

Abstract
Dysfunction of primary cilia is related to dyshomeostasis, leading to a wide range of disorders. The ventromedial hypothalamus (VMH) is known to regulate several homeostatic processes, but those modulated specifically by VMH primary cilia are not yet known. In this study, we identify VMH primary cilia as an important organelle that maintains energy and skeletal homeostasis by modulating the autonomic nervous system. We established loss-of-function models of primary cilia in the VMH by either targeting IFT88 (IFT88-KOSF-1) using steroidogenic factor 1–Cre (SF-1–Cre) or injecting an adeno-associated virus Cre (AAV-Cre) directly into the VMH. Functional impairments of VMH primary cilia were linked to decreased sympathetic activation and central leptin resistance, which led to marked obesity and bone-density accrual. Obesity was caused by hyperphagia, decreased energy expenditure, and blunted brown fat function and was also associated with insulin and leptin resistance. The effect of bone-density accrual was independent of obesity, as it was caused by decreased sympathetic tone resulting in increased osteoblastic and decreased osteoclastic activities in the IFT88-KOSF-1 and VMH primary cilia knockdown mice. Overall, our current study identifies VMH primary cilia as a critical hypothalamic organelle that maintains energy and skeletal homeostasis.
Keywords: Endocrinology, Metabolism
Keywords: Homeostasis, Leptin, Obesity

Introduction
Homeostatic regulation in the CNS is a complex process that is critical for systemic physiological balance. Therefore, identifying the brain systems integrating internal and external signals is paramount to understanding how the CNS maintains body homeostasis under diverse conditions. The hypothalamus is a vital center for body functions, including energy and skeletal homeostasis. Its functions include receiving and integrating complex peripheral signals and responding to them using humoral factors, autonomic nerves, and neuronal and behavioral components. Recently, hypothalamic primary cilia, a solitary antenna-like extension of the plasma membrane, was suggested as an important organelle for the regulation of body homeostasis (1, 2). Although these studies suggested the role of primary cilia in the arcuate nucleus of the hypothalamus (ARC) and melanocortin pathway–mediated weight regulation, the physiological roles of primary cilia in different hypothalamic nuclei remain poorly understood. Moreover, much less is known about the mechanism by which hypothalamic primary cilia mediate diverse homeostatic functions.
The ventromedial hypothalamus (VMH) is an oval-shaped hypothalamic nucleus located directly above the ARC that has been identified as a site for regulation of body weight and glucose homeostasis (3). Early studies have shown that VMH lesions induced parasympathetic tone, leading to hyperinsulinemia and obesity (4, 5). Later, electrical stimulation experiments confirmed that the VMH is involved in the regulation of not only parasympathetic, but also sympathetic nerve activity (6). Furthermore, research has shown that the VMH is engaged in other various homeostatic regulations, such as skeletal homeostasis, mood behaviors, and reproductive function (7–9). Interestingly, a recent report showed that primary cilia are expressed in the VMH and that the length of VMH primary cilia is dynamically changed by metabolic conditions in which shorter cilia are dominant in metabolically unfavorable conditions such as obesity and leptin resistance (10). Though these dynamic changes suggest that VMH primary cilia might play key roles in the maintenance of body homeostasis, there is no report yet on functional roles of VMH primary cilia in regulating body homeostasis.
The intraflagellar transport 88 (IFT88) is a core member of IFT machinery (IFT B complex) that carries ciliary building blocks along microtubules during the assembly and maintenance of the cilium (11). Previously, conditional deletion of IFT88 was used to address cell- or tissue-specific primary cilia function (1, 12). In the present study, in order to gain insight into the functional roles of primary cilia in the VMH, we generated VMH-specific primary cilia KO mice by crossing steroidogenic factor 1–Cre (SF-1–Cre) and floxed IFT88 (IFT88fl/fl) mice and investigated the homeostatic role of primary cilia in this nucleus. We also administrated bilateral adeno-associated virus Cre (AAV-Cre) in IFT88fl/fl mice, specifically targeting the VMH, to overcome any potential peripheral effect of the SF-1–Cre line.
We found that normal primary cilia function in the VMH is required for maintaining normal sympathetic nerve activity. Specific deletion of the primary cilia using the Cre-loxP system or bilateral AAV-Cre injection into the VMH caused metabolic dysregulations and abnormal skeletal homeostasis. Thus, this study suggests that VMH primary cilia play critical roles in regulating energy and skeletal homeostasis through modulation of the sympathetic nervous system (SNS).
Results
Specific deletion of primary cilia in the VMH.
To examine the physiological role of primary cilia in the VMH, we used the conditional KO system and disrupted intraflagellar transport (IFT) machinery by deleting the Ift88 gene, an essential factor for primary cilia formation, specifically in the SF-1 neurons of the VMH. Breeding IFT88fl/fl with SF-1–Cre mice resulted in mice with deficiency of primary cilia solely in those neurons (Figure 1, A and B) (13, 14). While the number of neurons expressing primary cilia and the length of the cilia were comparable in the paraventricular nucleus of the hypothalamus (PVN), dorsomedial hypothalamus (DMH), lateral hypothalamus (LH), and ARC between WT and IFT88-KOSF-1 mice, these factors were markedly reduced in the VMH of IFT88-KOSF-1 mice (Figure 1, A and B, and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI138107DS1).
Figure 1. Deletion of primary cilia in the VMH leads to obesity.

(A) Primary cilia were visualized by ACIII (green) in the VMH of WT and IFT88-KOSF-1 mice. Scale bar: 10 μm. (B) Length and percentage of the VMH cilia shown in A. **P < 0.01; ***P < 0.001, Student’s t test. (C) Left: weekly body weight of WT and IFT88-KOSF-1 mice fed on NC. Right: representative photographs of 18-week-old male mice on NC diet. *P < 0.05, 2-way ANOVA. (D) Left: fat and lean mass were measured by NMR. Right: representative DEXA image. **P < 0.01, Student’s t test. (E) Representative s.c.WAT and gWAT from WT and IFT88-KOSF-1 mice. Scale bars: 0.5 mm. (F and G) H&E staining (F) and adipocyte cell surface (G) of gWAT. Adipocyte cell surface measured by ImageJ. Scale bar: 50 μm. ***P < 0.001, 2-way ANOVA. (H) Serum leptin levels in WT and IFT88-KOSF-1 mice at 14 week of age. **P < 0.01, Student’s t test. (I) Left: schematic showing bilateral viral injection into VMH. Right: schematic for AAV2-GFP and AAV2-Cre-GFP constructs used for viral injection (upper) and validation images of primary cilia KD in VMH (lower). Scale bars: 0.1 mm (yellow); 10μm (white). (J) Weekly body weight after injection with AAV2-GFP or AAV2-Cre-GFP into the VMH. ***P < 0.001, 2-way ANOVA. (K) Left: body compositions 10 weeks after injection of AAV2-GFP or AAV2-Cre-GFP. Right: representative DEXA images. ***P < 0.001, Student’s t test. Number of animals examined is expressed in parentheses in each graph. Results are expressed as mean ± SD.
Deletion of IFT88 did not affect overall brain morphology, including brain width and length (Supplemental Figure 2, A and B). Neuronal cytoarchitecture of the VMH and expression of SF-1 in the VMH were also intact in IFT88-KOSF-1 mice (Supplemental Figure 2, C–E). In addition, the numbers and soma size of SF-1 neurons were comparable between WT and IFT88-KOSF-1 mice (Supplemental Figure 2, F–H). Together, these results indicate that the VMH of IFT88-KOSF-1 mice was intact and the neurons were viable. Next, we performed a coexpression analysis of primary cilia and SF-1 via immunohistochemistry on peripheral tissues, which express EGFP under the regulatory elements of the SF-1 gene through the BAC transgene (15). For this method, we used the antibodies somatostatin receptor 3 (SSTR3), acetylated tubulin (Ac-Tub), and adenylyl cyclase III (ACIII), which are all well-known markers of primary cilia. Consistent with results from previous studies (16–18), primary cilia in the SF-1–expressing peripheral cells, such as the pituitary gonadotrophs, adrenal cortex, ovary theca, and testis Leydig cell, were not detected (Supplemental Figure 3, A–D). The pituitary and adrenal glands, ovary, and testis were anatomically intact, and circulating corticosterone, aldosterone, and estradiol (E2) levels were also comparable between WT and IFT88-KOSF-1 mice (Supplemental Figure 4, A–H) (19). Furthermore, comparable litter size and litter per month, coupled with no differences in organ structure and steroid hormone levels, confirmed that the IFT88-KOSF-1 mice have normal hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-gonadal (HPG) axes (Supplemental Table 1). These results indicate that the deletion of IFT88 in the SF-1 neurons of the VMH using SF-1–Cre leads to specific primary cilia deletion only in those neurons, without developmental or detectable hormonal changes in the brain and the periphery.
Primary cilia in the VMH are required to maintain normal body weight.
Although the VMH has been identified as a brain site for energy homeostasis (19–21), it is not known whether VMH primary cilia play a role in the regulation of whole-body energy homeostasis. To address this question, body weight was monitored weekly in male and female IFT88-KOSF-1 mice. Normal chow–fed (NC-fed) IFT88-KOSF-1 mice displayed significantly increased body weight compared with WT littermates (Figure 1C and Supplemental Figure 5A). Similarly, IFT88-KOSF-1 male mice fed a high-fat diet (HFD) showed an obese phenotype (Supplemental Figure 6A). Dual-energy X-ray absorptiometry (DEXA) analysis revealed a distinct fat mass increment in IFT88-KOSF-1 mice (Figure 1D and Supplemental Figures 5B and 6B). In accordance with these findings, the subcutaneous white adipose tissue (s.c.WAT) and gonadal white adipose tissue (gWAT) areas of the IFT88-KOSF-1 mice were significantly bigger than those of WT mice (Figure 1E). In addition, IFT88-KOSF-1 mice exhibited increases in the number of hypertrophic adipocytes, as revealed by histological analysis (Figure 1, F and G). Serum leptin levels were also increased in the IFT88-KOSF-1 mice (Figure 1H).
