

Abstract
Among Alzheimer's disease (AD) brain hallmarks, the presence of reactive astrocytes was demonstrated to correlate with neuronal loss and cognitive deficits. Evidence indeed supports the role of reactive astrocytes as mediators of changes in neurons, including synapses. However, the complexity and the outcomes of astrocyte reactivity are far from being completely elucidated. Another key role in AD pathogenesis is played by alterations in brain cholesterol metabolism. Oxysterols (cholesterol oxidation products) are crucial for brain cholesterol homeostasis, and we previously demonstrated that changes in the brain levels of various oxysterols correlate with AD progression. Moreover, oxysterols have been shown to contribute to various pathological mechanisms involved in AD pathogenesis. In order to deepen the role of oxysterols in AD, we investigated whether they could contribute to astrocyte reactivity, and consequently impact on neuronal health. Results showed that oxysterols present in mild or severe AD brains induce a clear morphological change in mouse primary astrocytes, accompanied by the upregulation of some reactive astrocyte markers, including lipocalin-2 (Lcn2). Moreover, astrocyte conditioned media analysis revealed a significant increase in the release of Lcn2, cytokines, and chemokines in response to oxysterols. A significant reduction of postsynaptic density protein 95 (PSD95) and a concurrent increase in cleaved caspase-3 protein levels have been demonstrated in neurons co-cultured with oxysterol-treated astrocytes, pointing out that mediators released by astrocytes have an impact on neurons. Among these mediators, Lcn2 has been demonstrated to play a major role on synapses, affecting neurite morphology and decreasing dendritic spine density. These data demonstrated that oxysterols present in the AD brain promote astrocyte reactivity, determining the release of several mediators that affect neuronal health and synapses. Lcn2 has been shown to exert a key role in mediating the synaptotoxic effect of oxysterol-treated astrocytes.
Keywords: Oxysterols, Astrocytes, Astrocyte reactivity, Lipocalin-2, Synaptotoxicity, Alzheimer's disease
Abbreviations: α-EPOX, 5α,6α-epoxycholesterol; β-EPOX, 5β,6β-epoxycholesterol; 24-OHC, 24-hydroxycholesterol; 27-OHC, 27-hydroxycholesterol; 7-KC, 7-ketocholesterol; 7α-OHC, 7α-hydroxycholesterol; 7β-OHC, 7β-hydroxycholesterol; Aβ, Amyloid-β; ACM, Astrocyte conditioned media; AD, Alzheimer's disease; ApoE, Apolipoprotein E; CCL, C–C motif chemokine; CXCL, C-X-C motif chemokine; DIV, Days in vitro; GC-MS, Gas chromatography-mass spectrometry; G-CSF, Granulocyte colony-stimulating factor; GFAP, Glial fibrillary acidic protein; Iba1, Ionized calcium binding adaptor molecule 1; IL, Interleukin; Lcn2, Lipocalin-2; LDH, Lactate dehydrogenase; LPS, Lipopolysaccharide; NFTs, Neurofibrillary tangles; NGAL, Neutrophil gelatinase-associated lipocalin; NMDAR, N-methyl-d-aspartate receptor; PSD, Postsynaptic density; PSD95, Postsynaptic density protein 95; SerpinA3N, Serine protease inhibitor A3N; sICAM-1, Soluble intercellular adhesion molecule-1; siRNA, Small interfering RNA; TNF-α, Tumor necrosis factor-α
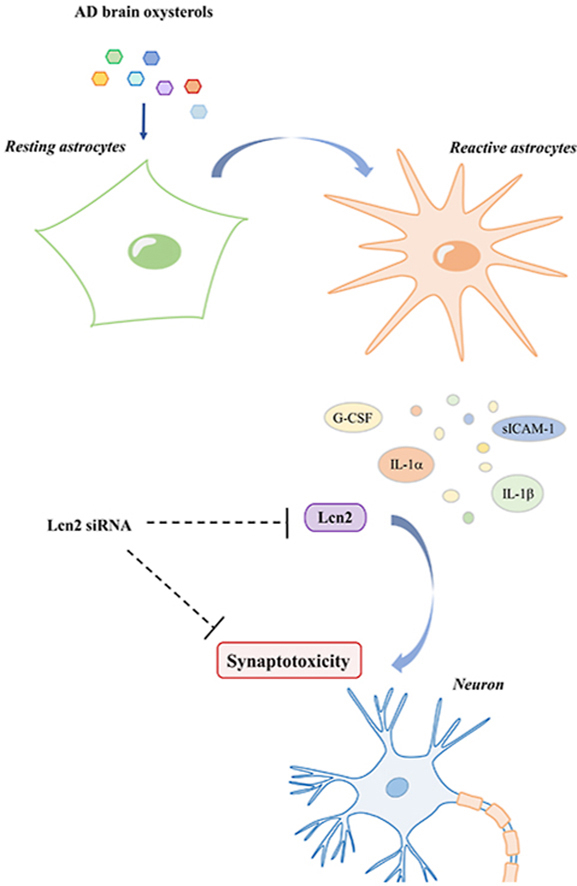
Graphical abstract
Graphical representation of AD brain oxysterols’ effects on astrocytes and neurons.
Highlights
-
•
AD brain oxysterols upregulate astrocyte reactivity markers, including Lcn2.
-
•
Oxysterol-treated astrocytes release Lcn2, cytokines, and chemokines.
-
•
Lcn2 released by oxysterol-treated astrocytes affects neuronal health and synapses.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder, that represents the most common form of dementia and affects millions of people worldwide [1]. The pathogenic hallmarks of AD are extracellular deposits of amyloid-β (Aβ) peptides in the form of senile plaques and intracellular neurofibrillary tangles (NFTs) made of hyperphosphorylated tau protein. These lesions are typically accompanied by gliosis, especially surrounding senile plaques [2]. Neuronal death and synapse loss are also observed; in particular, synaptic loss exceeds neuronal loss, meaning that remaining neurons also lose synapses. Synapse loss is indeed the best correlate of cognitive decline [3].
Astrocytes play a crucial role in maintaining brain homeostasis and their reaction to different kinds of insults leads to a heterogeneous range of changes, known as “astrocyte reactivity” [4]. An increase in the number of reactive astrocytes is a typical histopathological feature of AD brain, and it correlates with cognitive decline and neuronal loss also in transgenic mouse models [5,6]. Besides morphological changes and the upregulation of common markers of reactivity (e.g. the glial fibrillary acidic protein, GFAP), reactive astrocytes show significant alterations in gene expression and functions, depending on the specific stimulus [7,8]. However, the outcome of astrocyte reactivity is still somewhat controversial. Several studies highlighted that reactive astrocytes lose neuroprotective functions, including their ability to promote neurite growth, neuronal survival, and synapse formation [9,10]. One of the mechanisms through which astrocytes could have an impact on neurons is by the altered release of different kinds of molecules including cytokines, chemokines, growth factors, and neurotransmitters. For instance, an increase in the release of inflammatory mediators by reactive astrocytes has been shown to affect neuronal viability, tau phosphorylation [9,11], and synaptic function [12,13]. Another mediator released by reactive astrocytes is lipocalin-2 (Lcn2), a protein identified as a pan-reactive astrocyte marker [8]. Lcn2 is a member of the lipocalin protein family and plays important roles in the immune response, cell migration and proliferation [14]. Increased levels of Lcn2 have been found in the entorhinal cortex and hippocampus of AD brain [15]. Emerging evidence indicates that Lcn2 in the brain is synthetized and secreted as an inducible factor by activated microglia, reactive astrocytes, neurons, and endothelial cells in response to inflammatory stimuli, infections or other insults [16,17].
In addition to amyloid plaques and NFTs, Alois Alzheimer originally described the presence of “adipose inclusions” in the glial cells of AD brains, suggesting a malfunctioning of lipid metabolism [18]. A clear link between AD and lipid metabolism was established later by the identification of the ε4 allele of apolipoprotein E (ApoE) as a strong genetic risk factor for AD [19]. Moreover, in the last few decades, much other evidence supports a role for lipids, in particular cholesterol, in AD pathogenesis [[20], [21], [22]]. As the main lipid component of neuronal and glial membranes, as well as a key constituent of myelin, cholesterol plays an essential role in synapse formation, maintenance, and function [23,24]. Astrocytes are the main producers of cholesterol in the brain, that is delivered to neurons loaded into ApoE-containing lipoproteins. Brain cholesterol homeostasis is closely controlled by pathways regulating cholesterol biosynthesis, storage, and elimination; in particular, the main process responsible for cholesterol elimination is enzymatic oxidation to oxysterols that are able to cross the blood-brain barrier. Enzymatically produced oxysterols are synthetized by both neurons and glial cells. Excess brain cholesterol is indeed essentially oxidized to 24-hydroxycholesterol (24-OHC) mainly by neurons. Another enzymatically produced oxysterol, 27-hydroxycholesterol (27-OHC), is mainly produced by glial cells [25] and various oxysterols deriving from cholesterol non-enzymatic oxidation are also present [26,27]. Oxysterols are not only cholesterol metabolites but they also play many regulatory functions, such as modulating cholesterol biosynthesis, inflammatory pathways, and the immune response [28]. We have previously demonstrated a correlation between changes in brain levels of various oxysterols and AD progression. Through analysis of the oxysterol composition in AD and aged-matched control cortex, we observed a significant increase in the levels of various enzymatic (e.g. 27-OHC) and non-enzymatic oxysterols (e.g. 7α-hydroxycholesterol, 7α-OHC; 7β-hydroxycholesterol, 7β-OHC; 7-ketocholesterol, 7-KC; 5α,6α-epoxycholesterol, α-EPOX; 5β,6β-epoxycholesterol, β-EPOX). In contrast, 24-OHC levels were markedly decreased compared to control brains, probably as a result of neuronal loss [27]. The effects of oxysterols are still controversial but growing evidence suggests that some of them (e.g. 27-OHC, 7-KC, 7α- and 7β-OHC) may play a role in AD pathogenesis [29,30] by inducing oxidative stress, inflammation [31], Aβ formation and accumulation [32,33], tau hyperphosphorylation [34], synaptic dysfunction [35], and cell death [36,37].
