

Abstract
Genomewide association studies (GWAS) across psychiatric phenotypes have shown that common genetic variants generally confer risk with small effect sizes (OR<1.1) that additively contribute to polygenic risk. Summary statistics derived from large discovery GWAS can be used to generate polygenic risk scores (PRS) in independent, target datasets to examine correlates of polygenic disorder liability (e.g., does genetic liability to schizophrenia predict cognition). The intuitive appeal and generalizability of PRS have led to their widespread use and new insight into mechanisms of polygenic liability. However, presently, when applied across traits they account for small effects (less than 3% of variance) and are relatively uninformative for clinical treatment and, in isolation, provide no insight into molecular mechanisms. Larger GWAS are needed to increase their precision and novel approaches integrating various data sources (e.g., multi-trait analysis of GWAS) may improve the utility of current PRS.
Keywords: polygenic, PRS, GWAS, candidate, schizophrenia, psychopathology
It has long been recognized that psychiatric phenotypes travel in families (Schulze et al., 2004). By the 1820s, the first dedicated psychiatric hospital in the world, Bethlem, routinely asked whether presenting illnesses were hereditary (Plomin et al., 2013). In the early 20th century, family- and twin-based designs were used to empirically examine the latent genetic architecture of psychiatric phenotypes (Kendler, 2015). For example, in the first large scale systematic modern psychiatric genetics study, Rüdin examined rates of schizophrenia, known as dementia praecox at the time, among 2,733 siblings of 725 diagnosed probands (Rudin, 1916). Family and twin studies conducted over the subsequent century have largely confirmed early observations that, much like other complex traits, psychiatric disorders and related phenotypes (e.g., personality, brain volume) are generally moderately to highly heritable with a complex non-Mendelian polygenetic architecture that is importantly moderated by experience (Polderman et al., 2015). Clinically, the results from early family-based latent genetic research were initially used to enact eugenics programs (e.g., forced sterilization) in egregious and ill-fated efforts to reduce severe mental illness (Schulze et al., 2004) before informing our understanding of disease etiology, comorbidity, and nosology (Gilbertson et al., 2002, Posthuma and Polderman, 2013). The direct clinical application of latent genetic research is inherently limited; it cannot inform genetic risk beyond simple assessments of familial history, detect actionable therapeutic pathways (e.g., specific molecular pathways through which latent genetic risk is manifested), or lead to generalizable individually tailored treatments.
Advances in molecular genetic knowledge and technology facilitating the examination of specific measured genetic variation generated considerable enthusiasm that clinically-relevant and actionable results would soon be obtained [e.g., mechanistic insight, identification of treatment targets, personalized medicine (Craddock and Owen, 1996)]. Early hypothesis-driven (candidate gene) efforts adopted a bottom-up approach by studying variants with putative functional implications [e.g., COMT rs4680 (Egan et al., 2001), ADH1B rs1229984 (Thomasson et al., 1991), 5-HTTLR within SLC6A4 (Lesch et al., 1996, Caspi et al., 2003), MAOA promoter variant (Caspi et al., 2002)] within pathways (e.g., serotonin, dopamine, alcohol metabolism) known to be associated with psychiatric expression. Complementary studies probed biological [e.g., neural phenotypes (Hariri et al., 2002, Egan et al., 2001)] and behavioral [e.g., working memory (Goldberg et al., 2003)] mechanisms through which such putative risk may manifest (Bogdan et al., 2017) as well as how genetic variation may impact treatment response (Perlis et al., 2010).
In the past decade, technological advancements and related cost reductions, combined with the realization of the candidate gene research limitations (Duncan and Keller, 2011) and unprecedented collaborative team science have encouraged the transition to genomewide association studies (GWAS) and the use of a top down scientific approach (i.e., from associations with clinical manifestations to more basic biological mechanisms)(Kendler, 2013, Visscher et al., 2017)]. GWAS results have validated the role of some candidate loci [e.g., rs1229984 for alcohol dependence (Gelernter et al., 2014)] and genes [e.g., SIRT1 (CONVERGE, 2015), DRD2 (Schizophrenia Working Group of the Psychiatric Genomics, 2014)], but not others [e.g., COMT rs4680 and schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics, 2014); but see also meta-analytic work supporting this association with evidence of recessive mode of inheritance (Gonzalez-Castro et al., 2016)], and led to the discovery of replicable associations with novel variants [e.g., CACNA1C (Ferreira et al., 2008)]. More broadly, results from GWAS have profoundly reshaped our understanding of the genetic architecture of mental illness in two important ways. First, our hypotheses regarding the nature of biological contributors to disease were inadequate. Not only was our focus on specific pathways (e.g., dopamine for addictions; serotonin for depression) relatively naïve, but our estimates of the effect size associated with single variants was far too optimistic. We now know that, with few exceptions [e.g., APOE, rs429358 and rs7412 haplotypes and Alzheimer’s Disease (Corder et al., 1993)], individual common variants are associated with small effects [e.g., odds ratios < 1.1 (Schizophrenia Working Group of the Psychiatric Genomics, 2014)]. Our second lesson is more encouraging - the additive effects of independent and commonly occurring variants, when weighted by their association with a disorder or related construct in an adequately powered GWAS (Figure 1), are reliably predictive of variance in the same and related phenotypes (Figure 2; Tables 1–6). These additive scores, which we refer to as polygenic risk scores (PRS),1 along with other polygenic approaches (e.g., pathway-based analyses, machine learning, LD score regression) hold tremendous potential to contribute to our etiologic understanding of psychopathology and quantification of individual risk that may ultimately contribute to advances in the characterization and treatment of mental illness.
Figure 1. Practicalities of PRS computation.
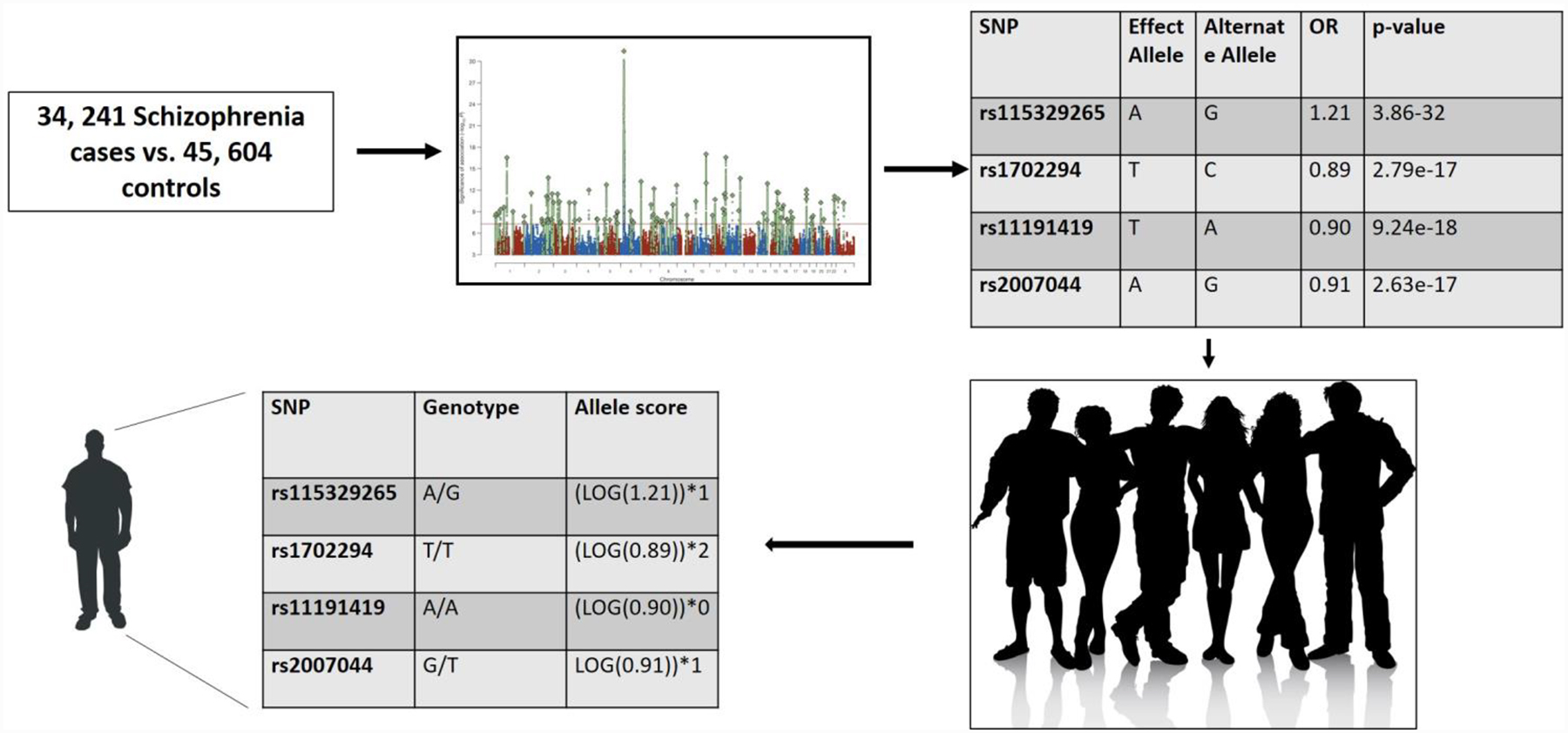
Here, we exemplify using statistics from the most recent Psychiatric Genetics Consortium GWAS of schizophrenia (Schizophrenia Working Group, 2014) in which a discovery GWAS of 34,241 schizophrenia cases and 45,604 controls (initial discovery not including replication due to shared summary statistics). The effect allele and effect size for each SNP from the discovery GWAS is applied to each individual in a target dataset to create a unique person-specific polygenic risk score. We illustrate the approach using 4 SNPs. Summary statistics may be obtained here: https://www.med.unc.edu/pgc/results-and-downloads
Figure 2. Schizophrenia PRS Effect Sizes Across 6 domains.
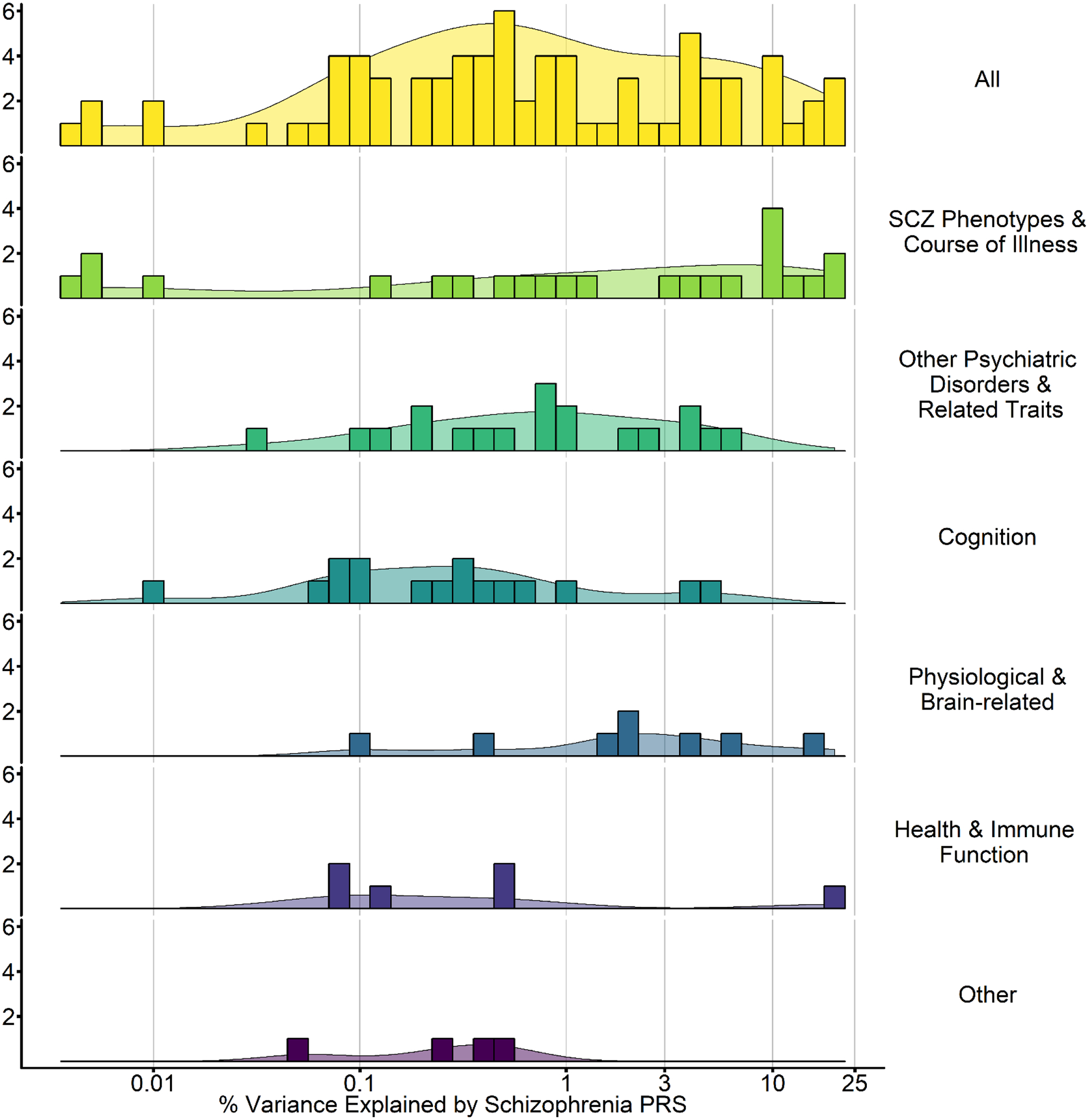
Plotted here is the range of the maximal percentage of variance explained by PGC SCZ2 PRS (Schizophrenia Working Group of the Psychiatric Genomics, 2014) in each study. Studies using other summary statistics are not included. Notably, as these are the maximal amount of variance explained and many studies reporting null effects did not report effect sizes, these estimates should be viewed as optimistic. The percentage of variance explained by Schizophrenia PRS in related traits ranges from 0.001 to 40% with 0.5–1.0% being most common. All = all identified Schizophrenia PRS studies. Course of illness = studies assessing course of illness. Psychiatric disorders and traits = studies evaluating other psychaitic disorders (e.g., bipolar disorder) or traits (e.g., neuroticism). Cognition = cognitive phenotypes (e.g., working memory). Brain-related = imaging phenotypes (e.g., cortical thickness), Health and immune function = immune (CRP levels) and reported health (e.g., physical health). Other = various other phenotypes related to schizophrenia (e.g., urbanicity). Individual studies contributing to this are summarized and * in Tables 1–6.
Table 1.
SCZ PRS and SCZ Phenotypes and Course of Illness.
| Domain | Phenotype & References | N | Association and Effect size* |
|---|---|---|---|
| SCZ Diagnosis, Symptoms, and Course | SCZ Diagnosis (Schizophrenia Working Group of the Psychiatric Genomics, 2014)* | 2638 SCZ; 2482 CON | Increased in SCZ: 0.184 |
| SCZ symptoms, age of onset (Stepniak et al., 2014)* | Diagnosis: 1067 SCZ, 1169 CON; other phenotypes: 502 Males. | Increased SCZ risk: 0.14. NS associations with age, symptoms, or suicidality. | |
| SCZ and x Family History (Agerbo et al., 2015)* |
866 SCZ, 871 CON | Increased SCZ: 0.034. Interaction between PRS and family history (p=0.03) - 17.4% of the increased SCZ conferred by family history accounted for by PRS. | |
| (Goes et al., 2015)* | Sample 1: 592 SCZ, 505 CON Sample 2: 913 SCZ, 1,640 CON Both Ashkenazi |
Increased SCZ risk: 0.097 | |
| Childhood-onset SCZ (Ahn et al., 2016)* | 120 COS probands, 103 healthy siblings | Increased PRS among COS probands relative to healthy siblings: Up tp 0.1852 | |
| Adolescent symptom expression (Jones et al., 2016)* | 3,676–5,444 general population | Psychotic experiences, OR: 0.001, NS. Negative symptoms, age 16.5: 0.007. Anxiety disorder, age 15.5: 0.005. No association with depressive disorder, age 15.5: 0.00005, NS. | |
| SCZ Diagnosis and Clinical Course Correlates (Meier et al., 2016)* | Sample 1: 876 SCZ, 871 CON Sample 2: 821 SCZ, 832 CON Sample 3: 1773 SCZ, 2161 CON |
Increased SCZ: 0.016–0.053; Increased in- or out-patient hospital admissions: rs=0.022–0.113; Longer length of hospital stay: rs=0.069–0.090; More likely to live in supportive housing: 0.006; | |
| First episode (FE) psychosis (SCZ-PRS x childhood adversity) (Trotta et al., 2016) | 80 FE SCZ; 110 CON | Increased SCZ, adjusted B = 7.68 (interaction with childhood adversity not significant) | |
| Symptom dimensions in FE psychosis (Sengupta et al., 2017)* | 241 FE SCZ | Baseline: Increased general psychopathology, PANSS total scores, negative symptom scores, and anxiety: 0.0256–0.058. NS: positive symptoms, GAF, depression, social and occupational functioning. Follow-up 1–24 months: lack of association following treatment. | |
| Psychosis in Alzheimers Disease (AD+P) (DeMichele-Sweet et al., 2017)* | Discovery: 1761 AD + P, 1115 AD-P Replication: 734 AD+P; 1460 AD-P |
AD+P: 0.0025 – 0.0032 (101 loci) | |
| Neurodevelopment, salience attribution, emotion regulation (van Os et al., 2017) | First degree relatives of SCZ: 871 siblings, 812 parents; 523 CON | PRS healthy < PRS relatives. Relatives: Total (negative and positive) and depressive symptoms B=0.11–0.21. NS for positive and negative symptoms independently. Controls: Total (negative and positive) and positive and negative symptoms: B=0.11–0.22. | |
| First episode (FE) psychosis (Vassos et al., 2017)* | 445 FE SCZ, 265 CON (EA), 828 CON (AA) | Increased SCZ risk in EA: 0.094. AA: 0.011 NS. Predicted SCZ development relative to other psychotic disorders among EA: 0.092. |
|
| SCZ Treatment Response | (Frank et al., 2015) | 434 Clozapine Tx History, 370 Clozapine No History; 78 responders vs. 45 non-responders | Higher PRS among those with a history of Clozapine treatment (P=0.02) |
| Trend for higher PRS among Clozapine nonrsponders relative to those who responded positively (P=0.06); affect potentiated in those with poor premorbid social function. | |||
| Dosage (Hettige et al., 2016)* | 83 SCZ | No association with antipsychotic dosage: <0.0001 | |
| Response to Lurasidone (Li et al., 2017)* | 171 EA, 131 AA | Less responsive to treatment, total symptoms, up to 0.101 | |
| (Wimberley et al., 2017a) | 862 SCZ (181 treatment resistant) | No association between PRS and treatment resistence, HR 1.13. |
Studies only include those using summary statistics from (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Unless otherwise noted, effect size is represented as Nagelkerke’s Pseudo R2 (multiply by 100 to obtain %R2); r = correlation, OR = Odds-ration, B = beta coefficient, AUC = Area Under the Curve. EA = European ancestry. AA = African ancestry. NS= non significant. When pearson’s r, OR, or d were reported, we estimated r2.
indicates study with % of variance explained included in Figure 2.
Table 6.
SCZ PRS and Other Phenotypes
| Phenotypes | Study Reference | N | Association and Effect Size* |
|---|---|---|---|
| Urbanicity | (Paksarian et al., 2017)* | 1,549 SCZ, 1,549 CON (matched) | Born urban: OR = 1.09: 0.00056; Living urban (15y): OR 1.19: 0.00229 |
| Reproductive fitness | (Mullins et al., 2017) | 631SCZ | No sig. assoc. (# children, age at 1st child) |
| Risky Sexual Behavior | (Wang et al., 2017a)* | 2,379 | Antagonistically pleiotropic SCZ HIV SNP PRS associated with risky sexual behavior: up to ~ 0.005. |
| Longitudinal Study Participation | (Martin et al., 2016)* | 7,867 children and 7,850 mothers | Nonparticipation/Noncompletion: 0.0014 – 0.0042 |
Studies only include those using summary statistics from (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Unless otherwise noted, effect size is represented as Nagelkerke’s Pseudo R2 (multiply by 100 to obtain %R2); r = correlation, OR = Odds-ration, B = beta coefficient, AUC = Area Under the Curve. EA = European ancestry. AA = African ancestry. NS= non significant. When pearson’s r, OR, or d were reported, we estimated r2.
indicates study with % of variance explained included in Figure 2.
The Elephant in the Room: Why do Genetic Research
To appreciate achievements in psychiatric genetics and to confront emerging challenges, we must, at the outset, examine whether studies aimed at identifying disease-related genetic variation are worthwhile. In particular, critics have argued that risk-attributable effect sizes are too modest, at least for common variants (e.g., OR < 1.1), to be therapeutically meaningful. From a public health perspective, only a few might argue that highly heritable traits, such as Autism Spectrum Disorders, Schizophrenia, Bipolar Disorder and Late-Onset Alzheimer’s Disease should not be interrogated using genetic approaches. However, some may propose that the pursuit of common variant association identification has either not yielded sufficiently new discoveries or, as for Schizophrenia, has reached an asymptote where the clinical relevance of every new discovery may not be as proportionally related to magnitudes of sample size, effort and continued funding, particularly as newly discovered variants are likely to be associated with increasingly smaller effect sizes. Similarly, there has been skepticism regarding investment of resources to gene discovery for disorders such as depression and addictions (Merikangas and Risch, 2003), that are less heritable. The skeptical argument is that such conditions are amenable to environmental intervention and therefore, building a better genetic model is more a source of intellectual self-gratification than of consequence to public health.
