

Abstract
Most neuropsychiatric disease risk variants are in noncoding sequences and lack functional interpretation. Because regulatory sequences often reside in open chromatin, we reasoned that neuropsychiatric disease risk variants may affect chromatin accessibility during neurodevelopment. Using human induced pluripotent stem cell (iPSC)-derived neurons that model developing brains, we identified thousands of genetic variants exhibiting allele-specific open-chromatin (ASoC). These neuronal ASoCs were partially driven by altered transcription-factor-binding, overrepresented in brain gene enhancers and expression quantitative-trait-loci, and frequently associated with distal genes through chromatin contacts. ASoCs were enriched for genetic variants associated with brain disorders, enabling identification of functional schizophrenia risk variants and their cis-target genes. This study highlights ASoC as a functional mechanism of noncoding neuropsychiatric risk variants, providing a powerful framework for identifying disease causal variants/genes.
One Sentence Summary
Allele-specific open chromatin informs functional disease variants
The difficulty of assigning molecular function to noncoding disease risk variants has hindered the identification of causal variants/genes of complex disorders. Noncoding regulatory sequences are marked by open chromatin, and open chromatin regions (OCRs) are enriched for single nucleotide polymorphisms (SNPs) associated with gene expression and complex disorders/traits (1, 2). For neuropsychiatric disorders, disease-associated variants are also enriched in OCRs found not only in adult brains, but also in developing brains and iPSC-derived neuronal (iN) cells (3–6), suggesting the importance of neurodevelopmental chromatin regulation. However, as not all OCR variants are functional, it remains largely unknown whether neuropsychiatric disease risk variants affect chromatin accessibility during neurodevelopment.
To tie risk alleles to function, we identified allele-specific open chromatin (ASoC) variants displaying allelic imbalance of chromatin accessibility in Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) at heterozygous SNP sites (Fig. S1) (7, 8). We focused on 20 individuals carrying heterozygous (i.e., informative for ASoC assay) GWAS index SNPs at ~70/108 schizophrenia (SZ) loci (Fig. S2, Table S1) (7, 9, 10).
We differentiated iPSCs of these 20 individuals into neural progenitor cells (NPC), glutamatergic (iN-Glut), GABAergic (iN-GA), and dopaminergic (iN-DN) neurons with high purity (75–90%) (Figs. 1A, S3) (7). We performed ATAC-seq and RNA-seq in each cell type for 8 lines (“core-8”), and ATAC-seq for 12 additional lines in NPCs and iN-Glut (Figs. 1A, S4–5; Table S2). Pooling the reads of “core-8” lines, we identified 256–337K OCRs (FDR<0.05) in each cell subtype (Fig. S6A–C; Table S3).
Fig. 1. Mapping ASoC variants.
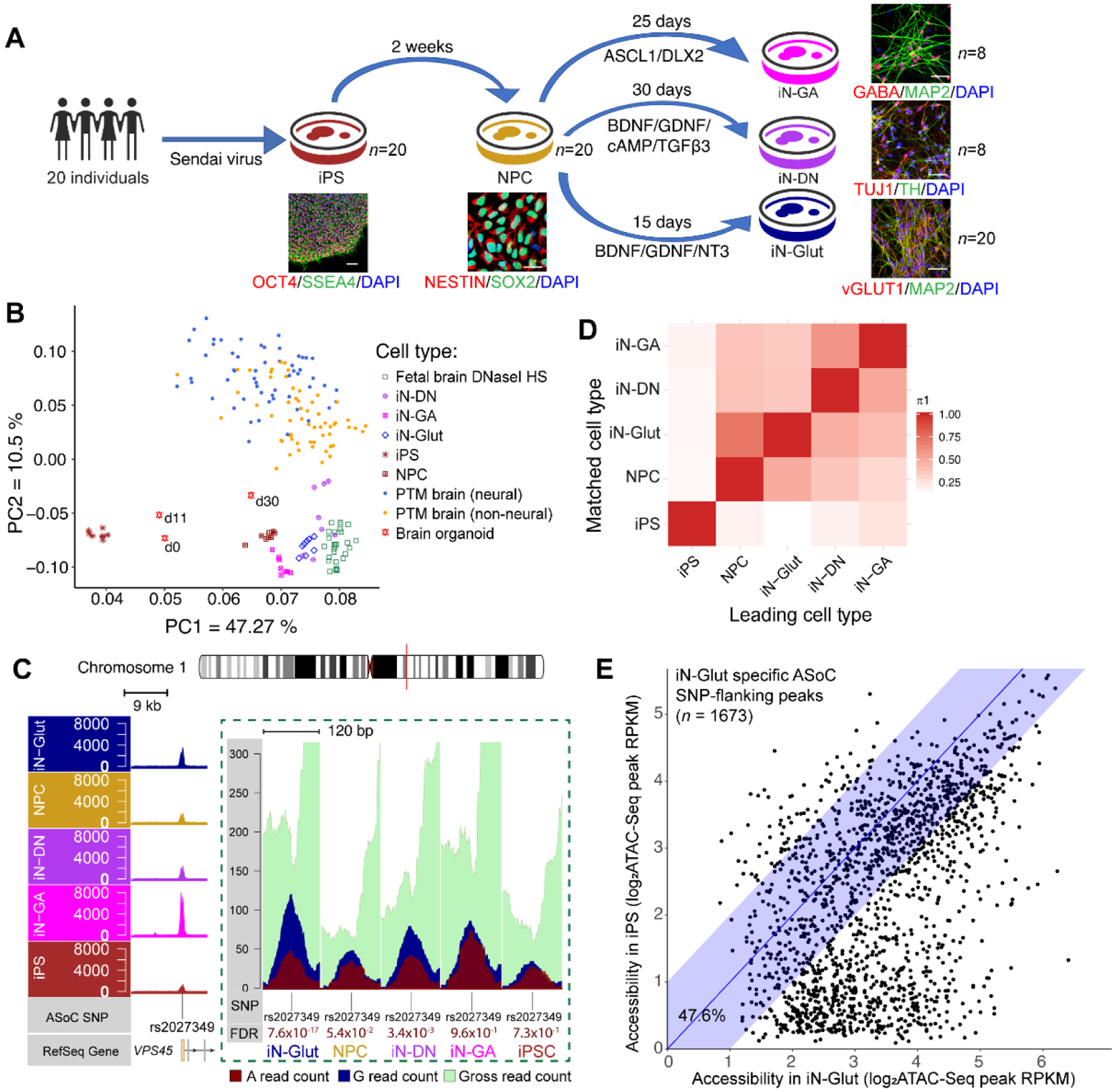
