

Abstract
Matrix metalloproteinases (MMPs) are synthesized by neurons and glia and released into the extracellular space, where they act as modulators of neuroplasticity and neuroinflammatory agents. Development of epilepsy (epileptogenesis) is associated with increased expression of MMPs, and therefore, they may represent potential therapeutic drug targets. Using quantitative PCR (qPCR) and immunohistochemistry, we studied the expression of MMPs and their endogenous inhibitors tissue inhibitors of metalloproteinases (TIMPs) in patients with status epilepticus (SE) or temporal lobe epilepsy (TLE) and in a rat TLE model. Furthermore, we tested the MMP2/9 inhibitor IPR-179 in the rapid-kindling rat model and in the intrahippocampal kainic acid mouse model. In both human and experimental epilepsy, MMP and TIMP expression were persistently dysregulated in the hippocampus compared with in controls. IPR-179 treatment reduced seizure severity in the rapid-kindling model and reduced the number of spontaneous seizures in the kainic acid model (during and up to 7 weeks after delivery) without side effects while improving cognitive behavior. Moreover, our data suggest that IPR-179 prevented an MMP2/9-dependent switch-off normally restraining network excitability during the activity period. Since increased MMP expression is a prominent hallmark of the human epileptogenic brain and the MMP inhibitor IPR-179 exhibits antiseizure and antiepileptogenic effects in rodent epilepsy models and attenuates seizure-induced cognitive decline, it deserves further investigation in clinical trials.
Keywords: Neuroscience, Therapeutics
Keywords: Epilepsy, Extracellular matrix, Seizures
Introduction
Currently, approximately 65 million people worldwide have epilepsy, and temporal lobe epilepsy (TLE) is the most common form in adults (1). Around 30% of patients inadequately respond to the available antiepileptic drugs. Therefore, novel therapeutic strategies are needed to control seizures. TLE is characterized by recurrent seizures, and several pathological alterations are observed in the brain of people with TLE, such as blood-brain barrier (BBB) dysfunction, aberrant synaptic plasticity, and reorganization of the extracellular matrix (ECM), all of which may contribute to epileptogenesis. Matrix metalloproteinases (MMPs), calcium-dependent endopeptidases, play an important role in the (dys)regulation of these processes (2), and therefore, inhibiting MMPs may be a potential therapeutic strategy for epilepsy.
MMPs are either membrane bound or secreted into the extracellular space. The propeptide domain covering the active site is cleaved off by active MMPs or other proteases initiating enzymatic activity. The MMP family contains at least 25 members, of which MMP2, MMP3, and MMP9 are among the most abundant in the brain (3). MMP enzymatic activity can be endogenously inhibited by tissue inhibitors of metalloproteinases (TIMPs). TIMPs are likely to be coreleased with MMPs and can noncovalently bind the active site of MMPs, making their activity transient (4). Proteinases, growth factors, cytokines, ECM, and cell adhesion molecules are among the many substrates that are proteolytically cleaved by MMPs. Therefore, MMPs are considered important players in diverse biological conditions, such as embryonic development, synaptic plasticity, and inflammation (5). Under normal physiological conditions, MMPs are involved in several processes, including the regulation of glutamatergic neurotransmission and hippocampal long-term potentiation (6, 7). MMPs have also been implicated in several pathological conditions, such as multiple sclerosis (8), Alzheimer’s disease (9, 10), and epilepsy (11). Implication of MMPs in epileptogenesis has been suggested by several studies using animal models of acute seizures or epileptogenesis (12–14). For example, it has been shown that MMP9 protein expression and enzymatic activity were increased after seizures and that MMP9 deficiency decreased sensitivity to electrically induced seizures (15). Importantly, previous studies already revealed that MMP9 mRNA and protein were upregulated within hours after kainic acid (KA) administration, which was followed several hours later by MMP9 enzymatic activation (16). Dendritic MMP9 immunoreactivity was already increased in the hippocampus 24 hours after seizure onset, peaked at 72 hours, and remained increased at 7 days after insult (15), indicating a window of opportunity for treatment.
Due to the potential key role of MMPs in a wide range of pathological conditions, several broad-spectrum inhibitors have been developed (17, 18). The majority of the first and second generation of MMP inhibitors, however, failed to succeed in clinical trials, showing a lack of specificity that led to severe side effects (17, 19). The third generation of more specific MMP inhibitors is currently being evaluated (20).
Since the expression of MMPs and TIMPs has not been systematically studied in the epileptogenic human brain or during epileptogenesis, we first investigated the expression of MMPs and TIMPs in the hippocampus of patients with drug-resistant TLE and in patients who died after status epilepticus (SE) as well as during epileptogenesis in a TLE rat model in which SE was evoked by electrical stimulation of the angular bundle. Since MMP9 and MMP2 are abundantly present in the brain and MMP9 has been suggested as playing an important role in epileptogenesis, we recently developed IPR-179 (also known as ACT-03), a BBB-permeable, selective gelatinase (MMP9 and MMP2, see Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI138332DS1) inhibitor (21). Here, we evaluate the effects of IPR-179 on seizure development in 2 animal models of epilepsy (the rapid-kindling rat model and the intrahippocampal KA mouse TLE model) and assessed whether it had antiseizure and/or antiepileptogenic effects.
Results
Increased expression of MMPs and TIMPs in the hippocampus of patients with SE or TLE.
Since MMP and TIMP expression have not been thoroughly studied in the epileptogenic human brain, we investigated the expression of MMPs and TIMPs in the hippocampus of patients who died shortly after SE (representing an acute phase of epileptogenesis) and in patients with drug-resistant TLE (representing a chronic phase of epileptogenesis).
Based on previous microarray studies in experimental TLE (22, 23), we first studied mRNA expression of MMP2, MMP3, MMP9, and MMP14 as well as TIMP1, TIMP2, TIMP3, and TIMP4 in fresh brain tissue (resected hippocampi) of patients with drug-resistant TLE. This could not be done for patients who died after SE, as freshly resected brain tissue was not available. Higher mRNA expression was observed in the hippocampus of patients with TLE for MMP2 (P < 0.0001; Figure 1A), MMP3 (P = 0.0401; Figure 1A), MMP14 (P = 0.0108; Figure 1A), TIMP2 (P = 0.0006; Figure 1B), and TIMP3 (P < 0.0001; Figure 1B) as compared with controls. The mRNA expression for MMP9 (Figure 1A), TIMP1, and TIMP4 (Figure 1B) did not change.
Figure 1. mRNA expression of MMPs and TIMPs in the human brain.

mRNA expression of MMP2, MMP3, MMP9, and MMP14 and TIMP1, TIMP2, TIMP3, and TIMP4 in hippocampi of autopsy controls (n = 13) and patients with TLE and HS (TLE – HS) (n = 14). *P < 0.05; ***P < 0.001, Mann-Whitney U test. Dots represent individual samples, while histograms indicate group mean + SEM.
Next, we studied protein expression for MMPs and TIMPs in fixed brain tissue (hippocampus) of patients with SE or TLE as well as in the hippocampus of controls. MMP2 was moderately expressed in neurons and showed no to weak expression in cells with glial morphology in control brain tissue (Supplemental Figure 1, A and B). In hippocampi from patients who died after SE, MMP2 expression was higher in neurons and glial cells (Supplemental Figure 1, C and D, and Supplemental Table 2) as compared with controls. In patients with TLE without hippocampal sclerosis (HS), MMP2 expression was higher in glia within CA1 as compared with controls (Supplemental Figure 1, E and F, and Supplemental Table 2). In patients with TLE with HS, MMP2 expression was higher in both neurons and glial cells within the dentate gyrus (DG) and in glial cells within CA1 as compared with controls (Supplemental Figure 1, G and H). The expression of MMP2 in glial cells within the hippocampus was higher in patients with TLE with HS as compared with those without HS (Supplemental Table 2).
In controls, MMP3 was moderately expressed in glial cells, while no to weak immunoreactivity was observed in neurons (Supplemental Figure 2, A and B). After SE, MMP3 expression was higher in pyramidal neurons of CA1 and in glial cells compared with those in controls (Supplemental Figure 2, C and D, and Supplemental Table 2). In patients with TLE, higher expression of MMP3 was observed in glial cells of the hippocampus as compared with controls. MMP3 expression in pyramidal cells of CA1 was higher in TLE with HS patients compared with both controls and TLE without HS patients (Supplemental Figure 2, E–H, and Supplemental Table 2).
In control hippocampal neurons, MMP9 was weakly expressed, while no expression was detected in glia (Figure 2, A and B). Higher MMP9 expression was observed in hippocampal neurons and glia of patients who died after SE (Figure 2, C and D, and Supplemental Table 2) compared with controls. In patients with TLE, expression in both neurons and glia was higher compared with that in controls (Figure 2, E–H, and Supplemental Table 2).
Figure 2. Expression of MMP9 protein in the DG and CA1 of the human hippocampus.

In controls, MMP9 was weakly expressed in neurons while it was not detected in glial cells (A and B). Higher MMP9 expression was observed in hippocampal neurons and glia of patients who died after SE (C and D) compared with controls. In patients with TLE, expression in both neurons and glia was increased compared with that in controls (E–H). Scale bar: 50 μm. Arrowheads indicate positive cells with glial morphology. Arrows indicate positive neurons. Insets depict double labeling of MMP9 in NeuN-positive cells (neurons) and GFAP-positive cells (astrocytes), but not with CR3/43-positive cells (microglia). Photographs are taken from representative cases of control (n = 8), SE (n = 5), TLE without HS (TLE – no HS) (n = 5), and TLE with HS (TLE – HS) (n = 10) patients.