We fundamentally disagree with the position that the search for common variants associated with psychopathology is no longer relevant. Gene discovery, for disorders with moderate and high heritability, continues to be essential from two perspectives: mechanistic insight, and treatment improvement. Indeed, GWAS would be less appealing if research were to cease at gene discovery. This is rarely the case. However, the progress from discovery and replication of a locus to the outline of its etiologic significance takes time, resources, and collaborative interdisciplinary science (International Schizophrenia et al., 2009, Sekar et al., 2016).
Mechanistic insight:
Systematic translational research inspired by genetic association has strengthened our understanding of gene-brain/body-behavior relationships and identified potential therapeutic pathways. We briefly illustrate with two examples:
Immune pathways to schizophrenia:
The most strongly linked common genetic variation to schizophrenia was identified through GWAS and resides within the major histocompatibility complex (MHC) region (e.g., rs115329265 OR=1.21, p≈2.48e−31) (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Seven years following the initial identification of this region for schizophrenia (International Schizophrenia et al., 2009), Sekar and colleagues (2016) distilled the MHC genetic signal, in part, to complex structural variation within the complement component 4 gene (C4) (Sekar et al., 2016). C4 is a component of the innate immune system’s non-specific defense against foreign pathogens, and cellular debris that has also been implicated in the pruning of synapses. These researchers also showed that C4 variation altered brain C4 expression proportional to schizophrenia risk and that C4 mediated synaptic pruning during postnatal development in mice. Collectively, these results suggest that the MHC genetic signal for schizophrenia may be partially attributable to variability in C4-related synaptic pruning, which ultimately may be leveraged for treatment and/or prevention.
HPA axis regulation and risk for stress-related disease:
Genes identified outside of GWAS have also produced promising insights. Work pioneered by Binder, Ressler, and colleagues has linked variation in FKBP5 to pleiotropic stress-related health effects (e.g., PTSD, depression, Alzheimer’s disease, and aging) and related biological correlates (e.g., cortisol response, amygdala function) (Zannas et al., 2016). FKBP5 is a critical regulator of the hypothalamic-pituitary-adrenal (HPA) axis; it is a co-chaperone of the glucocorticoid receptor (GR) complex that diminishes GR sensitivity to cortisol resulting in an impaired ability of the HPA axis to return to homeostasis and transcriptionally regulate the genome. Through a series of programmatic experiments, Klengel and colleagues (Klengel et al., 2013) have shown that increased GR-stimulated FKBP5 expression observed among individuals with at least one T allele at rs1360780 may arise from alterations in the 3D structure of FKBP5. The T allele brings a long range enhancer region in intron 2 into physical contact with the transcription start site (TSS) thereby allowing it to affect expression. Effects of these conformation changes can be further compounded by T-allele specific childhood stress-related demethylation of a functional glucocorticoid response element within intron 7, which also contacts the TSS. Demethylation here, which some evidence suggests may only be stable following stress exposure early in life, enhances FKBP5 expression in the context of GR stimulation, resulting in a further reduction of HPA axis negative feedback. It is thus possible that the 3D conformation changes lead to a prolonged stress response that, when coupled with early life stress exposure, results in lasting epigenetic changes further impairing HPA axis regulation including its ability to influence the transcriptome. While recent large-scale GWAS of stress-related disorders (e.g., PTSD and depression) have not identified variants in FKBP5 (Duncan et al., 2017, Stein et al., 2016, Wray and Sullivan, 2017), this is unsurprising given the highly interactional nature of these molecular mechanisms and trauma exposure during early life (i.e., without GWASxE such effects would not be expected).
These two examples demonstrate that analyses of common single genetic variants, from GWAS and other empirical research, remains relevant and incredibly important in our polygenic world by identifying potentially clinically informative and actionable mechanistic pathways. They also underscore the premise that moderately heritable traits (e.g. PTSD h2 ~40%; (Cornelis et al., 2010)) that impose considerable personal and public health burden (Friedrich, 2017) are worthy of genetic inquiry.
Treatment insights:
Accumulated wisdom cautions against dismissing the potential clinical relevance of loci that are associated with small effects as they may provide flags for potential therapeutic targets. Perhaps the most convincing example of how genetic associations with small or modest effect sizes can have great treatment potential comes when considering statins, the first line drug of choice for the treatment of high cholesterol. Statins, developed in the 1970s, work by competitively inhibiting HMG-CoA reductase within the cholesterol synthesis pathway. Subsequent candidate genetic and GWAS-based investigations linked common genetic variation within the HMG-CoA reductase gene (HMGCR) to cholesterol. However, the effect sizes of such common genetic variation on cholesterol and cardiovascular risk are miniscule (i.e., < 20×) when compared to the effect of statins (Willer and Mohlke, 2012, Barrett et al., 2015). For example, the HMGCR SNP rs12916 reduces LDL cholesterol levels by 2.5 mg/dl for each copy of the protective allele, with even smaller associations with coronary artery disease, while statins typically decrease LDL by 14–70 mg/dL. This example is not unique. Similar observations of large medication effects targeting proteins in which common genetic variation is more modestly associated with disease expression, have been observed across a host of phenotypes (e.g., bone density and estrogens (Hirschhorn and Gennari, 2008)). Indeed, a recent investigation finds that drug targets with evidence of disease association through GWAS are twice as likely to be met with success (Nelson et al., 2015). These results suggest that common genetic variation linked to intermediate phenotypes (e.g., cholesterol for coronary artery disease), and also with disease risk at modest effect sizes, have the potential for great therapeutic value. Similar expectations for psychiatric disorders are not unrealistic (Breen et al., 2016, Wendland and Ehlers, 2016). GWAS may be a particularly fruitful pathway to such discovery, especially within the field of psychiatry, which has thus far struggled to identify mechanistic pathways that may be leveraged for treatment outside of happenstance [e.g., the observed mood benefits of an agent developed to treat tuberculosis that inhibited monoamine oxidase and led to the development of modern antidepressant medications (Ramachandraih et al., 2011)].
In addition to identifying therapeutic targets, molecular genetic variation may also be used to personalize therapeutic options for patients. Such pharmacogenomics approaches have been extraordinarily successful in personalizing cancer care and elucidating treatment pathways (Wheeler et al., 2013). The genetic architecture of clinically relevant psychological traits and psychiatric disorders is more complex and any such diagnostic genetic panel for these highly polygenic conditions should be viewed with extreme caution. Nonetheless, genetic research is currently bearing clinically-applicable fruit - for example, rs16969968 within CHRNA5, is a common missense polymorphism strongly associated with tobacco smoking (b=1.03, p≈3×10−73 (Tobacco and Genetics, 2010). This locus moderates the efficacy of treatment and influence numbers needed to treat [NNT (Bierut et al., 2014)]; one study found that while 4 individuals with the high-risk smoking haplotype needed to be treated to benefit one individual (NNT), the corresponding estimate for those with the low-risk haplotype was >1000 (Chen et al., 2012). Further, inspired by associations between FKBP5 and stress-related psychopathology, initial trials of FKBP5 inhibitors in rodent models of stress have produced promising results (Gaali et al., 2015).
If we are convinced of the value of genetic research (more specifically, common variant discovery and mechanistic probing), but also cognizant of polygenicity, statistical power, and the inherent limitations of nosological boundaries (e.g., heterogeneity and comorbidity) and prior mechanistic knowledge, how might genetic research be leveraged to inform clinical psychology? Polygenic risk scores (PRS) may usefully contribute to the identification of disorder-related risk factors, our understanding of comorbidity, and begin to provide insights into the developmental trajectory of psychiatric phenotypes as well as their course, treatment, correlates, and underlying pathophysiologies.
Polygenic Risk Scores: A Bridge Between Population Variation and Individual Differences
What are PRS
In simple terms, a polygenic risk score is a cumulative index of measured genetic liability to a disorder. PRS are similar to “heritability” (h2) in that they represent, to some degree, the aggregate and additive effects of segregating loci of small effect (see also section: Differences from other Heritability Metrics). Similar to traditional twin estimates of narrow heritability, PRS are additive in nature, consistent with numerous illustrations that support the widespread role of additivity in complex genetics (Hill et al., 2008). Several excellent reviews, both of a technical/practical and interpretive nature, have been written about the utility of PRS (Wray et al., 2014, Chatterjee et al., 2016, Bogdan et al., 2017, Plenge et al., 2013, Dudbridge, 2016). Here, we provide a brief theoretical and practical overview, before exemplifying how PRS approaches have begun to inform the field of clinical psychology and considering ways forward.
PRS Practicalities
Figure 1 illustrates the basic process of deriving a PRS, the steps to which are briefly described here:
STEP 1: Identify a well-powered discovery GWAS in which your sample is not included and obtain summary statistics from that analysis.
The remarkable popularity of PRS is in part attributable to the increasing availability of full summary statistics from discovery GWAS, a data-sharing approach spearheaded by the PGC (https://www.med.unc.edu/pgc/results-and-downloads), who not only share the summary statistics of their GWAS but also do so prior to formal publication and host results files from other consortia and investigators. PRS are derived by applying the “effect” alleles and their corresponding effect sizes (odds-ratios or beta coefficients) from a discovery GWAS to an independent, target sample (Figure 1). Because the effect size is used to weight allelic risk, its reliability is critically important. Hence, the better powered the discovery GWAS is, which is undoubtedly related to sample size, but also, arguably, to phenotypic relevance and strength of assessment, as well as sample composition (CONVERGE, 2015), the greater the confidence we might have in the resulting effect sizes. In Figure 1, we use the example of the current largest GWAS of Schizophrenia which includes 36,989 cases and 113,075 controls (Schizophrenia Working Group of the Psychiatric Genomics, 2014). While it is not necessary for the discovery GWAS to have identified replicable genomewide significant loci (i.e., association p-values < 5 × 10−8), the ability for a GWAS to identify such loci may be seen as evidence for statistical power and its potential utility for PRS analyses.
STEP 2: Establish commonality between your target dataset and the discovery GWAS.
PRS are amenable to differences in SNP content across the discovery and target samples; it is preferable to begin by working with imputed data in both the discovery and target dataset to maximize convergence. First, SNPs in the target dataset that overlap with SNPs in the discovery GWAS are extracted. Second, the target data are aligned to the discovery dataset (i.e., individual genotypes are oriented to the same strand of DNA and strand-ambiguous SNPs - A/T or G/C - are either excluded or closely evaluated). These steps are critical to ensure that effect sizes from the discovery GWAS are being accurately applied to the target sample. Typically, sex chromosomes are also removed. Traditional quality control indices should be applied to the target dataset including minor allele frequency cutoffs, Hardy-Weinberg-equilibrium testing, missingness by individual and marker exclusion, cryptic relatedness exclusion, sex check, ancestral outliers, and imputation quality.
STEP 3: Eliminate SNPs in high LD.
Correlated variants can represent non-independent association signals that, if ignored, could overweight PRS in favor of loci in high LD, by essentially counting a single signal multiple times. Indeed, not thinning a PRS according to LD can reduce their precision (Wu et al., 2013). As a result, it is recommended that variants be clumped so that the LD statistic, r2, is no greater than 0.10. When there are correlated SNPs, it is recommended to select the SNP in your target dataset based on the strength of association in the discovery GWAS (i.e., p-value-informed clumping). For example, if there are 80 SNPs forming an LD block, the SNP selected to represent this cluster should be the one with the largest effect size in the discovery GWAS. Not clumping data may limit the polygenic interpretability of PRS, especially at more significant p-value thresholds where an entire PRS could be driven by a series of correlated variants; but see Improvements in PRS estimation for alternatives. Studies also commonly exclude areas of complex linkage structure (e.g., MHC region) or retain one representative SNP across such regions.
STEP 4: Calculate PRS for each individual in your sample.
For each individual in the target sample, multiply the effect size (if ORs, the log-transformed allelic OR) observed in the discovery GWAS by the number of effect alleles for that variant that the individual possesses. In our example (Figure 1), for each polymorphism, this product term represents an individual’s liability to schizophrenia that is attributable to the alleles present at that single variant. These individual allele scores can then be averaged across the genome to form an overall polygenomic liability, or PRS, for each individual. Typically, an investigator creates multiple PRS that represent degrees of type II error/alpha (i.e., varying p-value thresholds) in the discovery GWAS. Therefore, one might aggregate all variants that were significant at p<1.0, p < .50, p < 0.30, p < .10, < 0.05, p < 5 × 10–3, p < 5 × 10–5 and p < 5 × 10–8 in the discovery GWAS, generating a person-specific PRS for schizophrenia at each p-value threshold. To limit multiple testing, one could select a p-value threshold that is most predictive of variability of the GWAS phenotype or related phenotype in an independent sample or use permutation based testing to assess whether the overall pattern of associations across p-value thresholds is more than expected by chance within the target dataset (Carey et al., 2016b). PRS can then be used as continuous variables within traditional analytic settings (e.g., regression). Typical covariates in further analyses include factors that confound gene - outcome analyses, such as ancestry differences, age, and sex.
Software programs including PLINK (Purcell et al., 2007, Chang et al., 2015) and integration with PRSice (Euesden et al., 2015b) facilitate PRS calculation.
Technical Considerations.
Sample size:
This review underscores the rational range of effect sizes that should be considered when determining sample sizes that would be amenable to PRS analyses (Figure 2). There are two issues to consider here: how predictive is a PRS, and how large of a target sample will usefully detect the effects of that PRS on an outcome. Several technical papers discuss the predictive utility of PRS - So and Sham (So and Sham, 2017) found that SCZ PRS (Schizophrenia Working Group of the Psychiatric Genomics, 2014) had the greatest predictive power of all psychiatric disorders (max Area Under the Curve, AUC = 0.82), which was even higher than some health-related traits [e.g., Type 2 Diabetes and Coronary Artery Disease max AUC = 0.61 (So and Sham, 2017)]. Predictive power for psychiatric disorder PRS appears to increase at more inclusive p-value thresholds, unlike metabolic traits, which is consistent with a greater degree of expected polygenicity. Further, psychiatric PRS are modestly superior in performance when drawn from studies with greater phenotypic precision. Other expositions on the topic have considered R2 estimation in the context of discovery GWAS sample size (N), the number of presumed independent causal loci (M~70,000) and SNP-h2 (Daetwyler et al., 2008, Dudbridge, 2013). Simulations from these studies are sobering. For instance, in their GWAS meta-analysis of educational attainment, Rietveld and colleagues (Rietveld et al., 2013) concluded that a discovery GWAS of 1 million individuals for this phenotype might generate a polygenic score that explained 15% of the variance in the same phenotype in a new sample. The second consideration is whether a target sample size is large enough to rule out false negatives or positives. PRS analyses are often simple regressions, and typically account for 0.01% (conservatively) to 3% (optimistically) of variance in cross-trait prediction (e.g., schizophrenia PRS predicting task performance among healthy individuals; Figure 2, Tables 1–6). As such with optimistic estimates, target data sample sizes of at least 300 are needed to detect 3% of variation with 80% power. Samples of 800, 8,000, and 80,000 would be needed to account for 1%, 0.1%, and 0.01% of variance with 80% power, respectively (computed using GPower; (Faul et al., 2007).
Improvements in PRS estimation:
Method improvement for PRS estimation is an active area of research, with several new approaches only recently proposed - here, we briefly highlight two. One approach, LDpred (Vilhjalmsson et al., 2015), takes into account the LD between markers, thus eliminating the need for LD-thinning, which may limit the variance explained by PRS in some cases. LDpred estimates posterior mean effect sizes based on LD patterns from a reference genome, by specifying an LD radius (i.e., number of SNPs that are accounted for on either side of another SNP), and has been found to have better accuracy and calibration. For example, in the same target sample, LDpred explains 25% of the variance in schizophrenia compared to 18% from traditional PRS. Another approach, MTAG (Multi-Trait Analysis of GWAS; (Turley et al., 2017)), which may be thought of as an extension of traditional meta-analysis, enables the joint analysis of several traits, resulting in improved PRS precision. MTAG takes advantage of the increasing number of publicly-available GWAS summary statistics, and the development of techniques which can estimate trait heritability and genetic correlations from summary statistics [LDSR; (Bulik-Sullivan et al., 2015), see also related user-friendly online interfaces/databases (e.g., LD Hub; http://ldsc.broadinstitute.org/ (Zheng et al., 2017)]. Indeed, early analyses have successfully applied MTAG to the study of several psychiatrically relevant traits, resulting in prediction accuracy improvements of 25–50% and increased numbers of genomewide significant hits ((Hill et al., 2017, Turley et al., 2017). Beyond these two examples, several approaches incorporating Bayesian estimation (Mak et al., 2016, So and Sham, 2017) and machine-learning (Pare et al., 2017, Shi et al., 2016), have also been proposed.
Improvements in Discovery GWAS:
In addition to increasing the sample size of discovery GWAS and using new tools to improve the utility of PRS available from current GWAS summary statistics, improving discovery GWAS phenotyping may also produce more reliable, applicable, and precise PRS. Indeed, relying on GWAS of psychiatric disorders may be problematic for refining nosology and mechanistic understanding because many, if not all, psychiatric disorders represent heterogeneous amalgamations of symptoms and their correlates. Such heterogeneity may dilute observed genetic effects or result in the identification of broad and generalized genetic associations that may not be applicable to more specific phenotypes. For example, many patients with MDD do not exhibit the cardinal symptom of anhedonia (Treadway and Zald, 2011). Thus, genetic associations with heterogeneous MDD not predicated on the presence of anhedonia, may not be informative for reward-related neural or behavioral phenotypes (Bogdan et al., 2013). The potential implications of GWAS phenotyping may be observed when contrasting two GWAS of depression. In the larger study, which contained 9,240 MDD cases and 9,519 controls, no genomewide significant loci were detected (Major Depressive Disorder Working Group of the Psychiatric et al., 2013). The second study which consisted of Han Chinese women with recurrent, primarily melancholic, depression (n=5,303) or without MDD (n=5,337), identified 2 genomewide significant loci (CONVERGE, 2015), including one within a candidate gene, SIRT1, which has been previously linked to reward-related neural and behavioral phenotypes in non-human animal models, as well as anxiety and age-related disease and mortality (Libert et al., 2011, Kishi et al., 2010, Kuningas et al., 2007). Arguably, the phenotypic and population homogeneity of the CONVERGE effort resulted in relatively improved power (Sullivan, 2015). Notably, however, other large scale GWAS of even more heterogenous phenotypes (e.g., those self-reporting a depression diagnosis through 23andMe) have also been met with success (Hyde et al., 2016). Further, other evidence suggests that traditionally conceptualized intermediate phenotypes (e.g., brain volume), may not be associated with larger effects related to common genetic variation suggesting potential limitations to “deeper” phenotyping (Franke et al., 2016). Going forward it will be important to not only have large studies, but also studies with deep phenotyping so that both heterogenous and more precise phenotypes could be leveraged and their predictive utility for cross-trait association contrasted. Indeed, the genetic architecture of a disorder may be partially or even fully distinct from the genetic architecture underlying related pathophysiology, its response to current treatment (see also Treatment Relevance), or its interplay with the environment (see also Other Applications). Lastly, for some disorders, which require exposure (e.g., substance use disorders, PTSD), it may be important to restrict controls of discovery GWAS to exposed individuals who have not been diagnosed with the disorder to identify sources of disorder liability. Such an approach was recently successful when considering opioid dependence when contrasting with opioid-exposed controls who did not progress to daily injection (Nelson et al., 2016).
Cross-ancestry PRS prediction:
Because they rely on LD patterns, which vary ancestrally, PRS derived from a discovery GWAS of Europeans might not be an informative predictor in another ancestral group. Even within ancestral groups, inclusion of indices of nuanced ancestral variability derived from GWAS data, can significantly improve prediction in PRS analyses by eliminating such spurious admixture effects from the weighted PRS (Chen et al., 2015). Studies have begun to probe whether PRS generated from one ancestral population may be predictive of the same phenotype in another. For example, Bigdeli and colleagues (Bigdeli et al., 2017) combined results from the aforementioned CONVERGE Han Chinese cohort with those from a large GWAS meta-analysis of Europeans to report a trans-ancestry genetic correlation of ~0.30 – 0.40. However, LDpred PRS-based analyses were only predictive of recurrent depression, but not other depression phenotypes, after correction for multiple testing (R2=0.002). A similar study found that PRS derived from a GWAS of neuroticism in 170,910 individuals participating in the European UK Biobank (Okbay et al., 2016) predicted variance in both depression (R2=0.001) and neuroticism (R2=0.083) in Han Chinese women (Docherty et al., 2016). Thus, with caution, one could experiment with projecting effect sizes from population A to population B. Of course, the most valuable resource in this regard would be large well-powered discovery GWAS conducted in homogenous populations from different ancestral origins, which are becoming more common (Duncan et al., 2017, Meyers et al., 2017, Xu et al., 2015). Related approaches combining results from large and readily available European or mixed-ethnicity cohorts with smaller training datasets of another ancestry to produce ancestry-adjusted PRS have also been proposed ((Marquez-Luna and Price, 2016, Coram et al., 2017).
