

Abstract
This review covers some of the recent progress in the field of peptide antibiotics with a focus on compounds with novel or established mode of action and with demonstrated efficacy in animal infection models. Novel drug discovery approaches, linear and macrocyclic peptide antibiotics, lipopeptides like the polymyxins as well as peptides addressing targets located in the plasma membrane or in the outer membrane of bacterial cells are discussed.
Keywords: Antibiotic, antimicrobial peptide, antimicrobial resistance, OMPTA (outer membrane targeting antibiotic), peptide antibiotic, lipopeptide
Abbreviations: ADMET, absorption, distribution, metabolism and excretion – toxicity in pharmacokinetics; AMP, antimicrobial peptide; AMR, antimicrobial resistance; ATCC, ATCC cell collection; BAM, β-barrel assembly machinery; bid, bis in die (two times a day); CC50, cytotoxic concentration to kill 50% of cells; CD, circular dichroism; CFU, colony forming unit; CLSI, clinical and laboratory standards institute; CMS, colistin methane sulfonate; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; ESKAPE, acronym encompassing six bacterial pathogens (often carrying antibiotic resistance): Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp; FDA, U. S. Food and Drug Administration; HABP, hospital acquired bacterial pneumonia; HDP, host-defense peptide; HEK293, human embryonic kidney 293 cells; HepG2, human hepatocellular carcinoma cell line; HK-2, human kidney 2 cells (proximal tubular cell line); Hpg, 4-hydroxy-phenyl glycine; i.p., intraperitoneal; i.v., intravenous; ITC, isothermal titration calorimetry; KPC, Klebsiella pneumoniae metallo-β-lactamase C resistant; LPS, lipopolysaccharide; LptA, lipopolysaccharide transport protein A; LptC, lipopolysaccharide transport protein C; LptD, lipopolysaccharide transport protein D; MDR, multidrug-resistant; MIC, minimal inhibitory concentration; mITT population, minimal intend-to-treat population; MH-I, Müller-Hinton broth I; MH-II, Müller-Hinton broth II (cation adjusted); MoA, mechanism (mode) of action; MRSA, methicilline-resistant S. aureus; MSSA, methicilline-sensitive S. aureus; NDM-1, New Delhi metallo-β-lactamase resistant; NOAEL, no adverse effect level; ODL, odilorhabdin; OMPTA, outer membrane targeting antibiotic; Omp, outer membrane protein; PBMC, peripheral mononuclear blood cell; PBP, penicillin-binding protein; PBS, phosphate-buffered saline; PK, pharmacokinetics; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPG, 2-oleoyl-1-palmitoyl-sn-glycero-3-phospho-(1-glycerol); PrAMPs, polyproline antimicrobial peptides; RBC, red blood cell; SAR, structure-activity relationship; s.c., subcutaneous; SPase I, signal peptidase I; SPR, surface plasmon resonance; VABP, ventilator associated bacterial pneumonia; VIM-1, beta-lactamase 2 (K. pneumoniae); VISA, vancomycin-intermediate S. aureus; VRE, vancomycin-resistant enterococcus; WHO, World Health Organization; WT, wild type; WTA, wall teichoic acid; XDR, extremely drug-resistant
1. Introduction
The Infectious Disease Society of America (IDSA) report “Bad bugs, no drugs: no ESKAPE” published in 2009 (1) has urged equally health care organizations, politicians, doctors, and scientists to come up with a clear strategy to fight the global problem of antimicrobial resistance (AMR) (2,3). The global emergence of pan-resistant bacterial strains against essentially all currently available classes of standard of care antibiotics in combination with a dry pipeline ([4], [5], [6]) of novel classes of antibiotics with novel mechanisms of action (MoA) (7), require urgent and concerted actions to fight a global AMR crisis. The COVID-19 outbreak with its devastating consequences for people and economies, and its potential to further worthen the AMR problem (8), should be enough motivation to take action before it is too late.
The WHO has issued a global priority pathogens list (PPL) of antibiotic resistant bacteria (2) which currently constitute a severe threat to society. WHO priority 1 pathogens include carbapenem-resistant P. aeruginosa, A. baumannii, and third-generation cephalosporin and carbapenem-resistant Enterobacteriaceae, belonging all to the Gram-negative bacteria. Problematic Gram-positive strains include methicillin-resistant S. aureus and vancomycin-resistant enterococci. There is an urgent need of novel classes of antibiotics with novel mechanisms of action (MoA) against these pathogens (4,6,7).
In this review, we describe some of the most recent progress in the field of peptide antibiotics. This vast class of antibiotics comprises ribosomally and/or nonribosomally synthesized linear and cyclic peptides, and semisynthetic and fully synthetic linear and macrocyclic peptides in many forms and shapes (9). Many important classes of peptide antibiotics such as cyclopeptides (lysobactin, katanosin, pristinamycin, or enopeptins), glycopeptides (e.g., vancomycin, telavancin, dalbavancin, or ramoplanin), and lipopeptides (e.g., daptomycin and colistin) (7) are macrocyclic natural peptide antibiotics and some representatives are under clinical evaluation (10) or have been approved. The classical antimicrobial peptides (AMPs) or host-defense peptides (HDPs) are important constituents of the innate immunity (9,11,12) and form probably the most abundant class of peptide antibiotics. While many AMPs and HDPs target rather unspecifically the bacterial membrane, there is an increasing number of reports of peptide antibiotics that act via specific inhibition of targets located in the bacterial cell wall or membrane (13). In particular, compounds that inhibit specifically key components of the outer membrane assembly machines of Gram-negative bacteria (14), such as LptD (15), LptA (16), and BamA (17,18) were discovered recently. In addition, peptide antibiotics can also address successfully intracellular antimicrobial targets (19); the odilorhabdins (20) are examples.
Despite the fact that AMPs often show limited safety and efficacy profiles, they can provide interesting scaffolds and serve as excellent starting points for peptide medicinal chemistry programs. Our focus in this review will be on peptide antibiotics for which novel or established mechanisms of action and targets have been identified, and antimicrobial efficacy has been broadly demonstrated at least in animal infection models. As a prerequisite, such natural peptide-derived hits or leads should allow for a peptide-based medicinal chemistry approach in order to modulate and improve activity and ADMET properties.
2. Drug discovery approaches to identify new antibiotic lead structures
Antibiotic discovery constitutes a unique challenge as bacteria have developed powerful tools to resist the action of antibiotics by having developed hardly permeable cell walls and efficient resistance and efflux mechanisms that make penetration of antibiotics to the site of action very difficult. In particular, Gram-negative bacteria have developed a formidable shield against lipophilic antibiotics (21) by having a classical inner and an asymmetric outer membrane with lipopolysaccharide located at the outer and regular phospholipids at the inner leaflet (21).
It is therefore not surprising that most of the successful antibiotic classes to date have been discovered by isolation of natural products from extracts of soil microbes from actinomycetes mainly, and by phenotypic screening approaches (4,21,22), while target-based approaches have unfortunately not really been successful (23). Next generations of semisynthetic analogues derived from the original natural antibiotic classes (such as β-lactams, glycopeptides, aminoglycosides, tetracyclines, and macrolides) with improved antimicrobial spectrum and resistance profiles were successfully developed (4). However, the AMR crisis asks for novel antimicrobial drug discovery strategies and novel antibiotic classes with novel targets and/or mechanisms of action, for which the current pipeline is scarce.
Since to this date less than 1% of environmental microbes have been cultured and analyzed for their antibiotic natural product content, novel approaches and technologies of tapping into the universe of uncultured bacteria have significantly progressed and are generating first promising results (21,24). On the novel antibiotic class-novel target front, there have been recently some positive developments (13,21).
It is quite remarkable that among the new antibiotic classes with novel targets and/or MoA described recently, many belong to macrocyclic peptides. They were either isolated as unmodified natural products or made semi- or fully synthetically starting from a natural product scaffold (7).
3. Linear peptide antibiotics addressing diverse targets
3.1. Odilorhabdins
The discovery of Odilorhabdins (ODLs) resulted from the screening of Xenorhabdus and Photorhabdus strains from the Enterobacteriae family that are symbiotic bacteria of entomopathogenic nematodes. These bacteria produce a large diversity of antifungal and antimicrobial compounds (25). Their particular genomes with enriched nonribosomal peptide synthetase and polyketide synthase genes allow generation of secondary metabolites with high chemical diversity. The evaluation of the metabolites allowed the isolation of ODLs as a novel class of AMPs against Gram-positive and Gram-negative pathogens (20,26). The first ODL structures were characterized using NMR and LC/MS/MS fragmentation and confirmed by resynthesis are summarized in Figure 1 (NOSO-95A, NOSO-95B, and NOSO-95C). These ODLs are short linear cationic peptides comprising between 9 and 11 residues and containing natural and up to six unnatural amino acids, that is, aminothreonine or dehydroarginine. The mode of action of the ODLs was investigated first by assessing their effects on macromolecular synthesis pathways (protein, RNA, DNA, and peptidoglycan synthesis) by standard incorporation of radiolabeled precursors. The ODLs interfere with protein biosynthesis and were shown to bind to the small 30S ribosome subunit leading to the inhibition of protein translation (20).
Figure 1.

Structures of Odilorhabdin derivatives.
After isolating the natural compounds, a structure–activity relationship’s study was developed to understand which residues are important for membrane crossing and inhibition of the bacterial translation resulting in the best antibacterial activity against Enterobacteriaceae strains, for example, Escherichia coli, K. pneumoniae, or Enterobacter cloacae including carbapenem-resistant strains (i.e., KPC-3, NDM-1, or VIM-1) (27). In particular, NOSO-95179 (2) showed good activity (MIC 4–8 μg/mL for E. coli 2–8 μg/mL for K. pneumoniae) with low-resistance propensity (spontaneous resistance frequencies in the 10−9 range). In addition, this compound showed lower cytotoxicity against mammalian cells (HepG2; IC50~250 μM) and low hemolysis (0.36% at 100 μM). In vivo, NOSO-95179 showed a 2 and 3 log CFU reduction of bacterial burden at 40 and 80 mg/kg, respectively, in a K. pneumoniae ATCC 13883 lung efficacy model after subcutaneous injection in mice (27). Currently, a new derivative NOSO-502 (3, Figure 1) coming from the lead optimization phase is in preclinical phase and developed by Nosopharm.
3.2. Proline-rich antimicrobial peptides
Proline-rich antimicrobial peptides (PrAMPs) were first isolated from mammalian and insect cells for which they serve as host-defence peptides against Gram-negative bacteria ([28], [29], [30]). Their characteristics are a high content of proline and cationic amino acid residues (e.g., arginine) in repeated motifs (see Table 1 ). Despite a content of positive charges, PrAMPs do not exhibit an unspecific membranolytic mechanism of action typically observed for other cationic AMPs, which show antimicrobial activity against both Gram-positive and Gram- negative bacteria (30). In contrast, PrAMPS show good activity against Gram-negative bacteria without membrane disruption. The full D-enantiomer of PrAMP Bac7(1–35) does not display the same activity as the initial L-version pointing to a specific nonmembranolytic mechanism of action of Bac7(1–35), which contrasts to lytic AMPs where the D-enantiomers show the same activity (31). It was shown that PrAMPs can translocate actively the membrane through permease/transporter uptake and have an intracellular mode of action (28,29). One of the first identified intracellular targets of PrAMPs was the bacterial heat-shock protein DnaK. DnaK inhibition leads to protein misfolding and aggregation and ultimately to bacterial death. X-ray structure determination and modeling studies of PrAMPs with E. coli DnaK identified two binding sites ([32], [33], [34], [35]). Subsequently, the 70S ribosome was identified as a new target for PrAMPs (i.e., Api88, Api137, Onc72, and Onc112 (36,37)). Binding to the 70S ribosomal subunit by PrAMPs leads to inhibition of bacterial protein translation. The crystal structure of Onc112 (see Table 1) in complex with Thermus thermophilus 70S ribosome subunit has been solved (37). Onc112 binds to the initiation complex preventing entry into the elongation phase and therefore the protein translation. The same binding mode with other PrAMPs was shown using X-Ray crystallography with Bac71–35, Pyrrhocoricin, Metalnikowin, and shorter oncocin derivatives OncΔ15-19 and OncΔVD (see Table 1) (38). Roy et al. (39) showed that Onc112 binds to the 70S ribosome subunit around 50-fold stronger than to DnaK, highlighting the 70S ribosome as the main target of PrAMPs (39,40).
Table 1.
Amino Acid Sequences of Proline-Rich Antimicrobial Peptides
| Name | Sequence | Length | Ref |
|---|---|---|---|
| Apidaecin-Ib | G-N-N-R-P-V-Y-I-P-Q-P-R-P-P-H-P-R-L | 18 | (28) |
| Api137 | TMG-Orn-N-N-R-P-V-Y-I-P-R-P-R-P-P-H-P-R-L-OH | 18 | (41,45) |
| Api88 | TMG-Orn-N-N-R-P-V-Y-I-P-R-P-R-P-P-H-P-R-L-NH2 | 18 | (41,45) |
| Bac7(1–35) | R-R-I-R-P-R-P-P-R-L-P-R-P-R-P-R-P-L-P-F-P-R-P-G-P-R-P-I-P-R-P-L-P-F-P | 35 | (46) |
| Metalnikowin-I | V-D-K-P-D-Y-R-P-R-P-R-P-P-N-M | 15 | (47) |
| Pyrrhocoricin | V-D-K-G-S-Y-L-P-R-P-T-P-P-R-P-I-Y-N-R-N | 20 | (48,49) |
| Onc112 | V-D-K-P-P-Y-L-P-R-P-R-P-P-R-r-I-Y-N-r-NH2 | 19 | (50) |
| Onc72 | V-D-K-P-P-Y-L-P-R-P-R-P-P-R-Orn-I-Y-N-Orn-NH2 | 19 | (50,51) |
| OncΔ15–19 | V-D-K-P-P-Y-L-P-R-P-R-P-P-R | 14 | (38) |
| OncΔVD | K-P-P-Y-L-P-R-P-R-P-P-R-R-I-Y-N-R | 17 | (38) |
r = D-arginine; Orn = ornithine; TMG = N,N,N’,N’-tetramethylguanidino; NH2 = C-terminal primary amide.
Derivatives of Apidaecins (Api88 and Api137) and Oncocins (Onc 72 and Onc112) were tested in vivo using different routes of administration, that is i.p., s.c., or i.v. Pharmacokinetic studies showed a low residence time for the different compounds with high clearance through the kidney ([41], [42], [43]). The compounds have low stabilities in plasma, which explains their low bioavailability. Interestingly, Onc72 showed an unexpected better in vivo activity than meropenem, despite its 44-fold lower in vitro activity (44). This discrepancy between in vitro and in vivo activity was attributed to a negative effect on the in vitro activity by the MH-II media used according to the CLSI standard antimicrobial activity tests. At present, the high doses used for achieving in vivo efficacy in different models and the lack of safety data require significant additional work for this interesting family of antibiotics to progress for further development.
4. Lipopeptides
4.1. Introduction
Within the vast structural diversity of antimicrobial peptides, lipopeptides are of special importance highlighted by daptomycin and the polymyxins, which are valuable commercial antibiotics of therapeutic use.
Lipopeptides are characterized by the existence of a mostly macrocyclic peptidic core to which a hydrocarbon lipid tail (usually > C6) is linked via the N-terminus and may include hydroxy groups and unsaturation(s). The lipid tail interacts with lipids of bacterial and potentially mammalian cell membranes generally enhancing the bactericidal effect of lipopeptide antibiotics. The macrocyclic peptidic core is important for specific interactions of the lipopeptide with lipid and/or protein targets. Nevertheless, the lipophilic hydrocarbon tail does not generally improve desirable drug properties like good aqueous solubility and is prone to generate unspecific binding to phospholipid bilayers, thus increasing potential off-target effects.
In the last decades, a large number of antimicrobial lipopeptides of natural source has been discovered. Besides the mentioned daptomycin and polymyxins, there are many additional interesting lipopeptides, for example, the actinocarbasin (arylomycin D; 19, Figure 4), globomycin, enopeptins, or friulimicin (11,52).
Figure 4.

The arylomycin family of natural products comprises nonglycosylated and glycosylated lipopeptides A–D, with a common tripeptide macrocyclic scaffold. Structure of the synthetic actinocarbasin derivative M131 with a β-alanine moiety present in the lipophilic tail.
Within this part of the review, we would like to focus only on a few selected examples of lipopeptide antibiotics that stand out based on their novel MoA, have available in vivo efficacy data, and/or moved into preclinical or clinical development.
4.2. Tridecaptins
Tridecaptins (Figure 2 ) are nonribosomally synthesized natural products within the class of linear cationic tridecapeptides, which have been first isolated in 1978. Tridecaptins display good antimicrobial activity against Gram-negative bacteria with single-digit MICs [μg/mL] on Enterobacteriaceae and weaker activity against A. baumannii and moderate activity against P. aeruginosa (53,54). The antimicrobial activity against Gram-positive bacteria is generally moderate. SAR and MoA studies of one member of this family, Tridecaptin A1 (TriA1; 5, Figure 2) isolated from Paenibacillus terrae, have been described in more detail ([54], [55], [56]). Cochrane et al. have observed that the structure of the lipid tail of TriA1 could be varied without loss of antimicrobial activity (55). The analogue OctTriA1 (6) containing an octanoyl lipid chain fully retained its activity against all organisms (55). In a first step, TriA1, and also the derivatives Ent-TriA1 (enantiomer), Oct-TriA1 , and H-TriA1 (unacylated TriA1) bind with a similar affinity to lipopolysaccharide located in the outer membrane of Gram-negative bacteria (54). However, the unacylated H-TriA1 (4) as well as analogues with shorter lipid tail (<C6) are significantly less active, pointing to the important contribution of the lipid chain to the antimicrobial activity by interaction with phospholipids. TriA1 is a membrane-targeting peptide but does not act by generic membrane lysis mechanism like many other AMPs. The enantiomer of TriA1 (Ent-TriA1) is fourfold less active than the natural peptide, suggesting that TriA1 interacts with a chiral target. Indeed, TriA1 binds to lipid II, the monomeric intermediate in the peptidoglycan biosynthesis, located on the inner surface of the plasma membrane. Interestingly, it could be shown that TriA1 binds much weaker to the lipid II of Gram-positive bacteria (structure cf. Figure 9), which explains the lower activity on Gram-positive bacteria. In line with the postulated mechanism, Cochrane et al. nicely elucidated by ITC binding studies that the enantiomer of TriA1 (Ent-TriA1) lacks binding to lipid II (54). In addition to the interaction with lipid II, disruption of the proton motive force, a vital process to produce bacterial ATP within the bacterial membrane, seems to be affected and could explain the antibacterial activity of TriA1 (54). Another member of the tridecaptin class, tridecaptin M (7), exhibited good in vivo efficacy at 10 mg/kg bid s.c. in a neutropenic mouse thigh infection model against a colistin-resistant strain of K. pneumonia (57). Although reported to be not hemolytic on rabbit erythrocytes at 100 μg/mL, hemolysis of 50% of cells at 200 μg/mL has been observed (57). Therefore, possible future lead optimization based on the tridecaptin scaffold should focus on minimizing hemolysis and cytotoxicity (58). The design of synergistic dual-acting bifunctional antimicrobial conjugates (chimeric antibiotics) has become a popular approach to increase antimicrobial activity (59). Vederas and colleagues synthesized antibiotics conjugates starting from H-TriA1 (unacylated TriA1) linked via copper(I)-mediated click chemistry to vancomycin, rifampicin, and erythromycin analogues (60). These chimeric compounds showed enhanced but not synergistic activity in vitro in checkerboard studies. In vivo, survival rates were compared to both parent drugs in combination and alone in a K. pneumonia murine survival experiment, all conjugates and both parent compounds being administered intravenously (tail vein). Only the conjugate H-triA1-erythromycin led to a 2-fold higher survival rate compared to both parent drugs in combination (60). However, surprisingly, in this study, vancomycin alone, which has no activity on Gram-negative bacteria, showed an 80% survival rate in this model.
Figure 2.

