

Abstract
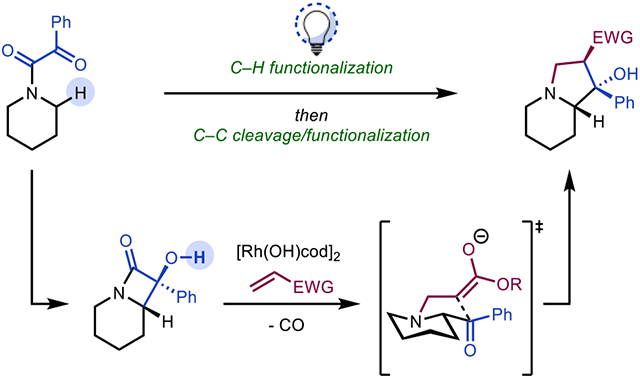
Herein we report the synthesis of substituted indolizidines and related N-fused bicycles from simple saturated cyclic amines through sequential C─H and C─C bond functionalizations. Inspired by the Norrish–Yang Type II reaction, C─H functionalization of azacycles is achieved by forming α-hydroxy-β-lactams from precursor α-ketoamide derivatives under mild, visible light conditions. Selective cleavage of the distal C(sp2)─C(sp3) bond in α-hydroxy-β-lactams using a Rh-complex leads to α-acyl intermediates which undergo sequential Rh-catalyzed decarbonylation, 1,4-addition to an electrophile, and aldol cyclization, to afford N-fused bicycles including indolizidines. Computational studies provide mechanistic insight into the observed positional selectivity of C─C cleavage, which depends strongly on the groups bound to Rh trans to the phosphine ligand.
Graphical Abstract
INTRODUCTION
Indolizidine-type alkaloids, three examples of which are shown in Figure 1 (castanospermine, gephyrotoxin, and rhazinilam), display a wide array of biological activity1 including, but not limited to, antiviral,2 CNS modulation,3 and anticancer4 properties. In addition, other N-fused saturated bicycles, featuring for example an azepane ring, occur in other bioactive natural products such as the stemona alkaloid family (see stemoamide) which display antitussive properties.5
Figure 1.

Indolizidine and N-fused bicycle-containing natural products.
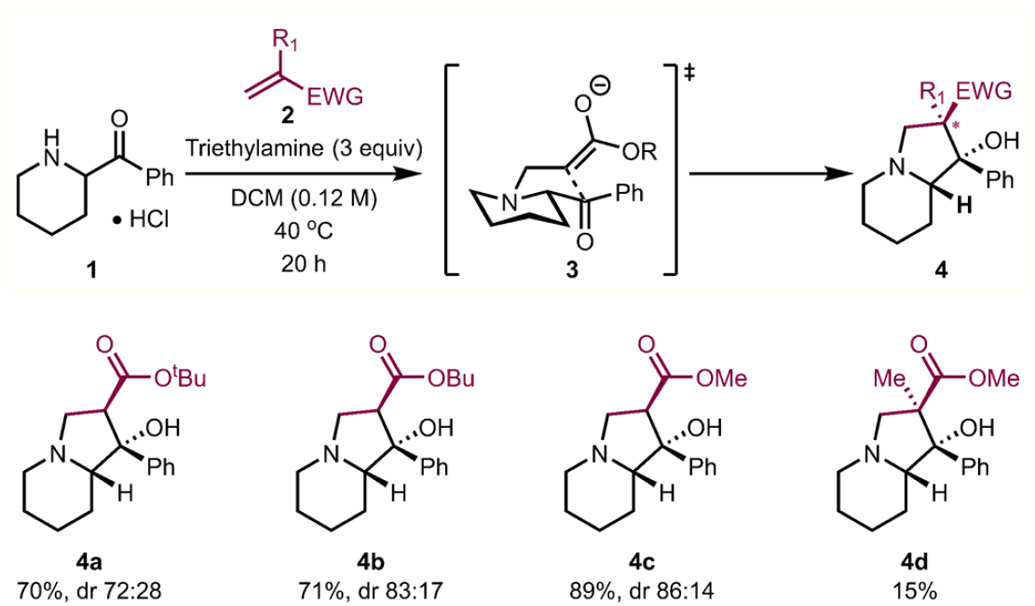
As a part of a broad program aimed at the general preparation of N-fused bicycles,6 we questioned whether the seemingly straightforward addition of α-acyl saturated azacycles (e.g., 1, Table 1) to activated, electron-deficient alkenes (i.e., 2) could be employed for this purpose. We anticipated that under an optimal set of conditions, amphoteric molecules7 such as 1 could be preferentially engaged in a conjugate addition (aza-Michael addition) with 2 followed by an aldol reaction of the resulting enolate with the ketone moiety (see proposed intermediate 3). Indeed, subjecting a mixture of the HCl salt of 1 (1·HCl) and tert-butyl acrylate (3 equiv) in methylene chloride to triethylamine (2 equiv) over 20 h smoothly led to the formation of 4a (70% yield, 72:28 dr). As shown in Table 1, this transformation works well for the small subset of acrylates (see 4b, 4c) and only moderately for methacrylate derivatives (4d) that were investigated in a preliminary study. However, despite the seemingly straightforward nature of this transformation, we quickly came to recognize several challenges that needed to be overcome to make this process more practical and widely useful.
Table 1.
Preliminary Study of Indolizidine Bicycle Formation from α-Acyl Piperidine Salt 1a
 |
All yields reported are isolated yields. Reaction was performed with 1·HCl (0.092 mmol) and acrylate 2 (3 equiv). dr was determined by comparison of isolated yields.
Notably, as we sought to expand the scope of the annulation reaction to include other α-acylated saturated heterocycles, we found that their reported preparations involved multiple steps as outlined in Scheme 1 that would not be compatible with their generation from a saturated azacycle precursor, especially at a late stage. For example, as shown in Scheme 1a, the use of a strong base for the α-lithiation in the Beak preparation of α-acylated azacycles8 severely limits its generality due to the functional group intolerance of these conditions.9 This limitation also holds for other elegant emerging methods for α-functionalization of saturated azacycles that also rely on the use of strong bases.10 Another reported approach to α-acylated saturated azacycles involves hydrogenation of pyridine rings bearing an α-acyl group, followed by oxidation (Scheme 1b)11. This approach is limited to the preparation of six-membered saturated azacycles from a pyridine precursor. As a result, the prior art for the preparation of α-acylated saturated azacycles is especially limiting if one seeks to prepare them at a late stage from an already resident saturated azacycle such as a piperidine. Piperidines are especially interesting as starting materials for derivatization given that they are the most commonly encountered saturated heterocycle in pharmaceuticals and also feature heavily in numerous agrochemicals.12 A vast array of natural products also possess a piperidine nucleus, which would serve as an excellent starting point for diversification.13
Scheme 1.

