

Abstract
Over the past 3 decades, there has been significant progress in refining gene therapy technologies and procedures. Transduction of hematopoietic stem cells ex vivo using lentiviral vectors can now create a highly effective therapeutic product, capable of reconstituting many different immune system dysfunctions when reinfused into patients. Here, we review the key developments in the gene therapy landscape for primary immune deficiency, from an experimental therapy where clinical efficacy was marred by adverse events, to a commercialized product with enhanced safety and efficacy. We also discuss progress being made in preclinical studies for challenging disease targets and emerging gene editing technologies that are showing promising results, particularly for conditions where gene regulation is important for efficacy.
Introduction
Primary immune deficiencies (PIDs) are inherited life-threatening diseases, characterized by susceptibility to infection, increased risk of malignancy, autoimmunity, and inflammation. They arise due to abnormalities in over 300 genes governing the development or function of a range of immune subsets of both the innate and adaptive immune system.1 Globally they are rare diseases, occurring at a rate of 1:10 000 births,2 although this can be 20-fold greater in countries with a higher rate of consanguinity,3 or populations with founder mutations.4-6
Symptoms often arise in childhood and historically treatment options have been limited, focused on supportive care with hematopoietic stem cell transplant (HSCT), the only curative approach. This technique has evolved over time and the associated morbidity and mortality have dramatically reduced in some settings. However, success is still largely based on the availability of good human leukocyte antigen (HLA)-matched donor, with reduced survival in the mismatch setting arising from graft-versus-host disease (GvHD), infection, and graft rejection. Autologous gene-corrected stem cell therapy offers an attractive alternative where a suitable HLA-matched donor is unavailable, with the possibility of avoiding GvHD and often the ability to use less toxic and immunosuppressive conditioning regimens.
As the founders of the immune system, hematopoietic stem cells (HSCs) offer a relatively accessible therapeutic target through either direct bone marrow harvest or, more recently, the preferred option of leukapheresis. Following granulocyte-colony stimulating factor (G-CSF) and plerixafor-mediated mobilization from the bone marrow into the periphery, harvesting through apheresis and CD34+ cell selection, HSC can be manipulated with gene corrective tools ex vivo, before returning to the patient to engraft and restore a fully functioning system. Retroviruses have become the tool of choice, due to their ability to irreversibly recombine their genome into host cell DNA and packaging capacity large enough to carry the complementary DNA (cDNA) of most genes. Primary immune deficiencies have been at the forefront of gene therapy research using viral vectors, and the success that is now being seen in the clinic for many diseases represents the culmination of decades of symbiotic research between clinicians, research scientists, and industrial partners, which has advanced the understanding of disease pathology, stem cell biology, virology, and molecular genetics.
Originally pathogenic viruses of animal and man (eg, Moloney murine leukemia virus [MoLV], human immunodeficiency virus [HIV]), these viruses have undergone a vectorization process to remove the ability to self-replicate, leaving only a single-stranded RNA genome and the proteins required for genome integration, packaged within a capsid, matrix proteins, and a lipid membrane coat (Figure 1A).
Figure 1.
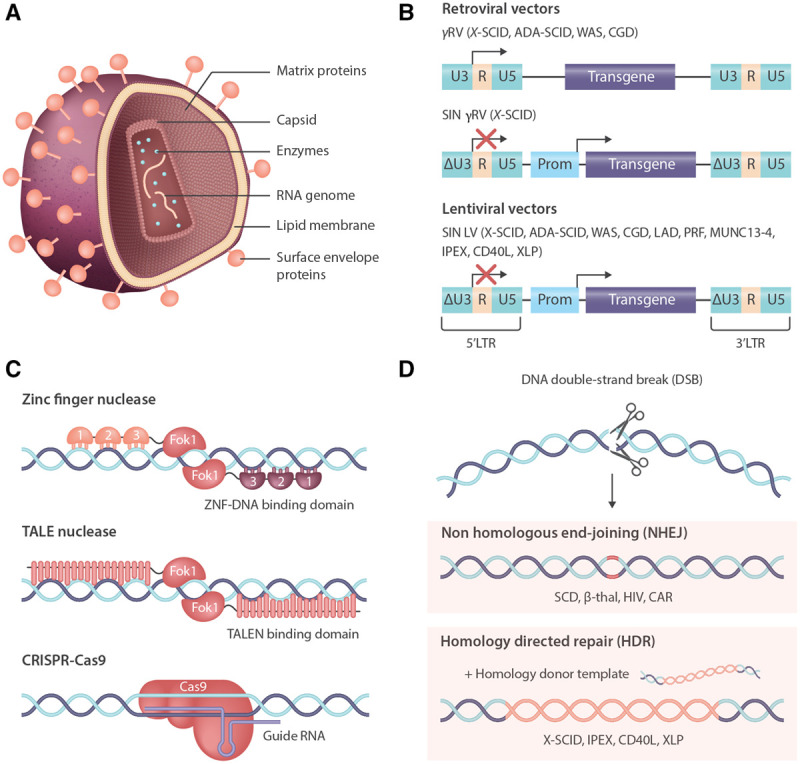
Gene therapy and gene editing technologies for correction of primary immunodeficiency. (A), Schematic of a retroviral particle, showing the viral protein architecture packaging the RNA genome. (B), Retroviral genomes used in gene therapy clinical trials, progressing in safety from the original wild type LTR-driven γRV vectors that have been associated with adverse events in several disease settings, to the SIN γRV and more widely used LVs that use chimeric LTRs and mammalian internal promoters to drive gene expression, providing an enhanced safety profile. (C), Emerging gene technologies are becoming a clinical reality due to highly active site-specific nucleases, most notably ZFNs, TALENs, and CRISPR/Cas9. (D), Nucleases create DNA DSBs that are repaired by several different pathways: NHEJ creates small INDELs, often leading to KO of genes—advantageous for therapies such as CCR5 KO T cells or HSC for HIV, or TCR/checkpoint inhibitor KO for improved CAR T cells—or enhancer elements, such as the erythroid enhancer for BCL11a, to promote γ-globin production for amelioration of SCD and β-thalassemia. In the presence of a repair template, HDR can occur, leading to precise insertion of therapeutic sequences, including whole gene cDNA—this approach is being explored for several immunodeficiencies, including X-SCID, IPEX, CD40L, and XLP. ADA-SCID = adenosine deaminase severe combined immunodeficiency; BCL11a = BAF chromatin remodeling complex subunit; CAR = chimeric antigen receptor; CCR5 = C-C chemokine receptor type 5; CD40L = CD40 ligand; cDNA = complementary DNA; CGD = chronic granulomatous disease; CRISPR/Cas9 = clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9; DSB = double-strand break; HDR = homology-directed repair; HIV = human immunodeficiency virus; HSC = hematopoietic stem cell; INDELs = insertions and deletions; IPEX = immune dysregulation, polyendocrinopathy, enteropathy, X-linked; KO = knock out; LAD = leukocyte adhesion defect; LTR = long terminal repeat; LV = lentiviral vector; MUNC13-4 = protein unc-13 homolog D; NHEJ = nonhomologous end-joining; PRF = perforin; SCD = sickle cell disease; SIN = self-inactivating; TALENs = TALE nucleases; TCR = T cell receptor; WAS = Wiskott-Aldrich syndrome; XLP = X-linked lymphoproliferative; X-SCID = X-linked severe combined immunodeficiency; ZFNs = zinc-finger nucleases; γRV = gammaretrovirus.