Structures of Tridecaptins; structures of Malacidin A and B. Amino acids of tridecaptin M (7) are annotated in brackets.
Figure 9.

Teixobactin, depsipeptide antibiotic isolated from E.terrae, inhibits cell-wall biosynthesis (105,113). Structure of lipid II; precursor of the cell wall biosynthesis; dissociation constants of teixobactin determined by Chiorean et al. (117) applying isothermal calorimetry. Structure of lipid III; precursor of the wall teichoic acids.
4.3. Malacidins
As mentioned before, it is assumed that less than 1% of all environmental microbes have been cultured in laboratory settings as a source to discover new antimicrobial molecules (4). Through applying emerging technologies such as environment-mimicking cultivation and genome mining of Biosynthetic Gene Clusters (BGCs), Brady and colleagues have discovered malacidin A and B (8a, 8b; Figure 2) as members of a new class of calcium-dependent macrocyclic lipopeptide antibiotics (61,62). Malacidin A consists of a macrocyclic nonapeptide, containing four nonproteinogenic amino acids, and an unsaturated C9-fatty chain acylated to an exocyclic β-methylaspartic acid. The total synthesis of malacidin A has been reported by Sun et al. and the absolute configurations of its five nonproteinogenic amino acid residues have been elucidated (63). Malacidin A exhibits broad activity against Gram-positive bacteria including methicillin-resistant S. aureus (MRSA) and vancomycin-resistant Enterococcus (VRE) and has potent antimicrobial activity (MIC range 0.2–2 μg/mL) in the presence of Ca2+ ions. Malacidin A was also able to clear S. aureus infection in a skin infection rat model. The MoA of malacidins involves binding to lipid II as described for other known antibiotics like vancomycin and teixobactin. However, since no cross resistance to vancomycin has been observed, binding of malacidin A to lipid II must be different from the binding mode of vancomycin.
4.4. Lipopeptides of the polymyxin/colistin/octapeptin family
The polymyxin lipopeptide antibiotic family was discovered in 1947 and is characterized by its potent, specific activity against Gram-negative bacteria (64). Polymyxin B (Polymyxin B1, 9, Figure 3 ), polymyxin E (colistin, 10), and colistin methanesulfonate (CMS, 11) were used clinically. However, they were subsequently gradually withdrawn from clinical practice in the 1960s after several reports of partially severe nephrotoxicity and rather mild neurological effects in a large number of patients (65,66). CMS (11) is a colistin prodrug that is applied parenterally and by inhalation (67). With the urgent need of new antibiotics due to increased emergence of multidrug-resistant bacteria and new and more stringent dosing regimens (68), colistin (10) has experienced a revitalization in the clinical use as a “last resort” antibiotic against P. aeruginosa, A. baumannii, and K. pneumoniae (69). Due to conservative use of polymyxins in the last century, resistance development has been rare and mostly based on modifications of lipid A (70). However, with their late use as last-line treatment against MDR infections and for their massive overuse in agriculture and poultry, resistance in clinical pathogens is on a global rise, especially the worrisome plasmid-borne mcr containing strains (71,72).
Figure 3.

Structures of the polymyxin/colistin and octapeptin antibiotics.
The mode of action of these rapidly bactericidal antibiotics is still not completely understood. Presumably, in an initial step, the positively charged polymyxins bind to the negatively charged lipid A part of lipopolysaccharides (LPS) followed by displacement of Ca2+ ions (73). The peptides cross the outer membrane through a “self-promoted uptake” mechanism and then interact with the cytoplasmic membrane to inhibit cellular energization, and possibly cause inhibition of cell division and/or cytoplasmic membrane permeabilization and subsequent cell death (74). In this context, the lipid tail of the polymyxins is crucial for activity as a shortened variant of polymyxin without the hydrophobic N-terminal fatty-acyl chain exhibits much reduced activity (75). In the context of the revival of their use, further research in their mechanism of resistance and action led to the proposal of new alternative mechanisms. In particular, research groups have explored the ability of polymyxins to bind to ribosomes, prevent cell division, and inhibit bacterial respiration (74).
The high need for new antibiotics in particular against Gram-negative bacteria (last new class were the Quinolones over 50 years ago (21)) and probably also the relative straightforward chemical accessibility has spurred the search for novel synthetic polymyxin derivatives and more recently also for the closely related octapeptin analogues (12, Figure 3) (64), with the potential to overcome polymyxin resistances and reduced renal toxicity (76).
Modifications within the cyclic heptapeptide ring, variation of the exocyclic amino acids and with particular focus, the replacement of the fatty-acyl chain lead to equally potent derivatives compared to the polymyxins B and E and with some derivatives partially overcoming polymyxin resistance. Many of these next-generation polymyxins have been extensively reviewed elsewhere (76,77). However, designing and optimizing compounds with reduced renal toxicity in animal settings translating reliably into human is still a challenge. So far, none of the new active analogues have shown improvement of the therapeutic index compared to polymyxins particularly regarding nephrotoxicity.
Recently, Brown et al. have described an interesting approach to tackle the problem of nephrotoxicity (76). SPR206 (14; Figure 3) is a polymyxin B analogue where the lipid tail was replaced by (S)-4-amino-3-(3-chlorophenyl)butanoic acid. SPR206 has been selected as a result of an intriguing medicinal chemistry effort based on the understanding of structure–activity relationship of antimicrobial activity, in vitro cytotoxicity against human kidney proximal tubular epithelial cell line (HK-2), and kidney exposure (78). The exact mechanism of renal toxicity of polymyxins is not completely understood, but it has been shown that polymyxins are reabsorbed through the proximal tubular cells and accumulate to exert renal toxicity (79). SPR206 showed 12 times lower cytotoxicity toward the HK-2 cell line compared to polymyxin B1 resulting in lower nephrotoxicity in an acute (24 hours) mouse in vivo model based on biomarkers and kidney histopathology assessment at comparable dose. Interestingly, kidney exposure of SPR206 in mice was similar compared to polymyxin B1. Recently, SPR206 has finished phase-I trial (NCT037992308). It will be interesting to see if SPR206 can successfully further progress in the clinic.
In the search of polymyxin derivatives with more favorable antimicrobial activity on colistin-resistant strains and nephrotoxicity profiles, derivatives of octapeptins (12, e.g., octapeptin C4 and octapeptin B5 (13; Figure 3) have been investigated (80,81). Battacin (13) shows interesting antimicrobial activity (MICs = 2–4 μg/mL) against some multidrug-resistant clinical strains of E. coli and P. aeruginosa (64). The overall positive charge together with the N-terminal fatty-acyl chain was considered to be a main contributor to the renal toxicity within the polymyxin class (82). Interestingly, octapeptins contain at least one cation less compared to polymyxins. However, octapeptin C4 shows a fourfold higher toxicity compared to polymyxin B on HK-2 cell line (80). Recently, a group at the University of Queensland, Australia, has received significant funding to further develop new octapeptin derivatives (83).
Different approaches were explored by Vaara and coworkers (84). By reducing the number of positive charges and altering of the exocyclic amino acids of polymyxin B, they obtained potentially less nephrotoxic derivatives. Polymyxin nonapeptide (PMBN) is a polymyxin derivative lacking the N-terminal fatty-acyl chain and the last exocyclic Dab-residue. It has no antibiotic activity; however, it binds to LPS, permeabilizes the outer membrane of Gram-negative bacteria, and thereby potentiates synergistically the antimicrobial activity of antibiotics like rifampin, clarithromycin, and azithromycin when applied in combination. NAB741 (SPR741) is a PMBN-derivative with three positive charges (at physiological conditions) and has completed two phase-I trials envisaged to be used as potentiator in combination with other antibiotics. In future clinical studies, SPR741 will be combined with other antibiotics using the i.v. route to show hopefully a clinical benefit (6).
5. Peptide antibiotics addressing targets located at the inner membrane
5.1. Arylomycin
Arylomycin A2 (15; Figure 4 ) and the synthetic derivative arylomycin A-C16 (16) are macrocyclic lipo-hexapeptide representatives of the arylomycin class of antibiotics (85,86). Arylomycin A2 was isolated from Streptomyces extracts (87) and is formed by nonribosomal peptide synthesis (86,88). The peptide scaffold has the sequence D-MeSer, D-Ala, Gly, L-MeHpg, L-Ala, and L-Tyr where the N-methyl-4-hydroxy-phenylglycin (L-MeHpg for biosynthesis see (89)) residue is covalently linked by a 3,3-biaryl bridge with the C-terminal tyrosine forming a 14-membered macrocyclic ring. In addition, a fatty acid is attached to the N-terminal D-MeSer residue forming a tertiary amide bond. Arylomycins A2 (15) and B2 (17) were reported to exist as a mixture of cis/trans isomers around the tertiary amide bond in MeOH and DMSO solution, with the trans isomer predominating (87). In DMSO solution, atropisomers around the biaryl bond were observed in addition (90). Likewise, the macrocyclic model compound 21 was found to exist as a 4:1 mixture of isomers in DMSO. The major form was also observed in arylomycin A2 in complex with the target protein (86,90). Peters et al. (91) recently used Cu-mediated oxidative phenol coupling to convert the linear tripeptide 22 into the macrocycle 23 (60% on 5 g scale; Figure 5 ). C-H functionalization catalyzed by a P450 enzyme is responsible for the biaryl bond formation in the biosynthesis of arylomycins (88).
Figure 5.

Cu-mediated oxidative phenol coupling mimicking the biosynthesis; reported by Peters et al. (91).
Arylomycin targets the bacterial protein secretion by inhibition of signal peptidase I (SPase I) of Gram-positive and Gram-negative bacteria (85,86,92). The E. coli enzyme is the best characterized signal peptidase I (85) and is a plasma membrane-bound protein (two helical transmembrane segments) with the catalytically active site residing in the large periplasmic region. The catalytic nucleophile Ser91 is activated by Lys146. Ser89 contributes to transition-state stabilization. SPase I cleaves off the signal peptide from secreted proteins. Signal peptides (Figure 6 ) have an amino terminal region (n-region), followed by hydrophobic residues (h-region) and the C-terminal region (c-region), which contains the recognition and cleavage site. Small aliphatic residues are found in P1 and the P3 positions (Ala-X-Ala substrate specificity). The c-region of the signal peptide binds in the extended β-strand conformation (85).
Figure 6.
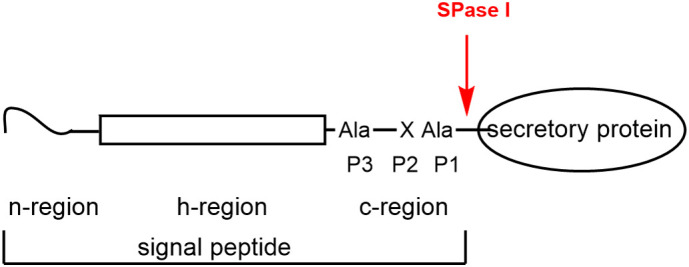
Signal peptides: N-terminal region (termed n-region) 1–5 amino acids; h-region 7–15 hydrophobic amino acids; c-region 3–7 amino acids; consensus sequence for the cleavage site consists of small aliphatic residues in P1 and P3 positions.
Gram-positive SPase I differs in its extra-cytoplasmic region from the periplasmic region of Gram-negative SPase I and has only a single transmembrane segment (85). Crystal structures of the S. aureus SPhase I protein SpsB in complex with cleavable and inhibitory peptides have been described by Ting et al. (93).
A cocrystal structure of a complex of arylomycin A2 (15) and the catalytically active fragment of E. coli SPase I (SPase Δ2-75; construct of E. coli signal peptidase lacking the transmembrane segments and the cytoplasmic region) was reported by Paetzel et al. (86). Arylomycin A2 is a noncovalently binding inhibitor. Previously, by crystal structure determination, a β-lactam type inhibitor was found to covalently bind to the γ-oxygen atom of the nucleophilic Ser of SPase Δ2-75 (94). Arylomycin A2 was reported to bind in parallel β-sheet fashion with the C-terminal carboxylate exhibiting hydrogen bonds with the catalytic Ser90 and Lys145 residues and with the oxyanion stabilizing Ser88 (Figure 7 ). The methyl group of Ala of arylomycin A2 occupies the P3 position and the D-Ala methyl group, the P5 position. All hydrogen bond donors and acceptors of the macrocyclic ring system make hydrogen bonds with SPase atoms either directly or through water molecules; the tail of the inhibitor contributes two additional hydrogen bonding interactions. The phenolic OH groups are solvent exposed. The fatty acid chain was discussed by Paetzel et al. (86) to possibly contribute to the inhibitor binding by presenting the ligand in the correct orientation to the enzyme within a lipid bilayer. Biophysical analyses were reported to be most consistent with a two-step binding mechanism involving rapid binding mode followed by a slow isomerism to the final bound state.
Figure 7.

Hydrogen bonding interactions between arylomycin A2 and E. coli SPase I (Δ2-75) (86). Pro84 (Pro83)⁎ in the E. coli enzyme is preventing a in the Ser mutant available hydrogen bonding interaction with the N-terminal fatty acid carbonyl group of arylomycin.
⁎SPase numbering used in SPase structures discussed is reported different by one residue, due to an error in the originally reported sequence of the E. coli protein, c.f. (95).
Despite inhibiting an essential target, arylomycins exhibit no activity against many pathogens, including S. aureus, E. coli, and P. aeruginosa. Insufficient penetration of the outer membrane was proposed to explain the lack of activity against Gram-negative bacteria (90,96). This hypothesis is supported by the comparison of the activity of arylomycin A-C16 (16) against a hyper-permeable strain of E. coli (with an LptD mutation; MIC = 16 μg/mL) with that against E. coli MG1655 (MIC of > 64 μg/mL) (97). The penetration hypothesis, however, would not explain the lack of activity against Gram-positive bacteria as pointed out by Smith et al. (98), who studied the resistance development of the sensitive organism Staphylococcus epidermidis to arylomycin A-C16 and identified two-point mutations in SPase I, among them Ser to Pro in position 29, with marked increase in arylomycin resistance. In the SPase I sequence of resistant S. aureus, E. coli and in one of two SPases I of P. aeruginosa a Pro is found in the corresponding positions (Pro29 in Gram-positive S. aureus SPase I; Pro84 in Gram-negative E. coli and P. aeruginosa SPase I). Mutant strains of S. aureus, E. coli, and P. aeruginosa with Pro replaced by Ser exhibited increased sensitivity to arylomycin A-C16 (Table 2 ). The sensitivity of the latter two organisms demonstrates that arylomycin penetrates the outer membrane. Biological consequences of SPase I inhibition and mechanisms of resistance have been recently reviewed and discussed by Craney and Romesberg (99).
Table 2.
MICs of Arylomycin A-C16 for Wild-Type and Mutant Gram-Positive and Gram-Negative Bacterial Strains; Taken from Ref. (98)
| Bacterial strain | SPase | MIC [μg/mL] |
|---|---|---|
| S. epidermidis RP62A | WT | 0.25 |
| S. epidermidis PAS9001 | S29P | 8 |
| S. aureus NTCT8325 | WT | >128 |
| S. aureus PAS8001 | P29S | 2 |
| E. coli MG1655 | WT | >128 |
| E. coli PAS0232 | P84S | 4 |
| P. aeruginosa PAO1 | WT | >128 |
| P. aeruginosa PAS2006 | P84S | 8 |
The Pro residue, which confers resistance, interacts with the lipopeptide tail of arylomycin A2. Initial efforts to optimize the arylomycins focused on derivatives with altered lipopeptide tails (100). Of particular interest was compound 24 (Figure 8 ) with improved activity against WT S. aureus NCTC 8325 compared to arylomycin A-C16 (16). No gain of activity was, however, observed against Gram-negative E. coli MG1655 and P. aeruginosa PAO1. The D-MeSer of the natural product is replaced in 24 by a homolog inserting additional methylene groups in the peptide tail, increasing its flexibility. Later, based on a screening for compounds, which synergize with β-lactams against methicillin-resistant S. aureus (MRSA), scientists at Merck identified actinocarbasin (19, also called arylomycin D, Figure 4) and derived thereof the synthetic derivative M131 (20, Figure 4), both with a β-alanine moiety present in the lipopeptide tail (101). Remarkably, cotreatment with M131 (20), a potent SpsB inhibitor with an IC50 of 10 nM, restores β-lactam sensitivity of MRSA in vitro and in vivo probably by preventing signal-peptidase mediated secretion of resistance-conferring proteins (99,101).
Figure 8.