Previously Reported Preparations of α-Acylated Piperidines
On the basis of these considerations, we envisioned an approach to N-fused azacycles that would begin with the in situ generation of α-acyl saturated azacycles under mild conditions from saturated azacycles. Additionally, we sought to couple the formation of these desired α-acyl saturated azacyclic addends with the generation of a N-based nucleophile that would obviate the need for isolating the free base of amphoteric molecules such as 1. We hypothesized that these requirements could be met using sequential C─H and C─C bond functionalization reactions. These transformations could be conducted at a late stage in a synthesis campaign under mildly basic conditions, broadening the functional group tolerance of methods to N-fused bicycles.
The direct site-selective functionalization of C─H bonds to form C─C or C─X bonds represents a powerful approach for diversification in organic synthesis.14 In addition, C─C functionalization reactions, which continue to emerge, provide direct access to synthetically complex products from readily available precursors.15 We theorized that by combining C─H and C─C bond functionalization reactions under mild conditions, α-acylations of saturated azacycles could be achieved, concomitant with the generation of a nitrogen nucleophile, setting the stage for annulations of saturated azacycles (Figure 2). Specifically, we envisioned that derivatization of saturated azacycles to α-ketoamides such as 5 would set the stage for a Norrish–Yang Type II reaction16 to form α-hydroxy-β-lactam 6, which we have shown occurs at room temperature using blue LEDs (400–450 nm) following the precedent of Aoyama (Figure 2; see Supporting Information for details on the preparation of 6 and related α-hydroxy-β-lactams).17 Here, to access α-acyl derivatives, we anticipated that a regioselective C─C bond cleavage of the distal bond of the α-hydroxy-β-lactam (see 7) could be achieved through Rh-catalyzed β-carbon elimination, in line with the precedent of Murakami.18 Decarbonylation19 of acyl rhodium intermediate 8 would then lead to rhodated intermediate 9, which would serve as a functional equivalent to amphoteric molecules such as 1. In this case, we expected that some level of stabilization may be achieved through a five-membered chelate (see 9), which would avoid the deleterious decomposition pathways that 1 undergoes upon forming its free base. From rhodacycle 9, binding of an alkene (such as acrylate) followed by conjugate addition would lead to an enolate that would undergo a subsequent intramolecular aldol reaction to furnish functionalized indolizidine 10.
Figure 2.

A C─H/C─C bond functionalization approach to synthesis of N-fused heterocycles from saturated azacycles.
RESULTS AND DISCUSSION
Reaction Discovery and Optimization.
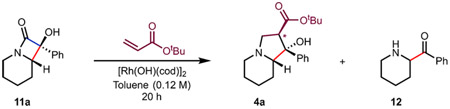
Our initial studies to convert α-hydroxy-β-lactams related to 6 to substituted indolizidine products such as 10 centered on the hypothesis that by tuning the electronic and steric properties of the ligand on the Rh metal center, positional selectivity of the β-carbon elimination could be achieved by cleaving either the proximal C(sp3)─C(sp3) bond (highlighted in blue, Figure 2) or the distal C(sp2)─C(sp3) bond (highlighted in red) to the azacyclic ring. Because our previous studies have shown that Pd complexes effect proximal cleavage of α-hydroxy-β-lactams,17 we focused on Rh complexes, which are also known to effect cyclobutanol openings but could offer opportunities for different binding modes and selectivities.20 The ring opening of 11a was examined under various reaction conditions as outlined in Table 2. In the presence of a rhodium complex such as hydroxy(cyclooctadiene) rhodium-(I) dimer (i.e., [Rh(OH)(cod)]2) with potassium carbonate as an exogenous base at 100 °C, the desired distal bond cleaved product (12) was formed, albeit in low yield (Table 2, entry 1). The addition of phosphine ligands facilitated the ring-opening process and led to higher conversions to 12 (Table 2, entry 2). In the presence of tert-butyl acrylate, the desired indolizidine product (4a) was formed in 73% yield as determined by NMR (Table 2, entry 3). Triphenylphosphine proved to be a ligand that led to higher conversions to 4a (Table 2, entry 4). Of note, the addition of water (5 equiv) to the reaction mixture either prevented cyclization and/or led to a retroaldol reaction of the product (4a) and β-elimination, effectively reversing the 1,4-addition reaction and giving a low yield of 4a (Table 2, entry 5). When the reaction was carried out at 70 °C, starting material was recovered (Table 2, entry 6), and on the basis of control experiments, potassium carbonate alone did not lead to C─C bond cleavage of 11a (Table 2, entry 7).
Table 2.
Initial Screening for Ring Opening Reactiona
 |
|||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst (mol %) | PPh3 (mol %) | K2CO3 (equiv) | temp (°C) | tert-butyl acrylate (equiv) | conversionb (%) | yieldb |
| 1 | 15 | – | 1.5 | 100 | – | 29 | 12, 25% |
| 2 | 5 | 15 | 1.1 | 100 | – | 69 | 12, 64% |
| 3 | 5 | – | 1.1 | 100 | 5 | 53 | 4a, 40%, dr 43:57 |
| 4 | 5 | 15 | 1.1 | 100 | 5 | >95 | 4a, 71%, dr 61:39 |
| 5c | 5 | 15 | 1.1 | 100 | 5 | >95 | 4a, 23%, dr 17:83 |
| 6 | 6 | – | – | 70 | – | <5 | |
| 7 | – | – | 1.1 | 100 | – | <5 | |
Reaction was performed with lactam 11a (0.092 mmol).
1H NMR conversion of 11a using trimethoxybenzene as internal standard. dr was determined by 1H NMR integration of resonances corresponding to diastereomers in the crude mixture for NMR.
5 equiv of H2O was added.
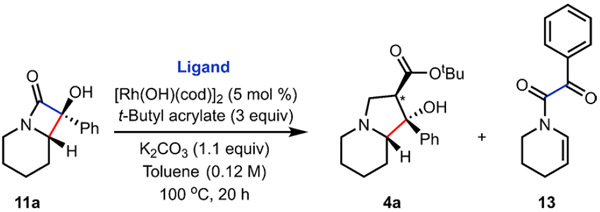
With the crucial components for an effective reaction using Rh catalysis on α-hydroxy-β-lactam 11a established, we focused on ligands that would facilitate better conversion and lead to a more efficient overall process (Table 3). XPhos, RuPhos, and t-BuDavePhos in combination with [Rh(OH)(cod)]2 led to the desired distal bond-selective cleavage. However, the subsequent Rh-catalyzed 1,4-addition and aldol cyclization reactions were inefficient under these conditions, resulting in lower overall yields of 4a (Table 3, entries 1–3). From our studies, we observed that position-selective distal C─C cleavage and the subsequent Rh-catalyzed cascade 1,4-addition/aldol cyclization reaction was most efficiently achieved using Xantphos as ligand (Table 3, entry 7). The yields did not improve when the reaction solvent was changed from toluene to dioxane (Table 3, entries 5, 6). Notably, the ligands DPPE (Table 3, entry 9) and (R)-DTBM-SEGPHOS (Table 3, entry 10) gave mixtures of proximal and distal cleavage products (i.e., indolizidine 4a and enamide 13, the latter resulting from β-hydride elimination of an alkyl Rh intermediate; Table 3, entry 10). The formation of 4a and 13 using (R)-DTBM-SEGPHOS as a ligand highlights the importance of not only the nature of the metal, but also the ligand of the complex in driving the regioselectivity of the C─C bond cleavage (i.e., proximal versus distal). A comprehensive ligand screen carried out using HTE screening in a collaboration with Merck and Co., Inc. confirmed the identification of Xantphos as the optimal ligand for this process (see Supporting Information for more details on the HTE screen). To gain insight into the Rh-catalyzed regioselective opening of the α-hydroxy-β-lactam, as well as the overall proposed mechanism, a computational study was undertaken as described below.