(A) Schematic of iPSC differentiation strategies. (B) PCA of OCRs from iNs, fetal brains/day-30 organoids and postmortem (PTM) adult brain. (C) Cell-type-specific ASoC rs2027349 at the SZ-associated VPS45 locus. The dashed box shows the ATAC-seq read pileups flanking rs2027349 and cell-type-specific allelic ratios. (D) Pairwise π1 estimation of ASoC sharing across cell types (7). ASoC SNPs were ascertained in the leading cell type, and π1 was estimated in the matched one. (E) Chromatin accessibility in iN-Glut vs iPSC for ASoC SNP-flanking peaks. The shaded area shows comparable (< 2-fold difference) accessibility between cell types.
The OCRs of cell-type-specific marker genes were confirmed (Fig. S6D–E) and principal component analysis (PCA) of ATAC-seq and RNA-seq samples showed cell-type-specific clustering (Figs. 1B, S7A–D). OCRs of iPSC-derived neural cells were more similar to fetal brains (11) and cortical organoids (12) than to adult brains (3) (Fig. 1B, S7C–D). Neuronal OCRs overlapped with ~53% of the 117,935 adult brain OCR peaks from PsychENCODE (3); however, the 62,813 overlapping peaks only accounted for ~20% of our neuronal OCRs (Table S3), consistent with the observation that the fetal cerebral cortex possesses more active enhancers than the adult brain (12).
We then tested ASoC variants in each iPSC-derived cell type and examined cell-type-specificity (Fig. S8) (7). We used a sample pooling approach (8, 13) after confirming the inter-individual concordance of the allelic imbalance directions of chromatin accessibility (Fig. S9A). We identified 920–2,392 ASoC SNPs (FDR<0.05) in each “core-8” cell type, and 5,611 and 3,547 ASoC SNPs in iN-Glut-20 and NPC-20 cells (n=20 lines each), respectively (Figs. S9B–E, Tables S4–11). These ASoC SNPs showed cell-type-specific patterns (Figs. 1C, S9B). Using Storey’s π1 analysis (7), we found a low percentage of estimated pairwise ASoC SNP sharing between neurons and iPSCs (10~20%; Fig. 1D). Even within neuronal cell types, ASoCs differed substantially (30–70% sharing) (Fig. 1D), suggesting cell-type-specificity of ASoC.
Cell-type-specific ASoC may be driven by either OCRs or different SNP effects (allelic ratios) across cell types. About half of the ASoC SNPs in neurons exhibited low or undetectable OCRs (read-depth<20 at SNP sites) in iPSCs (Fig. S10). On the other hand, about 50% of neuron-specific ASoC SNPs showed comparable or more accessible OCR in iPSCs (Figs.1E, S11), suggesting that variations in OCR read-depth and SNP effect size both contribute to cell-type-specific ASoC. Thus, cell-type-specific regulation likely occurs even in the absence of chromatin state changes.
To examine the cell-type-specific regulatory potential of ASoC SNPs, we compared the genomic/epigenomic features of cell-type-specific vs shared ASoC variants (7). ASoC SNPs were found enriched in both brain promoters and enhancers (Figs. 2A, S12A) (14). However, compared to shared ASoC SNPs, cell-type-specific SNPs showed stronger enrichment (2232-fold) in enhancers (Figs. 2A, Fig. S12B–E). This indicates that ASoC SNPs are associated with brain regulatory sequences and likely exhibit cell-type-specific enhancer activity.
Fig. 2. Characteristics of ASoC SNPs.