Optimization of arylomycins afforded G0775 (31) (102,103) a novel compound with potent broad spectrum of activity against Gram-negative organisms, acting on a new antibiotic target (SPase I).
Other approaches comprised modifications of the arylomycin C-terminus. The introduction of phosphonate groups to mimic the transition state of the acyl enzyme intermediate formation (e.g., compounds 25 and 26, Figure 8) went along with loss of activity for SPase I (97). Compound 27, however, with a glycyl aldehyde function—thought to form a covalent bond with the catalytic Ser-OH in the position of the scissile bond of the peptide substrate—was found to be more potent than arylomycin A-C16 against the E. coli and S. aureus enzyme, demonstrating that the presence of the C-terminal carboxylate is not required for binding. The position of the electrophilic group is important as the homologue aldehyde 28 was reported to be inactive (97). The increase in biochemical activity translated in an increased activity against MRSA (USA300) but did—like arylomycin A-C16—not exhibit activity against S. aureus 8325 or E. coli MG1655 (97).
Attempts to improve target accessibility in whole cells led to the synthesis of the arylomycin A-C16 analogue 29 (Figure 8), which retained biochemical activity against E. coli and S. aureus SPase I (97). Compound 29 retained activity against sensitive bacterial strains and gained weak activity against MRSA, which was explained by Liu et al. (97) by an interaction of the positively charged amine with the negatively charged teichoic acids of the Gram-positive cell wall.
In compound 30, modification of the N-terminal peptide and introduction of positively charged groups were combined providing activity against Gram-negative pathogens (K. pneumoniae MIC = 0.5 μg/mL; P. aeruginosa MIC = 8 μg/mL) (91).
The molecule G0775 (31; Figure 8), disclosed by Genentech scientists, integrates C-terminal electrophilic group, positively charged substituents, and an altered lipopeptide tail (102). A cocrystal structure of G0775 with Gram-negative SPase I revealed that the inhibitor binds like arylomycins and that the amino acetonitrile warhead forms a covalent bond not with Ser91 (the catalytic nucleophile) but with the Lys146 (the catalytic base) through amidine bond formation. The activity against laboratory strains of Gram-negative ESKAPE pathogens was reported (102) to be markedly improved (MICs of 0.125–2 μg/mL) and maintained against MDR clinical isolates of E. coli and K. pneumoniae and MDR strains of A. baumannii and P. aeruginosa. The uncharged analogue G8126 (32, Figure 8) (102) showed reduced activity against Gram-negative and Gram-positive strains indicating that the amines contribute to the potency of G0775 beyond facilitating the entry into the periplasma.
The in vivo efficacy of G0775 was demonstrated in a murine neutropenic thigh infection model (> 2-log decrease in colony-forming units (CFU) against E. coli ATCC 25922 at 1 mg/kg; K. pneumoniae ATCC 43816 at 5 mg/kg; and A. baumannii ATCC 17987 at 40 mg/kg; > 1 log decrease in CFU against P. aeruginosa ATCC 27853 at 40 mg/kg). In a lung infection model using the MDR strain K. pneumoniae CDC 0106, the compound showed a bacteriostatic effect at 2 mg/kg and a bactericidal effect at 20 mg/kg. In addition, G0775 was reported to protect mice in a peritonitis model from a lethal challenge of K. pneumoniae Z strain ATCC 43816 at 5 mg/kg delivered subcutaneously twice (2 and 11 h after initiation of the infection) with 100% of the mice viable after 84 h (102).The compound exhibited IC50 values > 50 μM against the mammalian cell lines A549 (human lung cancer), HEK-293T (human embryonic kidney cell line), Jurkat (immortalized human T lymphocyte cell line), and H23 (human lung cancer cell line) (102).
Arylomycins were suggested by Romesberg and colleagues (99,104) to be “latent antibiotics,” which were in the past potent and had a broad-spectrum activity. Selection of resistance (and not poor intrinsic properties) then narrowed their spectrum by a single point mutation in SPase I. Producer strains in turn start to synthesize variants of arylomycins (such as actiocarbasin/arylomycin D) to regain activity. This “coevolution hypothesis for producer and susceptible organisms” (104) describes a situation comparable to that of clinically used antibiotics with resistance compromised spectrum, leading to the development of next-generation compounds (100).
Optimization of arylomycins resulted in the novel compound G0775 with potent broad-spectrum activity against Gram-negative organisms, acting on a new antibiotic target. Serial passage experiments demonstrated that spontaneous resistance evolves with moderate frequency in the presence of low concentration (4 x MIC) of G0775, whereas with higher concentrations (8–16 x MIC), the frequency was found to be very low (102).
5.2. Teixobactin
The discovery of teixobactin (33; Figure 9 ), published in 2015 (105), got a lot of attention both in academia (106,107) and in the press (108) as it represented a new class of antibiotics with a novel MoA isolated by a new technique. New antibiotics are found only on rare occasions and teixobactin was discussed “to challenge the dogma of inevitable resistance” (109) as it was not possible to generate resistance in the laboratory (105). Discovery of teixobactin and development of analogues was recently reviewed by Iyer et al. (110) and by McCarthy (111). Karas et al. (112) summarized synthesis routes providing the natural product and analogues, structure–activity relationship (SAR) as well as structural studies.
Teixobactin (33) is produced by the soil-dwelling Gram-negative bacterium Eleftheria terrae by nonribosomal peptide synthesis. Using a novel cultivation technique by isolation chip (iChip), the natural product antibiotic was produced by a bacterium that could previously not be cultivated by traditional methods. The iChip is a diffusion chamber allowing to isolate and grow bacteria in soil in the presence of natural nutrients and growth factors. Teixobactin was identified by screening of extracts from 10’000 (!) bacterial strains (105,113,114).
Teixobactin (33) is a twofold positively charged depsipeptide with a molecular mass of 1242 Da, consisting of 11 amino acids. It contains four D-configured amino acids, among them at the N-terminal position N-methyl-D-phenylalanine and in position 5 D-allo-isoleucine. In addition, the uncommon amino acid L-allo-enduracididine was found in position 10. Amino acids 1–7 form the N-terminal tail, whereas the C-terminal macrolactone comprising 13 ring atoms is closed between the β-hydroxy group of D-Thr8 and the carboxylic acid group of Ile11 most probably mediated by thioesterase (105).
Teixobactin was reported by Lewis, Schneider, and colleagues (105) to exhibit potent activity against Gram-positive organisms, including S. aureus (MRSA; MIC: 0.25 μg/mL), Mycobacterium tuberculosis (MIC: 0.125 μg/mL), and C. difficile (MIC: 0.005 μg/mL). The potency against S. aureus (MSSA; MIC: 0.25 μg/mL) was retained in the presence of serum. (Ramchuran et al. (115) reported that MIC values of teixobactin analogues against S. aureus ATCC 29213 and Bacillus subtilis ATCC 6051 were not significantly affected by the presence of 50% human serum. Pamar et al. (116) also observed no serum shift of the MIC values of two analogues tested against MRSA ATCC 33591 (Table 3 )). Teixobactin has bactericidal activity against S. aureus but was found to be inactive against Gram-negative bacteria like P. aeruginosa, K. pneumoniae, or E. coli (MIC: 25 μg/mL). However, the strain E. coli asmB1 with defective outer membrane permeability barrier is sensitive to teixobactin (MIC: 2.5 μg/mL), indicating that the outer membrane protects the (Gram-negative) producer bacterium against it.
Table 3.
| Compound | MIC [μg/mL] | MIC (10% human serum) [μg/mL] | Reference |
|---|---|---|---|
| Teixobactin (33) | 0.25 | – | (116,132) |
| Arg(10)-teixobactin (34) | 2 | – | (116,132) |
| Lys(10)-teixobactin (35) | 1 | – | (132) |
| Ala(10)-teixobactin (36) | 1–2 | – | (116,132) |
| Leu(10)-teixobactin (38) | 0.25 | 0.25 | (116,132) |
| Ile(10)-teixobactin (39) | 0.25 | 0.25 | (116,132) |
| D-Arg(4)-Leu(10)-teixobactin (40) | 0.125 | – | (132) |
D-Arg(4)-Leu(10)-teixobactin (40; structure not shown) was evaluated in vivo in a topical instillation in a mouse-eye model of S. aureus ATCC 29213 (MIC < 0.0625 μg/mL) keratitis. As compared to untreated mice, the peptide 40 was found to reduce bacterial bioburden (by >99%) and to decrease corneal edema (132).
Teixobactin was not cytotoxic against mammalian NIH/3T3 and HepG2 cells at 100 μg/mL, not hemolytic, and did not exhibit genotoxicity. The compound showed minimal inhibition of the hERG channel and CYP isoenzymes, a half-life of 190 min in human plasma and good microsomal stability (105). The aqueous solubility, however, was found to be poor as a 1:20 dilution of a 10 mg/mL DMSO stock solution with PBS buffer at pH 7.4 led to gelation (118). A positive correlation of good antibiotic activity and propensity to form gels was reported by Nowick and colleagues (118,119) for teixobactin analogues.
Mode of action studies (105,113) revealed that teixobactin simultaneously inhibits the peptidoglycan and wall teichoic acid (WTA) biosynthesis triggering synergistic effects. It binds to the surface-exposed peptidoglycan precursor lipid II (teixobactin/lipid II 2:1 stoichiometric complex) and to the WTA precursor lipid III. Binding to both targets relies on the interaction of teixobactin with the pyrophosphate and the first sugar moieties (Figure 9). Teixobactin was also found to bind to undecaprenyl pyrophosphate. The apparent absence of resistance development is in line with the targets being lipid molecules and not proteins. Lipid II is targeted by several other antibiotics, including the glycopeptide antibiotic vancomycin (120). Vancomycin resistance occurred after 30 years of use in hospitals (4,114) probably resulting from of horizontal gene transfer (114,121).
Vancomycin interacts with the pentapeptide moiety of lipid II (specific interaction with the D-Ala-D-Ala motif) and is inactivated by binding to mature peptidoglycan. However, teixobactin does not bind to mature peptidoglycan (which does not have a pyrophosphate moiety). It is therefore active against vancomycin-intermediate S. aureus (VISA) strains (113) with modified cell walls including thicker peptidoglycan layers, and lower degree of cross linking, exposing more D-Ala-D-Ala binding sites to trap vancomycin (122). Teixobactin was furthermore found to be active against vancomycin-resistant enterococci (VRE) with modified lipid II (lipid II D-Ala-D-Lac or lipid II D-Ala-D-Ser instead of lipid II D-Ala-D-Ala) (105).
Pharmacokinetic parameters were determined in mice. After i.v. administration of a single dose of 20 mg/kg teixobactin, a serum level above MIC was found at 4 h post administration with a plasma half-life of 4.7 h, a C0 of 27.2 μg/mL, and an AUC0–24h of 57.8 μg.h/mL (105).
In vivo efficacy was demonstrated in a mouse septicemia model (MRSA) by intraperitoneal administration of a single dose of 0.5 mg/kg teixobactin 1 h post infection resulting in 100% survival of the animals (105). In a thigh infection model (neutropenic mice, MRSA), teixobactin was reported to be efficacious at 2.5 and 5 mg/kg doses (105). In a lung infection model (S. pneumoniae) with immunocompetent mice, treatment with 5 and 10 mg/kg (i.v., 24 and 36 h post infection) caused a CFU/mL reduction of six log units in the lung 48 h after infection, showing an effect comparable to amoxicillin at 10 mg/kg (105).
The research groups around Payne (123), Li (124), Nowick (118,125), Su (126), Albericio (127), Singh (128), and Brimble (129) developed syntheses of native teixobactin and/or analogues. Zhang and colleagues described a thioesterase-based chemo-enzymatic approach for the synthesis of teixobactin analogues (130). A gram-scale synthesis of teixobactin, applying a convergent strategy, was recently reported by Rao and colleagues (131). Many teixobactin analogues have been evaluated to establish a structure–activity relationship (SAR). These studies as well as the synthetic access to the parent natural product and analogues have been summarized in detail by Karas et al. (112). Therefore, only selected findings of the SAR studies are listed below.
Residues of the tail region and of the macrocycle were found to contribute to antibacterial potency of teixobactin, which is abrogated by substitution of nonpolar amino acids with lysin of appropriate configuration (127). The lysine scan was performed with Arg10-teixobactin (34) as a reference, where the hardly accessible L-allo-enduracididine residue in position 10 was replaced by arginine (Figure 10 ). The L-allo-End residue in position 10 can be replaced by positively charged amino acids maintaining similar activity than the natural product. Lys10-teixobactin (35) exhibited comparable potency against Gram-positive bacteria than Arg10-teixobactin (34) (125,126). Compound 35 was found to be nonhemolytic and not cytotoxic (RBCs and PBMCs showed a viability >90% at 64 μg/mL) (115).
Figure 10.

Teixobactin analogues obtained by substitution of L-allo-enduracididine in position 10.
A systematic alanine scan on Lys10-teixobactin (35) revealed that replacement of Lys10 by alanine surprisingly results in the still moderately active compound Ala10-teixobactin (36), which illustrates that a positive charge in this position is not essential (118). In line with this, also the L-citruline10 analogue 37 described by Schumacher et al. (129) showed considerably reduced but not completely abrogated activity. Remarkably hydrophobic substitution leading to Leu10- and Ile10-teixobactin was tolerated; the corresponding analogues 38 and 39 were reported by Singh and colleagues to be in vitro against MRSA as potent as teixobactin (Table 3) (116,132).
Substitution of NMe-D-Phe1 by D-biphenylalanine led to 41 (Figure 11 ), which exhibited excellent potency against MRSA and VRE. Further replacement of D-Thr8 by 2(R),3(S)-diaminobutanoic acid gave the twofold more potent lactam 42. The more readily available lactone 41 (dosed i.v. at 1–5 mg/kg) showed high efficacy in a mouse model of S. pneumoniae septicemia (133).
Figure 11.

Substitution D-Thr8 of lacton 41 by 2(R),3(S)-diaminobutanoic acid gave the twofold more potent lactam 42 (133). The disulfide macrocycle 43 (ring size increase by one atom) however was inactive (135). While the diastereomers 44 and 45 lost activity, the enantiomer conserved the activity of 34 against four Gram-positive organisms tested (125). The X-ray crystal structure of the truncated analogue 46 revealed that the amide groups of the depsipeptide ring residues Arg10 and Ile11 and those of Ser7 and D-Thr8 and the guanidinium group of Arg10 form a chloride anion-binding site (134).
Nowick and colleagues (125) compared the activity of two diastereomers 44 and 45 (Figure 11) of Arg10-teixobactin (34) and the enantiomer ent-Arg10-teixobactin (ent-34). While the diastereomers lost activity, the enantiomer conserved the activity of 34 against four Gram-positive organisms tested. The authors concluded that the NH groups of the macrolactone ring may undergo hydrogen-bonding interactions with the target pyrophosphate group. This view is corroborated by the X-ray crystal structure of Ac-Δ1–5-Arg10-teixobactin (46, Figure 11), a truncated analogue with the tail residues 1–5 replaced by an acetyl group, which crystallized as hydrochloride salt (even in presence of inorganic pyrophosphate anions and HCl) rather than forming a gel. The two amide groups of the depsipeptide ring residues Arg10 and Ile11 as well as the amide groups of Ser7 and D-Thr8 and the guanidinium group of Arg10 create a cavity into which the chloride anion is accommodated (134).
Furthermore, the Nowick group described the X-ray structure of the Lys10-teixobactin homologue 47 (MIC against S. aureus 16 μg/mL; Figure 12 ) with an α-N-methyl substituent at D-Gln4, which crystallized in the presence of sulfate ions as a hydrogen-bonded dimer (119). The NH-groups of the depsipeptide macrocycle (D-Thr8, Ala9, Lys10, and Ile11) of one molecule and the N-terminal methylammonium group of the other molecule form sulfate-binding sites. The tail regions of the two molecules form an amphipathic antiparallel β-sheet with the lipophilic side chains creating a hydrophobic surface. These β-sheet dimers form a larger β-sheet assembly, which consist of 16 molecules of homologue 47. Two such larger β-sheets form a double helix containing 32 molecules (119). Unlike 47, teixobactin and its gel-forming analogues are thought to form large β-sheet fibrils of this type (119). Based on these observations, Nowick and colleagues proposed that dimers or higher order assemblies of the antibiotic form pyrophosphate binding sites and adhere to the membrane surface.
Figure 12.

The X-ray crystal structure of the teixobactin homologue 47 solved by Nowick and colleagues (119) showed that β-sheet dimers are the subunits of larger assemblies. The NH-groups of the depsipeptide macrocycle of one molecule and the N-terminal methylammonium group of the other molecule create binding sites for oxy anions.
Efforts made to prepare teixobactin analogues with improved activity against Gram-negative bacteria, for example by increasing the number of positively charged side chains (Figure 13 ), unfortunately gave so far minimal results only (115,136). Synergy between teixobactin analogues and outer membrane disrupting peptides has been reported by Ng et al. (122) and Chiorean et al. (117).
Figure 13.

The threefold positively charged teixobactin analogue 48 exhibits only weak activity against Gram-negatives (115). In the presence of colistin, which disrupts the outer membrane, analogue 49 showed increased activity against P. aeruginosa PAO1 (MIC > 256 to 32 μg/mL) (122).
5.3. Ramoplanin
Ramoplanin A1–A3 are macrocyclic glycolipodepsipeptides produced by Actinoplanes through nonribosomal peptide synthesis (for details cf. Hoertz et al. (137)). Ramoplanins consist of 17 amino acid residues of which 13 are nonproteinogenic and ten of the amino acid residues have β-branched side chains. The 49-membered macrocyclic ring (which is essential for activity) is formed by 16 amino acid residues with a lactone bond between the β-hydroxy substituent of the asparagine residue in position 2 (HAsn2) and the C-terminus of the chloro-hydroxyphenylglycine in position 17. A disaccharide moiety is attached to the phenol OH group of hydroxyphenylglycine (Hpg) in position 11. The N-terminal exocyclic Asn1 is acylated with fatty acids. Ramoplanin A1–A3 differ only by this lipid substituent. Ramoplanin A2 (50; Figure 14 ) is most abundant and was evaluated in clinical trials. The structure elucidation, chemistry, and biology of ramoplanin including identification of the cellular target have been discussed and reviewed by Walker, Boger, and colleagues (138,139).
Figure 14.

Structure of ramoplanin A2 (50) (138) and schematic representation of the ramoplanin A2 dimer (147). Structure of the lipid II analogue 52 and the ramoplanin derivative NAI-603 (51) (148,149).
Ramoplanin is active against Gram-positive bacteria including S. aureus, B. subtilis, and E. faecalis strains and has no activity against Gram-negative bacteria as it probably cannot cross the outer membrane. The antibiotic is bactericidal and active against vancomycin-resistant enterococci, methicillin-resistant staphylococci, and vancomycin-intermediate-resistant C. difficile (138,140). Ramoplanin inhibits the cell-wall synthesis, leading to bacterial death. The intracellular enzyme MurG, catalyzing the conversion of lipid I into lipid II, has been proposed as target based on biochemical experiments using permeabilized bacterial cells and ramoplanin was thought to bind to the substrate (138,141). In the presence of an intact membrane, ramoplanin was proposed to block the polymerization of lipid II catalyzed by the bifunctional (transglycosylase, transpeptidase) penicillin-binding proteins PBP (138) by binding to the substrate, which is, as well as the enzyme, found on the external surface of the bacterial cellular membrane and thus accessible (138,142). Upon titration with the lipid II analogue 52 (Figure 14), ramoplanin complexes were found to polymerize to form fibrils, as evidenced by NMR and CD spectra and electron microscopy (142).
Ramoplanin was reported to exhibit good aqueous solubility (>100 mg/mL) (143). A solution structure of ramoplanin A2 monomer has been determined by NMR methods in water/DMSO-d6 4:1 (144) but did not allow to understand how the molecule binds lipid II. Lo et al. (145) observed in D2O spectrum of ramoplanin A2 sharp resonances of a single species. In CD3OD, two distinct species were observed, with concentration-dependent relative signal intensities. Lo et al. (145) found the data to be consistent with the presence of a monomer and a C2-symmetric dimer (as both species had only one set of signals for each ramoplanin proton). The interface of the dimer comprises amino acid residues 10–14 of each monomer and is stabilized by four hydrogen bonds. Based on their observations, Lo et al. (145) proposed ramoplanin A2 to exist as monomer in aqueous environment and to self-associate in proximity of the cellular membrane, creating lipid II binding sites. Enzyme-kinetic studies with E. coli PBP1b revealed a rapid rate increase with substrate (heptaprenyl lipid II) concentrations being more than half of the ramoplanin concentration which is in line with the antibiotic binding in 2:1 ratio to lipid II (146).
More recently, Hamburger et al. (147) reported a crystal structure of ramoplanin A2. Crystals were obtained in the presence of 1-hexadecyl-trimethylammoium bromide, with this detergent acting as membrane mimetic. Ramoplanin A2 was found to form a C2-symmetric amphipathic dimer. The hydrophobic face of the dimer is formed from Phe9, Leu15, D-Ala16, and the Asn1-attached acyl substituent. The U-shaped monomers interact with residues 9 to 15 with each other in antiparallel orientation, forming six intermolecular backbone hydrogen bonds and a hydrogen bond between D-Orn10 δ-NH and Asn1 γ-C O stabilizing the dimer. The peptide backbone of each monomer forms an antiparallel β-sheet connected at the ends by turns; the side chains of most residues are oriented outward (on the convex side of the U-shaped sheet). The two β-strands of each monomer are stabilized by eight (intramolecular) C O to backbone NH hydrogen bonds.
Boger and colleagues (150) performed an alanine scan for Dap2 analogue of ramoplanin A2 aglycon (53; Figure 15 ). The lactam 53 rather than the lactone was chosen for stability reasons (rapid hydrolysis of the lactone (151)) and its easiest synthesis. The antibacterial activity of 53 against S. aureus ATCC 25923 is comparable to that of ramoplanin A2 aglycon (150,151). In particular, D-Orn10 and then also D-Hpg3, D-Hpg7, and D-Orn4 were shown to be most critical for activity against the S. aureus strain (Table 4 ).
Figure 15.

