

Abstract
The use of in vitro 3D organoid/colony forming assay (CFA); which mimics the in vivo environment have provided insight into the mechanisms by which lung stem cells maintain and repair the lung. In recent years, the use of CFA has markedly expanded. However, variations among laboratories in lung cell isolation methods, media used, type, origin, and processing methods of mesenchymal cells used as feeders for the epithelial colonies, and terms utilized to describe and quantify the growing colonies, have caused difficulty in reproducing results among different labs. In this study, we compared several previously described methods for lung cell isolation and culture media, to identify their influence on retrieved cells and growing colonies. We also characterized the effect of freeze/thaw, and propagation of fibroblasts on their ability to support epithelial colonies. Importantly, we suggested markers to identify fibroblast subtypes that offer the best support to alveolar stem cell proliferation. Then, we used our optimized assay to confirm the in vitro identity of recently described epithelial progenitors. We also tested the effect of hyperoxia on lung stem cells, and examined the expression of the receptors for the SARS-COV-2 virus’s entry into epithelial cells, on our organoids. In summary, our findings facilitate CFA standardization, help understand how niche cell variations influence growing colonies, and confirm some of the recently described lung stem cells.
Keywords: Lung organoids, Colony-forming assay, Niche cells, Lung fibroblast subtypes
Introduction
Lung epithelial cells are predominantly quiescent during homeostasis, but demonstrate an astonishing reparative capacity after injury. However, the repair process is occasionally disturbed, leading to the development of fibrosis and/or formation of non-functional tissue[1]. The repair ability of the lung after injury is maintained through the activity of several lung-resident stem cell types. Lung stem cells, like other tissue stem cells, respond to various signals from the surrounding niche by proliferating, differentiating, or signaling back to other cells in their vicinity[2–4]. Our understanding of the mechanisms underlying lung stem cell interactions with their niche has remarkably advanced recently due to the combination of in vivo techniques such as lineage tracing, the development of mice with knockdown or over-expression of specific genes in certain cell types, and in vitro techniques, namely the lung 3D colonyforming assay (CFA) also called “organoid assay”[5–7]. Although the CFA does not completely mimic the complex cellular interactions occurring in the lung, it is extremely useful to understand the specific interactions between stem and niche cells, as it allows the examination of one factor at a time, e.g. a ligand, a receptor, a nutrient, or a niche cell type/subtype[3, 8, 9]. The use of similar in vitro 3D systems initiated in brain and intestinal cells has markedly improved our understanding of these organs’ stem cell behavior, gene functions, disease development, and possible therapeutic options[10, 11]. Previously, we have described a simple and easily reproducible CFA for the analysis of lung stem cells and their various niche components. We characterized the different types of colonies that grew in this assay and their baseline differentiation profiles. We also demonstrated how the assay can be used to identify various potential niche components and the effect of their modulation on lung stem cell activation, proliferation, and differentiation[8]. In this follow up study, we first examined several aspects of the stem and niche cell collection and culture methods, aiming to increase the efficiency and physiological aspects of the assay. We then used the optimized assay to characterize various lung fibroblast subtypes with regards to their in vitro ability to support alveolar stem cell proliferation and differentiation in light of the recent in vivo characterizations[12, 13]. Furthermore, we utilized the assay to identify aspects of the hyperoxia effect on lung stem cells. Then, we examined the similarity between our in vitro-grown colonies, and the previously described lineage-negative epithelial progenitors (LNEP), and alveolar epithelial progenitors (AEP)[14, 15]. Finally, in light of the current COVID-19 pandemic, and the urgent need to conduct more basic research to understand various aspects of the function of the causative virus; COV-SARS-2, and the lung epithelial cells response to it, we examined the expression of the virus’s entry and priming receptors, Ace2 and Tmprss2 respectively, on our organoids.
Materials and methods
Mice
Mice expressing green fluorescent protein (GFP) in surfactant protein C (Sftpc)-positive cells (CBA/Ca × C57BL6J) were obtained from Brigid Hogan, Duke University. C57BL6J mice were used, when GFP fluorescence was not required. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at Keio University, Tokyo, Japan.
Lung digestion with different protocols and collection of different types of lung cells
Lungs were collected from three mice per group using four different digestion protocols. Subsequently, lungs were processed utilizing various enzymes and incubated for diverse periods of time with or without intratracheal enzyme instillation.
Protocol 1. Dispase only: Lungs were inflated through a tracheal cannula with 1.5 mL 25 U dispase (BD) and placed in phosphate buffered saline (PBS). The inflated lungs were incubated at 37 °C for 30 min. Next, lung lobes were dissected from the trachea and mediastinal tissues, minced, and placed in 0.25% trypsin/EDTA at 37 °C for 20 min.
Protocol 2. Elastase only: Lungs were inflated with 1 mL 4 U elastase (EPC) solution, placed in PBS, and incubated at room temperature for 5 min. Subsequently, 3 x 0.5 mL additional elastase injections were introduced every 5 min through the tracheal cannula. Finally, lung lobes were dissected from the trachea and mediastinal tissues, minced, and placed in 0.25% trypsin/EDTA at 37 °C for 20 min, with brief vortexing every 5 min.
Protocol 3. Collagenase type IV, elastase, and dispase enzyme mixture: Lungs were inflated with 1 mL enzyme mixture containing collagenase type IV (400 U/mL), elastase (4 U/mL), and dispase (5 U/mL). Next, lung lobes were dissected from the trachea and mediastinal tissues, minced, and incubated in 2 mL of the above enzyme mixture at 37 °C for 25 min with frequent vortexing. Cells were washed with DMEM and incubated in 0.1% trypsin/EDTA at 37 °C for 20 min, with brief vortexing every 5 minutes.
Protocol 4. Liberase (a purified Collagenase I and Collagenase II) and dispase enzyme mixture: Lungs were collected without inflation. Lobes were dissected from the trachea and mediastinal tissues, minced, and incubated in 10 mL of the following enzyme mixture: liberase (Roche, 50 μg/mL) and dispase (5 U/mL) at 37 °C in a water bath with rocking movement for 10 min.
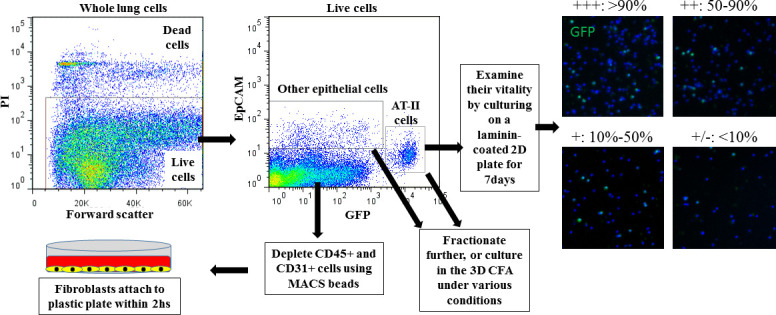
All cell solutions from the different protocols were passed four times through a 21 G needle and filtered using a 100 μM mesh. DNase (Worthington) was added to the mesh to facilitate the release of entangled cells. Red blood cells were lysed utilizing ACK lysing buffer (Thermo Fisher Scientific). We term this cell solution “whole lung cells, WLCs”. WLCs collected with the different digestion protocols were examined for the percentage of viable cells, the identification of percentages of lung cell types retrieved, and for vitality of the alveolar type (AT)-II cells. For estimation of endothelial cell retrieval, an aliquot of WLCs were stained for CD31 and examined by flow cytometry. For fibroblast retrieval, assessment and collection, hematopoietic and endothelial cells were depleted by staining the WLCs with CD31 and CD45 MACS beads and sorting on an AutoMACS machine (Miltenyi Biotec). CD45-CD31- cells were incubated in RPMI (Invitrogen) supplemented with 10% FBS at 37 °C in T75 flasks for 2 h. The attached cells were the fibroblasts. The percentage of AT-II cells were assessed, then sorted on a MoFlo XDP (Beckman Coulter) based on their GFP+ expression. Whole lung non- AT-II epithelial cells were collected by sorting on a MoFlo XDP for Ep-CAM+/GFP- cells. These cells were either cultured or further fractioned based on their staining for Sca-1+CD24+, integrin a6+CD200+CD14+, and GFP+/AT-II cells were sorted into Tm4sf1+ and Tm4sf1- as described in the results section. Feeder cells were prepared by treating cultured fibroblasts with 10 μg/mL mitomycin-C (Sigma) for 2 h. Cells were frozen at -80 °C using Cell Banker 1 (ZENOAQ). To examine the vitality of sorted AT-II cells, cells were cultured in in vitro 2D culture as previously described[16]. Sorted GFP+ cells (2 × 104 cells/well) were cultured in RPMI on laminincoated polycarbonate transwells. The ability of cells to attach and proliferate were monitored by fluorescence and bright-light microscopy (Keyence BZ-8000). Each experiment was repeated at least 3 times.
Fibroblast phenotyping and sorting of different subtypes using FACS
Freshly collected, and seven days-propagated fibroblasts were stained with rat anti-CD44 (BioLegend), anti-CD166-FITC (eBioscience), anti-CD90 (Thy1.1)-APC/Cy7 (BioLegend), anti- Sca1-APC/Cy7 (BioLegend), or anti-PDGFRa-PE (Abcam) antibody. Next, cells were examined/analyzed on a Gallios (BD) or sorted on a MoFlo (Beckman Coulter).
