

Abstract
The aorta and the large conductive arteries are immunoprivileged tissues and are protected against inflammatory attack. A breakdown of immunoprivilege leads to autoimmune vasculitis, such as giant cell arteritis, in which CD8+ Treg cells fail to contain CD4+ T cells and macrophages, resulting in the formation of tissue-destructive granulomatous lesions. Here, we report that the molecular defect of malfunctioning CD8+ Treg cells lies in aberrant NOTCH4 signaling that deviates endosomal trafficking and minimizes exosome production. By transcriptionally controlling the profile of RAB GTPases, NOTCH4 signaling restricted vesicular secretion of the enzyme NADPH oxidase 2 (NOX2). Specifically, NOTCH4hiCD8+ Treg cells increased RAB5A and RAB11A expression and suppressed RAB7A, culminating in the accumulation of early and recycling endosomes and sequestering of NOX2 in an intracellular compartment. RAB7AloCD8+ Treg cells failed in the surface translocation and exosomal release of NOX2. NOTCH4hiRAB5AhiRAB7AloRAB11AhiCD8+ Treg cells left adaptive immunity unopposed, enabling a breakdown in tissue tolerance and aggressive vessel wall inflammation. Inhibiting NOTCH4 signaling corrected the defect and protected arteries from inflammatory insult. This study implicates NOTCH4-dependent transcriptional control of RAB proteins and intracellular vesicle trafficking in autoimmune disease and in vascular inflammation.
Keywords: Autoimmunity, Immunology
Keywords: Adaptive immunity, T cells, Vasculitis

Introduction
The aorta and its major side branches are nonredundant for host survival and are protected against inflammatory attack; an immune privilege broken in the case of vasculitis (1). In giant cell arteritis (GCA), CD4+ T cells and macrophages form granulomatous lesions in the vascular wall (2). Inflammation-induced remodeling leads to wall vascularization and lumen-occluding intimal hyperplasia, causing tissue ischemia, stroke, blindness, and aortic arch syndrome (1). Several mechanisms contribute to the breakdown of the artery’s immunoprivilege; e.g., aberrant expression of the NOTCH ligand Jagged1 on vasa vasora (3) and leakiness of the microvascular basement membrane (4), rendering the tissue accessible to T cells and macrophages. Other immune defects in GCA include the deficiency of the PD-1/PD-L1 checkpoint (5), excess CD28-dependent signaling (6), and induction of long-lived tissue-resident memory T cells that sustain unabated tissue inflammation (7). Defective Treg cell function in GCA has been assigned to the CD8+ T cell compartment. CD8+FOXP3+CCR7+CD39+CD26– T cells suppress the activation of neighboring CD4+ T cells (8). CD8+ Treg cells function by restraining the overall size of the CD4+ T cell compartment. The molecular defect underlying the failure of CD8+ Treg cells is unknown.
A key feature of GCA T cells is the expression of the oncogene NOTCH1 (3). Aberrant NOTCH signaling is tumorigenic; more than 50% of patients with T cell acute lymphoblastic leukemia (TALL) have gain-of-function mutations in the NOTCH1 locus (9, 10). NOTCH signaling is required for T cell development (11) and NOTCH1 knockout in the bone marrow results in complete T cell loss (12). In Treg cells, NOTCH may function as a survival factor, protecting from cell death (13). In GCA, NOTCH signaling is vasculitogenic, costimulating CD4+ T cell activation, inducing mTORC1 activity, and biasing lineage commitment to proinflammatory Th1 and Th17 cells (3, 14). In all members of the NOTCH receptor family (NOTCH1, -2, -3, and -4), ligand binding triggers proteolytic receptor cleavage, releasing the NOTCH intracellular domain, which is translocated to the nucleus to activate NOTCH target genes (15).
A distinguishing feature of CD8+ Treg cells is the expression of NADPH oxidase 2, a sextameric complex in which the membrane-integrated proteins gp91phox (NOX2) and p22phox combine with several cytosolic subunits to react electrons with molecular oxygen to generate ROS. Defects in the NOX2 complex are involved in immunodeficiency and inflammatory disease, including chronic granulomatous disease (16). The primary purpose of the generated superoxide is to kill extracellular and phagocytosed pathogens, but also to mediate redox signaling (17, 18). ROS release must be strictly compartmentalized, e.g., toward the extracellular side of the plasma membrane or into the phagosomal lumen. In professional phagocytes, NOX2 is internalized by clathrin-coated pits, contained in an intracellular storage compartment, and upon activation is transferred to the phagosome or to the cell surface (19).
The small GTPase RAB proteins play an indispensable role in intracellular vesicle traffic and participate in the formation, movement, and fusion of endosomal compartments (20, 21). RAB5 marks the early endosome (22), and RAB5 dysfunction has been reported in patients with Alzheimer disease (23). RAB7 participates in the formation of the late endosome and lysosome and is required for exosome biogenesis (24, 25). RAB7 mutations result in Charcot-Marie-Tooth type 2 neuropathies (26). RAB11 facilitates the formation and fusion of recycling endosomes to the plasma membrane (27), contributes to host innate immunity in bacterial and viral infections (28), and is associated with neurodegenerative disease (29). Overall, RAB proteins regulate immunity by controlling intracellular signaling networks, pathogen uptake, and cytokine secretion, but have not been implicated in autoimmunity (30).
Here, we have identified aberrant NOTCH4 signaling as the cause of defective CD8+ Treg cell function, promoting uncontrolled and destructive arteritis. Specifically, NOTCH4 expression in CD8+ Treg cells from GCA patients was linked to normal survival but functional failure in vitro and in vivo. Molecular studies revealed abnormal subcellular distribution of the immunosuppressive enzyme NOX2 in NOTCH4hiCD8+ Treg cells, with diversion of NOX2 into intracellular vesicular storage sites. Disruption of NOTCH4 signaling in patient-derived CD8+ Treg cells reassigned NOX2 to exosomal release and restored immunosuppressive function. We identified the NOTCH4 target genes RAB5A, RAB7A, and RAB11A as central regulators of NOX2 transport and secretion. NOTCH-induced upregulation of RAB5A and RAB11A sequestered NOX2 in early and recycling endosomes, while HES5-dependent RAB7A repression diverted NOX2 away from the multivesicular body sorting machinery and prevented exosomal release. Therapeutic blockade of pathogenic NOTCH signaling restored protein trafficking in the endosomal system and successfully treated vascular inflammation.
Results
Functional deficiency in CD8+ Tregs from GCA patients.