At present, data regarding the impact of oxysterols on astrocytes are limited and mostly related to the ability of some of them to affect brain cholesterol synthesis and transport in various astrocytic cell lines [38,39]. 27-OHC was also observed to induce oxidative stress and to downregulate the antioxidant response in C6 glioma cells, leading to cell toxicity [40]. 24-OHC has been shown to affect redox homeostasis in human glial cells, although its impact may depend on the concentration [41], and 7-KC and 7β-OHC exhibited cytotoxic effects in mixed glial murine primary cultures [37]. High concentrations of 7β-OHC have also been observed to induce toxic effects and morphological changes in an in vitro model of reactive astrocytes [42].
Since growing data support the involvement of oxysterols in several aspects of AD pathology and given the presence of reactive astrocytes in the disease, we wanted to investigate whether these compounds could impact on astrocyte reactivity, potentially compromising neuronal health. With the aim of mimicking the oxysterol composition in human AD brain, we used two oxysterol mixtures, both including the main seven oxysterols previously quantified in cortical AD brain samples and representative of early or late stages of the disease [27], to investigate the effect of oxysterols in mouse cortical astrocytes and neuronal cultures.
2. Materials and methods
2.1. Composition of oxysterol mixtures
Cell cultures were treated with two oxysterol mixtures, whose compositions represent oxysterol amounts previously quantified in mild (Early AD mixture) or severe (Late AD mixture) AD brain samples [27]. Both oxysterol mixtures consisted of the same seven oxysterols but in different proportions. The Early AD mixture composition was: 24-OHC (52.9%), 27-OHC (3%), 7-KC (9.2%), 7α-OHC (4.5%), 7β-OHC (19.2%), α-EPOX (3%), and β-EPOX (8.2%). The Late AD mixture composition was: 24-OHC (33.4%), 27-OHC (5.8%), 7-KC (12.7%), 7α-OHC (5.4%), 7β-OHC (23.8%), α-EPOX (4.9%), and β-EPOX (14%). Oxysterols were dissolved in absolute ethanol.
Oxysterols were provided as described: 24-OHC (5275, Medical Isotopes, Pehlam, NH, USA), 27-OHC (700061P), 7-KC (700015P), 7α-OHC (700034P), 7β-OHC (700035P) (Avanti Polar Lipids, Alabaster, AL, USA), α-EPOX (C4130-000), and β-EPOX (C5030-000) (Steraloids, Newport, RI, USA).
2.2. Primary cultures and co-cultures
All animal procedures were in accordance with the European Communities Council Directive (86/609/EEC and 2010/63/EU), the UK Animals (Scientific Procedures) Act 1986, and the Italian Law for Care and Use of Experimental Animals (26/2016), with agreement from both the King's College London (Denmark Hill) Animal Welfare and Ethical Review Board and the Ethical Committee of the University of Turin.
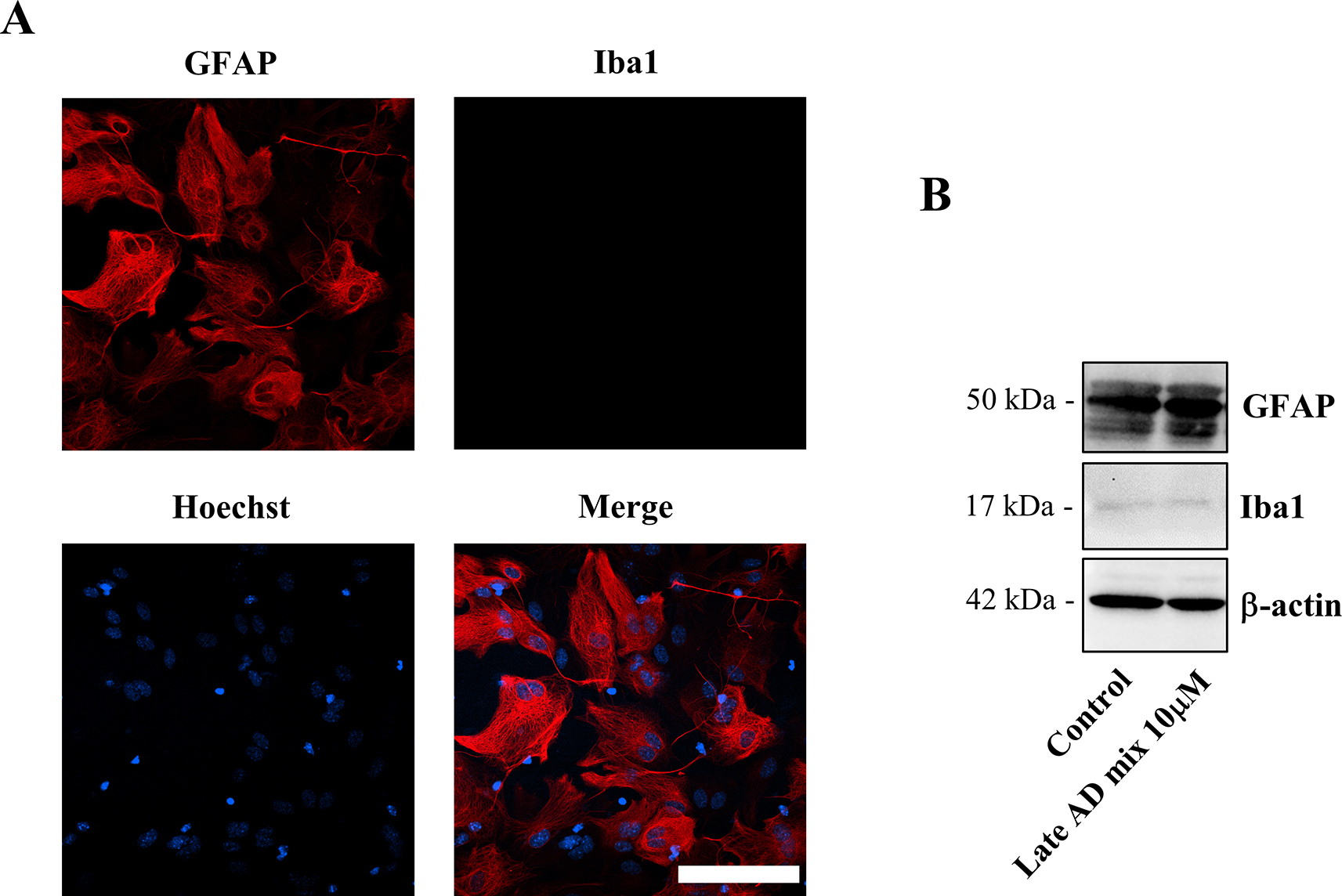
Primary astrocyte cultures were obtained from cerebral cortex of wild type CD1 mice on postnatal day 1–3 as previously described [43]. Briefly, after dissociation of the cortices, cells were re-suspended in growing medium (DMEM high glucose with glutaMAX, sodium pyruvate, 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin) and seeded into T75 flasks, previously coated with poly-d-lysine (10 μg/ml) for at least 1 h at 37 °C. Astrocytes were cultured for 7–14 days in a humidified 5% CO2 incubator at 37 °C, shaking them at 200 rpm overnight on days 3 and 7 to remove microglia and oligodendrocytes. The absence of microglial contamination was assessed by immunocytochemistry and Western blotting using antibodies that recognize the ionized calcium binding adaptor molecule 1 (Iba1, microglial marker) (Supplemental Fig. 1). Astrocyte-enriched cultures were then re-plated into 12-well plates using trypsin and the medium was changed to Neurobasal serum-free medium (supplemented with 2% B-27, 2 mM glutaMAX, 100 U/ml penicillin, 100 μg/ml streptomycin) 24 h before treatment. Astrocyte cultures were treated with the oxysterol mixtures (Early or Late at different concentrations) up to 24 h. Vehicle (ethanol, 34.2 mM) was added to control astrocytes.
Primary neurons were obtained from cerebral cortex of wild type CD1 mouse embryos at embryonic day 15, according to a previously published protocol [44]. Briefly, after dissociation, cortices were washed twice in HBSS (without Ca2+ and Mg2+) and homogenized in 1 ml of Neurobasal serum-free medium (supplemented with 2% B-27, 2 mM glutaMAX, 100 U/ml penicillin, 100 μg/ml streptomycin). The suspension was then filtered using a 40 μm cell strainer, live cells were counted and neurons were plated to a density 5 × 105 viable cells per well of 6-well plates previously coated with poly-d-lysine (10 μg/ml) for at least 1 h at 37 °C. After 1 h, neuron adhesion was checked and the medium was changed. Cultures were maintained at 37 °C with 5% CO2 in Neurobasal serum-free medium. Neuronal cultures were treated at 13–14 days in vitro (DIV). For neurite and spine analysis, neurons were transfected at 5–7 DIV with the plasmid peGFP-N1 (Clontech, Mountain View, CA, USA) using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and imaged live 24 h after treatment at 14 DIV using an Opera Phenix microscope (PerkinElmer, Waltham, MA, USA).
For co-culture experiments, primary astrocytes were plated into cell-culture inserts (0.4 μm pore membrane, Falcon, Corning, Corning, NY, USA), that allow the passage of small molecules in culture medium. Astrocytes were pre-treated with the Late AD mixture (10 μM) or vehicle (ethanol) for 12 h, medium was changed to remove oxysterols, and inserts on which astrocytes were grown were added to neuron cultures for a further 24 h.