Table 3.
Ligand Effects on the Ring Opening Reactiona
 | |||
|---|---|---|---|
| entry | ligand | 4a yieldb | 13 yield (%)b |
| 1 | XPhos | 59%, dr 67:33 | – |
| 2 | RuPhos | 64%, dr 67:33 | – |
| 3 | tBuDavePhos | 32%, dr 52:48 | – |
| 4 | (rac)-BINAP | 85%, dr 60:40 | – |
| 5 | DavePhos | 86%, dr 60:40 | – |
| 6c | DavePhos | 63%, dr 63:37 | – |
| 7 | Xantphos | 95%, dr 70:30 | – |
| 8d | Xantphos | 78%, dr 73:27 | – |
| 9 | DPPE | 50%, dr 60:40 | 7 |
| 10 | (R)-DTBM-SEGPHOS | 64%, dr 64:36 | 25 |
Reaction was performed with lactam 11a (0.092 mmol), ligand (15 mol %).
1H NMR yield using trimethoxybenzene as internal standard. dr was determined by 1H NMR integration of resonances corresponding to diastereomers in the crude NMR mixture.
Dioxane (0.12 M) solvent.
K2CO3 was not added.
COMPUTATIONAL STUDIES
Regioselectivity of C─C Bond Cleavage.
Density functional theory (DFT) calculations were performed with Xantphos as the prototypical ligand using azacycle 11a and methyl acrylate as the substrate and electrophile, respectively. Figure 3 shows the reaction energy profile starting from the Rh─OH complex (A1) through to the formation of the indolizidine complex A8, which we propose to be the final intermediate prior to product release. In our proposed mechanism, α-hydroxy-β-lactam 11a first binds to the three-coordinate (Xantphos)Rh(I)─OH complex A1 to form the four-coordinate adduct A2, which undergoes dehydration to yield alkoxide intermediate A3dis. The Rh(I) center in A3dis can ring-open the β-lactam through electrophilic attack at the carbonyl-carbon (C2) to cleave the distal C─C bond or at C4, leading to ring opening at the proximal C─C bond.
Figure 3.

Free energy reaction profile of the proposed mechanism.
Our calculations indicate that cleavage of the distal C─C bond is associated with a barrier of 12.8 kcal/mol, traversing transition state A3dis-TS. The alternative transition state (A3prox-TS) at a relative energy of 9.3 kcal/mol above the starting point, leads to a barrier of 18.6 kcal/mol. Thus, our calculations suggest a kinetic preference of 5.8 kcal/mol for the ring opening of the distal C─C bond, which is in good agreement with the experimental observation that the distal C─C bond is preferentially cleaved using Xantphos as a ligand (Table 3, entry 9).
To account for the selectivity during the C─C bond cleavage, a preliminary computational study was carried out to analyze the characteristics of the substrate itself. The anionic form of the α-hydroxy-β-lactam (A9−, Figure 4a) was reoptimized and analyzed using a natural bond orbital (NBO) calculation. Calculations show that the distal (C1─Cx) bond is shorter at 1.61 Å and has a higher bond order of 0.89, compared to the proximal C1─C8a bond, computed to be 1.71 Å long and with a 0.77 bond order. The proximal C─C bond was therefore expected to be easier to break. Consistent with this expectation, our previous work showed that Pd complexes selectively cleave the proximal C─C bond.17,20 On the basis of this precedent, the Rh-mediated distal C─C bond cleavage observed here is not consistent with the inherent substrate selectivity. The proximal C─C bond breaking was modeled, and the related reactant and product, A3prox and A4prox (see Figure 3), are both computed to be higher in energy than the respective distal analogues, A3dis and A4dis. The energy difference between A3dis and A3prox arises from the difference in coordination of the Rh complex and the resulting electronic structure (see Figure 4b). Rh complex A3dis has a stable 16 e− four-coordinate square planar structure. To engage the proximal C─C bond, however, the rhodium complex has to rearrange via dissociation of the coordinated lactam-carbonyl to give the three-coordinate 14 e− complex A3prox, which we found to be 2.1 kcal/mol higher in energy than A3dis as shown in red in Figure 3.
Figure 4.

(a) Optimized structure of free anionic azacycle, A9−. Bond orders are obtained from the Mayer–Mulliken bond order calculation implemented in the NBO calculation. (b) Atomic charge and P─Rh bond length in indicated structures. Atomic charges are obtained from the electrostatic potential. (c) Optimized structures of A3dis, A3prox, A3dis-TS, A4dis, A3prox-TS, and A4prox. White, red, blue, orange, and purple balls represent carbon, oxygen, nitrogen, phosphorus, and rhodium, respectively. Hydrogen atoms are omitted for clarity.
On the other hand, the energy difference between the related products, A4dis and A4prox, results from the different trans influence of the ligands. Along the reaction coordinate, the transition state A3dis-TS ultimately leads to a Rh─amide acyl bond, which imposes a stronger trans influence than the Rh─alkyl bond that results from traversing A3prox-TS. The Rh─amide acyl bond in A4dis likely tolerates the strong trans influence of the phosphine ligand, thus stabilizing the complex without significant structural change. Analysis of the electronic structures (Figure 4c) clearly supports this assertion, as atomic charges on the phosphine centers in A3dis were computed to be −0.91 and −0.72, whereas those on the phosphine centers in A4dis were computed to be reduced to −0.49 and −0.52, respectively. On the contrary, the alkyl ligand in A4prox has a weaker trans influence and is, therefore, less capable of supporting the strongly trans-directing phosphine ligand, leading to greater distortion of A4prox from A3prox. As a result, A4prox adopts a trigonal pyramidal structure with the alkyl ligand occupying the axial position. In this conformation, the substrate cannot effectively distribute the electron density imparted by the phosphines, resulting in atomic charges of −0.59 and −0.80 on each phosphine. This also leads to average P─Rh bond lengths that are 0.06 Å longer in A4dis. In summary, the ligand strength difference between the amide acyl and alkyl groups attached to the Rh center engenders structural differences between A4dis and A4prox, making A4prox 6.4 kcal/mol higher in energy as compared to A4dis. On the basis of the Hammond postulate,21 A3prox-TS should have a higher associated barrier compared to A3dis-TS. Because both C─C bond cleaving steps were computed to be essentially irreversible, the distal C─C bond breaking pathway should be favored as experimentally observed.