MTS assay
Fibroblast subpopulations (3,000 cells), sorted based on their PDGFRa and/or CD90 expression, were seeded in 100 μL into a 96- well culture plate. Each sample was analyzed in triplicate, and the assay was repeated three times. Cells were allowed to settle for a couple of hours. Cell proliferation was evaluated by adding 20 μL MTS CellTiter 96® AqQueous One Solution Reagent (Promega, Madison WI, USA) to the cells at indicated time points (day 1, 3, 5, and 7). Plates were incubated at 37 °C for 1 h and read at a wavelength of 490 nm on a plate reader.
In Vitro 3D Organoid Colony-Forming Assay
Lung fibroblasts (1.0 × 105 cells) were co-cultured with 1.0 × 105 sorted Ep-CAMhigh, Ep-CAMhigh/GFPhigh, or Ep-CAMhigh/GFP- cells in a 2:1 growth factor-reduced Matrigel® (BD Biosciences, San Jose, CA) with 150 μL placed in each transwell. Duplicate or triplicate wells were used for all experiments. MTEC/Plus medium (600 μL) was added to the lower chamber and replaced every other day. Some wells were treated with either the Notch inhibitor DBZ (20 μM) (Sigma) or the Notch activator DL-Sulforaphane (20 μM) (Sigma). The number of colonies per insert was counted on day 14 to 16. Both fluorescence and phase-contrast images were obtained using a Leica DMi6000B microscope. The type, number and size of colonies were quantified by visual counting as previously described[8]. In brief, the A, B, and C colony types were differentiated based on their morphological characteristics: Type A are rounded with big lumen and thin walls, with minimal GFP/Sftpc expression. Type B are irregular shaped with no or small lumen but with thick walls. They might partially express GFP/Sftpc. Type C are round or oval shaped with no or small lumen. Importantly, they express GFP/Sftpc in most cells. Collection of the Matrigel 3D colonies and processing for histological examination were performed as previously described[8]. Paraffin-embedded colonies were cut into 6-μm sections and stained for hematoxylin and eosin (H&E) or with goat and rabbit anti-Sftpc (Santa Cruz, Millipore), rabbit and mouse anti-Aqp5 (Abcam, Santa Cruz), anti-vimentin (Cell Signaling Technology), or mouse anti-Ecadherin (BD) antibody.
Immunofluorescent staining
The paraffin-embedded colony blocks were sectioned (6 μm) and stained with cell-type specific antibodies. The primary antibodies used were mouse anti-E-cadherin (BD), rabbit anti-vimentin (Cell Signaling Technologies), rabbit anti-cytokeratin-5 (K5) (Covance), mouse anti-PCNA (Santa Cruz) and mouse anti-acetylated alpha tubulin (Sigma). Nuclei were counterstained with hematoxylin or DAPI (Vector Labs, Burlingame, CA, USA) and examined. For cytospin slides, anti-alpha smooth muscle actin (aSMA) and anti-rat CD45 antibodies were also used.
Real-time qPCR
Total RNA was extracted from fibroblasts utilizing the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. cDNA was synthesized from total RNA with the High Capacity RNA to c-DNA kit (ThermoFisher Scientific). Real-time qPCR was carried out using the SYBR FAST ABI Prism qPCR Kit (Kapa Biosystems) according to the manufacturer’s protocol. Gene expression levels were analyzed with the QuantStudio 5 Real-Time PCR System (ThermoFisher Scientific). Mouse beta actin was utilized as an endogenous normalization control.
The following primers were used:
IL-6 Fwd: 5'-GAGGATACCACTCCCAACAGACC-3'
Rev: 5'-AAGTGCATCATCGTTGTTCATACA-3'
Stat 3 Fwd: 5'-ACCCAACAGCCGCCGTAG-3'
Rev: 5'-CAGACTGGTTGTTTCCATTCAGAT-3'
Grem2 Fwd: 5'-CGGGATCCAGTCAGCTGTGTGATGTCTGCTC-3'
Rev: 5'-GGAATTCTACAGAAAGACCTCGGTGTGTGTTTG-3'
Fgf7 Fwd: 5'-TTTGGAAAGAGCGACGACTT-3'
Rev: 5'-GGCAGGATCCGTGTCAGTAT-3'
Ace2 Fwd: 5'-TCCATTGGTCTTCTGCCATCC
Rev: 5'-AACGATCTCCCGCTTCATCTC
Tmpress2 Fwd: 5'-GAGAACCGTTGTGTTCGTCTC
Rev: 5'-GCTCTGGTCTGGTATCCCTTG
Statistical analysis
Data are expressed as means ± standard error of the mean (SEM). Data from ≥3 independent samples/experiments were averaged and either presented as a percentage of the total or compared using twotailed Student’s t-test. Results were considered statistically significant at P < 0.05.
Results and Discussion
Selection of a lung cell digestion protocol for efficient retrieval of various cell types (epithelial, mesenchymal, endothelial, and hematopoietic) with reasonable viability
To isolate a specific cell type from a tissue, cells should be detached from the extracellular matrix holding them together. This is typically achieved by exposing the tissue to a combination of proteolytic enzymes for variable time periods, usually in combination with gentle agitation. The type and concentration of enzyme(s) used, the period and temperature of incubation, and the method of physical tissue dissociation and enzyme exposure can all influence the type, number, and viability of retrieved cells[17]. Previous studies have used different protocols to digest lungs into single cell suspensions, which complicates the comparison of their findings, as the retrieved cell populations may differ significantly. Various types and concentrations of collagenases, elastase, dispase, and trypsin (with and without EDTA) have been used[8, 18–22]. We compared four common lung digestion protocols for the enrichment of specific cell types and viability of the retrieved cells. A collagenase/dispase protocol retrieved all cell types with satisfactory viability but impaired the vitality of AT-II cells (Tables 1, S1, and figure S1). We used the word “vitality” to describe the ability of viable AT-II cells to attach and proliferate on a matrix-coated 2D plate. Interestingly, a dispase only protocol was the most efficient method to retrieve AT-II and other lung epithelial cells with high viability and vitality; however, very few endothelial and mesenchymal cells were retrieved (Tables 1, S1, and figure S1).
Cell type identification: AT-II cells: GFP+ and/or Sftpc+, other epithelial cells: EpCAM+/GFP-. Fibroblasts: cells that attach to the bottom of a plastic culture plate when CD45/CD31-depleted lung cells are cultured in complete medium for 2 h. Endothelial cells: CD31+ cells. Viability was determined as the percentage of Propidium iodide (PI) negative WLCs (see also figure S1-A).
Comparative quantification: Each parameter is compared among the 4 protocols row-wise: +++: Digestion protocol with the highest number of cells retrieved, or with more than 66% of the cell number retrieved in the highest cell number retrieval digestion protocol.
++: number of cells retrieved is between 34%-66% of the cells retrieved by the highest cell number retrieval digestion protocol.
+: number of cells retrieved is between 10%-33% of the cells retrieved by the highest cell number retrieval digestion protocol.
+/-: no cells, or less than 10% of cells retrieved by the highest cell number retrieval digestion protocol.
Comparing AT-II vitality: AT-II cells seeded on laminin-coated 2D culture plates were examined for percentage of confluence on day 7 (see figure S1 for representative images):
+++: monolayer is more than 90% confluent on day 7, ++: 50-90% confluent on day 7, +: 10%-50% confluent on day 7, +/-: no cells, or cells covering less than 10% of the bottom of the well.
Furthermore, physical mincing and needle passage shearing before/after enzyme incubation increased the total number of retrieved cells, but reduced their viability. In the collagenase-using protocols, intratracheal instillation of the enzyme mixture markedly reduced epithelial cell viability and vitality. The use of cell sorting techniques (manual MACS, autoMACS, and FACS) to deplete, enrich, or sort specific cell types also reduced the viability and vitality of epithelial cells; manual MACS was the gentlest method but delivered the lowest purity, whereas FACS provided the lowest vitality, but highest purity. Importantly, repeated additions of up to 100 μL DNase to the cell suspension during the filtration steps released numerous cells that would have otherwise remained trapped in the sticky DNA commonly observed on the filter (released from dead inflammatory cells), thus resulting in a considerable increase of the total cell number retrieved per lung. We elected to use protocol 4., in all subsequent analyses, as it retrieved enough number of fibroblasts, usable as feeder cells, produced satisfactory numbers of non-AT-II epithelial cells, while also producing AT-II cells with acceptable vitality.
MTEC/Plus versus distal lung culture médium
Cell culture medium influences cell fate and behavior in in vitro studies. This is particularly relevant for stem cell culture due to the need to balance cell self-renewal and differentiation[23]. MTEC/Plus medium was originally optimized for the culture and differentiation of mouse trachea epithelial cells (MTEC) in an air-liquid interface (ALI) assay[24] and was later used for the culture of 3D CFA for MTEC (tracheo-spheres)[5, 25] and for CFAs for distal lung epithelium and AT-II cells[6, 8]. MTEC/Plus medium is supplemented with fresh retinoic acid (RA), bovine pituitary extract (BPE), epidermal growth factor (EGF), cholera toxin (CT), insulin, and transferrin. However, several studies used culture medium that did not include RA, BPE, EGF, or CT but included insulin, transferrin, and supplemented selenium (also named lung epithelium medium)[18]. When we compared MTEC/Plus with lung epithelium medium for CFA of whole lung epithelial cells (including both AT-II and non-AT-II cells), A and B colonies[8] growing in the MTEC/Plus medium were larger, with a more complex shape (i.e. differentiating and forming organoids). However, alveolar C-type colonies[8] were more abundant in the culture wells fed with lung epithelium medium though smaller (Figure 1A-D). When the growing colonies were examined for the various lung cell differentiation markers, we found that within the A/B colonies from both media types, the ratio of the various differentiated cell types were not different. Similarly, for the C colonies, the ratio of Aqp5 and SPC was not different, even though the size and number varied by media type (Figure S2)[8], indicating that the effect of medium type is on the number of stem cells that get activated from each type and its rate of proliferation, but not on its differentiation profile. This indicates that the MTEC/Plus medium is more efficient in activating various lung epithelial stem cell proliferation and differentiation, whereas lung epithelium medium is biased towards activating alveolar stem cell proliferation. Therefore, we chose to use the MTEC/Plus medium in all our in vitro CFA studies to obtain colonies that are more representative of the various lung stem cells.