MMP14 was moderately expressed in neurons and weakly expressed in glial cells of control brain tissue (Supplemental Figure 3, A and B). In patients with SE, high MMP14 expression was seen in hippocampal neurons and moderate expression in glial cells; this expression was higher compared with that in controls (Supplemental Figure 3, C and D, and Supplemental Table 2). In patients with TLE without HS, MMP14 expression was higher in glia within CA1 as compared with that in controls (Supplemental Figure 3, E and F). MMP14 expression was higher in glia within the DG and CA1 as well as in CA1 pyramidal cells in patients with TLE with HS as compared with controls (Supplemental Figure 3, G and H, and Supplemental Table 2).
TIMP1 expression was weak in both neurons and glial cells of controls (Supplemental Figure 4, A and B). Hippocampi of patients who died after SE had a moderate expression of TIMP1 in neurons and higher expression in glial cells compared with controls (Supplemental Figure 4, C and D). In patients with TLE without HS, TIMP1 expression was weak in neurons and glial cells (Supplemental Figure 4, E and F). In TLE patients with HS, however, TIMP1 expression was higher in CA1 neurons and in glial cells within the DG and CA1 (Supplemental Figure 4, G and H, and Supplemental Table 3) compared with that in controls.
TIMP2 was moderately expressed in neurons of controls, but not expressed or weakly expressed in glial cells (Supplemental Figure 5, A and B). Patients who died after SE showed higher expression in both neurons and glial cells compared with controls (Supplemental Figure 5, C and D). Patients with TLE without HS had higher expression in DG neurons and glia within the CA1 compared with controls (Supplemental Figure 5, E and F, and Supplemental Table 3). In TLE with HS patients, glial expression in DG and CA1 and neuronal expression in CA1 were higher compared with that in controls. Moreover, neuronal and glial expression in the CA1 area was higher compared with that in TLE without HS patients (Supplemental Figure 5, G and H, and Supplemental Table 3).
In controls, TIMP3 was weakly to moderately expressed in neurons, while it could not be detected in glial cells (Supplemental Figure 6, A and B). In patients who died after SE, weak to moderate TIMP3 expression was observed in neurons and higher expression was seen in glia of the DG (Supplemental Figure 6, C and D) compared with controls. In patients with TLE, TIMP3 expression was higher in both neurons and glia within the DG and compared with CA1 of controls (Supplemental Figure 6, E–H, and Supplemental Table 3).
Weak expression of TIMP4 was observed in neurons and glia of control hippocampi (Supplemental Figure 7, A and B). In patients who died after SE, higher TIMP4 expression was seen in both neurons and glia compared with controls (Supplemental Figure 7, C and D, and Supplemental Table 3). The same accounts for TLE patients; both neurons and glia showed higher expression of TIMP4 compared with that in controls, although the expression in glia within the CA1 was higher in patients with TLE with HS as compared with that in those with TLE without HS (Supplemental Figure 7, E–H, and Supplemental Table 3).
Fluorescent double labeling was performed to investigate the cellular localization of MMPs and TIMPs in patients with TLE with HS (See insets in Supplemental Figures 1–7 and Figure 2). All investigated MMPs and TIMPs colocalize with the neuronal marker NeuN. Colocalization of MMP2, MMP3, MMP9, MMP14, and TIMP1-4 was also observed with glial fibrillary acidic protein (GFAP), an astrocytic marker. Colocalization of MMP3 and TIMP3 was observed with the microglial markers CR3/43 and IBA-1, respectively.
Increased MMP expression in the electrical post-SE TLE rat model.
Since increased expression of MMPs was evident in the human epileptogenic brain in the acute phase (shortly after SE) as well as during the chronic phase (in patients with drug-resistant TLE), we also studied the expression of MMPs during epileptogenesis in the electrical post-SE rat model in the hippocampus as well as in the parahippocampal cortex (PHC). We first assessed mRNA expression at 3 different time points after SE, each corresponding to the various phases of epileptogenesis: the acute phase (1 day after SE), the latent phase (1 week after SE, absence of electrographic seizures), and the chronic phase (3.5 months after SE, recurrent spontaneous electrographic seizures are evident). Mmp2 expression was higher 1 day after induction of SE (acute phase; in the DG, CA1, and PHC; P = 0.032, 0.008, 0.019, respectively), 1 week after SE (latent phase; in the DG, CA1, and PHC; P = 0.008, 0.008, 0.010, respectively), and 3.5 months after SE (chronic phase; in the DG and CA1; P = 0.032, 0.016, respectively; Figure 3A) compared with that in controls, with a peak during the latent phase. Mmp3 expression was higher compared with that in controls during the acute phase (in the DG, CA1, and PHC, P = 0.016, 0.008, 0.029, respectively), the latent phase (in the PHC, P = 0.010), and the chronic phase (in the DG and CA1; P = 0.016, 0.016, resp.; Figure 3B). Mmp9 expression was higher during the acute phase (DG, CA1, PHC; P = 0.016, 0.008, 0.029, respectively) and the chronic phase (DG, CA1, PHC; P = 0.016, 0.008, 0.016, respectively; Figure 3C) compared with that in controls. During the latent phase, Mmp9 mRNA expression was lower than during acute and chronic phases, though still higher compared with that in controls (in the DG and CA1; P = 0.008, 0.032, respectively). Mmp14 expression was higher (P < 0.05) compared with that in controls during the acute and latent phases in all regions investigated (DG, P = 0.016, 0.032, respectively; CA1, P = 0.008, 0.008, resp.; PHC, P = 0.029, 0.010, respectively; Figure 3D).
Figure 3. mRNA expression of MMPs in rat brain.

mRNA expression of Mmp2 (A), Mmp3 (B), Mmp9 (C), and Mmp14 (D) in the DG, CA1, and PHC of post-SE rats sacrificed at different time points after SE, each corresponding to the phases of epileptogenesis: the acute phase (1 day after SE, n = 5), the latent phase (1 week after SE, absence of electrographic seizures, n = 6) and the chronic phase (3.5 months after SE, recurrent spontaneous electrographic seizures are evident, n = 5).*P < 0.05; **P < 0.01, Mann-Whitney U test. Dots represent the individual rats, while histograms indicate group mean + SEM.
Next, we studied MMP9 protein expression during epileptogenesis. In control rats, weak MMP9 protein expression was observed in neurons, while it could not be detected in glial cells (Figure 4, A and B). During the acute phase, moderate MMP9 expression was seen in neurons and the immunoreactivity score (IRS) for glial cells within CA1 was higher compared with that in controls (P = 0.018; Supplemental Table 4 and Figure 4, C and D). In the latent phase, the MMP9 IRS was higher compared with controls in neurons of the DG (P = 0.012) and glial cells of both DG and CA1 (P = 0.027, P = 0.012, resp.; Figure 4, E and F). The IRS was also higher during the chronic phase in neurons of the DG and CA1 compared with controls (chronic progressive: P = 0.002, P = 0.01, respectively; chronic nonprogressive: P = 0.02, P = 0.024, respectively; Figure 4, G–J).
Figure 4. Expression of MMP9 in the dentate gyrus and CA1 of the rat hippocampus.
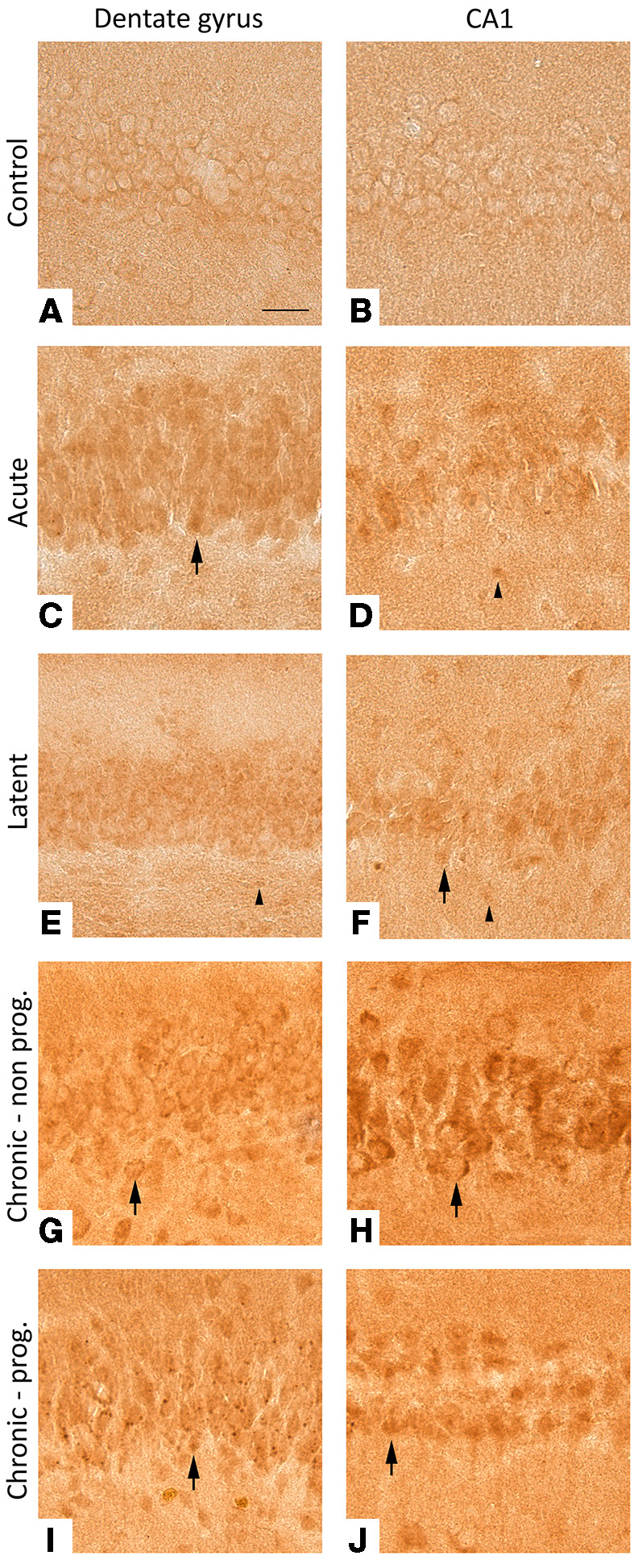
In controls rats, MMP9 was weakly expressed in neurons while it could not be detected in cells with glial morphology (A and B). In acute and latent phases, moderate MMP9 expression was seen in neurons and weak expression was seen in glia (C–F). During the chronic phase, moderate to high MMP9 expression was observed in neurons, while MMP9 could hardly be detected in cells with glial morphology (G–J). Scale bar: 50 μm. Arrowheads indicate positive cells with glial morphology. Arrows indicate positive neurons. Photographs are taken from representative cases of control (n = 5), acute (n = 5), latent (n = 6), chronic nonprogressive (n = 5), and chronic progressive (n = 6) animals.