Unsurprisingly, the products A4dis and A4prox containing strong Rh─carbon bonds are much lower in energy (at −33.9 and −27.5 kcal/mol, respectively) than the lactam adducts. Interestingly, our calculations show that the amide carbonyl binds in an η2-fashion to the rhodium center, instead of forming a six-membered metallacycle using the carbonyl oxygen functionality. In the productive direction, intermediate A4dis undergoes decarbonylation19b to form intermediate A5 (Figure 3), a piperidine-bound Rh(I)─carbonyl complex. This step is associated with a barrier of 25.3 kcal/mol, which should be accessible under the experimental conditions employed. Our calculations estimate the barrier for the liberation of carbon monoxide to be relatively high at 34.6 kcal/mol. Given the low solubility of CO in the solvents employed,22 the CO release to give three-coordinate Rh(I)─piperidine complex A6 is likely irreversible, as indicated in Figure 3.
Diastereoselectivity in the Aldol-like Cyclization.
The final sequence envisioned in the overall annulation to afford the N-fused bicyclic products consists of a conjugate addition of the Rh-bound acylpiperidine (A7 or A7′, Figure 5) and methyl acrylate followed by an aldol cyclization to produce the indolidizine. The phenyl group and the hydrogen at the ring-fusion carbon are syn-disposed in the experimentally observed major diastereomer product (A8). Our calculations show that the conjugate addition and aldol cyclizations are concerted and not stepwise processes, passing through a single transition state (A7-TS at 3.1 kcal/mol) resulting in a barrier of 17.5 kcal/mol from A6. To rationalize the stereochemical outcome of this transformation, we considered the energetics of the two diastereomeric possibilities, as illustrated in Figure 5. The energetically most favorable binding of the acylpiperidine is to engage the axial N-lone pair, leading to intermediate A7.
Figure 5.

(a) Free energy profile for determining C1 position. (b) Distortion–interaction analysis of 7 and 7′. (c) Overlapped structures of A7-[Rh+] and A7′-[Rh+]. Structures of (d) A7-[N−] and (e) A7′-[N−]. Unimportant hydrogen atoms are omitted for clarity.
Alternatively, in A7′ (6.8 kcal/mol higher in energy) the equatorial N-lone pair binds the metal. This energy difference is propagated and enhanced in the cyclization transition states, leading to a kinetic preference of 11.7 kcal/mol for the experimentally observed diastereomer that arises from A7. A distortion–interaction analysis was carried out by dividing the intermediates into [Rh+] and [N−] fragments, as shown in Figure 5b-e. The structures of metal-containing A7-[Rh+] and A7′-[Rh+] fragments are almost superimposable (Figure 5c) and unlikely to contribute to the energy difference. The main factor for the energy difference appears to be the orientation of the benzoyl moiety in [N−], increasing the distortion in A7′ to ~5.7 kcal/mol higher than in A7. The stronger interaction in A7′ of about 2.0 kcal/mol cannot compensate for the larger distortion energy and gives a final electronic energy difference of 3.8 kcal/mol between A7′ and A7. Entropy and solvation energy differences contribute 3 kcal/mol. Thus, the diastereoselectivity is mainly determined by the orientation of the benzoyl group as the metal binds the piperidine. Despite the low solubility of the potassium salts employed in this study in the optimal solvents, a formal cycloaddition reaction involving a potassium amido generated from A5 and the electron-deficient alkene coupling partner cannot be ruled out. Further mechanistic studies are ongoing (see Supporting Information for details).
Substrate Scope for the N-Fused Heterocycle Formation.
Following our computational mechanistic interrogation, the substrate scope for various electrophiles was investigated (Table 4). A wide range of α,β-unsaturated carbonyl compounds underwent the Rh-catalyzed cascade reaction to yield the corresponding indolizidine products. Specifically, tert-butyl-, butyl-, and methyl acrylate, as well as methyl methacrylate, underwent the cascade sequence with similar efficiencies (4a–d). Fluorine-substituted indolizidines (4e) could be accessed using an α-fluoro-substituted acrylate. When β-substituted electrophiles (e.g., methyl crotonate, methyl cinnamate, and dimethyl fumarate) were used, only one diastereomer was observed (4f–h). Likely, developing 1,3-diaxial interactions in the transition state dictate the diastereoselectivity and lead to the high observed dr for alkenes bearing substituents at the β position (see Supporting Information for more details on a computational model). Interestingly, indolizidine 4f was obtained by using methyl 3-butenoate as an electrophile. Presumably, the β,γ-unsaturated ester underwent base-promoted isomerization to methyl crotonate prior to reaction with Rh-amido intermediate 9.23 In addition, a diverse array of electrophiles were viable substrates for this process. A variety of electron-withdrawing groups including cyanide, amide, ketone, sulfone, and phosphonate on the electrophile were successfully engaged to give the corresponding indolizidines (4i–n). Due to the instability and separation challenges associated with the amide products under the purification conditions (SiO2 or neutral Al2O3), 4j and 4k gave relatively low isolation yields of the corresponding indolizidine products. Fused pyrrole 4o and fused tricycle (epi-4p) were obtained by using an electrophilic alkyne (phenyl-substituted methyl propiolate) or cyclopentenone as electrophiles, respectively. However, when cyclopentenone was used, epi-4p was formed as the major product presumably due to developing 1,3 diaxial interactions between the cyclopentanone ring and the ring fusion hydrogen and phenyl groups.
Table 4.
Scope of Indolizidine Formation with Respect to Electrophilesa
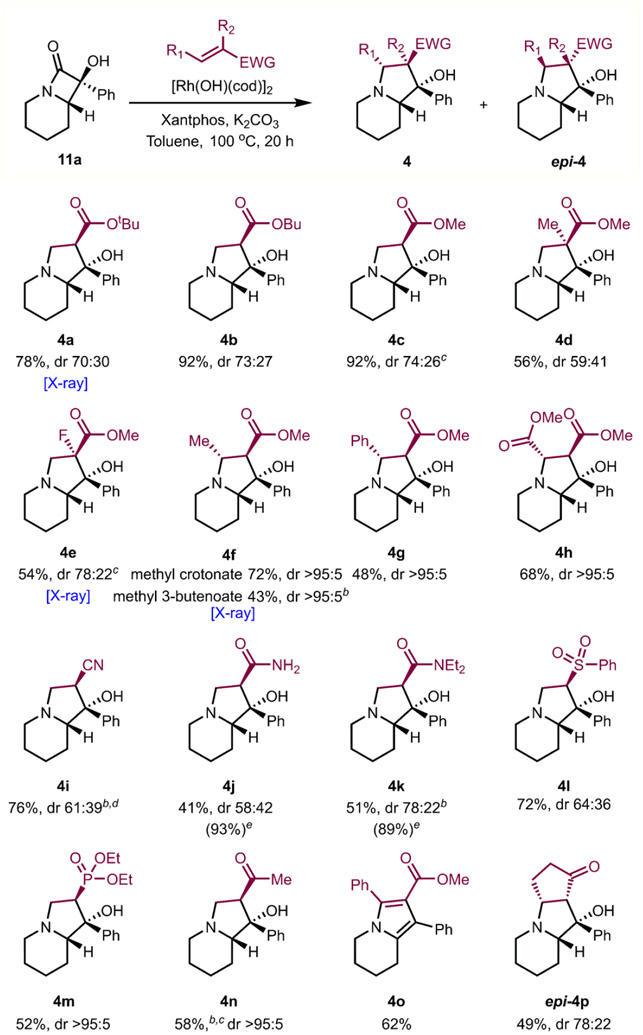 |
Reaction was performed with lactam 11a (0.092 mmol), [Rh(OH)(cod)]2 (5 mol %), Xantphos (15 mol %), K2CO3 (1.1 equiv), acrylates (3 equiv), and toluene (0.11 M) at 100 °C for 20 h. dr was determined by 1H NMR integration of resonances corresponding to diastereomers in the crude mixture for NMR.