Structures of lactam analogues 53 and 54 of ramoplanin A2 aglycon and structure of analogue 57 with truncated acyl chain (150,151). Structures of the hydrolysis products 55 and 56 of ramoplanin A2 (1) and ramoplanin A2 aglycon (152).
Table 4.
| Compound | MIC S. aureus ATCC 25923 [μg/mL] | MIC S. aureus ATCC 29213 [μg/mL] |
|---|---|---|
| Ramoplanin A2 aglycon | 0.11 | – |
| [Dap2]ramoplanin aglycon (53) | 0.07 | 0.4 |
| D-Hpg3 → D-Ala3-53 | 5.2 | 13 |
| D-Orn4 → D-Ala4-53 | 3.1 (44-fold) | 31 (78-fold) |
| D-Hpg7 → D-Ala7-53 | 3.7 | 13 |
| aThr8 → Ala8-53 | 2.5 | 38 |
| D-Orn10 → D-Ala10-53 | 38 (540-fold) | >50 (>125-fold) |
| D-aThr12 → D-Ala12-53 | 0.7 | 33 |
Based on their crystal structure, Hamburger et al. (147) explained these results: D-Orn10 is positioned at the interface between hydrophilic and hydrophobic regions of the ramoplanin A2 dimer and is thought to interact with the pyrophosphate moiety of lipid II. Walker et al. (138) compiled a list of lipid I analogues or fragments, which bind to ramoplanin and reported the presence of the pyrophosphate to be essential. D-Hpg3 was found to play a role in structure stabilization, D-Hpg7 and D-Orn4 in ligand recognition (147). The following (simplified) model for lipid II recognition by ramoplanin II was proposed by Hamburger et al. (147): Ramoplanin anchors (mediated by the N-acyl substituent) in the cellular membrane. The membrane stabilizes the dimer. The pyrophosphate group of lipid II forms a salt bridge with D-Orn10, lipid II polar head groups interact with the solvent exposed hydrophilic face of the dimer. The disaccharide (not important for activity (152)) serves a solubility mediator. The 2:1 stoichiometry was explained by formation of ligand-bridged ramoplanin dimers.
The alanine analogues of 53 have been tested later against S. aureus ATCC 29213 and among the four most affected positions were again D-Orn10 and D-Orn4 besides aThr8 and D-aThr12 (Table 4). Fang et al. (139) reported along with MIC values also the impact to bind a fluorescent lipid II analogue (and thus to inhibit the enzyme-mediated glycan polymerization). The D-Orn 10 → D-Ala analogue has the highest MIC and exhibits also a strongly decreased (ca 200-fold) binding of the lipid II analogue; the aThr8 → Ala analogue however showed an even larger increase (260-fold) in KD.
The hydrolysis products 55 and 56 (Figure 15) of ramoplanin A2 and ramoplanin A2 aglycon, respectively, were found to be markedly less potent (>250–500-fold against S. aureus ATCC 25923) than the macrocyclic natural product (152). Comparison of [Dap2]ramoplanin aglycon (53) with [Dab2]ramoplanin aglycon (54) also showed that the macrocycle is critical for antibacterial activity and introduction of the additional methylene group caused a > 100-fold loss in activity against S. aureus (151). The N-terminal lipid substituent contributes to potency, which was 16-fold reduced upon replacement by an acetyl group (→ 57) (151). The primary amide group of HAsn2, which is not present in 53, seems not to be required for activity.
Compound 54 showed in vitro inhibition of the E. coli PBP1b-catalyzed transglycosylation reaction comparable to 53 or ramoplanin A2 (50). The reduced potency observed in the antimicrobial assay resulted from an increased tendency of the compound to self-aggregate (153) rather than from reduced capability to inhibit the enzymatic reaction. Similarly, replacement of the lipid substituent of ramoplanin A2 aglycon by an acetyl group resulted in MIC value increase (reduced antimicrobial potency) without influencing the in vitro inhibition of the transglycosylase. This is in line with the key role of the lipid substituent in targeting the bacterial membrane (153). Ramoplanin was demonstrated by Cheng et al. (143) applying SPR methods to bind in concentration dependent manner to anionic (“bacterial”) membranes preferentially over zwitterionic (“mammalian”) ones.
Ramoplanin is not orally absorbed and in the plasma the lactone was described to be rapidly hydrolyzed (150,151,154). Jabes et al. (148), however, reported a half-life of 3.8 h in rat plasma. The local tolerability is low if injected i.v. (swelling and progressive necrotization at injection site, hemolysis revealed by urine discoloration within 24 h if dosed to rats at 10 mg/kg) (149,154). These properties are limiting the therapeutic application. Orally administered ramoplanin A2 was in clinical trials (10,155,156) and was approved by the FDA as antibiotic against gut colonization by vancomycin-resistant enterococcus (VRE) and against C. difficile (13). In 2017 and in 2019, no clinical trials were ongoing with ramoplanin (157,158) or there was no recent information.
Disease recurrence is observed in ca 25% of the patients after standard treatment against C. difficile and is thought to be at least in part a consequence of spores that remain in the gut lumen, tolerating the treatment of the acute infection. In an in vitro model, ramoplanin showed activity against C. difficile spores. Spore counts were reported by Kraus et al. (159) to be below the level of detection for 28 days after 30 minutes exposure to ramoplanin at 300 μg/mL (concentration found in feces). Vancomycin (500 μg/mL) or metronidazole (10 μg/mL) exposure however did not suppress spore counts. Kraus et al. postulated that ramoplanin adheres to the exosporium (an outer surface layer protecting the spores and preventing the release of inflammation triggering signals) ready to attack germinating cells. Ramoplanin might thus help to reduce relapse rates. Nanotherapeutics seemingly planned in 2016 a clinical investigation of the antispore properties of ramoplanin (155).
Attempts to overcome the limitation of ramoplanin to gastrointestinal and topical applications and to gain access to systemic use included the development of protecting formulations and the synthesis of analogues and derivatives with better stability/tolerability properties.
Ramoplanin administered in emulsions containing intralipid (160) as well as cyclodextrin-based formulations (154) have been reported to improve tolerability (in animals) while maintaining antimicrobial effectiveness (in vivo or in vitro).
Modification of the lipid substituent led to compound 51 (Figure 14; NAI-603; selected out of a set > 50 derivatives) with an improved pharmacological profile (148,149). Compound 51, while exhibiting potent in vitro antibacterial activity (spectrum comparable to that of the parent compound), caused no hematuria if dosed i.v. to rats at 20 mg/kg. No lesions at injection site or suffering was observed (148,149). The compound was further evaluated in vivo. The plasma half-life (3.27 h) was reported by Jabes et al. (148) to be comparable to that of ramoplanin, the Cmax (207 μg/mL, measured 3 min after dosing) of 51 was 2.6-fold higher after i.v. administration at a dose of 20 mg/kg. S.c. administration of the same dose gave a bioavailability of 52%. The in vivo potency of the compound was demonstrated in a rat granuloma pouch model (induced by MRSA). NAI-603 (51) induced a 2–3 log10-reduction in viable bacteria, if dosed at 40 mg/kg i.v.
6. Peptide antibiotics addressing targets located at the outer membrane
6.1. Thanatin
Isolated from hemipteran insect Podisus maculiventris (spined soldier bug), the 21-amino acid peptide Thanatin (G1SKKPVPIIYC11NRRTGKC18QRM, with a disulfide bond between Cys11 and Cys18, 58; Figure 16 ) was first described in 1996 (161). Thanatin showed antibacterial activity against some Gram-positive and Gram-negative bacteria, and also antifungal activity. The all-D thanatin enantiomer was significantly less active, in particular against E. coli. The MoA was not known until recently (16,162,163).
Figure 16.
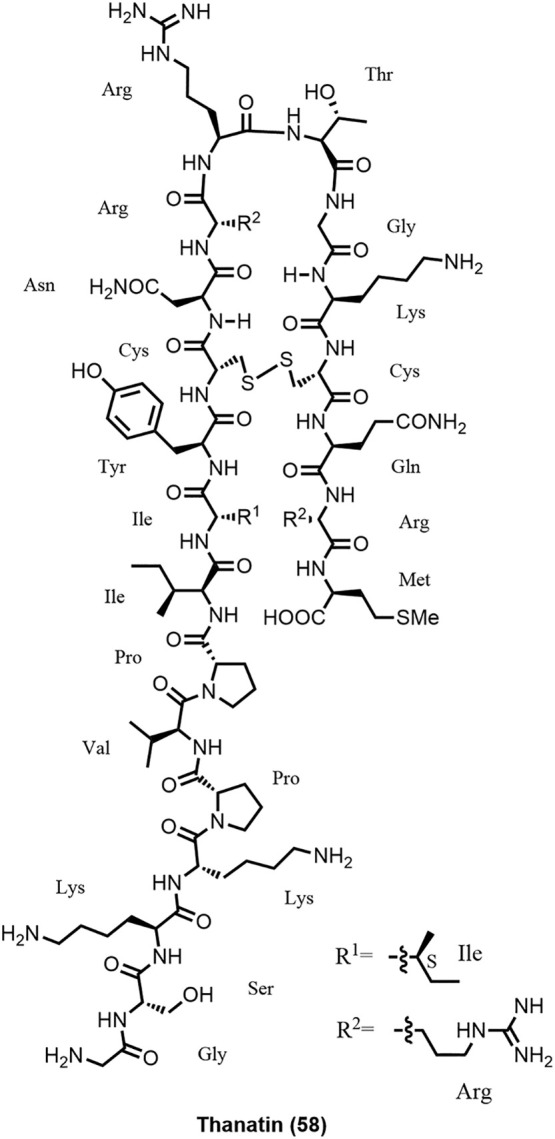
Structure of thanatin.
A close analogue of thanatin, S-Thanatin (Thr15 replaced by Ser), binds to lipopolysaccharide (LPS) and shows good in vivo efficacy in a mouse model of septic shock (164) (ED50 = 7 mg/kg). Alanine-scanning studies showed that residues in the disulfide loop seem critical for antimicrobial activity (165). S-Thanatin was well tolerated in mice (166). As shown by NMR, thanatin forms in solution a β-hairpin-like structure from residue 8 to the C-terminus stabilized by the disulfide loop and three C-terminal residues (167). The hairpin structure has been confirmed by an NMR solution structure in zwitterionic micelles (168). The hairpin conformation seems to be relevant for activity, especially against Gram-negative bacteria (165).
Several MoA hypotheses have been postulated. In Gram-negative bacteria, thanatin binds to LPS and leads to membrane permeabilization, however, at concentrations that are well above the observed MICs. Recently, it was postulated that thanatin disrupts the outer membrane of NDM-1-producing bacteria by competitively displacing divalent cations on the outer membrane and inducing release of LPS. In addition, it was shown that thanatin binds to the NDM-1 enzyme by displacing zinc ions from the active site, and reverses carbapenem resistance in NDM-1 producing bacteria in vitro and in vivo (163). However, this MoA alone does not explain the potent Gram-negative antimicrobial activity and spectrum of thanatin.
Robinson et al. showed conclusively by various biochemical, biophysical, structural biology, and genetic experiments (16) that the promising Gram-negative antimicrobial activity of thanatin was mainly driven by the interaction with lipopolysaccharide transport protein A (LptA), a crucial component of the LPS export machinery (169). These results were recently fully confirmed by another group showing that thanatin breaks the LptA-LptA and LptA-LptC interactions leading to inhibition of LPS transport across the periplasm (162).
In summary, MoA studies show that thanatin has some membrane permeabilization characteristics, which explains its relatively weak antimicrobial activity against Gram-positive bacteria and fungi. However, additional studies show that the promising Gram-negative activity of thanatin is mainly driven by its binding to LptA by inhibition of LPS export.
The thanatin example is a very interesting show case highlighting that AMPs can exert different MoAs. On one hand, thanatin targets rather unspecifically bacterial and fungal membranes, whereas in Gram-negative bacteria, it addresses a specific protein target.
6.2. Murepavadin
Naturally occurring antimicrobial peptides make interesting starting points for the design and synthesis of biologically active peptide mimetics. Phenotypic screening of a library of protein epitope mimetics (PEMs) (170,171 [176]), inspired by antimicrobial peptides such as protegrin I, on a panel of Gram-negative bacteria gave initial hits with interesting antimicrobial activity against P. aeruginosa, an important pathogen causing ventilator- and hospital-associated/acquired bacterial pneumonia (VABP and HABP). A significant lead optimization effort led to highly potent and selective antibiotics such as POL7001 and POL7080 (59; Figure 17 ) with excellent efficacy in various mouse infection models (15). POL7080 (Murepavadin) was selected as a clinical candidate for further development (172).
Figure 17.
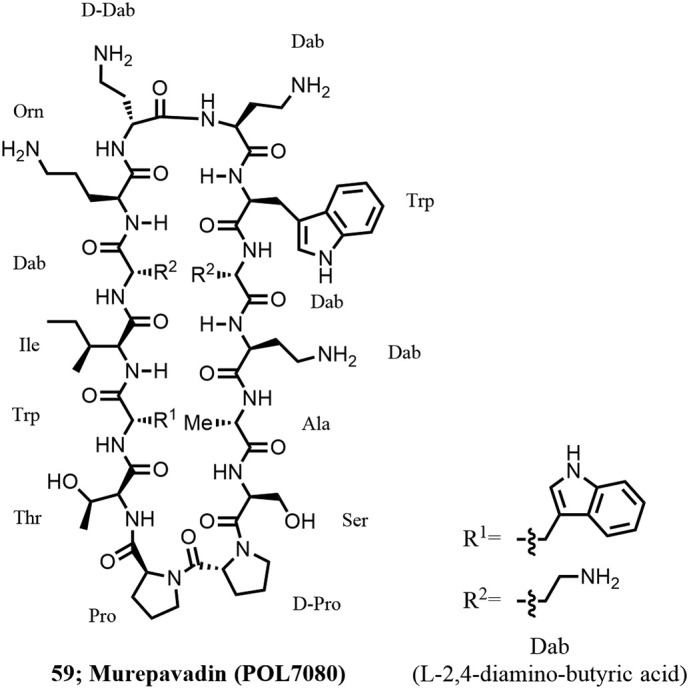
Structure of Murepavadin.
By various biochemical, biophysical, and structural biology experiments, it was shown that POL7080 (Murepavadin) binds to the periplasmatic jellyroll domain of lipopolysaccharide transport protein D (LptD) of Pseudomonas, which significantly differs from LptD of other Gram-negative bacteria, blocking translocation of LPS from the periplasm to the outer membrane (15,173,174). Murepavadin was the first antibiotic to target specifically an essential outer membrane protein (Omp). The in vitro and in vivo pharmacology, pharmacokinetics, and clinical pharmacology were recently described (172). In a phase II open-label study in VABP (NCT02096328), 25 VABP patients received murepavadin of which twelve patients had a microbiologically documented infection due to P. aeruginosa (mITT population), nine of which caused by a multidrug-resistant or extensively resistant isolate. Clinical cure at test of cure was achieved in 10 out of 12 (83%) patients with confirmed P. aeruginosa (mITT population) and the 28 days all-cause mortality in this population was very low (8%), far below the expected 20–40% mortality rate. Based on these promising results and albeit a small sample size, two phase III trials, PRISM-MDR (NCT03409679) and PRISM-UDR (NCT03582007), in HABP/VABP were initiated. Due to elevated incidences of acute kidney injury compared to the comparator arm, the trials were temporarily halted in May 2019.
Murepavadin is currently also investigated as an inhaled formulation for treatment of people with cystic fibrosis (CF). Initial in vitro and in vivo feasibility studies were done with POL7001, a close analogue of murepavadin (POL7080) (175), showing excellent antimicrobial activity on a large panel of a P. aeruginosa clinical CF strain collection. PK studies in mice showed a high compound exposure in the lung with low exposure in plasma by intratracheal administration. The excellent exposure in the lung translated in good efficacy in murine models of P. aeruginosa-RP73 acute and chronic infection. Pulmonary administration also resulted in body weight recovery and reduced inflammatory markers (175).
6.3. OMPTA-BamA
By linking two macrocyclic pharmacophores against two essential bacterial targets, LPS and BamA, located in proximity at the outer membrane of Gram-negative bacteria, a novel class of antibiotics with potent activity against carbapenem-resistant P. aeruginosa and A. baumannii and carbapenem and third-generation cephalosporin-resistant Enterobacteriaceae (WHO priority-1 pathogens) was discovered. First hits (e.g., 60; Figure 18 ) with good activity against MDR-XDR strains were discovered by phenotypic screening of a library constituted of 14-amino acid macrocyclic β-hairpin mimetics (7) derived from active peptide sequences of antimicrobial peptides, such as protegrin-1 or murepavadin (15). While 60 shows structural similarity to murepavadin in the basic loop region (LPS binding region), it differs significantly in the key β-hairpin region (Omp binding region) suggesting a different MoA from murepavadin. While MICs of 60 were in the range of 1–4 μg/mL in MH-II broth, there was a significant and problematic serum deactivation effect observed requiring further structural improvements.
Figure 18.
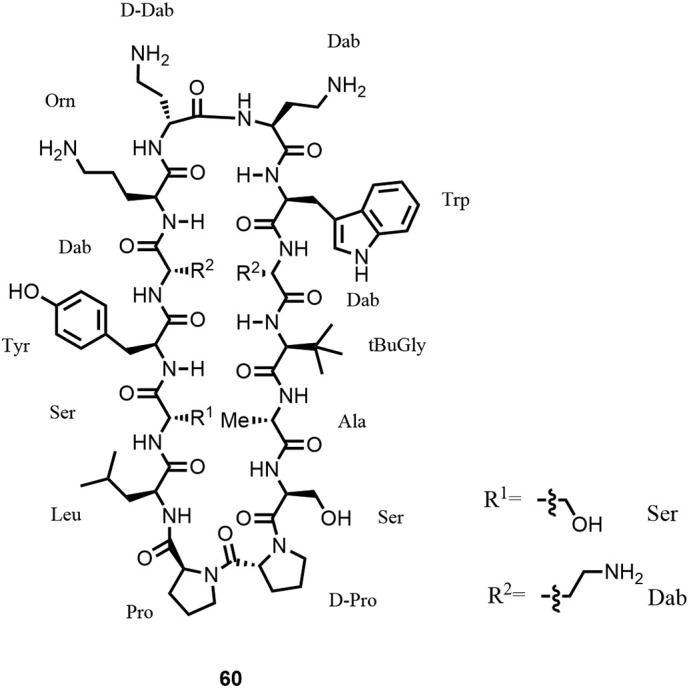
Structure of β-hairpin mimetic 60, corresponding to compound 2 in Ref. (17).
By linking a 7-amino acid macrocycle derived from colistin, known to have affinity towards the lipid A part of LPS, a favorable binding of such a chimeric antibiotic to LPS and neighboring essential β-barrel outer membrane proteins such as LptD and BamA was expected. After a significant library synthesis effort optimizing the β-hairpin and linker sequences as well as finding the optimal positioning of the linkage to the 7-amino acid macrocycle derived from colistin, chimeric antibiotics such as compound 61 (Figure 19 ) with potent in vitro antimicrobial activity against all priority-1 Gram-negative bacteria were discovered (17,177,178). The compounds were selective against Gram-negative bacteria with no residual activity against Gram-positive bacteria or fungi. In a substantial medicinal chemistry effort, initial hits were then optimized to obtain molecules with potent in vivo activity in various murine infection models and with appropriate ADMET properties. The chimeric antibiotics are bactericidal, not hemolytic, only moderately cytotoxic, and show a very low propensity to generate resistance. Excellent in vitro antimicrobial activity was obtained on a large panel of WHO priority-1 strains (MIC range 0.06–0.25 μg/mL).
Figure 19.
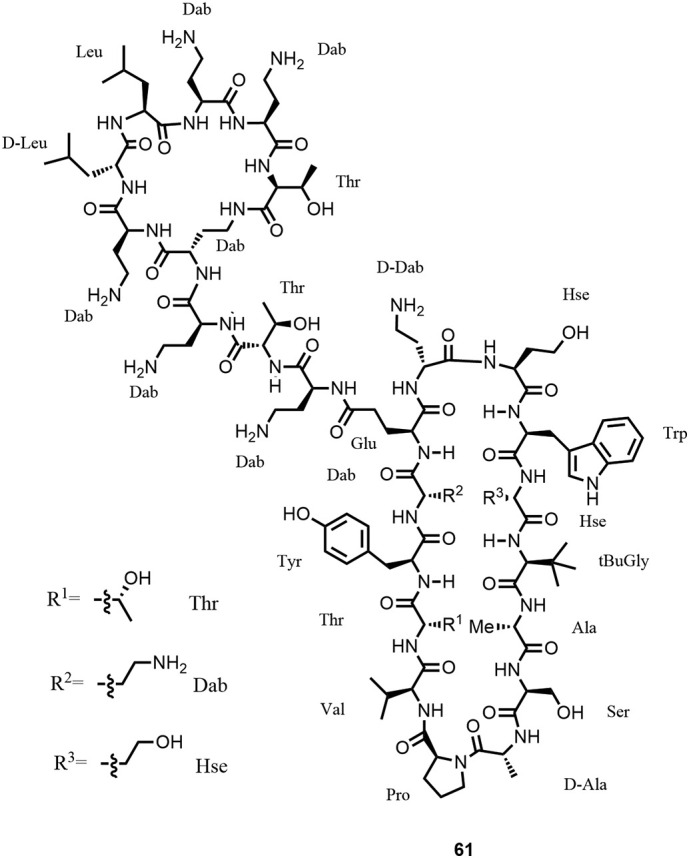
Structure of chimera 61, corresponding to compound 3 in Ref. (17).
After further optimization of ADMET properties such as cytotoxicity on different cell lines, a new scaffold was found where the hairpin structure was stabilized by a disulfide bond as represented by chimera 62 (Figure 20 ), which showed an excellent in vitro and in vivo profile. Potent in vivo efficacy was for example obtained in murine infection models against extensively drug-resistant A. baumannii NCTC 13301, resistant NDM-1 metallo-β-lactamase containing E. coli ATCC BAA2469 and colistin-resistant (mcr-1) E. coli AF45. POL7306, a close analogue of 62, was tested against a total of 891 clinical isolates collected by the SENTRY Antimicrobial Surveillance Program, focusing on MDR/XDR/colistin-resistant priority-1 Gram-negative pathogens, and demonstrated potent in vitro activity (MIC50/90, 0.12/0.25 μg/mL) (179). By combining biochemical, biophysical, and genetic studies as well as high-resolution NMR experiments, the mechanism of action could be identified and confirmed the initial hypothesis of LPS and BamA interaction. BamA is a bacterial β-barrel protein and an essential part of the BAM (β-barrel assembly machinery) complex, which is critical for proper folding and insertion of outer membrane protein precursors into the outer membrane (180). Optimized lead compounds are currently under preclinical evaluation.
Figure 20.
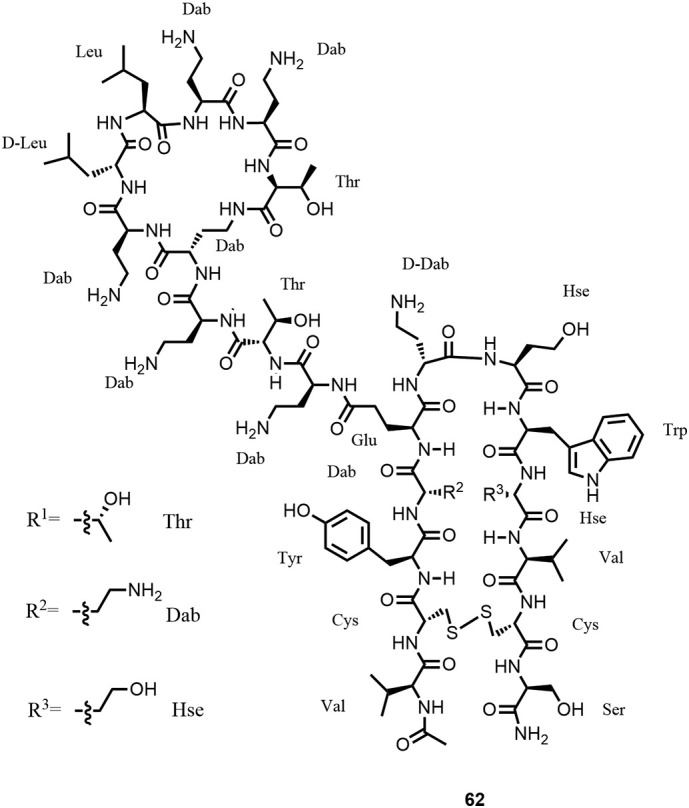
Structure of chimera 62, corresponding to compound 7 in Ref. (17).
6.4. Darobactin
Whereas most of the large successful antibiotic classes were discovered from soil microorganisms (mainly Actinomycetes), darobactin was isolated from Photorhabdus symbionts of nematodes (18,177,178). Darobactin (63; Figure 21 ) is a ribosomally synthesized peptide and features an unusual structure with two fused macrocyclic rings containing two substituted tryptophan indole rings forming a compact hairpin-like structure.
Figure 21.
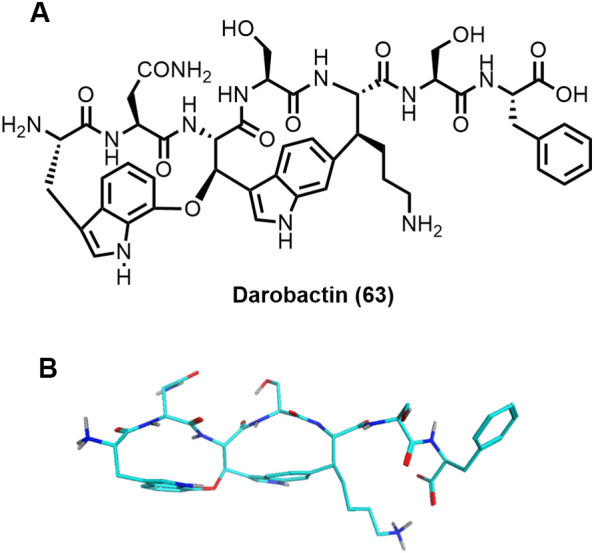
The structure of darobactin was generated performing a conformational search using low-mode molecular dynamics simulation in implicit water as implemented in MOE from the Chemical Computing Group (Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite#910, Montreal, QC, Canada, H3A 2R7, 2020).
Darobactin has a molecular weight of 965 Da and shows a good to moderate activity against a range of Gram-negative bacteria such as E. coli and K. pneumoniae, including colistin-resistant, extended spectrum β-lactamase-resistant (ESBL), and carbapenem-resistant clinical isolates in the 2 μg/mL range. Low activity was observed against A. baumannii and E. cloacae. Bactericidal activity was observed at concentrations around 8 μg/mL. The promising in vitro activity was confirmed in vivo in mouse thigh infection and septicemia models with three doses of 25 mg/kg per day given intraperitoneally (18). By a series of experiments, the mode of action and the target of darobactin were identified. Darobactin causes blebbing of the membrane, and eventual swelling and lysis of the cells indicating a membranolytic effect. However, mutational studies unambiguously showed several mutations in the gene encoding for BamA. Transferring the BamA mutations from resistant strains into a clean E. coli background by allelic replacement conferred resistance to darobactin and confirmed BamA as the main target. Binding of darobactin to BamA was also confirmed by NMR studies (18). BamA is an essential protein and a member of the BAM complex critically involved in folding and insertion of β-barrel outer membrane proteins (180).
6.5. Arenicin-3, NZ17074, and AA-139
Arenicin-1 and -2 are small antimicrobial peptides isolated from coelomocytes of marine lugworm Arenicola marina (Figure 22 ; Table 5 ; (181)) consisting of one disulfide bond and are rich in arginine and hydrophobic amino acids. Arenicin-3, originally isolated from the same lugworm, contains nine hydrophobic residues, four positively charged arginine residues, and two disulfide bonds (Cys3-Cys20 and Cys7-Cys16), which stabilize an amphipathic β-hairpin structure. NMR solution structures (deposited as PDB5v0y and BMRB30259) revealed that the two β-strands are connected by a type I’β-turn, formed by Asn11 and Gly12, and the β-sheet is further stabilized by four hydrogen bonds (182). Arenicin-3 shows good activity against a variety of Gram-negative bacteria (182,183), however, shows high protein binding (99%) to serum components (183), and was discussed to be hemolytic and cytotoxic (182).
Figure 22.

