

Inflammation is an immune response to something. Decades’ worth of mechanistic investigations have begun to peel back the complex layers of inflammatory triggers of arteriosclerosis and cardiovascular disease (CVD) – in time (phases of immune response), space (tissue bed-specific responses), and host (patient).1 Yet, fundamentally, models of inflammation-associated CVDs can be broken down into triggers and immune responses to these triggers. Essentially, antigens and/or physiologic perturbances interact with underlying host factors and comorbidities to drive immune cell biasing and phenotype-switching.2 These interactions ultimately dictate the balance between inflammation and resolution thereof, with effects on subclinical and overt clinical CVDs.
In experimental models, triggers and host factors may be cleanly manipulated to probe relationships between immunity, comorbidities, and CVDs. In humans, however, variable underlying host factors and comorbidities add barriers and complexity to the precise definition of these relationships. Rigorous approaches to investigating interactions are therefore necessary. In the manuscript by Stein et al in this issue of ATVB,3 the authors’ thoughtful approach to interactions between HIV and renal impairment with respect to endothelial function yields hypothesis-generating findings of potential mechanistic relevance. Leveraging harmonized participant-level data from 9 studies, the authors observed differences in the association of human immunodeficiency virus (HIV) serostatus with brachial artery flow-mediated dilation (FMD) depending on serum creatinine level.
Statistically, as the authors discuss, this is an example of negative confounding: a confounding variable (in this case, serum creatinine) biased the apparent association between exposure (HIV) and outcome (FMD) toward the null, and the significant association was found only after accounting for serum creatinine. This is the opposite of more commonly observed positive confounding, in which a confounding variable makes an association between exposure and outcome that may not be truly significant appear significant (at least, until the confounder is adjusted for). Informative statistical concepts observed in this study do not end at negative confounding, however, as there was also a significant interaction between HIV and renal impairment. In light of this interaction, the authors appropriately stratified by serum creatinine levels, which is the observational study analogy to subgroup analyses from a clinical trial. Only by doing this stratification were the authors able to find that there was a significant association of HIV with FMD at higher serum creatinine levels but not lower levels. This interaction and the implied biological relevance – that HIV’s putative effects on endothelial dysfunction may be more marked in the setting of renal impairment – warrant further discussion.
Does this statistical interaction reflect a true biological interaction with mechanistic implications? Well, maybe. As with subgroup analyses in a clinical trial, the different associations of HIV with FMD by creatinine subgroup observed by Stein et al. do not provide definitive evidence. Rather, they generate interesting hypotheses with biologic plausibility that merit further study. One such hypothesis is that a pro-inflammatory immunologic bias4 – in the setting of HIV viral reservoirs/reactivation, chronic immune activation, and impaired regulatory/inflammation-resolving immunity – makes people with HIV especially vulnerable to “second hits” driving persistent inflammation and tissue damage. Given well-described mechanisms of immune activation, inflammation, and endothelial dysfunction in HIV5 and chronic kidney disease6 (CKD), this is plausible. According to this hypothesis, conditions such as HIV and CKD would create a mutually reinforcing positive feedback loop of endothelial inflammation, damage, and dysfunction (Figure). This study raises this possibility, although far more mechanistic and clinical work needs to be done to define the veracity of this hypothesis.
Figure.
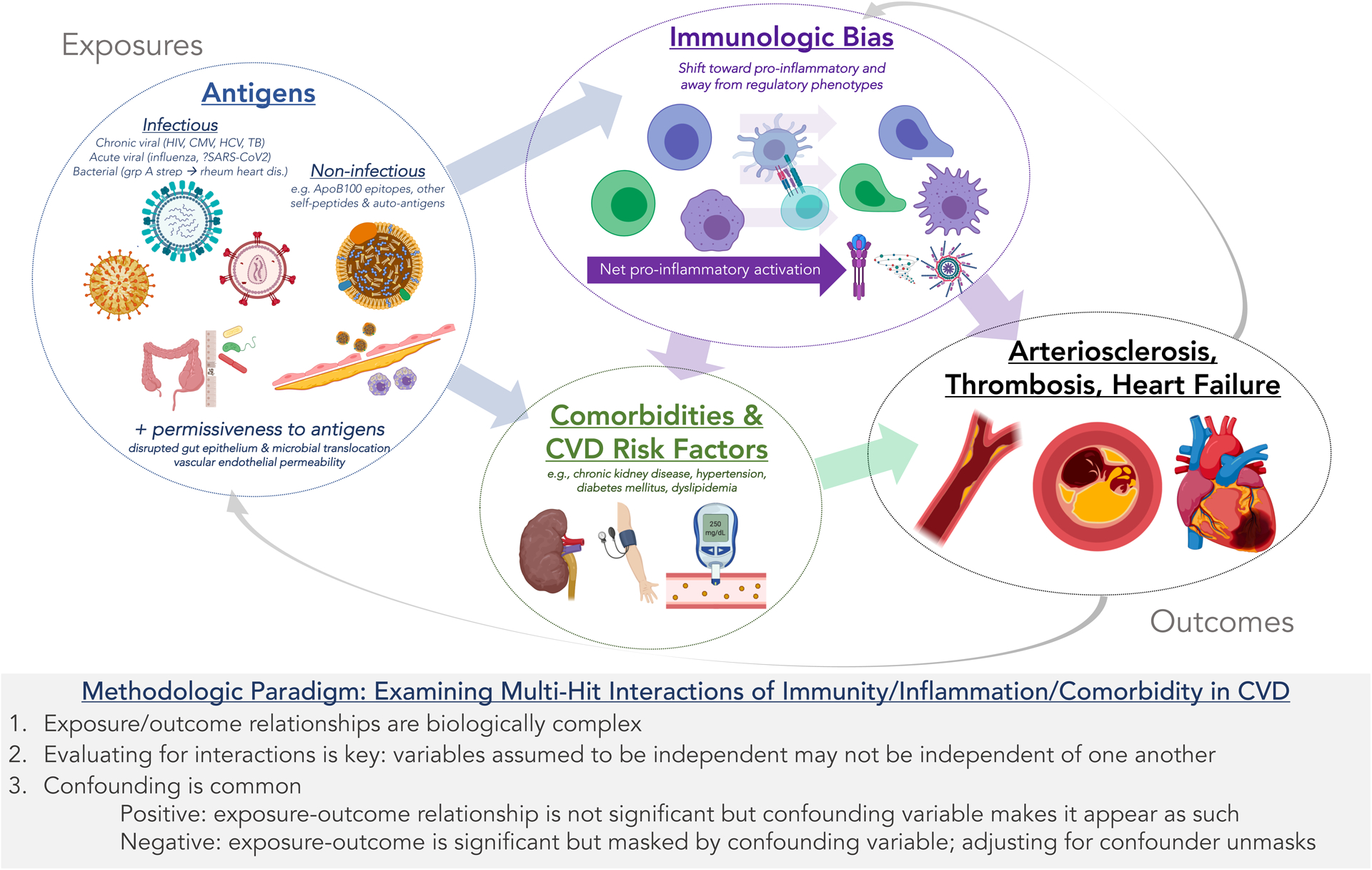
Multi-Hit Paradigm of Immune Response, Comorbidity, and Cardiovascular Diseases: Conceptual Model and Methodologic Implications. A complex interaction exists between (1) infectious and non-infectious antigens; (2) immune differentiation and signaling in response to these antigens; and (3) cardiovascular comorbidities and risk factors with respect to (4) cardiovascular disease pathogenesis. These relationships are neither linear nor unidirectional. Accordingly, studies that do not employ clearly delineated experimental models (e.g. observational studies in humans) require thoughtful consideration of confounding variables and interactions.