Early trials took advantage of the powerful promoter enhancer elements of the gammaretrovirus (γRV) long terminal repeat (LTR) sequences to drive robust expression of the therapeutic cDNA. However, the propensity of γRV vectors to integrate around the transcription start site of genes7 led to LTR-mediated proto-oncogene activation and leukemia transformation events in patients across several trials.8-10 The vector constructs were modified to a self-inactivating (SIN) configuration by deleting and replacing the wildtype viral sequences that exerted long-range promoter/enhancer effects and instead used internal mammalian promoters to drive transgene expression (Figure 1B).11-14 Although SIN γRV vectors have not been associated with severe adverse events, it was later realized that lentiviral vectors (LVs) based on HIV-1 had a safer integration profile, largely integrating within actively transcribed genes, therefore keeping exogenous promoters contained in vectors away from regulatory regions.15,16 SIN LV vectors are now the most widely used vectors and have excellent safety track record—more than 150 primary immune deficiency patients have been treated over the past decade without developing leukemia or myelodysplasia.17-19
The burgeoning interest in gene and engineered cell therapies using viral vectors has driven the optimization of good manufacturing practice (GMP) compliant large-scale suspension serum-free production systems and packaging cell lines. These processes reduce the amount of handling associated with adherent cell culture and reduce dependency on costly animal-derived products that carry a contamination risk and ethical concerns.20-22 Furthermore, interest has focused on the development of novel transduction enhancers that reduce the amount of high-cost virus required23-25 and cell culture media components that aim to retain HSC potency in culture.26 These technologies will lower the associated cost of gene therapy procedures and improve access, particularly when coupled with automated cell culture devices.27,28
Severe combined immunodeficiencies
Severe combine immunodeficiencies represent the most lethal PIDs and occur in an estimated 1:50 000-100 000 births.29 They are characterized by genetic faults leading to a block in T cell development programs, combined with deficiencies in numbers or function of natural killer (NK) or B cells resulting in both cellular and humoral immune abnormalities. These conditions often present in infancy with overwhelming infection and require urgent HSCT. Outcome following transplant can be negatively impacted by active infection alongside poorly matched donor status.30,31 Many countries around the globe have recently introduced newborn screening programs for severe combined immunodeficiency (SCID) to improve outcome through early diagnosis.32
SCID caused by a lack of adenosine deaminase enzyme (ADA-SCID) accounts for 10% of SCID diagnoses. Ubiquitous and highly conserved, ADA is a key enzyme in purine metabolism, responsible for safely converting adenosine to inosine; in its absence, toxic metabolites including adenosine, 2′-deoxyadenosine, and deoxyadenosine triphosphate (dATP) accumulate, leading to profound reduction in the numbers of circulating T, B, and NK cells. Suffering with a severe lack of cellular and humoral immunity, 85% of patients present to clinic in the first year of life due to a failure to thrive and high risk of opportunistic bacterial and fungal infections, alongside systemic abnormalities affecting the lungs and skeletal system. Neurological impairments, including deafness, developmental delay, and behavioral issues, are common.33 In contrast to the limited treatment options for most types of SCID, ADA deficient patients can receive enzyme replacement therapy (ERT) in the form of polyethylene glycol-conjugated bovine enzyme (PEG-ADA), which can successfully reduce the levels of metabolites and improve lymphocyte numbers. However, this is not curative, and long-term use is associated with reduced efficacy and significant cost. HSCT offers a curative therapy for patients with overall survival (OS) of 86% for those with matched sibling donors (MSD) and 81% in the matched-related donor (MRD) setting in a 106 patient cohort.34 However, in the mismatched donor setting survival falls to 66% for matched unrelated donors (MUD) and 43% for haploidentical donor transplants.34,35
As a monogenic disease, ADA-SCID was an attractive candidate for gene therapy. The first trials began in the 1990s, using γRV vectors to transduce and infuse T cells,36-38 umbilical cord blood cells,39 and bone marrow cells40 but failed to show long-term efficacy. Patients did not receive a preconditioning regimen, under the rationale that corrected cells would have a significant survival advantage despite continuing to receive PEG-ADA. Subsequent γRV trials incorporated both myeloreductive conditioning (busulfan, melphalan) and cessation of ERT, observing restoration of lymphocyte number, reduced rates of infection, and 100% survival of over 40 treated patients.41-45 In 2016, this protocol and vector was licensed as Strimvelis (GSK2696273), the first ex vivo gene therapy product to treat a primary immune deficiency licensed in Europe.46,47
To date, there has been no evidence of viral-mediated genotoxicity in this disease, despite evidence of integration sites near proto-oncogenes (LMO2, BCL2, CCND2) that have driven malignancy in other diseases.48 However, in line with safety improvements in the wider field, SIN LV vector approaches have been pursued, using the mammalian elongation factor 1α short (EFS) promoter to drive ADA expression. Murine models indicated that this vector was able to restore gene expression and restore immune function comparable to the γRV vector while demonstrating a significant reduction in transformation potential in vitro.49 In addition, studies in the same model revealed that while conditioning significantly improved engraftment, withdrawing PEG-ADA was less important, and that it may be preferable to maintain ERT to maintain cellularity in the bone marrow and reduce the period of lymphopenia post-transplant.50 Current trials now include pharmacokinetic (PK)-adjusted busulfan conditioning, maintained ERT until 30 days after infusion and increasingly the use of cryopreserved products allowing for more extensive testing release criteria to be completed before the product is infused (NCT02999984/NCT01380990/NCT02022696/NCT01852071) (Table 1).
Table 1.
Current and Historical Gene Therapy Trials for Primary Immune Deficiencies.
| Disease | Vector | Center | Trial Number | Cryopreservation | Participants | References |
|---|---|---|---|---|---|---|
| ADA-SCID | γRV | Italy, Madrid | NCT00599781/NCT00598481 | 22 | 42,46,51 | |
| United States | NCT03478670 | 5 | 52 | |||
| United States | NCT00018018 | 10 | 53 | |||
| United States | NCT00794508 | 10 | 44 | |||
| United Kingdom, London | NCT01279720 | 8 | 43 | |||
| SIN LV | United Kingdom, London | NCT01380990 | 20 | |||
| United States, UCLA | NCT01852071 | 20 | 54 | |||
| United States, UCLA | NCT02999984 | Yes | 10 | 54 | ||
| United Kingdom, London | NCT03765632 | Yes | 10 | |||
| United States | NCT02022696 | 1 | ||||
| X-SCID | γRV | France, Paris | NA | 10 | 8 | |
| United Kingdom, London | NA | 10 | 55 | |||
| SIN γRV | France/United Kingdom/United States | NCT01410019/NCT01175239/NCT01129544 | 14 | 56,57 | ||
| SIN LV | United States, NIH | NCT01306019 | 5 | 58 | ||
| United Kingdom, London | NCT03601286 | Yes | 10 (est recruitment) | |||
| United States, NIH | NCT03315078 | 13 (est recruitment) | ||||
| United States, St Jude | NCT01512888 | Yes | 8 (28 est recruitment) | 59 | ||
| United States, Boston | NCT03311503 | Yes | 10 (est recruitment) | |||
| China, Shenzhen | NCT03217617 | 10 (est recruitment) | ||||
| China, Chongqing | NCT04286815 | 10 (est recruitment) | ||||
| Artemis-SCID | SIN LV | United States, UCSF | NCT03538899 | Yes | 5 (15 est recruitment) | 60 |
| WAS | γRV | Germany, Hannover | DRKS00000330 | 10 | 9 | |
| SIN LV | United States, Boston | NCT01410825 | 5 | 61,62 | ||
| Italy, Milan | NCT01515462 | 8 | 63,64 | |||
| Italy, Milan | NCT03837483 | Yes | 6 | |||
| United Kingdom, London | NCT01347242 | 7 | 65 | |||
| France, Paris | NCT01347346 | 5 | 66 | |||
| CGD | γRV | Germany, Frankfurt | NCT00564759 | 2 | 10,67 | |
| Switzerland, Zurich | NCT00927134 | 2 | 68 | |||
| Korea, Seoul | NCT00778882 | 2 | 69 | |||
| United States, NIH | NCT00394316 | 3 | 70 | |||
| SIN γRV | Germany, Frankfurt | NCT01906541 | 5 (est recruitment) (adults) | |||
| LV | United Kingdom, London/United States, UCLA | NCT01855685/NCT02234934 | Part* | 9 | 71 | |
| France, Paris | NCT02757911 | 2 | 52 | |||
| China, Shenzhen | NCT03645486 | 10 (est recruitment) | ||||
| LAD-1 | γRV | United States, Boston | NCT00023010 | 2 | 72 | |
| LV | United States, UCLA | NCT03812263 | Yes | 9 (est recruitment) | ||
| United Kingdom, London | NCT03812263 | Yes | 3 (est recruitment) | |||
| Spain, Madrid | NCT03825783 | Yes | 2 (est recruitment) |
ADA-SCID = adenosine deaminase severe combined immunodeficiency; CGD = chronic granulomatous disease; LAD-1 = leukocyte adhesion defect type 1; LV = lentiviral vector; NA = not available; NIH = National Institutes of Health; SCID = severe combined immunodeficiency; X-SCID = X-linked severe combined immunodeficiency; SIN = self-inactivating; UCLA = University of California, Los Angeles; UCSF = University of California San Francisco; WAS = Wiskott-Aldrich syndrome; γRV = gammaretrovirus; UCLA = University of California - Los Angeles; UCSF = University of California - San Francisco.
*Some patients received cryopreserved products.