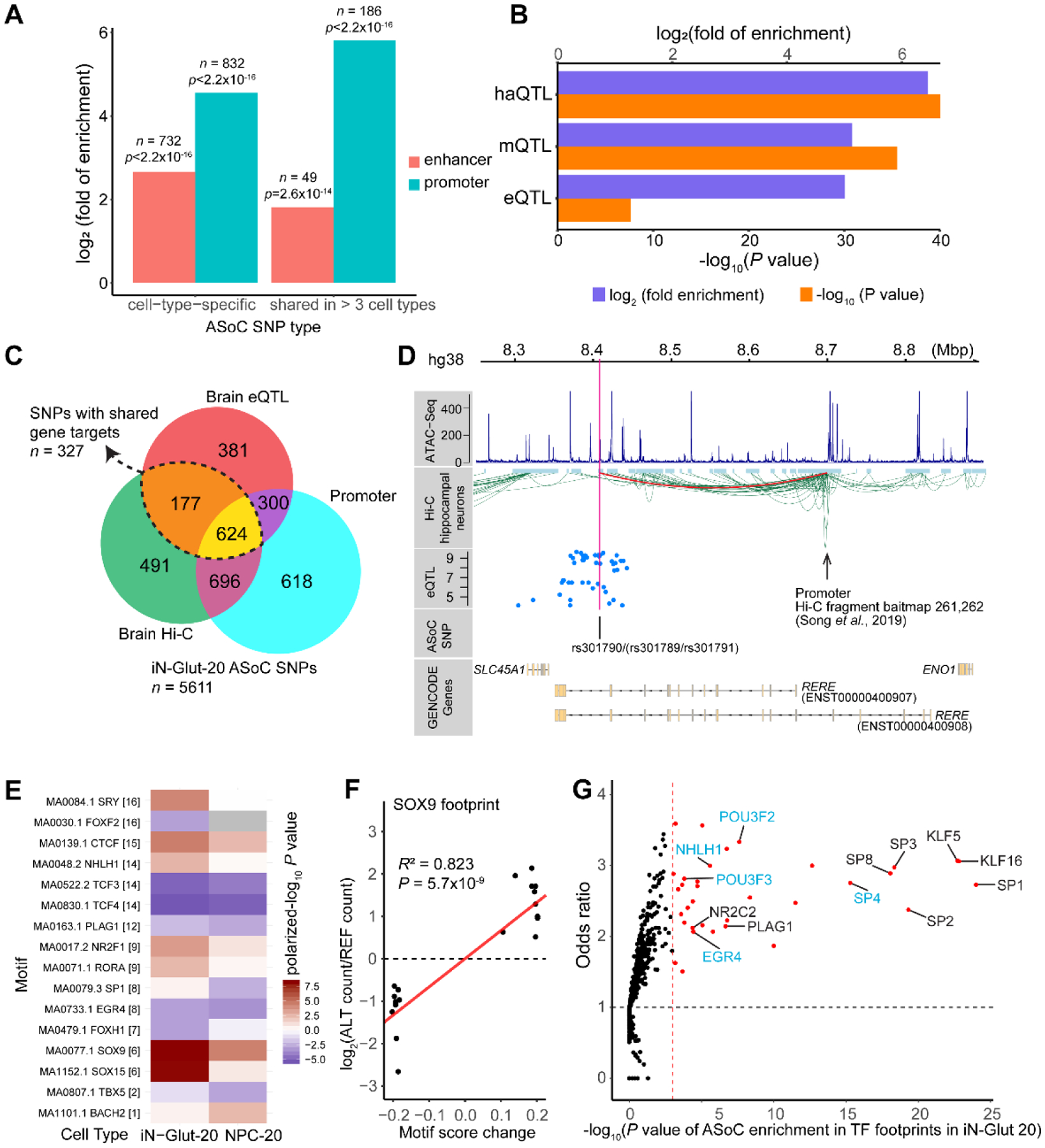
(A) Enrichment of ASoC SNPs (vs. genomic background) in the chromatin-state-based annotations of promoter and enhancer. (B) Enrichment of ASoC SNPs (vs genomic background) for brain QTL. (C) Venn diagram showing iN-Glut-20 ASoC SNPs mapped to cis-target genes through brain eQTL (17), brain/neural Hi-C (18), and promoter annotations. (D) An ASoC SNP mapped to RERE through eQTL and Hi-C. ATAC-seq peak: iN-Glut. (E) Selected TFs with significant correlations (FDR<0.05) of motif-disrupting scores and ASoC allelic imbalance. (F) SOX9 as an example of a putative chromatin activator showing positive correlation in (E). (G) TFs with binding-footprints enriched for neural ASoC SNPs (red dots; vs non-ASoC SNPs). Blue: neurodevelopmental TFs.
