

Abstract
Retiform and composite hemangioendotheliomas are both locally aggressive, rarely metastasizing vascular neoplasms characterized by arborizing vascular channels lined by endothelial cells with a hobnail morphology. Composite hemangioendothelioma (CHE) displays additional cytologic and architectural components, including often vacuolated epithelioid cells, solid areas or features reminiscent of well-differentiated angiosarcoma. Triggered by an index case of a soft tissue retiform hemangioendothelioma (RHE) which revealed a YAP1-MAML2 gene fusion by targeted RNA sequencing, we sought to investigate additional cases in this morphologic spectrum for this genetic abnormality. A total of 24 cases, 13 RHE and 11 CHE involving skin and soft tissue, were tested by FISH using custom BAC probes for rearrangements involving these genes. An additional visceral CHE with neuroendocrine differentiation was tested by targeted RNA sequencing. Among the soft tissue cohort, 5/13 (38%) RHE and 3/11 (27%) CHE showed YAP1 gene rearrangements, with 5 cases showing a YAP1-MAML2 fusion, including all 3 CHE. The single neuroendocrine CHE showed the presence of a PTBP1-MAML2 fusion. All YAP1-positive CHE lesions occurred in female children at acral sites, compared to fusion negative cases which occurred in adults, with a wide anatomic distribution. YAP1-positive RHE occurred preferentially in males and lower limb, compared to negative cases. These results suggest that RHE and CHE represent a morphologic continuum, sharing abnormalities in YAP1 and MAML2 genes. In contrast, the neuroendocrine CHE occurring in a 37-year-old male harbored a distinct PTBP1-MAML2 fusion, and showed aggressive clinical behavior (pancreatic mass with multiple liver and lung metastases). These preliminary findings raise the possibility that neuroendocrine CHE may be genetically distinct from the conventional RHE/CHE spectrum. Further studies are needed to investigate the pathogenetic relationship of fusion-negative cases with this subset and, less likely, with other members of the HE family of tumors.
Keywords: composite hemangioendothelioma, retiform hemangioendothelioma, fusion, YAP1, MAML2
INTRODUCTION
Retiform hemangioendothelioma (RHE) is a locally aggressive, rarely metastasizing, vascular lesion, characterized by distinctive arborizing blood vessels lined by endothelial cells with characteristic hobnail morphology, reminiscent of normal rete testis. The lesions occur preferentially in the skin and subcutaneous tissue of the distal extremities, particularly in the lower limb1. Similar to RHE, classic composite hemangioendothelioma (CHE) is also classified as a locally aggressive, rarely metastasizing, vascular neoplasm. CHE is characterized by an admixture of histologically distinct components, with retiform hemangioendothelioma being the most common architectural pattern, followed by epithelioid cell change, which may simulate epithelioid hemangioendothelioma (EHE)2. Most CHE cases occur in young adults, with a slight female predominance, often involving distal extremities, especially the hands and feet, or the head and neck. An aggressive form of CHE showing neuroendocrine marker expression has also been described, associated with metastatic disease in more than half of the cases3. No recurrent genetic abnormalities have been reported to date in either RHE or CHE, although PTBP1-MAML2 and EPC1-PCH2 gene fusions have been identified in single cases of neuroendocrine CHE3. In this study, prompted by an index case of RHE with a YAP1-MAML2 gene fusion, we have investigated a group of RHE and CHE for this genetic abnormality, in order to assess their pathogenetic relationship.
MATERIAL AND METHODS
Clinical data, including age, gender, and anatomic site were retrieved from pathology reports. Hematoxylin and eosin-stained slides from biopsy and resection specimens were re-reviewed by two of us (CRA, CDF). Pathologic features were recorded, including type of vasoformation (i.e. retiform vascular channels, hemangioma-like), degree of cellularity, cytomorphology, nuclear pleomorphism, mitotic activity, type of stromal component, amount of inflammatory infiltrate, etc. In all cases IHC for endothelial markers (CD31, ERG) were confirmed positive, and in a subset of cases immunostaining for podoplanin (D2–40) and synaptophysin was also performed. As most of the cases originated from personal consultations, the limited material available was prioritized for molecular studies. A diagnosis of RHE was based on the presence of the characteristic elongated and narrow arborizing vascular channels, lined by monomorphic endothelial cells with protuberant nuclei, showing a typical tombstone or hobnail appearance, reminiscent of normal rete testis. The cells showed scant cytoplasm and lack nuclear pleomorphism or increased mitotic activity, often blending with the underlying fibrotic stroma. Other architectural components were not present. In contrast, CHE was defined by at least two morphologically distinct vascular tumor elements, most often closely resembling RHE and epithelioid hemangioendothelioma (EHE). Other less common patterns include hemangioma-like or areas resembling low grade angiosarcomas. Similar to RHE, the degree of cytologic atypia was mild, with no increased mitotic activity or necrosis present. The study was approved by the Institutional Review Board.