IPR-179 reduces seizure severity in the rapid-kindling rat model.
Since we observed dysregulation of MMPs and TIMPs, including MMP2 and MMP9, in epileptogenic brain tissue, we evaluated the effects of the MMP2/MMP9 inhibitor IPR-179 on epileptogenesis. First, we studied the effects of IPR-179 on the development of epilepsy in the rapid-kindling rat model in the presence of IPR-179 (test phase) to test for the antiseizure effects of the drug as well as in the absence of IPR-179 (retest phase) to test for antiepileptogenic effects. Animals were randomly assigned to receive IPR-179 (n = 8), broad-spectrum MMP-inhibitor minocycline (n = 10), or vehicle (n = 8) intraperitoneally (i.p.) and were electrically stimulated 1 hour after injection 12 times a day on 3 consecutive days. The EEG was continuously recorded, and behavioral seizures were scored using Racine’s scale (24).
With the use of mixed-effects ordinal regression, an interaction between treatment group and stimulus number was observed for IPR-179 compared with vehicle (P = 0.017), while minocycline did not differ from vehicle. Taking a closer look at the individual stimulation numbers, IPR-179–treated rats showed less severe behavioral seizures in response to stimulation numbers 5, 8–10, 14, 23, 26, 29, and 31–34 (P = 0.033; 0.042; 0.042; 0.025; 0.005; 0.005; 0.033; 0.019; 0.025; 0.042; 0.042; 0.033, respectively) compared with vehicle-treated animals (Figure 5, A and B). Less severe behavioral seizures were seen in minocycline-treated rats compared with vehicle-treated rats in response to stimulation numbers 5 and 9 during the first day of stimulation (P = 0.028; Figure 5, A and C). During retest stimulations (in the absence of the drug and when IPR-179 could not be detected in the blood anymore), IPR-179–treated rats showed less severe behavioral scores in response to stimulation 1, 2, and 3 compared with vehicle-treated rats (P = 0.033; 0.033; 0.042, respectively), while minocycline-treated rats only showed less severe behavioral scores during stimulation 5 (P = 0.034; Figure 5, A–C).
Figure 5. The effects of IPR-179 on seizure development in the rapid-kindling rat model.

Percentages of behavioral scores according to Racine’s scale in rats treated with vehicle (A, n = 8), IPR-179 (B, n = 8), or minocycline (C, n = 10) during the test and retest phase. Mixed effects ordinal regression revealed a difference in the interaction between treatment and stimulus number was observed for IPR-179 compared with vehicle (P < 0.05). During kindling, IPR-179–treated rats (B) showed less severe behavioral seizures (stimulation 5, 8–10, 14, 23, 26, 29, 31–34; P < 0.05) compared with vehicle-treated animals (A). Less severe behavioral seizures were seen in minocycline-treated rats (C) compared with vehicle-treated rats (A) (stimulation 5 and 9; P < 0.05). In the absence of the drug, IPR-179–treated rats showed less severe behavioral scores during stimulation 1, 2, and 3 (B) compared with vehicle-treated rats (A), while minocycline-treated rats only showed less severe behavioral scores during stimulation 5 (C). IPR-179–treated rats had a tendency toward more stage 1 and fewer stage 2 and stage 5 seizures compared with vehicle-treated rats (D). During the kindling retest, there was a tendency toward more stage 1 and fewer stage 5 seizures in IPR-treated rats versus vehicle-treated rats (D). IPR-179–treated rats showed a higher relative expression of nectin-3 ratio of full protein over SPF (E). Data shown represent mean + SEMs. *P < 0.05; **P < 0.01, Mann-Whitney U test (A–C and E), 2-way RM ANOVA followed by Dunnett’s post hoc test (D).
During the initial 3 days of kindling, a trend toward more stage 1 and fewer stage 2 and stage 5 seizures was observed for IPR-179–treated rats compared with vehicle-treated rats, suggesting IPR-179 reduced seizure severity. During the kindling retest, a trend toward more stage 1 and fewer stage 5 seizures was observed for IPR-179–treated rats versus vehicle-treated rats (Figure 5D).
The weight of the body, brain, and liver of IPR-179–treated animals was not different between vehicle-treated and control animals (data not shown), and no other signs of behavioral toxicity were observed (this includes overall appearance, aggressiveness during manipulation, motor activity, and anxiety; see Supplemental Figure 11). Furthermore, morphological changes in organs were not noticed during necropsy examination of the animals. We therefore conclude that IPR-179 did not have serious adverse effects.
As a read-out for MMP9 activity, Western blot analysis was performed for nectin-3, which is targeted for degradation by MMP9, resulting in the formation of a small proteolytic fragment (SPF). The ratio of the full nectin-3 protein over the SPF was increased in IPR-179–treated kindled rats compared with vehicle-treated kindled rats, confirming that MMP9 activity was inhibited (P = 0.004; Figure 5E).
As it is known that both seizures and epilepsy can cause cell death, we were wondering whether IPR-179 treatment could ameliorate neuronal loss in the hippocampus. Using NeuN immunohistochemistry, neurons were counted in the hilus of the dorsal and ventral hippocampus (Supplemental Figure 15, A and B). A trend toward a lower neuronal density was observed in kindled, vehicle-treated rats compared with control animals. In contrast, a trend toward a higher neuronal density was observed in IPR-179–treated rats compared with vehicle-treated animals, while that was absent in minocycline-treated rats.
Antiseizure effects of IPR-179 in the intrahippocampal KA mouse model.
To evaluate the effects of IPR-179 on seizure development after SE, we used the intrahippocampal KA injection model and implanted C57BL/6J mice with electrodes for telemetric EEG recordings (Figure 6A). The implanted mice were randomly assigned to receive vehicle (n = 8) or IPR-179 (n = 10) i.p. after induction of SE. SE severity after KA injection did not differ between IPR-179 and vehicle groups in terms of the number of convulsion periods during the first hour after KA injection (P = 0.673, IPR-179: 11.2 ± 1.1; vehicle: 10.4 ± 1.5; Supplemental Figure 9A).
Figure 6. The effects of IPR-179 on seizure development in the intrahippocampal KA mouse model.

Experimental schedule with key milestones (A). Typical EEG recording of a seizure (B). Higher temporal resolution recordings of baseline (light blue), interictal (black), and ictal (red) activities outlined in B are shown (C, left). One second–long EEG epochs outlined by a red box (C, left) are further expanded (C, right). 2D representation of EEG-template library based on these epochs according to the power and coastline of recorded signals (D). Representations of epochs (C, right) are highlighted by circles of the same colors used to outline epochs. The log10 of epileptiform spike epoch counts on days 3, 9, and 17 (E), the percentage for epileptiform epochs (F), the total number of seizures (G), and average seizure durations (H) were reduced after IPR-197 treatment. Two-way RM ANOVA revealed a difference in distribution of epileptiform spike periods on day 3 (F1,14 = 5.127, P = 0.040), day 9 (F1,14 = 8.985, P = 0.010), and day 17 (F1,14 = 5.667, P = 0.032). Holm-Šidák post hoc test (E); unpaired t test was applied (H); Mann-Whitney U test due to non-Gaussian distribution of values (F and G). Data shown represent mean + SEMs; dots in histograms represent individual samples. Vehicle, n = 7; IPR-179, n = 9. *P < 0.05; **P < 0.01; ***P < 0.001.