2.3. Cytotoxicity assay
Cytotoxicity was assessed by measuring levels of lactate dehydrogenase (LDH) enzyme release into the media using Pierce LDH Cytotoxicity Assay Kit (Fisher Scientific, Thermo Fisher Scientific) according to the manufacturers’ instructions. Some cells were lysed with 0.5% Triton X-100 and LDH content in the medium was measured in order to evaluate the maximum LDH amount released from dead cells. LDH release was calculated as a percentage of total LDH released by lysed cells.
2.4. Gel electrophoresis and Western blotting
After treatments, the cell culture media were collected, cells were washed with PBS and directly lysed with PBS containing sample buffer (NuPAGE LDS Sample Buffer 4X, Invitrogen), reducing agent (NuPAGE Sample Reducing Agent 10X, Invitrogen), protease inhibitor (complete Mini EDTA-Free Protease Inhibitor Cocktail, Roche, Basel, CH) and phosphatase inhibitor (PhosSTOP, Roche) cocktails.
Equal amounts of protein samples (20 μg) were boiled, separated by electrophoresis using 10% precast gels (NuPAGE 10% Bis-Tris Protein Gels, Invitrogen) and then transferred to nitrocellulose membranes (Amersham Protran, GE Healthcare, Chicago, IL, USA). After blocking with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA) for 1 h at room temperature, membranes were incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: GFAP (Z0334, Dako, Agilent Technologies, Santa Clara, CA, USA), Lipocalin-2/NGAL (AF1857, R&D Systems, Minneapolis, MN, USA), SerpinA3N (AF4709, R&D Systems), cleaved caspase-3 (Asp175, 9661, Cell Signaling, Danvers, MA, USA), PSD95 (D74D3, Cell Signaling), Synapsin (6008–30, BioVision, Milpitas, CA, USA), Iba1 (019–19741, Wako Chemicals, Richmond, VA, USA) and β-actin (ab8226, Abcam, Cambridge, UK). After washing to remove unbound antibody with TBS-Tween20 0.05%, the appropriate fluorophore-coupled secondary antibody (1:10,000, LI-COR Biosciences) was added for 1 h at room temperature. Membranes were washed with TBS-Tween20 0.05% and scanned using an Odyssey infrared imaging system (LI-COR Biosciences). Band intensities were quantified using the Image Studio Software (LI-COR Biosciences) and normalized to the corresponding β-actin bands.
Similarly, to detect secreted Lcn2, 250 μl of culture media were concentrated using centrifugal filters (Amicon Ultra, Millipore, Merck, Darmstadt, DE); the protein concentration was calculated using the Bradford Assay (Protein Assay Dye Reagent Concentrate, Bio-Rad Laboratories, Hercules, CA, USA)and samples were prepared as above to perform Western blotting of cell culture media.
2.5. Immunocytochemistry
Astrocytes were plated on coverslips (12 or 18 mm diameter, No. 1.5) into 12- or 24-well plates. After treatment, cells were washed with PBS, fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, and then washed again twice with PBS. Cells were permeabilized and blocked (4% goat serum, 0.1% Triton in PBS) for 1 h at room temperature before incubating with the anti-GFAP (Z0334, Dako) and/or Iba1 (ab48004, Abcam) primary antibodies (4% goat serum in PBS) overnight at 4 °C. The following day, cells were incubated with the appropriate secondary antibody conjugated with fluorescent probes for 1 h at room temperature (Alexa Fluor 488 or 594, Invitrogen) and nuclei were stained with Hoescht 33258 (10 μg/ml in PBS, Sigma-Aldrich, St. Louis, MO, USA). Cells were imaged using an Eclipse Ti-E inverted Microscope (Nikon, Tokyo, JP) or LSM800 confocal microscope (Carl Zeiss, Oberkochen, DE).
2.6. Cytokine array
Astrocyte culture media were collected and stored at −20 °C prior to analysis of cytokine and chemokine content using Mouse Proteome Profiler arrays (Mouse Cytokine Array Panel A, R&D Systems), according to the manufacturers’ instructions. Briefly, array membranes were incubated in blocking buffer for 1 h at room temperature. Each sample of medium was incubated with the Detection Antibody Cocktail and this mix was then placed onto blocked membranes overnight at 4 °C. After washes, membranes were incubated with IRDye 800CW Streptavidin (1:2000, LI-COR Biosciences) for 30 min at room temperature. Membranes were then scanned using an Odyssey infrared imaging system and the spot intensities were quantified using the Image Studio Software (LI-COR Biosciences). Positive and negative control spots included in each membrane allowed quantitative analysis by densitometry and results were expressed as percentage change compared to control cultures.
2.7. Oxysterol quantification in astrocyte culture medium
Astrocyte culture medium (100 μl) was added to a screw-capped vial sealed with a Teflon septum together with D7-7α-hydroxycholesterol (50 ng), D7-7β-hydroxycholesterol (50 ng), D7-7-ketocholesterol (50 ng), D6-5α,6α-epoxycholesterol (50 ng), D6-5β,6β-epoxycholesterol, D6-24-hydroxycholesterol (250 ng), D6-27-hydroxycholesterol (50 ng) as internal standards, as well as 50 μl of butylated hydroxytoluene (5 g/l) and 50 μl of K3-EDTA (10 g/l) to prevent auto-oxidation. Each vial was then flushed with argon for 5 min to remove air. Alkaline hydrolysis, sterol extraction, and gas chromatography-mass spectrometry (GC-MS) analysis were performed as previously described [45].
2.8. RNA extraction and real-time RT-PCR
Total RNA was extracted using TriFast reagent (Eurogold TriFast, EuroClone, Pero, IT) following the manufacturers' instructions. RNA was dissolved in RNase-free water with RNase inhibitors (SUPERase-In RNase inhibitor, Invitrogen). The amount and purity of the extracted RNA were assessed by using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized by reverse transcription of 1 μg of RNA by using a commercial kit and random primers (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Thermo Fisher Scientific) following the manufacturers’ instructions.
Singleplex real-time RT-PCR was performed on 30 ng of cDNA by using TaqMan Gene Expression Assays for mouse Lcn2 (Mm01324470_m1) and β-actin (Mm02619580_g1), TaqMan Fast Universal PCR Master Mix, and a 7500 Fast Real-Time PCR System (Applied Biosystems). The PCR cycling parameters were set up as previously described [33]. The fractional cycle number (Ct) was determined for each gene considered and results were then normalized to β-actin expression. Relative quantification of target gene expression was achieved with a mathematical method [46].
2.9. Transient gene knockdown
The transient knockdown of Lcn2 gene was performed by using a Small Interfering RNA (siRNA) (Silencer Select Pre-designed siRNA S69122, Ambion, Thermo Fisher Scientific) following the manufacturers’ instructions. A non-targeting siRNA was used as a negative control (Silencer Select Negative Control #2 siRNA, Ambion). Briefly, astrocytes were plated into 12- or 24-well plates and respectively 50 μl or 100 μl of a mix containing siRNA and transfection agent (Lipofectamine RNAiMAX Reagent, Invitrogen) in a 1:1 ratio was added to each well, in order to reach a final siRNA concentration of 100 nM and 3 μl/ml of transfection agent. After 6 h transfection, the medium was changed and astrocytes were treated with the Late AD mixture (10 μM) for 12 h. After treatment, cells were washed with PBS and fresh medium was added. Astrocyte conditioned media were then collected after a further 24 h. Protein and RNA extraction, as well as real-time RT-PCR, were performed as described above. The silencing efficiency, validated by real-time RT-PCR, was approximately 85% (Supplemental Fig. 2).
2.10. Analysis of neuronal morphology
The neuronal morphology of 14 DIV neurons previously transfected with the plasmid peGFP-N1 (Clontech) on 5–7 DIV and treated on 13 DIV was assessed using high resolution digital images of live neurons taken using an Opera-Phenix microscope (PerkinElmer). Neurite complexity was analysed using Harmony software: total and maximum neurite length, and number of nodes and extremities were quantified and compared among groups. NeuronStudio software (CNIC, Mount Sinai School of Medicine) was used for dendritic spine analysis. Spine density was defined as number of spines per micrometer of dendrite length. Dendritic spine densities were calculated from 20 neurons/condition.
2.11. Statistical analysis
After performing a Shapiro-Wilk normality test, data were analysed using one-way ANOVA followed by Bonferroni post hoc test or Student's t-test (GraphPad Prism 7 Software, Graphpad Software, La Jolla, CA, USA). Results were considered statistically significant when P < 0.05. Data are represented as means ± standard deviation (SD).
3. Results
3.1. Oxysterol mixtures induce a morphological change in astrocytes
Astrocyte reaction to certain stimuli is defined by a wide range of heterogeneous responses, including changes in morphology [7,47]. To analyse the effect of oxysterols on astrocyte morphology and survival, cells were exposed to increasing concentrations of the Early and Late mixtures (1, 5 or 10 μM). Exposure of primary astrocytes to 5 or 10 μM of both oxysterol mixtures for 24 h induced a clear change in their morphology with astrocytes adopting a more stellate “brain-like” and reactive appearance rather than the large, flat fibroblast-like morphology typical of unstimulated dissociated astrocytes in culture (Fig. 1B). Importantly, this was not accompanied by cell death since there were no significant increases in the abundance of LDH released into culture medium following treatment when compared to control cultures (Fig. 1A). Time course treatments showed that astrocytes started to display the typical signs of reactivity (increase in the number of GFAP positive processes and stellate appearance) after 12 h of exposure to 10 μM of both Early and Late mixtures, with these changes becoming more evident 24 h after treatment (Fig. 2).
Fig. 1.