To provide further evidence in support of excluding potential peripheral effects and thus confirm that the obesity in IFT88-KOSF-1 mice came directly from the absence of primary cilia in the VMH, we bilaterally injected AAV2-GFP (control) or AAV2-Cre-GFP into the VMH of IFT88fl/fl mice (Figure 1I). Bilateral Cre virus injection induced VMH-specific knockdown (KD) of primary cilia (Figure 1I). The KD mice exhibited the obese phenotype seen in IFT88-KOSF-1 mice (Figure 1, J and K), suggesting the primary cilia in the VMH are critical organelles for maintaining normal energy homeostasis.
Decreased sympathetic tone is the major cause of obesity in IFT88-KOSF-1 mice.
To determine potential factors responsible for inducing obesity in IFT88-KOSF-1 mice, we monitored several metabolic parameters using indirect calorimetry (20, 22). Body weight–matched 12-week-old WT and IFT88-KOSF-1 littermates were maintained on a NC diet and subjected to metabolic cage studies (Figure 2A). Although food intake of IFT88-KOSF-1 mice was not markedly different from that of WT littermates, their VO2, VCO2, and energy expenditure (EE) were significantly lower, especially during the dark cycle (Figure 2, B–E). Under HFD-feeding conditions, the obesity phenotype of the IFT88-KOSF-1 mice was more apparent from early ages, with a significant increase in food intake and lower VO2, VCO2, and EE (Supplemental Figure 6, C–J). The expression of hypothalamic orexigenic Agrp significantly increased and anorexigenic Pomc decreased without changes in Npy expression (Figure 2F and Supplemental Figure 6K). Physical activity was comparable between WT and IFT88-KOSF-1 mice regardless of the type of diet (Figure 2G and Supplemental Figure 6, L and M). Together, these results highlight that primary cilia in the VMH are required for maintaining normal energy balance.
Figure 2. VMH primary cilia are important for regulation of EE and BAT function.

(A) Comparable body weights of WT and IFT88-KOSF-1 littermates at 12 week of age. (B) Cumulative daily food intake on NC diet. (C–E) Temporal changes of VO2 (C), VCO2 (D), and EE (E) in WT and IFT88-KOSF-1 littermates. *P < 0.05 (red asterisks), 2-way ANOVA; **P < 0.01, Student’s t test. (F) Hypothalamic Pomc, Agrp, and Npy expression. *P < 0.05, Student’s t test. (G) Temporal changes of movement in WT and IFT88-KOSF-1 littermates. (H) Morphology and H&E staining of the iBAT from WT and IFT88-KOSF-1 littermates. Scale bar, 0.5mm for yellow and 50μm for black. (I) Mitochondria DNA contents in iBAT. **P < 0.01, Student’s t test. (J) Immunohistochemistry for UCP1 in iBAT of WT and IFT88-KOSF-1 mice. Scale bar: 100μm. (K and L) Western blot (K) and relative fold changes of protein levels (L) for UCP1. ***P < 0.001, Student’s t test. (M) Relative gene expression levels in iBAT. *P < 0.05; **P < 0.01; ***P < 0.001, Student’s t test. (N) Age-dependent serum NE levels in WT and IFT88-KOSF-1 mice. *P < 0.05; **P < 0.01, 2-way ANOVA. (O) Correlation analysis between serum NE and body weight of WT and IFT88-KOSF-1 mice. Number of animals examined is expressed parentheses in each graph. Results are expressed as mean ± SD.
Interscapular brown adipose tissue (iBAT) plays an important roles in controlling basal metabolic rate, including EE, and the VMH is a brain region that regulates sympathetic tone to the iBAT (20, 22, 23). Uncoupling protein 1 (UCP1), a key regulator of thermogenesis in iBAT, is well known to be activated by sympathetic tone (24). Therefore, we determined whether the decreased EE in IFT88-KOSF-1 mice was linked to iBAT dysfunction. We found that the VMH primary cilia KO mice had hyperplasia in the iBAT, and H&E staining confirmed higher lipid infiltration in the IFT88-KOSF-1 mice with lower mitochondria contents (Figure 2, H and I). In addition, immunohistochemistry and Western blot results showed markedly decreased UCP1 expression in IFT88-KOSF-1 mice (Figure 2, J–L). Furthermore, thermogenic or mitochondrial function–related genes, such as Ucp1, peroxisome proliferator–activated receptor γ coactivator 1α (Pgc1α), type II iodothyronine deiodinase (Dio2), and mitochondrial transcription factor A (Tfam), were downregulated, whereas genes involved in de novo lipogenesis, including PPARγ and fatty acid synthase (FASN), were upregulated (Figure 2M). These results highly suggest that VMH primary cilia play an important role in maintaining normal iBAT function, potentially through sympathetic nerve activity. To directly confirm whether VMH primary cilia are involved in sympathetic regulation, we evaluated serum norepinephrine (NE) levels of mice age from 8 to 14 weeks, when the body weight of WT and IFT88-KOSF-1 mice were comparable. The IFT88-KOSF-1 mice showed a tendency of decreased NE levels from the first 8 weeks, with significantly different levels at 12 and 14 weeks (Figure 2N). In addition, correlation analysis between NE and body weight showed that the NE levels were inversely correlated with body weight only in IFT88-KOSF-1 mice (Figure 2O), indicating that decreased sympathetic activity would be one of the factors responsible for decreased EE and iBAT function.
If decreased sympathetic activity is a major cause of the metabolic phenotypes of IFT88-KOSF-1 mice, it is possible that the introduction of sympathomimetics would countervail the metabolic disorders. To this end, we administered mice daily with isoproterenol (ISO), a β-adrenoreceptor agonist, for 2 weeks at a concentration that does not disturb body weight for both WT and IFT88-KOSF-1 mice (Supplemental Figure 7A). The marked difference of the VO2, VCO2, and EE levels induced by VMH primary cilia KO as shown in Figure 2 was normalized by the ISO treatment without changing locomotor activity (Supplemental Figure 7, B–E). These results confirm that the metabolic phenotypes shown in IFT88-KOSF-1 mice primarily come from the decreased sympathetic activity.
Primary cilia are required for normal leptin sensitivity in the VMH.
In the hypothalamus, adipokine leptin suppresses food intake and increases sympathetic tone and EE (25, 26). The effects of increased food intake and decreased sympathetic tone and EE in IFT88-KOSF-1 mice led us to hypothesize that the VMH primary cilia may be important factors for leptin action. To address this proposition, we first monitored serum leptin levels of WT and IFT88-KOSF-1 mice at 8 to 14 weeks, a period when WT and IFT88-KOSF-1 littermates show comparable body weight. Surprisingly, the IFT88-KOSF-1 mice showed markedly higher leptin levels from 12 weeks along with a strong trend of increasing leptin levels even from 8 to 10 weeks, indicating that ciliary defects in the VMH may lead to primary leptin resistance (Table 1). Next, we examined leptin sensitivity either by measuring rebound food intake or by monitoring the feeding response to leptin injection after an 18-hour fast. Results indicated that the IFT88-KOSF-1 mice showed significantly increased rebound food intake and, following leptin administration, exhibited blunted response to the injected leptin, as demonstrated by significantly increased food intake (Figure 3, A and B).
Table 1. Serum leptin level in 8- to 14-week-old WT and IFT88-KOSF-1 mice.
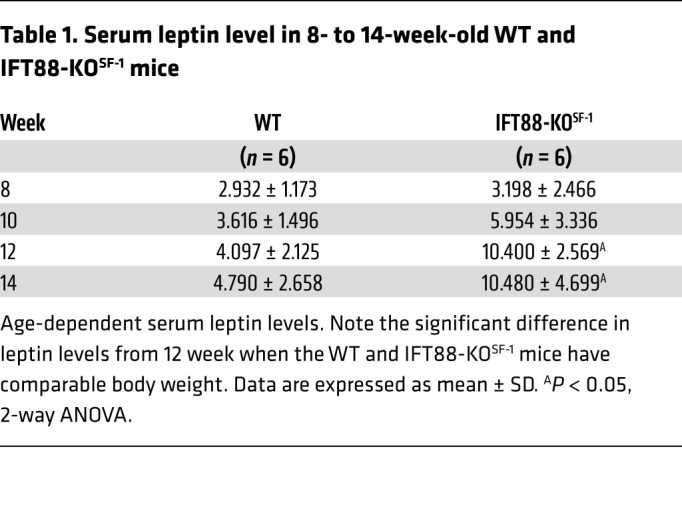
Figure 3. VMH primary cilia are required for leptin action.

(A) Cumulative rebound food intake of 10-week-old littermates after overnight fasting for 18 hours. *P < 0.05, 2-way ANOVA. (B) Rebound food intake of 12-week-old littermates after leptin administration. **P < 0.01, 2-way ANOVA. (C and D) Western blot (C) and relative fold changes of protein levels (D) for hypothalamic STAT3 in WT and IFT88-KOSF-1 mice. (E) Immunohistochemical analysis of p-STAT3 activation after leptin injection. Ten-week-old WT and IFT88-KOSF-1 mice were used for analysis. Note that p-STAT3–positive cells are specifically decreased in VMH. Scale bar: 100 μm. (F) Relative p-STAT3 expression in VMH or ARC. ***P < 0.001, Student’s t test. (G) Body weight for 12-week-old WT and IFT88-KOSF-1 littermates used for metabolic cage study shown in (H–K). (H) Cumulative food intake after leptin administration measured in metabolic chamber. **P < 0.01, 2-way ANOVA. (I–K) Temporal change of VO2 and its average (I), VCO2 and its average (J), and EE and its average (K). *P < 0.05, Student’s t test. Number of animals examined is expressed in parentheses in each graph. Data are expressed as mean ± SD.