Functional CD8+ Treg cells express NOX2 on the cell surface and constrain activation of nearby CD4+ T cells through exosome release (8). GCA patients have low frequencies of NOX2hiCD8+ Treg cells and fail to appropriately downregulate T cell immunity (8). In GCA patients and matched healthy controls, about 1.5% of circulating CD8+ T cells and about 3% of circulating CD4+ T cells had the Treg-associated FOXP3+ phenotype (Supplemental Figure 1, A and B). We sorted CD8+CD39+CD26– Treg cells from the blood of patients and controls and tested their functional competence in in vitro suppressor assays by quantifying ZAP70 phosphorylation in CD4+ T cells undergoing activation (8). The inhibitory efficiency of patient-derived CD8+ Tregs was significantly lower than that of controls (57% reduction, P = 0.011) (Figure 1A). We induced CD8+ Treg cells ex vivo by low-dose anti-CD3 stimulation combined with IL-15 and probed their function in CD4+/CD8+ Treg cocultures (Figure 1B). The inhibitory efficiency of induced GCA CD8+ Treg cells was significantly reduced compared with control CD8+ Treg cells (55% reduction, P = 0.0043) (Figure 1B). To determine whether the inhibition of CD4+ T cell activation had long-term consequences, we assessed the proliferative capacity of CD4+ T cells cultured with either freshly sorted or induced CD8+ Treg cells (Figure 1, C and D). Patient-derived CD8+ Treg cells were significantly less efficient than controls at slowing CD4+ T cell proliferation.
Figure 1. Functional deficiencies of CD8+ Treg cells in vasculitis.

(A) CD8+CD39+CD26– Tregs sorted from the peripheral blood of GCA patients and controls were mixed with CD4+ T cells and activated CD4+ pZAP70+ T cells were measured by flow cytometry after 15 minutes. Representative contour plots and results from 7 controls and 7 GCA patients. (B) CD8+ Tregs induced ex vivo from GCA patients and controls were mixed with CD4+ T cells, and pZAP70 was quantified as above. Representative contour plots and results from 6 controls and 6 GCA patients. (C and D) Freshly sorted and ex vivo–induced CD8+ Tregs were mixed with CFSE-labeled CD4+ T cells. Proliferation was measured 5 days later. Representative histograms from 6 controls and 6 GCA patients. (E and F) CD8+ Tregs were induced ex vivo, CFSE labeled, and mixed with heathy PBMCs before adoptive transfer into NSG mice. One control group received no CD8+ Tregs. (E) Absolute numbers of CD8+ Treg cells in the blood and spleen measured by flow cytometry. Results from 12 mice. (F) Absolute numbers of human CD4+ T cells in the spleen measured by flow cytometry. Results from 6 mice each. (G–I) Vasculitis was induced in human arteries engrafted into NSG mice. Ex vivo–induced CD8+ Tregs from patients and controls (HC) were coinjected. (G) Vessel wall–infiltrating T cells visualized by immunostaining for CD3 (green). Nuclei marked with DAPI. Representative images from the 3 study cohorts (n = 6 each). Scale bar: 20 μm. (H) Frequencies of vessel wall–infiltrating CD3+ T cells. Each dot represents 1 inflamed artery. (I) Tissue transcriptome of explanted arteries analyzed by RT-PCR; β-actin served as control. Each dot represents 1 artery specimen. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by 1-way ANOVA and post-ANOVA pairwise 2-group comparisons conducted with Tukey’s method (E, F, H, and I) or unpaired Mann-Whitney-Wilcoxon rank test (A and B).
Next, we examined healthy and patient-derived CD8+ Treg cells in vivo (Figure 1, E and F). CD8+ Treg cells were induced ex vivo, CFSE labeled, and adoptively transferred into immunodeficient NSG mice. CD8+ Treg cells were infrequent in the peripheral blood but homed to the spleen (Figure 1E). As a functional readout for their immunosuppressive function, we quantified the number of human CD4+ T cells accumulating in the spleen (Figure 1F). Healthy CD8+ Treg cells prevented CD4+ T cell expansion, reducing CD4+ T cell numbers by 70.4% (P = 0.0008; Figure 1F). In contrast, patient-derived CD8+ Treg cells failed to suppress homeostatic expansion of CD4+ T cells and the pool size of CD4+ T cells was similar to that in spleens that had not received Treg cells (Figure 1F).
To understand whether impaired CD8+ Treg cell function was functionally connected to the patients’ autoimmune disease, vascular inflammation was induced in human arteries engrafted into the NSG mice (Figure 1, G–I). In this model, adoptive transfer of patient-derived CD4+ T cells and macrophages resulted in robust vasculitis. Accordingly, arteries explanted from chimeric mice that had not received any Treg cells were densely infiltrated by human CD3+ T cells. Cotransfer of healthy CD8+ Treg cells prevented vessel wall invasion by CD3+ T cells. CD8+ Treg cells generated from GCA patients failed to suppress the inflammatory response (Figure 1G). In a series of 6 experiments, healthy CD8+ Treg cells essentially inhibited vasculitis by 83.2%, whereas patient-derived CD8+ Treg cells left vasculitic infiltrates unaffected (Figure 1H). Tissue transcriptome analysis of explanted arteries established a vasculitis-associated gene pattern: high expression of IFNG and IL17 transcripts combined with the transcription factors TBX21 and RORC (Figure 1I). Transcripts for the innate cytokines IL6, TNF, and IL1B were abundant in inflamed arteries. Injection of healthy CD8+ Treg cells consistently reduced gene expression of 8 inflammation-associated genes. In contrast, cotransfer of patient-derived CD8+ Treg cells had no effect on the gene expression profile (Figure 1I).
These experiments identified a functional and disease-relevant defect in patient-derived CD8+ Treg cells in vivo and in vitro. Defective CD8+ Treg cells failed to protect the artery from inflammatory attack.
NOTCH4 signaling underlies the defective CD8+ Treg function.
The NOTCH1 signaling pathway is strongly upregulated in GCA CD4+ T cells, rendering such T cells tissue-invasive and vasculitogenic (3). We screened patient-derived CD8+ Treg cells for the presence of NOTCH receptors (Figure 2A). Gene transcript analysis identified NOTCH4 as the dominant receptor on CD8+ Treg cells (Figure 2A). NOTCH4 protein expression was doubled on GCA CD8+ Treg cells (Figure 2B) and gene expression of the NOTCH target gene HES5 was significantly elevated (Figure 2C). Immunoblotting revealed higher concentrations of NOTCH4 intracellular domain (N4ICD) and HES5 in patient-derived CD8+ Treg cells (Figure 2, D and E), compatible with ongoing NOTCH pathway activation. Peripheral blood flow cytometry demonstrated significantly higher expression of NOTCH4 on CD8+CD39+CD26– cells in patients compared with controls (Figure 2F). HES5 transcripts were significantly higher in GCA CD8+CD39+CD26– Treg cells sorted from the peripheral blood (Figure 2G).