Fluorescence In Situ Hybridization
Subsequent to the results obtained from targeted RNA sequencing in the index case, all tumors were tested by fluorescence in situ hybridization (FISH) for YAP1 and MAML2 gene rearrangements. Custom probes made by bacterial artificial chromosomes (BAC) clones flanking the YAP1 and MAML2 genes of interest according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; https://bacpacresources.org/). DNA from each BAC was isolated according to the manufacturer’s instructions. The BAC clones were labeled with fluorochromes (fluorescent-labeled dUTPs, Enzo Life Sciences, New York, NY) by nick translation and validated on normal metaphase chromosomes. The 4 μm-thick FFPE slides were deparaffinized, pretreated, and hybridized with denatured probes. After overnight incubation, the slides were washed, stained with 4’,6-diamidino-2-phenylindole, mounted with an antifade solution, and then examined on a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) controlled by Isis 5 software (Metasystems). Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were omitted from the score.
Targeted RNA Sequencing
Two cases were analyzed by targeted RNA sequencing, using RNA extracted from FFPE tissue (cases #1,9). In the index case #1, the RNA was extracted with the Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA) and the fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-seq libraries were prepared using 20 to 100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA). Targeted RNA sequencing was performed on an Illumina MiSeq platform. For case # 9, RNA extracted by the automated Maxwell 16 Research extraction system (Promega, Madison, WI, USA) was analyzed on an Ion Torrent S5 sequencing platform using the Archer FusionPlex Sarcoma Assay4. Reads were independently aligned with STAR (version 2.3) against the human reference genome (hg19) and analyzed by STAR-Fusion.
RESULTS
Clinical and Pathologic Findings
Twenty-four cases of soft tissue hemangioendotheliomas from the authors consultation files were included in the study, subclassified as 13 RHE and 11 CHE, using well-defined WHO criteria and as described above1. The patients in both histologic groups had a wide age range at diagnosis, with 10–55 years (mean 29 years) for the RHE group and 7–68 (mean 29 years) for the CHE group. The anatomic distribution was also variable, with both RHE and CHE lesions occurring in the skin or superficial soft tissues of the lower or upper extremity, and less frequently in the head and neck. An additional patient with a CHE with neuroendocrine features was included in the analysis. This patient was a 37 year-old male, who presented with a pancreatic mass and multiple liver and lung lesions consistent with metastases. Regardless of histologic subtype, the lesions were ill-defined and showed infiltrative growth, often associated with a densely fibrotic stroma, and patchy lymphocytic infiltrates. None of the cases showed evidence of nuclear pleomorphism, increased mitotic activity or necrosis.
YAP1 gene rearrangements are detected in one-third of RHE.