In addition, leptin activates STAT3, and the phosphorylation of STAT3 (p-STAT3) in the brain is an indicator of leptin sensitivity. Thus, we examined the basal hypothalamic STAT3 level together with p-STAT3. Although there was no change in basal STAT3 expression, leptin-induced p-STAT3 activation was significantly impaired, specifically in the VMH where the primary cilia were removed (Figure 3, C–F). To further confirm the direct role of VMH primary cilia in leptin action, we used metabolic chambers to monitor metabolic parameters in body weight–matched WT and IFT88-KOSF-1 littermates after leptin administration (Figure 3G). The IFT88-KOSF-1 mice exhibited resistance to the reduction in food intake and increase in EE induced by leptin (Figure 3, H–K). Together, these results suggest that the primary cilia in SF-1 neurons of the VMH are necessary for normal leptin action and leptin resistance in IFT88-KOSF-1 mice is not a secondary consequence of obesity.
VMH-specific deletion of primary cilia leads to glucose and insulin intolerance.
We next determined whether the primary cilia expressed in SF-1 neurons of the VMH are involved in the regulation of glucose homeostasis and insulin sensitivity. IFT88-KOSF-1 mice exhibited significantly elevated serum glucose and insulin levels (Figure 4, A and B). Also, IFT88-KOSF-1 mice showed impaired tolerance of glucose and insulin, without significant changes in glucose-induced insulin secretion (Figure 4, C–E). These results indicate that primary cilia in the VMH are required for the regulation of normal glucose and insulin homeostasis.
Figure 4. Impaired VMH-primary cilia result in glucose and insulin intolerance.

(A and B) Blood glucose (A) and insulin (B) levels in WT and IFT88-KOSF-1 littermates at 12 week of age. *P < 0.05, Student’s t test. (C) Left: GTT between WT and IFT88-KOSF-1. Right: AUC from GTT. *P < 0.05; **P < 0.01, 2-way ANOVA. (D) Left: ITT in indicated genotypes. Right: AUC for the ITT. *P < 0.05; **P < 0.01; ***P < 0.001, 2-way ANOVA. (E) Glucose-stimulated insulin secretion (GSIS) during GTT experiments. (F) Relative expression of gluconeogenic genes in the liver. **P < 0.01; ***P < 0.001, Student’s t test. (G) H&E staining of liver from WT and IFT88-KOSF-1 mice. Scale bar: 50μm. (H) Relative expression of lipogenesis-related genes in the liver. *P < 0.05; **P < 0.01; ***P < 0.001, Student’s t test. (I) GTT (left) and its AUC (right) in viral-mediated VMH-specific IFT88 KD models. *P < 0.05, 2-way ANOVA. (J) ITT (left) and its AUC (right) in viral-mediated VMH-specific IFT88 KD models. *P < 0.05; **P < 0.01, 2-way ANOVA. Number of animals examined is expressed parentheses in each graph. Data are expressed as mean ± SD.
Gene expression analyses in liver showed increased expression of gluconeogenic glucose 6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) as well as decreased expression of glycolytic pyruvate kinase (Pklr), implying that aberrant gluconeogenesis might contribute to the high glucose and insulin insensitivity in IFT88-KOSF-1 mice (Figure 4F). In addition, liver from the IFT88-KOSF-1 mice exhibited evident steatosis along with a significant increment in hepatic lipogenic genes (Figure 4, G and H). The effects of both glucose and insulin intolerance were also distinct in AAV-Cre–mediated VMH-specific IFT88 KD mice, confirming that the VMH primary cilia play an important role for whole-body glucose homeostasis and insulin sensitivity (Figure 4, I and J).
Decreased sympathetic activity leads to high bone density in IFT88-KOSF-1 mice.
Evidence from animal and human studies demonstrated that changes in the SNS are linked to alterations in bone density. Moreover, the VMH is known to control bone homeostasis (27–30). Considering the decreased sympathetic tone and leptin resistance in IFT88-KOSF-1 mice, we thus determined whether the VMH primary cilia are important factors in maintaining skeletal homeostasis.
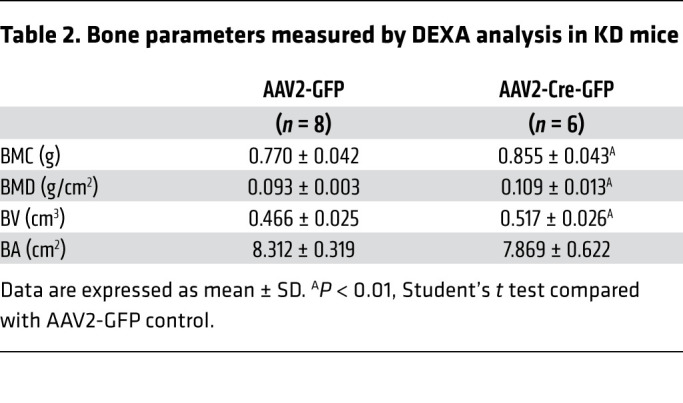
We performed age-dependent DEXA analysis to examine bone parameters. Interestingly, the long bones of the male IFT88-KOSF-1 mice showed increased bone mineral density (BMD), bone mineral content (BMC), bone volume (BV), and bone area (BA) compared with that of WT littermates, independent of the effect of obesity (Figure 5, A–D). Similarly, the female IFT88-KOSF-1 mice also exhibited increased BMD, BMC, BV, and BA (Supplemental Figure 8, A–D). The bone parameters examined were inversely correlated with serum NE only for IFT88-KOSF-1 mice (Figure 5, E–H). In addition, femur NE levels significantly decreased in IFT88-KOSF-1 mice, indicating that the decreased sympathetic activity may contribute to the increased bone density in these mice (Figure 5I). μCT analysis recapitulated the DEXA data and showed increased BMD and BV in addition to thicker trabeculae in the IFT88-KOSF-1 mice (Figure 5, J and K). To further confirm that the VMH primary cilia are directly involved in the regulation of bone mass, we analyzed viral-mediated VMH-specific IFT88 KD mice. Bilateral injection of AAV-Cre into the VMH of the IFT88fl/fl mice resulted in high bone density in the KD femur, indicating a critical role for VMH primary cilia in the regulation of bone homeostasis (Table 2 and Figure 5, L and M). Overall, these results identify normal VMH primary cilia as a requirement for bone mass homeostasis.
Figure 5. Deletion of VMH primary cilia leads to bone mass increment.

(A–D) DEXA analyses were performed at indicated ages and measured bone parameters. BMD (A), BMC (B), BV (C), and BA (D) levels of WT and IFT88-KOSF-1 mice. *P < 0.05; **P < 0.01, 2-way ANOVA. (E–H) Correlation between serum NE and BMD (E), BMC (F), BV (G), and BA (H) respectively. For correlation testing, linear regression models in R were utilized and R2 values were reported. (I) Femur NE level of WT and IFT88-KOSF-1 littermates at 16 weeks of age. ***P < 0.001, Student’s t test. (J) Representative μCT images of the femurs. The 12-week old WT and IFT88-KOSF-1 littermates were used for analyses. (K) BMD, BV/TV, BS, Tb.Th., Tb.N., and Tb.Sp. were analyzed using μCT from J. *P < 0.05; **P < 0.01, Student’s t test. (L) Representative μCT images of femurs in control (AAV2-GFP) and viral-mediated VMH-specific primary cilia KD (AAV2-Cre-GFP) mice. (M) BMD, BV/TV, BS, Tb.Th., Tb.N., and Tb.Sp. were analyzed using μCT from L. *P < 0.05, Student’s t test. Number of animals examined is expressed parentheses in each graph. Data are expressed as mean ± SD.
Table 2. Bone parameters measured by DEXA analysis in KD mice.
It has been shown that the effect of the SNS on bone homeostasis is mediated through β-adrenergic receptors (β-ARs), and several animal studies have demonstrated that β-blocker increased bone mass not only in normal, but also in ovariectomized conditions (29–31). To investigate whether chronic β-AR agonist treatment could offset the high bone mass shown in IFT88-KOSF-1 mice, we administered mice daily with ISO for 2 weeks and monitored bone parameters. The significantly increased basal BMD, BMC, BV, and BA shown in IFT88-KOSF-1 mice disappeared after ISO treatment (Figure 6, A–D). In addition, using μCT analysis, we confirmed the countervailing effect of ISO, as ISO treatment revealed no differences in BMD, bone volume/trabecular volume (BV/TV), bone surface (BS), trabecular thickness (Tb.Th.), trabecular number (Tb.N.), and trabecular separation (Tb.Sp.) between WT and IFT88-KOSF-1 mice (Figure 6, E and F). These results indicate that decreased sympathetic activity induced by primary cilia deletion in the VMH is a critical factor for the increased bone mass accrual in IFT88-KOSF-1 mice.
Figure 6. VMH primary cilia regulate bone homeostasis through SNS.

(A–D) Changes of bone parameters BMD (A), BMC (B), BV (C), and BA (D) in both WT and IFT88-KOSF-1 littermates before and after i.p. injection of ISO (15 mg/kg) for 2 weeks. Bone parameters measured using DEXA analysis. *P < 0.05; **P < 0.01, 2-way ANOVA. (E) Representative femur images of WT and IFT88-KOSF-1 littermates after ISO administration. (F) The BMD, BV/TV, BS, Tb.Th., Tb.N., and Tb.Sp. were analyzed using μCT in WT and IFT88-KOSF-1 mice given ISO. Number of animals examined is expressed in parentheses in each graph. Results are expressed as mean ± SD.
