteins. By contrast, replica-exchange titrations gave smooth titration curves for nearly all proteins. In addition to the reduction in statistical noise, our data also demonstrates that the pH replica-exchange protocol significantly accelerates pKa convergence, corroborating the previous findings by us40 and others using continuous and discrete constant pH methods.30–32 while the replica-exchange titrations gave converged pKa’s for all proteins at 4 ns per pH replica, the single-pH titrations for several proteins, e.g., A1AT, ACBPT17C, and HMCKS285A, did not converge even at 50 ns, due to persistent hydrogen bonds that resulted in a continued pKa decrease. Interestingly, the decreasing pKa’s of A1AT and ACBPT17C are associated with increasing deviations from experiment, whereas the decreasing pKa of HMCKS285A corresponds to an improved agreement with experiment. This explains why extending single-pH titrations from 10 ns to 50 ns did not reduce the RMSE of the calculated pKa’s, although the errors may start to decrease with a significantly longer simulation time, e.g., hundreds of nanoseconds. Contrasting the single-pH titrations, hydrogen bond life time is shorter in replica-exchange titrations, which resulted in faster convergence and lower statistical noise. Nonetheless, for computationally mutated structures, it remains to be seen if extending the replica-exchange titrations can further reduce errors. This issue will be addressed in a future study with the implementation of the asynchronous replica exchange scheme which allows users to perform replica-exchange CpHMD titrations on a single GPU card.
The benchmark data based on 21 proteins are encouraging; however, a caveat remains. For three phosphatase proteins, in which a deeply buried cysteine forms several strong hydrogen bonds (in the crystal structures), both CpHMD titrations (up to 50 ns in single pH mode or 4 ns in replica-exchange mode) correctly predicted the protonation state at physiological pH but failed to yield pKa values as the cysteine remained deprotonated in the entire pH range. This failure may be attributed to a known limitation of GB simulations, which tend to overstabilize hydrogen bonding and/or restrict the extent of conformational changes of buried groups.40,72 However, it is also possible that a large conformational change may take place in order to break the strong hydrogen bonds and allowing protonation of the deeply buried cysteine. Such a conformational change may involve a large kinetic barrier, which was not surmountable with the limited sampling time. This issue will be investigated in our future work using the GPU implementations of the fully explicit-solvent and the GB-based CpHMD methods with asynchronous replica exchange (Harris and Shen, ongoing work).
Our data demonstrates that CpHMD titrations outperform the conventional structure-based PB and empirical methods for cysteine pKa calculations and predictions of and thiolates or reactive cysteines at physiological pH. The analysis showed that the ability of thiolate to accept hydrogen bonds is the major driving force for the pKa downshifts, while solvent exclusion in the absence of hydrogen bonding is the major determinant of the pKa upshifts. The ability of CpHMD titrations to reproduce experimental pKa downshifts for cysteines that do not form hydrogen bonds in the crystal structures arises from the sampling of pH-dependent hydrogen bond formation, which is not accounted for by the structure-based methods. Our work also demonstrates that CpHMD titrations can reliably predict thiolates or reactive cysteines at physiological pH. These tasks are useful in preparing MD studies, understanding biological redox functions, and assisting covalent drug design.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge National Institutes of Health (R01GM098818) for funding.
Footnotes
Supporting Information Available
Supplemental tables and figures are included.