Differences from other heritability metrics:
Even though they represent the additive effects of segregating loci, PRS are distinct from twin-heritability and SNP-heritability. SCZ-PRS explain ~18% of the variance in the disorder in independent samples (Schizophrenia Working Group of the Psychiatric Genomics, 2014); family/twin-h2 and SNP-h2 are 80% and 41% respectively (Sullivan et al., 2017). Estimates of twin-h2 are based on latent sources of additive (and in some cases, non-additive) genetic factors and assume uniform effects of all segregating loci. Furthermore, they rely on certain assumptions, some of which can be reasonably questioned (e.g., random mating). PRS represent a portion of this genetic variance that is attributable to a set of uncorrelated loci, and are less assumption-laden. In fact, PRS approaches have been used to demonstrate the widespread occurrence of assortative, (non-random) mating - for instance, polygenic liability to educational attainment in one spouse accounts for 14–19% of the partner’s education outcomes (Hugh-Jones, Verweij et al. 2016). SNP-h2, calculated with a variety of methods [Genetic Complex Traits Analysis: GCTA (Yang et al., 2011); Linkage Disequilibrium Score Regression: LDScR; (Bulik-Sullivan et al., 2015)], may be used to examine the net effect of all genomewide variants, genotyped and imputed and is related to both twin-h2 and PRS. SNP-h2 is more similar to twin-h2 in that they harness effects of all variants, but only explain a proportion of twin-h2 (especially for behavioral traits), which are typically considered the denominator (perhaps, inaccurately) for such estimation. PRS at a threshold of p < 1.0 might reasonably be expected to approximate SNP-h2, however some have noted that the LD pruning step of PRS estimation may diminish its predictive utility (Vilhjalmsson et al., 2015), by removing correlated loci with independent effect. Nonetheless, PRS are only expected to predict variance in the same trait if SNP-h2 > 0, and in other traits if they are significantly genetically correlated (SNP-rg > 0); however, we view them to be philosophically different (although, SNP-h2 has attractive technical characteristics)2. Twin-h2 and SNP-h2 represent the total variance accounted for by latent genetic and measured common genomic variation, respectively. On the other hand, the essence of PRS is “prediction” in an independent sample, using a metric that is algebraically simple (weighted average), so that it might be viewed to have future generalizable clinical relevance.
PRS Application: The example of Schizophrenia
Undoubtedly, schizophrenia (SCZ), which was subjected to GWAS meta-analysis in 36,989 cases and 113,075 controls by the PGC [(Ripke et al., 2013) for other GWAS by the PGC, see (Sullivan et al., 2017)], is amongst the most frequently analyzed and robustly associated psychiatric PRS. Using these SCZ-PRS, in Figure 2; Tables 1–6, we illustrate the utility of the PRS approach in advancing our understanding of the genetic architecture of SCZ and its relationship with other traits and disorders. Broadly, two observations are immediately apparent. First, unlike GWAS, PRS may be examined in substantially smaller samples. Sample sizes range from <200 for certain clinical traits as well as neuroimaging outcomes (e.g., brain activation during probabilistic learning task) to > 100, 000 for population-based studies of common traits, such as self-rated health; how large a sample is sufficient is addressed above in Technical Considerations. Second, effect sizes are (mostly) modest with PRS explaining <1% of the variance for correlated traits [e.g., R2 for negative symptoms in the population is 0.007, or 0.7%; (Jones et al., 2016) or 0.3% for cognitive abilities (Riglin et al., 2017). These effect size estimates are sobering; even for highly correlated disorders, such as SCZ and Bipolar Disorder (Charney et al., 2017), PRS rarely account for more than 3% of variance in a trait [e.g., 2.5% for educational attainment PRS predicting cognitive function (Rietveld et al., 2013)]. Thus, studies accounting for >5% of variance in cross-trait prediction, or with fewer than several-hundred participants, should be viewed with some skepticism when using current GWAS summary statistics.
SCZ-PRS have primarily been applied to understand relationships across 6 broad domains: schizophrenia-related phenotypes (e.g., psychosis, treatment), other psychiatric disorders, cognition, brain-related phenotypes, immune and health-related and, finally, other phenotypes (e.g., urbancity). Effect sizes for these studies are graphically summarized in Figure 2 with individual study summaries provided in Table 1–6, and discussed (briefly) below.
Schizophrenia-related Phenotypes (Table 1):
SCZ-PRS have been repeatedly linked to various measures of psychosis, psychotic symptoms and experiences, within clinical and general population samples. Associations with negative and positive symptoms have also been noted. For instance, Jones et al (2016) report that SCZ-PRS are associated with 0.5–0.7% of the variance in psychotic experiences at ages 12–18. In addition, SCZ-PRS have been linked to chronicity and course of illness with one study finding SCZ-PRS to predict the number of hospital admissions, length of hospital stay and enrollment in supportive housing (Meier et al., 2016). Mixed evidence also exists for an effect of SCZ-PRS on treatment response (Wimberley et al., 2017b, Li et al., 2017). Interestingly, the predictive utility for antipsychotic response is improved by incorporating rare variants into such scores (Ruderfer et al., 2016), although null findings also exist (e.g., (Hettige et al., 2016)). Family history also accentuates risk conferred by SCZ-PRS (Agerbo et al., 2015).
Psychiatric disorders and traits (Table 2):
Table 2.
SCZ PRS and Other Psychiatric Disorders and Related Traits.
| Phenotypes | Phenotype & References | N | Effect size* |
|---|---|---|---|
| Development: Childhood Behavior and Psychopatholog y-related Traits | Early childhood behavioral problems, externalizing and internalizing (Jansen et al., 2017)* | 5,100 – 6,952 ages 4–9 | Increased internalizing and externalizing: up to ~0.008 |
| Anxiety, ADHD, and depression during childhood (Nivard et al., 2017) | 8,715 ages 6–16 | Positive association with childhood psychopathology: b=0.0182. Increasing from childhood to adolescence: r=0.10 to 0.25. | |
| (St Pourcain et al., 2017)* | 4,175–5,553 at ages 8, 11,14,17 | Elevated social communication difficulties: up to 0.0043 (increasing in effect size with increasing age). | |
| Social communication, emotion and conduct problems, (age 4–9) (Riglin et al., 2017)* | Worse communication, increased emotion and conduct problems (increasing from childhood to adolescence): up to 0.001 | ||
| Depression, Distress, Neuroticism, Loneliness | Distress and Neuroticism (SCZ-PRS x MDD) (Whalley et al., 2016)* | Sample 1: 2,587 MDD, 16,764 CON Sample 2: 6,049 MDD, 27,476 CON |
PRS x MDD interaction for neuroticism and personal distress (Bs>−0.04). Neuroticism: positive assoc in CON: 0.0017–0.0030; NS in MDD: 0.0000090.00003. |
| 2, 587 – 6,049 MDD cases; 16,764 – 27, 476 controls | Psych Distress: positive assoc in CON: .0015–.0022; NS in MDD. | ||
| Neuroticism (Gale et al., 2016)* | 108, 038 | Positive assoc.: 0.0012 | |
| Treatment response to anti-depressants (Garcia-Gonzalez et al., 2017)* | 3,756 | NS: 0.0006–0.018 | |
| Loneliness (CITE Gao et al., 2017)* | 6,924 ages ≥50 | Nominal evidence of a negative association, up to 0.0003 | |
| OCD | (Costas et al., 2016)* | 370 OCD, 443 CON | Increased OCD, up to: 0.037 |
| (Sumner et al., 2016)* | 1,293 trauma-exposed women | Increased PTSD risk, up to: 0.009 | |
| Substance Use | Alcohol, Cannabis, Cocaine Nicotine, Opioid Dependence (Carey et al., 2016a)* | 1,160 Alcohol Dependent, 1,413 CON ascertained from studies recruiting for alcohol dependence, cocaine dependence, and nicotine dependence | Surviving multiple test correction: Increased cannabis, cocaine, and nicotine dependence symptoms, up to 0.0109. Nominal positive associations: severe alcohol dependence symptoms, nonproblem cannabis use, nicotine use, opioid use and dependence symptoms.Associations with cannabis and cocaine survive correction for general substance liability. |
| Cannabis use prior to onset (Aas et al., 2017)* | Scz Cases: 381 Bipolar Disorder Spectrum: 220 Controls: 415 |
Patients with Schizophrenia or Bipolar who used cannabis weekly/daily prior to illness had elevated PRS: Cohen’s d = 0.3; up to 0.027. | |
| Substance Use Disorder (Hartz et al., 2017)* | Cases with a substance use disorder Study1: 918 Cases, 988 Controls Study 2: 643 Cases, 384 Controls Study 3: 210 Cases, 317 Controls |
SUD cases had elevated Scz PRS: up to 0.037. | |
| Cannabis use (Verweij et al., 2017)* | 6,931 | Increased lifetime, regular and quantity of use; up to 0.0049. | |
| Substance Use Disorders (Alcohol, Amphetamine, Cocaine, Cannabis, Opioid, Sedative, Early onset, Ever Smoking (Reginsson et al., 2017)* | 144,609 (10,036 admitted for inpatient addiction treatment, 35,754 smokers) | Increased substance use disorder, up to 0.0089. | |
| Suicide | Suicide attempts (Laursen et al., 2017)* | 1780 SCZ, 1768 CON | 2+ suicide attempts OR = 1.18 in full sample. Up to 0.0021. |
| Suicide attempt (Sokolowski et al., 2016)* | 660 | PRS of neurodevelopmental genes positively associated with suicide attempt: up to 0.052. | |
| Various Traits | 50 traits (Krapohl et al., 2016) | 3.152 16 year olds | NS. Nominal negative association with autism quotient: attention to detail. |
| Post-Concussive Symptoms | (Wang et al., 2017a) | 845 US Army soldiers who sustained traumatic brain injury | NS: up to 0.0089. |
Studies only include those using summary statistics from (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Unless otherwise noted, effect size is represented as Nagelkerke’s Pseudo R2 (multiply by 100 to obtain %R2); r = correlation, OR = Odds-ration, B = beta coefficient, AUC = Area Under the Curve. EA = European ancestry. AA = African ancestry. NS= non significant. When pearson’s r, OR, or d were reported, we estimated r2.
indicates study with % of variance explained included in Figure 2.
Genetic liability to SCZ has been linked to numerous other psychiatric conditions, including bipolar disorder, major depression, obsessive compulsive disorder, anorexia nervosa, posttraumatic stress disorder, alcohol dependence and attention deficit hyperactivity disorder (Anttila et al., 2016); however, a preponderance of these studies have calculated genetic correlations (SNP-rg). Nonetheless, studies employing the PRS approach find strong associations with a variety of psychiatric phenotypes. Of particular interest, SCZ-PRS successfully distinguish between clinical subtypes of Bipolar Disorder, with stronger associations with Bipolar I and schizoaffective forms (Charney et al., 2017). Substance use disorders have also been robustly linked to SCZ-PRS e.g., (Carey et al., 2016a). In particular, addressing the controversy regarding the role of cannabis use in the development of psychosis (Hall and Degenhardt, 2008), multiple studies have used SCZ-PRS to clearly demonstrate the role of shared genetic etiologies between these two phenotypes (e.g., R2=.47% for SCZ-PRS and ever using cannabis), thus challenging prior causal assertions and providing a new outlook on this relationship that was not possible to query with twin data (Power et al., 2014, Carey et al., 2016a, Verweij et al., 2017, Reginsson et al., 2017, Aas et al., 2017). Within the longitudinal framework, the correlation between SCZ-PRS and psychiatric traits (ADD, ODD/CD, Anxiety and Depression) increases from 0.10 at age 6–7 to 0.25 by age 16 (Nivard et al., 2017). This application of PRS for a clinically uncommon and severe discovery phenotype (i.e., SCZ) to a general population sample of longitudinally evaluated youth highlights the important role that PRS analyses play in bridging clinical psychiatry with developmental psychology and behavioral genetic research that was largely unattainable using latent genetic models of uncommon disorders (e.g., SCZ). One of the largest studies evaluating SCZ-PRS among other traits shows that it predicts a small amount of variance in neuroticism (0.12%) (Gale et al., 2016).
Cognition (Table 3):
Table 3.
SCZ PRS and Cognition.
| Phenotypes | Phenotypes and Study Reference | N | Association and Effect size* |
|---|---|---|---|
| IQ Metrics and Neurocognitive Function | Neurocognitive Performance (Hatzimanolis et al., 2015)* | 1,079 | Continuous performance and spatial N-back: ~0.00275. No significant association with IQ or verbal n-back. |
| Neurocognition and social cognition (Germine et al., 2016)* | 4303 (8 – 21y); Replication: 695 (18 – 35y) | Verbal reasoning and emotion identification speed: 0.003 – 0.005 NS: 36 other accuracy and speed indices. | |
| Cognitive function and Education (Hagenaars et al., 2016) | 36,035–112,067 | Negative associations with verbal-numerical reasoning, reaction time, memory: B=−0.006 to-0.062; positive association with educational attainment: B=0.025. | |
| 50 triats - personality, cognition, etc. (Krapohl et al., 2016) | No significant associations following multiple testing. But nominal positive correlations with English and negatively correlated with Autism Quotient: Attention to Detail. | ||
| Cognitive function in older adults (Liebers et al., 2016)* | 8, 616 | Total Cognitive Function, Attention/Language, Verbal memory: 0.0004–0.0008 | |
| Composite g and subfacets: memory digit, vocabulary, verbal, reaction time (SCZ-PRS x MDD) (Whalley et al., 2016)* | Sample 1: 16,764 CON; 2,587 MDD Sample 2: 27,476 CON; 6,049 MDD | Composite g: 0.0032 – 0.0062 (stronger in MDD); G Subfacets 0.0002 – 0.0109 |
|
| Cognition and learning, social and communication, emotion and mood regulation, behavior, age 4–9 (Riglin et al., 2017)* | 5,100 – 6,952 | Lower performance IQ and worse language intelligibility and fluency: up to 0.003 | |
| Executive Function (Benca et al., 2017)* | 386 | Positive correlation with update specific latent variable: r < 0.2 (nom. Sig.). Negatively associated with IQ,: up to 0.01. | |
| Emotion recognition (Coleman et al., 2017)* | 4,097 8 year old children | No association: r2=0.0001 – 0.0011 | |
| Psychosis cognitive and physiological endophenotypes (Ranlund et al., 2017)* | Total: 1,437–3,089 drawn from SCZ: 1,087 SCZ 1st-degree relative: 822 CON: 2,333 |
SCZ>SCZrelatives>CON. Worse performance on block design: 0.002.. NS: digit span, RAVLT. | |
| SCZ and Cog performance (Wang et al., 2017b)* | 1,130 Taiwanese SCZ | Not associated with SCZ: 0.0008. Lack of association PGC2 not used to test for association with cognitive phenotypes. Cognitive associations reported with PGC1. | |
| Creativity | Creativity (Power et al., 2015)* | 86, 292 | Increased creativity: up to 0.055 |
Studies only include those using summary statistics from (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Unless otherwise noted, effect size is represented as Nagelkerke’s Pseudo R2 (multiply by 100 to obtain %R2); r = correlation, OR = Odds-ration, B = beta coefficient, AUC = Area Under the Curve. EA = European ancestry. AA = African ancestry. NS= non significant. When pearson’s r, OR, or d were reported, we estimated r2.
indicates study with % of variance explained included in Figure 2.
SCZ-PRS have unequivocally established a link between genetic susceptibility to SCZ and cognitive deficits during the lifespan. SCZ-PRS are predictive of IQ at ages 7–9 (Riglin et al., 2017, Hubbard et al., 2016) and also to cognitive decline in later life (Liebers et al., 2016). One study suggests stronger effects on Wechsler IQ at age 70 but not age 11, with SCZ-PRS predicting 0.8% of the variance in this developmental decline. Collectively, this research suggests that cognition deficits observed in schizophrenia are attributable in part to common genetic variation, that can also be observed in the population.
Brain-related (Table 4):
Table 4.
SCZ PRS and Physiological and Brain-related Phenotypes.
| Phenotypes | Study References | N | Association and Effect size* |
|---|---|---|---|
| Electrophysiology | P300 (Hall et al., 2015)* | 271 SCZ or psychotic BIP, 128 CON | Whole group NS: P3 amplitude or latency; ASSR gamma, P50. However, in cases only reduced ASSR: 0.04. nteractions between PRS and case status (P=0.008). |
| Prepulse inhibition & baseline startle (Roussos et al., 2016)* | 1,493 male | Reduced PPI up to: 0.01175. NS: baseline startle. | |
| Acoustic startle, affective startle modulation, antisaccade, resting EEG Hz, skin conductance, P300 (Liu et al., 2017b) | 4,905 | NS after multiple testing correction. Nominal significance for overall startle (p=0,005). | |
| P300 amplitude and latency (Ranlund et al., 2017)* | 515 | NS: up to 0.019 | |
| Functional Magnetic Resonance Imaging | Probabilistic Learning (Lancaster et al., 2016a) | 83 | Reduced right frontal pole (whole brain; PFWE(whole brain)=0.037) and left ventral striatum (ROI: PFWE(ROI)=0.036) activation during shift > stay choice behavior, .NS: activation to choice outcome. |
| Emotion Processing, Episodic Memory, Reward Processing, Theory of Mind, Working Memory (Erk et al., 2017) | 578 | Emo: NS. Epi Mem and Theory of Mind: Increased perigenual anterior cingulate cortex during episodic memory recognition PFWE(ROI)<0.047. Rew: NS, ToM: Posterior cingulate/precuneus PFWE(ROI)<0.025. No other significant associations. | |
| Structural Magnetic Resonance Imaging | Hippocampal volume (Harrisberger et al., 2016) | 36 First Episode (FE) Psychosis; 42 At Risk Mental State (ARMS) | B = −0.42 |
| White matter integrity (Alloza et al., 2017) | 28 SCZ, 36 CON | SCZ diagnosis: 0.057; NS associations with network. | |
| Gray matter volume (Van der Auwera et al.,2015, Van der Auwera et al., 2017)* | Sample 1: 1,426 Sample 2: 1,299 |
NS: whole brain or ROI associations in amygdala, hippocampus, parahippocampus, NS: max ~0.001 from 2015. | |
| Hippocampus changes with 3 month exercise/cog intervention (Papiol et al., 2017)* | 20 cases & 23 con with interv. 21 cases with con interv. |
NS: total hippocampus. Less change in CA4/dentate gyrus: 0.358 (not plotted in figure) | |
| Lateral ventricular volume (Ranlund et al., 2017)* | 798 | NS: 0.004 | |
| Gray matter volume and white matter integrity (Reus et al., 2017) | Volume: 978 WM: 816 | NS. | |
| Cortical Thickness and Gyrification | Cortical thickness in context of cannabis use (French et al., 2015)* | Sample 1: 459 male adolescents Sample 2: 333 adolescents Sample 3: 295 male youth | SCZ-PRS x adolescent cannabis use predicted reduced cortical thickness across all 3 samples. Sample 1: 0.06. Main effects inconsistent. |
| Cortical gyrification (Liu et al., 2017a)* | 315, Replication: 357 | Lower gyrification in inferior parietal cortex, up to 0.021 | |
| (Neilson et al., 2017) | 43 SCZ/Bipolar cases; 32 CON | Reduced global cortical thickness in patients (F up to 4.54). NS in CON. |
Studies only include those using summary statistics from (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Unless otherwise noted, effect size is represented as Nagelkerke’s Pseudo R2 (multiply by 100 to obtain %R2); r = correlation, OR = Odds-ration, B = beta coefficient, AUC = Area Under the Curve. EA = European ancestry. AA = African ancestry. NS= non significant. When pearson’s r, OR, or d were reported, we estimated r2.
indicates study with % of variance explained included in Figure 2.
A variety of neuroimaging approaches have been used to evaluate potential neural substrates of schizophrenia (Kahn et al., 2015). However, with the exception of offspring and sibling-based studies (Chung and Cannon, 2015), it has been difficult to disarticulate whether such associations may be a consequence of medication (ubiquitous in most samples) or disorder expression as opposed to a marker of risk (Tost et al., 2010). SCZ-PRS can be leveraged to probe whether such neural metrics, or perhaps even novel ones (masked for example by medication usage), may be associated with genetic liability to schizophrenia within healthy populations unexposed to antipsychotic medication or disease course. Most commonly studied in this context are metrics of brain structure (e.g., cortical thickness, cortical gyrification, and gray matter volume), which have produced mixed evidence that increased schizophrenia genomic liability may be associated with reduced cortical thickness and volume (French et al., 2015, Van der Auwera et al., 2017). Other studies have begun to link SCZ PRS to functional imaging phenotypes such as reward- and working memory-related brain function; however results thus far have been mixed and are often derived from small samples (Lancaster et al., 2016b, Erk et al., 2017, Lancaster et al., 2016a). Overall, this evidence suggests that some neural phenotypes associated with schizophrenia may represent neural mechanisms of common polygenic liability. However, other evidence, drawn from large samples, suggests minimal genomic overlap between subcortical brain volume and schizophrenia (Franke et al., 2016). Lastly, a recent study reports that SCZ-PRS are associated with differentially methylated probes in postmortem tissues from the prefrontal cortex, striatum and cerebellum of schizophrenic and control decedents (Viana et al., 2017). The most significantly associated probe was within DISC1, a gene associated with schizophrenia. Such studies provide exciting new opportunities for linking static genomic variation, in their polygenic form, to epigenetic variation.
Immunity and Health (Table 5):
Table 5.
SCZ PRS and Health and Immune Function.
| Phenotypes | Study Reference | N | Association and Effect size* |
|---|---|---|---|
| Self-reported Health | (Harris et al., 2016) | 111, 749 | Reduced health: B=−0.028 |
| Self-reported Tiredness | (Deary et al., 2017) | 108,976 | Increased tiredness: B=0.028 |
| Amyotrophic Lateral Sclerosis | (McLaughlin et al., 2017)* | 12, 577 patients, 23, 475 CON | ALS have higher PRS: 0.0012 |
| Type 2 Diabetes | (Padmanabhan et al., 2016) | EA: 218 CON, 384 SCZ, SCZA, Psy BIP Probands AA: 104 CON, 257 SCZ, SCZA, Psy BIP Probands |
NS: positive association among probands and relatives, OR up to 1.41: 0.20. |
| Infection and infection-related SCZ | (Benros et al., 2016) | 1,692 SCZ, 1,724 CON | NS: infection or evidence or PRS x infection |
| Psoriasis | (Yin et al., 2016)* | Han Chinese: 1139 psoriasis cases, 1132 controls | AUC = 0.54 [0.51 – 0.58]: 0.0050 |
| Migraine | (Van der Auwera et al., 2016)* | 3, 973 | OR = 0.78 (based on 108 loci): 0.0047 |
Studies only include those using summary statistics from (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Unless otherwise noted, effect size is represented as Nagelkerke’s Pseudo R2 (multiply by 100 to obtain %R2); r = correlation, OR = Odds-ration, B = beta coefficient, AUC = Area Under the Curve. EA = European ancestry. AA = African ancestry. NS= non significant. When pearson’s r, OR, or d were reported, we estimated r2.
indicates study with % of variance explained included in Figure 2.