The very promising results seen in trials of gene therapy for ADA-SCID in terms of long-term immune recovery and safety have led to treatment guidelines suggesting the use of gene therapy rather than allogeneic HSCT from a matched unrelated donor (European Society for Blood and Marrow Transplantation Guidelines). Unfortunately, neither gene therapy nor HSCT can improve the nonimmune related disease manifestations seen in this condition.73
X-SCID is one of the more common forms of SCID, accounting for up to 40% of cases in some populations.74 Mutations in the IL2RG gene lead to an absence of the common gamma chain, a vital common component of the receptors for the cytokines interleukin (IL)-2, -4, -7, -9, -15, and -21. Gamma chain deficient lymphocytes are unable to receive the signals needed to develop, leading to an absence of circulating T and NK cells and dysfunctional B cells, resulting in severe immunodeficiency and susceptibility to severe and often opportunistic infection.75 HSCT was previously the only curative treatment, and while this procedure can be highly successful from a geno-identical donor (OS >90%), the outcome is less favorable for patients with mismatched donors, particularly when active infection is present.76,77
Gene therapy for X-SCID entered the clinic using a γRV vector in 1999. Clear clinical benefit was observed, with patients reconstituting functional T cells and, to a lesser extent, NK cells and reduced infections.78,79 However, 6 out of 20 patients enrolled developed T acute lymphoblastic leukemia as a result of insertional mutagenesis around proto-oncogenic loci and accumulated genetic abnormalities including deletion of tumor suppressor genes and translocation events.8,52,55
To address genotoxicity while retaining clinically efficacy, SIN γRV vectors were developed again employing the mammalian EFS promoter.12 T cell gene marking in treated patients was similar to the first trial, yet no severe adverse events relating to insertional mutagenesis have been recorded to date in the 9 surviving patients enrolled; the 1 death occurring due to existing viremia (NCT01410019/NCT01175239/NCT01129544).56 In both trials, the absence of a conditioning regimen prior to transplant contributed to suboptimal myeloid engraftment and humoral reconstitution, often requiring patients to stay on immunoglobulin therapy despite the survival advantage of corrected cells. More recently, LV vector trials for X-SCID have incorporated low-dose PK-adjusted Busulfan conditioning with the aim of improving efficacy; early results suggesting improved B cell reconstitution and normalization of immunoglobulin responses have recently been reported.58,59
SCID can also be caused by mutations in genes encoding proteins responsible for V(D)J rearrangement of T and B cell antigen receptors, such as DNA-dependent protein kinase (DNA-PKcs), catalytic subunit,80 DNA ligase 4 (LIG4),81 recombination activating gene 1 and 2 (RAG1/2),82 and Artemis.83 The latter three have long been identified as targets for gene therapy but have faced challenges in replicating the endogenous level of gene expression that is crucial for correct function.
While an absence of RAG1 or RAG2 causes a T– B– SCID phenotype, insufficient expression leads to Omenn syndrome, immune dysregulation, and autoimmunity, as seen in some patients with hypomorphic mutations.84-86 Preclinical gene therapy studies have struggled to obtain sufficiently high levels of gene expression from vectors that are suitable for clinical use,87-90 however, following successful outcomes in a murine model, an SIN LV vector using an MND promoter construct to drive RAG1 has now been selected for translation and a trial planned for the near future,91 while a Ubiquitous Chromatin Opening Element (UCOE) promoter has shown promising results for RAG292.
Several groups have generated LV vectors expressing the DCLRE1C gene that encodes the Artemis protein,93,94 however, toxicity was observed when expression levels were too high.95 A further study utilized the endogenous Artemis promoter and found it gave optimal reconstitution in Artemis knock out (KO) mice.96 Some preliminary results of a trial of 5 patients (NCT03538899) using this vector have indicated the efficacy of this approach, noting the reappearance of T cell subsets along with stable gene marking in T, B, NK, and myeloid cells, allowing the patients to leave isolation.60
SCIDs: paradigm for emerging therapies
For both X-SCID and ADA deficiency, the element of survival advantage in corrected lymphocytes makes these diseases attractive models for novel therapies. Techniques being tested include in vivo gene therapy, a minimally invasive technique to correct cells by direct infusion of gene transfer vectors. In vivo gene therapy has been attempted in ADA deficient mice and nonhuman primates using LV vectors, however, efficacy was limited outside the neonatal setting.97,98 In a recent study, premobilization of HSC and in vivo transduction using foamy virus vectors was corrective in a canine model of X-SCID.99 Foamy viruses are attractive gene transfer vectors for HSC, as they are resistant to serum inactivation, are able to transduce quiescent cells, and present a favorable integration profile; however, the safety, efficacy, and scalability of this approach remains a challenge for first-in-human studies.100
Gene editing offers the potential to provide therapeutic gene expression closest to the endogenous profile by inserting corrective sequences in situ. This technique has become clinically relevant due to the development of a series of highly site-specific nucleases, including zinc-finger nucleases (ZFNs), TALE nucleases (TALENs), and clustered regularly interspaced short palindromic repeats/CRISPR-associated protein systems (CRISPR/Cas) (Figure 1C).101 The creation of a DNA double-strand break provides a substrate for endogenous DNA repair pathways, which can be harnessed either to KO genes or to seamlessly insert therapeutic DNA by providing a suitable donor containing sequences homologous to the cleaved ends (Figure 1D). This placement conserves many of the native regulatory motifs surrounding a gene, many of which would be too large to fit into a LV vector and are often poorly defined.
All of these technologies have now entered the clinic, although so far, none using homology-directed repair (HDR). In the absence of a homology repair template, nonhomologous end-joining (NHEJ) occurs, creating small insertions and deletions (INDELs) which lead to gene KO. The first-in-man trial, initiated in 2009, used ZFN to create autologous C-C chemokine receptor type 5 (CCR5) KO T cell product for patients with HIV.102 With 1 severe adverse event out of 12 patients, unrelated to the editing procedure, this trial showed that editing tools can be safe, particularly in T cells; TALENs and CRISPR platforms have now been used extensively in immunotherapy products such as CAR T cells.103,104 In 2018, the first gene-edited HSC trials were announced for patients with sickle cell disease (SCD) and β-thalassemia (NCT03745287/NCT03655678), using CRISPR/Cas9 to disrupt the erythroid-specific enhancer of the BCL11A gene, aiming to increase γ-globin levels and ameliorate the disease (Figure 2).105
Figure 2.
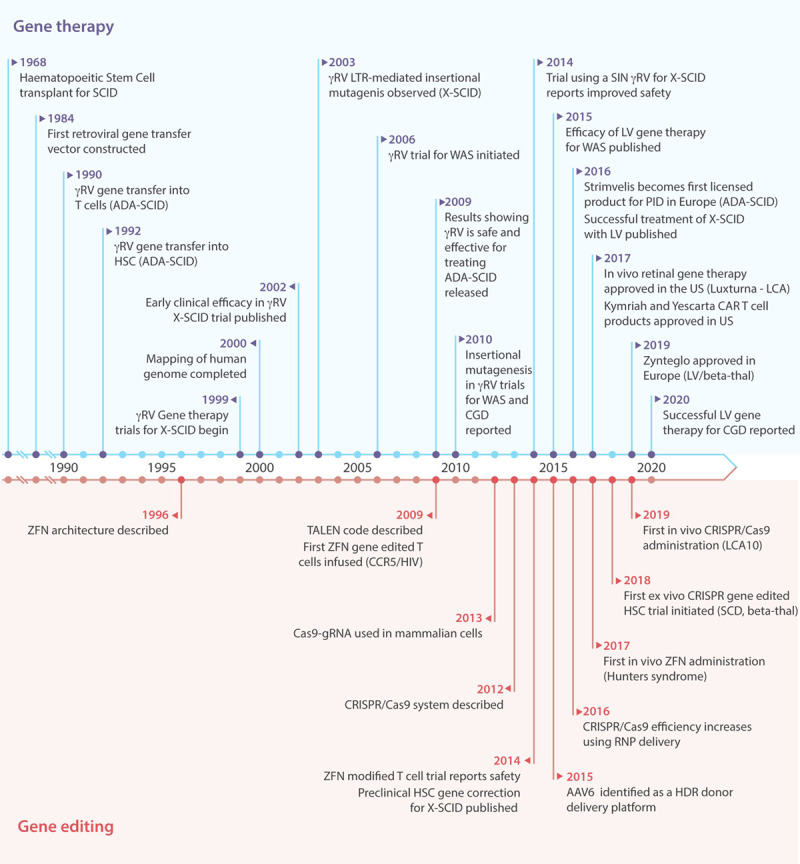
Timeline of major advances in gene therapy and gene editing therapeutics for primary immunodeficiency. AAV6 = adeno-associated virus serotype 6; ADA-SCID = adenosine deaminase severe combined immunodeficiency; CAR = chimeric antigen receptor; Cas9 = CRISPR associated protein 9; CCR5 = C-C chemokine receptor type 5; CGD = chronic granulomatous disease; CRISPR = clustered regularly interspaced short palindromic repeats; gRNA = guide RNA; HDR = homology-directed repair; HIV = human immunodeficiency virus; HSC = hematopoietic stem cell; LCA = leber congenital amaurosis; LTR = long terminal repeat; LV = lentiviral vector; PID = primary immunodeficiency; RNP = ribonucleoprotein; SCD = sickle cell disease; SCID = severe combined immunodeficiency; SIN = self-inactivating; TALENs = TALE nucleases; US = United States; WAS = Wiskott-Aldrich syndrome; X-SCID = X-linked severe combined immunodeficiency; ZFNs = zinc-finger nucleases; γRV = gammaretrovirus.