Among the 13 cases, 5 (38%) RHE showed the presence of YAP1 gene rearrangements. The index case (Case 1) of a 10-year-old with a superficial soft tissue mass on the knee (Fig 1) was investigated by targeted RNA sequencing and showed an intra-chromosomal inversion on 11q21-q22, resulting in a YAP1-MAML2 fusion, with YAP1 exon 5 fused to MAML2 exon 2 (Fig 2). An additional case (case 3) showed MAML2 gene rearrangement by FISH, in addition to YAP1 break-apart, while in the other 3 cases there were no abnormalities of MAML2 gene detected.
Figure 1. Morphologic features of RHE with YAP1 gene rearrangements.
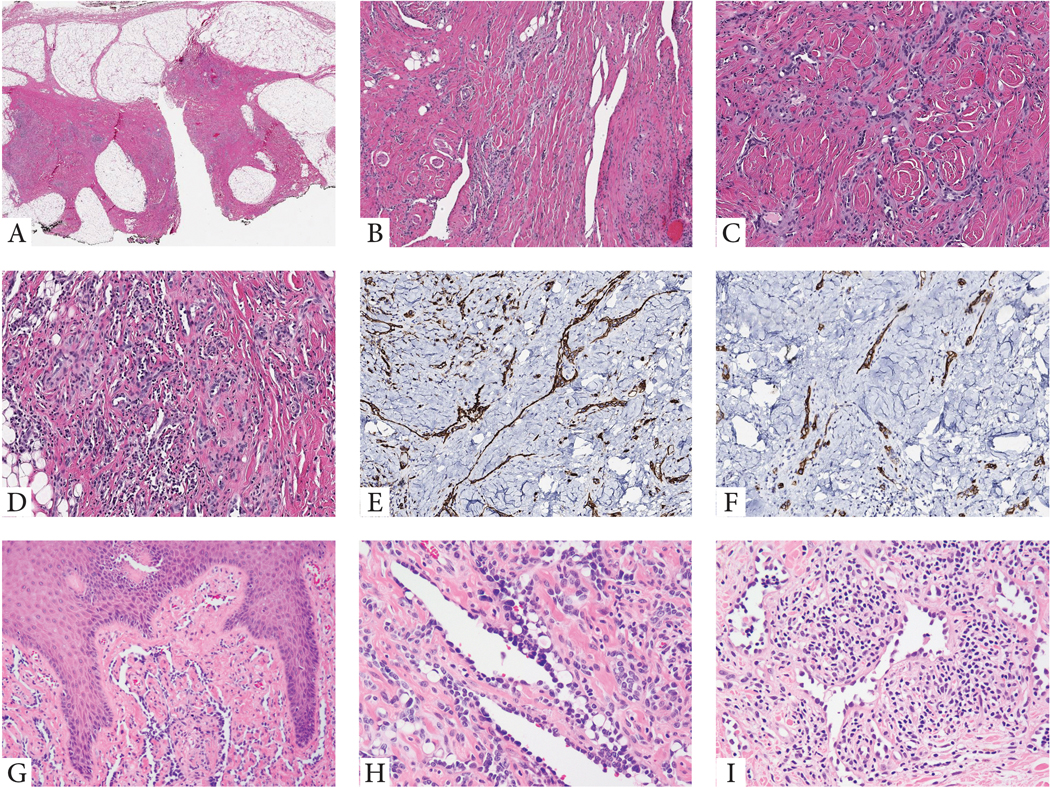
(A-F). Index case of a superficial soft tissue lesion from the knee in a 10 year-old male (case 1). Low power view showing a vague nodular and infiltrating growth pattern within subcutis which appears heavily fibrotic (A). Medium power shows densely sclerotic stromal background with either elongated, staghorn vessels (B), or shorter, retiform vascular network, lined by small endothelial cells with scant cytoplasm (C), with patchy prominent lymphocytic infiltrate, obscuring the vascular proliferation (D). Tumor cells typically co-express CD31 (E) and podoplanin (D2-40) (F). G-H. Cutaneous example showing elongated vascular channels infiltrating through the superficial dermis, lined by hobnailed endothelial cells (case 4, 10/M, buttock). I. Another RHE skin lesion in an adult (50/F, knee) showing angulated vessels with dilated lumina and protruding endothelial cells, some appearing detached and reminiscent of micropapillae. The vascular channels are embedded in a stroma with abundant lymphocytic infiltrate (case 5).
Figure 2. Diagrammatic representation of the YAP1-MAML2 and PTBP1-MAML2 fusions.

(A) Chromosomal location of YAP1 gene locus in 11q22.1-q22.2, PTBP1 in 19p13.3 and MAML2 in 11q21; red vertical lines depict the genomic breakpoint locus. Arrows show the direction of transcription of each gene. (B) Upper panel depicts the YAP1-MAML2 transcript, composed of the first 5 exons of YAP1 fused to most of MAML2 (exons 2-5). Middle and lower panels show the two transcript isoforms of PTBP1-MAML2, composed of either the first 2 exons or the first 10 exons of PTBP1 gene fused to MAML2 gene exons 2-5. The protein domains of each of the genes involved are also schematically depicted.
There was no significant age difference between the YAP1-positive versus YAP1-negative molecular groups of RHE, with a mean age of 25 years (range 10–50) versus a mean age of 33 years (range 14–55), respectively. However, all except one patient with a YAP1-positive RHE occurred in males on the lower extremity, while in the molecular-negative group the gender and anatomic site distribution was variable (Tables 1 &2).
Table 1.
Clinicopathologic features of RHE and CHE cases positive for YAP1 and MAML2 gene rearrangements.
| HE# | HE type | Age/Sex | Site | Genetic abnormality |
|---|---|---|---|---|
| 1 | RHE | 10/M | Knee | YAP1-MAML2α,β |
| 2 | RHE | 31/M | Shoulder | YAP1β |
| 3 | RHE | 23/M | 4th toe | YAP1-MAML2β |
| 4 | RHE | 10/M | Buttock | YAP1β |
| 5 | RHE | 50/F | Knee | YAP1β |
| 6 | CHE | 9/F | Foot | YAP1-MAML2β |
| 7 | CHE | 9/F | Heel | YAP1-MAML2β |
| 8 | CHE | 7/F | Middle finger | YAP1-MAML2β |
| 9 | NE-CHE | 37/M | Pancreas, Liver and Lung lesions | PTBP1-MAML2α |
HE, hemangioendothelioma; RHE, retiform hemangioendothelioma; CHE, composite hemangioendothelioma; NE-CHE, neuroendocrine CHE; F, female; M, male
, targeted RNA sequencing
, FISH
Table 2.
Clinicopathologic features of RHE and CHE cases negative for YAP1 and MAML2 gene abnormalities.