Immune-related vulnerabilities are commonly observed in those with SCZ (Muller et al., 2015). Maternal immune response to infectious agents have been causally implicated in individual risk for SCZ, particularly among individuals with a diathesis (Knuesel et al., 2014). Even ex-utero, exposure to infectious agents, such as Toxoplasmosis gondii, primarily transmitted to humans through fecal matter from infected carrier felines, precipitates onset of motor, psychotic, and cognitive symptoms (Torrey and Yolken, 2003, Sutterland et al., 2015). Antithetically, while individuals with schizophrenia are more likely to exhibit a pro-inflammatory phenotype (e.g., higher levels of cytokines), risk for rheumatoid arthritis, an autoimmune disorder is markedly reduced in those with schizophrenia and corresponds to a small, but significant, negative genetic correlation across the disorders as well (rg=−0.046) (Lee et al., 2015, Stringer et al., 2014, Euesden et al., 2015a). PRS may be particularly informative in studies exploring relationships with the immune system due to the overrepresentation of immune function loci in SCZ GWAS (Schizophrenia Working Group of the Psychiatric Genomics, 2014). For example, SCZ-PRS have been leveraged to test novel immune-related hypotheses with one study finding no evidence that SCZ-PRS moderates the relationship between infection and schizophrenia (Benros et al., 2016). Going forward, system-based PRS composition (i.e., forming a PRS from SNPs within biologically-defined pathways) may be particularly useful in this regard (see Improving PRS section below). Studies on health-related phenotypes also highlight several novel genetic relationships that transform our understanding of the relationship between schizophrenia, health and disease, ranging from self-rated health (Harris et al., 2016) to chronic and debilitating conditions such as migraines (Van der Auwera et al., 2016) and farther, to rare but serious progressive neurodegenerative disorders like Amyotrophic Lateral Sclerosis (ALS, Lou Gehrig’s Disease (McLaughlin et al., 2017). Prior to the development of the PRS approach, the notion that the genetic underpinnings of such health-related conditions could overlap with predisposition to psychiatric disorders seemed to be an insurmountable challenge to address, particularly for uncommon conditions (e.g., SCZ, ALS) that make family-based approaches impractical.
Others (Table 6):
The advent of the PRS approach has also led to unique answers to additional age-old questions. We now know that genetic liability to SCZ (i.e., PRS) is related to variance in living in urban environments and in neighborhood deprivation, both leading environmental contributors to schizophrenia. Notably, schizophrenia represents an evolutionary paradox (van Dongen and Boomsma, 2013) in that while risk alleles are at selective advantage, the disease is associated with lower reproductive fitness with older paternal age implicated as a risk factor [potentially, due to de novo mutations in the male germ line (Malaspina et al., 2001)]. Yet, SCZ-PRS when applied to a sample of 150, 656 individuals found no correlation with number of children or age at first birth. However, perhaps most sobering is evidence that individuals with higher SCZ-PRS are more likely to drop out of subsequent waves of longitudinal studies, suggesting that distributions of SCZ-PRS, and consequently, their range of related phenotypes, might not be fully represented in cohort data (Martin et al., 2016).
Other Applications
Gene x Environment Interaction (GxE):
Environmental factors, in particular, chronic and unpredictable stressors during childhood, are among the largest risk factors for the expression of psychopathology (Green et al., 2010). Such factors act independently and in concert with genetic liability through predisposing (i.e., gene - environment correlation, RGE) or moderating (i.e., GxE) mechanisms. PRS are beginning to be used to represent genomic liability in the context of stress. For example, five studies of depression PRS report that stressful life events or childhood trauma, as well as PRS, are independently associated with depression. However, much like the candidate single variant environment interaction literature (Duncan and Keller, 2011), these initial studies have provided conflicting evidence of their interactive effect (GxE) (Musliner et al., 2015, Mullins et al., 2016, Colodro-Conde et al., 2017). Of the 3 reports evaluating stressful life events, 2 report null interactions (Musliner et al., 2015, Mullins et al., 2016) with 1 (Colodro-Conde et al., 2017) suggesting that elevated PRS potentiates the depressogenic effects of stress. In the study with positive findings, the observed PRS x stressful life event interactions accounted for only 0.12% of variance in depression; however, their estimates suggest that it could potentially account for as much as 20% of variance in depression if PRS were derived from a larger GWAS. While significant PRS interactions with childhood trauma explained up to 1.9% of variance in depression, the shape of these interactions was inconsistent. One study found that childhood trauma was associated with increased depression risk among those with lower polygenic liability to depression while depression PRS were associated with elevated depression risk in the context of no childhood trauma (Mullins et al., 2016). In opposition to these findings, the other report found a significant interaction wherein elevated PRS was associated with increased childhood trauma-related depression (Peyrot et al., 2014). Multiple explanations may be evoked to explain these discrepancies such as the use of PRS derived from different GWAS and differences in phenotypic assessments, and sample composition and size. GWAS x E studies are needed (e.g., (Polimanti et al., 2017) as the genetic architecture supporting the development of psychopathology (e.g., depression) in the context of stress may be distinct from the genetic architecture conferring general disorder risk as assessed in the original GWAS from which PRS were derived.
Disorder Specificity:
It may be tempting to contrast the predictive value of PRS derived from discovery GWAS of different disorders to evaluate disorder specific or shared mechanisms that may ultimately inform nosology (e.g., do SCZ-PRS predict reward-related neural function better than depression-PRS?). We suggest that such comparisons should be interpreted with caution, for 2 reasons. First, much like recent unifactoral models of psychopathology (Lahey et al., 2012) and evidence of shared neural mechanisms (Hariri, 2009, Goodkind et al., 2015), cross-disorder GWAS (Cross-Disorder Group of the Psychiatric Genomics, 2013) suggest that common genetic variation conferring liability to various disorders may be shared. Thus, clear hypotheses are needed for expected disorder specificity which may be present for some psychiatric phenotypes, but not others. Second, the robustness of the discovery GWAS can make it challenging to compare PRS. For example, if a PRS for schizophrenia [derived from GWASs containing 36,989 cases and 113,075 controls and identifying 108 independent loci (Schizophrenia Working Group of the Psychiatric Genomics, 2014)] is associated with reward-related brain function, but a PRS derived for depression (derived from GWASs containing 9,240 cases and 9,519 controls identifying no genomewide significant SNPs (Major Depressive Disorder Working Group of the Psychiatric et al., 2013) is not, whether this is indicative of genetically-conferred disorder specificity or simply a confound of the power of the discovery GWAS cannot be disarticulated. However, as the field continues to advance and more well powered psychiatric GWAS are conducted across disorders, comparisons of PRS across disorders to detect shared and specific polygenic variability may prove informative.
Mendelian Randomization (MR):
MR is an extension of epidemiological causal models (for review see: (Davey Smith and Hemani, 2014, Holmes et al., 2014)). Assume that two traits are correlated - such as body mass index (A) and cardiovascular disease (B) - and that it is reasonable to hypothesize that A causes B. If one could identify genetic variants, including single loci, or multiple variants that comprise a PRS, that are robustly associated with A, but have no independent effects on B [although, methods that adjust for such pleiotropy exist (Bowden et al., 2015)] or on any covariate (e.g., smoking) related to B, then such a PRS might serve as a “genetic instrument” for MR analyses. If BMI PRS predict cardiovascular disease, indirectly through BMI, then such an association may be viewed as evidence in favor of causation (i.e., higher BMI results in cardiovascular disease) (Holmes et al., 2014). In an empirical test of this hypothesis, one study found that BMI PRS (discovery N = 108, 912) predicted cardiometabolic traits and outcomes, but not coronary artery disease, in an independent cohort of 34, 538 individuals. Each unit increase in the BMI-PRS corresponded to 1.08 kg/m2 increase in BMI, and was associated with inflammation (e.g., C-Reactive Protein: 12% increase), cardiometabolic traits (e.g., fasting insulin: 8.4% increase) and disorders (e.g., type 2 diabetes, OR = 1.29) suggesting that BMI may cause such outcomes. The use of PRS in the study of clinically relevant psychiatric phenotypes is limited, although some argue that cross-trait PRS predictions may, in some cases, be indicative of MR. Current tests of MR in clinical psychology rely on individual loci, arguing that they are less likely to have pleiotropic effect, or create a noisy instrument (e.g., cannabis initiation loci predicting SCZ (Gage et al., 2017)). However, as better powered GWAS of putative causal factors emerge, one may begin to craft PRS as genetic instruments, that may be used to test the plausibility of causality among phenotypes, as long as the potential of pleiotropic effects remains tractable (from a biological and statistical perspective). Several tools have been developed to facilitate MR analyses [e.g., MR-Base: (Hemani et al., 2016) and for gene expression: Summary-data-based Mendelian Randomization (SMR): (Zhu et al., 2016)]. However, it is unclear whether the assumptions of MR analyses (e.g., lack of assortative mating; no confounding pathways) can truly be overcome, and may become more concerning for PRS analyses in light of extensive shared polygenic pathways (Cross-Disorder Group of the Psychiatric Genomics, 2013) and even potential omnigenic undergirding (Boyle et al., 2017).
Treatment Relevance: A Prognostic and/or Diagnostic Tool?
In our opinion, current PRS are not clinically actionable, even though some studies have proposed to have identified marker sets (using other related approaches) that can be diagnostically implemented (Skafidas et al., 2014, Robinson et al., 2014, Belgard et al., 2014). Pleiotropic effects of PRS are evident from Figure 2 and Tables 1–6. How this overgeneralized liability can be harnessed to refine treatment currently remains unclear. At present, the utility of PRS might lie, not as harbingers of future diagnosis but as indicators of vulnerability, which may be modified by a variety of other mitigating or exacerbating influences (e.g., environment, modifier loci). For example, using traditional PRS analyses, SCZ PRS can account for up to 18% of the variance in liability to schizophrenia in an independent dataset with the upper decile being associated with an OR of 20. Such information may be leveraged to identify individuals at risk. While this likely would not currently impact the immediate treatment that one would receive, it would be intriguing to evaluate whether those most genomically vulnerable respond better or worse to early interventions, which may eventually have public health implications. Of note, PRS derived from GWAS of other psychopathologies (e.g., depression) do not currently account for nearly as much variance in disorder expression (Levine et al., 2014). However, it is expected that sample sizes for discovery GWAS will continue to increase, and with that, there will be greater precision in the effect sizes that are used as weights, and, consequently, the proportion of variance that they explain. Until then, it is critical that PRS are not construed as diagnostic tools but as ordinary predictors of risk, such as childhood trauma or family history, but with less explanatory power.
The current limited utility of PRS should not be taken to imply that there is no future for precision medicine in psychiatry. For some disorders, such as Autism Spectrum Disorders, highly penetrant mutations, copy number variants and chromosomal rearrangements have been compiled into genetic panels used for prediction (Vorstman et al., 2017). Even for other disorders, certain monogenic origins are routinely ruled out in clinical practice (e.g., a deletion on chromosome 22q that causes a schizophrenia-like syndrome) (Bassett and Chow, 2008). To improve and begin to personalize treatments, particularly for the broader range of psychiatric disorders characterized by such polygenicity and largely unknown mechanisms, it may be important to not rely on markers of genomic risk for disorder expression (which may usefully identify new therapeutic targets), but rather on markers of response to current treatments. Consistent with this speculation, one recent approach found no associations between depression and SCZ PRS and response to antidepressants (Garcia-Gonzalez et al., 2017) among 3756 treatment receiving patients. Further, pharmacogenomics GWAS (i.e., the study of treatment response) have consistently revealed larger effects (OR 2.5) than studies of disorder, even when comparing sample size-matched studies (Maranville and Cox, 2016). Thus, the genetics of treatment response, much like the genetics underlying interaction with the environment, may be, at least partially, distinct from the genetics of disorder liability and require its own PRS to have clinical implications.
A Summary of PRS Strengths and Limitations
Strengths
Consistent with their widespread use, PRS approaches offer many strengths. First, they model psychiatric disease liability within a polygenic framework. Second, they are easy to compute and analyze making them amenable to research extensions. Relatedly, their results are intuitive and readily interpretable by a large audience. Third, while PRS require large discovery samples, they can be tested within much smaller samples, making them highly appealing to studies with deeper phenotyping on fewer subjects (e.g., neuroimaging). How small is large enough? This will depend upon the precision of PRS and target sample phenotypes and questions. However, given that current observed effects from cross-trait associations suggest that PRS predict 0.01–3.00% of variance across traits, sample sizes would need to be >300 given optimistic estimates of effect size (3%), and in all likelihood, much larger (see Technical Considerations; Figure 2). However, the application of novel techniques such as LDpred and MTAG may increase their precision and allow for their application in smaller samples (Vilhjalmsson et al., 2015, Turley et al., 2017). Regardless, research using smaller samples should consider the possibility of false positive and/or negative results. Fourth, PRS can be extremely useful within general population samples. Indeed, the consequences of medication and disease expression are an important confound for identifying factors that confer disease risk, especially for severe conditions such as schizophrenia. By using PRS as tools to uncover genetically-driven individual differences in mechanistic phenotypes, the field can combat confounds of medication and disease expression. Fifth, unlike other measures of cumulative genomic influence (e.g., SNP-h2), PRS approaches can be generalized and applied across samples. For these reasons, PRS have accelerated the incorporation of genomically-informed analyses into various clinical psychology subspecialties.
Limitations
Despite their widespread application, unique strengths, and the formative insight generated by their application, PRS have numerous limitations that require careful consideration. First, PRS are constrained to the sample size and phenotyping of the discovery GWAS. Such phenotyping may have a completely or partially distinct genetic architecture of comorbidity, related pathophysiology, response to current treatment, or interplay with the environment. Thus, GWAS across a variety of phenotypes and attempts to harmonize results is becoming increasing important. Second, traditional PRS are uninformed by system-level knowledge or functional annotation and provide no insight into molecular mechanisms. Alternative approaches, such as biologically informed multilocus profiles (BIMPS) that additively combine candidate variants based on their functionality, or methods that combine SNPs into meaningful pathways or networks by liaising with mRNA expression data, and even more sophisticated machine learning methods may overcome this limitation of PRS, but also have their own challenges (Bogdan et al., 2017). Along these lines, PRS are not currently amenable to direct translational work in nonhuman animals, though multiplex CRISPR/Cas9 approaches are beginning to be employed (Cong et al., 2013, Wang et al., 2013), which may ultimately be used to model polygenic variability. Third, while PRS are more predictive than single common polymorphisms, they are not currently predictive of substantial variance, even in the same phenotype as originally probed (with the exception of schizophrenia; see Technical Considerations). As a result, for adequate power, they still require relatively large samples (>300 for optimistic estimates of cross trait assocaitions using traditional PRS). Fourth, PRS assume additivity. While this assumption has support (Hill et al., 2008), evidence of epistasis is also present (Gibert et al., 2017, Mitra et al., 2017, Hemani et al., 2014).
Improving PRS: Future Directions
How might PRS be improved? In addition to generating larger, well-phenotyped GWAS, and adopting refined approaches for PRS calculation, one potential immediate future pathway is to leverage additional data to enhance and refine meaningful signals. An example of how additional GWAS data may be leveraged in the context of GWAS-based summary statistics to inform PRS construction comes from our multisite study lead by Arloth and Binder (Arloth et al., 2015). Here, the GR agonist dexamethasone was administered to healthy and depressed participants who were genotyped. RNA expression was measured before and after (+3 h) dexamethasone administration. First, we identified SNPs that were associated with dexamethasone-related changes in gene expression. Second, we found that these SNPs were significantly enriched for nominal associations in GWAS of various psychiatric disorders, including depression. Third, we created a PRS of SNPs that are associated with both dexamethasone-induced gene expression and depression and found in independent samples that this profile was predictive of depression as well as an overgeneralized amygdala response. Thus, it is possible to use GWAS derived summary statistics alongside additional targeted results to prioritize SNPs with both evidence of mechanistic function and disease association that may be more powerfully predictive than disease association alone. In this context, it would be important to show that such biologically-constrained PRS are not merely reflective of global genomic risk and indeed provide greater predictive utility.
Such approaches could also be implemented without one’s own independent data using available databases of basal gene expression (e.g., GTEx (Consortium, 2013), Braineac (Hardy et al., 2009), CommonMind (Fromer et al., 2016)) and methylation to prioritize variants across the entire genome. Similar approaches have been used to functionally partition SNP-h2 using functional annotation. (Gusev et al., 2014, Finucane et al., 2015). Such integration may enhance the biological plausibility of disease associated variants and potentially enhance their power. However, such an approach becomes clearly dependent on the quality of data in both contributing datasets and currently available databases of biological phenotypes such as mRNA expression, which are typically composed of small samples with restricted age ranges and tissue types, as well as a lack of detailed phenotypic and exposure characterization. For instance, many psychiatric disorders are precipitated by stress. Given that stress responsive systems can have direct effects on gene transcription it may be important to consider stress-related gene transcription for such phenotypes (Arloth et al., 2015) and basal expression may not be as informative. Consistent with this notion, glucocorticoid-related gene expression outperforms baseline gene expression when predicting depression [though notably such data should be interpreted as preliminary given the relatively small current sample sizes; (Menke et al., 2012)].
As highlighted above (see Improvements in PRS estimation), several techniques can be leveraged to improve PRS in one’s own data, most of which do not require any, or only minimal, additional data. While they require additional work to compute, the potential pay-off cannot be overstated, as the improvement in accuracy will result in a substantially better-powered study. Here we describe one hypothetical workflow for how these techniques may be integrated into future analyses. First, MTAG may be used to generate summary statistics for polygenic risk scores by jointly analyzing the trait of interest with genetically-correlated traits (Turley et al., 2017). Instead of relying on summary statistics from a single GWAS study, MTAG may be used to integrate information from genetically-correlated phenotypes to improve the precision of summary statistics that can be leveraged for PRS construction. Importantly, summary statistic files can come from any study, as long as they are all of the same ethnicity - this includes partially or fully overlapping samples. The primary restriction on which studies can be included is that none of them should have substantially greater power than the study of the trait of interest, as this will increase false-positives.
Second, PRS could be generated using MTAG generated summary statistics by LDpred, which As mentioned above, leverages LD structure and results in improved PRS predictive ability relative to traditional PRS computation. Notably, there are several other computational approaches for improving PRS accuracy, both by adjusting effect-sizes and by modeling LD-structure. These approaches all require additional training data, and as yet a single approach which reliably out-performs all others has yet to emerge. Thus, the choice of adjustment algorithm will largely depend on the availability and nature of training data. LDpred (Vilhjalmsson et al., 2015) is the most well-vetted of these algorithms, and is thus the one we can most confidently recommend. It requires a moderately sized reference panel (N≥1,000) and is the most computationally intense, but importantly, only additional genetic reference panel data are required. This may prove to be particularly useful for studies examining less easily-obtained phenotypes. More recently developed approaches also hold great promise, though they need further validation. For example, GraBLD (Pare et al., 2017) is notable for requiring only a small training dataset (N≥200) that includes the trait of interest. It performs an adjustment for LD and updates effect sizes using a simple machine learning algorithm. Similarly, Lassosum (Mak et al., 2017) uses machine learning, penalized regression, to correct for LD structure and to update effect sizes. It is particularly notable as it can use cross-validation to replace an external training dataset.
Conclusions
The widespread use of PRS to represent generalizable polygenic disorder risk is beginning to provide tractable information about how polygenic liability leads to psychiatric expression as well as the structure of and mechanisms underlying psychiatric comorbidity. At present PRS have the most promise to improve our understanding of putative mechanisms of genetic liability. In this context, it is important to conceptualize such measures as indicators of vulnerability, rather than harbingers of future diagnosis. In fact, any current genetic prediction panel for psychiatric disorders that relies on common variation should be evaluated with considerable rigor for its purported clinical utility. Nonetheless, a unique strength to this approach is that PRS may be applied to large general population samples studying deeply phenotyped constructs, extending the range of psychiatric genetics research. Further, it will be important to consider that effect sizes are small and currently can reasonably be expected to account for only 0.01–3% of variance in related phenotypes when designing studies. In this context, it is critical for adequately powered research to be conducted so that false positive and false negative findings do not misguide research efforts.
As GWAS continue to expand and include large-scale studies of treatment response, it is possible that PRS results may be clinically applicable and potentially even usefully guide personalized treatment recommendations in the distant future. Further, as multiplex gene editing in preclinical models advance, it is plausible that polygenic effects across systems can be modeled to identify potentially influential, covarying, or even novel pathways for future treatment development. In addition to using novel analytic tools such as LDPred and MTAG to enhance the predictive utility of PRS, it will be important to increase the size and diversity of GWAS samples alongside the use of refined and diverse phenotyping. For example, it is possible that genetic loci associated with disorder expression are entirely or partially distinct from those associated with mechanistic pathways and treatment response. Lastly, integrating polygenic GWAS statistics with functional data (Arloth et al., 2015) and pathway analyses may help guide the identification of potential treatment pathways.
Central Points.
Polygenic risk scores (PRS) represent the additive effect of multiple common variants; they explain <5% of cross-trait variability.
Currently studies of polygenic risk require at least 300 participants to achieve adequate power for cross-trait associations, given optimistic estimates.
Clinical utility of PRS might necessitate discovery GWAS of specific traits (e.g., of treatment response).
Incorporating information from biological systems and/or summary statistics from GWAS of genetically-correlated traits (e.g., MTAG) may improve their predictive utility.