DavePhos as ligand.
5 equiv of electrophiles used.
10 equiv of acrylonitrile used.
NMR yield using trimethoxybenzene as internal standard.
We have also investigated the substrate scope with respect to the azacyclic ring of the β-lactams, which were readily accessed through Norrish–Yang II type reactions analogous to those employed for the formation of 11a (see Supporting Information for details). Morpholine led to the corresponding fused bicycle 14a, and saturated heterocycles of increasing ring size (azepane and azocane) also proved competent as substrates to form heterocycles 14b and 14c. Functionalized piperidines bearing gem-dimethyl (see 14d), cyclohexyl (14e), 1,3-dioxolane (14f), phenyl (14g), and nitrile groups (14h) at C4 also led to the desired products in moderate to good yields (Table 5).
Table 5.
Scope of Indolizidine Formation with Respect to the Azacycle Ring Sizea
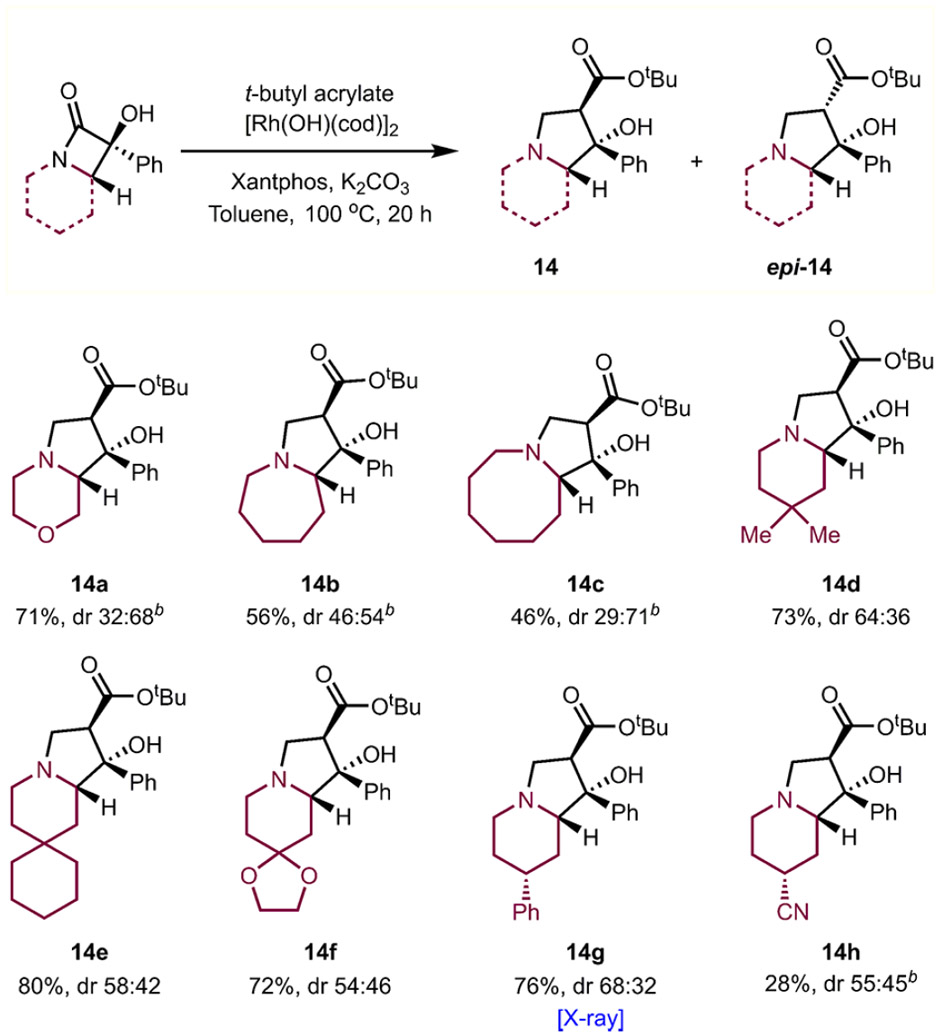 |
Reaction was performed with β-lactam (0.092 mmol), [Rh(OH)(cod)]2 (5 mol %), Xantphos (15 mol %), K2CO3 (1.1 equiv), tert-butyl acrylate (3 equiv), and toluene (0.11 M) at 100 °C for 20 h. dr was determined by 1H NMR integration of resonances corresponding to diastereomers in the crude NMR mixture.
DavePhos as ligand.
Monocyclic β-lactam 15, prepared from linear N,N-dibenzyl-2-oxo-2-phenylacetamide,24 also underwent C─C bond cleavage followed by Rh-mediated annulation to afford 16 (with methyl vinyl ketone), albeit in lower yield compared to that obtained using the bicyclic α-hydroxy-β-lactams (Scheme 2).
Scheme 2.

Pyrrolidine 16 from a Monocyclic β-Lactam
We have also explored the effect of varying the position of substitution on the aromatic group or varying the electronic properties of the arene group by installing electron-donating or electron-withdrawing groups.25 In most cases, electron-rich (17d, 17e) or electron-poor (17f, 17g) aryl groups on the α-hydroxy-β-lactam substrates gave the desired indolizidine products in similar yields and diastereoselectivities. α-Hydroxy-β-lactams bearing halide, methoxy, cyanide, methyl, and trifluoromethyl substituents (Table 6) were all competent substrates, providing the corresponding indolizidine products in moderate yields (17a–k). Notably, the halide-containing indolizidine products (i.e., 17b, 17c, 17i, 17j) may be further diversified through cross-coupling.
Table 6.