For several PIDs, gene editing using HDR may offer a safer therapy by avoiding aberrant gene expression from viral vectors, particularly useful for disorders where, for example, signaling molecules are affected and aberrant expression could be detrimental. Again, SCID was the first model in which proof of concept for this technology was shown. Genovese et al106 showed the feasibility of this approach; X-SCID HSC were corrected using ZFN and nonintegrating lentiviral vectors, which gave rise to functional lymphoid cells in an in vivo mouse model. In recent years, CRISPR/Cas9 and adeno-associated virus serotype 6 (AAV6) homology donors have risen to be the most promising tools, capable of correcting HSC to levels approaching 50% in vitro.107 Trials will determine the efficacy of these approaches in man.
Non-SCID immunodeficiencies
Following on from the successes in early trials of gene therapy for SCID, the approach was applied to more complex immune disorders where a survival advantage of gene-corrected cells may not have been so prominent.
Wiskott-Aldrich syndrome (WAS) is caused by an absence of WAS protein, a major regulator of the actin cytoskeleton in hematopoietic cells necessary for immune function and platelet production. The disease is associated with a spectrum of clinical presentations including immunodeficiency, thrombocytopenia, eczema, and increased risk of malignancy. Results for HSCT in WAS have improved over the years with the most recent report showing an OS of 90% regardless of donor source, if patients are treated in the first 5 years of life. For older patients, OS drops to 66%, and both acute and chronic GvHD is a significant risk (27%/17%, respectively).108 A clinical trial initiated in 2006 using γRV vectors showed clear clinical benefit and restoration of immune function.109 However, 7 out of the 9 patients that reconstituted immune function developed leukemia and integrations around LMO2/MDS1/EVI1 proto-oncogenic loci were later identified.9 To move forward, several centers chose a SIN LV vector that incorporated a 1.6 kb segment of the endogenous WAS promoter to drive WAS protein expression that had shown efficacy in preclinical models.110,111 A reduced intensity (busulfan/fludarabine) conditioning regimen was also standardized across centers.61,65,112 These trials are ongoing, but at the most recent published follow-up (up to 5.6 y post-procedure), collective data shows 90% survival and significant clinical improvement with sustained multi-lineage gene expression, correction of immune deficiency and eczema, and ability to stop immunoglobulin replacement therapy (NCT01515462/NCT01347242—1 death/15 treated patients).63,65 Post-procedure autoimmunity is seen in both HSCT and gene therapy cohorts.108,113 One major difference between the outcome of HSCT and gene therapy for WAS is the resolution of thrombocytopenia, which is superior following stem cell transplantation, although modest improvement in platelet count following gene therapy does prevent hemorrhagic events. The exact reason behind this is unclear and may relate to the number of gene-corrected cells infused114 but it is clear that gene-corrected platelets exhibit normal function.115
Chronic granulomatous disease (CGD) affects around 1:200 000 births and arises due to defects in the subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex. Expressed in phagocytic cells, this complex generates reactive oxygen species (including superoxide anion radical, hydrogen peroxide, and hypochlorite) and activates neutrophil proteases that kill the engulfed bacteria or fungi in phagocytic vacuoles. In its absence, severe infections and chronic inflammation result. Again, transplant results have improved with a recent report of 712 patients showing an OS of 85.7%, with reduced OS for adult patients (76%). However, GvHD remains a significant risk responsible for a third of the fatalities in this cohort.116
CYBB (cytochrome B-245 beta chain) mutations encoding Gp91phox (glycosylated 91-kDa glycoprotein) cause the most common X-linked form (65% cases), which has been the target for all trials to date.117 Despite a lack of survival advantage of corrected cells in this disease, low numbers of oxidase-positive neutrophils or residual levels of NADPH oxidase expression can confer a significantly increase survival,118,119 making X-CGD an appealing candidate for treatment with gene therapy. An early γRV trial, initiated in the mid-1990s, recruited 5 adult patients and was performed without conditioning. Although there were no severe adverse events, gene marking in the periphery was very low and transient.120 Subsequent trials in multiple centers incorporated a myeloablative conditioning regimen that increased engraftment and restoration of immune function; however, this effect was also transient, with most of the 12 patients losing NADPH oxidase expression after 3 months.67,69,121-123 Three patients that did achieve significant gene marking in neutrophils in a trial using a spleen focus forming virus (SFFV)-based LTR γRV were found to have integration events around proto-oncogenic loci (PRDM16 and MDS1/EVI1) and later developed myelodysplasia.10 Further studies revealed that LTR promoter elements were being methylated, leading to gene silencing, while the enhancer elements (and therefore mutagenic influence) were unaffected. To improve safety, efficacy, and longevity, SIN LV vectors were developed that aimed to provide preferential expression in myeloid cells and detarget expression from HSC. Studies have investigated myeloid-specific promoters,124,125 a minimal CYBB promoter coupled with myeloid-specific enhancers,126 and a myeloid-specific promoter used in parallel with HSC-expressed microRNA binding sites.127 However, the most widely adopted vector was constructed by fusing cathepsin G and c-Fes proximal regulatory sequences, with the aim of driving maximal expression during terminal myeloid differentiation.128 Murine studies confirmed NADPH oxidase expression from the vector closely mimicked the endogenous expression profile and gene silencing through methylation was not observed. Two trials using this vector have recently reported early findings for the 9 patients enrolled: while 2 patients succumbed to disease-related comorbidities, 6 out of the 7 patients alive had stable copy number and 16%-46% oxidase-positive neutrophils, with no evidence of transgene silencing or untoward clonal dominance, up to 3 years post-procedure (NCT01855685/NCT02234934).71 A similar approach has been adapted to another form of CGD caused by mutations in the NCF1 gene leading to abnormal P47phox expression with proof of concept demonstrated in a murine model.129 Clinical trials of this LV are anticipated to start in the near future.
Leukocyte adhesion defect type 1 (LAD-1) is characterized by severe life-threatening recurrent bacterial infections due to impaired migration of neutrophils to sites of infection arising as a result of defective membrane expression of CD18 integrin subunit encoded by the ITGB2 gene. Treatment with HSCT is necessary, as mortality rates are between 60% and 75% in infancy for the most severely affected patients.130,131 However, disease severity tightly correlates with the level of CD18 expression, indicating that even a low level of correction could significantly reduce mortality. An early trial using a γRV in 2 patients that did not receive a conditioning regimen failed, with no gene marked cells detectable in the periphery after 2 months.72 Subsequent studies using LV vectors in both murine132 and canine133 models have paved the way for a recently opened trial across Europe and the United States, using a busulfan conditioning regimen for patients without access to an HLA-identical sibling donor (NCT03825783, NCT03812263).