Figure 1: Effect of culture medium and various lung fibroblast manipulations on the in vitro colony formation of lung epithelial stem cells Whole lung epithelial cells from Sftpc-GFP mice were co-cultured with lung fibroblasts in the in vitro colony forming assay in either “MTEC/Plus” or “lung epithelial” culture medium. Colony types were identified based on their morphology and GFP expression as detailed in Methods: C-type colonies express GFP in most cells, A and B types express less to no GFP. Yellow arrows point to some A/B colonies. A, B) Representative images of the effect of the difference in the medium used on colony size and type. C, D) Quantification of the growing colonies revealed that the MTEC/Plus medium was more efficient in supporting the colonies, as they induced the formation of a higher number of A/B colonies that were bigger in size and more complex, whereas the lung epithelial medium contained a higher number of colonies but smaller GFP+ alveolar Ctype colonies. Lung epi med: Lung epithelial medium.E) Percentage of live lung fibroblasts and mitomycin-C-treated feeder cells without freezing and after freezing and thawing. F) Whole lung epithelial cells from Sftpc-GFP mice co-cultured with equal numbers of live lung fibroblasts and mitomycin-C-treated feeder cells without (fresh) and after freezing and thawing. Yellow arrows point to some A/B colonies. G) Quantification of the number of growing colonies showed that freeze/thaw did not significantly reduce the ability of fibroblasts to support lung epithelial stem cell colony formation but significantly reduced such ability in mitomycin-C-treated feeder cells. Fibro: fibroblasts, F/T: freeze/thaw, MMC: mitomycin-C-treated feeder cells. Data represent results from three independent experiments. * p<0.05. Scale bars = 500 μm. A), B), and F) are merged images of bright field and the green fluorescence channel to detect which colonies are expressing GFP.
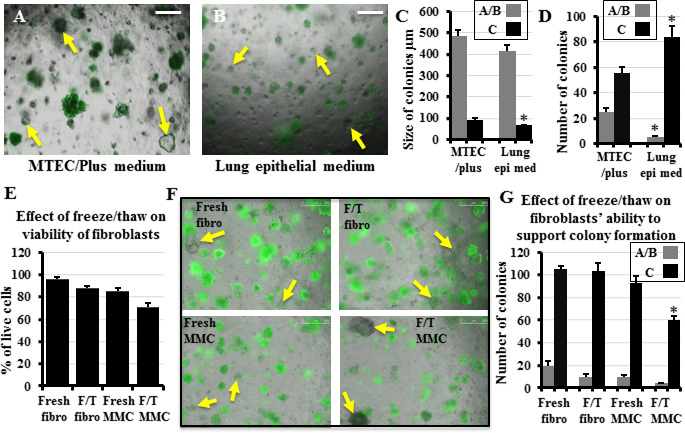
Effects of freezing lung fibroblasts and/or rendering them mitotically inactive on their viability and capacity to support colony formation
Previous studies have identified the necessity to co-culture lung epithelial cells with stromal cells (mostly fibroblasts, occasionally endothelial cells, and rarely mesenchymal stem cells[26]) to promote epithelial stem cell colony formation, using various subpopulations of lung fibroblasts. They utilized autologous lung EpCAM-/Sca-1+ cells[27], lung lipofibroblasts enriched from Pdgfra- H2B/GFP transgenic mice[6], or the neonatal mouse lung fibroblast cell line MLg[18, 28], whereas we used primary “whole lung” fibroblasts that were expanded for 7 days in culture (to increase their number) and treated with mitomycin-C (to render them mitotically inactive)[8]. Although the cryopreservation of mesenchymal stem cells from various species reduces their viability[29], it is almost technically unavoidable to pre-collect fibroblasts, freeze them for variable periods of time, and then thaw them for use in co-culture. Therefore, we examined the effect of freezing/thawing and mitomycin-C-treatment on whole lung fibroblasts with respect to their viability and ability to support colony formation. Lung fibroblasts that were collected from 10 different mice were split into two halves, one treated with mitomycin-C (to render them into feeder cells) and the other not treated. Then all the 20 samples were mixed with the “Cell Banker” freezing reagent and were stored at -80 °C for 3–30 days. Then cells were thawed, PI added, and examined by flow cytometry to identify the percentage of viable cells in comparison to freshly-collected fibroblasts, and freshlycollected, mitomycin-C-treated feeder cells. Freshly-collected fibroblasts showed >95% viability, untreated freeze/thaw fibroblasts had 80–90% viability, while freshly-collected mitomycin-C-treated feeders had 80–85% viability, and freeze/thaw mitomycin-C-treated feeder cells had 70-85% viability (Figure 1E). Next, based on the percentage of viable cells, equal numbers of viable mitomycin-Ctreated and untreated freshly-collected and freeze/thaw fibroblasts were co-cultured with whole lung epithelial cells in the CFA for two weeks. We found that freeze/thaw of the fibroblasts or treating them with mitomycin-C, then using them as feeders without freeze/thaw, did not significantly reduce their ability to support colony formation compared to the freshly collected fibroblasts. However, freeze/thaw of the mitomycin-C-treated fibroblast feeder cells significantly reduced their ability to support colony formation compared to freshly-collected fibroblasts (Figure 1F, G). Accordingly, we did not use mitomycin-C in subsequent experiments and used only fresh, or frozen/thawed fibroblasts.
Effects of fibroblast propagation in vitro on their phenotype and ability to support colony formation: Identifying the “niche” fibroblast subpopulations that are supportive and suppressive to lung stem cells
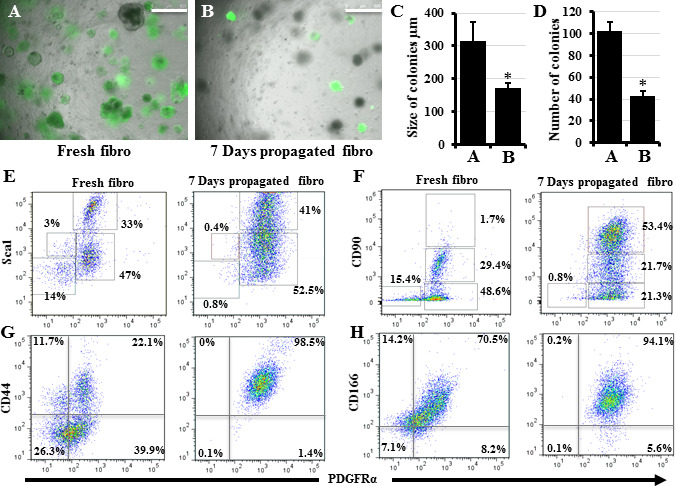
Various subpopulations of lung fibroblasts exist, and they all exhibit unique phenotypes and functions[30]. Previous studies have suggested that lung fibroblast subpopulations can switch between lipo- and myofibroblast phenotypes during lung development and in response to injury in adult lung[31, 32]. The presence of a lung fibroblast phenotype more closely associated with the lung alveolar stem cell niche has recently been suggested[6, 8, 13]. Many primary mesenchymal and stem cells undergo phenotype changes when expanded in vitro with both significant and non-significant impairments on their function after propagation[33–35]. Expanding a subpopulation of lung stromal cells (fibroblasts), sorted based on being Sca–1+ in vitro, decreased their ability to support stem cell colony formation probably due to a switch in their phenotype[36]. Accordingly, we first examined the effect of in vitro expansion on the unfractionated “whole” lung fibroblast ability to support colony formation. Lung epithelial cells co-cultured with freshly collected (day 0) lung fibroblasts produced colonies that were larger in size and number than those obtained when expanded (day 7) fibroblasts were used (Figure 2A-D). Therefore, we hypothesized that a subpopulation of lung fibroblasts exists that more efficiently supports lung stem cells, and that during in vitro expansion this more supportive subpopulation is taken over by a faster-expanding but less-supportive/inhibitory subpopulation. To identify these subpopulations, we studied the phenotype changes induced in “whole” lung fibroblasts by in vitro expansion. We examined them for changes in the expression of markers commonly associated with fibroblast identification and isolation, namely CD44, CD166, CD90 (Thy1.1), Sca1, and PDGFRa on day 0 (freshly-collected) and day 7. The expression of these markers in lung fibroblasts on day 0 ranged between 33-63%, whereas on day 7 their expression rose to 74-96% with considerable overlap among them (Figure 2E-H), indicating a marked phenotype shift within the 7 day propagation. However, previous reports have suggested that PDGFRa, Sca-1, and/or CD90- expressing lung fibroblasts are lipid-containing, stem cell-supportive niche cells[6, 28, 31]. Thus, the increase in percentage of fibroblasts expressing these markers on day 7 should have caused them to be more rather than less supportive for stem cell colonies. To resolve this controversy, we hypothesized that these markers are not exclusive for stem cell-supportive niche cells, and that there must be subgroups within the fibroblasts expressing these markers. Accordingly, we speculated that a majority subgroup is the supportive one and a minority subgroup assumes a non-supportive role, and that in vitro propagation favors the spread of the nonsupportive over the supportive phenotype. To identify such fibroblast subgroups, we compared subpopulations based on the expression of two markers, instead of a single marker, hoping to resolve the fibroblasts into several separate subpopulations. Plotting CD44 against PDGFRa showed separation into four different populations in fresh fibroblasts, but all cells were mostly double positive on day 7 (Figure 2G); therefore, CD44 was excluded. CD166 demonstrated almost complete overlap with PDGFRa in both fresh and day 7 fibroblasts (Figure 2H), and thus CD166 was also excluded. Plotting Sca-1 and CD90 against each other revealed a considerable overlap between Sca-1 and CD90-expressing cells both on day 0 and day 7 (data not shown). However, plotting PDGFRa against Sca1 or CD90 revealed that CD90 was more efficient in fractionating lung fibroblasts into four distinctive subpopulations with a considerable change in the percentage of each subpopulation between day 0 and day 7 (Figure 2E, F). On day 7, the CD90high cell population showed marked expansion in the CD90 plot. When this CD90high population was subgated to the Sca1 plot, it did not fit with the Sca1 high or low cells and contained a mixture of both; i.e., it is a unique population marked by CD90 and not by Sca1 (Figure 2E, F).