To follow the antiseizure effects of IPR-179, EEG recordings were performed in all mice for 24 hours per day every other day over a 3-week period. Ictal and interictal periods were automatically detected using a library of manually selected individual templates and customized software, as previously described (25, 26) (Figure 6, A–D). As shown in Figure 6F, IPR-179 reduced the number of epileptiform spike epochs with ictal and interictal activities (2-way repeated measures [RM] ANOVA, P = 0.045, Figure 6F; P = 0.020, Figure 6F). Furthermore, IPR-179 attenuated the number of long epileptiform spike epochs on days 3, 7, 9, 11, 15, and 17 (P = 0.040, 0.044, 0.010, 0.030, 0.023, 0.032, respectively) and showed a similar tendency on days 5, 19, and 21 (P = 0.061, 0.076, and 0.064, respectively; Figure 6E and Supplemental Figure 9B). In mice treated with IPR-179, only 2 of 9 analyzed animals showed electrographic seizures on day 21 after KA injection. In the vehicle group, 4 of 7 mice showed seizures on that day. Analysis of EEG seizure duration and frequency revealed that IPR-179 reduced the number of seizures for the whole recording period of 3 weeks (P = 0.012, Figure 6G) and for individual days (2-way RM ANOVA, P = 0.015, Figure 6G). Similarly, the mean duration of seizures during the whole recording period (P = 0.006, Figure 6H) and the duration of seizures on individual days (2-way RM ANOVA, P = 0.019, Figure 6H) were reduced by IPR-179. To evaluate the effects of IPR-179 during activity (dark) and inactivity (light) phases of circadian rhythm, we computed the percentages of ictal plus interictal events as a function of time after IPR-179/vehicle injection (Supplemental Figure 10). There was a difference between vehicle and IPR-179 groups (2-way RM ANOVA, P = 0.025; P = 0.021; Supplemental Figure 10B), with a stronger effect of IPR-179 during the activity phase.
Impaired learning and memory functions are recognized as typical comorbidities associated with epilepsy (27). Here, the novel object recognition (Figure 7A) and labyrinth (dry maze) (Figure 7D) tests were applied to evaluate the ability of mice to memorize objects and spatial configurations, and the open-field test served to control locomotor activity and general anxiety of the mice. In the open-field test, the distance traveled by IPR-179–treated mice was not different from that of the control group both before KA injection (P = 0.938, 43.2 ± 4.3 vs. 42.7 ± 4.1 m; Supplemental Figure 11A) and 3 weeks after injection (P = 0.991, 63.3 ± 6.1 vs. 63.1 ± 8.4 m; Supplemental Figure 11D). The time that IPR-179–treated mice spent in the central zone of observation chamber was also close to that of the control group before (P = 0.744, 184.4 ± 17.5 vs. 174.4 ± 25.3 s; Supplemental Figure 11B) and 3 weeks after KA injection (P = 0.822, 131.5 ± 11.4 vs. 135.6 ± 14.7 s; Supplemental Figure 11E). Since objects were placed in the central area of the open-field chamber, a potential anxiety bias in the performance of mice in the novel object recognition test was unlikely. In that same test, before KA injection, animals in both the IPR-179 group and control groups showed a normal ability to discriminate a novel object from a familiar one (P = 0.0103; 0.0459, respectively; Figure 7B); there was no difference in discrimination ratios between groups (P = 0.834, 15.2% ± 4.0% vs. 16.5% ± 4.6%; Figure 7B). Three weeks after KA injection, vehicle-treated animals could not discriminate a novel object (exploration time: 35.3 ± 4.1 s) from a familiar one (exploration time: 34.1 ± 9.5 s; discrimination ratio: 5.5% ± 7.1%, P > 0.1; Figure 7C), while IPR-179–treated mice spent more time with the novel object (45.9 ± 10.7 s) compared with the familiar one (23.3 ± 5.0 s, P = 0.0023) and thus could reliably discriminate novel objects (discrimination ratio: 26.5% ± 6.3%, P = 0.044; Figure 7C). In the labyrinth test, IPR-179–treated animals showed a tendency to improve their performance compared with the control group on both days 2 and 3 after initial presentation to the labyrinth (Figure 7, E and F), but the difference between treatments in terms of latency, distance, or mistakes made to approach the reward area did not reach the level of significance.
Figure 7. Effects of IPR-179 on memory deficits.

Effects of IPR-179 on novel object recognition memory (A–C) and spatial navigation (D–F) deficits associated with epileptogenesis in mice (vehicle, n = 7; IPR-179, n = 9). For the novel object recognition test (A), exploration time at familiar and novel objects and discrimination ratio before (B) and 3 weeks after KA injection (C) are shown. Data represent mean + SEMs; dots in histograms represent individual samples; box-and-whisker plots display median with 5–95 percentiles. *P < 0.05; **P < 0.01; t test (Mann-Whitney U test is applied for comparing exploration time). The design of the labyrinth (dry maze) and the typical navigation routes between the start and reward areas during sessions 1, 4, and 7 (D). The latency to approach the reward area (E) and numbers of entering nonreward corners (F) are shown for all training sessions. Two-way RM ANOVA did not reveal a difference between groups in the labyrinth test.
Antiepileptogenic effects of IPR-179 in the intrahippocampal KA mouse model.
After observing the strong antiseizure effects of chronically applied IPR-179, we asked whether treatment with IPR-179 starting 6 hours after induction of SE and lasting for 7 days would be sufficient to inhibit epileptogenesis and thus persistently reduce seizures during the 7-week period after IPR-179 treatment. The time course of the experiment is shown in Figure 8A, which includes 2 days of baseline EEG recording, 2 weeks of EEG recording sessions either directly after induction of SE or after 6 weeks, and behavioral experiments between and after the EEG sessions.
Figure 8. Effects of short-term IPR-179 treatment on epileptogenesis in the intrahippocampal KA mouse model.

Experimental schedule with key milestones (A). Two-way RM ANOVA revealed the effects of IPR-179 treatment on epileptiform spike epoch counts (F1,14 = 6.974, P = 0.019) (B) and total number of seizures (F1,14 = 9.281, P = 0.009) (C). The average seizure duration during the week of IPR-179 treatment was also reduced (F1,14 = 6.413, P = 0.024) (D). Epileptiform activity as a function of the circadian hour (E, left) and the circadian phase (E, right) was averaged for all EEG-monitoring days. Two-way RM ANOVA revealed differences between groups (F1,14 = 9.117, P = 0.009 and F1,14 = 8.862, P = 0.010, respectively). The effects of short-term IPR-179 treatment on memory deficits associated with epileptogenesis in mice were evaluated with the novel object recognition test (F) and novel object location test (G). Data are represented as mean + SEMs; dots in histograms represent individual samples; box-and-whisker plots display median with 5–95 percentiles. The log10 scale is used for B, C, and D. Vehicle, n = 7; IPR-179, n = 9. Two-way RM ANOVA on ranks was applied for panels B, C, and D due to non-Gaussian distribution of values. +P < 0.1; *P < 0.05; **P < 0.01; ***P < 0.001, Holm-Šidák post hoc test. A paired t test was applied to compare exploration times within the same treatment group. A nonpaired t test was used to compare the discrimination ratios between treatment groups.
The implanted C57BL/6J mice were randomly assigned to receive vehicle (n = 7) or IPR-179 (n = 9) after induction of SE. Comparison of the SE severity after KA injection showed no difference between IPR-179 and vehicle groups in terms of number of convulsion periods during the first hour after KA injection (P = 0.763, IPR-179: 12.4 ± 0.9; vehicle: 13.0 ± 1.7; Supplemental Figure 12, A and B) and occurrence of epileptiform epochs (F1,14 = 1.905, P = 0.189; Supplemental Figure 12C). SE was followed by a high number of electrographic seizures in vehicle-treated mice (Figure 8B and Supplemental Figure 13A), whereas IPR-179 reduced the number of epileptiform spike epochs during the whole EEG recording period (2-way RM ANOVA, F1,14 = 6.974, P = 0.019). Post hoc analysis revealed that IPR-179–treated animals had fewer epileptiform epochs on several days (Figure 8B). The effect of IPR-179 was significant during the treatment (F1,14 = 25.678, P = 0.0002) and the following week (F1,14 = 5.423, P = 0.0354), and the trend was preserved even 6–7 weeks after the end of IPR-179 treatment (F1,14 = 2.309, P = 0.151; Supplemental Figure 13, B–D). Analyzing the effects of IPR-179 over separate days revealed that IPR-179 persistently reduced the occurrence of long epileptiform epochs during IPR-179 treatment, but also afterward (examples are shown for days 2, 14, and 48 in Supplemental Figure 12D). Analysis of EEG seizure frequency also revealed that IPR-179 reduced the number of seizures for the whole recording period (F1,14 = 9.281, P = 0.009, Figure 8C) as well as for most of the days (P < 0.05, Figure 8C). The mean duration of seizures was reduced during the treatment week (F1,14 = 6.413, P = 0.024, Figure 8D), but not afterwards. Analysis of IPR-179 effects during activity (dark) and inactivity (light) phases of circadian rhythm revealed a stronger effect during the activity phase (data for whole recording period: F1,14 = 8.862, P = 0.010, Figure 8E). Performing analysis on a weekly basis showed that the epileptiform activity was reduced at both phases of circadian rhythm during IPR-179 injections, but that after drug withdrawal, the effect of IPR-179 was significant only during the activity phase (Supplemental Figure 13, B–D).