Gene therapy for primary immune deficiencies: future perspectives
Patients suffering from immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome exhibit severe autoimmunity due to mutation in the forkhead box P3 (FOXP3) gene. This transcription factor is considered a master regulator for successful development and function of regulatory T cells (Tregs), that are vital for maintaining immune tolerance to self-antigens.134 Studies have shown that effector T cells can be converted to Tregs by ectopic expression of FOXP3,135 and these cells exhibit suppressor function in vitro, and in mouse models of GvHD, offering the potential of a T cell therapy.136,137 However, generating sufficient numbers of Tregs for this purpose will be challenging and the life span of the cells in vivo is unknown.138 Correction at the level of HSC would provide a longer-lasting therapy, however, studies investigating this approach have noted that constitutive FOXP3 expression in HSC (where it is not usually expressed) had adverse effects on T cell differentiation and hematopoiesis.139 A recent study aiming to abrogate this effect by replicating the endogenous expression profile by harnessing 3 regulatory elements, the FOXP3 promoter and the 3′UTR (untranslated region) to regulate transgene expression, has shown promising results in vivo.140 Another recent study using gene editing tools to place FOXP3 cDNA under control of its native promoter reported partial correction of FOXP3 expression and suppressive function restored to within the lower range of healthy control cells, indicating this methodology could provide an alternative approach for IPEX patients.141
Deficiency of the T cell costimulatory molecule CD40 ligand (CD40L) gives rise to X-linked hyper-immunoglobulin M (hyper-IgM) syndrome (XHIGM). CD40L expression is upregulated after T cell activation and is essential for T cell: B cell interactions that induce immunoglobulin class switching and antibody affinity maturation in B cells.142,143 The resulting lack of humoral immunity leaves patients vulnerable to bacterial and opportunistic infections and increased risk of malignancy and autoimmunity.144 HSCT is used to treat patients, but OS after this procedure is suboptimal with latest figures suggesting a 5-year OS of 78%, indicating the need for a better therapy.145,146 HSC gene correction using γRV vectors was shown to restore humoral and cellular function in CD40L-deficient mice, however, the mice later developed thymic lymphoproliferation due to an unregulated expression profile.147,148 A LV vector utilizing a 1.3 kb fragment of the CD40L promoter was able to replicate the tissue specificity and activation dependency of CD40L transgene in vitro,149 although endogenous levels of gene expression post-activation were not achieved, potentially due to regulatory mechanisms in the 3′UTR that were not included in the construct. Gene editing has been investigated in both T cells and HSC (TALENs and CRISPR), with homology donors placing cDNA under the full native promoter and including the CD40L 3′UTR. Physiologically regulated gene expression and function was restored to T cells, and mice transplanted with gene-edited HSC showed no evidence of proliferation.150,151
X-linked lymphoproliferative (XLP) disease is caused by mutations and deletions in the SH2D1A gene that encodes the intracellular adapter SLAM-adapter protein (SAP). SAP is vital for correctly relaying signals in T, NK, and NKT cells, in its absence, the immune system is dysregulated leading to hypogammaglobulinemia, hemophagocytic lymphohistiocytosis (HLH), and lymphoma.152 SAP has a tightly controlled expression profile, undetectable in HSC, B cells, Tregs, or myeloid lineage cells, and levels change after T cell stimulation and within T cell subsets.153-155 While toxicity has not been observed in murine models of gene therapy using a constitutive promoter,156 concern remains that aberrant expression could lead to further dysregulation. Due to the most severe immune deficits arising due to lack of T cell function, a T cell gene therapy approach was pursued, which corrected many of the disease phenotypes in murine models.157 However, gene editing could offer therapeutic advantages and is being pursued in both T cells and HSC (B.C. Houghton and C. Booth, unpublished data, November 2020).
Familial hemophagocytic lymphohistiocytosis (FHLH) encompasses a number of genetic conditions with severe immune dysregulation that is fatal if not treated. FHLH types 2-5 occur due to genetic defects in T and NK cell granule-mediated cytotoxicity, rendering these cells unable to effectively kill their targets and creating a hyperinflammatory environment with uncontrolled proliferation and cytokine release. HSCT can be curative, but outcome is dependent on disease status at the time of transplant.158
The mutated gene in FHLH2 is PRF1, encoding perforin. In healthy individuals, perforin is released into the immune synapse to form a pore on target cells, thereby allowing entry to granzymes into the cytoplasm and initiating apoptosis. Mixed chimerism experiments in a mouse model had shown that low levels of wild type cells could restore immune regulation, indicating that gene therapy could be a suitable approach.159 LV vectors were constructed, employing either a constitutive PGK promoter, or a segment of the PRF1 promoter to drive transgene expression; both vectors were able to restore gene expression and cytotoxicity to NK and T cells in murine models.160 As an additional therapeutic intervention, a gene-corrected T cell strategy was also investigated, which could offer a bridge-to-transplant therapy for patients.161
FHLH3 has also been a target for gene therapy. It is caused by mutations in UNC13D, encoding protein unc-13 homolog D (Munc 13-4), which is essential for priming perforin-containing vesicles for exocytosis; cells without functional protein cannot degranulate properly giving rise to the cytotoxic defect. Several groups have investigated gene correction using SIN γRV,162 SIN alpharetrovirus,163 or LV vectors,164 noting functional restoration of degranulation activity in vitro and in vivo murine models. Gene-corrected T cells have also been investigated as an alternative therapeutic.165
X-linked agammaglobulinemia (XLA) is the most common PID and a good candidate for gene therapy due to the low level of corrected HSCs that are required to restore immunoglobulin production that arises due to the arrest of B cell development at the pre-B cell stage in the absence of an intact Bruton's tyrosine kinase (BTK) gene.166 γRV167 and LV vectors168,169 have been developed that have successfully restored B cell differentiation and function in murine models, but these remain at a preclinical stage. Current standard of care for XLA patients does not include HSCT as a curative therapy but relies on supportive care with lifelong immunoglobulin replacement; a significant burden that can impact on quality of life.170,171 As our collective gene therapy experience grows particularly in terms of safety, in conditions such as XLA where stem cell transplant is considered in very few cases, gene therapy may become a feasible treatment option, offsetting the lifelong complications (and economic cost) of both disease and treatment.
Conclusion
Autologous HSC gene therapy now offers therapeutic benefit across a range of conditions including immunodeficiencies, hematological, and metabolic disorders. The successes reported to date and increasing safety data generated through numerous clinical trials puts gene therapy back on an upward trajectory following earlier setbacks related to vector associated malignancy in gammaretroviral trials. Although enticing novel gene editing approaches are being investigated and indeed translated through to first-in-human trials, the infrastructure required to deliver lentiviral therapies widely and accessibility to treatment requires further effort. Commercial partners are becoming increasingly involved in gene therapy programs; while this has benefits in terms of driving preclinical studies more rapidly towards patient benefit, the logistic and economic challenges associated with delivering high cost, personalized drug products in rare disease remain. One must also look at the wider therapeutic landscape. Outcomes following HSCT, even in the mismatched and haploidentical donor setting, have improved over the past decade with the advent of more sophisticated graft manipulation techniques. As more long-term efficacy data emerges from gene therapy clinical trials, it will become easier for physicians to understand which patients may benefit from the different treatment options available.
Note added after acceptance
Since writing this manuscript, a patient treated with Strimvelis for ADA-SCID has been diagnosed with T cell leukaemia. Causality is under investigation. For more information: https://ir.orchard-tx.com/news-releases/news-release-details/orchard-statement-strimvelisr-gammaretroviral-vector-based-gene.
Sources of funding
This work is supported by Action Medical Research (AMR) and the National Institute of Health Research Great Ormond Street Hospital Biomedical Research Centre.
Disclaimer
The views expressed are those of the author(s) and not necessarily those of the National Health Service, the National Institute of Health Research or the Department of Health, or Action Medical Research.
Disclosures
The authors declare no competing interest.