Figure 2. Aberrant NOTCH4 signaling in defective CD8+ Treg cells.

(A–E) CD8+ Tregs were induced ex vivo from GCA patients and healthy controls as in Figure 1. (A) Expression of NOTCH1–4 receptor transcripts in CD8+ Tregs (RT-PCR, n = 11 patients). (B) Expression of NOTCH4 protein on control and patient-derived CD8+ Tregs (FACS, n = 11 samples each). (C) Transcripts for the NOTCH target gene HES5 (RT-PCR, n = 6 samples each). (D and E) Immunoblot analysis of the NOTCH4 intracellular domain (N4ICD) and HES5 in control and GCA CD8+ Tregs; β-actin served as loading control. N = 5 experiments. (F) Expression of NOTCH4 protein on CD8+CD39+CD26– Tregs sorted from controls and patients determined by FACS. FMO, fluorescence minus one. Representative histograms and results from n = 8 samples. (G) Transcripts for the NOTCH target gene HES5 quantified by q-PCR in sorted CD8+CD39+CD26– Tregs. Results from 10 patients and 10 controls. (H) Induced CD8+ Tregs were treated with DAPT or vehicle. Suppressive function was measured as in Figure 1A. Representative contour plots and results from 6 patients and 6 controls. (I) Induced CD8+ Tregs from GCA patients were transfected with NOTCH4 or control siRNA before suppressive function was measured as in Figure 1A. Representative contour plots. Results from 8 samples. (J) Induced CD8+ Tregs from healthy donors were transfected with an N4ICD-containing or control vector. Suppressive function was determined as in Figure 1A. Representative contour plots and results from 8 samples. (K and L) Induced CD8+ Tregs from GCA patients were transfected with NOTCH4 or control siRNA (K). Healthy CD8+ Tregs were transfected with a N4ICD-containing or control vector (L). Proliferation of CFSE-labeled CD4+ T cells was measured as in Figure 1C. Representative images from 6 experiments. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired Mann-Whitney-Wilcoxon rank test (B–F), paired Mann-Whitney-Wilcoxon rank test (H and I), or 1-way ANOVA and post-ANOVA pairwise 2-group comparisons conducted with Tukey’s method (A and G).
To confirm that NOTCH4 signaling controls CD8+ Treg function, we used the NOTCH signal inhibitor DAPT, a γ-secretase inhibitor that blocks the cleavage of NOTCH receptors. Healthy CD8+ Treg cells were more efficient in inhibiting CD4+ T cell activation after blocking NOTCH signaling with DAPT. In GCA CD8+ Treg cells, the effect was even more pronounced; the inhibitory efficiency of DAPT-treated cells was 2.5-fold higher than in vehicle-treated cells (P = 0.0022; Figure 2H). Similarly, knockdown of NOTCH4 in GCA CD8+ Treg cells markedly enhanced their inhibitory efficiency (2.9-fold, P = 0.0078) (Figure 2I). Conversely, enforced NOTCH signaling by N4ICD transfection into healthy CD8+ Tregs reduced immunosuppressive capacity (66% reduction, P = 0.0078) (Figure 2J). Transfection of GCA CD8+ Treg cells with siNOTCH4 restored their suppressive function and reduced proliferation of cocultured CD4+ T cells (Figure 2K). Transfection of healthy CD8+ Treg cells with an N4ICD-containing vector abrogated their suppressor activity, unleashing the proliferation of cocultured CD4+ T cells (Figure 2L).
These data linked the NOTCH4 signaling pathway to the functional fitness of CD8+ Treg cells.
NOTCH4 signaling promotes vascular inflammation.
To examine the effect of NOTCH4 in CD8+ Treg cells in autoimmune disease, we induced vasculitis in human arteries engrafted into NSG mice. After artery engraftment, chimeras were reconstituted with a mixture of GCA PBMCs and CD8+ Treg cells. Prior to the transfer, CD8+ Treg cells were transfected with an N4ICD overexpression plasmid or NOTCH4 siRNA. As a readout of autoimmune vasculitis, we quantified the T cell density within the human artery, the production of proinflammatory cytokines by tissue-residing T cells, and innate cytokines (IL1B, TNF, IL6) in the vasculitic lesions.
In the first series of experiments, healthy CD8+ Treg cells were transfected with an N4ICD-containing vector to amplify NOTCH4 signaling (Figure 3, A–F). Enhancement of NOTCH4 signaling selectively in CD8+ Treg cells increased the pool of CD4+ T cells in the spleen (2.7-fold; P = 0.0022) and in the engrafted artery (4.2-fold; P = 0.0022). IFN-γ–producing Th1 cells and IL-17–producing Th17 cells represent the most relevant effector T cell populations in the vasculitic lesions (31). Numbers of IFN-γ+ CD4+ T cells increased 4.5-fold (P = 0.0022) and IL-17+ Th17 cells were 4.1-fold higher (P = 0.0087) when the mice were reconstituted with N4ICD-transfected Tregs (Figure 3, A–D). IHC staining of tissue sections confirmed that tissue CD3+ T cells were more numerous after N4ICD overexpression in the CD8+ Treg cells (4.5-fold, P = 0.0020; Figure 3E). Tissue transcriptome analysis in explanted inflamed arteries assessed vasculitis-associated gene expression signatures (3) (Figure 3F). Enhanced NOTCH4 signaling in transferred CD8+ Tregs upregulated most of the genes, including the proinflammatory cytokine genes IFNG, IL17, IL6, TNF, and IL1B. The only gene unaffected by NOTCH4 signaling was GATA3, a transcription factor involved in Th2 cell induction.
Figure 3. NOTCH4 signaling promotes vascular inflammation.