Figure 2: Effect of propagating fibroblasts in vitro on their ability to support colony formation and on their marker expression A, B) Lung epithelial cells collected from Sftpc-GFP mice were co-cultured with freshly collected lung fibroblasts (A) or lung fibroblasts that were propagated in culture for 7 days (B). Quantification of colonies growing in A and B to detect differences in their size (C) and number (D). E-H) Changes in lung fibroblast phenotype as reflected by changes in the expression of major fibroblast markers. Freshly collected and 7 days propagated lung fibroblasts were stained for Sca1, CD90, CD44, CD166, and PDGFRa and examined by flow cytometry. Data represent results from three independent experiments. * p<0.05. Scale bars = 500 μm. A), and B) are merged images of bright field and the green fluorescence channel to detect which colonies are expressing GFP.
To identify whether any of these four subpopulations (resolved based on PDGFRa and CD90) is more/less supportive for lung stem cells, we sorted day 0 and day 7 lung fibroblasts into these 4 subpopulations (similar to the gates in Figure 2F) and co-cultured an equal number of each fibroblast subpopulation with equal numbers of lung epithelial cells in the CFA. The PDGFRα+/CD90high subpopulation (which is markedly expanded from day 0 to day 7, increasing from 5.8 to 53.4%) exhibited the lowest ability to support CFA (Figure 3A-K), whereas the PDGFRα+/CD90low and PDGFRα+/CD90medium subpopulations (which decrease in percentage in day 7 fibroblasts) demonstrated the highest capacity to support CFA. PDGFRα-/CD90- cells did not support CFA. Importantly, even though day 7 PDGFRα+/CD90low and PDGFRα+/CD90medium fibroblasts were more efficient in supporting CFA than other day 7 subpopulations, their colony numbers were still significantly lower than those of the colonies growing with their day 0 counterparts (Figure 3A-K). These findings imply the presence of additional factors, influencing the ability of each day 7 subpopulation to support the CFA. To identify whether the increase in the percentage of stem cell “suppressive” PDGFRα+/CD90high subpopulation is due to its intrinsic faster in vitro proliferative capacity or due to a culture-induced CD90 upregulation and phenotype transformation, the proliferation rates of the four sorted day 0 subpopulations were compared in an MTS assay. The PDGFRα+/CD90high cells were indeed the fastest proliferating subpopulation, and their proliferation rate increased over time. PDGFRα-/CD90- cells almost did not proliferate (Figure 3L).
Figure 3: Identification of the stem cell supportive and suppressive “niche” fibroblast sub-populations. A-J) Day 0 and day 7 lung fibroblasts were sorted based on their expression of PDGFRα and CD90 into four subpopulations and were then co-cultured with lung epithelial cells. K) Quantification of the number of colonies growing in A-J culture wells. Data represent results from three independent repeats. L) The sorted day 0 and day 7 lung fibroblasts subpopulations were examined in the MTS assay to compare their proliferation rates. M) The day 0 sorted four subpopulations were cultured separately for 7 days, then collected and examined for changes in their PDGFRα and CD90 expression profiles. Dot plots showing data compiled from three independent experiments. N, O) Day 0 and day 7 lung fibroblasts were cytospun and immune-stained for aSMA and vimentin. Arrows point to the aSMA+ cells. P) Representative immune-stained image showing the presence of a few contaminating hematopoietic and epithelial cells within the lung fibroblast populations. * p<0.05. Scale bars A-J = 500 μm, N-P: 20 μm. A) to J) are merged images of bright field and the green fluorescence channel to detect which colonies are expressing GFP.
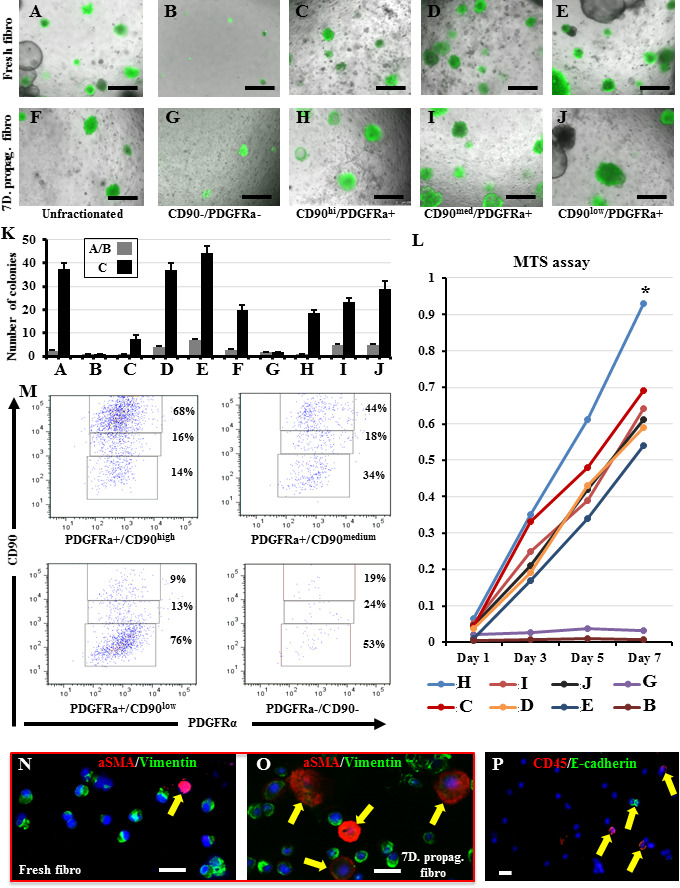
To identify whether the increase in PDGFRα+/CD90high cells is exclusively caused by their faster proliferation in vitro or whether a phenotype switch contributed to it, the four subpopulations sorted on day 0 were cultured/propagated separately for 7 days, and then cells were collected and examined for changes in their PDGFRα and CD90 expression. All populations showed some degree of change in their expression profile although to a various extent. PDGFRα-/CD90- cells did not proliferate, both the PDGFRα+/CD90low and PDGFRα+/CD90high cells mostly preserved their original profile, and the PDGFRα+/CD90medium cells clearly gave both PDGFRα+/CD90low and PDGFRα+/CD90high cells (Figure 3M, and Table S2), indicating that this PDGFRα+/CD90medium population possesses subtype plasticity and thus it is a major contributor to the increased number of PDGFRα+/CD90high cells observed on day 7. The conversion of one fibroblast subtype into another has previously been described during lung inflammation and fibrosis[36], where stem cell-supportive lipofibroblasts switch into stem cell-suppressive myofibroblasts[30]. To determine whether such conversion into myofibroblasts has occurred in the stem cell-less supportive day 7 fibroblasts, cytospin preparations from both day 0 and day 7 fibroblasts were stained for aSMA and vimentin (markers of myofibroblasts and fibroblasts). Indeed, day 7 fibroblasts contained a significantly higher number of myofibroblasts than day 0 fibroblasts (4.3% ± 1.3 vs. 27.4% ± 7.1, p < 0.01) (Figure 3N, O). In this staining, approximately 5–8% of cells were found to be vimentin-negative (i.e., non-fibroblasts/nonmesenchymal cells), indicating that our fibroblast isolation method also retrieved some non-fibroblast cells. To identify the identity of these contaminating non-fibroblast cells, we stained both day 0 and 7 fibroblasts for e-cadherin, CD31, and CD45 (markers of epithelial, endothelial, and hematopoietic cells). On day 0, we detected 4-7% and 1% hematopoietic and epithelial cells, whereas on day 7, we observed 5-8% hematopoietic cells with no detectable epithelial cells. No endothelial cell contamination was observed on day 0 or 7 (Figure 3P). Overall, we showed that within the freshly-collected lung fibroblasts a subpopulation marked as PDGFRα+/CD90medium demonstrated the highest ability to support alveolar stem cells. Furthermore, propagating lung fibroblasts in vitro resulted in upregulation of aSMA expression in all cells and marked expansion of the non-supportive PDGFRα+/CD90high subpopulation, leading to a considerable diminution of the stem cell supportive function of unfractionated fibroblasts.