| HE# | HE type | Age/Gender | Location |
|---|---|---|---|
| 1 | RHE | 34/F | elbow |
| 2 | RHE | 14/M | shoulder |
| 3 | RHE | 18/M | scalp |
| 4 | RHE | 46/F | knee |
| 5 | RHE | 17/M | forehead |
| 6 | RHE | 46/F | long finger |
| 7 | RHE | 30/F | thigh |
| 8 | RHE | 55/M | thigh |
| 9 | CHE | 19/M | hand |
| 10 | CHE | 56/F | forearm |
| 11 | CHE | 24/F | scalp |
| 12 | CHE | 36/F | scalp |
| 13 | CHE | 35/M | heel |
| 14 | CHE | 12/M | shoulder |
| 15 | CHE | 68/F | buttock |
| 16 | CHE | 42/F | finger |
HE, hemangioendothelioma, RHE, retiform hemangioendothelioma; CHE, composite hemangioendothelioma; F, female; M, male
Morphologically, no differences were noted between the two molecular groups (Figs 1, 4), or between tumors with YAP1-MAML2 fusions versus YAP1 gene abnormalities alone. Microscopically, RHE were composed of variably sized, angulated blood vessels, with either widely open lumina or more collapsed, narrow channels (Fig 1). There was no discernible lobular growth pattern, instead the vascular proliferation showed infiltrative growth, often extending to the inked margins. Most vascular lumens appeared empty, with only rare red blood cells. The endothelial lining typically displayed scant cytoplasm, with protruding, enlarged hobnailed nuclei. In some cases, the endothelial cells showed a moderate amount of eosinophilic cytoplasm, but vacuolated cells were relatively few (Fig 1). Mitotic figures were difficult to find (0–1MF/10HPFs). Nuclear pleomorphism and necrosis were absent in all cases. One case showed intra-luminal detached endothelial cells, reminiscent of micropapillary endothelial hyperplasia, however, none showed the fibrous cores, typically seen in papillary intralymphatic angioendothelioma (‘Dabska’s tumor’) (Fig 1). All cases were associated with a densely fibrotic stroma, infiltrating and encasing adipose tissue, as well as patchy lymphocytic infiltrates, in areas obscuring the vascular proliferation (Fig 1).
Figure 4. Histologic features of the molecular negative subset of RHE and CHE.
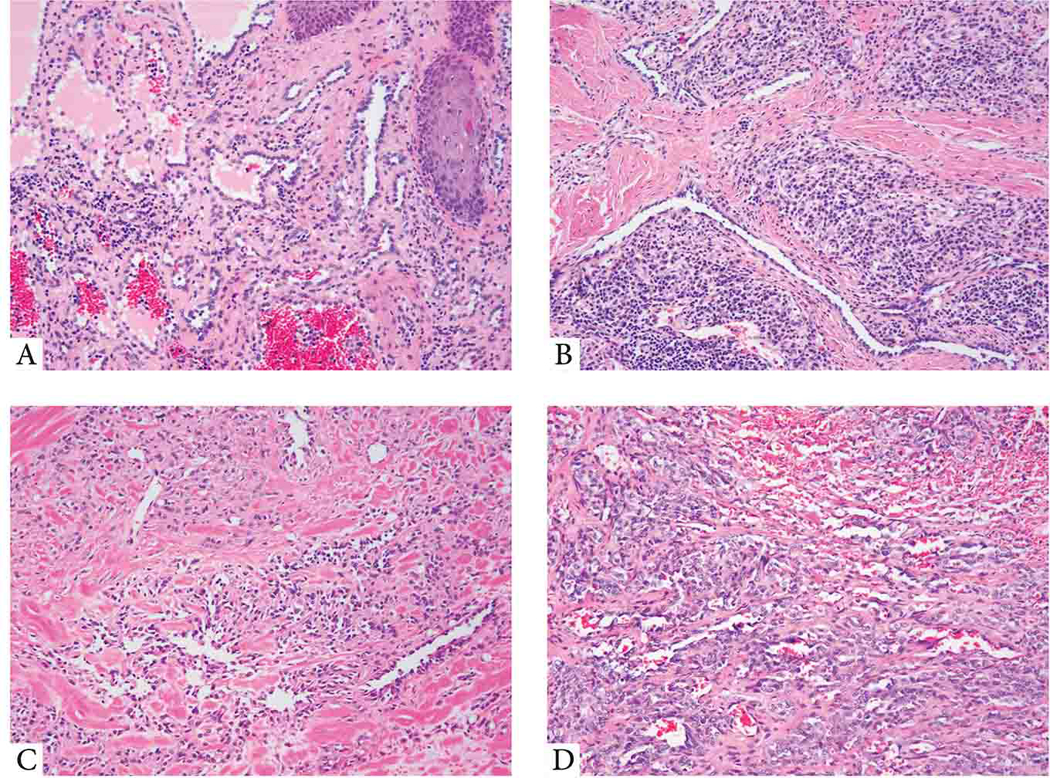
(A-B) YAP1-negative RHE. (A; 14/M, shoulder) Low power view showing dilated blood vessels within dermis lined by hobnail endothelial cells. (B; 55/M, thigh) Elongated, tubular vascular channels lined by cuboidal hyperchromatic endothelial cells, embedded in a stroma with abundant lymphocytic infiltrate. (C,D) CHE lacking YAP1 or MAML2 gene rearrangements. (C; 24/F, scalp) Predominant retiform areas with a focal epithelioid component arranged in single files. (D; 36/F, scalp) A variegated appearance showing solid, single cells as well as hemangioma-like areas.
In the index case, harboring YAP1-MAML2 fusion, material was available for further immunostaining, which showed positivity for podoplanin (D2–40), in addition to CD31 and ERG, while synaptophysin was negative (Fig 1).