Unresolved Issues.
Are PRS for disorders or traits useful for predicting treatment response or interactions with the environment?
Are PRS be clinically informative or prognostic?
How can PRS be modified to be informative from a molecular and system-level perspective?
Terms and Definitions.
Single nucleotide polymorphism, or SNP: Change in single base pair, A, T, G or C.
Linkage Disequilibrium, or LD: Genetic variation that is physically proximal to each other may be correlated. Knowing an individual’s genotype at one locus allows us to guess their genotype at a locus in high LD.
Admixture, or population stratification: Variations in allele frequency and LD across ancestral populations that can lead to spurious association signals if cases or controls have different proportions of individuals from each ancestral group.
GenomeWide Association Study, or GWAS: estimating the association between disease or behavioral trait and common SNPs across genome. Significance is p < 5 × 10−8, i.e., Bonferroni corrected for 1 million independent tests.
Haplotype: Set of alleles that are observed in a population, likely due to evolutionary selection, more often than expected by chance alone.
Imputation: Probabilistic estimation of an individual’s genotype, based on LD with known genotypes and comparison to a reference genome.
Major Histocompatibility Complex, or MHC: Large segment, spanning 4 × 10−6 base pairs on chromosome 6, with complex LD patterns across highly polymorphic SNPs spanning over 200 Human Leukocyte Antigen (HLA) genes involved in immune response.
Pleiotropy: The phenomenon of the same genetic variation contributing to different disorders or conditions.
Pharmacogenomics: Tailoring treatment based on an individual patient’s genetic composition.
Reference genome: Comprehensive sequence data of a reference set of individuals of a specific ancestry; freely available to compare with one’s study participant data.
Resources.
A variety of resources are available to facilitate PRS analyses.
LDHub (Zheng et al., 2017) allows for computation of SNP-h2 and inter-trait genetic correlation (SNP-rg) using summary statistics. Assists with identification of genetically correlated traits for MTAG: http://ldsc.broadinstitute.org/
MRBase (Hemani et al., 2016) run Mendelian Randomization analyses with summary statistics or data upload: http://www.mrbase.org/
PLINK Software (Chang et al., 2015, Purcell et al., 2007)and online user resources provide tools and information for PRS computation:https://www.cog-genomics.org/plink2 & http://zzz.bwh.harvard.edu/plink/
PRSice Software (Euesden et al., 2015b): Software package that works with plink to generate PRS scores: http://prsice.info/
Psychiatric Genomics Consortium Downloads contain summary statistics that can be used to compute PRS for a variety of psychiatric phenotypes for analyses from the PGC and other groups: https://www.med.unc.edu/pgc/results-and-downloads
Funding Acknowledgments:
RB was supported by NIA (NIA R01-AG045231) NICHD (R01-HD083614) and NIHAG (R01-AG052564). DB was funded by NSF (DGE-1745038). AA was funded by K02DA32573.
Footnotes
Scores are also referred to as genetic risk scores (GRS) or polygenic scores (PGS) within the broader literature.
SNP-h2, especially from LD Score Regression, does not require access to raw genotypes, can be used for quality control and can be automated, with less likelihood of user error. However, it requires large sample sizes (n > 3K).
Literature Cited
- AAS M, MELLE I, BETTELLA F, DJUROVIC S, LE HELLARD S, BJELLA T, RINGEN PA, LAGERBERG TV, SMELAND OB, AGARTZ I, ANDREASSEN OA & TESLI M 2017. Psychotic patients who used cannabis frequently before illness onset have higher genetic predisposition to schizophrenia than those who did not. Psychol Med, 1–7. [DOI] [PubMed] [Google Scholar]
- AGERBO E, SULLIVAN PF, VILHJALMSSON BJ, PEDERSEN CB, MORS O, BORGLUM AD, HOUGAARD DM, HOLLEGAARD MV, MEIER S, MATTHEISEN M, RIPKE S, WRAY NR & MORTENSEN PB 2015. Polygenic Risk Score, Parental Socioeconomic Status, Family History of Psychiatric Disorders, and the Risk for Schizophrenia: A Danish Population-Based Study and Meta-analysis. JAMA Psychiatry, 72, 635–41. [DOI] [PubMed] [Google Scholar]
- AHN K, AN SS, SHUGART YY & RAPOPORT JL 2016. Common polygenic variation and risk for childhood-onset schizophrenia. Mol Psychiatry, 21, 94–6. [DOI] [PubMed] [Google Scholar]
- ALLOZA C, BASTIN ME, COX SR, GIBSON J, DUFF B, SEMPLE SI, WHALLEY HC & LAWRIE SM 2017. Central and non-central networks, cognition, clinical symptoms, and polygenic risk scores in schizophrenia. Hum Brain Mapp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANTTILA V, BULIK-SULLIVAN B, FINUCANE HK, BRAS J, DUNCAN L, ESCOTT-PRICE V, FALCONE G, GORMLEY P, MALIK R, PATSOPOULOS N, RIPKE S, WALTERS R, WEI Z, YU D, LEE P, BREEN G, BULIK C, DALY M, DICHGANS M, FARAONE S, GUERREIRO R, HOLMANS P, KENDLER K, KOELEMAN B, MATHEWS C, SCHARF J, SKLAR P, WILLIAMS J, WOOD N, COTSAPAS C, PALOTIE A, SMOLLER J, SULLIVAN P, ROSAND J, CORVIN A & NEALE B 2016. Analysis of shared heritability in common disorders of the brain. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARLOTH J, BOGDAN R, WEBER P, FRISHMAN G, MENKE A, WAGNER KV, BALSEVICH G, SCHMIDT MV, KARBALAI N, CZAMARA D, ALTMANN A, TRUMBACH D, WURST W, MEHTA D, UHR M, KLENGEL T, ERHARDT A, CAREY CE, CONLEY ED, MAJOR DEPRESSIVE DISORDER WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., RUEPP A, MULLER-MYHSOK B, HARIRI AR, BINDER EB & MAJOR DEPRESSIVE DISORDER WORKING GROUP OF THE PSYCHIATRIC GENOMICS CONSORTIUM, P. G. C. 2015. Genetic Differences in the Immediate Transcriptome Response to Stress Predict Risk-Related Brain Function and Psychiatric Disorders. Neuron, 86, 1189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRETT JC, DUNHAM I & BIRNEY E 2015. Using human genetics to make new medicines. Nat Rev Genet, 16, 561–2. [DOI] [PubMed] [Google Scholar]
- BASSETT AS & CHOW EW 2008. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep, 10, 148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELGARD TG, JANKOVIC I, LOWE JK & GESCHWIND DH 2014. Population structure confounds autism genetic classifier. Mol Psychiatry, 19, 405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENCA CE, DERRINGER JL, CORLEY RP, YOUNG SE, KELLER MC, HEWITT JK & FRIEDMAN NP 2017. Predicting Cognitive Executive Functioning with Polygenic Risk Scores for Psychiatric Disorders. Behav Genet, 47, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENROS ME, TRABJERG BB, MEIER S, MATTHEISEN M, MORTENSEN PB, MORS O, BORGLUM AD, HOUGAARD DM, NORGAARD-PEDERSEN B, NORDENTOFT M & AGERBO E 2016. Influence of Polygenic Risk Scores on the Association Between Infections and Schizophrenia. Biol Psychiatry, 80, 609–16. [DOI] [PubMed] [Google Scholar]
- BIERUT LJ, JOHNSON EO & SACCONE NL 2014. A glimpse into the future - Personalized medicine for smoking cessation. Neuropharmacology, 76 Pt B, 592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIGDELI TB, RIPKE S, PETERSON RE, TRZASKOWSKI M, BACANU SA, ABDELLAOUI A, ANDLAUER TF, BEEKMAN AT, BERGER K, BLACKWOOD DH, BOOMSMA DI, BREEN G, BUTTENSCHON HN, BYRNE EM, CICHON S, CLARKE TK, COUVY-DUCHESNE B, CRADDOCK N, DE GEUS EJ, DEGENHARDT F, DUNN EC, EDWARDS AC, FANOUS AH, FORSTNER AJ, FRANK J, GILL M, GORDON SD, GRABE HJ, HAMILTON SP, HARDIMAN O, HAYWARD C, HEATH AC, HENDERS AK, HERMS S, HICKIE IB, HOFFMANN P, HOMUTH G, HOTTENGA JJ, ISING M, JANSEN R, KLOIBER S, KNOWLES JA, LANG M, LI QS, LUCAE S, MACINTYRE DJ, MADDEN PA, MARTIN NG, MCGRATH PJ, MCGUFFIN P, MCINTOSH AM, MEDLAND SE, MEHTA D, MIDDELDORP CM, MILANESCHI Y, MONTGOMERY GW, MORS O, MULLER-MYHSOK B, NAUCK M, NYHOLT DR, NOTHEN MM, OWEN MJ, PENNINX BW, PERGADIA ML, PERLIS RH, PEYROT WJ, PORTEOUS DJ, POTASH JB, RICE JP, RIETSCHEL M, RILEY BP, RIVERA M, SCHOEVERS R, SCHULZE TG, SHI J, SHYN SI, SMIT JH, SMOLLER JW, STREIT F, STROHMAIER J, TEUMER A, TREUTLEIN J, VAN DER AUWERA S, VAN GROOTHEEST G, VAN HEMERT AM, VOLZKE H, WEBB BT, WEISSMAN MM, WELLMANN J, WILLEMSEN G, WITT SH, LEVINSON DF, LEWIS CM, WRAY NR, FLINT J, SULLIVAN PF & KENDLER KS 2017. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl Psychiatry, 7, e1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOGDAN R, NIKOLOVA YS & PIZZAGALLI DA 2013. Neurogenetics of depression: a focus on reward processing and stress sensitivity. Neurobiol Dis, 52, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOGDAN R, SALMERON BJ, CAREY CE, AGRAWAL A, CALHOUN VD, GARAVAN H, HARIRI AR, HEINZ A, HILL MN, HOLMES A, KALIN NH & GOLDMAN D 2017. Imaging Genetics and Genomics in Psychiatry: A Critical Review of Progress and Potential. Biol Psychiatry, 82, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOWDEN J, DAVEY SMITH G & BURGESS S 2015. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol, 44, 512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOYLE EA, LI YI & PRITCHARD JK 2017. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell, 169, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREEN G, LI Q, ROTH BL, O’DONNELL P, DIDRIKSEN M, DOLMETSCH R, O’REILLY PF, GASPAR HA, MANJI H, HUEBEL C, KELSOE JR, MALHOTRA D, BERTOLINO A, POSTHUMA D, SKLAR P, KAPUR S, SULLIVAN PF, COLLIER DA & EDENBERG HJ 2016. Translating genome-wide association findings into new therapeutics for psychiatry. Nat Neurosci, 19, 1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BULIK-SULLIVAN BK, LOH PR, FINUCANE HK, RIPKE S, YANG J, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., PATTERSON N, DALY MJ, PRICE AL & NEALE BM 2015. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet, 47, 291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAREY CE, AGRAWAL A, BUCHOLZ KK, HARTZ SM, LYNSKEY MT, NELSON EC, BIERUT LJ & BOGDAN R 2016a. Associations between Polygenic Risk for Psychiatric Disorders and Substance Involvement. Front Genet, 7, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAREY CE, KNODT AR, CONLEY ED, HARIRI AR & BOGDAN R 2016b. Reward-Related Ventral Striatum Activity Links Polygenic Risk for Attention-Deficit/Hyperactivity Disorder to Problematic Alcohol Use in Young Adulthood. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASPI A, MCCLAY J, MOFFITT TE, MILL J, MARTIN J, CRAIG IW, TAYLOR A & POULTON R 2002. Role of genotype in the cycle of violence in maltreated children. Science, 297, 851–4. [DOI] [PubMed] [Google Scholar]
- CASPI A, SUGDEN K, MOFFITT TE, TAYLOR A, CRAIG IW, HARRINGTON H, MCCLAY J, MILL J, MARTIN J, BRAITHWAITE A & POULTON R 2003. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science, 301, 386–9. [DOI] [PubMed] [Google Scholar]
- CHANG CC, CHOW CC, TELLIER LC, VATTIKUTI S, PURCELL SM & LEE JJ 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHARNEY AW, RUDERFER DM, STAHL EA, MORAN JL, CHAMBERT K, BELLIVEAU RA, FORTY L, GORDON-SMITH K, DI FLORIO A, LEE PH, BROMET EJ, BUCKLEY PF, ESCAMILLA MA, FANOUS AH, FOCHTMANN LJ, LEHRER DS, MALASPINA D, MARDER SR, MORLEY CP, NICOLINI H, PERKINS DO, RAKOFSKY JJ, RAPAPORT MH, MEDEIROS H, SOBELL JL, GREEN EK, BACKLUND L, BERGEN SE, JUREUS A, SCHALLING M, LICHTENSTEIN P, ROUSSOS P, KNOWLES JA, JONES I, JONES LA, HULTMAN CM, PERLIS RH, PURCELL SM, MCCARROLL SA, PATO CN, PATO MT, CRADDOCK N, LANDEN M, SMOLLER JW & SKLAR P 2017. Evidence for genetic heterogeneity between clinical subtypes of bipolar disorder. Transl Psychiatry, 7, e993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHATTERJEE N, SHI J & GARCIA-CLOSAS M 2016. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat Rev Genet, 17, 392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN CY, HAN J, HUNTER DJ, KRAFT P & PRICE AL 2015. Explicit Modeling of Ancestry Improves Polygenic Risk Scores and BLUP Prediction. Genet Epidemiol, 39, 427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN LS, BAKER TB, PIPER ME, BRESLAU N, CANNON DS, DOHENY KF, GOGARTEN SM, JOHNSON EO, SACCONE NL, WANG JC, WEISS RB, GOATE AM & BIERUT LJ 2012. Interplay of genetic risk factors (CHRNA5-CHRNA3-CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry, 169, 735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG Y & CANNON TD 2015. Brain imaging during the transition from psychosis prodrome to schizophrenia. J Nerv Ment Dis, 203, 336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLEMAN JRI, LESTER KJ, KEERS R, MUNAFO MR, BREEN G & ELEY TC 2017. Genome-wide association study of facial emotion recognition in children and association with polygenic risk for mental health disorders. Am J Med Genet B Neuropsychiatr Genet, 174, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLODRO-CONDE L, COUVY-DUCHESNE B, ZHU G, COVENTRY WL, BYRNE EM, GORDON S, WRIGHT MJ, MONTGOMERY GW, MADDEN PAF, MAJOR DEPRESSIVE DISORDER WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., RIPKE S, EAVES LJ, HEATH AC, WRAY NR, MEDLAND SE & MARTIN NG 2017. A direct test of the diathesis-stress model for depression. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONG L, RAN FA, COX D, LIN S, BARRETTO R, HABIB N, HSU PD, WU X, JIANG W, MARRAFFINI LA & ZHANG F 2013. Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONSORTIUM, G. T. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet, 45, 580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONVERGE 2015. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature, 523, 588–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORAM MA, FANG H, CANDILLE SI, ASSIMES TL & TANG H 2017. Leveraging Multi-ethnic Evidence for Risk Assessment of Quantitative Traits in Minority Populations. Am J Hum Genet, 101, 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORDER EH, SAUNDERS AM, STRITTMATTER WJ, SCHMECHEL DE, GASKELL PC, SMALL GW, ROSES AD, HAINES JL & PERICAK-VANCE MA 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 261, 921–3. [DOI] [PubMed] [Google Scholar]
- CORNELIS MC, NUGENT NR, AMSTADTER AB & KOENEN KC 2010. Genetics of post-traumatic stress disorder: review and recommendations for genome-wide association studies. Curr Psychiatry Rep, 12, 313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTAS J, CARRERA N, ALONSO P, GURRIARAN X, SEGALAS C, REAL E, LOPEZ-SOLA C, MAS S, GASSO P, DOMENECH L, MORELL M, QUINTELA I, LAZARO L, MENCHON JM, ESTIVILL X & CARRACEDO A 2016. Exon-focused genome-wide association study of obsessive-compulsive disorder and shared polygenic risk with schizophrenia. Transl Psychiatry, 6, e768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRADDOCK N & OWEN MJ 1996. Modern molecular genetic approaches to psychiatric disease. Br Med Bull, 52, 434–52. [DOI] [PubMed] [Google Scholar]
- CROSS-DISORDER GROUP OF THE PSYCHIATRIC GENOMICS, C. 2013. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet, 381, 1371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAETWYLER HD, VILLANUEVA B & WOOLLIAMS JA 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS One, 3, e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVEY SMITH G & HEMANI G 2014. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet, 23, R89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEARY V, HAGENAARS SP, HARRIS SE, HILL WD, DAVIES G, LIEWALD DC, INTERNATIONAL CONSORTIUM FOR BLOOD PRESSURE, G., AGING, C. C., LONGEVITY, G., GROUP, C. C. I., MCINTOSH AM, GALE CR & DEARY IJ 2017. Genetic contributions to self-reported tiredness. Mol Psychiatry. [Google Scholar]
- DEMICHELE-SWEET MAA, WEAMER EA, KLEI L, VRANA DT, HOLLINGSHEAD DJ, SELTMAN HJ, SIMS R, FOROUD T, HERNANDEZ I, MORENO-GRAU S, TARRAGA L, BOADA M, RUIZ A, WILLIAMS J, MAYEUX R, LOPEZ OL, SIBILLE EL, KAMBOH MI, DEVLIN B & SWEET RA 2017. Genetic risk for schizophrenia and psychosis in Alzheimer disease. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOCHERTY AR, MOSCATI A, PETERSON R, EDWARDS AC, ADKINS DE, BACANU SA, BIGDELI TB, WEBB BT, FLINT J & KENDLER KS 2016. SNP-based heritability estimates of the personality dimensions and polygenic prediction of both neuroticism and major depression: findings from CONVERGE. Transl Psychiatry, 6, e926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUDBRIDGE F 2013. Power and predictive accuracy of polygenic risk scores. PLoS Genet, 9, e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUDBRIDGE F 2016. Polygenic Epidemiology. Genet Epidemiol, 40, 268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNCAN LE & KELLER MC 2011. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry, 168, 1041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNCAN LE, RATANATHARATHORN A, AIELLO AE, ALMLI LM, AMSTADTER AB, ASHLEY-KOCH AE, BAKER DG, BECKHAM JC, BIERUT LJ, BISSON J, BRADLEY B, CHEN CY, DALVIE S, FARRER LA, GALEA S, GARRETT ME, GELERNTER JE, GUFFANTI G, HAUSER MA, JOHNSON EO, KESSLER RC, KIMBREL NA, KING A, KOEN N, KRANZLER HR, LOGUE MW, MAIHOFER AX, MARTIN AR, MILLER MW, MOREY RA, NUGENT NR, RICE JP, RIPKE S, ROBERTS AL, SACCONE NL, SMOLLER JW, STEIN DJ, STEIN MB, SUMNER JA, UDDIN M, URSANO RJ, WILDMAN DE, YEHUDA R, ZHAO H, DALY MJ, LIBERZON I, RESSLER KJ, NIEVERGELT CM & KOENEN KC 2017. Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EGAN MF, GOLDBERG TE, KOLACHANA BS, CALLICOTT JH, MAZZANTI CM, STRAUB RE, GOLDMAN D & WEINBERGER DR 2001. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A, 98, 6917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERK S, MOHNKE S, RIPKE S, LETT TA, VEER IM, WACKERHAGEN C, GRIMM O, ROMANCZUK-SEIFERTH N, DEGENHARDT F, TOST H, MATTHEISEN M, MUHLEISEN TW, CHARLET K, SKARABIS N, KIEFER F, CICHON S, WITT SH, NOTHEN MM, RIETSCHEL M, HEINZ A, MEYER-LINDENBERG A & WALTER H 2017. Functional neuroimaging effects of recently discovered genetic risk loci for schizophrenia and polygenic risk profile in five RDoC subdomains. Transl Psychiatry, 7, e997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUESDEN J, BREEN G, FARMER A, MCGUFFIN P & LEWIS CM 2015a. The relationship between schizophrenia and rheumatoid arthritis revisited: genetic and epidemiological analyses. Am J Med Genet B Neuropsychiatr Genet, 168B, 81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUESDEN J, LEWIS CM & O’REILLY PF 2015b. PRSice: Polygenic Risk Score software. Bioinformatics, 31, 1466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAUL F, ERDFELDER E, LANG AG & BUCHNER A 2007. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods, 39, 175–91. [DOI] [PubMed] [Google Scholar]