Dose-response experiments to test the effect of the oxysterol mixtures on astrocyte viability and morphology. Primary astrocyte cultures were treated with the Early or Late AD oxysterol mixture (1, 5, or 10 μM), or vehicle (ethanol) for 24 h. (A) The bar graph shows the lactate dehydrogenase (LDH) release from treated astrocytes. Values are the proportion of LDH released into medium relative to total LDH in lysed cells normalized to values for control media. Data are expressed as mean ± SD from three different experiments (one-way ANOVA). (B) Astrocyte morphology was examined by immunocytochemistry using a glial fibrillary acidic protein (GFAP) antibody (red) and nuclei were stained with Hoechst 33258 (blue). Representative images from three experiments are shown. Cells were imaged using an LSM800 confocal microscope (Zeiss; 40× objective; scale bar: 100 μm).
Fig. 2.

Oxysterol mixtures induce a clear morphological change in astrocytes. Primary astrocytes were treated with 10 μM of the Early or Late AD oxysterol mixture for up to 24 h. Astrocyte morphology was examined by immunocytochemistry using a glial fibrillary acidic protein (GFAP) antibody (red) and nuclei were stained with Hoechst 33258 (blue). Representative images from three experiments are shown. Cells were imaged using an Eclipse Ti-E Microscope (Nikon; 20× objective; scale bar: 100 μm).
3.2. Oxysterol mixtures increase the synthesis of the pan-reactive astrocyte markers Lcn2 and SerpinA3N
To validate the induction of astrocytic reactivity by oxysterols and to confirm that the morphological alteration is accompanied by functional changes, we determined whether treatment with the Early or Late AD mixtures led to an increase in the levels of some pan-reactive astrocyte markers [8]. Exposure to both oxysterol mixtures (10 μM) resulted in a marked increase in Lcn2 and serine protease inhibitor A3N (SerpinA3N) protein levels 12 h (P < 0.0001 and P < 0.001) and 24 h (P < 0.0001) after treatment (Fig. 3), contemporaneous with morphological changes. These molecules are involved in the acute-phase response and were both identified as new reactive astrocyte markers [8,48]. Total levels of GFAP were only slightly increased (P < 0.05) or remained unchanged in response to the mixtures (Fig. 3), suggesting that the observed morphological changes are not only dependent on the expression of GFAP.
Fig. 3.

Oxysterol mixtures increase the synthesis of pan-reactive astrocyte markers. The glial fibrillary acidic protein (GFAP), serine protease inhibitor A3N (SerpinA3N), and lipocalin-2 (Lcn2) protein levels were determined by Western blotting of lysates from primary astrocytes treated with 10 μM of the Early or Late AD oxysterol mixture for up to 24 h. The amounts of proteins of interest were normalized to β-actin levels in the same sample and are represented as percentage of average control values. Data are expressed as mean values ± SD of three different experiments (one-way ANOVA). ****P < 0.0001, ***P < 0.001, *P < 0.05 vs. control.
3.3. Oxysterol-induced reactivity triggers the release of Lcn2, cytokines, and chemokines by astrocytes
Astrocyte reactivity is typically characterized by the release of cytokines, chemokines, and other mediators, that could have several effects on the surrounding cells [49]. Therefore, we investigated whether oxysterol-mediated astrocyte reactivity was accompanied by altered release of mediators into the cell culture medium.
Since Lcn2 is secreted by reactive astrocytes [17,50], we analysed its protein levels in astrocyte conditioned media (ACM) by Western blotting. We saw that the robust increase in intracellular levels of Lcn2 is accompanied by a significant increase in the secretion of Lcn2 after 24 h treatment with 10 μM of both Early and Late oxysterol mixtures (P < 0.0001) (Fig. 4A).
Fig. 4.

Oxysterol treatment increases the release of lipocalin-2 (Lcn2), cytokines and chemokines by astrocytes. Astrocyte cultures were treated with the Early or Late AD oxysterol mixture (10 μM) for 24 h. (A) Lcn2 protein levels in astrocyte conditioned media (ACM) were examined by Western blotting. Data are expressed as mean values ± SD from three different experiments and are shown as percentage change from average control values (one-way ANOVA). ****P < 0.0001 vs control. (B) Mouse cytokine profiler antibody arrays were used to detect the amounts of 40 mediators in ACM. Representative images of array membranes are shown, highlighted some of the cytokines found to be significantly increased in ACM from treated cells. The bar chart shows the cytokines significantly affected by oxysterol mixture treatments as percentage change from control. Data are expressed as mean values ± SD from three different experiments (one-way ANOVA). ***P < 0.001, **P < 0.01, and *P < 0.05 vs control.
The amounts of 40 cytokines and chemokines in ACM samples were next evaluated using Proteome Profiler antibody arrays. The analysis showed a significant increase in the release of several mediators by astrocytes treated for 24 h, especially with the Late AD oxysterol mixture (10 μM). Some of the cytokines found to be increased in the medium of astrocytes treated with this oxysterol mixture were interleukin-1β (IL-1β), soluble intercellular adhesion molecule-1 (sICAM-1) (P < 0.001), IL-1α, IL-5, granulocyte colony-stimulating factor (G-CSF), and C–C motif chemokine 12 (CCL12) (P < 0.01). In addition, the levels of CCL1, IL-7, IL-10, IL-13, IL-16, IL-17, and C-X-C motif chemokine 9 (CXCL9) were slightly but significantly increased compared to levels in control media (P < 0.05) (Fig. 4B and C). In contrast, when astrocytes were exposed to the Early AD oxysterol mixture (10 μM), only sICAM-1 (P < 0.01), IL-1α, and IL-1β (P < 0.05) were found to be significantly increased relative to levels in control ACM, highlighting a different impact of the two oxysterol mixtures on release of cytokines and chemokines by astrocytes (Fig. 4C).
3.4. Oxysterol-stimulated astrocytes compromise neuronal health
It has previously been shown that astrocyte reactivity depends on the specific stimulus and that heterogeneous responses of reactive astrocytes involve different signaling pathways. The consequences of these alterations for astrocytic support of neuronal health require further investigation [7,48].
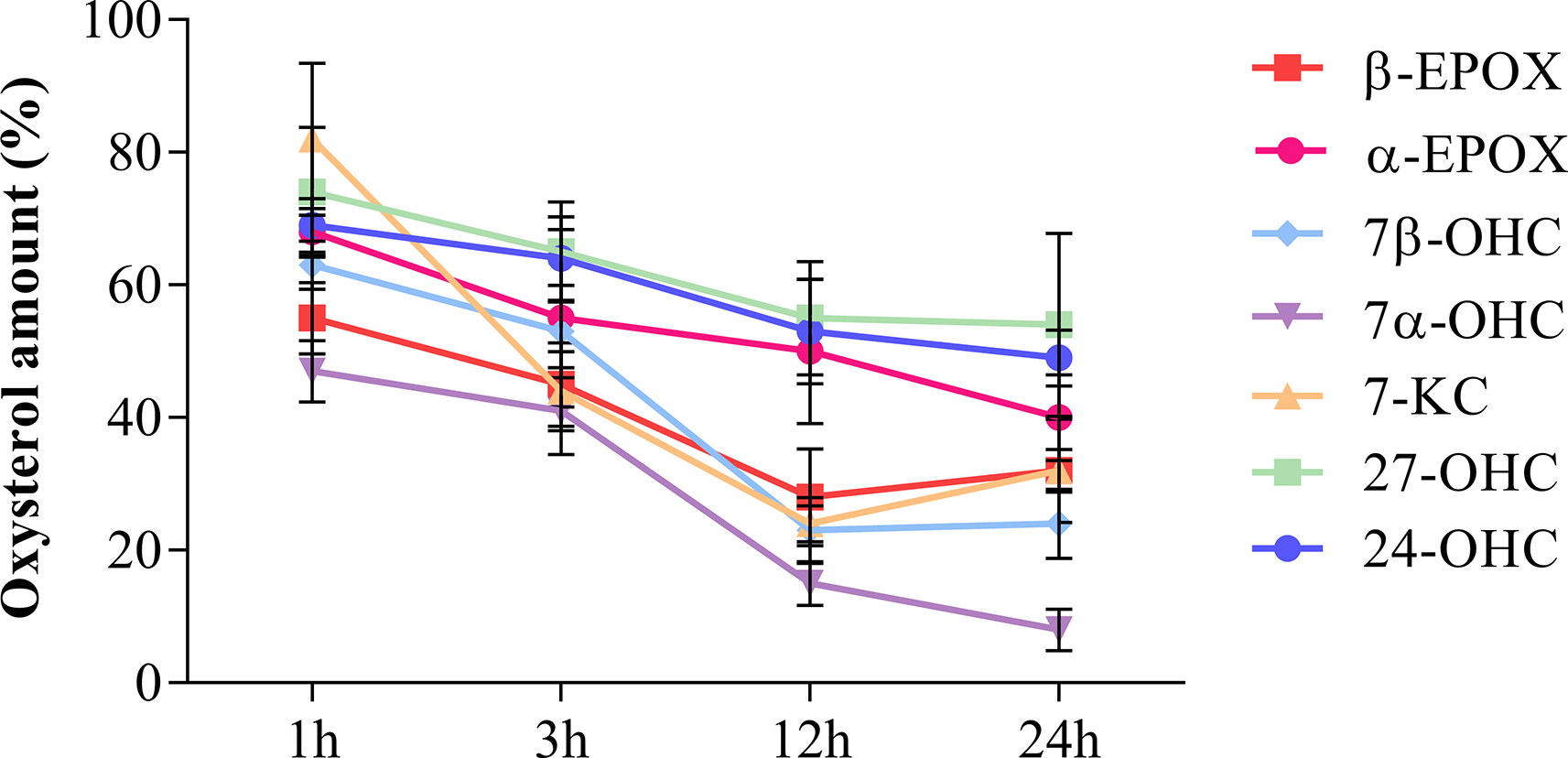
To test the effect of oxysterol-stimulated astrocytes on neuronal health, we performed co-culture experiments. GC-MS analysis of oxysterols in ACM showed that, although variable, a substantial proportion (8–54%) of some oxysterols remain in ACM 24 h after treatment (Supplemental Fig. 3). Since these oxysterols are known to directly affect neuron viability [33,35,51], we co-cultured neurons with astrocytes that had been pre-treated with oxysterols. As shown in Fig. 5A, astrocytes grown on cell culture inserts were pre-treated for 12 h with the Late AD oxysterol mixture (10 μM) and then transferred to culture plates containing neurons for a further 24 h: this allowed us to investigate the effect of mediators released from stimulated astrocyte on neurons, without transferring oxysterols that remained in ACM. In parallel, neuronal cultures were treated with the Late AD mixture or with ethanol for 24 h in the absence of astrocytes, in order to assess the direct effect of the same oxysterol mixture on neurons.