Increased osteoblastic and decreased osteoclastic activities in IFT88-KOSF-1 mice.
SNS activities have previously been linked to greater osteoclastic bone resorption by increasing osteoblastic RANKL expression (27). In addition, it has been shown that different hypothalamic nuclei influence SNS activity in distinct ways (32–34). To gain mechanistic insights into the observed bone density increment induced by primary cilia deletion in the VMH, we first examined genes involved in osteogenic- and osteoclastogenic-related signaling pathways. We found significantly decreased expression of Rankl, a key factor of osteoclast differentiation, but no change in Rank expression (Figure 7, A and B). In addition, the osteoclastic markers tartrate-resistant acid phosphatase (Trap) and dendrocyte-expressed 7 transmembrane proteins (Dc-stamp) were significantly decreased in IFT88-KOSF-1 mice (Figure 7, C and D). Consistent with these results, TRAP-positive cells were significantly reduced in IFT88-KOSF-1 femurs (Figure 7, E–G). In contrast, the osteogenic markers Sp7 transcription factor (Sp7) and alkaline phosphatase (Alp) were increased in femurs of IFT88-KOSF-1 mice (Figure 7, H and I). These results imply that the high bone mass phenotype in IFT88-KOSF-1 mice might be due to a concomitant increase in bone-formation factors and decrease in bone-resorption factors. Next, we measured bone turnover markers, including serum C-terminal telopeptide of type 1 collagen (CTX-1) and procollagen type 1 N-terminal propeptide (P1NP). We found that serum P1NP was elevated in IFT88-KOSF-1 mice, but serum CTX-1 did not change, indicating an increase in osteoblast differentiation (Figure 7, J and K). Finally, dynamic histomorphometry (calcein double labeling) analysis confirmed the increased bone formation and mineral apposition rate (MAR) in both cortical and trabecular bones of IFT88-KOSF-1 mice (Figure 8, A–E). Together, these results demonstrate that the high bone density in IFT88-KOSF-1 mice results from increased osteoblastic and decreased osteoclastic activities.
Figure 7. Bone parameter changes in VMH-specific primary cilia KO mice.

(A–D) Expression patterns of genes related to either osteoblast or osteoclast differentiation. (E) H&E (left) and TRAP chromogenic staining (right) were performed in trabecular bone of WT and IFT88-KOSF-1 littermates. Scale bars: 100 μm. (F) Number of osteoclasts per BS from E. (G) Amount of osteoclast surface per BS from E. (H and I) Relative expression of osteoblastogenic genes. (J) Serum P1NP levels of 12-week-old WT and IFT88-KOSF-1 mice. (K) Serum CTX-1 levels of 12-week-old WT and IFT88-KOSF-1 mice. Number of animals examined is expressed in parentheses in each graph. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001, Student’s t test.
Figure 8. Increment of bone growth in VMH-specific primary cilia KO mice.

(A) Calcein double labeling in cortical bone (upper) and trabecular bone (lower). Scale bar: 50 μm. (B and C), BFR (B) and MAR (C) of cortical bone of WT and IFT88-KOSF-1 mice. (D and E) BFR (D) and MAR (E) of trabecular bone of WT and IFT88-KOSF-1 mice. Ct, cortical bone; Tb, trabecular bone. Number of animals examined is expressed in parentheses in each graph. Data are expressed as mean ± SD. ***P < 0.001, Student’s t test.
Discussion
Deletion of VMH primary cilia by targeting the Ift88 gene using either SF-1–Cre or bilateral AAV-Cre injection resulted in metabolic dysregulations such as obesity, insulin/leptin resistance, impaired brown fat function, and increased fat mass. In addition, impaired primary cilia function in the VMH led to increased osteoblastic and decreased osteoclastic activities, resulting in high bone density. The VMH is known to regulate sympathetic activity, and decreased sympathetic activity is linked to reduction of metabolic rate and increase in bone density (6). Thus, we determined whether the VMH primary cilia play a critical role in controlling sympathetic activity. The IFT88-KOSF-1 mice exhibited several indications of impaired sympathetic activity, including decreased EE, blunted brown fat function, and leptin resistance. They also demonstrated markedly lower levels of serum NE independent of changes in body weight. These results indicate that the VMH primary cilia are critical for the regulation of normal sympathetic activity. Therefore, our current study identifies primary cilia expressed in the VMH as a key component of normal energy and skeletal homeostasis through the regulation of sympathetic nerve activity.
We found that mice lacking the Ift88 gene from SF-1 neurons of the VMH developed obesity with a calorimetric decrease in EE. The decreased EE in IFT88-KOSF-1 mice appeared to be a primary consequence of reduced sympathetic outflow rather than secondary to the obesity effect, considering that EE was already decreased when the mice were at comparable body weight (Figure 2, A and C–E). In addition to the decreased EE, the IFT88-KOSF-1 mice showed significantly increased Agrp and decreased Pomc expression in the hypothalamus; these genes potentially affect food intake (Figure 2F and Supplemental Figure 6K). Concomitantly, hyperphagia in the IFT88-KOSF-1 mice was evident in the HFD condition (Supplemental Figure 6C). As a contrast to the HFD setting, NC-fed IFT88-KOSF-1 mice showed comparable food intake with the WT littermates. Therefore, the decreased EE might play an important role in leading metabolic disorders of the IFT88-KOSF-1 mice, as the KO mice demonstrated an obese phenotype even with comparable NC consumption (Figure 1C and Figure 2B).
In addition to metabolic disorders, impaired primary cilia function in the VMH resulted in high bone density in the present study. Previously, it was suggested that circulating leptin that acted on bone cells stimulated bone growth and formation (35, 36). On the other hand, it was believed that leptin action in the CNS inhibits bone formation through activation of the SNS (37). While these findings suggest that leptin may regulate bone density differently either in the periphery or the brain, experimental approaches using various doses of leptin or supra physiological ob/ob and db/db mice may potentially influence other hormones affecting skeletal homeostasis (38–41). Indeed, systemic injection of leptin in ob/ob mice concomitantly altered steroid and pancreatic hormones, both factors influencing bone homeostasis (40). Therefore, careful attention must be paid to understanding leptin’s role in skeletal physiology in particular contexts.
The leptin receptor is widely expressed in several brain regions, including the hypothalamus and brain stem (42). Thus, it would be interesting to identify leptin’s region-specific roles in regulation of skeletal homeostasis. Unfortunately, however, early studies investigating brain region–specific functions of leptin are primarily focused on energy homeostasis (19, 43). As a result, only limited information is available for leptin’s brain site–specific role in the regulation of bone homeostasis. Specific leptin receptor deletion in the ARC or VMH did not affect bone density (19, 43). However, it was suggested that the leptin receptor expressed in Tph2-expressing neurons plays an important role in regulation of cancellous bone homeostasis, although some discrepancies exist in energy homeostasis regulation (44, 45). Despite these findings, mice chemically lesioned in the VMH using gold thioglucose (GTG) exhibited loss of VMH neurons and showed high bone-density phenotype, highly suggesting a possibility that bone homeostatic regulation through the VMH might require other components in addition to leptin, such as proper neuronal communication between the VMH and its targets (30). In the present study, the VMH-specific primary cilia KO mice showed primary leptin resistance, but this alone might not be enough to induce bone remodeling. Therefore, the high bone density observed in the IFT88-KOSF-1 mice emphasizes combined results of both leptin resistance and SNS decrement. Although we showed strong evidence of decreased sympathetic tone in IFT88-KOSF-1 mice, further examination of the direct mechanism mediating the primary cilia effects on skeletal homeostasis through neuronal elements would provide new insight into the function of neuronal primary cilia. In addition, it would also be interesting to investigate whether primary cilia located in extra VMH regions including the ARC, LH, or PVH are involved in regulation of skeletal homeostasis as well.
SF-1, a well-known marker of the VMH, is also expressed in pituitary gonadotrophs and the major steroidogenic tissues, such as the adrenal cortex and testicular Leydig cells (46). Surprisingly, primary cilia are not exhibited in SF-1–expressing endocrine cells, but are expressed in the pituitary somatotrophs and lactotrophs, adrenal capsule and medulla, and immature testis cells, implying that the primary cilia either would not or would minimally be affected by SF-1–Cre in the periphery organs (Supplemental Figure 3) (16–18). Intact structural integrities of the pituitary and adrenal glands, as well as the gonads, together with the functional HPA and HPG axes of IFT88-KOSF-1 mice support this notion (Supplemental Figure 4 and Supplemental Table 1). Consequently, the metabolic and skeletal phenotypes of IFT88-KOSF-1 mice might be the result of VMH primary cilia deletion.
To further rule out potential influence from peripheral primary cilia and to verify that the IFT88-KOSF-1 phenotypes indeed result from the VMH primary cilia, we injected AAV-Cre virus directly into the VMH of IFT88fl/fl mice and established VMH-specific primary cilia KD models, with the purpose of investigating energy and bone homeostasis (Figure 1, I–K, and Figure 4, I and J). Interestingly, the AAV-mediated KD mice phenocopied the metabolic and skeletal effects shown in IFT88-KOSF-1 mice (Figure 1 and Figure 5). These results confirm that the VMH primary cilia, but not the periphery primary cilia exhibited in SF-1–expressing cells, are responsible for the observed phenotypes, possibly through the regulation of SNS activity. One consideration is that the Cre virus will also be expressed in non–SF-1 neurons of the VMH. Therefore, it would be interesting to separate which neuronal populations in the VMH are mainly involved in the modulation of energy and skeletal homeostasis.