Scope Studies: Functionalization of Aromatic Ringa
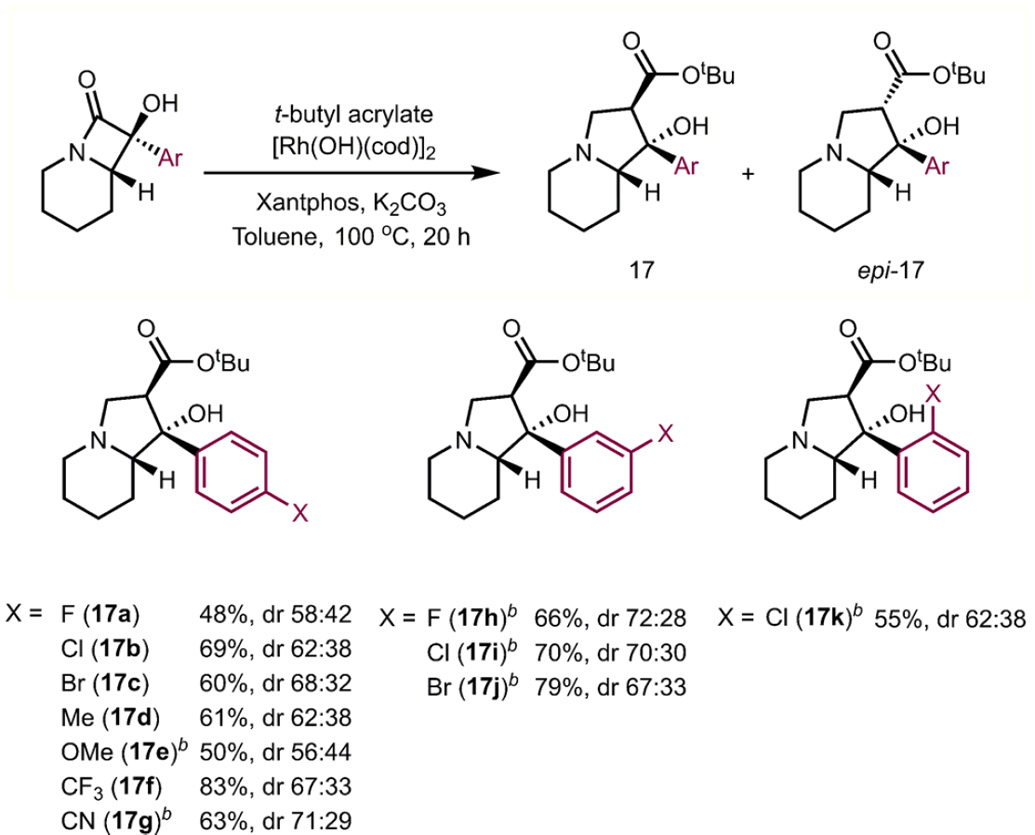 |
Reaction was performed with β-lactams (0.092 mmol), [Rh(OH)(cod)]2 (5 mol %), Xantphos (15 mol %), K2CO3 (1.1 equiv), tert-butyl acrylate (3 equiv), and toluene (0.11 M) at 100 °C for 20 h. dr was determined by 1H NMR integration of resonances corresponding to diastereomers in the crude mixture for NMR.
[Rh(OH)(cod)]2 (10 mol %), Xantphos (30 mol %) used.
Some additional studies that we have conducted provide even more insight into the proposed mechanism for the Rh-mediated bicyclizations described here. For example, heating diastereomeric indolizidines 4a/epi-4a at 100 °C under the reaction conditions (in the presence of rhodium complex [Rh(OH)(cod)]2, base, and tert-butyl acrylate), did not lead to epimerization at C2, supporting a kinetically driven diastereomeric outcome, likely arising from the aldol cyclization step (Figure 6a).
Figure 6.
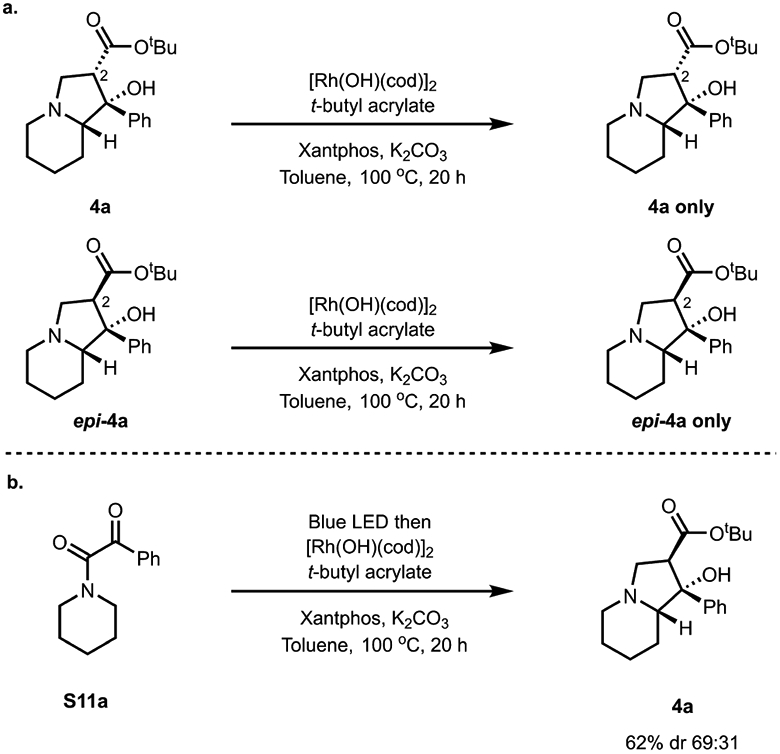
Additional insight into the bicyclization reaction.26(a) Lack of reversibility of observed diastereoselectivity. (b) One-pot indolizidine synthesis.
Finally (Figure 6b), the desired N-fused bicyclic products can also be obtained through a one-pot process from ketoamide S11a without isolation of the intermediate β-lactams. This latter observation enhances the synthetic practicality of this method to prepare N-fused bicycles from saturated azacycles.
CONCLUSION
In summary, we report a new method for the synthesis of indolizidines and related N-fused heterocycles from piperidines and other saturated cyclic amines. This process is initiated by a Rh-catalyzed C─C functionalization of α-hydroxy-β-lactams prepared through a Norrish–Yang Type II C─H functionalization of ketoamides derived from the saturated cyclic amine starting materials. This overall three-step sequence transforms readily available saturated cyclic amines into value-added N-fused heterocycles. A relatively wide range of N-fused bicyclic β-lactam precursors were accessed through the Norrish–Yang reaction. In the subsequent Rh-catalyzed C─C cleavage process, optimization of the ligand allowed for the selective cleavage of the distal C─C bond, followed by the formation of N-fused bicyclic products through a formal cycloaddition process. Computational studies were carried out to provide more insight into the factors that influence selectivity for distal C─C bond cleavage, unveiling the importance of trans influences in the rhodacycle intermediate.
Supplementary Material
ACKNOWLEDGMENTS
R.S. is grateful to the NSF under the CCI Center for Selective C─H functionalization (CHE-1700982) for partial support of the work reported herein (Berkeley). J.-S.H. is grateful to SK Innovation Co., Ltd. for support of his graduate studies, and J.B.R. is grateful for a BMS graduate fellowship. M.-H.B. acknowledges support from the Institute for Basic Science (IBS-R10-A1) in Korea. C.S.Y. is a Merck Disruptive Chemistry Fellow and is grateful to Shane W. Krska, Daniel DiRocco, Thomas W. Lyons, Dan Lehnherr, Louis-Charles Campeau, Alan Northrup, Nicholas K. Terrett, Emma R. Parmee, and Michael H. Kress (Merck) for support. We are grateful to Dr. Michaelyn Lux (Merck) for important insightful discussions. We are grateful to Merck and Co., Inc. for providing advanced building blocks to support the synthetic studies reported herein. We thank A.H. Christian and E. Miller (Berkeley) and Laura Wilson and Christina Kraml (Lotus Separations, LLC) for chiral resolution of 11a. We thank Lisa M. Nogle and Spencer McMinn (Merck) for large-scale chiral resolution of 11a and chiral analysis. We are grateful to Lisa M. Nogle, David A. Smith, Mark Pietrafitta, and Miroslawa Darlak (Merck) for help with reversed-phase purifications. We thank Josep Saurí (Merck) for help with 2D NMR analysis. We thank Dr. Hasan Celik and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in the CoC-NMR are supported in part by NIH S10OD024998.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04278.