Arenicins 1–3 and analogue NZ17074.
Table 5.
Physicochemical Properties of Arenicins and Analogues of Arenicin-3
| Peptide | Nr of aa | MW [Da] | S-S Bonds | calc. pI | Net Charge | Reference |
|---|---|---|---|---|---|---|
| Arenicin-1 | 21 | 2758.3 | 3–20 | +6 | (181) | |
| Arenicin-2 | 21 | 2772.3 | 3–20 | +6 | (181) | |
| Arenicin-3 | 21 | 2611.1 | 3–20; 7–16 | 11.17/9.25 | +4 | (182,184) |
| NZ-17074⁎ | 21 | 2538.0 | 3–20; 7–16 | 9.37 | +4 | ([182], [183], [184], [185]) |
| N2 | 21 | 2570.0 | 3–20; 7–16 | 9.38 | +4 | (184) |
| N6 | 21 | 2477.8 | 7–16 | 10.72 | +4 | (184) |
| AA-139 | 21 | 2548.1 | 3–20; 7–16 | 9.69 | +5 | (182) |
| NZ17143 | 21 | 2529.2 | 3–20; 7–16 | 10.45 | +5 | (182) |
| NZ17224 | 21 | 2550.1 | 3–20; 7–16 | 9.88 | +5 | (182) |
NZ17074 is an arenicin-3 variant, which was identified by Neve et al. from Novozyme by high throughput screening of yeast libraries (183,187). NZ17074 differs from arenicin-3 in positions 5 (Y→N) and 17 (Y→H). Based on the analysis of CD spectra, Wang et al. (186) reported NZ17074 to adopt in aqueous solution a secondary structure with β-sheet character; however, in lipophilic environment, in the presence of 10 mM sodium dodecyl sulfate, an α-helical structure was induced.
Synthetic NZ17074 was obtained by solid-phase peptide synthesis applying Fmoc chemistry (185). Recombinant NZ17074 (rNZ17074) was produced in yeast (Pichia pastoris) by expression as SUMO3-NZ17074 fusion protein, followed by purification, cleavage to release rNZ17074, and final purification. Recombinant NZ17074 has an additional Pro at the N-terminus; it was demonstrated to be active in antibacterial assays like NZ17074 (188). It is worth noting that recombinant 15N-labeled AA-139 (a further arenicin-3 analogue, see below) was expressed as His-tagged SUMO fusion protein in a bacterial system (E. coli) described by Zhang et al. (189).
Against Gram-negative bacteria, NZ17074 displays MIC values in the range 0.25–8 μg/mL (E. coli strains 0.25–1 μg/mL; Salmonella strains 0.25–0.5 μg/mL; P. aeruginosa strains 2–8 μg/mL) (183,186). Weaker activity (MICs of 2–32 μg/mL) has been reported against a selection of Gram-positive bacteria (186). In addition, NZ17074 exhibits antifungal activity against Candida albicans (MIC 4–16 μg/mL) (185,186) and low to moderate cytotoxicity against porcine intestinal epithelial cells and mouse peritoneal macrophages (186). As compared to arenicin-3, NZ17074 shows reduced serum protein binding (79% (183,187)) and was found to bind LPS as determined by MIC and probe displacement methods.
The antibacterial mechanism against E. coli cells proposed and investigated by Wang et al. (186) comprises binding to LPS, rapid penetration through the outer membrane, disruption of the plasma membrane and release of cell content, binding to DNA and induction of conformational changes, inhibition of DNA and RNA synthesis, and induction of apoptosis-like cell death.
In a mice peritonitis model (E. coli CVCC1515; i.p. injection of 2.5x108 CFU), survival of 100% of the animals was observed after i.p. injection of 1.25 and 2.5 mg/kg NZ17074 (administration at 0.5 h and 8 h after inoculation). Mice without treatment died within 24 h; a dose of 0.625 mg/kg polymyxin B resulted in 100% survival (186). Survival of the animals was monitored for 7 days. The activity of NZ17074 was also investigated in an endotoxemia model (i.p. injection of 30 mg/kg LPS in mice) where 100% of the mice treated at 0.5 and 8 h after inoculation with 5 mg/kg NZ17074 survived. In contrast, none of the mice treated with only 2.5 mg/kg NZ17074 survived in the same model (186).
Analogues N2, N6, and AA-139 starting from NZ17074 and arenicin-3, respectively, were obtained by modulation of lipophilicity/amphipathicity, optimizing cytotoxicity, charge distribution, and reducing the overall net positive charge content.
Yang et al. (184) reported the evaluation of seven analogues of NZ17074, which were prepared with the objective to reduce cytotoxicity. Essential for antimicrobial activity were the Val residues in positions 6, 8, 13, and 15 as well as Asn in positions 11 and 21 (184,187). Two analogues (Figure 23 ) with potent antibacterial activity were obtained: in N2, the Gly residues in positions 1 and 12 were replaced by Ala; in N6, the two Cys residues in positions 3 and 20 were replaced by Ala, leading to a molecule with only one disulfide bond.
Figure 23.

NZ17074 derived analogues (184).
Analogue N2 exhibits MIC values of 0.25–2 μg/mL against E. coli, Salmonella, and Pseudomonas strains and compound N6 was found to show equal or slightly reduced activity as compared to parent NZ17074 (184). Hemolysis against murine erythrocytes by N2 (0.4%) and N6 (0.04%) was reported to be lower than that of NZ17074 (3% against human RBC (185)) at 128 μg/mL. Compared to parent NZ17074, both analogues also exhibit lower cytotoxicity (determined as % survival at 64 and 128 μg/mL) against murine peritoneal RAW264.7 macrophage cells. Anti-fungal MICs were reported to be increased by one or two dilutions (C. albicans CGMCC2.2411: from 16 μg/mL for NZ17074 to 32 (N2) and 64 μg/mL (N6)) (184).
Like parent NZ17074, N2 and N6 were found to potently bind to LPS (comparable to polymyxin B) as determined by a probe displacement assay. Docking calculations suggest that the three molecules differ in the sites of interaction with lipid A (184).
In vivo activity of N2 and N6 was evaluated in mice peritonitis models (inoculation with E. coli CVCC1515 or with S. enteritidis CICC22956). In the E. coli model, 100% of the mice treated by i.p. injection of 2.5 mg/kg N2 0.5 h and 8 h after inoculation survived, whereas, however, only 66.7% of the mice receiving the same dose of N6 survived. In the S. enteritidis model, 100% survival was observed if mice were treated with 7.5 mg/kg N2 or N6; at lower dose, N2 administration resulted in higher survival rate than N6 administration. The LPS detoxifying activity of the two analogues was also demonstrated in vivo; N2 and N6 were reported to be more efficacious than polymyxin B (184).
AA-139 (Figure 24 ) is a synthetic analogue derived from arenicin-3, prepared by solid phase peptide synthesis. Val in positions 8 and 13 of arenicin-3 was replaced by Ala; Tyr in position 9 by Arg. These mutations reduced the lipophilicity and increased the overall charge (Figure 24, Table 5). The peptide was obtained as a member of a library, which was based on an alanine scan and removal or mutation of one to four residues at varying positions, while keeping the Cys residues to maintain integrity of the secondary structure (182).
Figure 24.