One interesting finding in this study is that the VMH primary cilia are necessary for proper leptin action. The relationship between leptin action and the functional involvement of primary cilia has long been questioned. Mechanistically, it was suggested that Bbs1, a component of the ciliary basal body, directly binds to the leptin receptor and is required for proper leptin receptor trafficking (47). Interestingly, leptin resistance developed independently of obesity only when Bbs1 was deleted in the leptin receptor–expressing neurons, suggesting that Bbs1 is required for proper leptin action (12). However, CAGG-CreER–mediated IFT88 deletion or Bbs4-KO animals showed leptin resistance secondary to obesity. Based on these results, the authors suggested that leptin signaling is not directly affected by both cilia loss (IFT88 KO) and defective cilia signaling (Bbs4 KO) (48). Later, the development of minimal leptin resistance was reported when cilia were removed from leptin receptor–expressing neurons (12). Together with our current results, these data suggest that the impact of primary cilia on leptin action may differ from neuron to neuron (Figure 3). Therefore, a future goal may be to identify distinct neuronal populations involved in the maintenance of whole-body leptin homeostasis.
Another intriguing finding in our study is that the VMH primary cilia are required for normal sympathetic nerve activity. It has been shown that VMH lesions affect sympathetic nervous activity and microinjection of glucose into the VMH increases the iBAT activity through the SNS (49, 50). In addition, genetic studies have also indicated that intact gene expression in the VMH is required for proper SNS regulation (13, 19, 22). These studies highlight that normal VMH function is necessary for intact SNS regulation. A key question regarding sympathetic activity regulation by the VMH primary cilia is the role that primary cilia play in this regulation. Since we ablated the signaling hub mediated by VMH-primary cilia, it is possible that gene expression might be changed in the neurons with deleted primary cilia. To investigate this possibility, we examined gene expression levels of SF-1, ObRb, and 5-HT2c, known factors involved in regulation of SNS activity (Supplemental Figure 9) (19, 22, 44). Although we could not detect a difference in expression levels of those genes, it is still possible to change transcriptomes in primary cilia–deleted neurons, which may be involved in the regulation of SNS activity. Approaches such as whole RNA-Seq after FACS sorting of labeled SF-1 neurons could be used to investigate the mechanism behind SNS modulation. In addition to the changing of transcriptomes, another possible regulator of SNS activity is the changing character of SF-1 neurons, for they lead to impairment in neuronal communication to the sympathetic preganglionic neurons. Indeed, Guo et. al. reported that primary cilia are required for proper neuronal circuit formation, especially those that affect interneuron morphology, connectivity, and synaptic integration. This suggests that cilia deletion in SF-1 neurons may influence local neuronal circuits (51). Thus, future studies could also consider whether neuronal primary cilia are involved in the formation of neural circuits specific to the SNS in order to determine the functional role of primary cilia in the regulation of SNS activity.
In summary, our study establishes the roles of VMH primary cilia in the regulation of energy and skeletal homeostasis through sympathetic nerve activity. It provides evidence that VMH ciliary signaling directly modulates metabolic parameters, such as leptin sensitivity, feeding behaviors, and brown fat function. We report that primary cilia are involved in bone homeostasis both locally and through neuronal signaling. Thus, proper VMH primary cilia are required and indispensable for energy and skeletal homeostasis.
Methods
All animals were maintained in a specific pathogen–free (SPF) facility. All mouse procedures were maintained on an inbred C57BL/6J genetic background and were housed under controlled room temperature (RT) (22–24°C) with a 12-hour light/12-hour dark cycle (light on at 8:00 am), with free access to either NC (PicoLab Rodent Diet 20, 0007688; 13% from fat, 3.03 kcal·g-1) or HFD (Research Diets, #D12492; 60% from fat, 5.24 kcal·g-1) and water.
Mouse models.
To generate experimental mice, IFT88-KOSF-1 male mice heterozygous for the SF-1–Cre transgene and homozygous for the floxed Ift88 allele (Ift88fl/fl) were crossed with female mice (Jackson Laboratory stock no. 022409) homozygous for the floxed Ift88 allele (Ift88fl/fl). The littermate mice homozygous for the floxed Ift88 allele (Ift88fl/fl) served as controls (WT).
The primers used for genotyping PCR were as follows: SF-1–Cre: forward (F), 5′-CTGAGCTGCAGCGCAGGGACAT-3′, reverse (R), 5′-TGCGAACCTCATCACTCGTTGCAT-3′; WT: F, 5′-GGTCAGCCTAATTAGCTCTGTCAT-3′, R, 5′-GATCTCCAGCTCCTCCTCTGTCT-3′; Ift88 floxed: F, 5′-GACCACCTTTTTAGCCTCCTG-3′, R, 5′-AGGGAAGGGACTTAGGAATGA-3′.
To generate VMH-specific primary cilia KD mice, 10- to 12-week old Ift88fl/fl male mice were used for stereotaxic surgery. Anesthetics were prepared by mixing Zoletil (30 mg/kg) and Rompun (10 mg/kg) in isotonic saline. Then the mixture was i.p. injected into the mice at 10 μL/g of body weight. Animals were placed into a stereotaxic apparatus (Kopf 1900) under sterile conditions. A small incision was made to expose the skull and drilled at the injection site. To target the injection site, a 3D MRI coordinate system was used as a reference. The coordinates for VMH were anterior-posterior: –4.80 mm, mediolateral: ±0.50 mm, and dorsal-ventral: –4.83 mm (500 nL injection). Viral constructs were injected at a rate of 50 nL/min for 10 minutes, and the injector remained in place for an additional 5 minutes before removal. After surgery, mice were placed on a heating pad for recovery. AAV expressing Cre-GFP (catalog 105545, AAV2-Cre-GFP) and GFP (catalog 105530, AAV2-GFP) were purchased from Addgene.
Validation of mouse models.
To validate the specific deletion or KD of primary cilia in the VMH, mice were perfused and their brains were collected. Mice were i.p. injected with Avertin (tribromoethanol, 25 mg/kg of body weight) for anesthesia, then transcardially perfused with 20 mL of PBS (pH7.4) and fixed with 20 mL of 10% neutral buffered formalin. Brains were dissected out and post-fixed in 10% neutral buffered formalin for 2 hours at RT, then were transferred to 20% of sucrose in PBS overnight at 4°C for cryoprotection. Brains were sliced into 20 μm sections using a sliding microtome (Leica SM2010, Leica Biosystems).
To visualize neuronal primary cilia, immunohistochemistry was performed using the free-floating method. Briefly, brain sections were washed in PBS and permeated with 0.25% Triton X-100 in PBS (v/v) for 30 minutes. After blocking with 3% goat serum prepared in PBS containing 0.25% Triton X-100 (PBT) for 1 hour at RT, the brain sections were incubated for 36 hours at 4°C with primary antibodies in PBT-azide containing 3% (v/v) goat serum. Then sections were rinsed with PBS 3 times and incubated for 2 hours in secondary antibodies diluted in PBT containing 3% (v/v) goat serum at RT. Afterwards, sections were rinsed and mounted on glass slides using mounting medium with DAPI (catalog H-1500, Vector Laboratories) and visualized by a confocal laser microscope (LSM700, Carl Zeiss AG). Further analysis was done using ZEN blue software (Zeiss) and ImageJ (NIH).
To evaluate any effects of peripheral primary cilia by SF-1–Cre, mice were perfused and the tissues known to express SF-1, including pituitary and adrenal glands, testis, and ovary, were collected. Post-fixed tissue samples were then paraffin embedded and cut into 4 to 5 μm slices. To visualize peripheral primary cilia, a regular immunohistochemistry method was performed using the paraffin-embedded tissue samples. After deparaffinization of the tissue samples, the slides were brought to a boil in 10 mM sodium citrate buffer (pH 6.0) for 10 minutes. Then they were washed in PBS, permeated with PBT for 30 minutes, and blocked by incubating the tissue sections with 3% BSA in PBT for 1 hour at RT. Primary antibodies diluted in PBT-azide containing 3% BSA were added, then slides were incubated overnight at 4°C. The following day, tissue samples were rinsed with PBS and incubated for 2 hours in secondary antibodies. Then the sections were rinsed and a coverslip was placed on the tissue sections using mounting medium with DAPI. Slides were visualized using a confocal laser microscope (LSM700), and analysis was processed using the ZEN blue software (Zeiss).
The primary antibodies used were as follows rabbit anti-ACIII (catalog sc-588, 1:2000, Santa Cruz Biotechnology Inc.), goat anti-SSTR3 (catalog sc-11617, 1:500, Santa Cruz Biotechnology Inc.), and mouse anti-Ac-Tub (acetylated α-tubulin, catalog T7451, 1:500, MilliporeSigma) for primary cilia and chicken anti-GFP (catalog GFP-1020, 1:10000, AvesLab) for either viral infection or SF-1–EGFP–positive cells.
Secondary antibodies used were as follows: Alexa Fluor 488–conjugated goat anti-rabbit antibody (catalog A21206, 1:1000), Alexa Fluor 594–conjugated goat anti-rabbit antibody (catalog A11012, 1:1000), Alexa Fluor 594–conjugated goat anti-mouse antibody (catalog A11005, 1:1000), Alexa Fluor 594–conjugated donkey anti-goat antibody (catalog A11058, 1:1000) from Invitrogen, and Alexa Fluor 488–conjugated donkey anti-chicken IgY†† (catalog 703-545-155, 1:5000, Jackson Immune Research).
Cresyl violet staining (Nissl staining) was performed to examine overall neuron structure. Briefly, brain sections were embedded in Superfrost Plus microscope slides (Fisher Scientific), and sections were left to dry at RT. After rinsing the sections in distilled water, slides were dipped in 0.1% cresyl violet solution for about 10 minutes. Then slides were rinsed in distilled water and differentiated in 90% and 95% ethanol for 3 minutes each. Afterwards, slides were dehydrated in absolute ethanol 2 times for 3 minutes each and were dipped in 100% xylene 3 times for 5 minutes each. Finally, slides were mounted with mounting media and checked under the microscope.