YAP1-MAML2 fusion-positive CHE occur with predilection for female children at acral sites
Among the 11 cases, 3 (27%) CHE showed the presence of YAP1 and MAML2 gene rearrangements, in keeping with a YAP1-MAML2 fusion. There was a significant difference in age at presentation in the molecular positive group, with a mean age of 8 years (range 7–9), compared to the negative cases, where the mean age was 34 (range 12–68) (Table 1). Moreover, all YAP1-MAML2 fusion-positive lesions occurred in females at acral sites, while the 8 negative cases had a relatively even gender and wide anatomic distribution, with only one case occurring in a distal extremity (heel) (Tables 1&2). Microscopically, the 3 fusion-positive CHE were characterized by variable amounts of RHE and epithelioid components. No other components were noted. The epithelioid cells were arranged in single cells, cords, solid nests or ill-formed vascular channels, reminiscent of EHE. Intracytoplasmic vacuoles were noted in all cases. However, all lesions were associated with a densely fibrotic stroma, lacking the myxochondroid matrix typically seen in EHE. The RHE component ranged from predominant to very focal (Fig 3), the angulated or elongated channels being lined by relatively small endothelial cells with hobnailed, uniformly hyperchromatic nuclei. All tumors showed low mitotic activity (0–1 MF/10 HPFs), lacking nuclear pleomorphism or necrosis. In one case (case 8), material was available for further immunostaining, showing podoplanin (D2–40) reactivity (Fig 3), while synaptophysin was negative.
Figure 3. Morphologic features of YAP1-MAML2 fusion-positive soft tissue CHE and PTBP1-MAML2-positive neuroendocrine CHE.
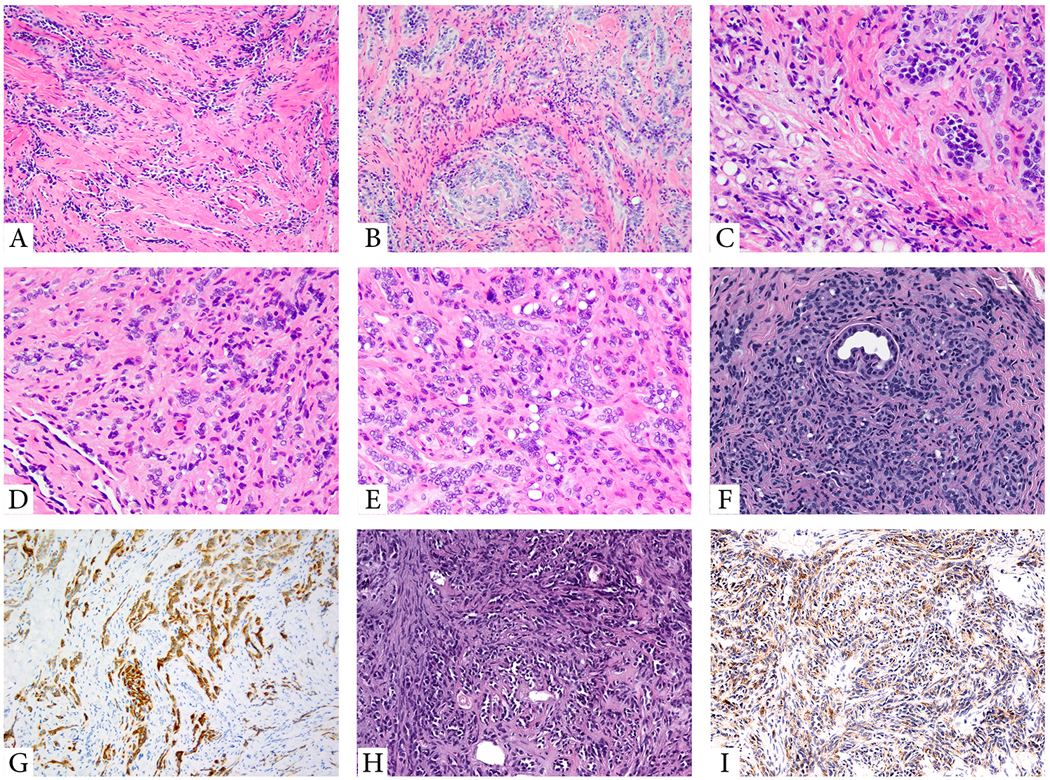
(A-C; Case 6, 9/F; foot). Low power showing a predominant retiform component (A), with other areas showing solid sheets or cords of epithelioid cells, embedded in a diffusely fibrotic stroma (B). High power showing epithelioid cells and vacuolated cells as well as small vessels lined by plump epithelioid cells, obstructing the lumina (C). (D-G; Case 9, 37/M, liver biopsy). Low power showing a small retiform area with predominantly epithelioid cells arranged in cords and single cells in a sclerotic stroma (D). Numerous vacuolated cells can be seen (E). The lesion lacks a lobular growth pattern, with cords and single tumor cells encasing adnexal structures (F). Tumor is positive for D2–40 (G) and ERG (not shown). (H-I; case 8, 7/F, middle finger). (H). Neuroendocrine CHE with a predominant retiform architecture lined by uniform hobnailed endothelial cells. (I) Tumor showed positivity for synaptophysin.
Although no significant microscopic differences were noted between the two CHE molecular groups, some of the fusion-negative cases showed more cellular solid growth and one showed a hemangioma-like pattern (Fig 4).