The effect of ASoC SNPs on enhancer/promoter activity was confirmed by reporter gene assay (7). In a massive parallel reporter assay (MPRA) SNP dataset from non-neural cells (15), 26~29% of neural ASoC SNPs altered reporter gene expression (~2-fold enrichment vs non-ASoC, p=0.001, Fisher’s exact test) (Fig. S13A–D, Tables S12–13). Using a reporter gene assay in NPCs, we found that 5 of 8 SZ-associated ASoC SNPs affected reporter gene expression (Fig. S13E–F, Table S14). These results suggest that a proportion of ASoC SNPs affect gene expression.
We then jointly analyzed our data with recent studies of genetic variants associated with brain gene expression (eQTL), histone modification (haQTL), and DNA methylation (meQTL) (7, 16). Putative causal variants underlying these QTL are enriched (> 30 fold) in iN-Glut-20 ASoC SNPs (Fig. 2B, Table S11). Intersecting with brain eQTL (17) and brain/neuronal Hi-C chromatin contact (18) annotations (7), we found that out of 5,611 iN-Glut-20 ASoC SNPs, 26% are also eQTL and 35% demonstrate chromatin contacts with distal promoters (Figs. 2C, S14A–B; Table S15). Together with promoter annotation, ~59% of ASoC SNPs were resolved to affect at least one cis-target gene (Figs. 2C, S14C). Integrating ASoC SNPs with brain eQTL and Hi-C data, we delineated putative causal eQTL SNPs and their affected cis-target genes (e.g.: Fig. 2D). These results support that many ASoCs affect distal genes through long-range chromatin contacts.
Mapping the transcription factor (TF)-binding footprints from ATAC-seq (7), we found that 32% of the 5,611 ASoC SNPs of iN-Glut-20 neurons were inside TF-binding sites (TFBSs), representing a 1.6-fold enrichment (vs non-ASoC SNPs, Fisher’s exact test, p=2.6×10−58) (Fig. S15A). Furthermore, most ASoC SNPs were within 200 bp of TF-binding footprints, and footprint analyses in other cell types gave similar results (Figs. S15B–G).
To test whether TF-motif disruptions by ASoC SNPs near footprints caused matched changes in chromatin accessibility, we performed a “motif-break analysis” (7). In iN-Glu-20 and NPC-20 cells, we identified 48 TFs with SNP motif-disruption scores correlated with allelic imbalances of ASoC (Figs. 2E–F, S16). Comparing our ASoC SNPs with an allele-specific TF-binding SNP dataset (19), we found that 70–80% of overlapping SNPs showed matched direction of allelic effects on chromatin accessibility and TF-binding (Fig. S17, Table S16). These results suggest that genetic variants that alter TF-binding result in ASoC.
We then performed TF-motif enrichment analysis for ASoC SNPs (±50 bp) and OCR (7), and identified cell-type-specific patterns of enriched TF motifs (Fig. S18; Tables S17–18). Restricting the ASoC analysis specifically to TF footprints gave similar patterns (Figs. 2G, S19) and included TCF4 and SP4, which are both SZ risk genes (10). Thus, ASoC SNPs may affect cell-type-specific binding of TFs important for cell fate commitment and neurodevelopment.
Given the brain-relevant regulatory effects of ASoC SNPs, we reasoned that ASoCs could help infer noncoding risk variants for neuropsychiatric disorders. Of the 5,611 iN-Glut-20 ASoC SNPs, 21 were SZ-associated SNPs (10) at 17 independent SZ loci (Fig. 3A, Tables S19–20). Accounting for the uncertainty of GWAS risk variants due to linkage disequilibrium (LD) with TORUS (7), neuronal ASoC SNPs showed ~70-fold enrichment for SZ risk variants, contrasting to ~3–6-fold enrichments in neuronal OCRs (Fig. 3B).
Fig. 3. Enrichment of ASoC variants for genetic risk of neuropsychiatric disorders/traits.