Sequence of the arenicin-3 analogue AA-139. NZ17143 combines the structural changes of NZ17074 (Y5N, Y17H) with the replacement with the Y9R replacement of AA-139. NZ17224 has like NZ17074 a Tyr replaced by His, combined with the V8A and Y9R mutations of AA-139.
AA-139 exhibited potent activity against a variety of Gram-negative bacteria, like the reference strain E. coli ATCC 25922 (MIC 0.125 μg/mL), the New Delhi metallo β-lactamase producing strain K. pneumoniae BAA-2146 (MIC 1 μg/mL), as well as MDR and XDR strains, including polymyxin-resistant P. aeruginosa clinical isolate (MIC 2 μg/mL) and XDR/PmxR A. baumannii clinical isolate (MIC 0.5 μg/mL). Lower activity, however, was observed against Gram-positive bacteria and yeasts. In the presence of serum or the lung surfactant Survanta®, a 2–16-fold loss of in vitro antibacterial activity was reported against a panel of Gram-negative control strains and clinical isolates (182) (Table 6 ). Compound NZ17143, combining the structural changes of NZ17074 (Y5N, Y17H) with the Y9R replacement of AA-139, shows comparable activity as well as NZ17224 with the Y5H, V8A, and Y9R mutations. (The compound combining all changes of NZ17074 and AA-139, so Y5N, V8A, Y9R, V13A, and Y17H was not reported).
Table 6.
Broth Microdilution MIC Values [μg/mL] of Arenicin-3 and Analogues in Mueller Hinton Broth (MHB); in the Presence of 50% Serum; in the Presence of 5% Survanta® (Human Lung Surfactant). Comparison with Colistin and Polymyxin B (PB); Data taken from (182) (incl. suppl. Information/Data)
|
E. coli ATCC 25922 |
P. aeruginosa ATCC 27853 |
P. aeruginosa FADDI-070 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| MHB | 50% Serum | 5% Survanta | MHB | 50% Serum | 5% Survanta | MHB | 50% Serum | 5% Survanta | |
| Arenicin-3 | 1 | 16 | 8 | 1 | 16 | 32 | 4 | ≥32 | ≥64 |
| AA-139 | 0.125 | 2 | 0.125 | 0.25 | 8 | 0.5 | 2 | 8 | 8 |
| NZ17143 | 0.25 | 1 | 0.5 | 1 | 8 | 1 | 8 | 16 | 8 |
| NZ17224 | 0.06 | 1 | 0.25 | 0.25 | 16 | 1 | 1 | 8 | 8 |
| Colistin | 0.25 | 0.125 | 64 | ||||||
| PB | 0.25 | 0.25 | 32 | ||||||
AA-139 as well as the analogues NZ17143 and NZ17224 showed higher CC50 values against three human cell lines (Table 7 ) than arenicin-3 and were less hemolytic. AA-139 was further reported to show an IC50 of 330 μg/mL against human primary hepatocytes.
Table 7.
Cytotoxicity of Arenicin-3 and Analogues against Human Cell Lines; as Reported by Elliott et al. (182)
| HEK293 CC50 [μg/mL] | HepG2 CC50 [μg/mL] | HK-2 CC50 [μg/mL] | |
|---|---|---|---|
| Arenicin-3 | 271 | >300 | 170 |
| AA-139 | >300 | >300 | >250 |
| NZ17143 | >300 | >300 | >250 |
| NZ17224 | >300 | >300 | >250 |
The mode of action of arenicin-3 and analogues was addressed in several additional experiments. Zhang et al. (189) used cyclized lipid bilayer nanodiscs as a membrane model to characterize the membrane binding properties of AA-139. Nanodiscs are self-assembled soluble particles consisting of two membrane scaffold proteins (MSP) wrapped around a phospholipid bilayer. In cyclized nanodiscs, the MSP C- and the N-termini are ligated. NMR methods and ITC were used to study the interaction of AA-139 with nanodiscs containing either zwitterionic (POPC) or anionic (POPC: POPG 4:1) lipids. They found that AA-139 interacts with lipophilic and positively charged residues at the termini of the β-hairpin structure with the anionic membrane (KD ca 1 μM) exclusively. No evidence was obtained for AA-139 disrupting the membrane in this model system.
Based on SPR experiments comparing the binding of arenicin-3 and analogues including AA-139 to DMPC and DMPC combined with E. coli lipid A (9:1 molar ratio), Elliott et al. (182) concluded that there was no binding to lipid A, as in the SPR experiment no significant difference in binding was observed. These authors also tested the permeabilization activity of arenicin-3 variants in several assays. The results were consistent with binding to and disruption of bacterial membranes, cell permeabilization, cytoplasmic leakage and cell death. Phospholipid transport was also associated with the mechanism of action of arenicin-3 in E. coli. Whole-genome sequencing of 20 days serial passage (forced resistance) isolates of E. coli ATCC 25922 in the presence of arenicin-3 led to a point mutation T32G in the mlaC gene, which translated into the L11R replacement in the protein. The mla operon (regulating the expression of the phospholipid transport system mlaABCDEF) maintains the outer membrane asymmetry (182).
Good efficacy for i.v. administered AA-139 was demonstrated in neutropenic murine peritonitis and thigh infection models (182). Aerolized AA-139, generated by a Hudson RCI Micro-mist nebulizer, was tested in a pneumonia model (P. aeruginosa ATCC 27853). When infected mice were exposed 2, 12, and 24 h after infection to aerosol of AA-139 (30 mg/mL) for 30 min, a 6.6 log reduction of bacterial burden in the lung was reported. To determine the exactly delivered dose was not possible. The PK profile of inhaled AA-139 showed high concentrations in epithelial lining fluid (ELF) and low concentrations in plasma (182). Finally, AA-139 was also demonstrated to be active in a urinary tract infection model; the analogue NZ17143 showed similar efficacy in this model (182).
Target organ toxicity was reported to affect the kidney with tubular nephropathy after i.v. dosing at 20, 25 or 30 mg/kg of AA-139 per day to minipigs for seven days. Severity increase was found to be dose related. A NOAEL of 20 mg/kg AA-139 per day (10 times the dose required for efficacy in murine infection models) by i.v. administration over 2 h once or twice daily was reported in mice. For inhaled AA-139, the NOAEL was 20 mg/kg per day in mice (182).
7. Summary and outlook
Natural products have been the cornerstone of antibiotic discovery and development. Among the different natural product classes, peptide-derived antibiotics play an increasingly important role. Within the peptide-derived antibiotics, AMPs and HDPs, as constituents of the innate immune system, have attracted a lot of attention as potential scaffolds for antibiotic drug design. It is clear now that AMPs and their derivatives are not simply membrane disrupting agents, but scientists start to uncover novel intriguing molecular targets and mechanisms of action.
We have focused in this short review on peptide-derived antibiotics, which have the potential to lead to novel antibiotic classes with novel MoA, which are eagerly awaited by patients, doctors, and healthcare organizations.
Examples such as the odilorhabdins (30S ribosomal subunit), tridecapeptins and malacidins (lipid II), arylomycins (SPase I), teixobactin (lipid II and lipid III), and ramoplanin (lipid II) are nice examples of promising peptide-derived antibiotics with well-described targets and MoAs. Recently, novel peptide-derived antibiotics have been discovered against so called “undruggable” essential targets located in the outer membrane of Gram-negative bacteria. Murepavadin (LptD), chimeric β-hairpin mimetics and darobactin (both BamA), and thanatin (LptA) are promising examples of natural products or nature-inspired peptide antibiotics that underscore the uniqueness and promise of peptide antibiotics to significantly contribute in the fight against antimicrobial resistance. An important success factor, however, will be the ability of medicinal chemists to optimize peptide scaffolds in order to achieve the appropriate required balance between in vivo antimicrobial activity and safety.
Author agreement
All authors have seen and approved the final version of the manuscript. Polyphor acknowledges funding from CARB-X and financial support from the REPAIR Impact Fund (Novo Holdings).
CRediT author statement
Gregory Upert: writing - original draft; writing review & editing. Daniel Obrecht: writing - original draft. Anatol Luther: writing - original draft. Philipp Ermert: writing - original draft; writing review & editing.
Funding Source
There is no funding source for this article.
Declaration of competing interest
Gregory Upert, Daniel Obrecht and Philipp Ermert are employees of Polyphor Ltd. Anatol Luther is an employee of Bachem AG.
Footnotes
In memory of Professor Sir Jack Baldwin
References
- 1.Boucher H.W., Talbot G.H., Bradley J.S., Edwards J.E., Gilbert D., Rice L.B., et al. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Tacconelli E., Carrara E., Savoldi A., Harbarth S., Mendelson M., Monnet D.L., et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- 3.O’Neill J. Antimicrobial resistance: tackling a crisis for the health and wealth of nations 2014. 2015. https://amr-review.org/sites/default/files/Report-52.15.pdf (Accessed October 10, 2020)
- 4.Wencewicz T.A. New antibiotics from Nature’s chemical inventory. Bioorg Med Chem. 2016;24:6227–6252. doi: 10.1016/j.bmc.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Theuretzbacher U., Outterson K., Engel A., Karlén A. The global preclinical antibacterial pipeline. Nat Rev Microbiol. 2020;18:275–285. doi: 10.1038/s41579-019-0288-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler M.S., Paterson D.L. Antibiotics in the clinical pipeline in October 2019. J Antibiot (Tokyo) 2020;73:329–364. doi: 10.1038/s41429-020-0291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luther A., Bisang C., Obrecht D. Advances in macrocyclic peptide-based antibiotics. Bioorg Med Chem. 2018;26:2850–2858. doi: 10.1016/j.bmc.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koehbach J., Craik D.J. The vast structural diversity of antimicrobial peptides. Trends Pharmacol Sci. 2019;40:517–528. doi: 10.1016/j.tips.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Koo H.B., Seo J. Antimicrobial peptides under clinical investigation. Pept Sci. 2019;111 doi: 10.1002/pep2.24122. [DOI] [Google Scholar]
- 11.Xue Y., Wang M., Zhao P., Quan C., Li X., Wang L., et al. Gram-negative bacilli-derived peptide antibiotics developed since 2000. Biotechnol Lett. 2018;40:1271–1287. doi: 10.1007/s10529-018-2589-1. [DOI] [PubMed] [Google Scholar]
- 12.Hancock R.E.W., Sahl H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 13.Naclerio G.A., Sintim H.O. Multiple ways to kill bacteria via inhibiting novel cell wall or membrane targets. Future Med Chem. 2020;12:1253–1279. doi: 10.4155/fmc-2020-0046. [DOI] [PubMed] [Google Scholar]
- 14.Choi U., Lee C.-R. Antimicrobial agents that inhibit the outer membrane assembly machines of gram-negative bacteria. J Microbiol Biotechnol. 2019;29:1–10. doi: 10.4014/jmb.1804.03051. [DOI] [PubMed] [Google Scholar]
- 15.Srinivas N., Jetter P., Ueberbacher B.J., Werneburg M., Zerbe K., Steinmann J., et al. Peptidomimetic antibiotics target outer-membrane biogenesis in P. aeruginosa. Science. 2010;327:1010–1013. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 16.Vetterli S.U., Zerbe K., Müller M., Urfer M., Mondal M., Wang S.-Y., et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in E. coli. Sci Adv. 2018;4 doi: 10.1126/sciadv.aau2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luther A., Urfer M., Zahn M., Müller M., Wang S.-Y., Mondal M., et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature. 2019;576:452–458. doi: 10.1038/s41586-019-1665-6. [DOI] [PubMed] [Google Scholar]
- 18.Imai Y., Meyer K.J., Iinishi A., Favre-Godal Q., Green R., Manuse S., et al. A new antibiotic selectively kills gram-negative pathogens. Nature. 2019;576:459–464. doi: 10.1038/s41586-019-1791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le C.-F., Fang C.-M., Sekaran S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob Agents Chemother. 2017;61 doi: 10.1128/AAC.02340-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pantel L., Florin T., Dobosz-Bartoszek M., Racine E., Sarciaux M., Serri M., et al. Odilorhabdins, antibacterial agents that cause miscoding by binding at a new ribosomal site. Mol Cell. 2018;70:83–94. doi: 10.1016/j.molcel.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Lewis K. The science of antibiotic discovery. Cell. 2020;181:29–45. doi: 10.1016/j.cell.2020.02.056. [DOI] [PubMed] [Google Scholar]
- 22.Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 23.Tommasi R., Brown D.G., Walkup G.K., Manchester J.I., Miller A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov. 2015;14:529–542. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 24.Roemer T., Boone C. Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol. 2013;9:222–231. doi: 10.1038/nchembio.1205. [DOI] [PubMed] [Google Scholar]
- 25.Dreyer J., Malan A.P., Dicks L.M.T. Bacteria of the genus Xenorhabdus, a novel source of bioactive compounds. Front Microbiol. 2018;9:3177. doi: 10.3389/fmicb.2018.03177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gualteri M., Villain-Guillot P., Givaudan A. 2013. Novel peptide derivatives as antibiotics. WO2013045600. [Google Scholar]
- 27.Sarciaux M., Pantel L., Midrier C., Serri M., Gerber C., Marcia de Figueiredo R., et al. Total synthesis and structure–activity relationships study of odilorhabdins, a new class of peptides showing potent antibacterial activity. J Med Chem. 2018;61:7814–7826. doi: 10.1021/acs.jmedchem.8b00790. [DOI] [PubMed] [Google Scholar]
- 28.Casteels P., Tempst P. Apidaecin-type peptide antibiotics function through a nonporeforming mechanism involving stereospecificity. Biochem Biophys Res Commun. 1994;199:339–345. doi: 10.1006/bbrc.1994.1234. [DOI] [PubMed] [Google Scholar]
- 29.Graf M., Mardirossian M., Nguyen F., Seefeldt A.C., Guichard G., Scocchi M., et al. Proline-rich antimicrobial peptides targeting protein synthesis. Nat Prod Rep. 2017;34:702–711. doi: 10.1039/C7NP00020K. [DOI] [PubMed] [Google Scholar]
- 30.Scocchi M., Tossi A., Gennaro R. Proline-rich antimicrobial peptides: converging to a non-lytic mechanism of action. Cell Mol Life Sci. 2011;68:2317–2330. doi: 10.1007/s00018-011-0721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podda E., Benincasa M., Pacor S., Micali F., Mattiuzzo M., Gennaro R., et al. Dual mode of action of Bac7, a proline-rich antibacterial peptide. Biochim Biophys Acta BBA - Gen Subj. 1760;2006:1732–1740. doi: 10.1016/j.bbagen.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Kragol G., Lovas S., Varadi G., Condie B.A., Hoffmann R., Otvos L. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry. 2001;40:3016–3026. doi: 10.1021/bi002656a. [DOI] [PubMed] [Google Scholar]
- 33.Rozgonyi F., Szabo D., Kocsis B., Ostorhazi E., Abbadessa G., Cassone M., et al. The antibacterial effect of a proline-rich antibacterial peptide A3-APO. Curr Med Chem. 2009;16:3996–4002. doi: 10.2174/092986709789352295. [DOI] [PubMed] [Google Scholar]
- 34.Zahn M., Berthold N., Kieslich B., Knappe D., Hoffmann R., Sträter N. Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK. J Mol Biol. 2013;425:2463–2479. doi: 10.1016/j.jmb.2013.03.041. [DOI] [PubMed] [Google Scholar]
- 35.Zahn M., Kieslich B., Berthold N., Knappe D., Hoffmann R., Strater N. Structural identification of DnaK binding sites within bovine and sheep bactenecin Bac7. Protein Pept Lett. 2014;21:407–412. doi: 10.2174/09298665113206660111. [DOI] [PubMed] [Google Scholar]
- 36.Krizsan A., Volke D., Weinert S., Sträter N., Knappe D., Hoffmann R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70 S ribosome. Angew Chem Int Ed. 2014;53:12236–12239. doi: 10.1002/anie.201407145. [DOI] [PubMed] [Google Scholar]
- 37.Seefeldt A.C., Nguyen F., Antunes S., Pérébaskine N., Graf M., Arenz S., et al. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat Struct Mol Biol. 2015;22:470–475. doi: 10.1038/nsmb.3034. [DOI] [PubMed] [Google Scholar]
- 38.Gagnon M.G., Roy R.N., Lomakin I.B., Florin T., Mankin A.S., Steitz T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016;44:2439–2450. doi: 10.1093/nar/gkw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy R.N., Lomakin I.B., Gagnon M.G., Steitz T.A. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat Struct Mol Biol. 2015;22:466–469. doi: 10.1038/nsmb.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng S., Yang M., Sun R.N., Liu Y., Wang W., Xi Q., et al. Mechanism of actions of Oncocin, a proline-rich antimicrobial peptide, in early elongation revealed by single-molecule FRET. Protein Cell. 2018;9:890–895. doi: 10.1007/s13238-017-0495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt R., Knappe D., Wende E., Ostorházi E., Hoffmann R. In vivo efficacy and pharmacokinetics of optimized apidaecin analogs. Front Chem. 2017;5 doi: 10.3389/fchem.2017.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knappe D., Schmidt R., Adermann K., Hoffmann R. Continuous subcutaneous delivery of proline-rich antimicrobial peptide Api137 provides superior efficacy to intravenous administration in a mouse infection model. Front Microbiol. 2019;10:2283. doi: 10.3389/fmicb.2019.02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt R., Ostorházi E., Wende E., Knappe D., Hoffmann R. Pharmacokinetics and in vivo efficacy of optimized oncocin derivatives. J Antimicrob Chemother. 2016;71:1003–1011. doi: 10.1093/jac/dkv454. [DOI] [PubMed] [Google Scholar]
- 44.Knappe D., Adermann K., Hoffmann R. Oncocin Onc72 is efficacious against antibiotic-susceptible K. pneumoniae ATCC 43816 in a murine thigh infection model: efficacy of Onc72 in susceptible K. pneumoniae ATCC 43816 in thigh infection model. Biopolymers. 2015;104:707–711. doi: 10.1002/bip.22668. [DOI] [PubMed] [Google Scholar]
- 45.Czihal P., Knappe D., Fritsche S., Zahn M., Berthold N., Piantavigna S., et al. Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant gram-negative pathogens. ACS Chem Biol. 2012;7:1281–1291. doi: 10.1021/cb300063v. [DOI] [PubMed] [Google Scholar]