- FERREIRA MA, O’DONOVAN MC, MENG YA, JONES IR, RUDERFER DM, JONES L, FAN J, KIROV G, PERLIS RH, GREEN EK, SMOLLER JW, GROZEVA D, STONE J, NIKOLOV I, CHAMBERT K, HAMSHERE ML, NIMGAONKAR VL, MOSKVINA V, THASE ME, CAESAR S, SACHS GS, FRANKLIN J, GORDON-SMITH K, ARDLIE KG, GABRIEL SB, FRASER C, BLUMENSTIEL B, DEFELICE M, BREEN G, GILL M, MORRIS DW, ELKIN A, MUIR WJ, MCGHEE KA, WILLIAMSON R, MACINTYRE DJ, MACLEAN AW, ST CD, ROBINSON M, VAN BECK M, PEREIRA AC, KANDASWAMY R, MCQUILLIN A, COLLIER DA, BASS NJ, YOUNG AH, LAWRENCE J, FERRIER IN, ANJORIN A, FARMER A, CURTIS D, SCOLNICK EM, MCGUFFIN P, DALY MJ, CORVIN AP, HOLMANS PA, BLACKWOOD DH, GURLING HM, OWEN MJ, PURCELL SM, SKLAR P, CRADDOCK N & WELLCOME TRUST CASE CONTROL, C. 2008. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet, 40, 1056–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINUCANE HK, BULIK-SULLIVAN B, GUSEV A, TRYNKA G, RESHEF Y, LOH PR, ANTTILA V, XU H, ZANG C, FARH K, RIPKE S, DAY FR, REPROGEN C, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., CONSORTIUM R, PURCELL S, STAHL E, LINDSTROM S, PERRY JR, OKADA Y, RAYCHAUDHURI S, DALY MJ, PATTERSON N, NEALE BM & PRICE AL 2015. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet, 47, 1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANK J, LANG M, WITT SH, STROHMAIER J, RUJESCU D, CICHON S, DEGENHARDT F, NOTHEN MM, COLLIER DA, RIPKE S, NABER D & RIETSCHEL M 2015. Identification of increased genetic risk scores for schizophrenia in treatment-resistant patients. Mol Psychiatry, 20, 913. [DOI] [PubMed] [Google Scholar]
- FRANKE B, STEIN JL, RIPKE S, ANTTILA V, HIBAR DP, VAN HULZEN KJE, ARIAS-VASQUEZ A, SMOLLER JW, NICHOLS TE, NEALE MC, MCINTOSH AM, LEE P, MCMAHON FJ, MEYER-LINDENBERG A, MATTHEISEN M, ANDREASSEN OA, GRUBER O, SACHDEV PS, ROIZ-SANTIANEZ R, SAYKIN AJ, EHRLICH S, MATHER KA, TURNER JA, SCHWARZ E, THALAMUTHU A, SHUGART YY, HO YY, MARTIN NG, WRIGHT MJ, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., CONSORTIUM, E., O’DONOVAN MC, THOMPSON PM, NEALE BM, MEDLAND SE & SULLIVAN PF 2016. Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat Neurosci, 19, 420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRENCH L, GRAY C, LEONARD G, PERRON M, PIKE GB, RICHER L, SEGUIN JR, VEILLETTE S, EVANS CJ, ARTIGES E, BANASCHEWSKI T, BOKDE AW, BROMBERG U, BRUEHL R, BUCHEL C, CATTRELL A, CONROD PJ, FLOR H, FROUIN V, GALLINAT J, GARAVAN H, GOWLAND P, HEINZ A, LEMAITRE H, MARTINOT JL, NEES F, ORFANOS DP, PANGELINAN MM, POUSTKA L, RIETSCHEL M, SMOLKA MN, WALTER H, WHELAN R, TIMPSON NJ, SCHUMANN G, SMITH GD, PAUSOVA Z & PAUS T 2015. Early Cannabis Use, Polygenic Risk Score for Schizophrenia and Brain Maturation in Adolescence. JAMA Psychiatry, 72, 1002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRIEDRICH MJ 2017. Depression Is the Leading Cause of Disability Around the World. JAMA, 317, 1517. [DOI] [PubMed] [Google Scholar]
- FROMER M, ROUSSOS P, SIEBERTS SK, JOHNSON JS, KAVANAGH DH, PERUMAL TM, RUDERFER DM, OH EC, TOPOL A, SHAH HR, KLEI LL, KRAMER R, PINTO D, GUMUS ZH, CICEK AE, DANG KK, BROWNE A, LU C, XIE L, READHEAD B, STAHL EA, XIAO J, PARVIZI M, HAMAMSY T, FULLARD JF, WANG YC, MAHAJAN MC, DERRY JM, DUDLEY JT, HEMBY SE, LOGSDON BA, TALBOT K, RAJ T, BENNETT DA, DE JAGER PL, ZHU J, ZHANG B, SULLIVAN PF, CHESS A, PURCELL SM, SHINOBU LA, MANGRAVITE LM, TOYOSHIBA H, GUR RE, HAHN CG, LEWIS DA, HAROUTUNIAN V, PETERS MA, LIPSKA BK, BUXBAUM JD, SCHADT EE, HIRAI K, ROEDER K, BRENNAND KJ, KATSANIS N, DOMENICI E, DEVLIN B & SKLAR P 2016. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci, 19, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAALI S, KIRSCHNER A, CUBONI S, HARTMANN J, KOZANY C, BALSEVICH G, NAMENDORF C, FERNANDEZ-VIZARRA P, SIPPEL C, ZANNAS AS, DRAENERT R, BINDER EB, ALMEIDA OF, RUHTER G, UHR M, SCHMIDT MV, TOUMA C, BRACHER A & HAUSCH F 2015. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol, 11, 33–7. [DOI] [PubMed] [Google Scholar]
- GAGE SH, JONES HJ, BURGESS S, BOWDEN J, DAVEY SMITH G, ZAMMIT S & MUNAFO MR 2017. Assessing causality in associations between cannabis use and schizophrenia risk: a two-sample Mendelian randomization study. Psychol Med, 47, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALE CR, HAGENAARS SP, DAVIES G, HILL WD, LIEWALD DC, CULLEN B, PENNINX BW, INTERNATIONAL CONSORTIUM FOR BLOOD PRESSURE GWAS, C. C. A., LONGEVITY G, BOOMSMA DI, PELL J, MCINTOSH AM, SMITH DJ, DEARY IJ & HARRIS SE 2016. Pleiotropy between neuroticism and physical and mental health: findings from 108 038 men and women in UK Biobank. Transl Psychiatry, 6, e791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARCIA-GONZALEZ J, TANSEY KE, HAUSER J, HENIGSBERG N, MAIER W, MORS O, PLACENTINO A, RIETSCHEL M, SOUERY D, ZAGAR T, CZERSKI PM, JERMAN B, BUTTENSCHON HN, SCHULZE TG, ZOBEL A, FARMER A, AITCHISON KJ, CRAIG I, MCGUFFIN P, GIUPPONI M, PERROUD N, BONDOLFI G, EVANS D, O’DONOVAN M, PETERS TJ, WENDLAND JR, LEWIS G, KAPUR S, PERLIS R, AROLT V, DOMSCHKE K, MAJOR DEPRESSIVE DISORDER WORKING GROUP OF THE PSYCHIATRIC GENOMIC, C., BREEN G, CURTIS C, SANG-HYUK L, KAN C, NEWHOUSE S, PATEL H, BAUNE BT, UHER R, LEWIS CM & FABBRI C 2017. Pharmacogenetics of antidepressant response: A polygenic approach. Prog Neuropsychopharmacol Biol Psychiatry, 75, 128–134. [DOI] [PubMed] [Google Scholar]
- GELERNTER J, KRANZLER HR, SHERVA R, ALMASY L, KOESTERER R, SMITH AH, ANTON R, PREUSS UW, RIDINGER M, RUJESCU D, WODARZ N, ZILL P, ZHAO H & FARRER LA 2014. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry, 19, 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERMINE L, ROBINSON EB, SMOLLER JW, CALKINS ME, MOORE TM, HAKONARSON H, DALY MJ, LEE PH, HOLMES AJ, BUCKNER RL, GUR RC & GUR RE 2016. Association between polygenic risk for schizophrenia, neurocognition and social cognition across development. Transl Psychiatry, 6, e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIBERT JM, BLANCO J, DOLEZAL M, NOLTE V, PERONNET F & SCHLOTTERER C 2017. Strong epistatic and additive effects of linked candidate SNPs for Drosophila pigmentation have implications for analysis of genome-wide association studies results. Genome Biol, 18, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILBERTSON MW, SHENTON ME, CISZEWSKI A, KASAI K, LASKO NB, ORR SP & PITMAN RK 2002. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci, 5, 1242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOES FS, MCGRATH J, AVRAMOPOULOS D, WOLYNIEC P, PIROOZNIA M, RUCZINSKI I, NESTADT G, KENNY EE, VACIC V, PETERS I, LENCZ T, DARVASI A, MULLE JG, WARREN ST & PULVER AE 2015. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet, 168, 649–59. [DOI] [PubMed] [Google Scholar]
- GOLDBERG TE, EGAN MF, GSCHEIDLE T, COPPOLA R, WEICKERT T, KOLACHANA BS, GOLDMAN D & WEINBERGER DR 2003. Executive subprocesses in working memory: relationship to catechol-O-methyltransferase Val158Met genotype and schizophrenia. Arch Gen Psychiatry, 60, 889–96. [DOI] [PubMed] [Google Scholar]
- GONZALEZ-CASTRO TB, HERNANDEZ-DIAZ Y, JUAREZ-ROJOP IE, LOPEZ-NARVAEZ ML, TOVILLA-ZARATE CA & FRESAN A 2016. The Role of a Catechol-O-Methyltransferase (COMT) Val158Met Genetic Polymorphism in Schizophrenia: A Systematic Review and Updated Meta-analysis on 32,816 Subjects. Neuromolecular Med, 18, 216–31. [DOI] [PubMed] [Google Scholar]
- GOODKIND M, EICKHOFF SB, OATHES DJ, JIANG Y, CHANG A, JONES-HAGATA LB, ORTEGA BN, ZAIKO YV, ROACH EL, KORGAONKAR MS, GRIEVE SM, GALATZER-LEVY I, FOX PT & ETKIN A 2015. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry, 72, 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREEN JG, MCLAUGHLIN KA, BERGLUND PA, GRUBER MJ, SAMPSON NA, ZASLAVSKY AM & KESSLER RC 2010. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: associations with first onset of DSM-IV disorders. Arch Gen Psychiatry, 67, 113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUSEV A, LEE SH, TRYNKA G, FINUCANE H, VILHJALMSSON BJ, XU H, ZANG C, RIPKE S, BULIK-SULLIVAN B, STAHL E, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., CONSORTIUM, S.-S., KAHLER AK, HULTMAN CM, PURCELL SM, MCCARROLL SA, DALY M, PASANIUC B, SULLIVAN PF, NEALE BM, WRAY NR, RAYCHAUDHURI S, PRICE AL, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C. & CONSORTIUM, S.-S. 2014. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet, 95, 535–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAGENAARS SP, HARRIS SE, DAVIES G, HILL WD, LIEWALD DC, RITCHIE SJ, MARIONI RE, FAWNS-RITCHIE C, CULLEN B, MALIK R, METASTROKE CONSORTIUM ICFBPG, SPIROMETA C, CHARGE CONSORTIUM PULMONARY GROUP, C. C. A., LONGEVITY G, WORRALL BB, SUDLOW CL, WARDLAW JM, GALLACHER J, PELL J, MCINTOSH AM, SMITH DJ, GALE CR & DEARY IJ 2016. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol Psychiatry, 21, 1624–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL MH, CHEN CY, COHEN BM, SPENCER KM, LEVY DL, ONGUR D & SMOLLER JW 2015. Genomewide association analyses of electrophysiological endophenotypes for schizophrenia and psychotic bipolar disorders: a preliminary report. Am J Med Genet B Neuropsychiatr Genet, 168B, 151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL W & DEGENHARDT L 2008. Cannabis use and the risk of developing a psychotic disorder. World Psychiatry, 7, 68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDY J, TRABZUNI D & RYTEN M 2009. Whole genome expression as a quantitative trait. Biochem Soc Trans, 37, 1276–7. [DOI] [PubMed] [Google Scholar]
- HARIRI AR 2009. The neurobiology of individual differences in complex behavioral traits. Annu Rev Neurosci, 32, 225–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARIRI AR, MATTAY VS, TESSITORE A, KOLACHANA B, FERA F, GOLDMAN D, EGAN MF & WEINBERGER DR 2002. Serotonin transporter genetic variation and the response of the human amygdala. Science, 297, 400–3. [DOI] [PubMed] [Google Scholar]
- HARRIS SE, HAGENAARS SP, DAVIES G, DAVID HILL W, LIEWALD DC, RITCHIE SJ, MARIONI RE, METASTROKE CONSORTIUM, I. C. F. B. P. G.-W. A. S. C. C. A., LONGEVITY GROUP, C. C. C. G., SUDLOW CL, WARDLAW JM, MCINTOSH AM, GALE CR & DEARY IJ 2016. Molecular genetic contributions to self-rated health. Int J Epidemiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRISBERGER F, SMIESKOVA R, VOGLER C, EGLI T, SCHMIDT A, LENZ C, SIMON AE, RIECHER-ROSSLER A, PAPASSOTIROPOULOS A & BORGWARDT S 2016. Impact of polygenic schizophrenia-related risk and hippocampal volumes on the onset of psychosis. Transl Psychiatry, 6, e868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARTZ SM, HORTON AC, OEHLERT M, CAREY CE, AGRAWAL A, BOGDAN R, CHEN LS, HANCOCK DB, JOHNSON EO, PATO CN, PATO MT, RICE JP & BIERUT LJ 2017. Association Between Substance Use Disorder and Polygenic Liability to Schizophrenia. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HATZIMANOLIS A, BHATNAGAR P, MOES A, WANG R, ROUSSOS P, BITSIOS P, STEFANIS CN, PULVER AE, ARKING DE, SMYRNIS N, STEFANIS NC & AVRAMOPOULOS D 2015. Common genetic variation and schizophrenia polygenic risk influence neurocognitive performance in young adulthood. Am J Med Genet B Neuropsychiatr Genet, 168B, 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEMANI G, SHAKHBAZOV K, WESTRA HJ, ESKO T, HENDERS AK, MCRAE AF, YANG J, GIBSON G, MARTIN NG, METSPALU A, FRANKE L, MONTGOMERY GW, VISSCHER PM & POWELL JE 2014. Detection and replication of epistasis influencing transcription in humans. Nature, 508, 249–53. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- HEMANI G, ZHENG J, WADE KH, LAURIN C, ELSWORTH B, BURGESS S, BOWDEN J, LANGDON R, TAN V, YARMOLINSKY J, SHIHAB HA, TIMPSON N, EVANS DM, RELTON C, MARTIN RM, DAVEY SMITH G, GAUNT TR & HAYCOCK PC 2016. MR-Base: a platform for systematic causal inference across the phenome using billions of genetic associations. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HETTIGE NC, COLE CB, KHALID S & DE LUCA V 2016. Polygenic risk score prediction of antipsychotic dosage in schizophrenia. Schizophr Res, 170, 265–70. [DOI] [PubMed] [Google Scholar]
- HILL WD, DAVIES G, MCINTOSH AM, GALE CR & DEARY IJ 2017. A combined analysis of genetically correlated traits identifies 107 loci associated with intelligence. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILL WG, GODDARD ME & VISSCHER PM 2008. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet, 4, e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRSCHHORN JN & GENNARI L 2008. Bona fide genetic associations with bone mineral density. N Engl J Med, 358, 2403–5. [DOI] [PubMed] [Google Scholar]
- HOLMES MV, LANGE LA, PALMER T, LANKTREE MB, NORTH KE, ALMOGUERA B, BUXBAUM S, CHANDRUPATLA HR, ELBERS CC, GUO Y, HOOGEVEEN RC, LI J, LI YR, SWERDLOW DI, CUSHMAN M, PRICE TS, CURTIS SP, FORNAGE M, HAKONARSON H, PATEL SR, REDLINE S, SISCOVICK DS, TSAI MY, WILSON JG, VAN DER SCHOUW YT, FITZGERALD GA, HINGORANI AD, CASAS JP, DE BAKKER PI, RICH SS, SCHADT EE, ASSELBERGS FW, REINER AP & KEATING BJ 2014. Causal effects of body mass index on cardiometabolic traits and events: a Mendelian randomization analysis. Am J Hum Genet, 94, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUBBARD L, TANSEY KE, RAI D, JONES P, RIPKE S, CHAMBERT KD, MORAN JL, MCCARROLL SA, LINDEN DE, OWEN MJ, O’DONOVAN MC, WALTERS JT & ZAMMIT S 2016. Evidence of Common Genetic Overlap Between Schizophrenia and Cognition. Schizophr Bull, 42, 832–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HYDE CL, NAGLE MW, TIAN C, CHEN X, PACIGA SA, WENDLAND JR, TUNG JY, HINDS DA, PERLIS RH & WINSLOW AR 2016. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet, 48, 1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INTERNATIONAL SCHIZOPHRENIA C, PURCELL SM, WRAY NR, STONE JL, VISSCHER PM, O’DONOVAN MC, SULLIVAN PF & SKLAR P 2009. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460, 748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANSEN PR, POLDERMAN TJC, BOLHUIS K, VAN DER ENDE J, JADDOE VWV, VERHULST FC, WHITE T, POSTHUMA D & TIEMEIER H 2017. Polygenic scores for schizophrenia and educational attainment are associated with behavioural problems in early childhood in the general population. J Child Psychol Psychiatry. [DOI] [PubMed] [Google Scholar]
- JONES HJ, STERGIAKOULI E, TANSEY KE, HUBBARD L, HERON J, CANNON M, HOLMANS P, LEWIS G, LINDEN DE, JONES PB, DAVEY SMITH G, O’DONOVAN MC, OWEN MJ, WALTERS JT & ZAMMIT S 2016. Phenotypic Manifestation of Genetic Risk for Schizophrenia During Adolescence in the General Population. JAMA Psychiatry, 73, 221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAHN RS, SOMMER IE, MURRAY RM, MEYER-LINDENBERG A, WEINBERGER DR, CANNON TD, O’DONOVAN M, CORRELL CU, KANE JM, VAN OS J & INSEL TR 2015. Schizophrenia. Nat Rev Dis Primers, 1, 15067. [DOI] [PubMed] [Google Scholar]
- KENDLER KS 2013. What psychiatric genetics has taught us about the nature of psychiatric illness and what is left to learn. Mol Psychiatry, 18, 1058–66. [DOI] [PubMed] [Google Scholar]
- KENDLER KS 2015. A joint history of the nature of genetic variation and the nature of schizophrenia. Mol Psychiatry, 20, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KISHI T, YOSHIMURA R, KITAJIMA T, OKOCHI T, OKUMURA T, TSUNOKA T, YAMANOUCHI Y, KINOSHITA Y, KAWASHIMA K, FUKUO Y, NAITOH H, UMENE-NAKANO W, INADA T, NAKAMURA J, OZAKI N & IWATA N 2010. SIRT1 gene is associated with major depressive disorder in the Japanese population. J Affect Disord, 126, 167–73. [DOI] [PubMed] [Google Scholar]
- KLENGEL T, MEHTA D, ANACKER C, REX-HAFFNER M, PRUESSNER JC, PARIANTE CM, PACE TW, MERCER KB, MAYBERG HS, BRADLEY B, NEMEROFF CB, HOLSBOER F, HEIM CM, RESSLER KJ, REIN T & BINDER EB 2013. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci, 16, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNUESEL I, CHICHA L, BRITSCHGI M, SCHOBEL SA, BODMER M, HELLINGS JA, TOOVEY S & PRINSSEN EP 2014. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol, 10, 643–60. [DOI] [PubMed] [Google Scholar]
- KRAPOHL E, EUESDEN J, ZABANEH D, PINGAULT JB, RIMFELD K, VON STUMM S, DALE PS, BREEN G, O’REILLY PF & PLOMIN R 2016. Phenome-wide analysis of genome-wide polygenic scores. Mol Psychiatry, 21, 1188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUNINGAS M, PUTTERS M, WESTENDORP RG, SLAGBOOM PE & VAN HEEMST D 2007. SIRT1 gene, age-related diseases, and mortality: the Leiden 85-plus study. J Gerontol A Biol Sci Med Sci, 62, 960–5. [DOI] [PubMed] [Google Scholar]
- LAHEY BB, APPLEGATE B, HAKES JK, ZALD DH, HARIRI AR & RATHOUZ PJ 2012. Is there a general factor of prevalent psychopathology during adulthood? J Abnorm Psychol, 121, 971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANCASTER TM, IHSSEN N, BRINDLEY LM, TANSEY KE, MANTRIPRAGADA K, O’DONOVAN MC, OWEN MJ & LINDEN DE 2016a. Associations between polygenic risk for schizophrenia and brain function during probabilistic learning in healthy individuals. Hum Brain Mapp, 37, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANCASTER TM, LINDEN DE, TANSEY KE, BANASCHEWSKI T, BOKDE AL, BROMBERG U, BUCHEL C, CATTRELL A, CONROD PJ, FLOR H, FROUIN V, GALLINAT J, GARAVAN H, GOWLAND P, HEINZ A, ITTERMANN B, MARTINOT JL, PAILLERE MARTINOT ML, ARTIGES E, LEMAITRE H, NEES F, ORFANOS DP, PAUS T, POUSTKA L, SMOLKA MN, VETTER NC, JURK S, MENNIGEN E, WALTER H, WHELAN R, SCHUMANN G & CONSORTIUM I 2016b. Polygenic Risk of Psychosis and Ventral Striatal Activation During Reward Processing in Healthy Adolescents. JAMA Psychiatry, 73, 852–61. [DOI] [PubMed] [Google Scholar]