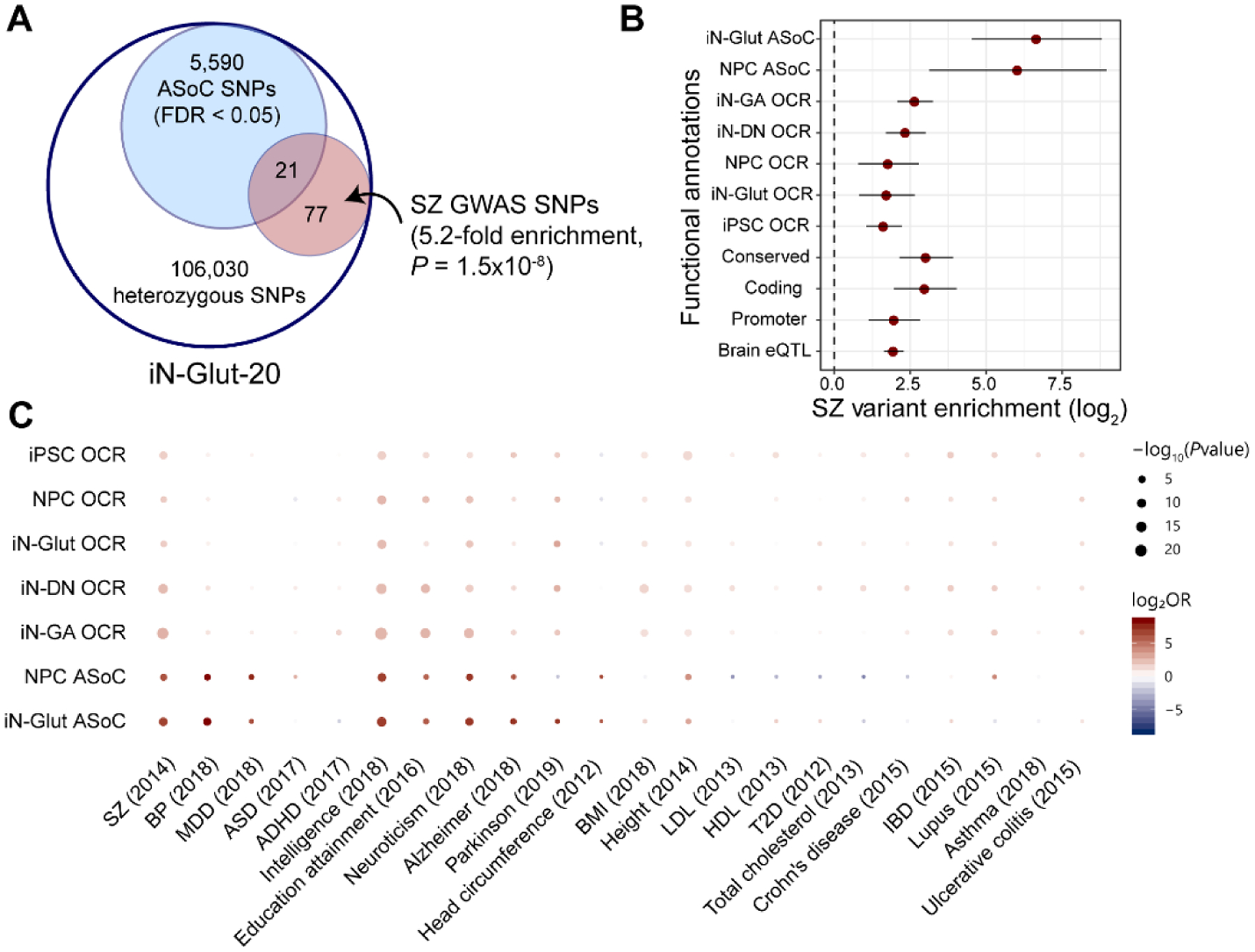
(A) iN-Glut-20 ASoC SNPs were enriched for SZ GWAS SNPs (Fisher’s exact test; vs. non-ASoC SNPs). (B) Enrichment of SZ risk variants in ASoC SNPs, OCRs and other functional annotations (vs genomic non-ASoC SNPs). Bar: 95% confidence interval. (C) Enrichment of risk variants of 22 disorders/traits in ASoC SNPs and OCRs (vs genomic non-ASoC SNPs). ADHD: Attention-Deficit/Hyperactivity Disorder, BMI: Body mass index, LDL: low-density lipoprotein, HDL: high-density lipoprotein, T2D: type 2 diabetes, IBD: Inflammatory bowel disease.
Expanding this analysis to 9 other brain disorders/traits (7), we observed enrichment of GWAS variants for bipolar disorder (BP), major depressive disorder (MDD), intelligence, educational attainment, and neuroticism in ASoC variants (iN-Glut and NPC) (Figs. 3C, S20; Table S21). Enrichment of ASoC SNPs (only iN-Glut) was also seen for Alzheimer’s disease (AD), but not Parkinson’s disease (PD) (Figs. 3C). In contrast, 12 control disorders/traits showed little or no enrichment in neuronal ASoC SNPs or OCRs (Fig. 3C), although this may reflect limited statistical power. These results suggest that neuronal ASoCs are linked to neuropsychiatric disorders and highlight the value of utilizing ASoCs in prioritizing studies of noncoding risk variants.
We selected the top 20 ASoC SNPs associated with SZ (Tables S19–20) to assess the regulatory potential of their tagged ASoC sequences and cis-target genes. We used a modified CRISPR droplet sequencing (CROP-seq) approach (20) (Fig. 4A) in NPCs stably expressing dCas9/Krüppel associated box (KRAB) which represses possible cis-genes in specific ASoC sites targeted by gRNAs (Fig. S21A–C, Table S22) (7). We analyzed 4,099 cells, of which 2,522 were assigned to unique gRNAs (Fig. S21D–F). We identified putative cis-targets as differentially expressed genes (permutation p<0.05) for a given gRNA/ASoC sequence (Figs. 4B, S22; Table S23), and then validated by independent CRISPRi/qPCR (Fig. S23; Table S24) (7). In total, CROP-seq identified cis-targets of 10 ASoC sequences, of which four share targets with brain eQTL and brain/neural Hi-C contacts (18, 21, 22) (Fig S24; Table S25).
Fig. 4. Functional characterization of SZ-associated ASoC SNPs.