Behavioral analysis revealed no effects of IPR-179 on the control locomotor activity and general anxiety (time spent in the center) of mice in the open field test in this cohort of animals 30 days after SE (Supplemental Figure 14), which was similar to what we observed for the first cohort. Five weeks after KA injection, vehicle-treated animals could not discriminate a novel object (exploration time: 26.6 ± 2.9 s) from the familiar one (exploration time: 28.4 ± 1.7 s; discrimination ratio: –4.4% ± 5.6%, P = 0.587; Figure 8F), while IPR-179–treated mice spent more time at the novel object (35.4 ± 1.7 s) compared with the familiar one (21.5 ± 3.0 s). Thus, IPR-179 could improve recognition of novel objects (discrimination ratio: 27.6% ± 7.1%, P = 0.002; Figure 8F) even 1 month after the end of treatment. As strong effects of chronic IPR-179 treatment were not observed in the labyrinth (data not shown), we decided to switch to another spatial learning test, the novel object location. Nine weeks after KA injection, vehicle-treated animals could not discriminate between an object moved to a new location (exploration time: 20.2 ± 2.8 s) and an object that was not relocated (exploration time: 20.3 ± 1.9 s; discrimination ratio: –2.6% ± 7.5%, P = 0.961; Figure 8G). In contrast, IPR-179–treated mice spent more time at the object at the new location (28.0 ± 4.3 s) compared with the object that was not relocated (18.8 ± 1.9 s) and thus could reliably discriminate novel object location (discrimination ratio: 16.6% ± 6.4%, P = 0.0489; Figure 8G). Thus, these data indicate that inhibition of MMP2/9 during the first week after SE resulted in a 2-month reduction in seizure rate and improved behavioral performance in cognitive tasks.
Discussion
We demonstrated increased mRNA and protein expression of several MMPs and TIMPs in the epileptogenic hippocampus of patients shortly after SE and in patients with drug-resistant TLE. Increased MMP expression was also observed shortly after SE (before the occurrence of spontaneous seizures) and throughout epileptogenesis in a post-SE rat model. Furthermore, treatment with the MMP inhibitor IPR-179 had antiseizure as well as antiepileptogenic effects and attenuated seizure-induced cognitive decline.
Increased MMP and TIMP expression in the human brain after SE and in TLE.
Since the expression of MMPs and TIMPs has not been systematically studied in the epileptogenic human brain, we studied their mRNA expression and showed higher expression of MMP2, MMP3, and MMP14 and TIMP2 and TIMP3 in hippocampi of patients with TLE compared with controls. We also confirmed that protein expression of these MMPs and TIMPs along with MMP9, TIMP1, and TIMP4 was higher in the hippocampus of these patients. A likely explanation for the discrepancy between gene and protein findings is that protein expression was studied at the cellular level using immunohistochemistry, while gene expression was determined in hippocampal homogenates using quantitative reverse-transcriptase PCR (RT-qPCR) analysis, in which subtle cell type–specific changes may have been obscured.
A limited number of studies have focused on the expression of MMPs and TIMPs in patients with TLE. Li et al. showed by Western blot that in a small cohort of TLE patients, both expression of the inactive propeptide and the active MMP9 protein were higher compared with that in controls (28). Higher activity of MMP2 and MMP9 were observed in the epileptogenic and perilesional area of patients with TLE (29). Additionally, in a small cohort of TLE patients, Acar and colleagues showed pronounced expression of TIMP1 and TIMP2 protein (30). In other epilepsy-related disorders, such as focal cortical dysplasia (FCD) and tuberous sclerosis complex (TSC), malformations of cortical developmental that are also characterized by epileptic episodes, increased MMP9 protein expression has been observed in the brain (11, 28). Moreover, MMP2, MMP3, and TIMP2 expression were higher in the dysplastic cortex of adult FCD patients compared with controls (11). In addition, we recently found higher expression of MMP2, MMP3, MMP9, and MMP14 and TIMP1, TIMP2, TIMP3, and TIMP4 in cortical tubers of TSC patients (31), which is associated with BBB dysfunction, as shown by albumin extravasation. In the present study, we compared TLE patients with and without HS and showed that increased protein expression for MMPs and TIMPs was most evident in patients with HS, suggesting that hippocampal pathology, characterized by neuronal cell loss and astrogliosis, is involved in the dysregulation. This is further supported by the fact that MMPs are known to be increased during inflammatory processes (32) and found to be activated by several cytokines, such as IL-1β, TNF-α, IL-6, and IFN-γ (33, 34), which are more evident in patients with TLE with HS compared with patients with TLE without HS.
We showed that the expression of MMPs and TIMPs was not only higher during the chronic epileptic phase in the human brain compared with control, but also shortly after SE. This suggests that changes in the expression of these molecules after an insult may contribute to epileptogenesis. We further investigated this in the post-SE TLE rat model.
Higher MMP expression during epileptogenesis in the rat brain after SE.
Using RT-qPCR analysis, we found higher Mmp2, Mmp3, Mmp9, and Mmp14 mRNA expression at different stages of epileptogenesis in the post-SE rat model. Furthermore, we observed higher MMP9 protein expression in both neurons and glial cells of the hippocampus during epileptogenesis.
In a large-scale microarray study, we showed that increased MMP expression occurred in a specific pattern: Mmp2 peaked during the latent phase; Mmp3 was higher during the acute, latent, and chronic phases; Mmp9 peaked during the chronic phase; and Mmp14 peaked during the acute phase in the post-SE TLE rat model (23). These coordinated expression patterns were confirmed by our present findings using RT-qPCR analysis. The involvement of the MMP proteolytic system in the development of epilepsy has been suggested in several experimental models of epilepsy (13). After KA-induced seizures, MMP3 and MMP9 expression were higher in the hippocampus (14, 35). Furthermore, Kim et al. showed, by Western blot and immunohistochemistry, an increase in MMP9 expression in mice 3 days after pilocarpine-induced SE (36). In addition, Zhang et al. observed increased activity of both MMP2 and MMP9 in the hippocampus after KA-induced SE in rats (37). More recently, it was found that total MMP activity was increased in the hippocampus 48 hours and 1 week after pilocarpine-induced SE in rats (38). We corroborated these findings, since we found increased expression of MMP2, MMP3, MMP9, and MMP14 shortly after SE (before the occurrence of spontaneous seizures). In addition, we showed that increased expression was evident during the latent and chronic phases, indicating a persistent dysregulation of MMPs throughout epileptogenesis.
MMPs are involved in several processes that play a role in the development of epilepsy, such as seizure-induced cell death, BBB dysfunction, neuroinflammation, and aberrant synaptic plasticity. For example, intracellular increase of MMP activity in the nucleus facilitates oxidative injury in neurons during early ischemia. DNA repair enzymes such as PARP-1 and XRCC1 are substrates of MMPs, and cleavage of these enzymes could contribute to the neuronal apoptosis present in the epileptic brain (39). Yang et al. have shown that MMPs target the tight junction proteins claudin-5 and occludin, which are responsible for the maintenance of restricted BBB permeability (40). In isolated brain capillaries of animals with pilocarpine-induced SE, the expression of MMP2, MMP9, TIMP1, and TIMP2 was increased, as well as the activity of MMP2 and MMP9, which was modulated via glutamate and coincided with a decreased expression of tight junction proteins (41). It is well known that MMPs can destabilize tight junction proteins and the basement membrane of blood vessels, leading to disruption of the BBB (40, 42). Several studies have confirmed the correlation between increased MMP activity and BBB dysfunction in neurological disorders, including epilepsy (43).
Taken together, our data indicate that the MMP proteolytic system is involved in epilepsy, and since higher expression of MMPs is already found in early stages after SE, it is likely to play a role in epileptogenesis. Hence, we studied the effects of small BBB-permeable MMP inhibitors in 2 animal models of epileptogenesis.
IPR-179 has antiseizure and antiepileptogenic effects.
In this study, we used the recently developed MMP inhibitor IPR-179, which has a high affinity for MMP9, but also for MMP2, although to a lesser extent (21), and tested its effects on seizure development in 2 rodent epilepsy models. In the rapid-kindling rat model, IPR-179 had antiseizure effects, since IPR-179–treated animals showed less severe behavioral seizures compared with vehicle-treated animals. After a washout period of a week, when IPR-179 could not be detected in blood anymore, IPR-179–treated animals still had less severe behavioral seizures compared with vehicle treated animals, suggesting an antiepileptogenic effect. Although our activity assays indicate that IPR-179 is a specific inhibitor for MMP2 and MMP9, we cannot fully exclude that the observed effects of IPR-179 are due to off-target effects. This needs to be evaluated in detail in future studies.
The BBB permeable broad-spectrum inhibitor minocycline was less effective in the rapid kindling model, which is in line with Arisi et al., who showed that minocycline did not affect the development or severity of spontaneous seizures after pilocarpine-induced SE (44). However, minocycline did increase the latency to stage 4 seizures, decreased the duration of stage 4 and 5 seizures in the mouse pentylenetetrazole kindling model (45), and had anticonvulsant effects in that rat amygdala kindling model (46), particularly at a high dose (50 mg/kg). This suggests that the dosage of 45 mg/kg minocycline used in our experiment was not sufficient. However, a dose of 50 mg/kg had side effects (reduced balance duration in rotarod test) in rats (46), and further increasing the dose is known to be toxic (47, 48). Therefore, we conclude that IPR-179 is more potent than minocycline, which was confirmed by in vitro data showing that IPR-179 is able to inhibit MMP9 and MMP2 in the nanomolar range (21) while minocycline has half-maximum inhibitory concentrations of around 200 μM (20). Furthermore, it was confirmed by our Western blot data, which indicated that minocycline did not efficiently inhibit MMP9, in contrast to IPR-179.