Fig. 5.

Oxysterol-stimulated astrocytes compromise neuronal health. Primary neurons (13 DIV) were treated with the Late AD oxysterol mixture (10 μM) for 24 h or they were co-cultured for 24 h with astrocytes grown on cell culture inserts that had been previously treated with the same oxysterol mixture for 12 h. (A) Graphical representation of co-culture experiments. (B) The protein levels of the postsynaptic density protein 95 (PSD95), synapsin, and cleaved caspase-3 were determined by Western blotting. Data were normalized to the corresponding β-actin levels. Data are expressed as mean values ± SD of three different experiments as percentage change from respective control (Student's t-test). **P < 0.01 and *P < 0.05 vs control.
Interestingly, Western blotting analysis showed a significant reduction of postsynaptic density protein 95 (PSD95) levels (P < 0.05), a scaffold protein important for postsynaptic density structure and function, in neurons cultured with astrocytes that had been previously exposed to oxysterols (Fig. 5B). This reduction was accompanied by a significant increase in the amount of cleaved (active) caspase-3 (P < 0.01) (Fig. 5B), that was demonstrated to play also various non-apoptotic roles in neurons, including to affect synaptic functions [52]. A mild but not significant reduction of the pre-synaptic marker synapsin was also observed. Moreover, when neurons were directly treated with oxysterols, PSD95 (P < 0.05), cleaved caspase-3 (P < 0.01), and synapsin levels (P < 0.05) were all significantly altered (Fig. 5B), confirming previous results showing that oxysterols directly compromise neuron health [33,35,53]. Importantly, all these results were obtained in the absence of significant neuronal death, as assessed by LDH release (Supplemental Fig. 4). Overall, these data suggest that factors released by astrocytes in response to the oxysterol mixture, whose composition is similar to AD brain oxysterol content, compromise synaptic and neuronal health without causing overt neurotoxicity.
3.5. Lcn2 secreted by oxysterol-stimulated astrocytes affects neurite complexity and decreases dendritic spine density
Lcn2 has previously been shown to affect synapses, neuronal health and morphology [17,54,55]. To investigate whether Lcn2 could play a role in mediating the effect of oxysterol-stimulated astrocytes on neurons, Lcn2 expression was silenced in astrocytes prior to determining the impact of ACM on neuron morphology. To further investigate the effects of ACM on synapses under these conditions, dendritic spine density was also evaluated.
For this purpose, astrocytes were transfected for 6 h with Lcn2 or scrambled siRNA, and the medium removed prior to treatment with the Late AD mixture (10 μM) for 12 h. The medium was removed and fresh medium was added for a further 24 h, to obtain ACM without oxysterols. Both real-time RT-PCR (Fig. 6A) and Western blotting results (Fig. 6B) confirmed that siRNA-mediated Lcn2 gene silencing prevents the increase in Lcn2 expression, synthesis, and release into the medium induced by the Late AD oxysterol mixture, compared to control conditions (scrambled siRNA). Interestingly, oxysterol-induced morphological changes were not prevented by Lcn2 gene silencing, suggesting that the pathway by which oxysterols induce astrocyte alterations is not dependent upon the presence of Lcn2 (Fig. 6C).
Fig. 6.

Lipocalin-2 (Lcn2) gene silencing prevents Lcn2 upregulation and release into the medium. Astrocyte cultures were transfected for 6 h with Lcn2 or scrambled siRNA and then treated with the Late AD oxysterol mixture 10 μM (Mix) for 12 h. After treatment, the medium was changed and astrocytes were incubated with fresh medium for 24 h. Transient Lcn2 gene knockdown was evaluated by (A) real-time RT-PCR and (B) Western blotting of both lysates and astrocyte conditioned media (ACM) samples. Data were normalized to the corresponding β-actin levels. Data are expressed as mean values ± SD from three different experiments as percentage change from control (one-way ANOVA). ****P < 0.0001, ***P < 0.001, and *P < 0.05 vs control; ####P < 0.0001 vs oxysterol treated. (C) Astrocyte morphology was examined by immunocytochemistry using a glial fibrillary acidic protein (GFAP) antibody (red) and nuclei were stained with Hoechst 33258 (blue). Representative images from three experiments are shown. Cells were imaged using an LSM800 confocal microscope (Zeiss, 40× objective; scale bar: 100 μm).
Next, ACM was added to cultured neurons to determine the impact of secreted Lcn2 on neuron morphology and dendritic spines. Medium from oxysterol-activated astrocytes transfected with the scrambled siRNA compromises neurite complexity, as shown by reduction of total and maximum neurite length, and the number of nodes, automatically identified by Harmony software (P < 0.05) (Fig. 7A). Importantly, the addition of ACM was also found to substantially reduce dendritic spine density (P < 0.001) (Fig. 7B), further indicating a synaptotoxic effect of proteins secreted by oxysterol-stimulated astrocytes. All of these effects were prevented by silencing Lcn2, confirming a key role of Lcn2 in mediating the impact of oxysterol-stimulated astrocytes on neurite complexity and synaptic health (Fig. 7).
Fig. 7.

Lipocalin-2 (Lcn2) secreted by oxysterol-stimulated astrocytes affects neurite complexity and decreases dendritic spine density. Primary neurons were incubated for 24 h with conditioned media from astrocytes transfected for 6 h with Lcn2 or scrambled siRNA and then treated with the Late AD oxysterol mixture 10 μM for 12 h. After treatment, the medium was changed and astrocytes were incubated with fresh medium for 24 h. High resolution digital images of live neurons were obtained using an Opera-Phenix microscope (PerkinElmer). (A) Neurite complexity was analysed using Harmony software. Total and maximum neurite length, number of nodes and extremities were quantified and compared among groups. Representative images are shown (scale bar: 100 μm). Data are expressed as mean values ± SD from three different experiments and were obtained from analysis of 20 neurons/condition (one-way ANOVA). *P < 0.05 vs control. (B) NeuronStudio software was used for dendritic spine analysis. Spine density was defined as number of spines per micrometer of dendrite length and was obtained from analysis of 20 neurons/condition. Representative images are shown (scale bar: 25 μm). Data are expressed as mean values ± SD from three different experiments (one-way ANOVA). ***P < 0.001 vs control.
4. Discussion
The pathophysiology of AD is still not fully understood, but evidence supports the involvement of many factors in sporadic AD onset, including inflammation and impaired cholesterol metabolism [56]. Microglia and astrocytes play a role in the neuroinflammation observed in AD and they are found in their activated state in affected brain regions, particularly surrounding amyloid plaques. Astrocyte reactivity is not an exclusive feature of AD brain but the association of reactive astrocytes with amyloid plaques is considered functionally significant. Indeed, post mortem neuropathological studies have shown that the number of reactive astrocytes increases with disease progression [6,57], and activated astrocytes and microglia correlate with dementia in AD [5].
Astrocyte reactivity leads to a heterogeneous range of phenotypic, transcriptomic, and functional changes, depending on the specific environmental stimulus [7,8], including those related to ageing and disease [10,58]. Here, we observed that application of various oxysterols, in the same proportions as those identified in early and late stage AD brain, distinctly altered astrocyte morphology after 24 h treatment. In particular, astrocytes adopted a more stellate appearance with many long and branched processes evident. In support of these data, 7β-OHC has previously been shown to induce morphological changes, characterized by process elongation, in cultured reactive astrocytes [42]. An increase in the number of processes is considered a reliable feature of reactivity, demonstrated in several cell culture models of reactive astrocytes [59,60].
Reactive astrocytes were conventionally identified by GFAP immunoreactivity; however, it is noteworthy that different stimuli can lead to similar degrees of GFAP upregulation while causing substantially different changes in transcriptome profile and cell functions [7]. Furthermore, unlike brain astrocytes, most astrocytes in culture are GFAP reactive as a result of the in vitro conditions, mainly the absence of their usual environment and the loss of inhibitory influences of neurons [42]. Astrocytes treated with Early or Late AD oxysterol mixture up to 24 h showed a significant but mild increase in GFAP protein levels despite the extensive morphological changes observed. The morphological changes we observed in response to oxysterol mixtures were, however, accompanied by a marked upregulation of Lcn2 and SerpinA3N, both proteins identified as “pan-reactive” astrocytes markers [8], as well as by increased release of Lcn2 into culture medium. Lcn2, also known as neutrophil gelatinase-associated lipocalin (NGAL), is an acute phase protein with several roles [14] that is secreted by different kinds of cells, including reactive astrocytes [16,17]. For instance, it has recently been shown that Aβ induces Lcn2 production in astrocytes [61]. It can act as an autocrine mediator of reactive astrogliosis, since it is able to induce morphological changes in primary astrocytes [59]. In addition, several studies showed that Lcn2 exerts synaptotoxic and neurotoxic effects, affecting neuronal viability, dendritic spine density and morphology, and ultimately cognitive functions [17,50,54,55]; for this reason, we decided to focus mainly on the effects mediated by astrocyte-produced Lcn2 on neuronal health. SerpinA3N, also known as alpha1-antichymotrypsin, is a protease inhibitor involved in the acute phase response and reactive and ageing astrocytes are its main producers in the brain [8,58]. Immunohistochemical analysis of AD brain samples showed that SerpinA3N co-localizes with amyloid plaques, reactive microglia and astrocytes [62].