Neuroendocrine CHE variant is associated with aggressive clinical behavior and a distinctive PTBP1-MAML2 fusion.
A 37 year-old male presented with a pancreatic mass and multiple liver lesions. A liver biopsy was obtained which showed a predominantly a retiform pattern of elongated vascular channels lined by enlarged, hobnailed endothelial cells (Fig 3). A focal epithelioid component was also present. The lesional cells showed diffuse hyperchromasia, mild cytologic atypia, but no discernible increase in mitotic activity. Large areas of infarction were noted. By immunohistochemistry the tumor cells were positive for CD31, ERG and synaptophysin, but negative for chromogranin. Based on the clinical presentation and morphologic appearance a diagnosis of low grade angiosarcoma was suspected, and tissue was submitted for targeted RNA sequencing. The results showed the presence of a t(11;19) resulting in a PTBP1-MAML2 fusion. The fusion reads showed the presence of 2 isoforms from alternative splicing, with MAML2 being fused to either exon 2 or exon 10 of PTPB1 (Fig. 2).
DISCUSSION
Both retiform hemangioendothelioma (RHE) and composite hemangioendothelioma (CHE) are currently classified as locally aggressive, rarely metastasizing, vascular lesions in the latest WHO classification, occurring preferentially in the skin and soft tissue of the extremities, particularly in the lower limb1. RHE has a wider age range at diagnosis, often affecting children and young adults, with equal gender distribution, while CHE occur chiefly in adults, with very rare pediatric or congenital cases, and a slight female predilection1,5. Interestingly, in our cohort all YAP1-positive CHE occurred in female children in acral sites, while fusion-negative CHE cases occurred in adults of both genders, and a wide anatomic distribution, including extremities and head and neck (Tables 1&2). In contrast, RHE patients had a wide age range at diagnosis and anatomic distribution regardless of the YAP1 gene rearrangement results.
RHE is characterized by distinctive arborizing blood vessels lined by endothelial cells with characteristic hobnail morphology, reminiscent of normal rete testis. Remarkably, CHE contains at least two morphologically distinct vascular tumor elements, most often closely resembling retiform hemangioendothelioma (RHE) and epithelioid hemangioendothelioma (EHE). Although CHE was previously defined as ‘an admixture of histologically benign, intermediate and malignant components’ 6, the subsequent 2013 WHO classification revised its defining histologic features as a vascular neoplasm ‘containing an admixture of histologically distinct components’, thus removing the designation of various risks of malignancy 1. This revised definition also reflects the typically favorable clinical course of patients with classic CHE, who, despite a high rate of local recurrence (~50%)7, likely a consequence of its infiltrative growth and incomplete excision, have a low risk of lymph node (6%) or distant (<1%) metastases2,8. Of note the behavior of RHE mirrors that of CHE, showing a high local recurrence rate due to incomplete removal and a low rate of locoregional metastases7,9,10. A recent study of 11 CHE cases exhibiting neuroendocrine differentiation highlighted a preference for deep soft tissue location and a significantly more aggressive behavior compared to the typical forms of CHE, developing distant metastases in half of patients with follow-up, including to bone, lung, liver, or brain3.
Although three of these cases were initially regarded as ‘angiosarcoma,’11, the authors reclassified them as CHE in a subsequent study due to their ‘composite’ constellation of histologic features, characterized by a distinctive admixture of retiform, nested, and solid/epithelioid areas, somewhat reminiscent of EHE. Thus, in their updated larger series including 11 patients, all cases showed morphologic features characteristic of CHE, including elongated, retiform vascular channels or ‘hemangioma-like’ foci lined by hyperchromatic ‘hobnail’ endothelial cells and solid growth of uniform epithelioid cells3. Moreover, the tumors showed low mitotic activity, lacking necrosis or areas of conventional angiosarcoma. Interestingly, whole transcriptome analysis performed in 5 of the cases showed one case with PTBP1-MAML2 and one case with EPC1-PHC2 fusion transcripts; fusion transcripts were not identified in the remaining cases. Moreover, FISH testing for gene rearrangements in MAML2 and EPC1 genes were not identified in the negative cases. Of note, the authors conclude that this unusual hemangioendothelioma with neuroendocrine features likely represents a clinically aggressive variant of CHE, although they also raise the possibility of an unusual variant of RHE, or even an altogether distinct entity. Our single case of visceral neuroendocrine CHE included in this series is thus the second case reported with a PTBP1-MAML2 fusion, with an identical transcript3, suggesting a recurrent event which is distinct from the conventional CHE spectrum. As their second neuroendocrine CHE case showed an EPC1-PHC2 fusion, we have tested our YAP1 and MAML2-negative cohort (Table 2) for EPC1 gene rearrangements by FISH but none were found, once again suggesting that the genetic abnormalities seen in their cohort might be different from the regular CHE/RHE spectrum.