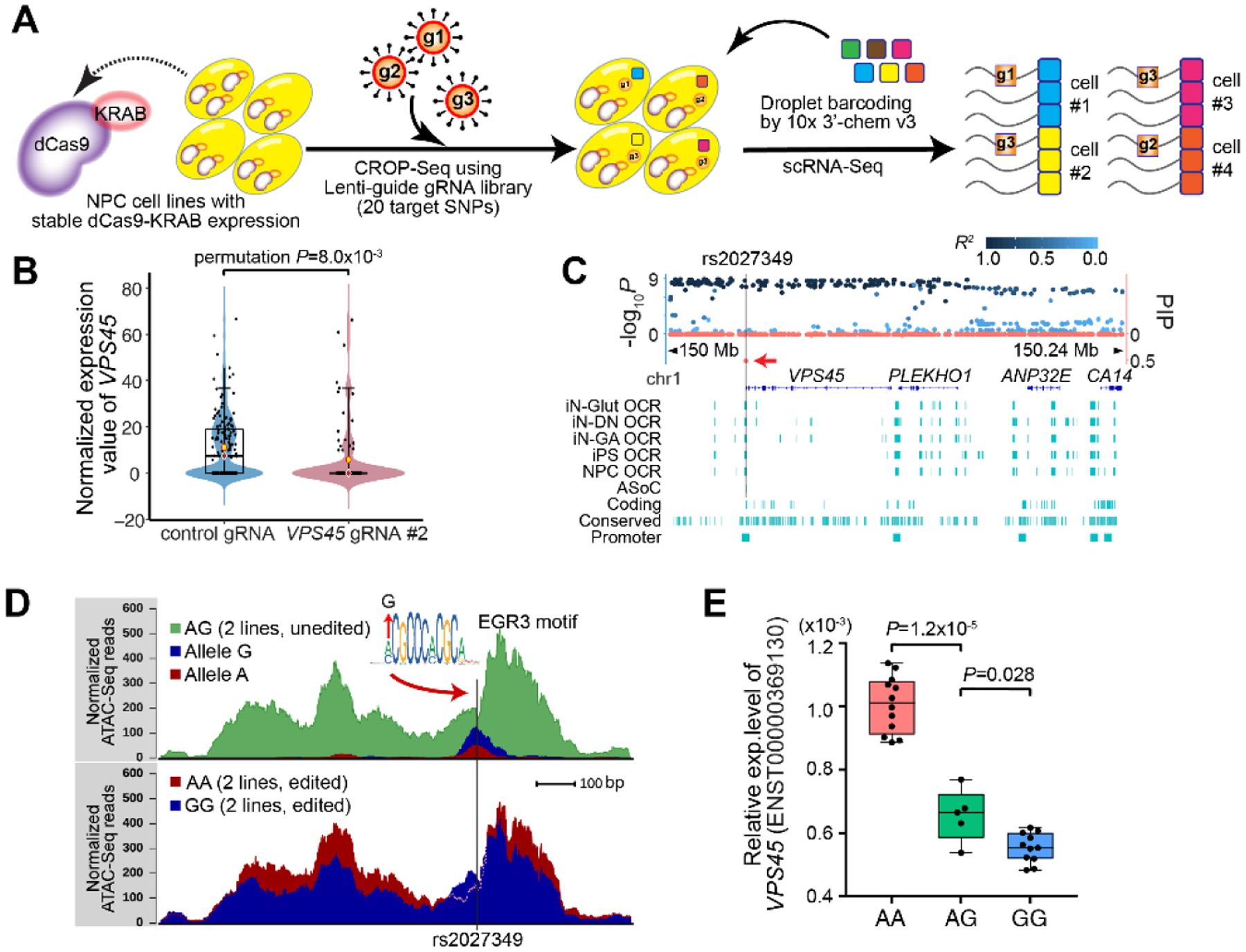
(A) Schematic of the modified CROP-seq (7) to screen cis-targets of the 20 ASoC sites. (B) Reduced VPS45 expression in single NPCs expressing gRNAs targeting rs2027349. (C) Fine-mapping the VPS45 (rs2027349) locus. Left y-axis: −log10 p-values from SZ GWAS (10) (points above the x-axis); right y-axis: PIPs (points below the x-axis, red). Vertical bar: rs2027349. (D) CRISPR-editing rs2027349 altered local OCR accessibility in isogenic NPCs (AA vs GG; 2 lines/genotype). The G allele disrupts the EGR3 motif; note that AA lines showed higher (more accessible) peak but with a lower ATAC-seq signal at the EGR3-binding site likely due to A allele’s stronger EGR3-binding that prevents transposase from accessing. (E) Editing rs2027349 altered VPS45 expression in NPCs (major transcript ENST00000369130; by qPCR). VPS45 expression was normalized to GAPDH. Two iPSC lines (AG at rs2027349) were edited to isogenic lines with AA or GG genotype, each with 2–3 clones (n=2–3 independent cultures). Student’s t-test (Welch’s correction) was used.