- 46.Gennaro R., Skerlavaj B., Romeo D. Purification, composition, and activity of two bactenecins, antibacterial peptides of bovine neutrophils. Infect Immun. 1989;57:3142–3146. doi: 10.1128/IAI.57.10.3142-3146.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chernysh S., Cociancich S., Briand J.-P., Hetru C., Bulet P. The inducible antibacterial peptides of the Hemipteran insect Palomena prasina: identification of a unique family of proline-rich peptides and of a novel insect defensin. J Insect Physiol. 1996;42:81–89. doi: 10.1016/0022-1910(95)00085-2. [DOI] [Google Scholar]
- 48.Cociancich S., Dupont A., Hegy G., Lanot R., Holder F., Hetru C., et al. Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus. Biochem J. 1994;300:567–575. doi: 10.1042/bj3000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taniguchi M., Ochiai A., Kondo H., Fukuda S., Ishiyama Y., Saitoh E., et al. Pyrrhocoricin, a proline-rich antimicrobial peptide derived from insect, inhibits the translation process in the cell-free E. coli protein synthesis system. J Biosci Bioeng. 2016;121:591–598. doi: 10.1016/j.jbiosc.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 50.Knappe D., Piantavigna S., Hansen A., Mechler A., Binas A., Nolte O., et al. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): a novel antibacterial peptide optimized against gram-negative human pathogens. J Med Chem. 2010;53:5240–5247. doi: 10.1021/jm100378b. [DOI] [PubMed] [Google Scholar]
- 51.Knappe D., Fritsche S., Alber G., Kohler G., Hoffmann R., Muller U. Oncocin derivative Onc72 is highly active against E. coli in a systemic septicaemia infection mouse model. J Antimicrob Chemother. 2012;67:2445–2451. doi: 10.1093/jac/dks241. [DOI] [PubMed] [Google Scholar]
- 52.Heinzelmann E., Berger S., Puk O., Reichenstein B., Wohlleben W., Schwartz D. A glutamate mutase is involved in the biosynthesis of the lipopeptide antibiotic friulimicin in actinoplanes friuliensis. Antimicrob Agents Chemother. 2003;47:447–457. doi: 10.1128/AAC.47.2.447-457.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shoji J., Hinoo H., Sakazaki R., Kato T., Wakisaka Y., Mayama M., et al. Isolation of tridecaptins A, B and C. Studies on antibiotics from the genus Bacillus. XXIII. J Antibiot (Tokyo) 1978;31:646–651. doi: 10.7164/antibiotics.31.646. [DOI] [PubMed] [Google Scholar]
- 54.Cochrane S.A., Findlay B., Bakhtiary A., Acedo J.Z., Rodriguez-Lopez E.M., Mercier P., et al. Antimicrobial lipopeptide tridecaptin A1 selectively binds to Gram-negative lipid II. Proc Natl Acad Sci. 2016;113:11561–11566. doi: 10.1073/pnas.1608623113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cochrane S.A., Lohans C.T., Brandelli J.R., Mulvey G., Armstrong G.D., Vederas J.C. Synthesis and structure–activity relationship studies of N-Terminal analogues of the antimicrobial peptide tridecaptin A1. J Med Chem. 2014;57:1127–1131. doi: 10.1021/jm401779d. [DOI] [PubMed] [Google Scholar]
- 56.Lohans C.T., van Belkum M.J., Cochrane S.A., Huang Z., Sit C.S., McMullen L.M., et al. Biochemical, structural, and genetic characterization of tridecaptin A1, an antagonist of Campylobacter jejuni. ChemBioChem. 2014;15:243–249. doi: 10.1002/cbic.201300595. [DOI] [PubMed] [Google Scholar]
- 57.Jangra M., Kaur M., Tambat R., Rana R., Maurya S.K., Khatri N., et al. Tridecaptin M, a new variant discovered in mud bacterium, shows activity against colistin- and extremely drug-resistant Enterobacteriaceae. Antimicrob Agents Chemother. 2019;63 doi: 10.1128/AAC.00338-19. /aac/63/6/AAC.00338-19.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ballantine R.D., Li Y.-X., Qian P.-Y., Cochrane S.A. Rational design of new cyclic analogues of the antimicrobial lipopeptide tridecaptin A1. Chem Commun. 2018;54:10634–10637. doi: 10.1039/C8CC05790G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klahn P., Brönstrup M. Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat Prod Rep. 2017;34:832–885. doi: 10.1039/C7NP00006E. [DOI] [PubMed] [Google Scholar]
- 60.Cochrane S.A., Li X., He S., Yu M., Wu M., Vederas J.C. Synthesis of tridecaptin–antibiotic conjugates with in vivo activity against gram-negative bacteria. J Med Chem. 2015;58:9779–9785. doi: 10.1021/acs.jmedchem.5b01578. [DOI] [PubMed] [Google Scholar]
- 61.Hover B.M., Kim S.-H., Katz M., Charlop-Powers Z., Owen J.G., Ternei M.A., et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant gram-positive pathogens. Nat Microbiol. 2018;3:415–422. doi: 10.1038/s41564-018-0110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lam Y.C., Crawford J.M. Discovering antibiotics from the global microbiome. Nat Microbiol. 2018;3:392–393. doi: 10.1038/s41564-018-0135-5. [DOI] [PubMed] [Google Scholar]
- 63.Sun Z., Shang Z., Forelli N., Po K.H.L., Chen S., Brady S.F., et al. Total synthesis of malacidin A by β-hydroxyaspartic acid ligation-mediated cyclization and absolute structure establishment. Angew Chem Int Ed. 2020 doi: 10.1002/anie.202009092. anie.202009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cochrane S.A., Vederas J.C. Lipopeptides from bacillus and paenibacillus spp.: a gold mine of antibiotic candidates. Med Res Rev. 2014;36:4–31. doi: 10.1002/med.21321. [DOI] [PubMed] [Google Scholar]
- 65.Brown J.M., Dorman D.C., Roy L.P. Acute renal failure due to overdosage of colistin. Med J Aust. 1970;2:923–924. doi: 10.5694/j.1326-5377.1970.tb63262.x. [DOI] [PubMed] [Google Scholar]
- 66.Koch-Weser J. Adverse effects of sodium colistimethate: manifestations and specific reaction rates during 317 courses of therapy. Ann Intern Med. 1970;72:857. doi: 10.7326/0003-4819-72-6-857. [DOI] [PubMed] [Google Scholar]
- 67.Biagi M., Butler D., Tan X., Qasmieh S., Wenzler E. A breath of fresh air in the fog of antimicrobial resistance: inhaled polymyxins for gram-negative pneumonia. Antibiotics. 2019;8:27. doi: 10.3390/antibiotics8010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Falagas M.E., Fragoulis K.N., Kasiakou S.K., Sermaidis G.J., Michalopoulos A. Nephrotoxicity of intravenous colistin: a prospective evaluation. Int J Antimicrob Agents. 2005;26:504–507. doi: 10.1016/j.ijantimicag.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Falagas M.E., Rafailidis P.I., Matthaiou D.K., Virtzili S., Nikita D., Michalopoulos A. Pandrug-resistant K. pneumoniae, P. aeruginosa and A. baumannii infections: characteristics and outcome in a series of 28 patients. Int J Antimicrob Agents. 2008;32:450–454. doi: 10.1016/j.ijantimicag.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 70.Baron S., Hadjadj L., Rolain J.-M., Olaitan A.O. Molecular mechanisms of polymyxin resistance: knowns and unknowns. Int J Antimicrob Agents. 2016;48:583–591. doi: 10.1016/j.ijantimicag.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 71.Davies M., Walsh T.R. A colistin crisis in India. Lancet Infect Dis. 2018;18:256–257. doi: 10.1016/S1473-3099(18)30072-0. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y.-Y., Wang Y., Walsh T.R., Yi L.-X., Zhang R., Spencer J., et al. Emergence of plasmid-mediated colistin resistance mechanism mcr-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16:161–168. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 73.Pacheco T., Bustos R.-H., González D., Garzón V., García J.-C., Ramírez D. An approach to measuring colistin plasma levels regarding the treatment of multidrug-resistant bacterial infection. Antibiotics. 2019;8:100. doi: 10.3390/antibiotics8030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trimble M.J., Mlynárčik P., Kolář M., Hancock R.E.W. Polymyxin: alternative mechanisms of action and resistance. Cold Spring Harb Perspect Med. 2016;6:a025288. doi: 10.1101/cshperspect.a025288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Velkov T., Thompson P.E., Nation R.L., Li J. Structure−activity relationships of polymyxin antibiotics. J Med Chem. 2010;53:1898–1916. doi: 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown P., Dawson M.J. Development of new polymyxin derivatives for multi-drug resistant Gram-negative infections. J Antibiot (Tokyo) 2017;70:386–394. doi: 10.1038/ja.2016.146. [DOI] [PubMed] [Google Scholar]
- 77.Velkov T., Roberts K.D., Thompson P.E., Li J. Polymyxins: a new hope in combating Gram-negative superbugs? Future Med Chem. 2016;8:1017–1025. doi: 10.4155/fmc-2016-0091. [DOI] [PubMed] [Google Scholar]
- 78.Brown P., Abbott E., Abdulle O., Boakes S., Coleman S., Divall N., et al. Design of next generation polymyxins with lower toxicity: the discovery of SPR206. ACS Infect Dis. 2019;5:1645–1656. doi: 10.1021/acsinfecdis.9b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abdelraouf K., Chang K.-T., Yin T., Hu M., Tam V.H. Uptake of polymyxin B into renal cells. Antimicrob Agents Chemother. 2014;58:4200–4202. doi: 10.1128/AAC.02557-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Becker B., Butler M.S., Hansford K.A., Gallardo-Godoy A., Elliott A.G., Huang J.X., et al. Synthesis of octapeptin C4 and biological profiling against NDM-1 and polymyxin-resistant bacteria. Bioorg Med Chem Lett. 2017;27:2407–2409. doi: 10.1016/j.bmcl.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Velkov T., Gallardo-Godoy A., Swarbrick J.D., Blaskovich Mark A.T., Elliott A.G., Han M., et al. Structure, function, and biosynthetic origin of octapeptin antibiotics active against extensively drug-resistant gram-negative bacteria. Cell Chem Biol. 2018;25:380–391. doi: 10.1016/j.chembiol.2018.01.005. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Velkov T., Roberts K.D. In: Polymyxin Antibiot. Lab. Bench Bedside. Li J., Nation R.L., Kaye K.S., editors. Springer International Publishing; Cham: 2019. Discovery of novel polymyxin-like antibiotics; pp. 343–362. [DOI] [Google Scholar]
- 83.Carb-X Funds University of Queensland. Headl Press Releaselink; 2020. https://carb-x.org/carb-x-news/carb-x-funds-university-of-queensland-to-accelerate-the-development-of-a-new-class-of-last-resort-antibiotics-to-treat-deadly-superbug-infections/ (Accessed August 12, 2019) [Google Scholar]
- 84.Vaara M. Novel derivatives of polymyxins. J Antimicrob Chemother. 2013;68:1213–1219. doi: 10.1093/jac/dkt039. [DOI] [PubMed] [Google Scholar]
- 85.Paetzel M. In: Bact. Cell Walls Membr. Kuhn A., editor. Vol. 92. Springer International Publishing; Cham: 2019. Bacterial signal peptidases; pp. 187–219. [DOI] [Google Scholar]
- 86.Paetzel M., Goodall J.J., Kania M., Dalbey R.E., Page M.G.P. Crystallographic and biophysical analysis of a bacterial signal peptidase in complex with a lipopeptide-based inhibitor. J Biol Chem. 2004;279:30781–30790. doi: 10.1074/jbc.M401686200. [DOI] [PubMed] [Google Scholar]
- 87.Höltzel A., Schmid D.G., Nicholson G.J., Stevanovic S., Schimana J., Gebhardt K., et al. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by streptomyces sp. Tue 6075. II. Structure elucidation. J Antibiot (Tokyo) 2002;55:571–577. doi: 10.7164/antibiotics.55.571. [DOI] [PubMed] [Google Scholar]
- 88.Liu W.-T., Kersten R.D., Yang Y.-L., Moore B.S., Dorrestein P.C. Imaging mass spectrometry and genome mining via short sequence tagging identified the anti-infective agent arylomycin in Streptomyces roseosporus. J Am Chem Soc. 2011;133:18010–18013. doi: 10.1021/ja2040877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hubbard B.K., Thomas M.G., Walsh C.T. Biosynthesis of L-p-hydroxyphenylglycine, a non-proteinogenic amino acid constituent of peptide antibiotics. Chem Biol. 2000;7:931–942. doi: 10.1016/S1074-5521(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 90.Roberts T.C., Smith P.A., Cirz R.T., Romesberg F.E. Structural and initial biological analysis of synthetic arylomycin A2. J Am Chem Soc. 2007;129:15830–15838. doi: 10.1021/ja073340u. [DOI] [PubMed] [Google Scholar]
- 91.Peters D.S., Romesberg F.E., Baran P.S. Scalable access to arylomycins via C–H functionalization logic. J Am Chem Soc. 2018;140:2072–2075. doi: 10.1021/jacs.8b00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith P.A., Romesberg F.E. Mechanism of action of the arylomycin antibiotics and effects of signal peptidase I inhibition. Antimicrob Agents Chemother. 2012;56:5054–5060. doi: 10.1128/AAC.00785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ting Y.T., Harris P.W.R., Batot G., Brimble M.A., Baker E.N., Young P.G. Peptide binding to a bacterial signal peptidase visualized by peptide tethering and carrier-driven crystallization. IUCrJ. 2016;3:10–19. doi: 10.1107/S2052252515019971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Paetzel M., Dalbey R.E., Strynadka N.C.J. Crystal structure of a bacterial signal peptidase in complex with a β-lactam inhibitor. Nature. 1998;396:186–190. doi: 10.1038/24196. [DOI] [PubMed] [Google Scholar]
- 95.Liu J., Luo C., Smith P.A., Chin J.K., Page M.G.P., Paetzel M., et al. Synthesis and characterization of the arylomycin lipoglycopeptide antibiotics and the crystallographic analysis of their complex with signal peptidase. J Am Chem Soc. 2011;133:17869–17877. doi: 10.1021/ja207318n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kulanthaivel P., Kreuzman A.J., Strege M.A., Belvo M.D., Smitka T.A., Clemens M., et al. Novel lipoglycopeptides as inhibitors of bacterial signal peptidase I. J Biol Chem. 2004;279:36250–36258. doi: 10.1074/jbc.M405884200. [DOI] [PubMed] [Google Scholar]
- 97.Liu J., Smith P.A., Steed D.B., Romesberg F. Efforts toward broadening the spectrum of arylomycin antibiotic activity. Bioorg Med Chem Lett. 2013;23:5654–5659. doi: 10.1016/j.bmcl.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith P.A., Roberts T.C., Romesberg F.E. Broad-spectrum antibiotic activity of the arylomycin natural products is masked by natural target mutations. Chem Biol. 2010;17:1223–1231. doi: 10.1016/j.chembiol.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Craney A., Romesberg F.E. The inhibition of type I bacterial signal peptidase: biological consequences and therapeutic potential. Bioorg Med Chem Lett. 2015;25:4761–4766. doi: 10.1016/j.bmcl.2015.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roberts T.C., Schallenberger M.A., Liu J., Smith P.A., Romesberg F.E. Initial efforts toward the optimization of arylomycins for antibiotic activity. J Med Chem. 2011;54:4954–4963. doi: 10.1021/jm1016126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Therien A.G., Huber J.L., Wilson K.E., Beaulieu P., Caron A., Claveau D., et al. Broadening the spectrum of β-Lactam Antibiotics through inhibition of signal peptidase type I. Antimicrob Agents Chemother. 2012;56:4662–4670. doi: 10.1128/AAC.00726-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith P.A., Koehler M.F.T., Girgis H.S., Yan D., Chen Y., Chen Y., et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature. 2018;561:189–194. doi: 10.1038/s41586-018-0483-6. [DOI] [PubMed] [Google Scholar]
- 103.Heise C.E., Koehler M.F.T., Murray J., Smith P.A. 2020. Peptide antibiotic complexes and methods of use thereof. US20200262869. [Google Scholar]
- 104.Tan Y.X., Peters D.S., Walsh S.I., Holcomb M., Santos-Martins D., Forli S., et al. Initial analysis of the arylomycin D antibiotics. J Nat Prod. 2020 doi: 10.1021/acs.jnatprod.9b01174. acs.jnatprod.9b01174. [DOI] [PubMed] [Google Scholar]
- 105.Ling L.L., Schneider T., Peoples A.J., Spoering A.L., Engels I., Conlon B.P., et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sandle T. Teixobactin: a new class of antibiotic. SOJ Microbiol Infect Dis. 2015;3:1–2. doi: 10.15226/sojmid/3/1/00128. [DOI] [Google Scholar]
- 107.von Nussbaum F., Süssmuth R.D. Multiple attack on bacteria by the new antibiotic teixobactin. Angew Chem Int Ed. 2015;54:6684–6686. doi: 10.1002/anie.201501440. [DOI] [PubMed] [Google Scholar]
- 108.Grady Denise. New antibiotic stirs hope against resistant bacteria. N Y Times. 2015;13 [Google Scholar]
- 109.Lewis K. New approaches to antimicrobial discovery. Biochem Pharmacol. 2017;134:87–98. doi: 10.1016/j.bcp.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 110.Iyer A., Madder A., Singh I. Teixobactins: a new class of 21st century antibiotics to combat multidrug-resistant bacterial pathogens. Future Microbiol. 2019;14:457–460. doi: 10.2217/fmb-2019-0056. [DOI] [PubMed] [Google Scholar]
- 111.McCarthy M.W. Teixobactin: a novel anti-infective agent. Expert Rev Anti Infect Ther. 2019;17:1–3. doi: 10.1080/14787210.2019.1550357. [DOI] [PubMed] [Google Scholar]
- 112.Karas J.A., Chen F., Schneider-Futschik E.K., Kang Z., Hussein M., Swarbrick J., et al. Synthesis and structure−activity relationships of teixobactin. Ann N Y Acad Sci. 2020;1459:86–105. doi: 10.1111/nyas.14282. [DOI] [PubMed] [Google Scholar]
- 113.Homma T., Nuxoll A., Gandt A.B., Ebner P., Engels I., Schneider T., et al. Dual targeting of cell wall precursors by teixobactin leads to cell lysis. Antimicrob Agents Chemother. 2016;60:6510–6517. doi: 10.1128/AAC.01050-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wright G. An irresistible newcomer. Nature. 2015;517:442–444. doi: 10.1038/nature14193. [DOI] [PubMed] [Google Scholar]
- 115.Ramchuran E.J., Somboro A.M., Abdel Monaim S.A.H., Amoako D.G., Parboosing R., Kumalo H.M., et al. In vitro antibacterial activity of teixobactin derivatives on clinically relevant bacterial isolates. Front Microbiol. 2018;9:1535. doi: 10.3389/fmicb.2018.01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parmar A., Iyer A., Prior S.H., Lloyd D.G., Leng Goh E.T., Vincent C.S., et al. Teixobactin analogues reveal enduracididine to be non-essential for highly potent antibacterial activity and lipid II binding. Chem Sci. 2017;8:8183–8192. doi: 10.1039/C7SC03241B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chiorean S., Antwi I., Carney D.W., Kotsogianni I., Giltrap A.M., Alexander F.M., et al. Dissecting the binding interactions of teixobactin with the bacterial cell-wall precursor lipid II. ChemBioChem. 2020;21:789–792. doi: 10.1002/cbic.201900504. [DOI] [PubMed] [Google Scholar]