To induce vasculitis, NSG mice were engrafted with human arteries and reconstituted with a mixture of PBMCs and CD8+ Tregs as in Figure 1. Prior to the adoptive transfer, NOTCH4 signaling in healthy, induced CD8+ Tregs was enhanced by transfecting them with an N4ICD-containing vector (A–F). Alternatively, NOTCH4 signaling in patient-derived CD8+ Tregs was suppressed by siRNA transfection (G–L). Human arteries were explanted and examined by IHC staining, tissue transcriptome analysis, and flow cytometry of extracted T cells. (A and B) Numbers of human CD4+ T cells in the spleen and in the engrafted artery analyzed by FACS. (C and D) Tissue-infiltrating T cells were isolated from the inflamed arteries, and IFN-γ and IL-17 production were assessed by intracellular staining and flow cytometry. (E) Tissue-infiltrating T cells in the arteries analyzed by immunostaining for CD3 (green). Nuclei marked with DAPI. Representative images from 6 grafts. Scale bars: 20 μm. (F) Tissue transcriptome in explanted arteries analyzed by RT-PCR. (G and H) Numbers of human CD4+ T cells in the spleen and in the human arteries analyzed by FACS. (I and J) Flow cytometric staining of intracellular IFN-γ and IL-17 in T cells extracted from explanted human arteries. (K) Tissue-infiltrating T cells in the arteries analyzed by immunostaining for CD3 (green). Nuclei marked with DAPI. Representative images from 6 grafts. Scale bars: 20 μm. (L) Tissue transcriptome analysis in explanted arteries by RT-PCR. All data are mean ± SEM from 6 samples. *P < 0.05; **P < 0.01 by paired Mann-Whitney-Wilcoxon rank test.
These data identified NOTCH4 signaling in CD8+ Treg cells as a critical pathway in determining the severity of autoimmune vascular inflammation.
Alternatively, GCA CD8+ Treg cells were rendered NOTCH4 signaling incompetent. Mice carrying human arteries received GCA CD8+ Treg cells that had been transfected with NOTCH4 or control siRNA. In a series of 6 experiments, vessel wall–infiltrating CD4+ T cells (P = 0.0087) and spleen-residing CD4+ T cells (P = 0.0022) declined markedly. Similarly, IFN-γ–producing Th1 cells and IL-17–producing Th17 cells isolated from the inflamed arteries were multifold reduced (P = 0.0087 and P = 0.0022; respectively) (Figure 3, G–J). Immunostaining visualized the powerful immunosuppressive effects induced by disabling NOTCH4 signaling in the transferred CD8+ Treg cells. If the CD8+ Tregs were rendered competent by inhibiting NOTCH4 signaling, the arteries contained only a fraction of vasculitogenic CD3+ T cells (Figure 3K). Also, disrupting the NOTCH4 signaling pathway in transferred CD8+ Treg cells resulted in significant reduction of IFNG, IL17, IL6, TNF, and IL1B transcripts (Figure 3L).
Taken together, these findings showed that NOTCH4 was a critical regulator of CD8+ Treg cells in vivo: enhanced NOTCH4 signaling promoted vascular inflammation and NOTCH signaling blockade protected the artery from inflammatory attack.
NOTCH4 signaling regulates the surface translocation of NOX2.
A typical feature of CD8+ Treg cells is the expression of NOX2 on the cell surface (8). Quantification of cell surface NOX2 expression on peripheral blood CD8+FOXP3+ Treg cells and on ex vivo–induced CD8+CD45RA+CCR7+ Treg cells confirmed that reduced function was associated with reduced NOX2 surface expression (Supplemental Figure 2, A and B). A mechanistic link between cell surface NOX2 and suppressive activity was established by forced overexpression of NOX2 in GCA Treg cells (Supplemental Figure 2C). Reconstitution of surface NOX2 expression normalized the suppressive activity of GCA CD8+ Treg cells (P = 0.015; Supplemental Figure 2C). Flow cytometry confirmed that NOX2 overexpression was sufficient to bring the enzyme to the cell surface (Supplemental Figure 2D).
The lack of surface NOX2 on GCA CD8+ Treg cells was not a consequence of FOXP3 instability. Flow cytometric analysis of CD8+ Treg populations expressing low or high levels of NOX2 (Supplemental Figure 3A) demonstrated similar FOXP3 expression. Also, FOXP3 expression was indistinguishable in control and GCA CD8+ Treg cells (Supplemental Figure 3B).
Considering the importance of NOTCH4 in CD8+ Treg function, we investigated the role of NOTCH4 signaling in NOX2 expression. In CD8+ Treg cells, NOTCH4 and NOX2 cell membrane expression were inversely related (Figure 4A). DAPT treatment increased the NOX2 surface density in both healthy and GCA CD8+ Tregs (Figure 4B). Knockdown of NOTCH4 in GCA CD8+ Treg cells improved NOX2 surface density (46% upregulation, P = 0.0098) (Figure 4C), while enforced NOTCH signaling by N4ICD transfection into healthy CD8+ Treg cells resulted in decreased surface NOX2 (42% reduction, P = 0.0005) (Figure 4D). The effect of N4ICD transfection in changing surface NOX2 was unrelated to FOXP3 stability (Supplemental Figure 3C).
Figure 4. NOTCH4 signaling regulates the surface translocation of NOX2.

(A) Surface expression of NOTCH4 and NOX2 on CD8+ Tregs determined by flow cytometry. Each dot represents 1 sample. Linear regression curve generated by GraphPad Prism software. P value calculated by F test. (B) CD8+ Tregs were treated with the NOTCH signal inhibitor DAPT or vehicle. Cell surface NOX2 expression determined by flow cytometry. Result from 8 controls and 7 patients. (C and D) CD8+ Tregs were transfected as indicated. (C) NOTCH4 or control siRNA. (D) N4ICD-containing or control vector. Cell surface NOX2 measured by flow cytometry. Representative histograms and results from 10–12 samples. (E) Healthy CD8+ Tregs were transfected with an N4ICD-containing vector and treated with actinomycin D or vehicle. Cell surface NOX2 was measured at the indicated times. Representative histograms and results from 4 samples. (F) NOX2 transcript concentrations in control and GCA CD8+ Tregs (RT-PCR, n = 7 samples each). (G) NOX2 protein determined by immunoblots in isolated cell membranes and in whole cell lysate from control and GCA CD8+ Tregs. CD8 was used as a loading control for cell membranes and β-actin for whole cell lysates. Data from 5 samples. (H–J) CD8+ Tregs were treated or transfected as indicated. NOX2 protein was quantified in isolated cell membranes and in whole cell lysates as above. CD8+ Treg cells were induced ex vivo. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired Mann-Whitney-Wilcoxon rank test (F and G), paired Mann-Whitney-Wilcoxon rank test (C and D), or 1-way ANOVA and post-ANOVA pairwise 2-group comparisons conducted with Tukey’s method (B).