As no specific marker to sort whole lung fibroblasts exists currently, several researchers used Sca-1 and PDGFRa as markers to sort presumably pure populations of lung fibroblasts[6, 27, 28]. However, Sca-1+ lung cells were a heterogeneous population that besides a subpopulation of fibroblasts included also other mesenchymal cells[27]. Furthermore, PDGFRα, although acting as an enrichment marker for lung alveolar stem cell niche fibroblasts, was also expressed in smooth muscle cells surrounding the major airways and blood vessels of adult mouse lung[12, 33]. Similarly, our method for collecting whole lung fibroblasts, based on their unique ability to quickly attach to plastic surfaces[8], yielded fibroblasts contaminated with non-fibroblast cells (as shown in Figure 3P). Thus, based on our results, we propose that sorting PDGFRα+/CD90medium and/or PDGFRα+/CD90low from freshly collected plastic-attached lung fibroblasts is a simple and quick method to collect a stem cellsupportive “niche” fibroblast subpopulation.
Similarity between in vitro-collected and in vivo-characterized stem cell-supportive “niche” fibroblast subpopulations
Zepp et al have described a mesenchymal alveolar niche cell (MANC), which demonstrates an enhanced ability to support alveolar growth and regeneration in vivo[12]. MANCs express higher levels of FGF7, IL-6, Stat3, and GREM2 than other lung fibroblasts[12]. To examine whether our in vitro-collected PDGFRα+/CD90medium fibroblast subpopulation overlaps with the in vivo-identified MANCs, we examined all fibroblast subpopulations for the expression of FGF7, IL-6, Stat3, and GREM2. Interestingly, GREM2 was almost absent in all day 7 fibroblast subpopulations and showed highest expression in a freshly-collected CD90 medium subpopulation (Figure 4A). Furthermore, although all fibroblast subpopulations on day 0 and day 7 expressed some FGF7, IL-6, and Stat3, the levels of FGF7 and Stat3 were consistently higher on day 0 in PDGFRα+/CD90medium fibroblasts. Only IL-6 showed higher expression in PDGFRα+/CD90low fibroblasts (Figure 4B-D). This suggests that day 0 PDGFRα+/CD90medium fibroblasts bear some genotypic similarity to MANCs.
Figure 4: Expression of genes proposed as markers for the stem cell supportive “niche” fibroblasts in our fibroblast subpopulations and effect of mild hyperoxia on colony formation efficiency A-E) Day 0 and day 7 lung fibroblasts were sorted based on their expression of PDGFRα and CD90 into four subpopulations; next, RNA was collected and examined for the expression of Grem2, Fgf7, Stat-3, IL-6, and Wnt5a. F) Lung epithelial cells from B6 mice were co-cultured with unfractionated fibroblasts under normoxic or hyperoxic conditions for 12 days. On day 14, cells in the hyperoxia chamber were transferred to normoxia and observed for an additional 12 days. G) Quantification of colonies growing in F. H) MTECs were cultured in the colony formation assay under normoxic or hyperoxic conditions for 12 days. On day 14, cells in the hyperoxia chamber were transferred to normoxia and observed for an additional 12 days. I) Quantification of colonies growing in H. Data represent results from three independent experiments. * p<0.05 compared to the normoxia, # p<0.05 compared to the hyperoxia. Scale bars = 100 μm.
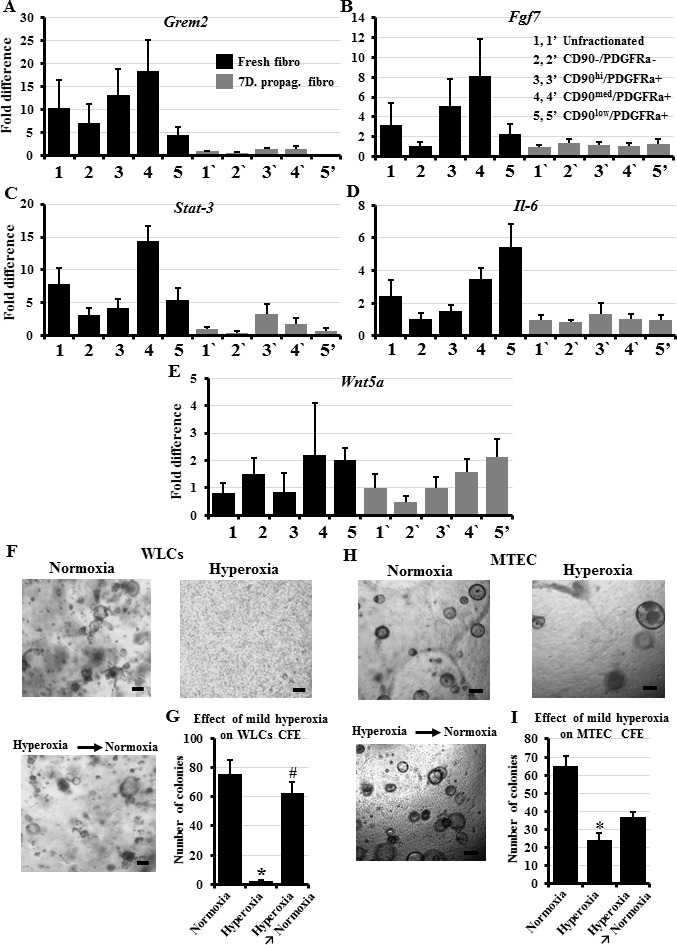
On the other hand, Nabhan et al showed that the presence of a Wnt signaling cross talk between a fibroblast and its nearby AT-II cells defines the “niche” fibroblast and its alveolar “stem” cell[13]. These “niche” fibroblasts specifically expressed Wnt5a, whereas other “regular” fibroblasts did not. Thus, we examined our in vitro fractionated fibroblast subpopulations for the expression of Wnt5a and found no significant differences among them (Figure 4E). This probably indicates that the presence of Wnt cross talk is a short-lived “functional” marker, which is precisely regulated by multiple signaling interactions among various lung cells, and that dissociating the lung fibroblasts by enzyme digestion, and exposing them to various physical stresses during staining, washing, and sorting abolishes the Wnt “identity” of these cells. In addition, Nabhan et alshowed that injured lungs would not rely on the niche fibroblast Wnt signaling and would increase the number of active alveolar progenitors (through a switch/recruitment of non-progenitor AT-IIs) by upregulating Wnt5a in these AT-II cells in an autocrine manner[13].
Mild hyperoxia significantly suppresses lung epithelial stem cell colony-forming efficiency
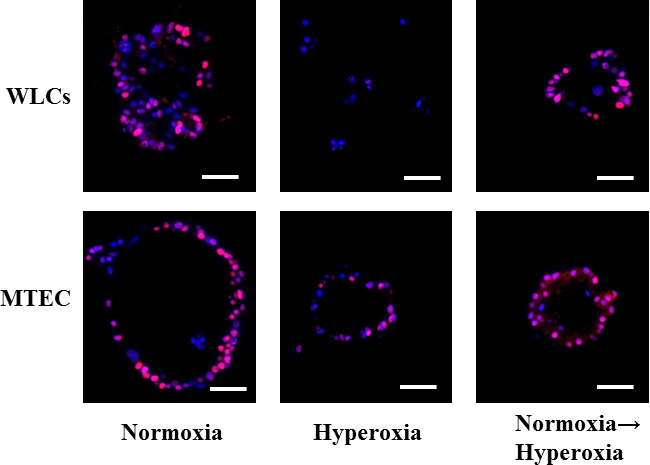
Whereas numerous studies have determined the harmful or beneficial effects of severe hyperoxia (>90% oxygen exposure) on the lung, few investigations have examined the influence of mild hyperoxia (40%–60% oxygen). Contradictory results were reported for the effect of hyperoxia on fibroblasts, showing both induction and inhibition of proliferation[38, 39]. Mild hyperoxia apparently caused minimal harmful effects to alveolar cells due to the protection offered by the glutathione-based antioxidant system[40]. The recent trend of using mild hyperoxia as an adjuvant therapy in cancer treatment[41] led us to explore its potential influence on lung stem cell proliferation exploiting our optimized CFA. Surprisingly, exposing whole lung cells to 50% oxygen for up to two weeks in the in vitro CFA completely suppressed their colony forming efficiency (Figure 4F, G). We confirmed that the failure to form colonies is due to the suppression of their activation/proliferation and not due to oxygen “toxicity” by collecting the lung cells that failed to form colonies after two weeks in culture, and examining them for apoptosis/necrosis using the annexin v/PI assay. Cells exposed to the standard 20% oxygen exhibited 99.6% ± 0.18 viability, whereas the cells exposed to the 50% oxygen demonstrated 93.7% ± 1.4 viability. The minimal decrease in viability in response to mild hyperoxia confirms that the 50% oxygen rather suppressed alveolar stem cell activation/proliferation and did not cause cell death. To identify whether the hyperoxia-induced suppression of WLCs stem cell activity is permanent or can be reversed, hyperoxia-exposed culture wells that did not grow any colonies were transferred into standard 20% oxygen for an additional two weeks. Many colonies grew in the well, indicating that the hyperoxia-induced suppression of lung stem cell activation/proliferation is reversible (Figure 4F, G). To explore whether this extreme oxygen sensitivity is lung stem cellspecific, we also cultured tracheal epithelial cells in the CFA under 50% oxygen for 2 weeks. Exposure of the tracheal stem cells to the same 50% oxygen level did not completely suppress, but still significantly reduced their colony forming efficiency (Figure 4H, I). When oxygen was restored to normoxia and the culture wells were observed for a further week, a few new colonies started to grow (Figure 4H, I). The colonies and cells were also collected from the various wells and stained for the proliferation marker, PCNA.