- 118.Chen K.H., Le S.P., Han X., Frias J.M., Nowick J.S. Alanine scan reveals modifiable residues in teixobactin. Chem Commun. 2017;53:11357–11359. doi: 10.1039/C7CC03415F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang H., Wierzbicki M., Du Bois D.R., Nowick J.S. X-ray crystallographic structure of a teixobactin derivative reveals amyloid-like assembly. J Am Chem Soc. 2018;140:14028–14032. doi: 10.1021/jacs.8b07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Breukink E., de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5:321–332. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 121.Marshall C.G., Lessard I.A.D., Park I.-S., Wright G.D. Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob Agents Chemother. 1998;42:2215–2220. doi: 10.1128/AAC.42.9.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ng V., Kuehne S.A., Chan W.C. Rational design and synthesis of modified teixobactin analogues: in vitro antibacterial activity against S. aureus, Propionibacterium acnes and P. aeruginosa. Chem A Eur J. 2018;24:9136–9147. doi: 10.1002/chem.201801423. [DOI] [PubMed] [Google Scholar]
- 123.Giltrap A.M., Dowman L.J., Nagalingam G., Ochoa J.L., Linington R.G., Britton W.J., et al. Total Synthesis of Teixobactin. Org Lett. 2016;18:2788–2791. doi: 10.1021/acs.orglett.6b01324. [DOI] [PubMed] [Google Scholar]
- 124.Jin K., Po K.H.L., Wang S., Reuven J.A., Wai C.N., Lau H.T., et al. Synthesis and structure-activity relationship of teixobactin analogues via convergent Ser ligation. Bioorg Med Chem. 2017;25:4990–4995. doi: 10.1016/j.bmc.2017.04.039. [DOI] [PubMed] [Google Scholar]
- 125.Yang H., Chen K.H., Nowick J.S. Elucidation of the teixobactin pharmacophore. ACS Chem Biol. 2016;11:1823–1826. doi: 10.1021/acschembio.6b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wu C., Pan Z., Yao G., Wang W., Fang L., Su W. Synthesis and structure–activity relationship studies of teixobactin analogues. RSC Adv. 2017;7:1923–1926. doi: 10.1039/C6RA26567G. [DOI] [Google Scholar]
- 127.Abdel Monaim S.A.H., Jad Y.E., Ramchuran E.J., El-Faham A., Govender T., Kruger H.G., et al. Lysine scanning of Arg 10 –teixobactin: deciphering the role of hydrophobic and hydrophilic residues. ACS Omega. 2016;1:1262–1265. doi: 10.1021/acsomega.6b00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Parmar A., Iyer A., Vincent C.S., Van Lysebetten D., Prior S.H., Madder A., et al. Efficient total syntheses and biological activities of two teixobactin analogues. Chem Commun. 2016;52:6060–6063. doi: 10.1039/C5CC10249A. [DOI] [PubMed] [Google Scholar]
- 129.Schumacher C.E., Harris P.W.R., Ding X.-B., Krause B., Wright T.H., Cook G.M., et al. Synthesis and biological evaluation of novel teixobactin analogues. Org Biomol Chem. 2017;15:8755–8760. doi: 10.1039/C7OB02169K. [DOI] [PubMed] [Google Scholar]
- 130.Mandalapu D., Ji X., Chen J., Guo C., Liu W.-Q., Ding W., et al. Thioesterase-mediated synthesis of teixobactin analogues: mechanism and substrate specificity. J Org Chem. 2018;83:7271–7275. doi: 10.1021/acs.joc.7b02462. [DOI] [PubMed] [Google Scholar]
- 131.Zong Y., Fang F., Meyer K.J., Wang L., Ni Z., Gao H., et al. Gram-scale total synthesis of teixobactin promoting binding mode study and discovery of more potent antibiotics. Nat Commun. 2019;10:3268. doi: 10.1038/s41467-019-11211-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Parmar A., Lakshminarayanan R., Iyer A., Mayandi V., Leng Goh E.T., Lloyd D.G., et al. Design and syntheses of highly potent teixobactin analogues against S. aureus, methicillin-resistant S. aureus (MRSA), and vancomycin-resistant enterococci (VRE) in vitro and in vivo. J Med Chem. 2018;61:2009–2017. doi: 10.1021/acs.jmedchem.7b01634. [DOI] [PubMed] [Google Scholar]
- 133.Zong Y., Sun X., Gao H., Meyer K.J., Lewis K., Rao Y. Developing equipotent teixobactin analogues against drug-resistant bacteria and discovering a hydrophobic interaction between lipid II and teixobactin. J Med Chem. 2018;61:3409–3421. doi: 10.1021/acs.jmedchem.7b01241. [DOI] [PubMed] [Google Scholar]
- 134.Yang H., Du Bois D.R., Ziller J.W., Nowick J.S. X-ray crystallographic structure of a teixobactin analogue reveals key interactions of the teixobactin pharmacophore. Chem Commun. 2017;53:2772–2775. doi: 10.1039/C7CC00783C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Malkawi R., Iyer A., Parmar A., Lloyd D., Leng Goh E., Taylor E., et al. Cysteines and disulfide-bridged macrocyclic mimics of teixobactin analogues and their antibacterial activity evaluation against methicillin-resistant S. aureus (MRSA) Pharmaceutics. 2018;10:183. doi: 10.3390/pharmaceutics10040183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abdel Monaim S.A.H., Ramchuran E.J., El-Faham A., Albericio F., de la Torre B.G. Converting teixobactin into a cationic antimicrobial peptide (AMP) J Med Chem. 2017;60:7476–7482. doi: 10.1021/acs.jmedchem.7b00834. [DOI] [PubMed] [Google Scholar]
- 137.Hoertz A.J., Hamburger J.B., Gooden D.M., Bednar M.M., McCafferty D.G. Studies on the biosynthesis of the lipodepsipeptide antibiotic Ramoplanin A2. Bioorg Med Chem. 2012;20:859–865. doi: 10.1016/j.bmc.2011.11.062. [DOI] [PubMed] [Google Scholar]
- 138.Walker S., Chen L., Hu Y., Rew Y., Shin D., Boger D.L. Chemistry and biology of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. Chem Rev. 2005;105:449–475. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 139.Fang X., Nam J., Shin D., Rew Y., Boger D.L., Walker S. Functional and biochemical analysis of a key series of ramoplanin analogues. Bioorg Med Chem Lett. 2009;19:6189–6191. doi: 10.1016/j.bmcl.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.de la Cruz M., González I., Parish C.A., Onishi R., Tormo J.R., Martín J., et al. Production of ramoplanin and ramoplanin analogs by actinomycetes. Front Microbiol. 2017;8:343. doi: 10.3389/fmicb.2017.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Somner E.A., Reynolds P.E. Inhibition of peptidoglycan biosynthesis by ramoplanin. Antimicrob Agents Chemother. 1990;34:413–419. doi: 10.1128/AAC.34.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lo M.-C., Men H., Branstrom A., Helm J., Yao N., Goldman R., et al. A new mechanism of action proposed for ramoplanin. J Am Chem Soc. 2000;122:3540–3541. doi: 10.1021/ja000182x. [DOI] [Google Scholar]
- 143.Cheng M., Huang J.X., Ramu S., Butler M.S., Cooper M.A. Ramoplanin at bactericidal concentrations induces bacterial membrane depolarization in S. aureus. Antimicrob Agents Chemother. 2014;58:6819–6827. doi: 10.1128/AAC.00061-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kurz M., Guba W. 3D structure of ramoplanin: a potent inhibitor of bacterial cell wall synthesis. Biochemistry. 1996;35:12570–12575. doi: 10.1021/bi961017q. [DOI] [PubMed] [Google Scholar]
- 145.Lo M.-C., Helm J.S., Sarngadharan G., Pelczer I., Walker S. A new structure for the substrate-binding antibiotic ramoplanin. J Am Chem Soc. 2001;123:8640–8641. doi: 10.1021/ja011080p. [DOI] [PubMed] [Google Scholar]
- 146.Hu Y., Helm J.S., Chen L., Ye X.-Y., Walker S. Ramoplanin inhibits bacterial transglycosylases by binding as a dimer to lipid II. J Am Chem Soc. 2003;125:8736–8737. doi: 10.1021/ja035217i. [DOI] [PubMed] [Google Scholar]
- 147.Hamburger J.B., Hoertz A.J., Lee A., Senturia R.J., McCafferty D.G., Loll P.J. A crystal structure of a dimer of the antibiotic ramoplanin illustrates membrane positioning and a potential Lipid II docking interface. Proc Natl Acad Sci. 2009;106:13759–13764. doi: 10.1073/pnas.0904686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jabes D., Brunati C., Candiani G., Riva S., Romanó G., Maffioli S., et al. Pharmacological properties of NAI-603, a well-tolerated semisynthetic derivative of ramoplanin. Antimicrob Agents Chemother. 2014;58:1922–1929. doi: 10.1128/AAC.01620-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ciabatti R., Maffioli S.I., Panzone G., Canavesi A., Michelucci E., Tiseni P.S., et al. Synthesis and preliminary biological characterization of new semisynthetic derivatives of ramoplanin. J Med Chem. 2007;50:3077–3085. doi: 10.1021/jm070042z. [DOI] [PubMed] [Google Scholar]
- 150.Nam J., Shin D., Rew Y., Boger D.L. Alanine scan of [l - Dap2] ramoplanin A2 aglycon: assessment of the importance of each residue. J Am Chem Soc. 2007;129:8747–8755. doi: 10.1021/ja068573k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rew Y., Shin D., Hwang I., Boger D.L. Total synthesis and examination of three key analogues of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. J Am Chem Soc. 2004;126:1041–1043. doi: 10.1021/ja039671y. [DOI] [PubMed] [Google Scholar]
- 152.Jiang W., Wanner J., Lee R.J., Bounaud P.-Y., Boger D.L. Total synthesis of the ramoplanin A2 and ramoplanose aglycon. J Am Chem Soc. 2003;125:1877–1887. doi: 10.1021/ja0212314. [DOI] [PubMed] [Google Scholar]
- 153.Chen L., Yuan Y., Helm J.S., Hu Y., Rew Y., Shin D., et al. Dissecting ramoplanin: mechanistic analysis of synthetic ramoplanin analogues as a guide to the design of improved antibiotics. J Am Chem Soc. 2004;126:7462–7463. doi: 10.1021/ja047879t. [DOI] [PubMed] [Google Scholar]
- 154.Pelzer S., Vente Andreas. 2008. New lipoglycodepsipeptide compositions. EP1994938A1. [Google Scholar]
- 155.Bassères E., Endres B.T., Dotson K.M., Alam M.J., Garey K.W. Novel antibiotics in development to treat Clostridium difficile infection. Curr Opin Gastroenterol. 2017;33:1–7. doi: 10.1097/MOG.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 156.Petrosillo N., Granata G., Cataldo M.A. Novel antimicrobials for the treatment of clostridium difficile infection. Front Med. 2018;5:96. doi: 10.3389/fmed.2018.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.World Health Organization . World Health Organization; Geneva: 2017. Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, including tuberculosis. [Google Scholar]
- 158.World Health Organization . World Health Organization; Geneva: 2019. Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline. [Google Scholar]
- 159.Kraus C.N., Lyerly M.W., Carman R.J. Ambush of clostridium difficile spores by ramoplanin: activity in an in vitro model. Antimicrob Agents Chemother. 2015;59:2525–2530. doi: 10.1128/AAC.04853-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Parenti Francesco, Candiani Gianpaolo, Ciabatti Romeo, Cavaleri Marco. 2000. New injectable formulations containing ramoplanin. WO2000006119A1. [Google Scholar]
- 161.Fehlbaum P., Bulet P., Chernysh S., Briand J.P., Roussel J.P., Letellier L., et al. Structure-activity analysis of thanatin, a 21-residue inducible insect defense peptide with sequence homology to frog skin antimicrobial peptides. Proc Natl Acad Sci. 1996;93:1221–1225. doi: 10.1073/pnas.93.3.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Moura E.C.C.M., Baeta T., Romanelli A., Laguri C., Martorana A.M., Erba E., et al. Thanatin impairs lipopolysaccharide transport complex assembly by targeting LptC–LptA interaction and decreasing LptA stability. Front Microbiol. 2020;11:909. doi: 10.3389/fmicb.2020.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma B., Fang C., Lu L., Wang M., Xue X., Zhou Y., et al. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-β-lactamase. Nat Commun. 2019;10:3517. doi: 10.1038/s41467-019-11503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu G., Wu H., Fan X., Zhao R., Li X., Wang S., et al. Selective toxicity of antimicrobial peptide S-thanatin on bacteria. Peptides. 2010;31:1669–1673. doi: 10.1016/j.peptides.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 165.Lee M.-K., Cha L.-N., Lee S.-H., Hahm K.-S. Role of amino acid residues within the disulfide loop of thanatin, a potent antibiotic peptide. BMB Rep. 2002;35:291–296. doi: 10.5483/BMBRep.2002.35.3.291. [DOI] [PubMed] [Google Scholar]
- 166.Wu G., Deng X., Wu P., Shen Z., Xu H. Subacute toxicity of antimicrobial peptide S-thanatin in ICR mice. Peptides. 2012;36:109–113. doi: 10.1016/j.peptides.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 167.Mandard N., Sodano P., Labbe H., Bonmatin J.-M., Bulet P., Hetru C., et al. Solution structure of thanatin, a potent bactericidal and fungicidal insect peptide, determined from proton two-dimensional nuclear magnetic resonance data. Eur J Biochem. 1998;256:404–410. doi: 10.1046/j.1432-1327.1998.2560404.x. [DOI] [PubMed] [Google Scholar]
- 168.Sinha S., Ng W.J., Bhattacharjya S. NMR structure and localization of the host defense antimicrobial peptide thanatin in zwitterionic dodecylphosphocholine micelle: Implications in antimicrobial activity. Biochim Biophys Acta BBA - Biomembr. 1862;2020:183432. doi: 10.1016/j.bbamem.2020.183432. [DOI] [PubMed] [Google Scholar]
- 169.Owens T.W., Taylor R.J., Pahil K.S., Bertani B.R., Ruiz N., Kruse A.C., et al. Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature. 2019;567:550–553. doi: 10.1038/s41586-019-1039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Luther A., Moehle K., Chevalier E., Dale G., Obrecht D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr Opin Chem Biol. 2017;38:45–51. doi: 10.1016/j.cbpa.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 171.Obrecht D., Chevalier E., Moehle K., Robinson J.A. β-Hairpin protein epitope mimetic technology in drug discovery. Drug Discov Today Technol. 2012;9:e63–e69. doi: 10.1016/j.ddtec.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 172.Martin-Loeches I., Dale G.E., Torres A. Murepavadin: a new antibiotic class in the pipeline. Expert Rev Anti Infect Ther. 2018;16:259–268. doi: 10.1080/14787210.2018.1441024. [DOI] [PubMed] [Google Scholar]
- 173.Werneburg M., Zerbe K., Juhas M., Bigler L., Stalder U., Kaech A., et al. Inhibition of lipopolysaccharide transport to the outer membrane in P. aeruginosa by peptidomimetic antibiotics. ChemBioChem. 2012;13:1767–1775. doi: 10.1002/cbic.201200276. [DOI] [PubMed] [Google Scholar]
- 174.Andolina G., Bencze L.-C., Zerbe K., Müller M., Steinmann J., Kocherla H., et al. A peptidomimetic antibiotic interacts with the periplasmic domain of LptD from P. aeruginosa. ACS Chem Biol. 2018;13:666–675. doi: 10.1021/acschembio.7b00822. [DOI] [PubMed] [Google Scholar]
- 175.Cigana C., Bernardini F., Facchini M., Alcalá-Franco B., Riva C., De Fino I., et al. Efficacy of the novel antibiotic POL7001 in preclinical models of P. aeruginosa pneumonia. Antimicrob Agents Chemother. 2016;60:4991–5000. doi: 10.1128/AAC.00390-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Obrecht D., Robinson J., Bernardini F., Bisang C., DeMarco S., Moehle K., et al. Recent progress in the discovery of macrocyclic compounds as potential anti-infective therapeutics. Curr Med Chem. 2009;16:42–65. doi: 10.2174/092986709787002844. [DOI] [PubMed] [Google Scholar]
- 177.Sousa M.C. New antibiotics target bacterial envelope. Nature. 2019;576:389–390. doi: 10.1038/d41586-019-03730-x. [DOI] [PubMed] [Google Scholar]
- 178.McLaughlin M.I., van der Donk W.A. The fellowship of the rings: macrocyclic antibiotic peptides reveal an anti-gram-negative target. Biochemistry. 2020;59:343–345. doi: 10.1021/acs.biochem.9b01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sader H.S., Rhomberg P.R., Duncan L.R., Locher H.H., Dale G.E., Flamm R.K. Antimicrobial activity of POL7306 tested against clinical isolates of gram-negative bacteria collected worldwide. J Antimicrob Chemother. 2020;75:1518–1524. doi: 10.1093/jac/dkaa020. [DOI] [PubMed] [Google Scholar]
- 180.Tomasek D., Rawson S., Lee J., Wzorek J.S., Harrison S.C., Li Z., et al. Structure of a nascent membrane protein as it folds on the BAM complex. Nature. 2020;583:473–478. doi: 10.1038/s41586-020-2370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ovchinnikova T.V., Aleshina G.M., Balandin S.V., Krasnosdembskaya A.D., Markelov M.L., Frolova E.I., et al. Purification and primary structure of two isoforms of arenicin, a novel antimicrobial peptide from marine polychaeta Arenicola marina. FEBS Lett. 2004;577:209–214. doi: 10.1016/j.febslet.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 182.Elliott A.G., Huang J.X., Neve S., Zuegg J., Edwards I.A., Cain A.K., et al. An amphipathic peptide with antibiotic activity against multidrug-resistant Gram-negative bacteria. Nat Commun. 2020;11:3184. doi: 10.1038/s41467-020-16950-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Neve . 2011. NZ17074: An Arenicin-3 variant found by HTS screening of yeast libraries. Poster F1-2070. [Google Scholar]
- 184.Yang N., Liu X., Teng D., Li Z., Wang X., Mao R., et al. Antibacterial and detoxifying activity of NZ17074 analogues with multi-layers of selective antimicrobial actions against E. coli and Salmonella enteritidis. Sci Rep. 2017;7:3392. doi: 10.1038/s41598-017-03664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wang X., Wang X., Teng D., Zhang Y., Mao R., Xi D., et al. Candidacidal mechanism of the arenicin-3-derived peptide NZ17074 from Arenicola marina. Appl Microbiol Biotechnol. 2014;98:7387–7398. doi: 10.1007/s00253-014-5784-6. [DOI] [PubMed] [Google Scholar]
- 186.Wang X., Teng D., Mao R., Yang N., Hao Y., Wang J. Combined systems approaches reveal a multistage mode of action of a marine antimicrobial peptide against pathogenic E. coli and its protective effect against bacterial peritonitis and endotoxemia. Antimicrob Agents Chemother. 2017;61 doi: 10.1128/AAC.01056-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hoegenhaug Hans-Henrik Kristensen, Myging Per Holse, Kruse Thomas, Segura Raventos, Dorotea Sandvang, Hoj Dorthe, et al. 2011. Antimicrobial peptide variants and polynucleotides encoding same. US20110306750. [Google Scholar]
- 188.Wang X.J., Wang X.M., Teng D., Zhang Y., Mao R.Y., Wang J.H. Recombinant production of the antimicrobial peptide NZ17074 in Pichia pastoris using SUMO3 as a fusion partner. Lett Appl Microbiol. 2014;59:71–78. doi: 10.1111/lam.12246. [DOI] [PubMed] [Google Scholar]
- 189.Zhang A.H., Edwards I.A., Mishra B.P., Sharma G., Healy M.D., Elliott A.G., et al. Elucidating the lipid binding properties of membrane-active peptides using cyclised nanodiscs. Front Chem. 2019;7:238. doi: 10.3389/fchem.2019.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]