To evaluate whether low NOX2 expression on GCA CD8+ Treg cells was an intrinsic defect or the result of the culture environment, we mixed GCA PBMCs (CellTrace Violet labeled) and healthy PBMCs and induced CD8+ Treg cells per standard protocol. Healthy PBMCs did not repair NOX2 expression, localizing the defect to the patients’ CD8+ Treg cells (Supplemental Figure 4).
To better understand how NOTCH4 signaling regulates NOX2, we examined the kinetics of the effect and probed dependence on transcriptional activity with actinomycin D. Monitoring of surface NOX2 on CD8+ Treg cells transfected with N4ICD demonstrated a delayed effect with maximal downregulation of surface NOX2 reached after 24 hours (Figure 4E). Actinomycin D treatment largely counteracted NOTCH-dependent NOX2 loss, confirming the requirement for transcriptional activity (Figure 4E).
We compared the amount of NOX2 transcripts and the subcellular distribution of the enzyme in functional and impaired CD8+ Treg cells. NOX2 transcripts were equally abundant in control and patient-derived CD8+ Tregs (Figure 4F), excluding a direct effect of NOTCH signaling on NOX2 transcription and pointing toward an effect involving NOX2 protein trafficking. To proceed with subcellular distribution studies, cell membranes were isolated from CD8+ Tregs, and NOX2 protein concentrations on the cell surface versus whole cell extracts were compared. Whole cell lysates contained similar amounts of NOX2 in healthy and GCA CD8+ Treg cells (Figure 4G), with a trend toward a higher NOX2 pool in patient-derived cells. Conversely, cell surface–residing NOX2 was barely detectable in GCA CD8+ Treg cells and was 3.7-fold higher in control CD8+ Treg cells (P = 0.0079; Figure 4G). Imaging studies confirmed that NOX2 was primarily localized on the cell surface in healthy CD8+ Tregs and in the cytoplasm of GCA Tregs (Supplemental Figure 6A). To examine the NOTCH4 signaling dependence of the surface localization of NOX2, NOTCH4 signaling in patient-derived CD8+ Treg cells was manipulated through 3 strategies: NOTCH4 knockdown, DAPT-mediated blocking of NOTCH4 processing, and N4ICD overexpression (Figure 4, H–J). In all experiments, NOX2 protein concentrations were compared in whole cell lysates versus isolated cell membranes, and confocal microcopy was applied to visualize surface-localized NOX2 versus cytoplasmic NOX2. None of the interventions had a significant effect on the overall amount of NOX2 expressed within the cells, nor on NOX2 transcript concentrations (Figure 4, H–J and Supplemental Figure 5). Conversely, NOX2 cell surface expression was highly dependent on NOTCH4 signaling. NOTCH4 knockdown brought NOX2 back to the cell surface (Figure 4H and Supplemental Figure 6B). Similarly, blocking NOTCH4 signaling with DAPT markedly increased the fraction of NOX2 on the cell membrane (Figure 4I and Supplemental Figure 6C). Also, enhanced NOTCH4 signaling after N4ICD overexpression reduced membrane-integrated NOX2 (Figure 4J and Supplemental Figure 6D).
Together, these data excluded NOTCH4 as a transcriptional regulator of NOX2, but implicated the transcription factor in regulating cellular trafficking of NOX2.
NOTCH signaling regulates the endocytic pathway.
NOX2 is a plasma membrane–localized enzyme able to produce extracellular superoxide anions. NOX2 has been localized to early endosomes and recycling endosomes, where it participates in cellular redox signaling (32, 33). Thus, we hypothesized that NOTCH4 signaling controls NOX2 trafficking within endocytic vesicles.
We first quantified endosomal compartments in CD8+ Treg cells. In confocal microscopy studies as well as flow cytometry studies, expression of the early endosome protein EEA1 and the recycling endosome protein RAB11 were significantly expanded in patient-derived cells (Supplemental Figure 7, A–D). Healthy CD8+ Treg cells contained low amounts of EEA1 and RAB11 and in colocalization studies, NOX2 protein mapped to the cell surface (Figure 5, A and B). In contrast, the cytoplasm of patient-derived CD8+ Treg cells was filled with EEA1+ and RAB11+ vesicles (Supplemental Figure 7, A–D) and NOX2 was almost exclusively localized to these cytoplasmic compartments (Figure 5, A and B).
Figure 5. NOTCH4 signaling regulates the endocytic pathway.

(A) Confocal microscopy of CD8+ Tregs stained for the early endosome marker EEA1 (green), NOX2 (red), and DAPI (blue). Scale bars: 5 μm. Intensities of EEA1/NOX2 overlap for 10 cells in each group are shown. (B) Confocal microscopy of CD8+ Tregs stained for the recycling endosome marker RAB11 (green), NOX2 (red), and DAPI (blue). Scale bars: 5 μm. Intensities of RAB11/NOX2 overlap in 10 cells from each group are shown. (C and D) CD8+ Tregs were incubated with FITC-labeled transferrin (Tfn) for the indicated times. Internalized Tfn-FITC was determined by flow cytometry. Kinetic measurements (C) for 4 control and 4 GCA Tregs. Tfn uptake at 10 minutes (D) from 9 samples each. (E) CD8+ Tregs were incubated with Tfn-FITC and visualized by confocal microscopy (10 minutes). Scale bars: 10 μm. (F and G) GCA CD8+ Tregs were treated with the endocytosis inhibitor Dynasore (F) or transfected with a RAB5DN-containing or control vector (G). Cell surface NOX2 protein quantified by flow cytometry. Representative histograms and results from 8 patients each. (H) Tfn uptake was quantified in GCA CD8+ Tregs treated with DAPT/vehicle. Flow cytometry data (10 minutes) from 8 samples. Internalized Tfn was visible by confocal microscopy. Scale bars: 10 μm. (I) Tfn uptake was quantified in GCA CD8+ Tregs transfected with NOTCH4 or control siRNA. Flow cytometry data (10 minutes) from 8 patients. (J) Tfn uptake was quantified by FACS in healthy CD8+ Tregs transfected with a NOTCH4 ICD-containing or empty vector. Results from 10 samples. (K) HC CD8+ Tregs transfected with empty vector or NOTCH4 ICD-containing plasmid were treated with Dynasore or vehicle. Cell surface NOX2 protein was determined by flow cytometry. Representative histograms and results from 9 samples. CD8+ Treg cells were induced ex vivo. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired Mann-Whitney-Wilcoxon rank test (A, B, and D), paired Mann-Whitney-Wilcoxon rank test (E–I), or 1-way ANOVA and post-ANOVA pairwise 2-group comparisons conducted with Tukey’s method (J).