CROP-seq screening of cis-targets of an ASoC SNP-flanking sequence did not inform SNP allelic effect. To link SNP risk allelic function to a cis-target gene, we carried out precise SNP editing by CRISPR/Cas9 (7). We prioritized an ASoC SNP for editing based on the support from ASoC mapping, CROP-seq screening, and the Posterior Inclusion Probability (PIP) (7), the statistical evidence of an SNP being a disease-causal variant based on fine-mapping (23). Among the 20 targeted ASoC SNPs, 9 had PIP>0.10 (Table S26) of which 6 had at least one CROP-seq cis-gene (Table S23). Of these, rs2027349 at the VPS45 locus showed the strongest ASoC in NPC-20 (Table S20), exhibited a repressive effect on VPS45 in CROP-seq (p=0.008, permutation; Fig. 4B), and was the only local SNP with a high PIP (0.45, Fig. 4C). rs2027349 is within a TF-binding footprint, with the G allele predicted to disrupt the motif of EGR3/4 (Fig. 4D), a TF involved in neurodevelopment and SZ (24). rs2027349 was thus prioritized for CRISPR/Cas9 editing.
We edited two iPSC lines heterozygous for rs2027349 from A/G to A/A and G/G (Fig. S25A–B, Table S27) and characterized the SNP allelic effect on VPS45 expression in NPCs. Allele A was associated with increased expression of VPS45, especially its major transcript isoform (Figs. 4E, S25C–D). This is consistent with the allelic effect of rs2027349 on VPS45 in PsychENCODE brain RNA-seq data (Fig. S25E). In addition, rs2027349-flanking OCR in CRISPR-edited A/A lines showed higher chromatin accessibility (vs G/G) (Figs. 4D). We did not observe similar effects on two other cis-genes (ANP32E and SF3B4) nominated by CROP-seq (Fig. S22A–B). Taken together, we identified rs2027349 as a likely SZ risk variant that affects the expression of VPS45, a gene potentially relevant to SZ given its involvement in vesicle-mediated protein trafficking and neurotransmitter release (25).
We also performed CRISPR/Cas9 editing of rs12895055 (C→T) (Fig. S26), an SZ-associated SNP that did not have a CROP-seq cis-target, but showed the strongest ASoC in iN-Glut (Table S19) and a high probability of being a causal SZ variant (PIP=0.33) (Table S26). Because rs12895055 is in an intron of BCL11B (a deep-layer neuronal marker) with undetectable expression in NPCs (Table S23), we examined its allelic effect on BCL11B in CRISPR-edited iN-Glut cells (Fig. S26, Table S28). The risk allele T was associated with reduced expression of BCL11B and is predicted to alter binding-motifs of multiple TFs (Fig. S26). Notably, rs12895055 is in perfect LD with the BP-associated rs11624408 (p=5×10−8) (26). BCL11B is thus a candidate risk gene for both SZ and BP, warranting follow-up.
In summary, we have found that genetic variation commonly affects chromatin accessibility and exhibits cell-type-specific ASoC in iPSC-based neurodevelopmental model. The enrichments of neuronal ASoC SNPs for brain enhancers, TFBSs, and brain QTL suggest mechanistic links between chromatin accessibility and gene expression. Our study identified ASoC variants that have plausible causal associations with SZ, and demonstrates that some neuropsychiatric disease risk variants alter chromatin accessibility.
Supplementary Material
Acknowledgements:
We thank Dr. W. J. Greenleaf for helping on ATAC-seq. Funding: R01AA023797 (ZP); R01MH110531 (XH); R01MH106575, R01MH116281, and R01AG063175 (JD).
Footnotes
Competing interests: The authors declare no conflicts of interest.
Data and materials availability: All sequencing data are available at NCBI GEO as GSE129017 and GSE132757. Codes have been deposited at http://doi.org/10.5281/zenodo.3728037.
Supplementary Materials List:
Materials and Methods
Figs. S1 to S26;
Tables S1 to S28;
References 27–85.
Reference and Notes
- 1.Maurano MT et al. , Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finucane HK et al. , Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryois J et al. , Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat Commun 9, 3121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Torre-Ubieta L et al. , The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304 e218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forrest MP et al. , Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 21, 305–318 e308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fullard JF et al. , An atlas of chromatin accessibility in the adult human brain. Genome research 28, 1243–1252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Materials and methods are available as supplementary materials.