Experimental procedures, computational details, and compound characterization (PDF)
X-ray data for epi-4a (CIF)
X-ray data for 4a (CIF)
X-ray data for epi-4e (CIF)
X-ray data for 4e (CIF)
X-ray data for 4f (CIF)
X-ray data for 14g (CIF)
X-ray data for 16 (CIF)
The authors declare no competing financial interest.
Contributor Information
Jin Su Ham, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Bohyun Park, Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea; Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea.
Mina Son, Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea; Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea.
Jose B. Roque, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Justin Jurczyk, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Charles S. Yeung, Disruptive Chemistry Fellow, Department of Discovery Chemistry, Merck & Co., Inc., Boston, Massachusetts 02115, United States.
Mu-Hyun Baik, Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea; Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea.
Richmond Sarpong, Department of Chemistry, University of California, Berkeley, California 94720, United States.
REFERENCES
- (1).Michael JP Simple Indolizidine and Quinolizidine Alkaloids In The Alkaloids: Chemistry and Biology; Knölker H-J, Ed.; Academic Press, 2016; Vol. 75, pp 1–498. [DOI] [PubMed] [Google Scholar]
- (2).Whitby K; Pierson TC; Geiss B; Lane K; Engle M; Zhou Y; Doms RW; Diamond MS Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol 2005, 79, 8698–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Varkhedkar R; Dogra S; Tiwari D; Hussain Y; Yadav PN; Pandey G Discovery of Novel Muscarinic Receptor Modulators by Integrating a Natural Product Framework and a Bioactive Molecule. ChemMedChem 2018, 13, 384–395. [DOI] [PubMed] [Google Scholar]
- (4).(a) Baudoin O; Guénard D; Guéritte F The Chemistry and Biology of Rhazinilam and Analogues. Mini-Rev. Org. Chem 2004, 1, 333–341. [Google Scholar]; (b) Bowie AL; Hughes CC; Trauner D Concise Synthesis of (±)-Rhazinilam through Direct Coupling. Org. Lett 2005, 7, 5207–5209. [DOI] [PubMed] [Google Scholar]
- (5).Yoritate M; Takahashi Y; Tajima H; Ogihara C; Yokoyama T; Soda Y; Oishi T; Sato T; Chida N Unified Total Synthesis of Stemoamide-Type Alkaloids by Chemoselective Assembly of Five-Membered Building Blocks. J. Am. Chem. Soc 2017, 139, 18386–18391. [DOI] [PubMed] [Google Scholar]
- (6).(a) Chastanet J; Roussi G N-Methylpiperidine N-Oxide as a Source of Nonstabilized Ylide: A New and Efficient Route to Octahydroindolizine Derivatives. J. Org. Chem 1985, 50, 2910–2914. [Google Scholar]; (b) Pearson WH; Stoy P; Mi Y A Three-Component, One-Pot Synthesis of Indolizidines and Related Heterocycles Via the [3 + 2] Cycloaddition of Nonstabilized Azomethine Ylides. J. Org. Chem 2004, 69, 1919–1939. [DOI] [PubMed] [Google Scholar]; (c) Bhat C; Tilve SG Recent Advances in the Synthesis of Naturally Occurring Pyrrolidines, Pyrrolizidines and Indolizidine Alkaloids Using Proline as a Unique Chiral Synthon. RSC Adv. 2014, 4, 5405–5452. [Google Scholar]; (d) Quevedo-Acosta Y; Jurberg ID; Gamba-Sánchez D Activating Imides with Triflic Acid: A General Intramolecular Aldol Condensation Strategy Toward Indolizidine, Quinolizidine, and Valmerin Alkaloids. Org. Lett 2020, 22, 239–243. [DOI] [PubMed] [Google Scholar]
- (7).He Z; Zajdlik A; Yudin AK Air- and Moisture-Stable Amphoteric Molecules: Enabling Reagents in Synthesis. Acc. Chem. Res 2014, 47, 1029–1040. [DOI] [PubMed] [Google Scholar]
- (8).(a) Beak P; Zajdel WJ Dipole-Stabilized Carbanions: The α′ Lithiation of Piperidides. J. Am. Chem. Soc 1984, 106, 1010–1018. [Google Scholar]; (b) Beak P; Basu A; Gallagher DJ; Park YS; Thayumanavan S Regioselective, Diastereoselective, and Enantioselective Lithiation–Substitution Sequences: Reaction Pathways and Synthetic Applications. Acc. Chem. Res 1996, 29, 552–560. [Google Scholar]
- (9).(a) Treatment of heteroatom-containing azacycles (e.g., morpholine) with strong bases results in β-elimination to give ring-opened products, instead of α-fuctionalization.Babudri F; Florio S; Reho A; Trapani G Lithiation α to Heteroatoms: Eliminative Ring Fission of Heterocycles. J. Chem. Soc., Perkin Trans 1 1984, 1949–1955.