Differential staining patterns for EEA1 and RAB11 in functional and impaired CD8+ Treg cells suggested differences in intracellular trafficking patterns of NOX2. To compare endocytic function, we dynamically detected internalized transferrin by flow cytometry. The cell surface and intracellular pool of the transferrin receptor CD71 was similar in control and patient cells (Supplemental Figure 7, E and F). Compared with healthy CD8+ Treg cells, GCA CD8+ Tregs absorbed distinctly more transferrin, with both cell types reaching peak performance at 10 minutes (Figure 5C). Transferrin uptake in CD8+ Treg cells from 9 healthy individuals and 9 GCA patients showed 38% higher uptake rates in the patients’ cells (P = 0.0012; Figure 5D). Results were confirmed by confocal microscopy imaging of the internalized transferrin (Figure 5E). Further evidence for intracellular vesicular transport regulating NOX2 turnover came from experiments utilizing Dynasore, an inhibitor that blocks endosome formation, and from manipulating RAB5, a protein indispensable for early endosome formation (22). Dynasore treatment increased NOX2 concentration on the plasma membrane of CD8+ Treg cells (P = 0.0078; Figure 5F). Similarly, transfection of dominant-negative RAB5 (RAB5DN), known to block endocytic protein transport, doubled the frequency of CD8+ Treg cells with high surface expression of NOX2 (Figure 5G).
To examine whether NOTCH4 has a role in regulating the endocytic pathway, we manipulated NOTCH4 signaling through 3 approaches: blocking with DAPT, NOTCH4 knockdown, and N4ICD overexpression (Figure 5, H–J). Endocytic capacity was assessed by transferrin internalization. DAPT treatment of patient-derived CD8+ Treg cells reduced transferrin uptake by 26% (P = 0.0078) and minimized the amount of cytoplasmic transferrin detected by imaging (Figure 5H). After NOTCH4 knockdown, GCA CD8+ Treg cells internalized only 54% of transferrin compared with controls (Figure 5I). Conversely, after N4ICD overexpression, 50% more transferrin was taken up (P = 0.0039; Figure 5J). Transfection of healthy CD8+ Treg cells with an N4ICD-containing plasmid reduced NOX2 cell surface density by 38% (P = 0.0039; Figure 5K). Disrupting endocytic vesicle formation with Dynasore enhanced the surface density of NOX2 and completely counteracted the effect of N4ICD-induced NOTCH signaling (Figure 5K).
These data assigned intracellular NOX2 to early and recycling endosomes, implicated the endocytic pathway in controlling NOX2 cell membrane localization, and identified NOTCH4 as a positive regulator of the protein internalization, sorting, and trafficking process.
NOTCH4 signaling regulates RAB5A and RAB11A transcription.
As transcription factors, NOTCH receptors function primarily through transcriptional control of target genes. To discover how NOTCH4 signaling regulates intracellular vesicle transport, we screened for the expression of endosome-related genes in healthy and dysfunctional CD8+ Treg cells (Supplemental Figure 8). Patient-derived CD8+ Tregs had abundant transcripts for RAB5A, a gene encoding a small GTPase required for plasma membrane fusion and early endosome formation (22), and for RAB11A, a marker for recycling endosomes (27). Conversely, nonfunctional GCA CD8+ Treg cells produced lower transcript concentrations for RAB7A, which is involved in early to late endosomal maturation (24). In a validation cohort, RAB5A and RAB11A transcripts were 1.5-fold and 1.3-fold higher, respectively, in GCA compared with healthy CD8+ Treg cells (P = 0.0001, P = 0.0039; Figure 6A).
Figure 6. NOTCH4 signaling regulates RAB5A and RAB11A transcription.

(A) CD8+ Tregs were induced from controls or GCA patients. RAB5A and RAB11A transcripts were determined by q-PCR. Mean ± SEM from 10 samples each. (B) Healthy CD8+ Tregs were transfected with empty vector or a NOTCH4 ICD overexpression plasmid, and RAB5A and RAB11A transcript levels were determined by q-PCR. Mean ± SEM from 6 control and 6 GCA samples. (C) GCA CD8+ Tregs were treated with DAPT or vehicle. RAB5A and RAB11A mRNA were quantified by q-PCR. Mean ± SEM from 10 samples. (D) GCA CD8+ Tregs were transfected with control or NOTCH4 siRNA. RAB5A and RAB11A transcript expression was determined by q-PCR. Mean ± SEM from 6 samples. (E) CD8+ Tregs were generated from GCA patients. ChIP assays targeting NOTCH4 or control IgG were performed on the promoter of RAB5A or a negative site. The signal was normalized to 10% of input. Mean ± SEM from 6 samples. (F) GCA CD8+ Tregs were treated with DAPT or vehicle. ChIP assays targeting NOTCH4 were performed on the promoter of RAB5A. The signal was normalized to 10% of input. Mean ± SEM from 6 samples. (G) In GCA CD8+ Tregs, ChIP assays targeting NOTCH4 or control IgG were performed on the promoter of RAB11A or a negative site. The signal was normalized to 10% of input. Mean ± SEM from 6 samples. (H) CD8+ Tregs were treated with DAPT or vehicle. The occupancy of NOTCH4 on the RAB11A promoter was determined by ChIP assay and normalized to 10% of input. Mean ± SEM from 6 samples. CD8+ Treg cells were induced ex vivo for all experiments. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired Mann-Whitney-Wilcoxon rank test (A) or paired Mann-Whitney-Wilcoxon rank test (B–D, F, and H).
To verify that both RAB5A and RAB11A are NOTCH4 target genes, transcript expression for the 2 genes was assessed after blocking (DAPT treatment, NOTCH4 knockdown) or enhancing (N4ICD overexpression) NOTCH signaling. Strengthening NOTCH4 signaling increased RAB5A (2.8-fold, P = 0.0023) and RAB11A (1.9-fold, P = 0.0043) transcript levels (Figure 6B), whereas dampening NOTCH4 signaling lowered transcript concentrations (DAPT treatment, RAB5A: 45% reduction, P = 0.002 and RAB11A: 49% reduction, P = 0.002; NOTCH4 knockdown, RAB5A: 86% reduction, P = 0.0022 and RAB11A: 52% reduction, P = 0.0260; Figure 6, C and D).