References
- (1).Roos G; Foloppe N; Messens J Understanding the pKa of Redox Cysteines: The Key Role of Hydrogen Bonding. Antioxid. Redox Signal 2013, 18, 94–127. [DOI] [PubMed] [Google Scholar]
- (2).Lu J; Holmgren A The Thioredoxin Superfamily in Oxidative Protein Folding. Antioxid. Redox Signal 2014, 21, 457–470. [DOI] [PubMed] [Google Scholar]
- (3).Kerksick C; Willoughby D The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr 2005, 2, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dröge W Oxidative stress and ageing: is ageing a cysteine deficiency syndrome? Phil. Trans. R. Soc. B 2005, 360, 2355–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Moosmann B; Behl C Mitochondrially encoded cysteine predicts animal lifespan. Aging Cell 2008, 7, 32–46. [DOI] [PubMed] [Google Scholar]
- (6).Marino SM; Gladyshev VN Analysis and Functional Prediction of Reactive Cysteine Residues. J. Biol. Chem 2012, 287, 4419–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bulaj G; Kortemme T; Goldenberg DP Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry 1998, 37, 8965–8972. [DOI] [PubMed] [Google Scholar]
- (8).Winterbourn CC; Metodiewa D Reactivity of Biologically Important Thiol Compounds with Super-oxide and Hydrogen Peroxide. Free Radic. Biol. Med 1999, 27, 322–328. [DOI] [PubMed] [Google Scholar]
- (9).Chaikuad A; Koch P; Laufer SA; Knapp S The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed 2018, 57, 4372–4385. [DOI] [PubMed] [Google Scholar]
- (10).Liu R; Yue Z; Tsai C-C; Shen J Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc 2019, 141, 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MBD; Bachovchin DA; Mowen K; Baker D; Cravatt BF Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; González-Páez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Soylu İnanç,; Marino, S. M. Cy-preds: An algorithm and a web service for the analysis and prediction of cysteine reactivity. Proteins 2016, 84, 278–291. [DOI] [PubMed] [Google Scholar]
- (14).Wang H; Chen X; Li C; Liu Y; Yang F; Wang C Sequence-Based Prediction of Cysteine Reactivity Using Machine Learning. Biochemistry 2018, 57, 451–460. [DOI] [PubMed] [Google Scholar]
- (15).Zhang T; Hatcher JM; Teng M; Gray NS; Kostic M Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation. Cell Chem. Biol 2019, 26, 1486–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Awoonor-Williams E; Walsh AG; Rowley CN Modeling covalent-modifier drugs. Biochim. Biophys. Acta 2017, 1865, 1664–1675. [DOI] [PubMed] [Google Scholar]
- (17).Awoonor-Williams E; Rowley CN Evaluation of Methods for the Calculation of the pKa of Cysteine Residues in Proteins. J. Chem. Theory Comput 2016, 12, 4662–4673. [DOI] [PubMed] [Google Scholar]
- (18).Anandakrishnan R; Aguilar B; Onufriev AV H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gunner MR; Zhu X; Klein MC MCCE analysis of the pKas of introduced buried acids and bases in staphylococcal nuclease. Proteins 2011, 79, 3306–3319. [DOI] [PubMed] [Google Scholar]
- (20).Wang L; Zhang M; Alexov E DelPhiPKa web server: predicting pKa of proteins, RNAs and DNAs. Bioinformatics 2016, 32, 614–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Søndergaard CR; Mats HM Olsson MR; Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- (22).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Aliev AE; Kulke M; Khaneja HS; Chudasama V; Sheppard TD; Lanigan RM Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins 2013, 82, 195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Alexov E; Mehler EL; Baker N; Baptista AM; Huang Y; Milletti F; Nielsen JE; Farrell D; Carstensen T; Olsson MHM; Shen JK; Warwicker J; Williams S; Word JM Progress in the prediction of pKa values in proteins. Proteins 2011, 79, 3260–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Baptista AM; Teixeira VH; Soares CM Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys 2002, 117, 4184–4200. [Google Scholar]