It is well known that in many cases astrocyte activation is accompanied by an increase in the release of several soluble factors, including reactive oxygen species and cytokines [11,49]. Astrocytes are both targets and effectors of cytokines and many other mediators that affect not only their immune and inflammatory functions but also their synapse-directed and neuronal functions [49]. Interestingly, 7α,25-dihydroxycholesterol has been shown to inhibit the lipopolysaccharide (LPS)-induced release of pro-inflammatory cytokines, including IL-17/tumor necrosis factor-α (TNF-α), in cultured human astrocytes [63]. Other oxysterols (e.g. 27-OHC) have been demonstrated to decrease LPS-induced expression of IL-6 and TNF-α mRNA in mouse primary glial cells [64]. However, there are no data on how oxysterols directly affect astrocyte secretion profile. Our cytokine array analysis showed that oxysterol treatment significantly increases the release of a wide range of cytokines and chemokines by primary astrocytes after 24 h. Interestingly, this analysis highlighted a differential impact of the two oxysterol mixtures; the majority of the significant increases were indeed observed in ACM from astrocytes treated with the Late AD oxysterol mixture, likely because of the different composition of this mixture. Moreover, even if both mixtures showed a similar effect on astrocyte morphology, the functional changes can differ, as suggested by several studies [7]. Among the released mediators, inflammatory cytokines (IL-1α, IL-1β, and IL-17), adhesion molecules (sICAM-1), growth factors and chemokines (IL-5, IL-7, IL-16, G-CSF, CCL1, CCL12, and CXCL9), immunoregulatory cytokines (IL-5 and IL-13), and anti-inflammatory cytokines (IL-10) were elevated. In particular, IL-1β and sICAM-1 were the most significantly increased. IL-1β has been previously shown to induce reactive astrogliosis [10], to be released by reactive astrocytes and astrocytes from 5xFAD AD mouse model [65,66], and to contribute to astrocyte-mediated neuronal death [67]; moreover, its levels are increased in AD brains [27,68]. ICAM-1 is a transmembrane glycoprotein belonging to the immunoglobulin family of adhesion molecules, expressed not only by endothelial and immune system cells [69] but also by astrocytes [8]. sICAM-1 is released by proteolytic cleavage of the transmembrane protein [66,70]; its levels are increased in AD cerebrospinal fluid and correlate with various AD markers including total tau, phosphorylated tau, and cortical thinning [71,72]. The data presented here provide the first evidence for a direct impact of oxysterols on astrocyte morphology, markers of reactivity, and secretion profile, and strongly suggest that oxysterol mixtures mimicking oxysterol composition in AD brain induce astrocyte reactivity.
The functional implications of astrocyte reactivity are various and very context-dependent. For instance, astrocytes have been shown to mediate Aβ-induced neurotoxicity and tau phosphorylation, as well as to induce a decrease of synaptic markers; interestingly, the release of pro-inflammatory cytokines from astrocytes seems to play a key role in some of these processes [11,73]. To investigate the consequences of oxysterol-mediated astrocytic reactivity on neurons, we exposed neurons to astrocytes that had previously been challenged with oxysterols. Our data indicated that neurons co-cultured with pre-treated astrocytes are characterized by a significant reduction in PSD95 protein levels and by a clear increase in cleaved caspase-3 protein levels, suggesting that oxysterol-induced reactivity is detrimental for neuronal health. Importantly, similar changes were obtained when neuronal cultures were directly treated with the Late AD mixture in the absence of astrocytes, confirming that oxysterols have a direct negative effect on neuronal health. Then, these results suggest that brain oxysterols could affect neuronal health both directly and by inducing astrocyte reactivity. PSD95 is a scaffold protein abundant in the postsynaptic density (PSD), that anchors N-methyl-d-aspartate receptor (NMDAR) as well as other proteins (e.g potassium channels, cell adhesion molecules) to the PSD. It is also involved in the assembly of specific signaling proteins which act downstream of NMDAR [74]. PSD95 is recognized to play an important role in synaptic plasticity and memory, and in-keeping with this its levels decrease in AD [5]. To the best of our knowledge, no studies have yet assessed the specific actions of oxysterol-treated astrocytes on PSD95 levels, however various mediators released by oxysterol-treated astrocytes have previously been shown to affect synapses, including Lcn2 and IL-1α/β [54,75,76]. Indeed, the binding of cytokines and chemokines to their receptors leads to the activation of diverse signaling pathways [77], some of which are involved in PSD95 regulation [78]. Concerning oxysterols, it has been demonstrated that 27-OHC reduces dendritic spine density and PSD95 levels in primary mouse hippocampal neurons [35], whereas 24-OHC affects synaptic plasticity via modulation of NMDAR [79]. Since oxysterols are able to directly modulate several pathways involved in PSD95 regulation [51], other oxysterols present into the mixture could be involved in the observed PSD95 reduction, even if their role needs further investigation. Moreover, the ability of oxysterols to alter membrane structure could affect transmembrane protein localization and function, thus modifying the integrity of the PSD structure [80,81]. The increase in the levels of cleaved caspase-3 fragment further indicates that oxysterol-activated astrocytes are detrimental to neurons. Besides being an effector of apoptosis, caspase-3 was demonstrated to play various non-apoptotic roles in neurons involved in both physiological processes and neurodegenerative diseases [52]. For instance, it is involved in the pathological cleavage of tau protein [82] and its activation correlates with dendritic spine loss and cognitive decline in the absence of neuronal cell death [83]. Interestingly, it has previously been shown that increase in neuronal cleaved caspase-3 levels in response to Aβ is exacerbated in the presence of reactive astrocytes, and that increased caspase-3 activity is reduced upon pretreatment with the anti-inflammatory molecule minocycline in vitro [11] and in vivo [84]. These and other data highlight that neuronal caspase-3 can be activated by extracellular inflammatory mediators. Caspase-3 can also be activated by oxysterols, mainly those oxidized at C7 position but also others [35,51,[85], [86], [87]].
In AD, synaptic loss is the parameter that best correlates with dementia [88,89]; therefore, elucidating how synapses are lost is of great importance. This can be examined in vitro by monitoring dendritic spine density and/or neurite complexity. The direct effect of oxysterols on synaptic health has been partially investigated as stated above, but we were interested in assessing how oxysterol-activated astrocytes affect synapses. Our results indicate that ACM from oxysterol-activated astrocytes is synaptotoxic, as shown by significant decreases in the number of dendritic spines and reduced complexity of the neurites in primary neurons, as well as by PSD95 reduction as described above. Since Lcn2 is secreted upon oxysterol-induced astrocyte activation and has proven to be neurotoxic [17,55], we tested whether we could revert the synaptotoxic effect of reactive astrocytes by suppressing Lcn2 expression and therefore reducing its secretion. Our results clearly demonstrate that dendritic spine density and neurites architecture are better preserved when Lcn2 expression is silenced prior to treating astrocytes with oxysterols. These data reveal a major role for Lcn2 in mediating the synaptotoxic effect of oxysterol-treated astrocytes. These findings extend previous studies showing that Lcn2 affects dendritic spine density and morphology [54], as well as contributes to hippocampal damage and cognitive impairment in a mouse model of vascular dementia [50].
5. Conclusion
Overall, these results describe the complexity of oxysterol effects on astrocytes and neurons. Of interest was our observation that astrocytes respond most strongly to the oxysterol mixture representative of late AD brain in comparison to early AD-mimicking mixture. These data add to an emerging consensus that astrocyte responses are differentially altered at specific disease stages in response to various changes in the local environment. Indeed, we demonstrated that oxysterols induce a clear morphological change in astrocytes, that is accompanied by an increase in some reactive astrocyte markers and the release of several mediators. We also demonstrated that oxysterols are detrimental to neuronal health but do not cause overt neurotoxicity. Importantly, we have shown that oxysterol-activated astrocytes induce synaptotoxicity that is mediated by Lcn2. This study thus reveals new aspects of brain oxysterol effects on astrocytes and neurons, confirming their potential ability to contribute to AD pathogenesis, and providing support for further investigations into a potential role for Lcn2 as a novel therapeutic target in AD.
Funding
This work was supported by Alzheimer's Research UK (ARUK-RF2014-2, ARUK-EG2013-B1, ARUK-PG2019A-004), the University of Turin (RILO 2019), and Compagnia di San Paolo (CST0167048). Erica Staurenghi was supported by Fondazione Adriano Buzzati-Traverso.
Declaration of competing interest
The authors declare that they have no competing interests.