To further evaluate the antiseizure and antiepileptogenic effects (49, 50) of IPR-179, we used the intrahippocampal KA mouse model, which is one of the widely used models of chemically induced TLE in mice (51). To minimize mortality related to SE, sustained seizures were terminated by a single application of the antiepileptic compound lorazepam 6 hours after KA injection (52, 53), and KA injection was performed into the neocortex just above dorsal hippocampus (54). This approach resulted in a mortality rate of 11.4% and a high frequency of spontaneous recurrent seizures. IPR-179 treatment rapidly and persistently reduced the number and duration of spontaneous seizures. We observed a stable reduction of epileptiform activity from days 3 to 21 after KA injection, and the effect of the drug was persistent during the day, without any tendency to decline at the end of time interval between IPR-179 injections. Furthermore, IPR-179 improved performance in the novel object recognition task compared with vehicle-treated animals, indicating that it can reduce cognitive decline induced by SE. Importantly, we did not observe alterations in total body weight and morphology and weight of brain, liver, kidney, and lungs. Neither did we observe any alterations in motor activity and anxiety (Supplemental Figure 11), indicating that IPR-179 did not cause serious side effects.
Analysis of the antiepileptogenic effects of IPR-179 in the experiments when drug administration was terminated 1 week after SE revealed a reduction of epileptiform EEG activity and seizure frequency even 7 weeks after the end of treatment. Also, the performance in the novel object recognition and novel object location tests was substantially improved, i.e., IPR-179 has long-lasting antiepileptogenic effects. Interestingly, we noticed a clear circadian rhythm in epileptiform activity distribution in the control KA-injected group, with more ictal plus interictal spikes in the dark phase of the daily cycle (activity phase for mice). This is in line with another study in mice (55). In humans, the pattern of circadian distribution of seizures is brain region dependent: temporal lobe seizures maximally occur in the light phase (active phase of patients) of the daily light-dark cycle, while parietal lobe seizures occur nocturnally and out of phase with limbic seizures (56). Strikingly, IPR-179 inhibited activity-dependent upregulation in hippocampal seizures even after the termination of drug treatment, strongly pointing to the crucial role of MMP2/9 in specific epileptogenic mechanisms. These results suggest that there is a specific homeostatic mechanism normally restraining network excitability during the activity period, which is switched off in MMP2/9-dependent fashion during the first week after SE. IPR-179 is a drug that specifically prevents this switch. Acute application of IPR-179 has additional antiseizure effects on excitability, resulting in reduced seizure duration and epileptiform activity during the inactivity phase. Thus, our data provide an insight into mechanisms of epileptogenesis in addition to supporting a possible antiseizure and antiepileptogenic treatment.
Between SE and the beginning of spontaneous seizures (the latent period), reorganization of neuronal networks occurs. The latent period usually lasts weeks to years in patients (57). In the mouse model of KA-triggered epileptogenesis, this period can vary from a few hours to days and weeks (54, 58). In our experiments with KA injection, we noticed that spontaneous recurrent seizures appeared already 5 hours after SE, which is partially related to a rather low duration threshold (8 seconds) that we set for detection of seizures, although in some other studies even a 5-second threshold has been used (59, 60). Many additional factors might influence the length of the latent period, including but not limited to the animal strain used (61), the amount and routine of KA administration (62–64), the dorsal temporal coordinates as well as the subregion of hippocampus in which KA is injected (54, 58), the usage of an antiepileptic drug to stop SE (52, 62), and even the anesthesia protocol during KA injection (65).
Western blot analysis of the hippocampus from kindled rats confirmed that MMP9’s target nectin-3 (66) was less cleaved compared with that in vehicle-treated animals and thereby MMP9 activity was decreased in IPR-179–treated animals. Previous studies showed that MMP9 deficiency decreased the sensitivity to kindling (12, 15), suggesting that MMP9 has important effects on neuronal excitability. Therefore, MMP inhibitors may have a therapeutic potential in epilepsy. Interestingly, the general MMP inhibitor GM6001 attenuated induced capillary leakage ex vivo and in vitro by the prevention of tight junction protein degradation (41, 67). However, more than 50 MMP inhibitors have been tested during clinical trials, and they had low bioavailability and/or severe side effects (20). Here, we showed that IPR-179, a potent and BBB-permeable gelatinase inhibitor, has antiseizure as well as antiepileptogenic effects in 2 rodent models of epilepsy and attenuated seizure-induced cognitive decline, without severe side effects. Therefore, this recently developed MMP inhibitor deserves further investigation in clinical trials.
Methods
Study design.
The overall objective of this study was to investigate the expression of MMPs in experimental and human TLE and to examine antiseizure and antiepileptogenic effects of a recently developed MMP inhibitor, IPR-179, in 2 models of TLE. Samples sizes for TLE patients and age-matched autoptic controls used for RT-qPCR and IHC were based on the availability of material. Sample sizes for in vivo experiments were determined based on power calculation and experience from previous research using these models and are specified in the Supplemental Methods. For the rapid-kindling rat model, animals were randomly assigned to different treatment groups. Animals that did not have proper EEG signals were excluded from the study (n = 2 in vehicle and IPR-treated group). In the intrahippocampal KA mouse model, the injection order of animals was randomly assigned. Animals were allocated into 2 treatment groups to counterbalance the number of convulsions monitored during the first hour after KA application. The treatment information was blinded and decoded only after the analysis of behavioral and EEG data was completed. In the long-term antiseizure IPR-179 treatment experiment, 2 animals in the vehicle group died after KA injection (1 on the first day and 1 on the fourth day); 1 animal in the vehicle group and 1 animal in the IPR-179 group were excluded due to poor EEG signal. With the short-term antiepileptogenic IPR-179 treatment, 2 animals in the vehicle group died on the day of KA injection, and 1 animal in the IPR-179 group died on the fourth day after KA injection; 2 animals in the vehicle group and 1 animal in the IPR-179 group were excluded due to poor EEG signal; and 1 animal in the vehicle group had a damaged transmitter when it was removed from the headstage chamber. Endpoints, equal for all animals, were defined before the beginning of the experiments, clearly stated, and approved by the local ethics committees. Exact samples sizes, exclusion criteria, and use of replicates are further specified in the respective paragraphs.
Human subjects.
The cases included in this study were obtained from the archive of the Department of Neuropathology of the Amsterdam UMC and the Department of Neuropathology of the University of Bonn Medical Center. A total of 25 hippocampal specimens removed from patients undergoing surgery for drug-resistant TLE were examined. All cases were reviewed independently by 2 neuropathologists, and classification of HS was based on analysis of microscopic examination as described by the International League Against Epilepsy (68). In total, 20 patients with HS and 5 patients without HS were used. In addition, autopsy material of 5 patients who died after SE was used in this study. SE arose de novo, and pathological examination excluded structural abnormalities in the brain as well as encephalitis or meningitis. Control material was obtained during autopsy of age-matched individuals without a history of seizures or other neurological diseases (n = 16). Supplemental Table 5 summarizes the clinical characteristics of the patients and controls.
Animals.
Adult male Sprague-Dawley rats (ordered at 250 g, about 2 months of age, n = 74; Harlan Netherlands) were used for the electrical stimulation–induced SE model and the rapid kindling model. The rats were housed individually in a controlled environment (21 ± 2°C; humidity 55% ± 15%; lights on 08:00 am–8:00 pm; food and water available ad libitum).
Forty-four 3-month-old male C57BL/6J mice from the local DZNE Magdeburg colony were used for the intrahippocampal KA model. These mice were housed individually in a controlled environment (21 ± 2°C; lights on 9:00 pm–9:00 am; food and water available ad libitum). All animals were acclimatized to the local animal house for at least 7 days before experimental use.
Electrical SE rat model.
For electrode implantation, SE induction, EEG monitoring, and preparation of rat brain tissue, see Supplemental Methods.
Rapid-kindling rat model.
For kindling stimulation, see Supplemental Methods.
Drug treatment during kindling.
Rats were treated i.p. with 6 mg/kg IPR-179 (n = 8, Accure Therapeutics) once a day for 1 week; 5.0% Tween 80 in 0.9% saline was used as a vehicle, and the volume of administration was 10 mL/kg. After 1 week of washout, tail-vein blood was collected and tested for the presence of IPR-179. This dose was selected based on a previous dose-response study in which seizures were evoked using pentylenetetrazole in mice. Efficacy was not observed at 3 mg/kg. However, administration of 6 mg/kg and 18 mg/kg of IPR-179 resulted in a lower behavioral seizure severity. Since an MMP9 activity assay showed effective inhibition at a dose of 6 mg/kg, this dose was chosen for further experiments. As references, 2 groups were used: a vehicle group (5.0% Tween 80 in 0.9% saline, n = 8) and a drug group (minocycline hydrochloride, a broad-spectrum MMP inhibitor, 45 mg/kg/d, n = 10; Renno Tech).
Intrahippocampal KA mouse model.
For implantations, intrahippocampal KA injection, and EEG recording and analysis, see Supplemental Methods and Supplemental Figure 8.
Drug treatment after KA injection.
At the beginning of the dark phase (9:00 am), the body weights of animals were recorded. For the long-term antiseizure treatment, vehicle control or IPR-179 (6 mg/kg /10 mL) in 5.0% Tween 80 in 0.9% saline was injected i.p. once daily (10 mL/kg) starting 24 hours after KA injection and continuing for 30 days. For the experiment with short-term antiepileptogenic IPR-179 treatment, vehicle control of IPR-179 was i.p. administered at the same concentrations as above starting 6 hours after KA injection for 7 days.
Behavioral tests.
Immunohistochemistry.