Body weight and compositions.
The body weights of WT and IFT88-KOSF-1 mice were monitored weekly from weaning (4 weeks old). For the HFD study, WT and IFT88-KOSF-1 littermates were maintained on the regular chow diet until they were 8 weeks old, then were switched to HFD for an additional 12 to 16 weeks. Body weight measurement for AAV-injected KD mice started from 1 week after stereotaxic surgery. Body compositions of all mice were analyzed by nuclear magnetic resonance (NMR) (LF90 Minispec, Bruker Corp.).
Metabolic cage study.
For metabolic cage studies, weight-matched 12-week-old WT (30.10 ± 1.11 g) and IFT88-KOSF-1 (31.30 ± 1.33 g) male littermates fed NC were used and 16-week-old WT (46.63.10 ± 1.78 g) and IFT88-KOSF-1 (53.40 ± 2.21 g) male littermates fed HFD were used. Metabolic rates were assessed with an indirect calorimetry system (CaloSys Calorimetry System, TSE Systems Inc.), as previously described (20, 24). For environmental acclimation, first, the experimental mice were housed 5 days in metabolic cages individually and maintained in the same room where the metabolic analyses were performed. Then the mice were individually housed in the metabolic chambers and acclimated for 48 hours. After acclimation in the chamber, food intake, oxygen consumption (VO2), carbon dioxide production (VCO2), heat generation, and movement were measured and the relationship between metabolic rate and body mass was normalized using lean body mass. Diet (NC or HFD) and water were available ad libitum unless otherwise indicated.
To measure leptin sensitivity using metabolic cages, body weight matched (28.54 ± 1.39 g for WT and 29.33 ± 1.10 g for IFT88-KOSF-1) 12-week old mice were used. After 3 days of regular cage studies, chow was removed at 6:00 pm, and mice were fasted for 1 day. The following day, experimental mice were given leptin (i.p., 5 mg/kg of body weight) with food; then metabolic parameters, including food intake, VO2, VCO2, heat generation, and movement, were monitored during the experimental period.
To assess the metabolic effects of ISO (I6504, MilliporeSigma), an adrenoreceptor agonist, 16-week old WT and IFT88-KOSF-1 mice were used and were given ISO (15 mg/kg of body weight, daily i.p. injection) for 2 weeks.
Rebound food intake.
For the rebound feeding experiments, 10- to 12- week-old mice were fasted overnight for 18 hours with water provided ad libitum. The following day, mice were given the same amount of food; then food intake and body weight were recorded at the indicated time points. To assess leptin sensitivity, 12-week-old WT and IFT88-KOSF-1 mice were given either saline or leptin (5 mg/kg of body weight) via i.p. injection after an overnight fast. Refeeding food intake and body weight were recorded at the indicated time points.
Hypothalamic p-STAT3 staining.
For p-STAT3 immunohistochemistry, weight-matched 10-week-old WT and IFT88-KOSF-1 mice fasted for 24 hours were given leptin (5 mg/kg of body weight) or saline. After 1 hour, animals were transcardially perfused with 10% neutral buffered formalin. Rabbit anti–p-STAT3 antibody (catalog 9131, 1:1000, Cell Signaling Technology) was used and regular immunohistochemistry was followed, as described elsewhere.
Serum analyses.
To measure fed glucose levels, mice were acclimated for 2 hours in the experimental area before measurement. Then a small drop of blood was obtained from tail nick and measured with a glucometer (Contour TS, Ascensia Diabetes Care).
For insulin and leptin measurement, blood was collected from the tail-nick after 2 hours of acclimation in the experiment area. Serum was analyzed with specific ELISA kits (Morinaga Institute of Biological Science) in accordance with the manufacturer’s instructions.
For corticosterone, aldosterone, and E2 measurements, mice were either housed in a group or individually, and blood samples were collected at 2:00 pm. An acclimation period of 3 days was given to the mice caged individually. Corticosterone and aldosterone levels were measured using an ELISA kit obtained from Abcam (ab108821 and ab136933, respectively). Plasma E2 was measured using ELISA kits (3830, BioVision Inc.).
For serum P1NP and CTX-1 measurements, blood samples were collected from 12-week-old WT and IFT88-KOSF-1 male littermates at 2:00 pm. P1NP and CTX-1 were measured using commercial kits (AC-33F1 for P1NP, and AC-06F1 for CTX-1, Immunodiagnostic Systems) in accordance with the manufacturer’s instructions.
NE measurements.
For serum NE, blood was collected from the tail of 8- to 14-week old male mice at 2-week intervals. For femur NE, male mice were sacrificed and right femurs were dissected. Frozen femur powder was resuspended in glutathione tissue extraction buffer (0.01N HCl, 0.15 mM EDTA, 0.1% reduced l-glutathione) and neutralized with 1.0M Tris pH 8.0 (1/10 vol) prior to NE ELISA. Extracted serum and femur NE were acylated and then converted enzymatically before being quantitatively determined. Levels were determined by a competitive enzyme immunoassay method using commercial ELISA kits (BA-E-5200, Labor Diagnostika Nord GmbH & Co.) following the manufacturer’s instructions. Aliquot of femur suspended in extraction buffer was used to quantify protein concentration and to normalize femur NE measurements.
Glucose and insulin tolerance tests. For glucose tolerance test (GTT), 12-week old mice were fasted overnight for 18 hours and provided with water ad libitum. The next day, mice were housed in individual cages and allowed to acclimate for 2 hours followed by i.p. injection of 1.0 g kg−1 glucose (G8270, MilliporeSigma). For insulin tolerance test (ITT), mice were fasted for 2 hours in individual cages with free access to water. Insulin (0.8 U kg−1, Eli Lilly and Co.) was administered by i.p. injection. Blood samples were obtained from a tail nick, and blood glucose was measured at 0, 15, 30, 45, 60, 90, 120, and 150 minutes using a commercial glucometer (Contour TS, Ascensia Diabetes Care).
Bone densitometry (DEXA).
WT and IFT88-KOSF-1 male littermates were examined for whole-body fat and lean masses, BMD, BMC, BV, and BA using DEXA (InAlyzer, MEDIKORS) under light isoflurane anesthesia for about 1 minute. The detection sensitivity of the DEXA instrument was 0.001 g/cm2. A standard calibration block was used to calibrate the DEXA device before measurements in accordance with the operator’s manual. Software integrated to the scan was used for data analysis.
μCT)analysis.
The right femur from each mouse was dissected and fixed overnight in 10% neutral buffered formalin, loaded into 12.3 mm diameter scanning tubes, and imaged using a desk-top μCT scanner (SkyScan 1076, Bruker Micro CT). The scans were integrated into 3D voxel images and a region of interest (ROI) that consisted of 100 slices starting from about 0.5 mm proximal to growth plate, constituting 1.6 mm in length, was chosen for analysis. The following 3D indices in the defined ROI were analyzed, including total tissue volume, BV, BV/TV, Tb.N., Tb.Th., and Tb.Sp.
ISO treatment.
For ISO treatment, 14-week-old WT and IFT88-KOSF-1 male littermates were used. Prior to treatment, mice were examined for fat and lean mass, BMD, BMC, BV, and BA using DEXA. After 2 days of recovery, mice were given ISO (15 mg/kg of body weight, daily i.p. injection) for 2 weeks. After 2 weeks, mice were measured again for fat and lean mass and bone parameters.
Immunohistochemistry and H&E staining.
For SF-1 staining, brain sections were mounted on Superfrost Plus microscope slides (Fisher Scientific), then dried overnight. The following day, samples were pretreated with 0.3% hydrogen peroxide in PBS (pH 7.4) for 30 minutes at RT; then antigen retrieval was done for 5 minutes. Samples were blocked in 3% normal donkey serum, then incubated with rabbit anti–SF-1 antibody (22, 52) overnight at 4°C. After washing in PBS, sections were incubated in biotinylated donkey anti-rabbit IgG (065-152, 1:1000, Jackson ImmnuoResearch) for 2 hours at RT, followed by incubation for 1 hour in a solution of avidin-biotin complex (Vectastain Universal ABC Kit, PK-6200, Vector Laboratories). Sections were next washed in PBS and stained with DAB-peroxidase substrate solution (0.04% DAB and 0.01% H2O2) in PBS. The stained slides were visualized by a Nikon Digital Camera DXM1200 microscope system.
For UCP1 staining, iBAT was sectioned and paraffin embedded. The sections were incubated with primary UCP1 antibody (catalog ab10983, 1:1000, Abcam) at 4°C for 24 hours. Slides were washed 5 times with PBS and then incubated with secondary antibody for and additional 2 hours using the Vectastain Universal ABC Kit. After washing 3 times in PBS for 10 minutes each, slides were then stained with DAB-peroxidase substrate solution. Stained slides were visualized by a Nikon Digital Camera DXM1200 microscope system.
Peripheral tissues including WAT, BAT, liver, pituitary, adrenal gland, testis, ovary, and femurs were dissected and post-fixed overnight at 4°C. Tissue samples were then paraffin embedded and cut into 4 μm slices. Slices were stained with H&E following the standard H&E procedure.
Dynamic bone histomorphometry.
The left femur from each animal was dissected and fixed in 10% neutral buffered formalin. Samples were decalcified in ChelatorCal (BBC Biochemical) and embedded in paraffin, then cut into 4 μm slices. Femur sections were stained by either H&E or TRAP for osteoclast analysis. TRAP staining was carried out with the TRAP Stain Kit (TRAP/ALP Stain Kit, Wako Pure Chemical Industries Ltd.). Deparaffinized and rehydrated sections were incubated at 37°C for 30 minutes in TRAP substrate buffer, and then slides were mounted with VectaMount (Vector Laboratories). Images were visualized with an Olympus IX71/F22PH microscope and measurements performed with Bio-Quant software. The TRAP-positive osteoclast number (N. Oc) and surface (Oc.S) were normalized with BS.