Acknowledgements
We thank the Wohl Cellular Imaging Centre for assistance in microscopy.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101837.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Prince M.J., Wimo A., Guerchet M.M., Ali G.C., Wu Y.T., Prina A.M. World alzheimer report 2015: the global impact of dementia: an analysis of prevalence, incidence, cost and trends. Alzheimer’s Disease International. 2015 https://www.alzint.org/u/WorldAlzheimerReport2015.pdf [Google Scholar]
- 2.Lane C.A., Hardy J., Schott J.M. Alzheimer's disease. Eur. J. Neurol. 2018;25(1):59–70. doi: 10.1111/ene.13439. [DOI] [PubMed] [Google Scholar]
- 3.Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez-Nievas B.G., Serrano-Pozo A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018;10:114. doi: 10.3389/fnagi.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Nievas B.G., Stein T.D., Tai H.C., Dols-Icardo O., Scotton T.C., Barroeta-Espar I. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain. 2013;136(Pt 8):2510–2526. doi: 10.1093/brain/awt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vehmas A.K., Kawas C.H., Stewart W.F., Troncoso J.C. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol. Aging. 2003;24(2):321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 7.Anderson M.A., Ao Y., Sofroniew M.V. Heterogeneity of reactive astrocytes. Neurosci. Lett. 2014;565:23–29. doi: 10.1016/j.neulet.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zamanian J.L., Xu L., Foo L.C., Nouri N., Zhou L., Giffard R.G. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012;32(18):6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iram T., Trudler D., Kain D., Kanner S., Galron R., Vassar R. Astrocytes from old Alzheimer's disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol. Dis. 2016;96:84–94. doi: 10.1016/j.nbd.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garwood C.J., Pooler A.M., Atherton J., Hanger D.P., Noble W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167. doi: 10.1038/cddis.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavanagh C., Tse Y.C., Nguyen H.B., Krantic S., Breitner J.C., Quirion R. Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer's disease model. Neurobiol. Aging. 2016;47:41–49. doi: 10.1016/j.neurobiolaging.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Lian H., Yang L., Cole A., Sun L., Chiang A.C., Fowler S.W. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron. 2015;85(1):101–115. doi: 10.1016/j.neuron.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jha M.K., Lee S., Park D.H., Kook H., Park K.G., Lee I.K. Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci. Biobehav. Rev. 2015;49:135–156. doi: 10.1016/j.neubiorev.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Naude P.J., Nyakas C., Eiden L.E., Ait-Ali D., van der Heide R., Engelborghs S. Lipocalin 2: novel component of proinflammatory signaling in Alzheimer’s disease. FASEB J. 2012;26(7):2811–2823. doi: 10.1096/fj.11-202457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira A.C., Da Mesquita S., Sousa J.C., Correia-Neves M., Sousa N., Palha J.A. From the periphery to the brain: lipocalin-2, a friend or foe? Prog. Neurobiol. 2015;131:120–136. doi: 10.1016/j.pneurobio.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Bi F., Huang C., Tong J., Qiu G., Huang B., Wu Q. Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. U. S. A. 2013;110(10):4069–4074. doi: 10.1073/pnas.1218497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alzheimer A., Stelzmann R.A., Schnitzlein H.N., Murtagh F.R. An English translation of Alzheimer's 1907 paper, Uber eine eigenartige Erkankung der Hirnrinde. Clin. Anat. 1995;8(6):429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 19.Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 20.Di Paolo G., Kim T.W. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat. Rev. Neurosci. 2011;12(5):284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood W.G., Li L., Muller W.E., Eckert G.P. Cholesterol as a causative factor in Alzheimer's disease: a debatable hypothesis. J. Neurochem. 2014;129(4):559–572. doi: 10.1111/jnc.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gamba P., Testa G., Gargiulo S., Staurenghi E., Poli G., Leonarduzzi G. Oxidized cholesterol as the driving force behind the development of Alzheimer's disease. Front. Aging Neurosci. 2015;7 doi: 10.3389/fnagi.2015.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dason J.S., Smith A.J., Marin L., Charlton M.P. Cholesterol and F-actin are required for clustering of recycling synaptic vesicle proteins in the presynaptic plasma membrane. J Physiol. 2014;592(4):621–633. doi: 10.1113/jphysiol.2013.265447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Deijk A.F., Camargo N., Timmerman J., Heistek T., Brouwers J.F., Mogavero F. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia. 2017;65(4):670–682. doi: 10.1002/glia.23120. [DOI] [PubMed] [Google Scholar]
- 25.Gilardi F., Viviani B., Galmozzi A., Boraso M., Bartesaghi S., Torri A. Expression of sterol 27-hydroxylase in glial cells and its regulation by liver X receptor signaling. Neuroscience. 2009;164(2):530–540. doi: 10.1016/j.neuroscience.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 26.Iuliano L., Crick P.J., Zerbinati C., Tritapepe L., Abdel-Khalik J., Poirot M. Cholesterol metabolites exported from human brain. Steroids. 2015;99(Pt B):189–193. doi: 10.1016/j.steroids.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Testa G., Staurenghi E., Zerbinati C., Gargiulo S., Iuliano L., Giaccone G. Changes in brain oxysterols at different stages of Alzheimer's disease: their involvement in neuroinflammation. Redox Biol. 2016;10:24–33. doi: 10.1016/j.redox.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mutemberezi V., Guillemot-Legris O., Muccioli G.G. Oxysterols: from cholesterol metabolites to key mediators. Prog. Lipid Res. 2016;64:152–169. doi: 10.1016/j.plipres.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Zarrouk A., Vejux A., Mackrill J., O'Callaghan Y., Hammami M., O'Brien N. Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res. Rev. 2014;18:148–162. doi: 10.1016/j.arr.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Zarrouk A., Debbabi M., Bezine M., Karym E.M., Badreddine A., Rouaud O. Lipid biomarkers in Alzheimer’s disease. Curr. Alzheimer Res. 2018;15(4):303–312. doi: 10.2174/1567205014666170505101426. [DOI] [PubMed] [Google Scholar]
- 31.Testa G., Gamba P., Badilli U., Gargiulo S., Maina M., Guina T. Loading into nanoparticles improves quercetin's efficacy in preventing neuroinflammation induced by oxysterols. PloS One. 2014;9(5) doi: 10.1371/journal.pone.0096795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X., Xi Y., Yu H., An Y., Wang Y., Tao L. 27-hydroxycholesterol promotes Abeta accumulation via altering Abeta metabolism in mild cognitive impairment patients and APP/PS1 mice. Brain Pathol. 2019;29(4):558–573. doi: 10.1111/bpa.12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gamba P., Leonarduzzi G., Tamagno E., Guglielmotto M., Testa G., Sottero B. Interaction between 24-hydroxycholesterol, oxidative stress, and amyloid-beta in amplifying neuronal damage in Alzheimer's disease: three partners in crime. Aging Cell. 2011;10(3):403–417. doi: 10.1111/j.1474-9726.2011.00681.x. [DOI] [PubMed] [Google Scholar]
- 34.Marwarha G., Dasari B., Prasanthi J.R., Schommer J., Ghribi O. Leptin reduces the accumulation of Abeta and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J. Alzheimers Dis. 2010;19(3):1007–1019. doi: 10.3233/JAD-2010-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merino-Serrais P., Loera-Valencia R., Rodriguez-Rodriguez P., Parrado-Fernandez C., Ismail M.A., Maioli S. 27-Hydroxycholesterol induces aberrant morphology and synaptic dysfunction in hippocampal neurons. Cerebr. Cortex. 2019;29(1):429–446. doi: 10.1093/cercor/bhy274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang E.R., Lee C.S. 7-ketocholesterol induces apoptosis in differentiated PC12 cells via reactive oxygen species-dependent activation of NF-kappaB and Akt pathways. Neurochem. Int. 2011;58(1):52–59. doi: 10.1016/j.neuint.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Nury T., Samadi M., Zarrouk A., Riedinger J.M., Lizard G. Improved synthesis and in vitro evaluation of the cytotoxic profile of oxysterols oxidized at C4 (4alpha- and 4beta-hydroxycholesterol) and C7 (7-ketocholesterol, 7alpha- and 7beta-hydroxycholesterol) on cells of the central nervous system. Eur. J. Med. Chem. 2013;70:558–567. doi: 10.1016/j.ejmech.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 38.An Y., Zhang D.D., Yu H.L., Ma W.W., Lu Y.H., Liu Q.R. 27-Hydroxycholesterol regulates cholesterol synthesis and transport in C6 glioma cells. Neurotoxicology. 2017;59:88–97. doi: 10.1016/j.neuro.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Abildayeva K., Jansen P.J., Hirsch-Reinshagen V., Bloks V.W., Bakker A.H., Ramaekers F.C. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 2006;281(18):12799–12808. doi: 10.1074/jbc.M601019200. [DOI] [PubMed] [Google Scholar]
- 40.Ma W.W., Li C.Q., Yu H.L., Zhang D.D., Xi Y.D., Han J. The oxysterol 27-hydroxycholesterol increases oxidative stress and regulate Nrf2 signaling pathway in astrocyte cells. Neurochem. Res. 2015;40(4):758–766. doi: 10.1007/s11064-015-1524-2. [DOI] [PubMed] [Google Scholar]
- 41.Cigliano L., Spagnuolo M.S., Napolitano G., Iannotta L., Fasciolo G., Barone D. 24S-hydroxycholesterol affects redox homeostasis in human glial U-87MG cells. Mol. Cell. Endocrinol. 2019;486:25–33. doi: 10.1016/j.mce.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Bochelen D., Langley K., Adamczyk M., Kupferberg A., Hor F., Vincendon G. 7beta-hydroxysterol is cytotoxic to neonatal rat astrocytes in primary culture when cAMP levels are increased. J. Neurosci. Res. 2000;62(1):99–111. doi: 10.1002/1097-4547(20001001)62:1<99::AID-JNR11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Schildge S., Bohrer C., Beck K., Schachtrup C. Isolation and culture of mouse cortical astrocytes. JoVE. 2013;71 doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hudry E., Wu H.Y., Arbel-Ornath M., Hashimoto T., Matsouaka R., Fan Z. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer's disease. J. Neurosci. 2012;32(9):3176–3192. doi: 10.1523/JNEUROSCI.6439-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leoni V., Strittmatter L., Zorzi G., Zibordi F., Dusi S., Garavaglia B. Metabolic consequences of mitochondrial coenzyme A deficiency in patients with PANK2 mutations. Mol. Genet. Metabol. 2012;105(3):463–471. doi: 10.1016/j.ymgme.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 47.Sovrea A.S., Bosca A.B. Astrocytes reassessment - an evolving concept part one: embryology, biology, morphology and reactivity. J. Mol. Psychiatry. 2013;1:18. doi: 10.1186/2049-9256-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liddelow S.A., Barres B.A. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Sofroniew M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014;20(2):160–172. doi: 10.1177/1073858413504466. [DOI] [PubMed] [Google Scholar]