- (26).Lee MS; Salsbury FR Jr.; Brooks CL III Constant-pH molecular dynamics using continuous titration coordinates. Proteins 2004, 56, 738–752. [DOI] [PubMed] [Google Scholar]
- (27).Mongan J; Case DA; McCammon JA Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem 2004, 25, 2038–2048. [DOI] [PubMed] [Google Scholar]
- (28).Khandogin J; Brooks III, Constant CL pH molecular dynamics with proton tautomerism. Biophys. J 2005, 89, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Donnini S; Tegeler F; Groenhof G; Grubmüller H Constant pH molecular dynamics in explicit solvent with λ-dynamics. J. Chem. Theory Comput 2011, 7, 1962–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Itoh SG; Damjanović, Brooks BR pH replica-exchange method based on discrete protonation states. Proteins 2011, 79, 3420–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Swails JM; Roitberg AE Enhancing conformation and protonation state sampling of hen egg white lysozyme using pH replica exchange molecular dynamics. J. Chem. Theory Comput 2012, 8, 4393–4404. [DOI] [PubMed] [Google Scholar]
- (32).Swails JM; York DM; Roitberg AE Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput 2014, 10, 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Goh GB; Knight JL; Brooks CL Towards Accurate Prediction of Protonation Equilibrium of Nucleic Acids. J. Phys. Chem. Lett 2013, 4, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wallace JA; Wang Y; Shi C; Pastoor KJ; Nguyen B-L; Xia K; Shen JK Toward accurate prediction of pKa values for internal protein residues: the importance of conformational relaxation and desolvation energy. Proteins 2011, 79, 3364–3373. [DOI] [PubMed] [Google Scholar]
- (35).Goh GB; Laricheva EN; III CLB Uncovering pH-Dependent Transient States of Proteins with Buried Ionizable Residues. J. Am. Chem. Soc 2014, 136, 8496–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu J; Swails J; Zhang JZH; He X; Roitberg A A Coupled Ionization-Conformational Equilibrium Is Required To Understand the Properties of Ionizable Residues in the Hydrophobic Interior of Staphylococcal Nuclease. J. Am. Chem. Soc 2018, 140, 1639–1648. [DOI] [PubMed] [Google Scholar]
- (37).Huang Y; Yue Z; Tsai C-C; Henderson JA; Shen J Predicting Catalytic Proton Donors and Nucleophiles in Enzymes: How Adding Dynamics Helps Elucidate the Structure-Function Relationships. J. Phys. Chem. Lett 2018, 9, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wallace JA; Shen JK Predicting pKa values with continuous constant pH molecular dynamics. Methods Enzymol. 2009, 466, 455–475. [DOI] [PubMed] [Google Scholar]
- (39).Chen W; Morrow BH; Shi C; Shen JK Recent development and application of constant pH molecular dynamics. Mol. Simul 2014, 40, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wallace JA; Shen JK Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J. Chem. Theory Comput 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Huang Y; Chen W; Wallace JA; Shen J All-Atom Continuous Constant pH Molecular Dynamics With Particle Mesh Ewald and Titratable Water. J. Chem. Theory Comput 2016, 12, 5411–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kong X; Brooks III, λ-dynamics CL: A new approach to free energy calculations. J. Chem. Phys 1996, 105, 2414–2423. [Google Scholar]
- (43).Huang Y; Harris RC; Shen J Generalized Born Based Continuous Constant pH Molecular Dynamics in Amber: Implementation, Benchmarking and Analysis. J. Chem. Inf. Model 2018, 58, 1372–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Harris RC; Shen J GPU-Accelerated Implementation of Continuous Constant pH Molecular Dynamics in Amber: Predictions with Single-pH Simulations. J. Chem. Inf. Model 2019, 59, 4821–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nguyen H; Roe DR; Simmerling C Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham T III; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Huang Y; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 2018. 2018. [Google Scholar]