- LAURSEN TM, TRABJERG BB, MORS O, BORGLUM AD, HOUGAARD DM, MATTHEISEN M, MEIER SM, BYRNE EM, MORTENSEN PB, MUNK-OLSEN T & AGERBO E 2017. Association of the polygenic risk score for schizophrenia with mortality and suicidal behavior - A Danish population-based study. Schizophr Res, 184, 122–127. [DOI] [PubMed] [Google Scholar]
- LEE SH, BYRNE EM, HULTMAN CM, KAHLER A, VINKHUYZEN AA, RIPKE S, ANDREASSEN OA, FRISELL T, GUSEV A, HU X, KARLSSON R, MANTZIORIS VX, MCGRATH JJ, MEHTA D, STAHL EA, ZHAO Q, KENDLER KS, SULLIVAN PF, PRICE AL, O’DONOVAN M, OKADA Y, MOWRY BJ, RAYCHAUDHURI S, WRAY NR, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., RHEUMATOID ARTHRITIS CONSORTIUM, I., SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS CONSORTIUM, A., BYERLEY W, CAHN W, CANTOR RM, CICHON S, CORMICAN P, CURTIS D, DJUROVIC S, ESCOTT-PRICE V, GEJMAN PV, GEORGIEVA L, GIEGLING I, HANSEN TF, INGASON A, KIM Y, KONTE B, LEE PH, MCINTOSH A, MCQUILLIN A, MORRIS DW, NOTHEN MM, O’DUSHLAINE C, OLINCY A, OLSEN L, PATO CN, PATO MT, PICKARD BS, POSTHUMA D, RASMUSSEN HB, RIETSCHEL M, RUJESCU D, SCHULZE TG, SILVERMAN JM, THIRUMALAI S, WERGE T, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS CONSORTIUM, C., AGARTZ I, AMIN F, AZEVEDO MH, BASS N, BLACK DW, BLACKWOOD DH, BRUGGEMAN R, BUCCOLA NG, CHOUDHURY K, CLONINGER RC, CORVIN A, CRADDOCK N, DALY MJ, DATTA S, DONOHOE GJ, DUAN J, DUDBRIDGE F, FANOUS A, FREEDMAN R, FREIMER NB, FRIEDL M, GILL M, GURLING H, DE HAAN L, HAMSHERE ML, HARTMANN AM, HOLMANS PA, KAHN RS, KELLER MC, KENNY E, KIROV GK, KRABBENDAM L, KRASUCKI R, LAWRENCE J, LENCZ T, LEVINSON DF, LIEBERMAN JA, LIN DY, et al. 2015. New data and an old puzzle: the negative association between schizophrenia and rheumatoid arthritis. Int J Epidemiol, 44, 1706–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESCH KP, BENGEL D, HEILS A, SABOL SZ, GREENBERG BD, PETRI S, BENJAMIN J, MULLER CR, HAMER DH & MURPHY DL 1996. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science, 274, 1527–31. [DOI] [PubMed] [Google Scholar]
- LEVINE ME, CRIMMINS EM, PRESCOTT CA, PHILLIPS D, ARPAWONG TE & LEE J 2014. A polygenic risk score associated with measures of depressive symptoms among older adults. Biodemography Soc Biol, 60, 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI J, YOSHIKAWA A, BRENNAN MD, RAMSEY TL & MELTZER HY 2017. Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr Res. [DOI] [PubMed] [Google Scholar]
- LIBERT S, POINTER K, BELL EL, DAS A, COHEN DE, ASARA JM, KAPUR K, BERGMANN S, PREISIG M, OTOWA T, KENDLER KS, CHEN X, HETTEMA JM, VAN DEN OORD EJ, RUBIO JP & GUARENTE L 2011. SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell, 147, 1459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEBERS DT, PIROOZNIA M, SEIFFUDIN F, MUSLINER KL, ZANDI PP & GOES FS 2016. Polygenic Risk of Schizophrenia and Cognition in a Population-Based Survey of Older Adults. Schizophr Bull, 42, 984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU B, ZHANG X, CUI Y, QIN W, TAO Y, LI J, YU C & JIANG T 2017a. Polygenic Risk for Schizophrenia Influences Cortical Gyrification in 2 Independent General Populations. Schizophr Bull, 43, 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU M, MALONE SM, VAIDYANATHAN U, KELLER MC, ABECASIS G, MCGUE M, IACONO WG & VRIEZE SI 2017b. Psychophysiological endophenotypes to characterize mechanisms of known schizophrenia genetic loci. Psychol Med, 47, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAJOR DEPRESSIVE DISORDER WORKING GROUP OF THE PSYCHIATRIC, G. C., RIPKE S, WRAY NR, LEWIS CM, HAMILTON SP, WEISSMAN MM, BREEN G, BYRNE EM, BLACKWOOD DH, BOOMSMA DI, CICHON S, HEATH AC, HOLSBOER F, LUCAE S, MADDEN PA, MARTIN NG, MCGUFFIN P, MUGLIA P, NOETHEN MM, PENNINX BP, PERGADIA ML, POTASH JB, RIETSCHEL M, LIN D, MULLER-MYHSOK B, SHI J, STEINBERG S, GRABE HJ, LICHTENSTEIN P, MAGNUSSON P, PERLIS RH, PREISIG M, SMOLLER JW, STEFANSSON K, UHER R, KUTALIK Z, TANSEY KE, TEUMER A, VIKTORIN A, BARNES MR, BETTECKEN T, BINDER EB, BREUER R, CASTRO VM, CHURCHILL SE, CORYELL WH, CRADDOCK N, CRAIG IW, CZAMARA D, DE GEUS EJ, DEGENHARDT F, FARMER AE, FAVA M, FRANK J, GAINER VS, GALLAGHER PJ, GORDON SD, GORYACHEV S, GROSS M, GUIPPONI M, HENDERS AK, HERMS S, HICKIE IB, HOEFELS S, HOOGENDIJK W, HOTTENGA JJ, IOSIFESCU DV, ISING M, JONES I, JONES L, JUNG-YING T, KNOWLES JA, KOHANE IS, KOHLI MA, KORSZUN A, LANDEN M, LAWSON WB, LEWIS G, MACINTYRE D, MAIER W, MATTHEISEN M, MCGRATH PJ, MCINTOSH A, MCLEAN A, MIDDELDORP CM, MIDDLETON L, MONTGOMERY GM, MURPHY SN, NAUCK M, NOLEN WA, NYHOLT DR, O’DONOVAN M, OSKARSSON H, PEDERSEN N, SCHEFTNER WA, SCHULZ A, SCHULZE TG, SHYN SI, SIGURDSSON E, SLAGER SL, et al. 2013. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry, 18, 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAK TS, KWAN JS, CAMPBELL DD & SHAM PC 2016. Local True Discovery Rate Weighted Polygenic Scores Using GWAS Summary Data. Behav Genet, 46, 573–82. [DOI] [PubMed] [Google Scholar]
- MAK TSH, PORSCH RM, CHOI SW, ZHOU X & SHAM PC 2017. Polygenic scores via penalized regression on summary statistics. Genet Epidemiol, 41, 469–480. [DOI] [PubMed] [Google Scholar]
- MALASPINA D, HARLAP S, FENNIG S, HEIMAN D, NAHON D, FELDMAN D & SUSSER ES 2001. Advancing paternal age and the risk of schizophrenia. Arch Gen Psychiatry, 58, 361–7. [DOI] [PubMed] [Google Scholar]
- MARANVILLE JC & COX NJ 2016. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J, 16, 388–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARQUEZ-LUNA C & PRICE AL 2016. Multi-ethnic polygenic risk scores improve risk prediction in diverse populations. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN J, TILLING K, HUBBARD L, STERGIAKOULI E, THAPAR A, DAVEY SMITH G, O’DONOVAN MC & ZAMMIT S 2016. Association of Genetic Risk for Schizophrenia With Nonparticipation Over Time in a Population-Based Cohort Study. Am J Epidemiol, 183, 1149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLAUGHLIN RL, SCHIJVEN D, VAN RHEENEN W, VAN EIJK KR, O’BRIEN M, KAHN RS, OPHOFF RA, GORIS A, BRADLEY DG, AL-CHALABI A, VAN DEN BERG LH, LUYKX JJ, HARDIMAN O, VELDINK JH, PROJECT MIN, E. G. C. & SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C. 2017. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat Commun, 8, 14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEIER SM, AGERBO E, MAIER R, PEDERSEN CB, LANG M, GROVE J, HOLLEGAARD MV, DEMONTIS D, TRABJERG BB, HJORTHOJ C, RIPKE S, DEGENHARDT F, NOTHEN MM, RUJESCU D, MAIER W, MOO DSSCZC, WERGE T, MORS O, HOUGAARD DM, BORGLUM AD, WRAY NR, RIETSCHEL M, NORDENTOFT M, MORTENSEN PB & MATTHEISEN M 2016. High loading of polygenic risk in cases with chronic schizophrenia. Mol Psychiatry, 21, 969–74. [DOI] [PubMed] [Google Scholar]
- MENKE A, ARLOTH J, PUTZ B, WEBER P, KLENGEL T, MEHTA D, GONIK M, REX-HAFFNER M, RUBEL J, UHR M, LUCAE S, DEUSSING JM, MULLER-MYHSOK B, HOLSBOER F & BINDER EB 2012. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology, 37, 1455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERIKANGAS KR & RISCH N 2003. Genomic priorities and public health. Science, 302, 599–601. [DOI] [PubMed] [Google Scholar]
- MEYERS JL, ZHANG J, WANG JC, SU J, KUO SI, KAPOOR M, WETHERILL L, BERTELSEN S, LAI D, SALVATORE JE, KAMARAJAN C, CHORLIAN D, AGRAWAL A, ALMASY L, BAUER L, BUCHOLZ KK, CHAN G, HESSELBROCK V, KOGANTI L, KRAMER J, KUPERMAN S, MANZ N, PANDEY A, SEAY M, SCOTT D, TAYLOR RE, DICK DM, EDENBERG HJ, GOATE A, FOROUD T & PORJESZ B 2017. An endophenotype approach to the genetics of alcohol dependence: a genome wide association study of fast beta EEG in families of African ancestry. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITRA I, LAVILLAUREIX A, YEH E, TRAGLIA M, TSANG K, BEARDEN CE, RAUEN KA & WEISS LA 2017. Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders. PLoS Genet, 13, e1006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER N, WEIDINGER E, LEITNER B & SCHWARZ MJ 2015. The role of inflammation in schizophrenia. Front Neurosci, 9, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLINS N, INGASON A, PORTER H, EUESDEN J, GILLETT A, OLAFSSON S, GUDBJARTSSON DF, LEWIS CM, SIGURDSSON E, SAEMUNDSEN E, GUDMUNDSSON OO, FRIGGE ML, KONG A, HELGASON A, WALTERS GB, GUSTAFSSON O, STEFANSSON H & STEFANSSON K 2017. Reproductive fitness and genetic risk of psychiatric disorders in the general population. Nat Commun, 8, 15833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLINS N, POWER RA, FISHER HL, HANSCOMBE KB, EUESDEN J, INIESTA R, LEVINSON DF, WEISSMAN MM, POTASH JB, SHI J, UHER R, COHEN-WOODS S, RIVERA M, JONES L, JONES I, CRADDOCK N, OWEN MJ, KORSZUN A, CRAIG IW, FARMER AE, MCGUFFIN P, BREEN G & LEWIS CM 2016. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder. Psychol Med, 46, 759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUSLINER KL, SEIFUDDIN F, JUDY JA, PIROOZNIA M, GOES FS & ZANDI PP 2015. Polygenic risk, stressful life events and depressive symptoms in older adults: a polygenic score analysis. Psychol Med, 45, 1709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEILSON E, BOIS C, GIBSON J, DUFF B, WATSON A, ROBERTS N, BRANDON NJ, DUNLOP J, HALL J, MCINTOSH AM, WHALLEY HC & LAWRIE SM 2017. Effects of environmental risks and polygenic loading for schizophrenia on cortical thickness. Schizophr Res, 184, 128–136. [DOI] [PubMed] [Google Scholar]
- NELSON EC, AGRAWAL A, HEATH AC, BOGDAN R, SHERVA R, ZHANG B, AL-HASANI R, BRUCHAS MR, CHOU YL, DEMERS CH, CAREY CE, CONLEY ED, FAKIRA AK, FARRER LA, GOATE A, GORDON S, HENDERS AK, HESSELBROCK V, KAPOOR M, LYNSKEY MT, MADDEN PA, MORON JA, RICE JP, SACCONE NL, SCHWAB SG, SHAND FL, TODOROV AA, WALLACE L, WANG T, WRAY NR, ZHOU X, DEGENHARDT L, MARTIN NG, HARIRI AR, KRANZLER HR, GELERNTER J, BIERUT LJ, CLARK DJ & MONTGOMERY GW 2016. Evidence of CNIH3 involvement in opioid dependence. Mol Psychiatry, 21, 608–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON MR, TIPNEY H, PAINTER JL, SHEN J, NICOLETTI P, SHEN Y, FLORATOS A, SHAM PC, LI MJ, WANG J, CARDON LR, WHITTAKER JC & SANSEAU P 2015. The support of human genetic evidence for approved drug indications. Nat Genet, 47, 856–60. [DOI] [PubMed] [Google Scholar]
- NIVARD MG, GAGE SH, HOTTENGA JJ, VAN BEIJSTERVELDT CE, ABDELLAOUI A, BARTELS M, BASELMANS BM, LIGTHART L, POURCAIN BS, BOOMSMA DI, MUNAFO MR & MIDDELDORP CM 2017. Genetic Overlap Between Schizophrenia and Developmental Psychopathology: Longitudinal and Multivariate Polygenic Risk Prediction of Common Psychiatric Traits During Development. Schizophr Bull. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKBAY A, BEAUCHAMP JP, FONTANA MA, LEE JJ, PERS TH, RIETVELD CA, TURLEY P, CHEN GB, EMILSSON V, MEDDENS SF, OSKARSSON S, PICKRELL JK, THOM K, TIMSHEL P, DE VLAMING R, ABDELLAOUI A, AHLUWALIA TS, BACELIS J, BAUMBACH C, BJORNSDOTTIR G, BRANDSMA JH, PINA CONCAS M, DERRINGER J, FURLOTTE NA, GALESLOOT TE, GIROTTO G, GUPTA R, HALL LM, HARRIS SE, HOFER E, HORIKOSHI M, HUFFMAN JE, KAASIK K, KALAFATI IP, KARLSSON R, KONG A, LAHTI J, VAN DER LEE SJ, DELEEUW C, LIND PA, LINDGREN KO, LIU T, MANGINO M, MARTEN J, MIHAILOV E, MILLER MB, VAN DER MOST PJ, OLDMEADOW C, PAYTON A, PERVJAKOVA N, PEYROT WJ, QIAN Y, RAITAKARI O, RUEEDI R, SALVI E, SCHMIDT B, SCHRAUT KE, SHI J, SMITH AV, POOT RA, ST POURCAIN B, TEUMER A, THORLEIFSSON G, VERWEIJ N, VUCKOVIC D, WELLMANN J, WESTRA HJ, YANG J, ZHAO W, ZHU Z, ALIZADEH BZ, AMIN N, BAKSHI A, BAUMEISTER SE, BIINO G, BONNELYKKE K, BOYLE PA, CAMPBELL H, CAPPUCCIO FP, DAVIES G, DE NEVE JE, DELOUKAS P, DEMUTH I, DING J, EIBICH P, EISELE L, EKLUND N, EVANS DM, FAUL JD, FEITOSA MF, FORSTNER AJ, GANDIN I, GUNNARSSON B, HALLDORSSON BV, HARRIS TB, HEATH AC, HOCKING LJ, HOLLIDAY EG, HOMUTH G, HORAN MA, et al. 2016. Genome-wide association study identifies 74 loci associated with educational attainment. Nature, 533, 539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN JL, NANDA P, TANDON N, MOTHI SS, BOLO N, MCCARROLL S, CLEMENTZ BA, GERSHON ES, PEARLSON GD, SWEENEY JA, TAMMINGA CA & KESHAVAN MS 2016. Polygenic risk for type 2 diabetes mellitus among individuals with psychosis and their relatives. J Psychiatr Res, 77, 52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAKSARIAN D, TRABJERG BB, MERIKANGAS KR, MORS O, BORGLUM AD, HOUGAARD DM, MCGRATH JJ, PEDERSEN CB, MORTENSEN PB & AGERBO E 2017. The role of genetic liability in the association of urbanicity at birth and during upbringing with schizophrenia in Denmark. Psychol Med, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPIOL S, POPOVIC D, KEESER D, HASAN A, SCHNEIDER-AXMANN T, DEGENHARDT F, ROSSNER MJ, BICKEBOLLER H, SCHMITT A, FALKAI P & MALCHOW B 2017. Polygenic risk has an impact on the structural plasticity of hippocampal subfields during aerobic exercise combined with cognitive remediation in multi-episode schizophrenia. Transl Psychiatry, 7, e1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARE G, MAO S & DENG WQ 2017. A machine-learning heuristic to improve gene score prediction of polygenic traits Short title: Machine-learning boosted gene scores. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERLIS RH, FIJAL B, DHARIA S, HEINLOTH AN & HOUSTON JP 2010. Failure to replicate genetic associations with antidepressant treatment response in duloxetine-treated patients. Biol Psychiatry, 67, 1110–3. [DOI] [PubMed] [Google Scholar]
- PEYROT WJ, MILANESCHI Y, ABDELLAOUI A, SULLIVAN PF, HOTTENGA JJ, BOOMSMA DI & PENNINX BW 2014. Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry, 205, 113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLENGE RM, SCOLNICK EM & ALTSHULER D 2013. Validating therapeutic targets through human genetics. Nat Rev Drug Discov, 12, 581–94. [DOI] [PubMed] [Google Scholar]
- PLOMIN R, DEFRIES JC, KNOPIK VS & NEIDERHISER JM 2013. Behavioral genetics : a primer, New York, Worth Publishers. [Google Scholar]
- POLDERMAN TJ, BENYAMIN B, DE LEEUW CA, SULLIVAN PF, VAN BOCHOVEN A, VISSCHER PM & POSTHUMA D 2015. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet, 47, 702–9. [DOI] [PubMed] [Google Scholar]
- POLIMANTI R, KAUFMAN J, ZHAO H, KRANZLER HR, URSANO RJ, KESSLER RC, GELERNTER J & STEIN MB 2017. A genome-wide gene-by-trauma interaction study of alcohol misuse in two independent cohorts identifies PRKG1 as a risk locus. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POSTHUMA D & POLDERMAN TJ 2013. What have we learned from recent twin studies about the etiology of neurodevelopmental disorders? Curr Opin Neurol, 26, 111–21. [DOI] [PubMed] [Google Scholar]
- POWER RA, STEINBERG S, BJORNSDOTTIR G, RIETVELD CA, ABDELLAOUI A, NIVARD MM, JOHANNESSON M, GALESLOOT TE, HOTTENGA JJ, WILLEMSEN G, CESARINI D, BENJAMIN DJ, MAGNUSSON PK, ULLEN F, TIEMEIER H, HOFMAN A, VAN ROOIJ FJ, WALTERS GB, SIGURDSSON E, THORGEIRSSON TE, INGASON A, HELGASON A, KONG A, KIEMENEY LA, KOELLINGER P, BOOMSMA DI, GUDBJARTSSON D, STEFANSSON H & STEFANSSON K 2015. Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat Neurosci, 18, 953–5. [DOI] [PubMed] [Google Scholar]
- POWER RA, VERWEIJ KJ, ZUHAIR M, MONTGOMERY GW, HENDERS AK, HEATH AC, MADDEN PA, MEDLAND SE, WRAY NR & MARTIN NG 2014. Genetic predisposition to schizophrenia associated with increased use of cannabis. Mol Psychiatry, 19, 1201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PURCELL S, NEALE B, TODD-BROWN K, THOMAS L, FERREIRA MA, BENDER D, MALLER J, SKLAR P, DE BAKKER PI, DALY MJ & SHAM PC 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 81, 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMACHANDRAIH CT, SUBRAMANYAM N, BAR KJ, BAKER G & YERAGANI VK 2011. Antidepressants: From MAOIs to SSRIs and more. Indian J Psychiatry, 53, 180–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANLUND S, CALAFATO S, THYGESEN JH, LIN K, CAHN W, CRESPO-FACORRO B, DE ZWARTE SMC, DIEZ A, DI FORTI M, GROUP, IYEGBE C, JABLENSKY A, JONES R, HALL MH, KAHN R, KALAYDJIEVA L, KRAVARITI E, MCDONALD C, MCINTOSH AM, MCQUILLIN A, PEIC, PICCHIONI M, PRATA DP, RUJESCU D, SCHULZE K, SHAIKH M, TOULOPOULOU T, VAN HAREN N, VAN OS J, VASSOS E, WALSHE M, WTCCC, LEWIS C, MURRAY RM, POWELL J & BRAMON E 2017. A polygenic risk score analysis of psychosis endophenotypes across brain functional, structural, and cognitive domains. Am J Med Genet B Neuropsychiatr Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REGINSSON GW, INGASON A, EUESDEN J, BJORNSDOTTIR G, OLAFSSON S, SIGURDSSON E, OSKARSSON H, TYRFINGSSON T, RUNARSDOTTIR V, HANSDOTTIR I, STEINBERG S, STEFANSSON H, GUDBJARTSSON DF, THORGEIRSSON TE & STEFANSSON K 2017. Polygenic risk scores for schizophrenia and bipolar disorder associate with addiction. Addict Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REUS LM, SHEN X, GIBSON J, WIGMORE E, LIGTHART L, ADAMS MJ, DAVIES G, COX SR, HAGENAARS SP, BASTIN ME, DEARY IJ, WHALLEY HC & MCINTOSH AM 2017. Association of polygenic risk for major psychiatric illness with subcortical volumes and white matter integrity in UK Biobank. Sci Rep, 7, 42140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIETVELD CA, MEDLAND SE, DERRINGER J, YANG J, ESKO T, MARTIN NW, WESTRA HJ, SHAKHBAZOV K, ABDELLAOUI A, AGRAWAL A, ALBRECHT E, ALIZADEH BZ, AMIN N, BARNARD J, BAUMEISTER SE, BENKE KS, BIELAK LF, BOATMAN JA, BOYLE PA, DAVIES G, DE LEEUW C, EKLUND N, EVANS DS, FERHMANN R, FISCHER K, GIEGER C, GJESSING HK, HAGG S, HARRIS JR, HAYWARD C, HOLZAPFEL C, IBRAHIM-VERBAAS CA, INGELSSON E, JACOBSSON B, JOSHI PK, JUGESSUR A, KAAKINEN M, KANONI S, KARJALAINEN J, KOLCIC I, KRISTIANSSON K, KUTALIK Z, LAHTI J, LEE SH, LIN P, LIND PA, LIU Y, LOHMAN K, LOITFELDER M, MCMAHON G, VIDAL PM, MEIRELLES O, MILANI L, MYHRE R, NUOTIO ML, OLDMEADOW CJ, PETROVIC KE, PEYROT WJ, POLASEK O, QUAYE L, REINMAA E, RICE JP, RIZZI TS, SCHMIDT H, SCHMIDT R, SMITH AV, SMITH JA, TANAKA T, TERRACCIANO A, VAN DER LOOS MJ, VITART V, VOLZKE H, WELLMANN J, YU L, ZHAO W, ALLIK J, ATTIA JR, BANDINELLI S, BASTARDOT F, BEAUCHAMP J, BENNETT DA, BERGER K, BIERUT LJ, BOOMSMA DI, BULTMANN U, CAMPBELL H, CHABRIS CF, CHERKAS L, CHUNG MK, CUCCA F, DE ANDRADE M, DE JAGER PL, DE NEVE JE, DEARY IJ, DEDOUSSIS GV, DELOUKAS P, DIMITRIOU M, EIRIKSDOTTIR G, ELDERSON MF, ERIKSSON JG, et al. 2013. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science, 340, 1467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIGLIN L, COLLISHAW S, RICHARDS A, THAPAR AK, MAUGHAN B, O’DONOVAN MC & THAPAR A 2017. Schizophrenia risk alleles and neurodevelopmental outcomes in childhood: a population-based cohort study. Lancet Psychiatry, 4, 57–62. [DOI] [PubMed] [Google Scholar]