To determine bone-formation rate (BFR) and mineralization, 6-week-old male mice were injected with 10 mg/kg calcein (MilliporeSigma) dissolved in 2.0% sodium bicarbonate (pH 7.0) 2 times a day for 7 days. Bones were fixed in 4% formalin. Femurs were embedded in paraffin blocks and sectioned to be 4 μm thick. Before histomorphometric analysis, femur metaphyseal sections were viewed with a fluorescent Nikon Digital Camera DXM1200 Microscope System, and 5 digital images per section were taken. The distance between the calcein lines (MAR) and the length of the calcein lines (mineralized surface) along the BS were measured to calculate BFR using ImageJ.
Western blot analysis.
Tissues samples were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) containing protease and phosphatase inhibitors (Roche). Protein extracts were obtained by centrifugation at 13,000g for 15 minutes at 4°C. Protein concentration was measured using Bio-Rad Protein Assay reagent (500006, Bio-Rad). Equal amounts of protein were loaded and separated on SDS/PAGE gels and transferred to PVDF membranes (Millipore). Then membranes were incubated with specific antibodies in TBS with 0.1% Tween-20 (TBST) overnight at 4°C. Primary antibodies used were rabbit anti-UCP1 (catalog ab10983, 1:10000, Abcam), STAT3 (catalog 9139, 1:5000, Cell Signaling Technology), and rabbit anti-β actin (catalog GTX109639, 1:5000, GeneTex). Secondary antibodies conjugated to HRP were added for 1 hour at RT. Secondary antibody used was goat anti-rabbit IgG-HRP (catalog 31460, 1:5000, Thermo Fisher Scientific). Protein levels were detected using Pierce ECL Western blotting substrate following the standard Western blot procedure, and blots were visualized using the chemiluminescent image analyzer LAS 4000 (GE Healthcare Life Science).
Mitochondria contents in the iBAT.
For mitochondrial DNA content analysis, total DNA from iBAT was extracted using DNAzol Reagent (Invitrogen) according to the manufacturer’s instructions. Mitochondrial DNA was amplified using primers specific for the mitochondrial cytochrome C oxidase subunit 2 (COX2) gene and normalized to genomic DNA by amplification of the 40S ribosomal protein s18 (Rps18) nuclear gene (53). Primers used were as follows: mouse COX2: F, 5′-ATAACCGAGTCGTTCTGCCAAT-3′, R, 5′-TTTCAGAGCATTGGCCATAGAA-3′; Rps18: F, 5′-TGTGTTAGGGGACTGGTGGACA-3′, R, 5′-CATCACCCACTTACCCCCAAAA-3′.
RNA isolation and real-time qPCR.
Total RNA was extracted from tissue samples (hypothalamus, liver, iBAT, and femur) using TRIzol reagent (Life Technologies) following the manufacturer’s instructions; 1 μg of total RNA was used to synthesize cDNA using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO). An aliquot (1/100 vol) of the cDNA was then subjected to real-time quantitative PCR (qPCR) using iQ SYBR Green Supermix (Bio-Rad) in a 96-well real-time PCR machine (CFX96, Bio-Rad). Fold changes were calculated and determined using the 2-ΔΔCt method and expression levels normalized to the average of the housekeeping genes 18S or HRPT.
Primers used were as follows: mouse Pomc: F, 5′-CAGGTCCTGGAGTCCGAC-3′, R, 5′-CATGAAGCCACCGTAACG-3′; Npy: F, 5′-CTACTCCGCTCTGCGACACT-3′, R, 5′-AGTGTCTCAGGGCTGGATCTC-3′; Agrp: F, 5′-CGGCCACGAACCTCTGTAG-3′, R, 5′-CTCATCCCCTGCCTTTGC-3′; SF-1: F, 5′-CCCTTATCCGGCTGAGAATT-3′, R, 5′-CCAGGTCCTCGTCGTACGA-3′; ObRb: F, 5′-AACCCCAAGAATTGTTCCTGG-3′, R, 5′-GGAGACCATAGCTGCTGGGACC-3′; 5-HT2c: F, 5′-GCCAATGAACACCTTCTTTCC-3′, R, 5′-GCCAATTAGGTGCACAAGGAG-3′; Ucp1: F, 5′-GGCCCTTGTAAACAACAAAATAC-3′, R, 5′-GGCAACAAGAGCTGACAGTAAAT-3′; Pgc1α: F, 5′-AACCACACCCACAGGATCAGA-3′, R, 5′-TCTTCGCTTTATTGCTCCATGA-3′; Dio2: F, 5′-TTCTCCAACTGCCTCTTCCTG-3′, R, 5′-CCCATCAGCGGTCTTCTCC-3′; Tfam: F, 5′-AGGAGGCAAAGGATGATTCG-3′, R, 5′-CCTCAGGAGACAGATTTTTCCA-3′; Pklr: F, 5′-AGGAGTCTTCCCCTTGCTCTAC-3′, R, 5′-GGAGAGGCGTTTCAGGATATG-3′; PEPCK: F, 5′-CGCAAGCTGAAGAAATATGACAA-3′, R, 5′-TCGATCCTGGCCACATCTC-3′; G6Pase: F, 5′-TGGGCAAAATGGCAAGGA-3′, R, 5′-TCTGCCCCAGGAATCAAAAAT-3′; Ehhadh: F, 5′-CTTGGAATTCTGGATGTAG-3′, R, 5′-TGGGTTTACCTATAACCG-3′; PPARγ: F, 5′-CAAGAATACCAAAGTGCGATCAA-3′, R, 5′-GAGCTGGGTCTTTTCAGAATAATAAG-3′; Srebp1: F, 5′-GGAGCCATGGATTGCACATT-3′, R, 5′-GGCCCGGGAAGTCACTGT-3′; FASN: F, 5′-GGTGTGGTGGGTTTGGTGAATTGT-3′, R, 5′-TCACGAGGTCATGCTTTAGCACCT-3′; HSL: F, 5′-GCGCTGGAGGAGTGTTTTT-3′, R, 5′-CCGCTCTCCAGTTGAACC-3′; Rank: F, 5′-CCAGGAGAGGCATTATGAGCA-3′, R, 5′-ACTGTCGGAGGTAGGAGTGC-3′; Rankl: F, 5′-CAGCATCGCTCTGTTCCTGTA-3′, R, 5′-CTGCGTTTTCATGGAGTCTCA-3′; Trap: F, 5′-GCAACATCCCCTGGTATGTG-3′, R, 5′-GCAAACGGTAGTAAGGGCTG-3′; Dc-Stamp: F, 5′-GGGGACTTATGTGTTTCCACG-3′, R, 5′-ACAAAGCAACAGACTCCCAAAT-3′; Sp7: F, 5′-TCCCTGGATATGACTCATCCCT-3′, R, 5′-CCAAGGAGTAGGTGTGTTGCC-3′; Alp: F, 5′-CCTTGAAAAATGCCCTGAAA-3′, R, 5′-TTACTGTGGAGACGCCCATA-3′; 18S: F, 5′-AACCCGTTGAACCCCATT-3′, R, 5′-CCATCCAATCGGTAGTAGCG-3′; and HRPT: F, 5′-CTCATGGACTGATTATGGACAGGAC-3′, R, 5′-GCAGGTCAGCAAAGAACTTATAGCC-3′.
Statistics.
All statistical tests for data values were conducted using GraphPad Prism (version 5) and R software (version 3.6.0). Full details of each statistical test used are provided in each figure legend. P values of less than 0.05 were defined a statistically significant. Statistical significance was determined by unpaired 2-tailed Student’s t test to compare 2 groups and 2-way ANOVA with Bonferroni’s post test to compare multiple groups. For correlation testing, linear regression models in R were utilized, and R2 values were reported. The R package ggplot2 was used to plot the result.
Study approval.
All experimental and surgical protocols were approved by the IACUC of Yonsei University Health System (YUHS). All animals were maintained in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Author contributions
JSS and DJY performed experiments and analyzed data. KWK wrote the Abstract, Introduction, Results, and Discussion sections. DJY and JSS wrote Methods and the figure legends. AWK performed preliminary experiments. SGY and JKS performed metabolic cage studies. SJM, JK, DMS, YHC, and KWK conceptualized the research, analyzed data, and edited and finalized the manuscript. The order of co–first authorship was determined by workload, particularly effort in data acquisition and drafting the manuscript. JSS and DJY share first authorship, and the order in which they are listed was determined by workload.
Supplementary Material
Acknowledgments
We thank Joel K. Elmquist (University of Texas Southwestern Medical Center, Dallas, Texas, USA) for kindly providing us with the SF-1–Cre mice. We also thank Andrew Shin (Texas Tech University, Lubbock, Texas, USA), Alexandre Caron (University of Texas Southwestern Medical Center), and Jessica Hong (Brown University, Providence, Rhode Island, USA) for critical reading of the manuscript. This work was supported by the National Research Foundation, Korea (2016R1C1B3012748, 2020M3E5E2038221, and 2016R1A5A2008630 to KWK; 2015H1A2A1032009 to DJY; 2013M3A9D5072550 to JKS). This research was also supported by a grant from the Korean Diabetes Association (2018F-1 to KWK).
Version 1. 10/06/2020
In-Press Preview
Version 2. 01/04/2021
Print issue publication
Funding Statement
This work was supported by the National Research Foundation, Korea (2016R1C1B3012748, 2020M3E5E2038221, and 2016R1A5A2008630 for K.W.K).
For D.J.Y
For J.K.S
Footnotes
YHC’s present address is: Department of Internal Medicine, Division of Hypothalamic Research, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(1):e138107.https://doi.org/10.1172/JCI138107.