The diagnosis of RHE is often straightforward based on the characteristic elongated and narrow arborizing vascular channels, lined by monomorphic endothelial cells with prominent protuberant nuclei, with a typical tombstone or hobnail appearance. The cells have scant cytoplasm and lack nuclear pleomorphism or increased mitotic activity, often blending with the underlying fibrotic stroma. However, if the vascular channels are small or collapsed, the retiform architecture may be difficult to recognize. Focal solid areas composed of sheets of endothelial cells are often identified, which may suggest epithelioid hemangioendothelioma (EHE) or CHE. However, EHE is characterized by cords of epithelioid cells with relatively abundant eosinophilic, glassy cytoplasm and numerous vacuolated cells, which are embedded in a distinctive myxochondroid stroma, features not observed in RHE. Although rare intravascular papillae with hyaline collagenous cores may be occasionally seen as a focal finding in RHE, these are not as prominent as seen in papillary intralymphatic angioendothelioma (aka Dabska tumor)12. RHE is the most common component of CHE, and therefore any additional architectural patterns seen adjacent to an RHE are diagnostic of CHE. However, the distinction between these 2 entities appears somewhat arbitrary, especially in CHE cases with a predominant RHE component, while the remaining patterns might be focal and characterized by non-specific solid areas. Indeed, some discordance in assigning RHE versus CHE subtype was noted among the co-authors, suggesting the possibility of an overlapping morphologic spectrum, centered on the retiform growth, with minor components of other patterns. However, the designation of RHE versus CHE in the current study was performed blinded to the molecular results.
CHE presents as a poorly circumscribed, infiltrative lesion that is typically centered in the dermis and subcutis, although occasional cases are deep-seated or involve viscera. It comprises a complex admixture of different vascular components that vary greatly in their relative proportions, which include RHE, EHE-like, hemangioma-like, angiosarcoma-like areas, etc. Not all cases contain every component. Vacuolated, pseudolipoblastic endothelial cells are frequently present. The angiosarcoma-like areas are usually characterized by a low-grade angiosarcomatous appearance, composed of complex dissecting vascular channels with subtle endothelial atypia and relatively few mitotic figures. However, there is a single convincing reported case containing foci resembling high-grade epithelioid angiosarcoma13. In contrast, conventional angiosarcoma is characterized by high nuclear grade, brisk mitotic activity, and frequent necrosis. Neuroendocrine CHE is characterized by a distinctive admixture of RHE, EHE-like areas, and a component with a strikingly nested appearance, as well as by expression of neuroendocrine markers (most often synaptophysin).
Interestingly, both YAP1 and MAML2 related fusions have been reported previously in vascular lesions. First, YAP1-TFE3 fusion, resulting in oncogenic TFE3 upregulation, represents the driver alteration of a small subset of EHE, showing distinctive morphologic features from the classic WWTR1-CAMTA1 positive EHE, by displaying well-formed vascular channels lined by plump epithelioid cells with voluminous cytoplasm14,15. Second, WWTR1-MAML2 has been described recently in another subset of EHE, which has a predilection for the heart16. WWTR1 and its paralogue YAP1 encode the proteins TAZ and YAP, respectively, which are downstream effectors of the Hippo tumor suppressor pathway and function as regulators of TEAD-dependent transcription17. Other tumors harboring YAP1 gene fusions includes a subset of MUC4-negative sclerosing epithelioid fibrosarcoma (YAP1-KMT2A)18,19; recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma20; and more recently YAP1-MAML2 fusions have been reported in distinctive metaplastic thymomas 21. Moreover, MAML2 gene fusions occur in most mucoepidermoid carcinomas (MECT1-MAML2, CRTC1-MAML2)22.
Our molecular findings suggest a close relationship between RHE and CHE, sharing in a similar proportion (one-third of the cases) YAP1 gene rearrangements, often with YAP1-MAM2 gene fusions. We also report the second case of a neuroendocrine CHE with PTBP1-MAML2 fusion, which appears to define a clinical subset with aggressive clinical behavior. Further NGS studies are needed to investigate the genetic signatures of the fusion-negative cases of RHE, CHE and the aggressive, neuroendocrine subset of CHE.
Supplementary Material
Acknowledgements:
The authors thank and acknowledge the following referring pathologists who kindly provided case material: Dr. Robert S. Katz, Morristown, NJ, Dr. S. Sanders, Port Chester, NY, Dr. Debra M. Jih, Horsham, PA, Dr. Duane Barber, Calgary, AB, Dr. Liz Roberts, Nelson, New Zealand, Dr. Cher-Wei Liang, Taipei, Taiwan, Dr. Igor Shendrik, Tulsa, OK, Dr. Bostjan Luzar, Ljubljana, Slovenia, Dr. Montserrat Giles, Middlesbrough, U.K., Dr. Chitra Pushpanathan, St. John’s, NL, Dr. Yushan Chiou, New York, NY, Dr. Justin Hilson, Framingham, MA, Dr. Elen Blochin, New York, NY, Dr. Richard B. Hessler, Chattanooga, TN, Dr. Muammar A. Arida, Greensboro, NC, Dr. Sotirios Barbanis, Thessaloniki, Greece, Dr. Michael Zhang, Newton, MA, Dr. Ralph Winkler, Alpharetta, GA, Dr. Stephen Lyle, Portsmouth, NH; Dr. Joost van Gorp, Utrecht, The Netherlands and Dr. Giulio Rossi, Modena, Italy.