References
- 1.Picard C, Gaspar HB, Al-Herz W, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018; 38:96–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010; 125(2 suppl 2):S182–S194. [DOI] [PubMed] [Google Scholar]
- 3.Al-Muhsen S, Alsum Z. Primary immunodeficiency diseases in the Middle East. Ann N Y Acad Sci. 2012; 1250:56–61. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez JJ, Monaghan G, Børsting C, et al. Carrier frequency of a nonsense mutation in the adenosine deaminase (ADA) gene implies a high incidence of ADA-deficient severe combined immunodeficiency (SCID) in Somalia and a single, common haplotype indicates common ancestry. Ann Hum Genet. 2007; 71(pt 3):336–347. [DOI] [PubMed] [Google Scholar]
- 5.Morton DH, Morton CS, Strauss KA, et al. Pediatric medicine and the genetic disorders of the Amish and Mennonite people of Pennsylvania. Am J Med Genet C Semin Med Genet. 2003; 121C:5–17. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Moshous D, Zhou Y, et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002; 168:6323–6329. [DOI] [PubMed] [Google Scholar]
- 7.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003; 300:1749–1751. [DOI] [PubMed] [Google Scholar]
- 8.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008; 118:3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome - long-term efficacy and genotoxicity. Sci Transl Med. 2014; 6:227ra33. [DOI] [PubMed] [Google Scholar]
- 10.Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010; 16:198–204. [DOI] [PubMed] [Google Scholar]
- 11.Yu SF, von Rüden T, Kantoff PW, et al. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc Natl Acad Sci U S A. 1986; 83:3194–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thornhill SI, Schambach A, Howe SJ, et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol Ther. 2008; 16:590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schambach A, Bohne J, Chandra S, et al. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol Ther. 2006; 13:391–400. [DOI] [PubMed] [Google Scholar]
- 14.Zychlinski D, Schambach A, Modlich U, et al. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008; 16:718–725. [DOI] [PubMed] [Google Scholar]
- 15.Schröder ARW, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002; 110:521–529. [DOI] [PubMed] [Google Scholar]
- 16.Poletti V, Mavilio F. Interactions between retroviruses and the host cell genome. Mol Ther Methods Clin Dev. 2018; 8:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zufferey R, Dull T, Mandel RJ, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998; 72:9873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998; 72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cicalese MP, Aiuti A. New perspectives in gene therapy for inherited disorders. Pediatr Allergy Immunol. 2020; 31(suppl 24):5–7. [DOI] [PubMed] [Google Scholar]
- 20.Valkama AJ, Leinonen HM, Lipponen EM, et al. Optimization of lentiviral vector production for scale-up in fixed-bed bioreactor. Gene Ther. 2018; 25:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanber KS, Knight SB, Stephen SL, et al. Construction of stable packaging cell lines for clinical lentiviral vector production. Sci Rep. 2015; 5:9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauler M, Roberts JK, Wu CC, et al. Production of lentiviral vectors using suspension cells grown in serum-free media. Mol Ther Methods Clin Dev. 2020; 17:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schott JW, Leon-Rico D, Ferreira CB, et al. Enhancing lentiviral and alpharetroviral transduction of human hematopoietic stem cells for clinical application. Mol Ther Methods Clin Dev. 2019; 14:134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masiuk KE, Zhang R, Osborne K, Hollis RP, Campo-Fernandez B, Kohn DB. PGE2 and poloxamer synperonic F108 enhance transduction of human HSPCs with a β-globin lentiviral vector. Mol Ther Methods Clin Dev. 2019; 13:390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrillo C, Thorne LG, Unali G, et al. Cyclosporine H overcomes innate immune restrictions to improve lentiviral transduction and gene editing in human hematopoietic stem cells. Cell Stem Cell. 2018; 23:820–832.e829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tajer P, Pike-Overzet K, Arias S, Havenga M, Staal FJT. Ex vivo expansion of hematopoietic stem cells for therapeutic purposes: lessons from development and the niche. Cells. 2019; 8:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adair JE, Waters T, Haworth KG, et al. Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nat Commun. 2016; 7:13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Rivière I. Genetic engineering and manufacturing of hematopoietic stem cells. Mol Ther Methods Clin Dev. 2017; 5:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer A, Le Deist F, Hacein-Bey-Abina S, et al. Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol Rev. 2005; 203:98–109. [DOI] [PubMed] [Google Scholar]
- 30.Pai S-Y, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014; 371:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiercy JM. How to select the best available related or unrelated donor of hematopoietic stem cells? Haematologica. 2016; 101:680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Burg M, Mahlaoui N, Gaspar HB, et al. Universal newborn screening for severe combined immunodeficiency (SCID). Front Pediatr. 2019; 7:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitmore KV, Gaspar HB. Adenosine deaminase deficiency - more than just an immunodeficiency. Front Immunol. 2016; 7:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassan A, Booth C, Brightwell A, et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012; 120:3615–3624; quiz 3626. [DOI] [PubMed] [Google Scholar]
- 35.Flinn AM, Gennery AR. Adenosine deaminase deficiency: a review. Orphanet J Rare Dis. 2018; 13:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blaese RM, Culver KW, Miller AD, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995; 270:475–480. [DOI] [PubMed] [Google Scholar]
- 37.Bordignon C, Notarangelo LD, Nobili N, et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995; 270:470–475. [DOI] [PubMed] [Google Scholar]
- 38.Onodera M, Ariga T, Kawamura N, et al. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood. 1998; 91:30–36. [PubMed] [Google Scholar]
- 39.Kohn DB, Weinberg KI, Nolta JA, et al. Engraftment of gene–modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med. 1995; 1:1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoogerbrugge PM, van Beusechem VW, Fischer A, et al. Bone marrow gene transfer in three patients with adenosine deaminase deficiency. Gene Ther. 1996; 3:179–183. [PubMed] [Google Scholar]
- 41.Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002; 296:2410–2413. [DOI] [PubMed] [Google Scholar]
- 42.Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009; 360:447–458. [DOI] [PubMed] [Google Scholar]
- 43.Gaspar HB, Cooray S, Gilmour KC, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011; 3:97ra80. [DOI] [PubMed] [Google Scholar]
- 44.Shaw KL, Garabedian E, Mishra S, et al. Clinical efficacy of gene-modified stem cells in adenosine deaminase-deficient immunodeficiency. J Clin Invest. 2017; 127:1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cicalese MP, Ferrua F, Castagnaro L, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016; 128:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cicalese MP, Ferrua F, Castagnaro L, et al. Gene therapy for adenosine deaminase deficiency: a comprehensive evaluation of short- and medium-term safety. Mol Ther. 2018; 26:917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shahryari A, Saghaeian Jazi M, Mohammadi S, et al. Development and clinical translation of approved gene therapy products for genetic disorders. Front Genet. 2019; 10:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biasco L, Ambrosi A, Pellin D, et al. Integration profile of retroviral vector in gene therapy treated patients is cell-specific according to gene expression and chromatin conformation of target cell. EMBO Mol Med. 2011; 3:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carbonaro DA, Zhang L, Jin X, et al. Preclinical demonstration of lentiviral vector-mediated correction of immunological and metabolic abnormalities in models of adenosine deaminase deficiency. Mol Ther. 2014; 22:607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carbonaro DA, Jin X, Wang X, et al. Gene therapy/bone marrow transplantation in ADA-deficient mice: roles of enzyme-replacement therapy and cytoreduction. Blood. 2012; 120:3677–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohn DB, Hershfield MS, Puck JM, et al. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol. 2019; 143:852–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cavazzana M, Bushman FD, Miccio A, et al. Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat Rev Drug Discov. 2019; 18:447–462. [DOI] [PubMed] [Google Scholar]
- 53.Candotti F, Shaw KL, Muul L, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012; 120:3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kohn DB, Shaw KL, Garabedian E, et al. Lentiviral gene therapy with autologous hematopoietic stem and progenitor cells (HSPCs) for the treatment of severe combined immune deficiency due to adenosine deaminase deficiency (ADA-SCID): results in an expanded cohort. Blood. 2019; 134(supplement_1):3345–3345. [Google Scholar]
- 55.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008; 118:3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hacein-Bey-Abina S, Pai SY, Gaspar HB, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014; 371:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pai SY, Thrasher AJ. Gene therapy for X-linked severe combined immunodeficiency: historical outcomes and current status. J Allergy Clin Immunol. 2020; 146:258–261. [DOI] [PubMed] [Google Scholar]