Colonies exposed to normoxia for two weeks, and newly formed colonies after restoration of normoxia showed several proliferating cells per colony. On the other hand, the hyperoxia-exposed whole lung cells were solitary or in duplicates, and showed no positive staining for PCNA. The few MTEC colonies detected also showed few PCNA positive cells (Figure S3). These results indicate that hyperoxia acts at least in part through suppressing stem cell activation/proliferation, and although further expansion is required, highlight the need to balance the beneficial effects of oxygen as adjuvant to classic cancer therapy with the potential harmful influence on stem cells in the rest of the lung
Isolating and confirming the in vitro identity of lineage-negative epithelial progenitors (LNEP) and alveolar epithelial progenitors (AEP)
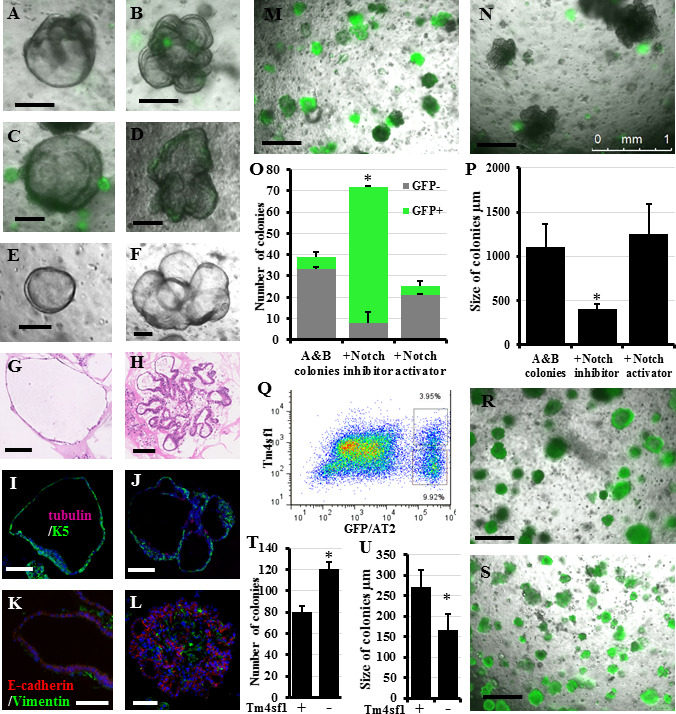
Early work has suggested the presence of rare epithelial progenitors in the lung, which can differentiate toward both the airway and alveolar lineages, and under special circumstances can contribute to lung healing and repair, in addition to the abundant facultative alveolar stem cells[42]. More recently, several markers have been suggested to isolate these rare progenitors from whole lung cell suspension using flow cytometry. These markers include, in addition to the pan-epithelial marker EpCAM, integrin a6, CD104, and CD24[43], or Sca-1 and CD24[44, 18]. We and others have described the morphological and differentiative characteristics of colonies growing from non-alveolar progenitors[8, 42, 18]. Recently, Vaughan et al described rare lung stem cells that are mostly quiescent but become activated after severe lung injury and termed them LNEPs. These LNEPs express proximal airway basal cell markers, integrin α6/β4, p63, and K5. Furthermore, LNEPs can be isolated based on their expression of integrin α6/β4, CD200, and CD14 and have the potential to differentiate into both airway and alveolar cells[14]. Differentiation toward the alveolar lineage after injury required Notch downregulation. Persistence of Notch signaling during the resolution stage results in failure of alveolar differentiation and prevalence of basal cell characteristics leading to “bronchiolization” of the lung[14]. To determine whether these various isolation methods and markers result in the same progenitor cells, we compared the colonies growing from the collected cells using different sets of markers. We also examined the effect of Notch signal inhibition and activation on their differentiation towards the airway lineage. We found that (after exclusion of alveolar cells) culturing whole EpCAM+GFP- cells (non-alveolar epithelial cells) (Figure 5A, B) or fractionating them further based on being Sca-1+CD24+ (Figure 5C, D) or integrin a6+CD200+CD14+ (Figure 5E, F) produced typically identical “A and B” colonies (Figure 5G, H)[8, 18] that expressed markers indicative mostly of proximal airways and rarely of alveolar differentiation (Figure 5I, J) in addition to the usual fibroblast and epithelial cell close association (Figure 5K, L)[8]. Treating these cultures with Notch inhibitor resulted in a marked shift of the differentiation toward the alveolar lineages, producing smaller GFP+ (i.e., Sftpc+) colonies. However, treating culture wells with a Notch activator reduced the overall colony number and preserved the differentiation bias toward the airway lineages and the large size of colonies (Figure 5M, N, quantified in O and P). Hence, our data suggests that other than the abundant alveolar cells, lung epithelial cells harbor another LNEP, which can be partially enriched by the use of the described markers. They form “A and B” colonies in vitro, and Notch expression controls their activation and differentiation toward the alveolar lineage. Recently, a subpopulation of alveolar cells was described to be more “stem” within the wider population of alveolar epithelial progenitors and thus was termed AEP. Isolation was based on the expression of the cell surface marker Tm4sf1[15]. We sorted the GFP+ (i.e., AT-II cells) into Tm4sf1+ and Tm4sf1– cells. Tm4sf1+ cells represented 26.4 ± 2.04% of alveolar cells. When cultured in CFA, and contrary to the original report, GFP+/Tm4sf1+ (AEP) cells formed fewer colonies than GFP+/Tm4sf1– alveolar cells, although AEP colonies were bigger in size, i.e., proliferating faster (Figure 5Q-S, quantified in T and U). This may be due to differences in culture medium, niche fibroblasts, or lung digestion and cell fractionation methods used by both studies. These findings highlight again the importance of identifying various technical and methodological differences among different labs, which should help reproduce the results and thus better understand the underlying biological phenomena.
Figure 5: Verifying the isolation methods for the LNEP and the AEP(A-F): Colonies growing when GFP–/EpCAM+ cells (non-AT-II lung epithelial cells) (A, B), or when GFP–/EpCAM+ cells were further purified based on their expression ofintegrin a6, CD104, and CD24 (C, D), or expression of Sca-1 and CD24 (E, F), they were sorted from whole lung epithelial cells from Sftpc-GFP mice and were co-culturedwith lung fibroblasts in the in vitro colony forming assay. (G, –H) Representative H&E stained colonies showing the typical characteristics of the “A and B” colonies (asdescribed in (8) and Methods) that were seen in in all the different cultures. I, J) Representative immune-stained colonies showing that most of them expressed basal andciliated cell markers. K, L) Representative immune-stained colonies showing the close association between fibroblasts and the epithelial colonies. Representative photo forthe effect of culture well treatment shown in A-F with Notch inhibitor M) or Notch activator N). O, P) Quantification of the effect of Notch manipulation on GFP (Sftpc)expression, colony number, and size. Q) Dot plot demonstrating the fractionation of GFP+/AT-II cells into Tm4sf1+ and Tm4sf1– AT-II cells. R, S) Representative imagesshowing the colonies growing in Tm4sf1+ and Tm4sf1– AT-II cell culture. T, U) Quantification of the number and size of colonies growing in R and S.Data represent results from three independent experiments. * p<0.05. Scale bars A-F = 200 μm, G-L: 100 μm, M, N, R, S: 500 μm.
Organoid Modeling of Respiratory Viral Infections
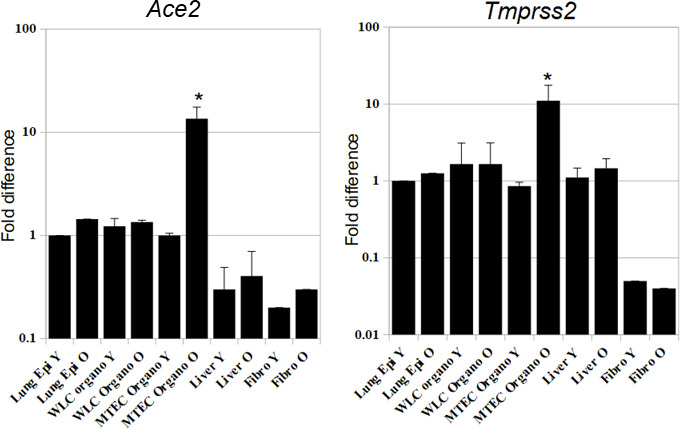
Beginning in late 2019 and through 2020, infection with a new coronavirus, termed SARS-COV2, has caused severe lung damage and multi-organ failure, a syndrome that was termed COVID-19, and was declared as a pandemic by the WHO (www.who.int/emergencies/diseases/novel-coronavirus-2019). There are still many unknowns regarding SARS-COV2, and having a relevant in vitro model that mimic the in vivo physiology and pathophysiology to study it might help in combating the highly anticipated recurrent waves of COVID-19 sicknesses over the coming months[45]. To identify whether our organoid system is usable for the study of SARS-COV2, we examined the expression of the virus’s entry and priming receptors, Ace2 and Tmprss2 respectively, on various organoid cells and control tissues. We also examined whether this expression varies between cells collected from young and old mice, as COVID-19 disease tends to be more severe in older individuals. We found that in young mice, both distal lung and proximal airway organoids express high levels of both receptors comparable to the level in naive lung epithelium. Interestingly, aging did not significantly influence the level of expression in either receptor in naive lung epithelium or distal lung organoids, but caused significant marked upregulation of both receptors in tracheal organoids. This is in line with recent reports that showed upper airway epithelium to be the main port of entry for the virus, and that receptor expression levels in children and young adults is lower than expression in older people[46, 47]. The liver had a high expression of Tmprss2, but variable low levels of expression of Ace2, and no effect of aging was detected. Fibroblasts had very low expression of Ace2 and almost no expression of Tmprss2, and no effect of aging was detected (Figure S4). This indicates that our organoid model is very relevant and suitable for conducting various pre-clinical experiments towards understanding the COVID-19 disease pathogenesis, such as studying cellular responses to the virus, age-induced changes, trial of new therapeutic agents, and factors that interact with stem cell repair etc.