Disclosures: Supported in part by: P50 CA 140146-01 (CRA), P50 CA217694 (CRA), P30 CA008748, Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA), EHE Foundation
Footnotes
Conflict of interests: none
REFERENCES
- 1.Rubin BP. Composite haemangioendothelioma In: Fletcher CD, Bridge JA, Pancras CW, et al. [eds]. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2013. [Google Scholar]
- 2.Nayler SJ, Rubin BP, Calonje E, et al. Composite hemangioendothelioma: a complex, low-grade vascular lesion mimicking angiosarcoma. Am J Surg Pathol. 2000;24:352–361. [DOI] [PubMed] [Google Scholar]
- 3.Perry KD, Al-Lbraheemi A, Rubin BP, et al. Composite hemangioendothelioma with neuroendocrine marker expression: an aggressive variant. Mod Pathol. 2017;30:1589–1602. [DOI] [PubMed] [Google Scholar]
- 4.Kirchner M, Neumann O, Volckmar AL, et al. RNA-Based Detection of Gene Fusions in Formalin-Fixed and Paraffin-Embedded Solid Cancer Samples. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shang Leen SL, Fisher C, Thway K. Composite hemangioendothelioma: clinical and histologic features of an enigmatic entity. Adv Anat Pathol. 2015;22:254–259. [DOI] [PubMed] [Google Scholar]
- 6.Rubin BP. Composite haemangioendothelioma In: Fletcher CD, Unni KK, Mertens F [eds]. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone, 3rd ed Lyon, France: IARC Press; 2002. [Google Scholar]
- 7.Calonje E, Fletcher CD, Wilson-Jones E, et al. Retiform hemangioendothelioma. A distinctive form of low-grade angiosarcoma delineated in a series of 15 cases. Am J Surg Pathol. 1994;18:115–125. [PubMed] [Google Scholar]
- 8.Requena L, Luis Diaz J, Manzarbeitia F, et al. Cutaneous composite hemangioendothelioma with satellitosis and lymph node metastases. J Cutan Pathol. 2008;35:225–230. [DOI] [PubMed] [Google Scholar]
- 9.Bhutoria B, Konar A, Chakrabarti S, et al. Retiform hemangioendothelioma with lymph node metastasis: a rare entity. Indian J Dermatol Venereol Leprol. 2009;75:60–62. [DOI] [PubMed] [Google Scholar]
- 10.Mentzel T, Stengel B, Katenkamp D. [Retiform hemangioendothelioma. Clinico-pathologic case report and discussion of the group of low malignancy vascular tumors]. Pathologe. 1997;18:390–394. [DOI] [PubMed] [Google Scholar]
- 11.Tessier Cloutier B, Costa FD, Tazelaar HD, et al. Aberrant expression of neuroendocrine markers in angiosarcoma: a potential diagnostic pitfall. Hum Pathol. 2014;45:1618–1624. [DOI] [PubMed] [Google Scholar]
- 12.Albertini AF, Brousse N, Bodemer C, et al. Retiform hemangioendothelioma developed on the site of an earlier cystic lymphangioma in a six-year-old girl. Am J Dermatopathol. 2011;33:e84–87. [DOI] [PubMed] [Google Scholar]
- 13.Leen SL, Clarke PM, Chapman J, et al. Composite Hemangioendothelioma of the Submandibular Region. Head Neck Pathol. 2015;9:519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer. 2013;52:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenbaum E, Jadeja B, Xu B, et al. Prognostic stratification of clinical and molecular epithelioid hemangioendothelioma subsets. Mod Pathol. 2020;33:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suurmeijer AJH, Dickson BC, Swanson D, et al. Variant WWTR1 gene fusions in epithelioid hemangioendothelioma-A genetic subset associated with cardiac involvement. Genes Chromosomes Cancer. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanas MR, Ma S, Jadaan FO, et al. Mechanism of action of a WWTR1(TAZ)-CAMTA1 fusion oncoprotein. Oncogene. 2016;35:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kao YC, Lee JC, Zhang L, et al. Recurrent YAP1 and KMT2A Gene Rearrangements in a Subset of MUC4-negative Sclerosing Epithelioid Fibrosarcoma. Am J Surg Pathol. 2020;44:368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puls F, Agaimy A, Flucke U, et al. Recurrent Fusions Between YAP1 and KMT2A in Morphologically Distinct Neoplasms Within the Spectrum of Low-grade Fibromyxoid Sarcoma and Sclerosing Epithelioid Fibrosarcoma. Am J Surg Pathol. 2020;44:594–606. [DOI] [PubMed] [Google Scholar]
- 20.Sekine S, Kiyono T, Ryo E, et al. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J Clin Invest. 2019;130:3827–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vivero M, Davineni P, Nardi V, et al. Metaplastic thymoma: a distinctive thymic neoplasm characterized by YAP1-MAML2 gene fusions. Mod Pathol. 2020;33:560–565. [DOI] [PubMed] [Google Scholar]
- 22.Behboudi A, Enlund F, Winnes M, et al. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer. 2006;45:470–481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.