To investigate whether NOTCH4 directly affected RAB5A and RAB11A transcription, NOTCH4 bound to the RAB5A and RAB11A promoters was immunoprecipitated. Compared with the IgG control, ChIP with anti-NOTCH4 antibody showed a much higher occupancy on both promotors (Figure 6, E–G). Neither control IgG nor anti-NOTCH4 yielded a signal on the negative site (Figure 6, E–G). DAPT treatment almost depleted all NOTCH4 occupancy on both promotors (Figure 6, F and H). We also evaluated the occupancy of NOTCH4 on the RAB7A promoter. ChIP with anti-NOTCH4 antibody yielded no evidence for NOTCH4 binding to the RAB7A promoter (Supplemental Figure 9A).
Thus, NOTCH4 is a transcriptional regulator of key genes in the endocytic pathway, enabling NOTCH family receptors to control subcellular protein distribution, including the translocation of NOX2 to the cell surface.
NOTCH4 signaling modulates exosome production.
Comparative transcriptome analysis in healthy and GCA CD8+ Treg cells indicated that the induction of RAB5A and RAB11A transcripts in the NOTCH4hiCD8+ Treg cells from patients co-occurred with the suppression of RAB7A transcription (Figure 7A). RAB7A is required for the maturation of late endosomes and exosome secretion (24, 25). We explored whether GCA CD8+ Treg cells have a defect in exosome production. Release of CD63+ exosomes was 49% reduced in GCA CD8+ Treg cells when compared with healthy cells (Figure 7B; P = 0.0002). Quantification of exosomal NOX2 revealed that exosomes from GCA CD8+ Treg cells contained less of the oxidase than control exosomes (40% reduction, P = 0.0002; Figure 7C).
Figure 7. NOTCH4 signaling regulates exosome production.

(A) Gene expression for the late endosome marker RAB7A in CD8+ Tregs (RT-PCR, n = 10). (B) Quantification of CD8+ Treg–produced exosomes (n = 10 controls, 7 patients). (C) NOX2 protein expressed on secreted exosomes. Representative histograms and FACS results from 8 samples each. (D–F) GCA CD8+ Tregs were transfected with NOTCH4 or control siRNA. (D) RAB7A transcripts. RT-PCR from 6 patients. (E) Secreted exosomes from n = 7 samples. (F) Exosomal NOX2 protein. Flow cytometry from 6 samples. (G–I) Healthy CD8+ Tregs were transfected with an N4ICD plasmid or empty vector. (G) RAB7A transcripts quantified by RT-PCR in 6 samples. (H) Secreted exosomes determined in 8 samples. (I) Exosomal NOX2 protein. Flow cytometry from 6 samples. (J and K) Control CD8+ Tregs (J) and GCA CD8+ Tregs (K) were transfected as indicated. (J) HES5-containing or empty vector; (K) HES5 or control siRNA. RT-PCR for RAB7A transcripts in n = 6 samples. (L) ChIP assays targeting HES5 or control IgG were performed on the promoter of RAB7A or a negative site. Signal normalized to 10% of input. Data from 6 GCA CD8+ Treg samples. (M) GCA CD8+ Tregs were transfected with HES5 or control siRNA. Occupancy of HES5 on the RAB7A promoter was examined by ChIP assay. Signal normalized to 10% of input. n = 6 samples. (N) GCA CD8+ Tregs were treated with the lysosomal inhibitors chloroquine (CQ) and leupeptin (Leu), the proteasome inhibitor MG132, or vehicle. Cell surface NOX2 was evaluated by FACS. Representative histograms and results from 5 samples. (O) GCA CD8+ Tregs were transfected with a RAB5DN-containing, a RAB7DN-containing, or control vector. Lysosome intensity was measured by LysoTracker. Representative histograms and FACS results from 6 samples. CD8+ Treg cells were induced ex vivo. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired (A–C) and paired (D–K and M) Mann-Whitney-Wilcoxon rank test, or ANOVA and post-ANOVA pairwise 2-group comparisons conducted with Tukey’s method (N and O).
Knockdown of NOTCH4 in GCA CD8+ Treg cells resulted in upregulation of RAB7A (P = 0.0022) and more than doubled exosomal release (P = 0.0156; Figure 7, D and E). Disrupting NOTCH4 signaling by transfecting NOTCH4 siRNA also improved the packaging of NOX2 into the exosomes (49% increase, P = 0.0313; Figure 7F). Enforced NOTCH4 signaling in healthy CD8+ Treg cells by transfecting N4ICD resulted in prompt suppression of RAB7A transcription (85% reduction, P = 0.0107; Figure 7G) and diminished exosome production (67% reduction, P = 0.0078; Figure 7H). Exosomes released from N4ICD-transfected CD8+ Treg cells contained significantly less NOX2 (P = 0.0313; Figure 7I).
We wanted to clarify how NOTCH4 could suppress RAB7A transcription, thereby restraining exosomal release. Since NOTCH proteins always act as transcriptional activators, NOTCH4 is unlikely to function as a transcriptional repressor of RAB7A. However, bHLH family proteins, known as classic NOTCH target genes, are transcription repressors. HES5, encoding the bHLH protein HES5, was expressed in CD8+ Treg cells and controlled by NOTCH4 (Supplemental Figure 10). We examined whether NOTCH4 mediates the RAB7A downregulation through HES5. HES5 overexpression inhibited RAB7A mRNA expression but did not affect RAB5A and RAB11A transcripts (Figure 7J and Supplemental Figure 11A). Conversely, HES5 siRNA-mediated knockdown increased RAB7A transcripts, while RAB5A and RAB11A mRNA levels remained unchanged (Figure 7K and Supplemental Figure 11B). ChIP assays confirmed HES5 occupancy on the RAB7A promoter (Figure 7L), identifying RAB7A as a HES5 target gene. Knockdown of HES5 via siRNA removed HES5 from the RAB7A promotor (Figure 7M), while no HES5 was detected at the RAB5A and the RAB11A promoter (Supplemental Figure 9, B and C).
We tested whether NOTCH4 signaling affected other late endosome–related genes, including VAMP8 and RAB27A (Supplemental Figure 13, A and B). While NOTCH4 signaling was highly effective in regulating RAB7A, VAMP8 and RAB27A transcripts remained unaffected after healthy CD8+ Tregs were transfected with N4ICD.
These data demonstrated that RAB7A is a HES5 target gene and that NOTCH4 controls exosome production through the NOTCH4/HES5/RAB7 axis.