In conclusion, despite the marked recent advances in our knowledge of lung stem cells and their niches, there is still insufficient information, especially with regards to the behavior of stem cells under various injuries/pathological conditions such as the effects of cigarette smoke, emphysema, and pulmonary fibrosis. CFA holds great potential to facilitate the understanding of the underlying pathology and to experiment on potential new therapeutic targets and drugs that will enhance endogenous repair. In the current study, we attempted to optimize and standardize the “organoid” assay and to render it more defined, reproducible, and physiological, so that data from one laboratory can be reproduced and expanded on by other laboratories.
Acknowledgments
The authors would like to thank Akihiro Tsutsumi (Keio University) and Fatma Y. Meligy (Asiut University) for their help with experimental procedures. The authors also acknowledge the contributions of the late Professor Tomoko Betsuyaku (Deceased September 1, 2018) in the planning and data interpretation of the early stages of this work.
Glossary
Abbreviations
- AEP:
Alveolar Epithelial Progenitors
- ALI:
Air-Liquid Interface
- aSMA:
Alpha Smooth Muscle Actin
- AT:
Alveolar Type
- CFA:
Colony Forming Assay
- COVID-19:
Coronavirus Disease 2019
- GFP:
Green Fluorescent Protein
- K5:
Cytokeratin-5
- LNEP:
Lineage-Negative Epithelial Progenitors
- MANC:
Mesenchymal Alveolar Niche Cell
- MTEC:
Mouse Trachea Epithelial Cells
- SARS-CoV2:
Pathogenic SARS-Coronavirus 2
- SEM:
Standard Error of the Mean
- Sftpc:
Surfactant Protein C
- WLC:
Whole Lung Cells
Potential Conflicts of Interests
None
Sponsors/Grants
This work was supported by JSPS Grant-in-Aid for Scientific Research (A.E.H. 20K08548). 5 Chome-3-1 Kōjimachi, Chiyoda City, Tokyo 102-0083
Additional Information
Supplementary Information accompanies this article. Supplementary information and figures are linked to the online version of the article.
Supplementary Information
Supplementary Table S1: Effect of various lung digestion protocols on the number of the different cell types retrieved and their viability (related to Table 1).
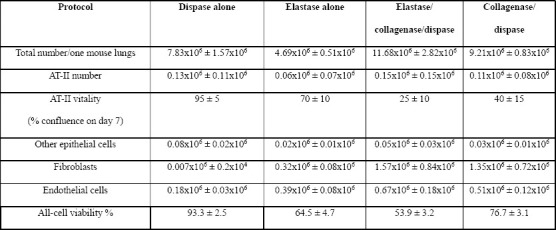
Table 1: Effect of various lung digestion protocols on the types of cells retrieved and their viability.
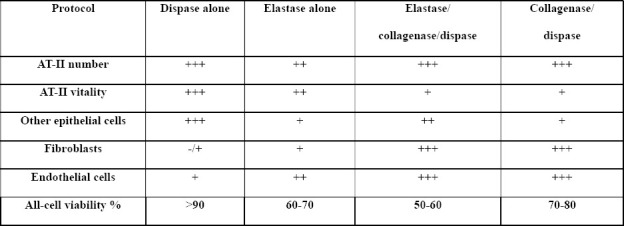
Actual numbers/percentages retrieved from each protocol, from at least 3 biological replicates, using at least 3 mice/protocol in each repeat. Data are expressed as means ± standard error of the mean.
Supplementary Table S2: Changes in the percentage of expression of various fibroblast markers between freshly-collected, and 7 day-propagated fibroblasts (related to Figure 2).
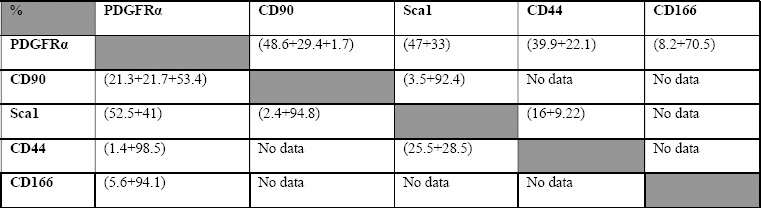
-Upper right half shows the double positive expressions on day 0, while the lower left half shows the expressions on day 7.
-Expression level is written as (Negative/low +medium +high).
Supplementary Figure S1: Summary of the processing of WLCs into the various cell types, and representative images for examination of AT-II cell vitality Figure S1: WLC solutions collected using the various enzyme digestion protocols are stained with PI and EpCAM. Live cells are gated (upper left, viability), then AT-II cells and other non-AT-II epithelial cells are sorted based on GFP and EpCAM expression (center). To examine vitality of collected AT-II cells, cells are seeded on laminin-coated plates, and examined by fluorescent microscope on D7 to assess percentage of confluence (right side photomicrographs). As most AT-II cells differentiate in culture into AT-I cells, only a few GFP+ cells are still visible at D7. Non-epithelial cells are also sorted then CD45+/CD31+ cells are depleted using AutoMACS. The CD45-/CD31- cells are cultured on uncoated plastic plates in complete medium for 2 h to isolate the fibroblasts (lower left).
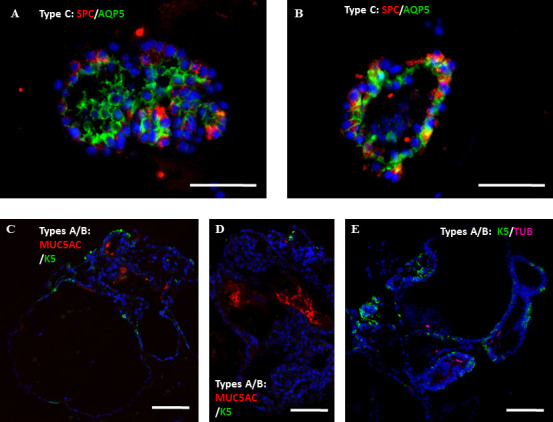
Supplementary Figure S2: Summary of the processing of WLCs into the various cell types, and representative images for examination of AT-II cell vitality Figure S2: Whole lung epithelial cells from Sftpc-GFP mice were co-cultured with lung fibroblasts in the in vitro colony forming assay in either “MTEC/Plus” or “lung epithelial” culture medium. The growing colonies were collected at 2 wks, immune-stained, and examined for the effect on their differentiation profile. A, B) Representative images showing staining with the ATII and AT-I cell markers, Sftpc and Aquaporin5, in type C colonies. C, D) Representative images showing staining of the A/B colonies with the markers of secretory and basal cells, MUC5AC and Keratin-5. E) Representative image showing staining of the A/B colonies with the markers of ciliated and basal cells, acetylated a-tubulin and Keratin-5. Scale bars A, B = 50 μm, C-E: 100 μm.
Supplementary Figure S3: Effect of mild hyperoxia on lung stem cells proliferation in vitro (related to Figure 4F-I) Figure S3: Whole lung epithelial cells and MTECs were co-cultured with lung fibroblasts in the in vitro colony forming assay in normoxia, hyperoxia of 50%, or hyperoxia followed by normoxia, as described in the results. The colonies and cells were collected, and immune-stained for the proliferation marker PCNA. Representative images showing one colony from each condition, except WLCs hyperoxia where no colonies were detected. Only solitary cells, which were negative for PCNA were detectable. Scale bars = 50 μm.
Supplementary Figure S4: Expression of the SARS-COV2 virus-related receptors on lung stem cell organoids and other control tissues from young and old mice Figure S4: RNA was collected from freshly-collected whole lung epithelial cells, WLC organoids, MTEC organoids, freshly collected liver tissue, and freshly collected fibroblasts, from young (8-12 wks) and old mice (1 year old), to detect the expression of these receptors, and to determine the influence of aging on the expression. Y-axis is in logarithmic scale for better comparison of the samples.