Contributor Information
Ji Su Sun, Email: SUNJISU@yuhs.ac.
Dong Joo Yang, Email: ydj1991@yuhs.ac.
Ann W. Kinyua, Email: annmendi@gmail.com.
Seul Gi Yoon, Email: sg.yoon@snu.ac.kr.
Je Kyung Seong, Email: snumouse@snu.ac.kr.
Juwon Kim, Email: juwon76@yonsei.ac.kr.
Seok Jun Moon, Email: SJMOON@yuhs.ac.
Dong Min Shin, Email: dmshin@yuhs.ac.
Yun-Hee Choi, Email: yun-hee.choi@utsouthwestern.edu.
Ki Woo Kim, Email: kiwoo-kim@yuhs.ac.
References
- 1.Davenport JR, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17(18):1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siljee JE, et al. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat Genet. 2018;50(2):180–185. doi: 10.1038/s41588-017-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78(4):149–172. [Google Scholar]
- 4.Powley TL. The ventromedial hypothalamic syndrome, satiety, and a cephalic phase hypothesis. Psychol Rev. 1977;84(1):89–126. doi: 10.1037/0033-295X.84.1.89. [DOI] [PubMed] [Google Scholar]
- 5.Sclafani A. Neural pathways involved in the ventromedial hypothalamic lesion syndrome in the rat. J Comp Physiol Psychol. 1971;77(1):70–96. doi: 10.1037/h0031574. [DOI] [PubMed] [Google Scholar]
- 6.Shimazu T. Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia. 1981;20(Suppl 1):343–356. doi: 10.1007/BF00254502. [DOI] [PubMed] [Google Scholar]
- 7.King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87(2):221–244. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Choi YH, Fujikawa T, Lee J, Reuter A, Kim KW. Revisiting the ventral medial nucleus of the hypothalamus: the roles of SF-1 neurons in energy homeostasis. Front Neurosci. 2013;7:71. doi: 10.3389/fnins.2013.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Idelevich A, Baron R. Brain to bone: What is the contribution of the brain to skeletal homeostasis? Bone. 2018;115:31–42. doi: 10.1016/j.bone.2018.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han YM, et al. Leptin-promoted cilia assembly is critical for normal energy balance. J Clin Invest. 2014;124(5):2193–2197. doi: 10.1172/JCI69395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol. 2011;12(4):222–234. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- 12.Guo DF, et al. The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet. 2016;12(2):e1005890. doi: 10.1371/journal.pgen.1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujikawa T, et al. P110β in the ventromedial hypothalamus regulates glucose and energy metabolism. Exp Mol Med. 2019;51(4):1–9. doi: 10.1038/s12276-019-0249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haycraft CJ, et al. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134(2):307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 15.Stallings NR, Hanley NA, Majdic G, Zhao L, Bakke M, Parker KL. Development of a transgenic green fluorescent protein lineage marker for steroidogenic factor 1. Mol Endocrinol. 2002;16(10):2360–2370. doi: 10.1210/me.2002-0003. [DOI] [PubMed] [Google Scholar]
- 16.Iwanaga T, Hozumi Y, Takahashi-Iwanaga H. Immunohistochemical demonstration of dopamine receptor D2R in the primary cilia of the mouse pituitary gland. Biomed Res. 2011;32(3):225–235. doi: 10.2220/biomedres.32.225. [DOI] [PubMed] [Google Scholar]
- 17.Iwanaga T, Miki T, Takahashi-Iwanaga H. Restricted expression of somatostatin receptor 3 to primary cilia in the pancreatic islets and adenohypophysis of mice. Biomed Res. 2011;32(1):73–81. doi: 10.2220/biomedres.32.73. [DOI] [PubMed] [Google Scholar]
- 18.Nygaard MB, Almstrup K, Lindbæk L, Christensen ST, Svingen T. Cell context-specific expression of primary cilia in the human testis and ciliary coordination of Hedgehog signalling in mouse Leydig cells. Sci Rep. 2015;5:10364. doi: 10.1038/srep10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhillon H, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49(2):191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 20.Kim KW, et al. FOXO1 in the ventromedial hypothalamus regulates energy balance. J Clin Invest. 2012;122(7):2578–2589. doi: 10.1172/JCI62848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong Q, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5(5):383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim KW, et al. Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci U S A. 2011;108(26):10673–10678. doi: 10.1073/pnas.1102364108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minokoshi Y, Saito M, Shimazu T. Sympathetic denervation impairs responses of brown adipose tissue to VMH stimulation. Am J Physiol. 1986;251(5 Pt 2):R1005–R1008. doi: 10.1152/ajpregu.1986.251.5.R1005. [DOI] [PubMed] [Google Scholar]
- 24.Doan KV, et al. FoxO1 in dopaminergic neurons regulates energy homeostasis and targets tyrosine hydroxylase. Nat Commun. 2016;7:12733. doi: 10.1038/ncomms12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantzoros CS. Leptin and the hypothalamus: neuroendocrine regulation of food intake. Mol Psychiatry. 1999;4(1):8–12. doi: 10.1038/sj.mp.4000497. [DOI] [PubMed] [Google Scholar]
- 26.Gautron L, Elmquist JK. Sixteen years and counting: an update on leptin in energy balance. J Clin Invest. 2011;121(6):2087–2093. doi: 10.1172/JCI45888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elefteriou F, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434(7032):514–520. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, et al. Dissociation of the neuronal regulation of bone mass and energy metabolism by leptin in vivo. Proc Natl Acad Sci U S A. 2008;105(51):20529–20533. doi: 10.1073/pnas.0808701106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khosla S, et al. Sympathetic β1-adrenergic signaling contributes to regulation of human bone metabolism. J Clin Invest. 2018;128(11):4832–4842. doi: 10.1172/JCI122151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeda S, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305–317. doi: 10.1016/S0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 31.Bonnet N, Laroche N, Vico L, Dolleans E, Benhamou CL, Courteix D. Dose effects of propranolol on cancellous and cortical bone in ovariectomized adult rats. J Pharmacol Exp Ther. 2006;318(3):1118–1127. doi: 10.1124/jpet.106.105437. [DOI] [PubMed] [Google Scholar]
- 32.Idelevich A, Sato K, Nagano K, Rowe G, Gori F, Baron R. Neuronal hypothalamic regulation of body metabolism and bone density is galanin dependent. J Clin Invest. 2018;128(6):2626–2641. doi: 10.1172/JCI99350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JG, et al. AgRP neurons regulate bone mass. Cell Rep. 2015;13(1):8–14. doi: 10.1016/j.celrep.2015.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allison SJ, et al. Conditional deletion of hypothalamic Y2 receptors reverts gonadectomy-induced bone loss in adult mice. J Biol Chem. 2006;281(33):23436–23444. doi: 10.1074/jbc.M604839200. [DOI] [PubMed] [Google Scholar]
- 35.Cornish J, et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002;175(2):405–415. doi: 10.1677/joe.0.1750405. [DOI] [PubMed] [Google Scholar]
- 36.Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept. 2000;92(1-3):73–78. doi: 10.1016/S0167-0115(00)00152-X. [DOI] [PubMed] [Google Scholar]
- 37.Ducy P, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100(2):197–207. doi: 10.1016/S0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 38.Hamrick MW, Pennington C, Newton D, Xie D, Isales C. Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone. 2004;34(3):376–383. doi: 10.1016/j.bone.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 39.Yano H, Ohya K, Amagasa T. Effects of insulin on in vitro bone formation in fetal rat parietal bone. Endocr J. 1994;41(3):293–300. doi: 10.1507/endocrj.41.293. [DOI] [PubMed] [Google Scholar]
- 40.Cornish J, Callon KE, Reid IR. Insulin increases histomorphometric indices of bone formation In vivo. Calcif Tissue Int. 1996;59(6):492–495. doi: 10.1007/BF00369216. [DOI] [PubMed] [Google Scholar]
- 41.Abrahamsen B, Rohold A, Henriksen JE, Beck-Nielsen H. Correlations between insulin sensitivity and bone mineral density in non-diabetic men. Diabet Med. 2000;17(2):124–129. doi: 10.1046/j.1464-5491.2000.00234.x. [DOI] [PubMed] [Google Scholar]
- 42.Scott MM, et al. Leptin targets in the mouse brain. J Comp Neurol. 2009;514(5):518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balthasar N, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42(6):983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Yadav VK, et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138(5):976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lam DD, et al. Leptin does not directly affect CNS serotonin neurons to influence appetite. Cell Metab. 2011;13(5):584–591. doi: 10.1016/j.cmet.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 47.Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet. 2009;18(7):1323–1331. doi: 10.1093/hmg/ddp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berbari NF, et al. Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice. Proc Natl Acad Sci U S A. 2013;110(19):7796–7801. doi: 10.1073/pnas.1210192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen FM, Nilsson P, Hustvedt BE, Nilsson-Ehle P, Løvø A. Significance of hyperinsulinemia in ventromedial hypothalamus-lesioned rats. Am J Physiol. 1983;244(3):E203–E208. doi: 10.1152/ajpendo.1983.244.3.E203. [DOI] [PubMed] [Google Scholar]
- 50.Sakaguchi T, Bray GA. The effect of intrahypothalamic injections of glucose on sympathetic efferent firing rate. Brain Res Bull. 1987;18(5):591–595. doi: 10.1016/0361-9230(87)90128-6. [DOI] [PubMed] [Google Scholar]
- 51.Guo J, et al. Primary cilia signaling shapes the development of interneuronal connectivity. Dev Cell. 2017;42(3):286–300.e4. doi: 10.1016/j.devcel.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao L, et al. Central nervous system-specific knockout of steroidogenic factor 1 results in increased anxiety-like behavior. Mol Endocrinol. 2008;22(6):1403–1415. doi: 10.1210/me.2008-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gomes AP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.