- (47).Im W; Lee MS; Brooks III, Generalized CL Born model with a simple smoothing function. J. Comput. Chem 2003, 24, 1691–1702. [DOI] [PubMed] [Google Scholar]
- (48).Nguyen H; Maier J; Huang H; Perrone V; Simmerling C Folding Simulations for Proteins with Diverse Topologies Are Accessible in Days with a Physics-Based Force Field and Implicit Solvent. J. Am. Chem. Soc 2014, 136, 13959–13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pahari S; Sun L; Alexov E PKAD: a database of experimentally measured pKa values of ionizable groups in proteins. Database 2019, 2019, baz204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Elliott PR; Pei XY; Dafforn TR; Lomas DA Topography of a 2.0 Å structure of α1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 2000, 9, 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Jensen KS; Pedersen JT; Winther JR; Teilum K The pKa Value and Accessibility of Cysteine Residues are Key Determinants for Protein Substrate Discrimination by Glutaredoxin. Biochemistry 2014, 53, 2533–2540. [DOI] [PubMed] [Google Scholar]
- (52).Perkins A; Nelson KJ; Williams JR; Parsonage D; Poole LB; Karplus PA The Sensitive Balance between the Fully Folded and Locally Unfolded Conformations of a Model Peroxiredoxin. Biochemistry 2013, 52, 8708–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wilson MA; Collins JL; Hod Y; Ringe D; Petsko GA The 1.1-Å resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson’s disease. Proc. Natl. Acad. Sci 2003, 16, 9256–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shen Y; Tang L; Zhou H; Lin Z Structure of human muscle creatine kinase. Acta Crystallogr. D 2001, 57, 1196–1200. [DOI] [PubMed] [Google Scholar]
- (55).Quillin ML; Arduini RM; Olson JS Jr.;, P. GN High-Resolution Crystal Structures of Distal Histidine Mutants of Sperm Whale Myoglobin. J. Mol. Biol 1993, 234, 140–155. [DOI] [PubMed] [Google Scholar]
- (56).Lim JC; Gruschus JM; Ghesquiere B; Kim G; Piszczek G; Tjandra N; Levine RL Characterization and Solution Structure of Mouse Myristoylated Methionine Sulfoxide Reductase A. J. Biol. Chem 2012, 287, 25589–25595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Daniels DS; Mol CD; Arvai AS; Kanugula S; Pegg AE; Tainer JA Active and alkylated human AGT structures: A novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000, 19, 1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Pickersgill RW; Harris GW; Garman E Structure of Monoclinic Papain at 1.60 Å Resolution. Acta Crystallogr. B 1992, 48, 59–67. [DOI] [PubMed] [Google Scholar]
- (59).Pickersgill RW; Rizkallah P; Harris GW; Goodenough PW Determination of the Structure of Papaya Protease Omega. Acta Crystallogr. B 1991, 47, 766–771. [Google Scholar]
- (60).Hasnain S; Hirama T; Tam A; Mort JS. Characterization of Recombinant Rat Cathepsin B and Nonglycosylated Mutants Expressed in Yeast. J. Biol. Chem 1992, 267, 4713–4721. [PubMed] [Google Scholar]
- (61).VanDemark AP; Hofmann RM; Tsui C; Pickart CM; Wolberger C Molecular Insights into Polyubiquitin Chain Assembly: Crystal Structure of the Mms2/Ubc13 Heterodimer. Cell 2001, 105, 711–720. [DOI] [PubMed] [Google Scholar]
- (62).Miura T; Klaus W; Ross A; Güntert P; Senn H Letter to the Editor: The NMR structure of the class I human ubiquitin-conjugating enzyme 2b. J. Biomol. NMR 2002, 22, 89–92. [DOI] [PubMed] [Google Scholar]
- (63).Lin Y; Hwang WC; Basavappa R Structural and Functional Analysis of the Human Mitotic-specific Ubiquitin-conjugating Enzyme, UbcH10. J. Biol. Chem 2002, 277, 21913–21921. [DOI] [PubMed] [Google Scholar]