Human brain tissue was stained using antibodies against MMP2, MMP3, MMP9, MMP12, TIMP1, TIMP2, TIMP3, and TIMP4, and post-SE rat brain tissue was stained using an antibody against MMP9. Rapid-kindling rat tissue was stained using an antibody against NeuN. For details, see Supplemental Methods.
PCR, Western blotting, and activity assays.
See Supplemental Methods.
Statistics.
Statistical analysis of immunohistochemistry data was performed using IBM SPSS Statistics 22 using the Kruskal-Wallis test followed by Mann-Whitney U tests. Data analysis of the kindling experiment was performed using RStudio (version 1.1.383) with the R package Ordinal in combination with the Cumulative Link Mixed Models (cclm2) function to perform a mixed-effects ordinal regression with random effect of animal and fixed effects of treatment group, stimulus number, and the interaction between treatment group and stimulus number. Statistical analysis of the results on intrahippocampal kainic injection model was performed with SigmaPlot 13.0. For data obtained from RM, a 2-way RM ANOVA with the Holm-Šidák or Dunnet’s post hoc test was applied. For data obtained from behavioral tests, a normality test (Shapiro-Wilk method) and an equal variance test (Brown-Forsythe method) were applied to determine which parametric test should be used (Supplemental Table 8). Thereafter, a 2-sided paired t test was applied for analysis of time in the novel object recognition and novel object location test; a 2-sided unpaired t test was applied for 2-group comparisons (discrimination ratio in the novel object recognition test, mean value of average seizure duration, travel distance, and time spent in the open-field test and convulsion number during the first hour after KA injection). For comparison of data sets with non-Gaussian distributions (mean value of percentage for epileptiform epochs and the total number of seizures, exploration time in the novel object recognition test 3 weeks after KA injection in mice with long-term IPR-179 treatment, time spent in central or peripheral area in mice with short-term treatment), the Mann-Whitney U test, Wilcoxon’s signed rank test, or 2-way RM ANOVA on ranks was applied. P < 0.05 was used to reject the null hypothesis. The t test and nonparametric tests provided very close P values for behavioral data (Supplemental Table 8), and in only 2 cases the nonparametric tests, which are supposed to have less power than parametric tests, detected a tendency, while the t test indicated a significant difference.
Study approval.
The patient tissue used for expression studies was obtained and used in accordance with the Declaration of Helsinki and the Amsterdam UMC Research Code provided by the Medical Ethics Committee. Animal experiments were conducted in accordance with the European Communities Council Directive (2010/63/EU) and approved by the local ethics committees. The Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines (https://www.nc3rs.org.uk/arrive-guidelines) were followed.
Author contributions
DWMB, A Bertran, SJ, RP, AD, and EAVV wrote the manuscript. JCB, SI, AJB, and EA provided human brain tissue. DWMB, AK, JJA, EA, and EAVV were involved in processing human brain tissue for RT-qPCR and IHC experiments. DWMB, WJW, JAG, and EAVV were involved in the planning and execution of experiments using the post-SE rat model and the rapid-kindling rat model. DWMB, A Bongaarts, and JDM performed statistical analysis of behavioral seizure data of the rapid-kindling rat model. A Bertran, JS, TT, and RP developed and provided the inhibitor IPR-179. SJ, A Bertran, OS, EC, and AD were involved in the planning and execution of experiments using the intrahippocampal KA model. SJ, A Bertran, and AD performed statistical analysis of EEG data and behavioral data of the intrahippocampal KA model. SJ, OS, and AD were involved in optimizing battery-exchangeable EEG transmitters as well as in designing the 3D-printed headstage and the 3D-printed labyrinth. Order of co–first authors was based on the following: DWMB conducted research and analyzed data of the experiments on the patient material, the post-SE rat model, and the rapid-kindling rat model. A Bertran developed and provided the inhibitor and conducted the acute experiments in the intrahippocampal KA model together with SJ. SJ also conducted the long-term experiments in the intrahippocampal KA model and analyzed all data emerging from these experiments.
Supplementary Material
Acknowledgments
We thank Leszek Kaczmarek (Nencki Institute, Warsaw, Poland) for the information about MMP9 activity assays and Matthew Walker (UCL Institute of Neurology, University College London, Queen Square, London, United Kingdom) for critically reading the manuscript. The research leading to these results has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Marie Sklodowska Curie grant agreement no. 642881 (ECMED; to A Bertran, SJ, AK, EC, EA, and AD), the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 602102 (EPITARGET; to EAVV, JAG, and EA), and the Dutch Epilepsy Foundation, project number 16-05 (to DWMB and EAVV).
Version 1. 11/03/2020
In-Press Preview
Version 2. 01/04/2021
Print issue publication
Footnotes
Conflict of interest: RP is an employee of Accure Therapeutics SL, which has the patent WO 2017/085034 A1, “Gelatinase inhibitors and use thereof” (JS, TT, and RP, authors).
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(1):e138332.https://doi.org/10.1172/JCI138332.
Contributor Information
Diede W.M. Broekaart, Email: d.w.broekaart@amsterdamumc.nl.
Alexandra Bertran, Email: sandra.bertran.junque@gmail.com.
Shaobo Jia, Email: Shaobo.Jia@dzne.de.
Anatoly Korotkov, Email: a.korotkov@amsterdamumc.nl.
Oleg Senkov, Email: neurobite@gmail.com.
Anika Bongaarts, Email: a.bongaarts@amsterdamumc.nl.
James D. Mills, Email: j.d.mills@amc.uva.nl.
Sander Idema, Email: s.idema@amsterdamumc.nl.
Elodie Chabrol, Email: elodiechabrol@gmail.com.
Jan A. Gorter, Email: j.a.gorter@uva.nl.
Eleonora Aronica, Email: e.aronica@amc.uva.nl.
Roger Prades, Email: rprades@accure.health.
Alexander Dityatev, Email: alexander.dityatev@dzne.de.
Erwin A. van Vliet, Email: E.A.vanVliet@uva.nl.
References
- 1.Blumcke I, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377(17):1648–1656. doi: 10.1056/NEJMoa1703784. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015;1623:30–38. doi: 10.1016/j.brainres.2015.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6(12):931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- 4.Pitkänen A, et al. Neural ECM and epilepsy. Prog Brain Res. 2014;214:229–262. doi: 10.1016/B978-0-444-63486-3.00011-6. [DOI] [PubMed] [Google Scholar]
- 5.Ethell IM, Ethell DW. Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res. 2007;85(13):2813–2823. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- 6.Conant K, Wang Y, Szklarczyk A, Dudak A, Mattson MP, Lim ST. Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience. 2010;166(2):508–521. doi: 10.1016/j.neuroscience.2009.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy V, et al. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci. 2006;26(7):1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Horssen J, et al. Matrix metalloproteinase-19 is highly expressed in active multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2006;32(6):585–593. doi: 10.1111/j.1365-2990.2006.00766.x. [DOI] [PubMed] [Google Scholar]
- 9.Horstmann S, et al. Matrix metalloproteinases in peripheral blood and cerebrospinal fluid in patients with Alzheimer’s disease. Int Psychogeriatr. 2010;22(6):966–972. doi: 10.1017/S1041610210000827. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8(2):205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 11.Konopka A, et al. Matrix metalloproteinase-9 (MMP-9) in human intractable epilepsy caused by focal cortical dysplasia. Epilepsy Res. 2013;104(1-2):45–58. doi: 10.1016/j.eplepsyres.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 12.Mizoguchi H, et al. Matrix metalloproteinase-9 contributes to kindled seizure development in pentylenetetrazole-treated mice by converting pro-BDNF to mature BDNF in the hippocampus. J Neurosci. 2011;31(36):12963–12971. doi: 10.1523/JNEUROSCI.3118-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizoguchi H, Yamada K. Roles of matrix metalloproteinases and their targets in epileptogenesis and seizures. Clin Psychopharmacol Neurosci. 2013;11(2):45–52. doi: 10.9758/cpn.2013.11.2.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J, Lim E, Kim Y, Li E, Park S. Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J Endocrinol. 2010;205(3):263–270. doi: 10.1677/JOE-10-0040. [DOI] [PubMed] [Google Scholar]
- 15.Wilczynski GM, et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J Cell Biol. 2008;180(5):1021–1035. doi: 10.1083/jcb.200708213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szklarczyk A, Lapinska J, Rylski M, McKay RD, Kaczmarek L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J Neurosci. 2002;22(3):920–930. doi: 10.1523/JNEUROSCI.22-03-00920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranger K, et al. Endogenous and synthetic MMP inhibitors in CNS physiopathology. Prog Brain Res. 2014;214:313–351. doi: 10.1016/B978-0-444-63486-3.00014-1. [DOI] [PubMed] [Google Scholar]
- 18.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3(3):207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 19.Fingleton B. MMPs as therapeutic targets — still a viable option? Semin Cell Dev Biol. 2008;19(1):61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13(12):904–927. doi: 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]
- 21.Bertran A, et al. Design and synthesis of selective and blood-brain barrier-permeable hydroxamate-based gelatinase inhibitors. Bioorg Chem. 2020;94:103365. doi: 10.1016/j.bioorg.2019.103365. [DOI] [PubMed] [Google Scholar]