- (10).(a) Chen W; Ma L; Paul A; Seidel D Direct α-C─H Bond Functionalization of Unprotected Cyclic Amines. Nat. Chem 2018, 10, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paul A; Seidel D α-Functionalization of Cyclic Secondary Amines: Lewis Acid Promoted Addition of Organometallics to Transient Imines. J. Am. Chem. Soc 2019, 141, 8778–8782. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen W; Paul A; Abboud KA; Seidel D Rapid functionalization of multiple C─H bonds in unprotected alicyclic amines. Nat. Chem 2020, 12, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Li C; Ji Y; Cao Q; Li J; Li B Concise and Facile Synthesis of (R,R)-Dexmethylphenidate Hydrochloride and Its Three Stereoisomers. Synth. Commun 2017, 47, 1301–1306. [Google Scholar]; (b) Yadav-Samudrala BJ; Eltit JM; Glennon RA Synthetic Cathinone Analogues Structurally Related to the Central Stimulant Methylphenidate as Dopamine Reuptake Inhibitors. ACS Chem. Neurosci 2019, 10, 4043–4050. [DOI] [PubMed] [Google Scholar]
- (12).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles Among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (13).(a) Sun H; Tawa G; Wallqvist A Classification of Scaffold-Hopping Approaches. Drug Discovery Today 2012, 17, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu Y; Stumpfe D; Bajorath J Recent Advances in Scaffold Hopping. J. Med. Chem 2017, 60, 1238–1246. [DOI] [PubMed] [Google Scholar]; (c) Roque JB; Kuroda Y; Gottemann LT; Sarpong R Deconstructive diversification of cyclic amines. Nature 2018, 564, 244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Roque JB; Kuroda Y; Gottemann LT; Sarpong R Deconstructive fluorination of cyclic amines by carbon-carbon cleavage. Science 2018, 361, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For reviews, see: (a) Dyker G Transition Metal Catalyzed Coupling Reactions under C─H Activation. Angew. Chem., Int. Ed 1999, 38, 1698–1712. [DOI] [PubMed] [Google Scholar]; (b) Bellina F; Rossi R Transition Metal-Catalyzed Direct Arylation of Substrates with Activated sp3 Hybridized C─H Bonds and Some of Their Synthetic Equivalents with Aryl Halides and Pseudohalides. Chem. Rev 2010, 110, 1082–1146. [DOI] [PubMed] [Google Scholar]; (c) Jazzar R; Hitce J; Renaudat A; Sofack-Kreutzer J; Baudoin O Functionalization of Organic Molecules by Transition-Metal-Catalyzed C(sp3)─H Activation. Chem. - Eur. J 2010, 16, 2654–2672. [DOI] [PubMed] [Google Scholar]; (d) Baudoin O Transition Metal-Catalyzed Arylation of Unactivated C(sp3)─H Bonds. Chem. Soc. Rev 2011, 40, 4902–4911. [DOI] [PubMed] [Google Scholar]; (e) Girard SA; Knauber T; Li CJ The Cross-Dehydrogenative Coupling of Csp3─H Bonds: A Versatile Strategy for C─C Bond Formations. Angew. Chem. Int. Ed 2014, 53, 74–100. [DOI] [PubMed] [Google Scholar]
- (15).For reviews, see: (a) Murakami M; Ito Y Cleavage of Carbon─Carbon Single Bonds by Transition Metals. Top. Organomet. Chem 1999, 3, 97–129. [Google Scholar]; (b) Rybtchinski B; Milstein D Metal Insertion into C─C Bonds in Solution. Angew. Chem., Int. Ed 1999, 38, 870–883. [DOI] [PubMed] [Google Scholar]; (c) Jun C-H; Moon CW; Lee DY Chelation-Assisted Carbon─Hydrogen and Carbon─Carbon Bond Activation by Transition Metal Catalysts. Chem. - Eur. J 2002, 8, 2422–2428. [DOI] [PubMed] [Google Scholar]; (d) Jun C-H Transition Metal-Catalyzed Carbon─Carbon Bond Activation. Chem. Soc. Rev 2004, 33, 610–618. [DOI] [PubMed] [Google Scholar]; (e) Seiser T; Cramer N Enantioselective Metal-Catalyzed Activation of Strained Rings. Org. Biomol Chem 2009, 7, 2835–2840. [DOI] [PubMed] [Google Scholar]; (f) Colby DA; Bergman RG; Ellman JA Rhodium-Catalyzed C─C Bond Formation via Heteroatom-Directed C─H Bond Activation. Chem. Rev 2010, 110, 624–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Aoyama H; Hasegawa T; Omote Y Solid State Photochemistry of N,N-Dialkyl-α-oxoamides. Type II Reactions in the Crystalline State. J. Am. Chem. Soc 1979, 101, 5343–5347. [Google Scholar]
- (17).Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts CA; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R C─C Cleavage Approach to C─H Functionalization of Saturated Aza-Cycles. ACS Catal. 2020, 10, 2929–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For representative examples of Rh-catalyzed ring opening of cyclobutanol, see: (a) Seiser T; Cramer N Rhodium-Catalyzed C─C Bond Cleavage: Construction of Acyclic Methyl Substituted Quaternary Stereogenic Centers. J. Am. Chem. Soc 2010, 132, 5340–5341. [DOI] [PubMed] [Google Scholar]; (b) Ishida N; Sawano S; Masuda Y; Murakami M Rhodium-Catalyzed Ring Opening of Benzocyclobutenols with Site-Selectivity Complementary to Thermal Ring Opening. J. Am. Chem. Soc 2012, 134, 17502–17504. [DOI] [PubMed] [Google Scholar]; (c) Ishida N; Nakanishi Y; Murakami M Reactivity Change of Cyclobutanols towards Isocyanates: Rhodium Favors c-Carbamoylation over o-Carbamoylation. Angew. Chem., Int. Ed 2013, 52, 11875–11878. [DOI] [PubMed] [Google Scholar]
- (19).(a) Murphy SK; Park J-W; Cruz FA; Dong VM Rh-Catalyzed C─C Bond Cleavage by Transfer Hydroformylation. Science 2015, 347, 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dermenci A; Whittaker RE; Gao Y; Cruz FA; Yu Z-X; Dong G Rh-Catalyzed Decarbonylation of Conjugated Ynones via Carbon─Alkyne Bond Activation: Reaction Scope and Mechanistic Exploration via DFT Calculations. Chem. Sci 2015, 6, 3201–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).See Supporting Information for more details of a qualitative comparison of Pd- and Rh-catalyzed C─C bond cleavage selectivities.
- (21).Hammond GS A Correlation of Reaction Rates. J. Am. Chem. Soc 1955, 77, 334–338. [Google Scholar]
- (22).Jáuregui-Haza UJ; Pardillo-Fontdevila EJ; Wilheim AM; Delmas H Solubility of hydrogen and carbon monoxide in water and some organic solvent. Lat. Am. Appl. Res 2004, 34, 71–74. [Google Scholar]
- (23).Alcock SG; Baldwin JE; Bohlmann R; Harwood LM; Seeman JI On the Conjugative Isomerizations of β,γ-Unsaturated Esters. Stereochemical Generalizations and Predictions for 1,3-Prototropic Shifts under Basic Conditions. J. Org. Chem 1985, 50, 3526–3535. [Google Scholar]
- (24).Aoyama H; Sakamoto M; Kuwabara K; Yoshida K; Omote Y Photochemical Reactions of α-Oxo Amides. Norrish Type II Reactions via Zwitterionic Intermediates. J. Am. Chem. Soc 1983, 105, 1958–1964. [Google Scholar]
- (25).The α-ketoamide precursors were synthesized by NIS-catalyzed oxidative amidation of acetophenone derivatives using TBHP according to the precedent of Prabhu. A subsequent Norrish–Yang reaction gave the desired β-lactams bearing diverse aryl groups. See Supporting Information for details.; Lamani M; Prabhu KR NIS-Catalyzed Reactions: Amidation of Acetophenones and Oxidative Amination of Propiophenones. Chem. - Eur. J 2012, 18, 14638–14642. [DOI] [PubMed] [Google Scholar]
- (26).When enantiopure lactam 11a was used as the starting material, erosion in ee of the product was observed (4a 73% ee, epi-4a 66% ee), pointing to racemization of α-carbonyl piperidine intermediate 9 prior to cyclization. These observations set the stage for a dynamic kinetic asymmetric annulative transformation, which is currently under investigation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.