- 50.Kim J.H., Ko P.W., Lee H.W., Jeong J.Y., Lee M.G., Kim J.H. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia. 2017;65(9):1471–1490. doi: 10.1002/glia.23174. [DOI] [PubMed] [Google Scholar]
- 51.Lordan S., Mackrill J.J., O'Brien N.M. Oxysterols and mechanisms of apoptotic signaling: implications in the pathology of degenerative diseases. J. Nutr. Biochem. 2009;20(5):321–336. doi: 10.1016/j.jnutbio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 52.D'Amelio M., Sheng M., Cecconi F. Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 2012;35(11):700–709. doi: 10.1016/j.tins.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 53.Yammine A., Zarrouk A., Nury T., Vejux A., Latruffe N., Vervandier-Fasseur D. Prevention by dietary polyphenols (resveratrol, quercetin, apigenin) against 7-ketocholesterol-induced oxiapoptophagy in neuronal N2a cells: potential interest for the treatment of neurodegenerative and age-related diseases. Cells. 2020;9(11) doi: 10.3390/cells9112346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mucha M., Skrzypiec A.E., Schiavon E., Attwood B.K., Kucerova E., Pawlak R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc. Natl. Acad. Sci. U. S. A. 2011;108(45):18436–18441. doi: 10.1073/pnas.1107936108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee S., Lee W.H., Lee M.S., Mori K., Suk K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J. Neurosci. Res. 2012;90(3):540–550. doi: 10.1002/jnr.22779. [DOI] [PubMed] [Google Scholar]
- 56.Karch C.M., Goate A.M. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatr. 2015;77(1):43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serrano-Pozo A., Mielke M.L., Gomez-Isla T., Betensky R.A., Growdon J.H., Frosch M.P. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am. J. Pathol. 2011;179(3):1373–1384. doi: 10.1016/j.ajpath.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boisvert M.M., Erikson G.A., Shokhirev M.N., Allen N.J. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018;22(1):269–285. doi: 10.1016/j.celrep.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee S., Park J.Y., Lee W.H., Kim H., Park H.C., Mori K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J. Neurosci. 2009;29(1):234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holtje M., Hoffmann A., Hofmann F., Mucke C., Grosse G., Van Rooijen N. Role of Rho GTPase in astrocyte morphology and migratory response during in vitro wound healing. J. Neurochem. 2005;95(5):1237–1248. doi: 10.1111/j.1471-4159.2005.03443.x. [DOI] [PubMed] [Google Scholar]
- 61.Dekens D.W., De Deyn P.P., Sap F., Eisel U.L.M., Naude P.J.W. Iron chelators inhibit amyloid-beta-induced production of lipocalin 2 in cultured astrocytes. Neurochem. Int. 2020;132:104607. doi: 10.1016/j.neuint.2019.104607. [DOI] [PubMed] [Google Scholar]
- 62.Styren S.D., Kamboh M.I., DeKosky S.T. Expression of differential immune factors in temporal cortex and cerebellum: the role of alpha-1-antichymotrypsin, apolipoprotein E, and reactive glia in the progression of Alzheimer's disease. J. Comp. Neurol. 1998;396(4):511–520. [PubMed] [Google Scholar]
- 63.Rutkowska A., Shimshek D.R., Sailer A.W., Dev K.K. EBI2 regulates pro-inflammatory signalling and cytokine release in astrocytes. Neuropharmacology. 2018;133:121–128. doi: 10.1016/j.neuropharm.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 64.Mutemberezi V., Buisseret B., Masquelier J., Guillemot-Legris O., Alhouayek M., Muccioli G.G. Oxysterol levels and metabolism in the course of neuroinflammation: insights from in vitro and in vivo models. J. Neuroinflammation. 2018;15(1):74. doi: 10.1186/s12974-018-1114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Gijsel-Bonnello M., Baranger K., Benech P., Rivera S., Khrestchatisky M., de Reggi M. Metabolic changes and inflammation in cultured astrocytes from the 5xFAD mouse model of Alzheimer's disease: alleviation by pantethine. PloS One. 2017;12(4) doi: 10.1371/journal.pone.0175369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi S.S., Lee H.J., Lim I., Satoh J., Kim S.U. Human astrocytes: secretome profiles of cytokines and chemokines. PloS One. 2014;9(4) doi: 10.1371/journal.pone.0092325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma V., Mishra M., Ghosh S., Tewari R., Basu A., Seth P. Modulation of interleukin-1beta mediated inflammatory response in human astrocytes by flavonoids: implications in neuroprotection. Brain Res. Bull. 2007;73(1–3):55–63. doi: 10.1016/j.brainresbull.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 68.Morales I., Guzman-Martinez L., Cerda-Troncoso C., Farias G.A., Maccioni R.B. Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014;8:112. doi: 10.3389/fncel.2014.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hubbard A.K., Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic. Biol. Med. 2000;28(9):1379–1386. doi: 10.1016/s0891-5849(00)00223-9. [DOI] [PubMed] [Google Scholar]
- 70.Lyons P.D., Benveniste E.N. Cleavage of membrane-associated ICAM-1 from astrocytes: involvement of a metalloprotease. Glia. 1998;22(2):103–112. doi: 10.1002/(sici)1098-1136(199802)22:2<103::aid-glia1>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 71.Trombetta B.A., Carlyle B.C., Koenig A.M., Shaw L.M., Trojanowski J.Q., Wolk D.A. The technical reliability and biotemporal stability of cerebrospinal fluid biomarkers for profiling multiple pathophysiologies in Alzheimer's disease. PloS One. 2018;13(3) doi: 10.1371/journal.pone.0193707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Janelidze S., Mattsson N., Stomrud E., Lindberg O., Palmqvist S., Zetterberg H. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867–e877. doi: 10.1212/WNL.0000000000006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allaman I., Gavillet M., Belanger M., Laroche T., Viertl D., Lashuel H.A. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J. Neurosci. 2010;30(9):3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheng M. The postsynaptic NMDA-receptor--PSD-95 signaling complex in excitatory synapses of the brain. J. Cell Sci. 2001;114(Pt 7):1251. doi: 10.1242/jcs.114.7.1251. [DOI] [PubMed] [Google Scholar]
- 75.Prieto G.A., Cotman C.W. Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev. 2017;34:27–33. doi: 10.1016/j.cytogfr.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khairova R.A., Machado-Vieira R., Du J., Manji H.K. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 2009;12(4):561–578. doi: 10.1017/S1461145709009924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner M.D., Nedjai B., Hurst T., Pennington D.J. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta. 2014;1843(11):2563–2582. doi: 10.1016/j.bbamcr.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 78.Mao L.M., Wang J.Q. Synaptically localized mitogen-activated protein kinases: local substrates and regulation. Mol. Neurobiol. 2016;53(9):6309–6315. doi: 10.1007/s12035-015-9535-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paul S.M., Doherty J.J., Robichaud A.J., Belfort G.M., Chow B.Y., Hammond R.S. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J. Neurosci. 2013;33(44):17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kulig W., Cwiklik L., Jurkiewicz P., Rog T., Vattulainen I. Cholesterol oxidation products and their biological importance. Chem. Phys. Lipids. 2016;199:144–160. doi: 10.1016/j.chemphyslip.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 81.Dias I.H., Borah K., Amin B., Griffiths H.R., Sassi K., Lizard G. Localisation of oxysterols at the sub-cellular level and in biological fluids. J. Steroid Biochem. Mol. Biol. 2019;193:105426. doi: 10.1016/j.jsbmb.2019.105426. [DOI] [PubMed] [Google Scholar]
- 82.Perez M.J., Vergara-Pulgar K., Jara C., Cabezas-Opazo F., Quintanilla R.A. Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol. Neurobiol. 2018;55(2):1004–1018. doi: 10.1007/s12035-017-0385-x. [DOI] [PubMed] [Google Scholar]
- 83.D'Amelio M., Cavallucci V., Middei S., Marchetti C., Pacioni S., Ferri A. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat. Neurosci. 2011;14(1):69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- 84.Noble W., Garwood C., Stephenson J., Kinsey A.M., Hanger D.P., Anderton B.H. Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J. 2009;23(3):739–750. doi: 10.1096/fj.08-113795. [DOI] [PubMed] [Google Scholar]
- 85.Ragot K., Delmas D., Athias A., Nury T., Baarine M., Lizard G. alpha-Tocopherol impairs 7-ketocholesterol-induced caspase-3-dependent apoptosis involving GSK-3 activation and Mcl-1 degradation on 158N murine oligodendrocytes. Chem. Phys. Lipids. 2011;164(6):469–478. doi: 10.1016/j.chemphyslip.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 86.Nakazawa T., Miyanoki Y., Urano Y., Uehara M., Saito Y., Noguchi N. Effect of vitamin E on 24(S)-hydroxycholesterol-induced necroptosis-like cell death and apoptosis. J. Steroid Biochem. Mol. Biol. 2017;169:69–76. doi: 10.1016/j.jsbmb.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 87.Kim A., Nam Y.J., Lee C.S. Taxifolin reduces the cholesterol oxidation product-induced neuronal apoptosis by suppressing the Akt and NF-kappaB activation-mediated cell death. Brain Res. Bull. 2017;134:63–71. doi: 10.1016/j.brainresbull.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 88.DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 89.Crimins J.L., Pooler A., Polydoro M., Luebke J.I., Spires-Jones T.L. The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer's disease. Ageing Res. Rev. 2013;12(3):757–763. doi: 10.1016/j.arr.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.