- (64).Barford D; Flint AJ; Tonks NK Crystal Structure of Human Protein Tyrosine Phosphatase 1B. Science 1994, 263, 1397–1404. [PubMed] [Google Scholar]
- (65).Stuckey JA; Schubert HL; Fauman EB; Zhang Z-Y; Dixon JE; Saper MA Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 Å and the complex with tungstate. Nature 1994, 370, 571–575. [DOI] [PubMed] [Google Scholar]
- (66).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TA; Rempfer C; Bordoli L; Lepore R; Schwede T SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Brooks BR; Brooks CL III; Mackerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR6; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Thurlkill RL; Grimsley GR; Scholtz JM; Pace CN pK values of the ionizable groups of proteins. Protein Sci. 2006, 15, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Platzer G; Okon M; McIntosh LP pH-dependent random coil 1H, 13C, and 15N chemical shifts of the ionizable amino acids: a guide for protein pKa measurements. J. Biomol. NMR 2014, 60, 109–129. [DOI] [PubMed] [Google Scholar]
- (71).Khandogin J; Brooks III, Toward CL the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry 2006, 45, 9363–9373. [DOI] [PubMed] [Google Scholar]
- (72).Shi C; Wallace JA; Shen JK Thermodynamic coupling of protonation and conformational equilibria in proteins: theory and simulation. Biophys. J 2012, 102, 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Guengerich FP; Fang Q; Liu L; Hachey DL; Pegg AE O6-Alkylguanine-DNA Alkyltransferase: Low pKa and High Reactivity of Cysteine 145. Biochemistry 2003, 42, 10965–10970. [DOI] [PubMed] [Google Scholar]
- (74).Wang P-F; McLeish MJ; Kneen MM; Lee G; Kenyon GL An Unusually Low pKa for Cys282 in the Active Site of Human Muscle Creatine Kinase. Biochemistry 2001, 40, 11698–11705. [DOI] [PubMed] [Google Scholar]
- (75).Witt AC; Lakshminarasimhan M; Remington BC; Hasim S; Pozharski E; Wilson MA Cysteine pKa Depression by a Protonated Glutamic Acid in Human DJ-1. Biochemistry 2008, 47, 7430–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Pinitglang S; Watts AB; Patel M; Reid JD; Noble MA; Gul S; Bokth A; Naeem A; Patel H; Thomas EW; Sreedharan SK; Verma C; Brocklehurst K A Classical Enzyme Active Center Motif Lacks Catalytic Competence until Modulated Electrostatically. Biochemistry 1997, 36, 9968–9982. [DOI] [PubMed] [Google Scholar]
- (77).Griffiths SW; King J; Cooney CL The Reactivity and Oxidation Pathway of Cysteine 232 in Recombinant Human α1-Antitrypsin. J. Biol. Chem 2002, 277, 25486–25492. [DOI] [PubMed] [Google Scholar]
- (78).Lim JC; Gruschus JM; Kim G; Berlett BS; Tjandra N; Levine RL A Low pKa Cysteine at the Active Site of Mouse Methionine Sulfoxide Reductase A. J. Biol. Chem 2012, 287, 25596–25601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Nelson KJ; Parsonage D; Hall A; Karplus PA; Poole LB Cysteine pKa Values for the Bacterial Peroxiredoxin AhpC. Biochemistry 2008, 47, 12860–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Tolbert BS; Tajc SG; Webb H; Snyder J; Nielsen JE; Miller BL; Basavappa R The Active Site Cysteine of Ubiquitin-Conjugating Enzymes Has a Significantly Elevated pK a : Functional Implications. Biochemistry 2005, 44, 16385–16391. [DOI] [PubMed] [Google Scholar]
- (81).Miranda JL Position-Dependent Interactions between Cysteine Residues and the Helix Dipole. Protein Sci. 2003, 12, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Pahari S; Sun L; Basu S; Alexov E DelPhiPKa: Including salt in the calculations and enabling polar residues to titrate. Proteins 2018, 86, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Dolinsky TJ; Czodrowski P; Li H; Nielsen JE; Jensen JH; Klebe G; Baker NA PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.