References
- 1.Zemans RL, Henson PM, Henson JE, Janssen WJ. Conceptual Approaches to Lung Injury and Repair. Ann Am Thorac Soc. 2015;12(Suppl 1):S9–15. doi: 10.1513/AnnalsATS.201408-402MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss DJ. Current Status of Stem Cells and Regenerative Medicine in Lung Biology and Diseases. Stem Cells. 2014;32((1)):16–25. doi: 10.1002/stem.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schilders KA, Eenjes E, van Riet S, Poot AA, Stamatialis D, Truckenmüller R, Hiemstra PS, Rottier RJ. Regeneration of the lung: Lung stem cells and the development of lung mimicking devices. Respir Res. 2016;17:44. doi: 10.1186/s12931-016-0358-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donne ML, Lechner AJ, Rock JR. Evidence for lung epithelial stem cell niches. BMC Dev Biol. 2015;15:32. doi: 10.1186/s12861-015-0082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, Randell SH, Hogan BL. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106((31)):12771–5. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123((7)):3025–36. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 2009;4((6)):525–34. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegab AE, Arai D, Gao J, Kuroda A, Yasuda H, Ishii M, Naoki K, Soejima K, Betsuyaku T. Mimicking the niche of lung epithelial stem cells and characterization of several effectors of their in vitro behavior. Stem Cell Res. 2015;15((1)):109–21. doi: 10.1016/j.scr.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BL. Lung organoids: current uses and future promise. Development. 2017;144((6)):986–997. doi: 10.1242/dev.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Lullo E, Kriegstein AR. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. 2017;18((10)):573–584. doi: 10.1038/nrn.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura T, Sato T. Sugimoto S, Sato T. Advancing Intestinal Organoid Technology Toward Regenerative Medicine. Cell Mol Gastroenterol Hepatol. 2017;5((1)):51–60. doi: 10.1016/j.jcmgh.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, Morrisey EE. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell. 2017;170((6)):1134–1148.e10. doi: 10.1016/j.cell.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359((6380)):1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA. Lineage-negative Progenitors Mobilize to Regenerate Lung Epithelium after Major Injury. Nature. 2015;517((7536)):621–5. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555((7695)):251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flodby P, Kim YH, Beard LL, Gao D, Ji Y, Kage H, Liebler JM, Minoo P, Kim KJ, Borok Z, Crandall ED. Knockout Mice Reveal a Major Role for Alveolar Epithelial Type I Cells in Alveolar Fluid Clearance. Am J Respir Cell Mol Biol. 2016;55((3)):395–406. doi: 10.1165/rcmb.2016-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molecular Biology of the Cell, 4th edition. Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter. New York: Garland Science; 2002.
- 18.Chen H, Matsumoto K, Brockway BL, Rackley CR, Liang J, Lee JH, Jiang D, Noble PW, Randell SH, Kim CF, Stripp BR. Airway Epithelial Progenitors Are Region Specific and Show Differential Responses to Bleomycin-Induced Lung Injury. Stem Cells. 2012;30((9)):1948–60. doi: 10.1002/stem.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demaio L, Tseng W, Balverde Z, Alvarez JR, Kim KJ, Kelley DG, Senior RM, Crandall ED, Borok Z. Characterization of mouse alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2009;296((6)):L1051–8. doi: 10.1152/ajplung.00021.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Driscoll B, Kikuchi A, Lau AN, Lee J, Reddy R, Jesudason E, Kim CF, Warburton D. Isolation and Characterization of Distal Lung Progenitor Cells. Methods Mol Biol. 2012;879:109–22. doi: 10.1007/978-1-61779-815-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108((52)):E1475–83. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aumiller V, Balsara N, Wilhelm J, Günther A, Königshoff M. WNT/β-catenin signaling induces IL-1β expression by alveolar epithelial cells in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49((1)):96–104. doi: 10.1165/rcmb.2012-0524OC. [DOI] [PubMed] [Google Scholar]
- 23.van der Sanden B, Dhobb M, Berger F, Wion D. Optimizing stem cell culture. J Cell Biochem. 2010;111((4)):801–7. doi: 10.1002/jcb.22847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol Lung Cell Mol Physiol. 2002;283((6)):L1315–21. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- 25.c
- 26.Leeman KT, Pessina P, Lee JH, Kim CF. Mesenchymal Stem Cells Increase Alveolar Differentiation in Lung Progenitor Organoid Cultures. Sci Rep. 2019;9((1)):6479. doi: 10.1038/s41598-019-42819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McQualter JL, Brouard N, Williams B, Baird BN, Sims-Lucas S, Yuen K, Nilsson SK, Simmons PJ, Bertoncello I. Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the sca-1 positive cell fraction. Stem Cells. 2009;27((3)):623–33. doi: 10.1634/stemcells.2008-0866. [DOI] [PubMed] [Google Scholar]
- 28.Teisanu RM, Lagasse E, Whitesides JF, Stripp BR. Prospective isolation of bronchiolar stem cells based upon immunophenotypic and autofluorescence characteristics. Stem Cells. 2009;27((3)):612–22. doi: 10.1634/stemcells.2008-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Xu X, Ma XH, Liu J, Cui ZF. Effect of various freezing solutions on cryopreservation of mesenchymal stem cells from different animal species. Cryo Letters. 2011;32((5)):425–35. [PubMed] [Google Scholar]
- 30.Fries KM, Blieden T, Looney RJ, Sempowski GD, Silvera MR, Willis RA, Phipps RP. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin Immunol Immunopathol. 1994;72((3)):283–92. doi: 10.1006/clin.1994.1144. [DOI] [PubMed] [Google Scholar]
- 31.Phipps RP, Penney DP, Keng P, Quill H, Paxhia A, Derdak S, Felch ME. Characterization of two major populations of lung fibroblasts: distinguishing morphology and discordant display of Thy 1 and class II MHC. Am J Respir Cell Mol Biol. 1989;1((1)):65–74. doi: 10.1165/ajrcmb/1.1.65. [DOI] [PubMed] [Google Scholar]
- 32.Gouveia L, Betsholtz C, Andrae J. Expression analysis of platelet -derived growth factor receptor alpha and its ligands in the developing mouse lung. Physiol Rep. 2017;5((6)):pii–e13092. doi: 10.14814/phy2.13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bara JJ, Richards RG, Alini M, Stoddart MJ. Concise review: Bone marrow-derived mesenchymal stem cells change phenotype following in vitro culture: implications for basic research and the clinic. Stem Cells. 2014;32((7)):1713–23. doi: 10.1002/stem.1649. [DOI] [PubMed] [Google Scholar]
- 34.Darling EM, Athanasiou KA. Rapid phenotypic changes in passaged articular chondrocyte subpopulations. J Orthop Res. 2005;23((2)):425–32. doi: 10.1016/j.orthres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Zhang CC, Lodish HF. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood. 2005;105((11)):4314–20. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McQualter JL, McCarty RC, Van der Velden J, O'Donoghue RJ, Asselin-Labat ML, Bozinovski S, Bertoncello I. TGF-β signaling in stromal cells acts upstream of FGF-10 to regulate epithelial stem cell growth in the adult lung. Stem Cell Res. 2013;11((3)):1222–33. doi: 10.1016/j.scr.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 37.El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, Szibor M, Kosanovic D, Schwind F, Schermuly RT, Henneke I, MacKenzie B, Quantius J, Herold S, Ntokou A, Ahlbrecht K, Braun T, Morty RE, Günther A, Seeger W, Bellusci S. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell. 2017;20((2)):261–273.e3. doi: 10.1016/j.stem.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Zglinicki T, Saretzki G, Döcke W, Lotze C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res. 1995;220((1)):186–93. doi: 10.1006/excr.1995.1305. [DOI] [PubMed] [Google Scholar]
- 39.Senavirathna LK, Huang C, Yang X, Munteanu MC, Sathiaseelan R, Xu D, Henke CA, Liu L. Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci Rep. 2018;8((1)):2709. doi: 10.1038/s41598-018-21073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aerts C, Wallaert B, Voisin C. In vitro effects of hyperoxia on alveolar type II pneumocytes: inhibition of glutathione synthesis increases hyperoxic cell injury. Exp Lung Res. 1992;18((6)):845–61. doi: 10.3109/01902149209031711. [DOI] [PubMed] [Google Scholar]
- 41.Stępień K, Ostrowski RP, Matyja E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med Oncol. 2016;33((9)):101. doi: 10.1007/s12032-016-0814-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121((6)):823–35. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 43.McQualter JL, Yuen K, Williams B, Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci U S A. 2010;107((4)):1414–9. doi: 10.1073/pnas.0909207107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zacharek SJ, Fillmore CM, Lau AN, Gludish DW, Chou A, Ho JW, Zamponi R, Gazit R, Bock C, Jäger N, Smith ZD, Kim TM, Saunders AH, Wong J, Lee JH, Roach RR, Rossi DJ, Meissner A, Gimelbrant AA, Park PJ, Kim CF. Lung stem cell self-renewal relies on BMI1-dependent control of expression at imprinted loci. Cell Stem Cell. 2011;9((3)):272–81. doi: 10.1016/j.stem.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ali I. COVID-19: Are We Ready for the Second Wave?. Disaster Med Public Health Prep. 2020. pp. 1–3. [DOI] [PMC free article] [PubMed]
- 46.Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, Talavera-López C, Maatz H, Reichart D, Sampaziotis F, Worlock KB, Yoshida M, Barnes JL. HCA Lung Biological Network. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26((5)):681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saheb Sharif-Askari N, Saheb Sharif-Askari F, Alabed M, Temsah MH, Al Heialy S, Hamid Q, Halwani R. Airways Expression of SARS-CoV-2 Receptor, ACE2, and TMPRSS2 Is Lower in Children Than Adults and Increases with Smoking and COPD. Mol Ther Methods Clin Dev. 2020;18:1–6. doi: 10.1016/j.omtm.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]