More broadly, what are the implications for understanding multi-hit interactions between underlying immune dysregulation and second-hit comorbidities/stressors in CVD pathogenesis? And how much of this immune dysregulation – a broadly defined entity considered in this discussion as imbalanced and inappropriate pro-inflammatory vs. inflammation-resolving immune responses – is truly “underlying,” versus triggered by comorbidities and exposures?
These are important questions without immediately clear answers. But recent studies provide some clues. We know that cells of the immune system (lymphoid and myeloid lineage) are the primary effectors of inflammation and resolution thereof.7 The composition of these cells, both in terms of surface phenotype and function, is profoundly varied between individuals;8 this variation is largely non-heritable but, rather, influenced by antigenic exposures.9 These exposures include epitopes derived from infectious4 and non-infectious10 materials and can dictate immune differentiation, chemotaxis, and function in systemic and tissue-specific manners. Additional comorbidity-driven “hits” (e.g. stressors occurring in CKD, diabetes, and hypertension among other comorbidities) may then produce highly divergent immune responses that vary in function (e.g. pro-inflammatory vs. inflammation-resolving), place (systemic vs. tissue-specific), and duration. This proposed multi-hit paradigm of immune vulnerability to comorbidity-driven triggers in CVD pathogenesis is outlined in the Figure, along with related methodologic and mechanistic considerations.
Disentangling these complex relationships between antigens, immune responses, and CVD pathogenesis requires complementary approaches across the translational spectrum. Observational studies in humans generate hypotheses that can be probed in further depth with detailed human immunophenotyping studies and experimental models. Because human observational studies are not performed in controlled experimental conditions, thoughtful approaches to confounding and interactions – as performed by Stein et. al in this issue – are needed to inform these hypotheses, define mechanisms of interest for future study, and ultimately lead to effective therapies.
Sources of Funding
American Heart Association (Fellow-to-Faculty Transition Award 16 FTF 31200010)
National Institutes of Health: National Heart, Lung, and Blood Institute (R01 HL 154862)
Footnotes
Disclosure
None
References
- 1.Libby P Inflammation in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolf D and Ley K. Immunity and Inflammation in Atherosclerosis. Circulation research. 2019;124:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein J Arteriosclerosis, thrombosis, and vascular biology. 2020. [Google Scholar]
- 4.Tracy RP, Doyle MF, Olson NC, Huber SA, Jenny NS, Sallam R, Psaty BM and Kronmal RA. T-helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: the Multi-Ethnic Study of Atherosclerosis. Journal of the American Heart Association. 2013;2:e000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feinstein MJ, Hsue PY, Benjamin LA, Bloomfield GS, Currier JS, Freiberg MS, Grinspoon SK, Levin J, Longenecker CT, Post WS, American Heart Association Prevention Science Committee of the Council on E, Prevention, Council on C, Stroke N, Council on Clinical C and Stroke C. Characteristics, Prevention, and Management of Cardiovascular Disease in People Living With HIV: A Scientific Statement From the American Heart Association. Circulation. 2019:CIR0000000000000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shang F, Wang SC, Hsu CY, et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J Am Soc Nephrol. 2017;28:3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inflammation Medzhitov R. 2010: new adventures of an old flame. Cell. 2010;140:771–776. [DOI] [PubMed] [Google Scholar]
- 8.Brodin P and Davis MM. Human immune system variation. Nat Rev Immunol. 2017;17:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, Maecker HT and Davis MM. Variation in the human immune system is largely driven by non-heritable influences. Cell. 2015;160:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura T, Kobiyama K, Winkels H, et al. Regulatory CD4(+) T Cells Recognize Major Histocompatibility Complex Class II Molecule-Restricted Peptide Epitopes of Apolipoprotein B. Circulation. 2018;138:1130–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]