- 22.Gorter JA, et al. Potential new anti-epileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26(43):11083–11110. doi: 10.1523/JNEUROSCI.2766-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorter JA, et al. Dynamic changes of proteases and protease inhibitors revealed by microarray analysis in CA3 and entorhinal cortex during epileptogenesis in the rat. Epilepsia. 2007;48 Suppl 5:53–64. doi: 10.1111/j.1528-1167.2007.01290.x. [DOI] [PubMed] [Google Scholar]
- 24.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 25.Wykes RC, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med. 2012;4(161):161ra152. doi: 10.1126/scitranslmed.3004190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright S, et al. Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain. 2015;138(pt 11):3159–3167. doi: 10.1093/brain/awv257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson JN, Schulz KM, Patel M. Specific alterations in the performance of learning and memory tasks in models of chemoconvulsant-induced status epilepticus. Epilepsy Res. 2014;108(6):1032–1040. doi: 10.1016/j.eplepsyres.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, et al. Increased expression of matrix metalloproteinase 9 in cortical lesions from patients with focal cortical dysplasia type IIb and tuberous sclerosis complex. Brain Res. 2012;1453:46–55. doi: 10.1016/j.brainres.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Quirico-Santos T, Nascimento Mello A, Casimiro Gomes A, de Carvalho LP, de Souza JM, Alves-Leon S. Increased metalloprotease activity in the epileptogenic lesion — lobectomy reduces metalloprotease activity urokinase-type uPAR circulating levels. Brain Res. 2013;1538:172–181. doi: 10.1016/j.brainres.2013.09.044. [DOI] [PubMed] [Google Scholar]
- 30.Acar G, Tanriover G, Acar F, Demir R. Increased expression of matrix metalloproteinase-9 in patients with temporal lobe epilepsy. Turk Neurosurg. 2015;25(5):749–756. doi: 10.5137/1019-5149.JTN.10738-14.0. [DOI] [PubMed] [Google Scholar]
- 31.Broekaart DWM, et al. Increased matrix metalloproteinases expression in tuberous sclerosis complex: modulation by microRNA 146a and 147b in vitro. Neuropathol Appl Neurobiol. 2020;46(2):142–159. doi: 10.1111/nan.12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Candelario-Jalil E, González-Falcón A, García-Cabrera M, León OS, Fiebich BL. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J Neurochem. 2007;100(4):1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- 33.Chandler S, et al. Matrix metalloproteinases, tumor necrosis factor and multiple sclerosis: an overview. J Neuroimmunol. 1997;72(2):155–161. doi: 10.1016/S0165-5728(96)00179-8. [DOI] [PubMed] [Google Scholar]
- 34.Gottschall PE, Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J Neurochem. 1995;64(4):1513–1520. doi: 10.1046/j.1471-4159.1995.64041513.x. [DOI] [PubMed] [Google Scholar]
- 35.Penkowa M, et al. Metallothionein reduces central nervous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J Neurosci Res. 2005;79(4):522–534. doi: 10.1002/jnr.20387. [DOI] [PubMed] [Google Scholar]
- 36.Kim GW, Kim HJ, Cho KJ, Kim HW, Cho YJ, Lee BI. The role of MMP-9 in integrin-mediated hippocampal cell death after pilocarpine-induced status epilepticus. Neurobiol Dis. 2009;36(1):169–180. doi: 10.1016/j.nbd.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Zhang JW, Deb S, Gottschall PE. Regional and differential expression of gelatinases in rat brain after systemic kainic acid or bicuculline administration. Eur J Neurosci. 1998;10(11):3358–3368. doi: 10.1046/j.1460-9568.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 38.Dubey D, et al. Increased metalloproteinase activity in the hippocampus following status epilepticus. Epilepsy Res. 2017;132:50–58. doi: 10.1016/j.eplepsyres.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikonomidou C. Matrix metalloproteinases and epileptogenesis. Mol Cell Pediatr. 2014;1(1):6. doi: 10.1186/s40348-014-0006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27(4):697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 41.Rempe RG, et al. Matrix Metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J Neurosci. 2018;38(18):4301–4315. doi: 10.1523/JNEUROSCI.2751-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011;44(2):130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 43.Rempe RG, Hartz AMS, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36(9):1481–1507. doi: 10.1177/0271678X16655551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arisi GM, Foresti ML, Montañez A, Shapiro LA. Minocycline ameliorates neuronal loss after pilocarpine-induced status epilepticus. J Neurol Disord Stroke. 2014;2(3):1055 [Google Scholar]
- 45.Ahmadirad N, Shojaei A, Javan M, Pourgholami MH, Mirnajafi-Zadeh J. Effect of minocycline on pentylenetetrazol-induced chemical kindled seizures in mice. Neurol Sci. 2014;35(4):571–576. doi: 10.1007/s10072-013-1552-0. [DOI] [PubMed] [Google Scholar]
- 46.Beheshti Nasr SM, Moghimi A, Mohammad-Zadeh M, Shamsizadeh A, Noorbakhsh SM. The effect of minocycline on seizures induced by amygdala kindling in rats. Seizure. 2013;22(8):670–674. doi: 10.1016/j.seizure.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Yang L, et al. Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res. 2003;74(2):278–285. doi: 10.1002/jnr.10709. [DOI] [PubMed] [Google Scholar]
- 48.Blum D, Chtarto A, Tenenbaum L, Brotchi J, Levivier M. Clinical potential of minocycline for neurodegenerative disorders. Neurobiol Dis. 2004;17(3):359–366. doi: 10.1016/j.nbd.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 49.Löscher W. The holy grail of epilepsy prevention: preclinical approaches to antiepileptogenic treatments. Neuropharmacology. 2020;167:107605. doi: 10.1016/j.neuropharm.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 50.Engel J, Pitkänen A. Biomarkers for epileptogenesis and its treatment. Neuropharmacology. 2020;167:107735. doi: 10.1016/j.neuropharm.2019.107735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jefferys J, Steinhäuser C, Bedner P. Chemically-induced TLE models: topical application. J Neurosci Methods. 2016;260:53–61. doi: 10.1016/j.jneumeth.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Brophy GM, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17(1):3–23. doi: 10.1007/s12028-012-9695-z. [DOI] [PubMed] [Google Scholar]
- 53.Kienzler-Norwood F, et al. A novel animal model of acquired human temporal lobe epilepsy based on the simultaneous administration of kainic acid and lorazepam. Epilepsia. 2017;58(2):222–230. doi: 10.1111/epi.13579. [DOI] [PubMed] [Google Scholar]
- 54.Bedner P, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. 2015;138(Pt 5):1208–1222. doi: 10.1093/brain/awv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein S, Bankstahl M, Löscher W. Inter-individual variation in the effect of antiepileptic drugs in the intrahippocampal kainate model of mesial temporal lobe epilepsy in mice. Neuropharmacology. 2015;90:53–62. doi: 10.1016/j.neuropharm.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 56.Quigg M, Straume M. Dual epileptic foci in a single patient express distinct temporal patterns dependent on limbic versus nonlimbic brain location. Ann Neurol. 2000;48(1):117–120. doi: 10.1002/1531-8249(200007)48:1<117::AID-ANA19>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 57.Reddy DS, Kuruba R. Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. Int J Mol Sci. 2013;14(9):18284–18318. doi: 10.3390/ijms140918284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gröticke I, Hoffmann K, Löscher W. Behavioral alterations in a mouse model of temporal lobe epilepsy induced by intrahippocampal injection of kainate. Exp Neurol. 2008;213(1):71–83. doi: 10.1016/j.expneurol.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 59.Lu X, Yang Y, Zhou R, Li Y, Yang Y, Wang X. Protrudin modulates seizure activity through GABAA receptor regulation. Cell Death Dis. 2019;10(12):897. doi: 10.1038/s41419-019-2118-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zub E, et al. The GR-ANXA1 pathway is a pathological player and a candidate target in epilepsy. FASEB J. 2019;33(12):13998–14009. doi: 10.1096/fj.201901596R. [DOI] [PubMed] [Google Scholar]
- 61.McKhann GM, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122(2):551–561. doi: 10.1016/S0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- 62.Bouilleret V, Ridoux V, Depaulis A, Marescaux C, Nehlig A, Le Gal La Salle G. Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience. 1999;89(3):717–729. doi: 10.1016/S0306-4522(98)00401-1. [DOI] [PubMed] [Google Scholar]
- 63.Puttachary S, et al. Immediate epileptogenesis after kainate-induced status epilepticus in C57BL/6J mice: evidence from long term continuous video-EEG telemetry. PLoS One. 2015;10(7):e0131705. doi: 10.1371/journal.pone.0131705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki F, Junier MP, Guilhem D, Sørensen JC, Onteniente B. Morphogenetic effect of kainate on adult hippocampal neurons associated with a prolonged expression of brain-derived neurotrophic factor. Neuroscience. 1995;64(3):665–674. doi: 10.1016/0306-4522(94)00463-F. [DOI] [PubMed] [Google Scholar]
- 65.Bar-Klein G, et al. Isoflurane prevents acquired epilepsy in rat models of temporal lobe epilepsy. Ann Neurol. 2016;80(6):896–908. doi: 10.1002/ana.24804. [DOI] [PubMed] [Google Scholar]
- 66.van der Kooij MA, et al. Role for MMP-9 in stress-induced downregulation of nectin-3 in hippocampal CA1 and associated behavioural alterations. Nat Commun. 2014;5:4995. doi: 10.1038/ncomms5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lischper M, Beuck S, Thanabalasundaram G, Pieper C, Galla HJ. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010;1326:114–127. doi: 10.1016/j.brainres.2010.02.054. [DOI] [PubMed] [Google Scholar]
- 68.Blümcke I, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2013;54(7):1315–1329. doi: 10.1111/epi.12220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.