- 8.Xu J et al. , Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat Genet 49, 377–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J et al. , Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schizophrenia C Working Group of the Psychiatric Genomics, Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein BE et al. , The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amiri A et al. , Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onuchic V et al. , Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science 361, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Handsaker RE et al. , Large multiallelic copy number variations in humans. Nat Genet, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tewhey R et al. , Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng B et al. , An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci 20, 1418–1426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fromer M et al. , Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19, 1442–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song M et al. , Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat Genet 51, 1252–1262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagih O, Merico D, Delong A, Frey BJ, Allele-specific transcription factor binding as a benchmark for assessing variant impact predictors. bioRxiv, 253427 (2018). [Google Scholar]
- 20.Datlinger P et al. , Pooled CRISPR screening with single-cell transcriptome readout. Nature methods 14, 297–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajarajan P et al. , Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Won H et al. , Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, Sarkar A, Carbonetto P, Stephens M, A simple new approach to variable selection in regression, with application to genetic fine-mapping. bioRxiv, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marballi KK, Gallitano AL, Immediate Early Genes Anchor a Biological Pathway of Proteins Required for Memory Formation, Long-Term Depression and Risk for Schizophrenia. Front Behav Neurosci 12, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dulubova I et al. , How Tlg2p/syntaxin 16 ‘snares’ Vps45. EMBO J 21, 3620–3631 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stahl EA et al. , Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 51, 793–803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levinson DF et al. , Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry 168, 302–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller FJ et al. , A bioinformatic assay for pluripotency in human cells. Nature methods 8, 315–317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weissbein U, Schachter M, Egli D, Benvenisty N, Analysis of chromosomal aberrations and recombination by allelic bias in RNA-Seq. Nat Commun 7, 12144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen Z et al. , Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y et al. , Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barretto N et al. , ASCL1- and DLX2-induced GABAergic neurons from hiPSC-derived NPCs. J Neurosci Methods 334, 108548 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez R et al. , Deriving dopaminergic neurons for clinical use. A practical approach. Sci Rep 3, 1463 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao Y, Smyth GK, Shi W, The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen KD, Irizarry RA, Wu Z, Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bray NL, Pimentel H, Melsted P, Pachter L, Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Hoffman GE et al. , Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat Commun 8, 2225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ring KL et al. , Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 11, 100–109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srikanth P et al. , Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep 12, 1414–1429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuhn RM, Haussler D, Kent WJ, The UCSC genome browser and associated tools. Brief Bioinform 14, 144–161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jun G et al. , Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet 91, 839–848 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li D et al. , Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell 21, 819–833 e816 (2017). [DOI] [PubMed] [Google Scholar]
- 43.McKenna A et al. , The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van de Geijn B, McVicker G, Gilad Y, Pritchard JK, WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nature methods 12, 1061–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storey JD, Tibshirani R, Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grundberg E et al. , Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 44, 1084–1089 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLean CY et al. , GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roadmap Epigenomics C et al. , Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heinz S et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visel A, Minovitsky S, Dubchak I, Pennacchio LA, VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res 35, D88–92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reilly SK et al. , Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ernst J, Kellis M, Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 33, 364–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yeo NC et al. , An enhanced CRISPR repressor for targeted mammalian gene regulation. Nature methods 15, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sung MH, Guertin MJ, Baek S, Hager GL, DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell 56, 275–285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gusmao EG, Allhoff M, Zenke M, Costa IG, Analysis of computational footprinting methods for DNase sequencing experiments. Nature methods 13, 303–309 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Davey Smith G, Hemani G, Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 23, R89–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McVicker G et al. , Identification of genetic variants that affect histone modifications in human cells. Science 342, 747–749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS, Quantifying similarity between motifs. Genome Biol 8, R24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen X, Molecular QTL discovery incorporating genomic annotations using Bayesian false discovery rate control. Ann Appl Stat 10, (2016). [Google Scholar]
- 60.Berisa T, Pickrell JK, Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie S, Duan J, Li B, Zhou P, Hon GC, Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol Cell 66, 285–299 e285 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Soneson C, Robinson MD, Bias, robustness and scalability in single-cell differential expression analysis. Nature methods 15, 255–261 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Ran FA et al. , Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wood AR et al. , Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46, 1173–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Locke AE et al. , Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu JZ et al. , Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morris AP et al. , Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44, 981–990 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demenais F et al. , Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet 50, 42–53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bentham J et al. , Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 47, 1457–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Willer CJ et al. , Discovery and refinement of loci associated with lipid levels. Nat Genet 45, 1274–1283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wray NR et al. , Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grove J et al. , Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Savage JE et al. , Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 50, 912–919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okbay A et al. , Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagel M et al. , Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet 50, 920–927 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Jansen IE et al. , Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iwaki H et al. , Genome-wide association study of Parkinson’s disease progression biomarkers in 12 longitudinal patients’ cohorts. bioRxiv, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taal HR et al. , Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat Genet 44, 532–538 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Obri A et al. , ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 505, 648–653 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Takata A, Matsumoto N, Kato T, Genome-wide identification of splicing QTLs in the human brain and their enrichment among schizophrenia-associated loci. Nat Commun 8, 14519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vidal C et al. , Amyloid beta perturbs elevated heme flux induced with neuronal development. Alzheimers Dement (N Y) 5, 27–37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ugur B et al. , The Krebs Cycle Enzyme Isocitrate Dehydrogenase 3A Couples Mitochondrial Metabolism to Synaptic Transmission. Cell Rep 21, 3794–3806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Le N et al. , Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci 8, 932–940 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Park SM, Littleton JT, Park HR, Lee JH, Drosophila Homolog of Human KIF22 at the Autism-Linked 16p11.2 Loci Influences Synaptic Connectivity at Larval Neuromuscular Junctions. Exp Neurobiol 25, 33–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haslinger D et al. , Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol Autism 9, 56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.