- 58.De Ravin SS, Wu X, Moir S, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2016; 8:335ra57–335ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med. 2019; 380:1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cowan MJ, Yu J, Facchino J, et al. Early outcome of a phase I/II clinical trial (NCT03538899) of gene-corrected autologous CD34+ hematopoietic cells and low-exposure busulfan in newly diagnosed patients with artemis-deficient severe combined immunodeficiency (ART-SCID). Biol Blood Marrow Transplant. 2020; 26:S88–S89. [Google Scholar]
- 61.Chu JI, Henderson LA, Armant M, et al. Gene therapy using a self-inactivating lentiviral vector improves clinical and laboratory manifestations of Wiskott-Aldrich syndrome. Blood. 2015; 126:260–260. [Google Scholar]
- 62.Labrosse R, Chu J, Armant M, et al. Outcome of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Blood. 2019; 134(supplement_1):4629–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019; 6:e239–e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferrua F, Marangoni F, Aiuti A, et al. Gene therapy for Wiskott-Aldrich syndrome: history, new vectors, future directions. J Allergy Clin Immunol. 2020; 146:262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hacein-Bey Abina S, Gaspar HB, Blondeau J, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015; 313:1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther. 2015; 26:210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006; 12:401–409. [DOI] [PubMed] [Google Scholar]
- 68.Siler U, Paruzynski A, Holtgreve-Grez H, et al. Successful combination of sequential gene therapy and rescue Allo-HSCT in two children with X-CGD - importance of timing. Curr Gene Ther. 2015; 15:416–427. [DOI] [PubMed] [Google Scholar]
- 69.Kang HJ, Bartholomae CC, Paruzynski A, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther. 2011; 19:2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kang EM, Marciano BE, DeRavin S, et al. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2011; 127:1319–1326; quiz 1327–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohn DB, Booth C, Kang EM, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med. 2020; 26:200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bauer TR, Jr, Hickstein DD. Gene therapy for leukocyte adhesion deficiency. Curr Opin Mol Ther. 2000; 2:383–388. [PubMed] [Google Scholar]
- 73.Hönig M, Albert MH, Schulz A, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood. 2007; 109:3595–3602. [DOI] [PubMed] [Google Scholar]
- 74.Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers. 2015; 1:1–18. [DOI] [PubMed] [Google Scholar]
- 75.Noguchi M, Yi H, Rosenblatt HM, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993; 73:147–157. [DOI] [PubMed] [Google Scholar]
- 76.Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999; 340:508–516. [DOI] [PubMed] [Google Scholar]
- 77.Gennery AR, Slatter MA, Grandin L, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010; 126:602–610.e1–e11. [DOI] [PubMed] [Google Scholar]
- 78.Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002; 346:1185–1193. [DOI] [PubMed] [Google Scholar]
- 79.Gaspar HB, Parsley KL, Howe S, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004; 364:2181–2187. [DOI] [PubMed] [Google Scholar]
- 80.van der Burg M, IJspeert H, Verkaik NS, et al. A DNA-PKcs mutation in a radiosensitive T–B– SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009; 119:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van der Burg M, van Veelen LR, Verkaik NS, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006; 116:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schwarz K, Gauss GH, Ludwig L, et al. RAG mutations in human B cell-negative SCID. Science. 1996; 274:97–99. [DOI] [PubMed] [Google Scholar]
- 83.Moshous D, Callebaut I, de Chasseval R, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001; 105:177–186. [DOI] [PubMed] [Google Scholar]
- 84.Villa A, Santagata S, Bozzi F, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998; 93:885–896. [DOI] [PubMed] [Google Scholar]
- 85.Notarangelo LD, Kim MS, Walter JE, et al. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016; 16:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delmonte OM, Villa A, Notarangelo LD. Immune dysregulation in patients with RAG deficiency and other forms of combined immune deficiency. Blood. 2020; 135:610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pike-Overzet K, Rodijk M, Ng Y-Y, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011; 25:1471–1483. [DOI] [PubMed] [Google Scholar]
- 88.van Til NP, Sarwari R, Visser TP, et al. Recombination-activating gene 1 (Rag1)–deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol. 2014; 133:1116–1123. [DOI] [PubMed] [Google Scholar]
- 89.Lagresle-Peyrou C, Yates F, Malassis-Séris M, et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood. 2006; 107:63–72. [DOI] [PubMed] [Google Scholar]
- 90.Yates F, Malassis-Séris M, Stockholm D, et al. Gene therapy of RAG-2-/- mice: sustained correction of the immunodeficiency. Blood. 2002; 100:3942–3949. [DOI] [PubMed] [Google Scholar]
- 91.Garcia-Perez L, van Eggermond M, van Roon L, et al. Successful preclinical development of gene therapy for recombinase-activating gene-1-deficient SCID. Mol Ther Methods Clin Dev. 2020; 17:666–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Til NP, de Boer H, Mashamba N, et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther. 2012; 20:1968–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benjelloun F, Garrigue A, Demerens-de Chappedelaine C, et al. Stable and functional lymphoid reconstitution in artemis-deficient mice following lentiviral artemis gene transfer into hematopoietic stem cells. Mol Ther. 2008; 16:1490–1499. [DOI] [PubMed] [Google Scholar]
- 94.Mostoslavsky G, Fabian AJ, Rooney S, et al. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci U S A. 2006; 103:16406–16411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Multhaup M, Karlen AD, Swanson DL, et al. Cytotoxicity associated with artemis overexpression after lentiviral vector-mediated gene transfer. Hum Gene Ther. 2010; 21:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Punwani D, Kawahara M, Yu J, et al. Lentivirus mediated correction of artemis-deficient severe combined immunodeficiency. Hum Gene Ther. 2017; 28:112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carbonaro DA, Jin X, Petersen D, et al. In vivo transduction by intravenous injection of a lentiviral vector expressing human ADA into neonatal ADA gene knockout mice: a novel form of enzyme replacement therapy for ADA deficiency. Mol Ther. 2006; 13:1110–1120. [DOI] [PubMed] [Google Scholar]
- 98.Carbonaro-Sarracino DA, Tarantal AF, Lee CCI, et al. Dosing and re-administration of lentiviral vector for in vivo gene therapy in rhesus monkeys and ADA-deficient mice. Mol Ther Methods Clin Dev. 2020; 16:78–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Humbert O, Chan F, Rajawat YS, et al. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Adv. 2018; 2:987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simantirakis E, Tsironis I, Vassilopoulos G. FV vectors as alternative gene vehicles for gene transfer in HSCs. Viruses. 2020; 12:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Porteus MH. A new class of medicines through DNA editing. N Engl J Med. 2019; 380:947–959. [DOI] [PubMed] [Google Scholar]
- 102.Tebas P, Stein D, Tang WW, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014; 370:901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017; 9:eaaj2013. [DOI] [PubMed] [Google Scholar]
- 104.Ashmore-Harris C, Fruhwirth GO. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin Transl Med. 2020; 9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016; 539:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Genovese P, Schiroli G, Escobar G, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014; 510:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pavel-Dinu M, Wiebking V, Dejene BT, et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun. 2019; 10:1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burroughs LM, Petrovic A, Brazauskas R, et al. Excellent outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome: a PIDTC report. Blood. 2020; 135:2094–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Boztug K, Schmidt M, Schwarzer A, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010; 363:1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marangoni F, Bosticardo M, Charrier S, et al. Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models. Mol Ther. 2009; 17:1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Galy A, Roncarolo M-G, Thrasher AJ. Development of lentiviral gene therapy for Wiskott Aldrich syndrome. Expert Opin Biol Ther. 2008; 8:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013; 341:1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood. 2011; 118:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fischer A. Platelets are the Achilles’ heel of Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2019; 144:668–670. [DOI] [PubMed] [Google Scholar]
- 115.Sereni L, Castiello MC, Di Silvestre D, et al. Lentiviral gene therapy corrects platelet phenotype and function in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2019; 144:825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiesa R, Wang J, Blok H-J, et al. Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood. 2020; 136:1201–1211. [DOI] [PubMed] [Google Scholar]
- 117.Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010; 38:3–10. [DOI] [PubMed] [Google Scholar]
- 118.Malech HL. Progress in gene therapy for chronic granulomatous disease. J Infect Dis. 1999; 179(suppl 2):S318–S325. [DOI] [PubMed] [Google Scholar]
- 119.Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010; 363:2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Malech HL, Maples PB, Whiting-Theobald N, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci U S A. 1997; 94:12133–12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grez M, Reichenbach J, Schwäble J, Seger R, Dinauer MC, Thrasher AJ. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther. 2011; 19:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim JG, Ahn HS, Kang HJ, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Blood. 2008; 112:2349–2349. [Google Scholar]