- RIPKE S, O’DUSHLAINE C, CHAMBERT K, MORAN JL, KAHLER AK, AKTERIN S, BERGEN SE, COLLINS AL, CROWLEY JJ, FROMER M, KIM Y, LEE SH, MAGNUSSON PK, SANCHEZ N, STAHL EA, WILLIAMS S, WRAY NR, XIA K, BETTELLA F, BORGLUM AD, BULIK-SULLIVAN BK, CORMICAN P, CRADDOCK N, DE LEEUW C, DURMISHI N, GILL M, GOLIMBET V, HAMSHERE ML, HOLMANS P, HOUGAARD DM, KENDLER KS, LIN K, MORRIS DW, MORS O, MORTENSEN PB, NEALE BM, O’NEILL FA, OWEN MJ, MILOVANCEVIC MP, POSTHUMA D, POWELL J, RICHARDS AL, RILEY BP, RUDERFER D, RUJESCU D, SIGURDSSON E, SILAGADZE T, SMIT AB, STEFANSSON H, STEINBERG S, SUVISAARI J, TOSATO S, VERHAGE M, WALTERS JT, MULTICENTER GENETIC STUDIES OF SCHIZOPHRENIA, C., LEVINSON DF, GEJMAN PV, KENDLER KS, LAURENT C, MOWRY BJ, O’DONOVAN MC, OWEN MJ, PULVER AE, RILEY BP, SCHWAB SG, WILDENAUER DB, DUDBRIDGE F, HOLMANS P, SHI J, ALBUS M, ALEXANDER M, CAMPION D, COHEN D, DIKEOS D, DUAN J, EICHHAMMER P, GODARD S, HANSEN M, LERER FB, LIANG KY, MAIER W, MALLET J, NERTNEY DA, NESTADT G, NORTON N, O’NEILL FA, PAPADIMITRIOU GN, RIBBLE R, SANDERS AR, SILVERMAN JM, WALSH D, WILLIAMS NM, WORMLEY B, PSYCHOSIS ENDOPHENOTYPES INTERNATIONAL C, ARRANZ MJ, BAKKER S, BENDER S, BRAMON E, COLLIER D, CRESPO-FACORRO B, et al. 2013. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet, 45, 1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON EB, HOWRIGAN D, YANG J, RIPKE S, ANTTILA V, DUNCAN LE, JOSTINS L, BARRETT JC, MEDLAND SE, MACARTHUR DG, BREEN G, O’DONOVAN MC, WRAY NR, DEVLIN B, DALY MJ, VISSCHER PM, SULLIVAN PF & NEALE BM 2014. Response to ‘Predicting the diagnosis of autism spectrum disorder using gene pathway analysis’. Mol Psychiatry, 19, 859–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROUSSOS P, GIAKOUMAKI SG, ZOURARAKI C, FULLARD JF, KARAGIORGA VE, TSAPAKIS EM, PETRAKI Z, SIEVER LJ, LENCZ T, MALHOTRA A, SPANAKI C & BITSIOS P 2016. The Relationship of Common Risk Variants and Polygenic Risk for Schizophrenia to Sensorimotor Gating. Biol Psychiatry, 79, 988–96. [DOI] [PubMed] [Google Scholar]
- RUDERFER DM, CHARNEY AW, READHEAD B, KIDD BA, KAHLER AK, KENNY PJ, KEISER MJ, MORAN JL, HULTMAN CM, SCOTT SA, SULLIVAN PF, PURCELL SM, DUDLEY JT & SKLAR P 2016. Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry, 3, 350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUDIN E 1916. Studien uber Vererbung und entstehung geistiger Storungen, I: Zur vererbung und neuentstehung der Dementia praecox (Studies on the Inheritance and Origin of Mental Illness, I: The Problem of the Inheritance and Primary Origin of Dementia Praecox). Monographien aus dem Gesamtgebiet der Neurologie und Psychiatrie, 12. [DOI] [PubMed] [Google Scholar]
- SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C. 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHULZE TG, FANGERAU H & PROPPING P 2004. From degeneration to genetic susceptibility, from eugenics to genethics, from Bezugsziffer to LOD score: the history of psychiatric genetics. Int Rev Psychiatry, 16, 246–59. [DOI] [PubMed] [Google Scholar]
- SEKAR A, BIALAS AR, DE RIVERA H, DAVIS A, HAMMOND TR, KAMITAKI N, TOOLEY K, PRESUMEY J, BAUM M, VAN DOREN V, GENOVESE G, ROSE SA, HANDSAKER RE, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS, C., DALY MJ, CARROLL MC, STEVENS B & MCCARROLL SA 2016. Schizophrenia risk from complex variation of complement component 4. Nature, 530, 177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENGUPTA SM, MACDONALD K, FATHALLI F, YIM A, LEPAGE M, IYER S, MALLA A & JOOBER R 2017. Polygenic Risk Score associated with specific symptom dimensions in first-episode psychosis. Schizophr Res, 184, 116–121. [DOI] [PubMed] [Google Scholar]
- SHI J, PARK JH, DUAN J, BERNDT ST, MOY W, YU K, SONG L, WHEELER W, HUA X, SILVERMAN D, GARCIA-CLOSAS M, HSIUNG CA, FIGUEROA JD, CORTESSIS VK, MALATS N, KARAGAS MR, VINEIS P, CHANG IS, LIN D, ZHOU B, SEOW A, MATSUO K, HONG YC, CAPORASO NE, WOLPIN B, JACOBS E, PETERSEN GM, KLEIN AP, LI D, RISCH H, SANDERS AR, HSU L, SCHOEN RE, BRENNER H, CONSORTIUM MG, GECCO, CONSORTIUM G-OTG, CONSORTIUM P, PANSCAN C, CONSORTIUM G-OE, STOLZENBERG-SOLOMON R, GEJMAN P, LAN Q, ROTHMAN N, AMUNDADOTTIR LT, LANDI MT, LEVINSON DF, CHANOCK SJ & CHATTERJEE N 2016. Winner’s Curse Correction and Variable Thresholding Improve Performance of Polygenic Risk Modeling Based on Genome-Wide Association Study Summary-Level Data. PLoS Genet, 12, e1006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SKAFIDAS E, TESTA R, ZANTOMIO D, CHANA G, EVERALL IP & PANTELIS C 2014. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol Psychiatry, 19, 504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SO HC & SHAM PC 2017. Exploring the predictive power of polygenic scores derived from genome-wide association studies: a study of 10 complex traits. Bioinformatics, 33, 886–892. [DOI] [PubMed] [Google Scholar]
- SOKOLOWSKI M, WASSERMAN J & WASSERMAN D 2016. Polygenic associations of neurodevelopmental genes in suicide attempt. Mol Psychiatry, 21, 1381–90. [DOI] [PubMed] [Google Scholar]
- ST POURCAIN B, ROBINSON EB, ANTTILA V, SULLIVAN BB, MALLER J, GOLDING J, SKUSE D, RING S, EVANS DM, ZAMMIT S, FISHER SE, NEALE BM, ANNEY RJ, RIPKE S, HOLLEGAARD MV, WERGE T, I P-SSIBAG, RONALD A, GROVE J, HOUGAARD DM, BORGLUM AD, MORTENSEN PB, DALY MJ & DAVEY SMITH G 2017. ASD and schizophrenia show distinct developmental profiles in common genetic overlap with population-based social communication difficulties. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEIN MB, CHEN CY, URSANO RJ, CAI T, GELERNTER J, HEERINGA SG, JAIN S, JENSEN KP, MAIHOFER AX, MITCHELL C, NIEVERGELT CM, NOCK MK, NEALE BM, POLIMANTI R, RIPKE S, SUN X, THOMAS ML, WANG Q, WARE EB, BORJA S, KESSLER RC, SMOLLER JW, ARMY STUDY TO ASSESS, R. & RESILIENCE IN SERVICEMEMBERS, C. 2016. Genome-wide Association Studies of Posttraumatic Stress Disorder in 2 Cohorts of US Army Soldiers. JAMA Psychiatry, 73, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPNIAK B, PAPIOL S, HAMMER C, RAMIN A, EVERTS S, HENNIG L, BEGEMANN M & EHRENREICH H 2014. Accumulated environmental risk determining age at schizophrenia onset: a deep phenotyping-based study. The Lancet Psychiatry, 1, 444–453. [DOI] [PubMed] [Google Scholar]
- STRINGER S, KAHN RS, DE WITTE LD, OPHOFF RA & DERKS EM 2014. Genetic liability for schizophrenia predicts risk of immune disorders. Schizophr Res, 159, 347–52. [DOI] [PubMed] [Google Scholar]
- SULLIVAN PF 2015. Genetics of disease: Associations with depression. Nature, 523, 539–40. [DOI] [PubMed] [Google Scholar]
- SULLIVAN PF, AGRAWAL A, BULIK C, ANDREASSEN OA, BORGLUM A, BREEN G, CICHON S, EDENBERG H, FARAONE SV, GELERNTER J, MATHEWS CA, NIEVERGELT CM, SMOLLER J & O’DONOVAN M 2017. Psychiatric Genomics: An Update and an Agenda. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUMNER JA, DUNCAN L, RATANATHARATHORN A, ROBERTS AL & KOENEN KC 2016. PTSD has shared polygenic contributions with bipolar disorder and schizophrenia in women. Psychol Med, 46, 669–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUTTERLAND AL, FOND G, KUIN A, KOETER MW, LUTTER R, VAN GOOL T, YOLKEN R, SZOKE A, LEBOYER M & DE HAAN L 2015. Beyond the association. Toxoplasma gondii in schizophrenia, bipolar disorder, and addiction: systematic review and meta-analysis. Acta Psychiatr Scand, 132, 161–79. [DOI] [PubMed] [Google Scholar]
- THOMASSON HR, EDENBERG HJ, CRABB DW, MAI XL, JEROME RE, LI TK, WANG SP, LIN YT, LU RB & YIN SJ 1991. Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. Am J Hum Genet, 48, 677–81. [PMC free article] [PubMed] [Google Scholar]
- TOBACCO & GENETICS, C. 2010. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet, 42, 441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORREY EF & YOLKEN RH 2003. Toxoplasma gondii and schizophrenia. Emerg Infect Dis, 9, 1375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOST H, BRAUS DF, HAKIMI S, RUF M, VOLLMERT C, HOHN F & MEYER-LINDENBERG A 2010. Acute D2 receptor blockade induces rapid, reversible remodeling in human cortical-striatal circuits. Nat Neurosci, 13, 920–2. [DOI] [PubMed] [Google Scholar]
- TREADWAY MT & ZALD DH 2011. Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci Biobehav Rev, 35, 537–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TROTTA A, IYEGBE C, DI FORTI M, SHAM PC, CAMPBELL DD, CHERNY SS, MONDELLI V, AITCHISON KJ, MURRAY RM, VASSOS E & FISHER HL 2016. Interplay between Schizophrenia Polygenic Risk Score and Childhood Adversity in First-Presentation Psychotic Disorder: A Pilot Study. PLoS One, 11, e0163319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURLEY P, WALTERS RK, MAGHZIAN O, OKBAY A, LEE JJ, FONTANA MA, NGUYEN-VIET TA, FURLOTTE NA, MAGNUSSON P, OSKARSSON S, JOHANNESSON M, VISSCHER PM, LAIBSON D, CESARINI D, NEALE B & BENJAMIN DJ 2017. MTAG: Multi-Trait Analysis of GWAS. bioRxiv. [Google Scholar]
- VAN DER AUWERA S, TEUMER A, HERTEL J, HOMUTH G, VOLKER U, LUCHT MJ, DEGENHARDT F, SCHULZE T, RIETSCHEL M, NOTHEN MM, JOHN U, NAUCK M & GRABE HJ 2016. The inverse link between genetic risk for schizophrenia and migraine through NMDA (N-methyl-D-aspartate) receptor activation via D-serine. Eur Neuropsychopharmacol, 26, 1507–15. [DOI] [PubMed] [Google Scholar]
- VAN DER AUWERA S, WITTFELD K, HOMUTH G, TEUMER A, HEGENSCHEID K & GRABE HJ 2015. No association between polygenic risk for schizophrenia and brain volume in the general population. Biol Psychiatry, 78, e41–2. [DOI] [PubMed] [Google Scholar]
- VAN DER AUWERA S, WITTFELD K, SHUMSKAYA E, BRALTEN J, ZWIERS MP, ONNINK AM, USBERTI N, HERTEL J, VOLZKE H, VOLKER U, HOSTEN N, FRANKE B & GRABE HJ 2017. Predicting brain structure in population-based samples with biologically informed genetic scores for schizophrenia. Am J Med Genet B Neuropsychiatr Genet, 174, 324–332. [DOI] [PubMed] [Google Scholar]
- VAN DONGEN J & BOOMSMA DI 2013. The evolutionary paradox and the missing heritability of schizophrenia. Am J Med Genet B Neuropsychiatr Genet, 162B, 122–36. [DOI] [PubMed] [Google Scholar]
- VAN OS J, VAN DER STEEN Y, ISLAM MA, GULOKSUZ S, RUTTEN BP, SIMONS CJ & INVESTIGATORS G 2017. Evidence that polygenic risk for psychotic disorder is expressed in the domain of neurodevelopment, emotion regulation and attribution of salience. Psychol Med, 1–17. [DOI] [PubMed] [Google Scholar]
- VASSOS E, DI FORTI M, COLEMAN J, IYEGBE C, PRATA D, EUESDEN J, O’REILLY P, CURTIS C, KOLLIAKOU A, PATEL H, NEWHOUSE S, TRAYLOR M, AJNAKINA O, MONDELLI V, MARQUES TR, GARDNER-SOOD P, AITCHISON KJ, POWELL J, ATAKAN Z, GREENWOOD KE, SMITH S, ISMAIL K, PARIANTE C, GAUGHRAN F, DAZZAN P, MARKUS HS, DAVID AS, LEWIS CM, MURRAY RM & BREEN G 2017. An Examination of Polygenic Score Risk Prediction in Individuals With First-Episode Psychosis. Biol Psychiatry, 81, 470–477. [DOI] [PubMed] [Google Scholar]
- VERWEIJ KJ, ABDELLAOUI A, NIVARD MG, SAINZ CORT A, LIGTHART L, DRAISMA HH, MINICA CC & INTERNATIONAL CANNABIS C 2017. Short communication: Genetic association between schizophrenia and cannabis use. Drug Alcohol Depend, 171, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIANA J, HANNON E, DEMPSTER E, PIDSLEY R, MACDONALD R, KNOX O, SPIERS H, TROAKES C, AL-SARAJ S, TURECKI G, SCHALKWYK LC & MILL J 2017. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum Mol Genet, 26, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILHJALMSSON BJ, YANG J, FINUCANE HK, GUSEV A, LINDSTROM S, RIPKE S, GENOVESE G, LOH PR, BHATIA G, DO R, HAYECK T, WON HH, SCHIZOPHRENIA WORKING GROUP OF THE PSYCHIATRIC GENOMICS CONSORTIUM, D. B., RISK OF INHERITED VARIANTS IN BREAST CANCER, S., KATHIRESAN S, PATO M, PATO C, TAMIMI R, STAHL E, ZAITLEN N, PASANIUC B, BELBIN G, KENNY EE, SCHIERUP MH, DE JAGER P, PATSOPOULOS NA, MCCARROLL S, DALY M, PURCELL S, CHASMAN D, NEALE B, GODDARD M, VISSCHER PM, KRAFT P, PATTERSON N & PRICE AL 2015. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genet, 97, 576–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VISSCHER PM, WRAY NR, ZHANG Q, SKLAR P, MCCARTHY MI, BROWN MA & YANG J 2017. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet, 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VORSTMAN JAS, PARR JR, MORENO-DE-LUCA D, ANNEY RJL, NURNBERGER JI JR. & HALLMAYER JF 2017. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet, 18, 362–376. [DOI] [PubMed] [Google Scholar]
- WANG H, YANG H, SHIVALILA CS, DAWLATY MM, CHENG AW, ZHANG F & JAENISCH R 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell, 153, 910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Q, POLIMANTI R, KRANZLER HR, FARRER LA, ZHAO H & GELERNTER J 2017a. Genetic factor common to schizophrenia and HIV infection is associated with risky sexual behavior: antagonistic vs. synergistic pleiotropic SNPs enriched for distinctly different biological functions. Hum Genet, 136, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG SH, HSIAO PC, YEH LL, LIU CM, LIU CC, HWANG TJ, HSIEH MH, CHIEN YL, LIN YT, CHANDLER SD, FARAONE SV, LAIRD N, NEALE B, MCCARROLL SA, GLATT SJ, TSUANG MT, HWU HG & CHEN WJ 2017b. Polygenic risk for schizophrenia and neurocognitive performance in patients with schizophrenia. Genes Brain Behav. [DOI] [PubMed] [Google Scholar]
- WENDLAND JR & EHLERS MD 2016. Translating Neurogenomics Into New Medicines. Biol Psychiatry, 79, 650–6. [DOI] [PubMed] [Google Scholar]
- WHALLEY HC, ADAMS MJ, HALL LS, CLARKE TK, FERNANDEZ-PUJALS AM, GIBSON J, WIGMORE E, HAFFERTY J, HAGENAARS SP, DAVIES G, CAMPBELL A, HAYWARD C, LAWRIE SM, PORTEOUS DJ, DEARY IJ & MCINTOSH AM 2016. Dissection of major depressive disorder using polygenic risk scores for schizophrenia in two independent cohorts. Transl Psychiatry, 6, e938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHEELER HE, MAITLAND ML, DOLAN ME, COX NJ & RATAIN MJ 2013. Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet, 14, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLER CJ & MOHLKE KL 2012. Finding genes and variants for lipid levels after genome-wide association analysis. Curr Opin Lipidol, 23, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIMBERLEY T, GASSE C, MEIER SM, AGERBO E, MACCABE JH & HORSDAL HT 2017a. Polygenic Risk Score for Schizophrenia and Treatment-Resistant Schizophrenia. Schizophr Bull. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIMBERLEY T, GASSE C, MEIER SM, AGERBO E, MACCABE JH & HORSDAL HT 2017b. Polygenic Risk Score for Schizophrenia and Treatment-Resistant Schizophrenia. Schizophr Bull, 43, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WRAY NR, LEE SH, MEHTA D, VINKHUYZEN AA, DUDBRIDGE F & MIDDELDORP CM 2014. Research review: Polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry, 55, 1068–87. [DOI] [PubMed] [Google Scholar]
- WRAY NR & SULLIVAN PF 2017. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU J, PFEIFFER RM & GAIL MH 2013. Strategies for developing prediction models from genome-wide association studies. Genet Epidemiol, 37, 768–77. [DOI] [PubMed] [Google Scholar]
- XU K, KRANZLER HR, SHERVA R, SARTOR CE, ALMASY L, KOESTERER R, ZHAO H, FARRER LA & GELERNTER J 2015. Genomewide Association Study for Maximum Number of Alcoholic Drinks in European Americans and African Americans. Alcohol Clin Exp Res, 39, 1137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG J, LEE SH, GODDARD ME & VISSCHER PM 2011. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet, 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YIN X, WINEINGER NE, WANG K, YUE W, NORGREN N, WANG L, YAO W, JIANG X, WU B, CUI Y, SHEN C, CHENG H, ZHOU F, CHEN G, ZUO X, ZHENG X, FAN X, WANG H, WANG L, LEE J, LAM M, TAI ES, ZHANG Z, HUANG Q, SUN L, XU J, YANG S, WILHELMSEN KC, LIU J, SCHORK NJ & ZHANG X 2016. Common susceptibility variants are shared between schizophrenia and psoriasis in the Han Chinese population. J Psychiatry Neurosci, 41, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZANNAS AS, WIECHMANN T, GASSEN NC & BINDER EB 2016. Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology, 41, 261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG J, ERZURUMLUOGLU AM, ELSWORTH BL, KEMP JP, HOWE L, HAYCOCK PC, HEMANI G, TANSEY K, LAURIN C, EARLY G, LIFECOURSE EPIDEMIOLOGY ECZEMA C, POURCAIN BS, WARRINGTON NM, FINUCANE HK, PRICE AL, BULIK-SULLIVAN BK, ANTTILA V, PATERNOSTER L, GAUNT TR, EVANS DM & NEALE BM 2017. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics, 33, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU Z, ZHANG F, HU H, BAKSHI A, ROBINSON MR, POWELL JE, MONTGOMERY GW, GODDARD ME, WRAY NR, VISSCHER PM & YANG J 2016. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet, 48, 481–7. [DOI] [PubMed] [Google Scholar]