- 123.Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009; 114:2619–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brendel C, Hänseler W, Wohlgensinger V, et al. Human miR223 promoter as a novel myelo-specific promoter for chronic granulomatous disease gene therapy. Hum Gene Ther Methods. 2013; 24:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brendel C, Müller-Kuller U, Schultze-Strasser S, et al. Physiological regulation of transgene expression by a lentiviral vector containing the A2UCOE linked to a myeloid promoter. Gene Ther. 2012; 19:1018–1029. [DOI] [PubMed] [Google Scholar]
- 126.Barde I, Laurenti E, Verp S, et al. Lineage- and stage-restricted lentiviral vectors for the gene therapy of chronic granulomatous disease. Gene Ther. 2011; 18:1087–1097. [DOI] [PubMed] [Google Scholar]
- 127.Chiriaco M, Farinelli G, Capo V, et al. Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol Ther. 2014; 22:1472–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Santilli G, Almarza E, Brendel C, et al. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther. 2011; 19:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schejtman A, Aragão-Filho WC, Clare S, et al. Lentiviral gene therapy rescues p47phox chronic granulomatous disease and the ability to fight Salmonella infection in mice. Gene Ther. 2020; 7:S2–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fischer A, Lisowska-Grospierre B, Anderson DC, et al. Leukocyte adhesion deficiency: molecular basis and functional consequences. Immunodefic Rev. 1988; 1:39–54. [PubMed] [Google Scholar]
- 131.Almarza Novoa E, Kasbekar S, Thrasher AJ, et al. Leukocyte adhesion deficiency-I: a comprehensive review of all published cases. J Allergy Clin Immunol Pract. 2018; 6:1418–1420.e10. [DOI] [PubMed] [Google Scholar]
- 132.Leon-Rico D, Aldea M, Sanchez-Baltasar R, et al. Lentiviral vector-mediated correction of a mouse model of leukocyte adhesion deficiency type I. Hum Gene Ther. 2016; 27:668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hunter MJ, Tuschong LM, Fowler CJ, et al. Gene therapy of canine leukocyte adhesion deficiency using lentiviral vectors with human CD11b and CD18 promoters driving canine CD18 expression. Mol Ther. 2011; 19:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol. 2007; 120:744–750; quiz 751–752. [DOI] [PubMed] [Google Scholar]
- 135.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003; 4:330–336. [DOI] [PubMed] [Google Scholar]
- 136.Passerini L, Rossi Mel E, Sartirana C, et al. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med. 2013; 5:215ra174. [DOI] [PubMed] [Google Scholar]
- 137.Honaker Y, Hubbard N, Xiang Y, et al. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci Transl Med. 2020; 12:eaay6422. [DOI] [PubMed] [Google Scholar]
- 138.Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009; 30:656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Santoni de Sio FR, Passerini L, Valente MM, et al. Ectopic FOXP3 expression preserves primitive features of human hematopoietic stem cells while impairing functional T cell differentiation. Scientific Reports. 2017; 7:15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Masiuk KE, Laborada J, Roncarolo MG, et al. Lentiviral gene therapy in HSCs restores lineage-specific Foxp3 expression and suppresses autoimmunity in a mouse model of IPEX syndrome. Cell Stem Cell. 2019; 24:309–317.e7. [DOI] [PubMed] [Google Scholar]
- 141.Goodwin M, Lee E, Lakshmanan U, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv. 2020; 6:eaaz0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lane P, Traunecker A, Hubele S, et al. Activated human T cells express a ligand for the human B cell-associated antigen CD40 which participates in T cell-dependent activation of B lymphocytes. Eur J Immunol. 1992; 22:2573–2578. [DOI] [PubMed] [Google Scholar]
- 143.Castle BE, Kishimoto K, Stearns C, et al. Regulation of expression of the ligand for CD40 on T helper lymphocytes. J Immunol. 1993; 151:1777–1788. [PubMed] [Google Scholar]
- 144.Levy J, Espanol-Boren T, Thomas C, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997; 131(1 pt 1):47–54. [DOI] [PubMed] [Google Scholar]
- 145.de la Morena MT, Leonard D, Torgerson TR, et al. Long-term outcomes of 176 patients with X-linked hyper-IgM syndrome treated with or without hematopoietic cell transplantation. J Allergy Clin Immunol. 2017; 139:1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ferrua F, Galimberti S, Courteille V, et al. Hematopoietic stem cell transplantation for CD40 ligand deficiency: results from an EBMT/ESID-IEWP-SCETIDE-PIDTC study. J Allergy Clin Immunol. 2019; 143:2238–2253. [DOI] [PubMed] [Google Scholar]
- 147.Brown MP, Topham DJ, Sangster MY, et al. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat Med. 1998; 4:1253–1260. [DOI] [PubMed] [Google Scholar]
- 148.Sacco MG, Ungari M, Catò EM, et al. Lymphoid abnormalities in CD40 ligand transgenic mice suggest the need for tight regulation in gene therapy approaches to hyper immunoglobulin M (IgM) syndrome. Cancer Gene Ther. 2000; 7:1299–1306. [DOI] [PubMed] [Google Scholar]
- 149.Romero Z, Torres S, Cobo M, et al. A tissue-specific, activation-inducible, lentiviral vector regulated by human CD40L proximal promoter sequences. Gene Ther. 2011; 18:364–371. [DOI] [PubMed] [Google Scholar]
- 150.Hubbard N, Hagin D, Sommer K, et al. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016; 127:2513–2522. [DOI] [PubMed] [Google Scholar]
- 151.Kuo CY, Long JD, Campo-Fernandez B, et al. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 2018; 23:2606–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Booth C, Gilmour KC, Veys P, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. 2011; 117:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Katz G, Voss K, Yan TF, et al. FOXP3 renders activated human regulatory T cells resistant to restimulation-induced cell death by suppressing SAP expression. Cell Immunol. 2018; 327:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mehrle S, Frank S, Schmidt J, et al. SAP and SLAM expression in anti-CD3 activated lymphocytes correlates with cytotoxic activity. Immunol Cell Biol. 2005; 83:33–39. [DOI] [PubMed] [Google Scholar]
- 155.Hale JS, Youngblood B, Latner DR, et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity. 2013; 38:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rivat C, Booth C, Alonso-Ferrero M, et al. SAP gene transfer restores cellular and humoral immune function in a murine model of X-linked lymphoproliferative disease. Blood. 2013; 121:1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Panchal N, Houghton B, Diez B, et al. Transfer of gene-corrected T cells corrects humoral and cytotoxic defects in patients with X-linked lymphoproliferative disease. J Allergy Clin Immunol. 2018; 142:235–245.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Henter J-I, Horne A, Aricò M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007; 48:124–131. [DOI] [PubMed] [Google Scholar]
- 159.Terrell CE, Jordan MB. Mixed hematopoietic or T-cell chimerism above a minimal threshold restores perforin-dependent immune regulation in perforin-deficient mice. Blood. 2013; 122:2618–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Carmo M, Risma KA, Arumugam P, et al. Perforin gene transfer into hematopoietic stem cells improves immune dysregulation in murine models of perforin deficiency. Mol Ther. 2015; 23:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ghosh S, Carmo M, Calero-Garcia M, et al. T-cell gene therapy for perforin deficiency corrects cytotoxicity defects and prevents hemophagocytic lymphohistiocytosis manifestations. J Allergy Clin Immunol. 2018; 142:904–913.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dettmer V, Bloom K, Gross M, et al. Retroviral UNC13D gene transfer restores cytotoxic activity of T cells derived from familial hemophagocytic lymphohistiocytosis type 3 patients in vitro. Hum Gene Ther. 2019; 30:975–984. [DOI] [PubMed] [Google Scholar]
- 163.Soheili T, Durand A, Sepulveda FE, et al. Gene transfer into hematopoietic stem cells reduces HLH manifestations in a murine model of Munc13-4 deficiency. Blood Adv. 2017; 1:2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Takushi MS, Paik MNY, Fedanov MA, et al. Lentiviral gene therapy for familial hemophagocytic lymphohistiocytosis type 3, caused by UNC13D genetic defects. Hum Gene Ther. 2020; 31:626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Soheili T, Rivière J, Ricciardelli I, et al. Gene-corrected human Munc13-4-deficient CD8+ T cells can efficiently restrict EBV-driven lymphoproliferation in immunodeficient mice. Blood. 2016; 128:2859–2862. [DOI] [PubMed] [Google Scholar]
- 166.Conley ME, Rohrer J, Minegishi Y. X-linked agammaglobulinemia. Clin Rev Allergy Immunol. 2000; 19:183–204. [DOI] [PubMed] [Google Scholar]
- 167.Yu PW, Tabuchi RS, Kato RM, et al. Sustained correction of B-cell development and function in a murine model of X-linked agammaglobulinemia (XLA) using retroviral-mediated gene transfer. Blood. 2004; 104:1281–1290. [DOI] [PubMed] [Google Scholar]
- 168.Ng YY, Baert MR, Pike-Overzet K, et al. Correction of B-cell development in Btk-deficient mice using lentiviral vectors with codon-optimized human BTK. Leukemia. 2010; 24:1617–1630. [DOI] [PubMed] [Google Scholar]
- 169.Sather BD, Ryu BY, Stirling BV, et al. Development of B-lineage predominant lentiviral vectors for use in genetic therapies for B cell disorders. Mol Ther. 2011; 19:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bryan BA, Battersby A, Shillitoe BM, et al. Respiratory health and related quality of life in patients with congenital agammaglobulinemia in the northern region of the UK. J Clin Immunol. 2016; 36:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Winkelstein JA, Conley ME, James C, et al. Adults with X-linked agammaglobulinemia: impact of disease on daily lives, quality of life, educational and socioeconomic status, knowledge of inheritance, and reproductive attitudes. Medicine (Baltimore). 2008; 87:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]