The cell surface expression of many receptor molecules is attenuated when ligand-induced endocytosis is followed by lysosomal or autophagosomal degradation instead of recycling to the plasma membrane. First, we determined NOX2’s subcellular localization in relation to 3 vesicular markers: the endosomal marker HRS, the lysosomal marker LAMP1, and the autophagosomal marker p62 (Supplemental Figure 12). In healthy CD8+ Treg cells, HRS had no overlap with LAMP1 or p62, but part of the NOX2 signal colocalized with HRS.
To understand whether the reduction of surface NOX2 on GCA CD8+ Treg cells resulted from lysosomal digestion, we mapped the colocalization of NOX2 with lysosomal vesicles and tested the effect of lysosomal inhibitors on surface translocation. The lysosomal inhibitors chloroquine (CQ) and leupeptin (Leu) had no effect on NOX2 surface expression. However, the proteasome inhibitor MG132 dramatically increased NOX2 plasma membrane expression on CD8+ Treg cells (Figure 7N), assigning NOX2 degradation to the proteasome and not the lysosome. Also, confocal imaging demonstrated very limited colocalization of NOX2 with the lysosomal marker LAMP1 in both control and GCA CD8+ Treg cells (Supplemental Figure 13C). Finally, we assessed how manipulating RAB5 and RAB7 affected lysosomal activity and NOX2 surface expression. Transfection of RAB5DN, targeting early endosomes, enhanced surface NOX2 (Figure 5G) while suppressing lysosome formation (Figure 7O). In contrast, transfection of RAB7DN, disrupting transport to late endocytic compartments, reduced the cells’ lysosomal load (Figure 7O) but left surface NOX2 expression unaffected (Supplemental Figure 13D).
To determine whether exosomal release was relevant for the overall pool of the oxidase in CD8+ Treg cells, we suppressed transport to late endosomes via RAB7DN transfection and measured intracellular NOX2 by flow cytometry. Halting exosome release, which mimicked conditions in GCA CD8+ Treg cells, resulted in intracellular retention of the protein (Supplemental Figure 13E).
To better define whether intracellular degradation determines NOX2 fate, we tested whether trafficking of the oxidase is affected by flux through macroautophagy (Supplemental Figure 14). CD8+ Treg cells from healthy individuals were treated with the autophagy activator A769662 (which also activates the energy sensor AMPK). A769662 treatment enhanced the cellular load of the autophagosome marker p62, but NOX2 cell surface expression was unaffected (Supplemental Figure 14A). Colocalization studies by confocal imaging confirmed that NOX2 did not map to the p62+ autophagosome (Supplemental Figure 14B).
Together, these data allocated NOX2 to an early endosome/recycling endosome storage compartment; implicated exosomal release as one pathway of NOX2 loss; and identified proteasomes, not lysosomes or autophagosomes, as the NOX2-degrading organelles.
Discussion
Treg cells function as universal protectors against autoimmune and autoinflammatory disease by exerting control on the numbers and activity of innate and adaptive immune cells (34). Here, we have implicated vesicular trafficking in the functional fitness of CD8+ Treg cells that safeguard the aorta and large arteries from destructive inflammation. CD8+ Treg cells utilize exosomal release of the oxidase NOX2 to suppress surrounding CD4+ T cells (8). Dysfunctional CD8+ Treg cells had high expression of NOTCH4 receptors and sustained NOTCH signaling activity and redirected intracellular protein trafficking by deviating the endosomal gene program. Instead of routing the oxidase NOX2 toward the multivesicular body sorting machinery and exosomal expulsion, defective CD8+ Treg cells retrieved cell surface NOX2 into early and slow recycling endosomes. Faulty sorting of NOX2 left nonfunctional CD8+ Treg cells deprived of their core immunosuppressive mechanism, the secretion of NOX2+ exosomes. Functional failure of CD8+ Treg cells was reversible and druggable, identifying hitherto unrecognized targets to restore tissue tolerance.
Treg cells utilize multiple processes to suppress immunity, e.g., release of antiinflammatory cytokines, induction of adenosine, and conversion of DCs into tolerogenic antigen-presenting cells (35). Mostly, such suppressor T cells are CD4+ T cells and are generated in the thymus or emerge in the periphery (36). The CD8+ Treg cells examined here live in secondary lymphoid organs and function by controlling the overall size of the CD4+ T cell compartment (8). They protect tissues by preventing the entrance of T cells and macrophages into immunoprivileged sites, such as the arterial wall. CD8+ Treg cells fail in older individuals, contributing to senoinflammation, and in patients with large vessel vasculitis. Data presented here add a new mechanism through which the immune system maintains tissue tolerance.
The pinnacle abnormality in dysfunctional CD8+ Treg cells was the aberrant activation of the NOTCH signaling pathway. Cell fate decisions, cellular proliferation, and cell death regulation are all NOTCH signaling dependent (37). Four different NOTCH receptors (NOTCH1–4) bind to 5 canonical ligands (Jagged1 and -2, and Delta-like protein 1, 3, and 4). Cleavage of the NOTCH receptor leads to the release of the intracellular domain that regulates target gene transcription, including the basic helix-loop-helix transcription factors HES and HEY (15, 38). NOTCH mutations lead to abnormalities in the vascular system, where NOTCH receptors and ligands are widely expressed (39, 40). NOTCH1 and -4 are typically found on endothelial cells (3). Mutations in NOTCH1, -2, or -3 contribute to a range of congenital cardiovascular syndromes. So far, NOTCH4 mutations have not been described (41), but NOTCH4 polymorphisms have been associated with risk of developing schizophrenia (42). NOTCH signaling is typically activated in lymphoid malignancies. Ligand-dependent and ligand-independent modes of usurping the signaling pathway contribute to supraphysiologic NOTCH activation (43). Interactions between NOTCH and NOTCH ligands are fundamentally involved in thymic T cell development and remain important as a costimulatory amplifier in adult T cells. NOTCH1 is aberrantly expressed on a subset of circulating CD4+ T cells in patients with GCA, where it mediates mTORC1 activation and T cell lineage commitment (3). Notably, aberrant NOTCH1 expression was limited to CD4+ T cells, while CD8+ T cells expressed NOTCH4, exposing both T cell populations to enhanced TCR signaling and accelerated differentiation. NOX2+CD8+ Treg cells strictly derive from naive CD8+ T cells, and NOTCH-induced differentiation of naive CD8+ T cells may be the cause of age-dependent decline in CD8+ Treg cell function (8).
NOTCH has been implicated in a broad range of cellular functions, but here we report that NOTCH signaling directly affects endocytic recycling. We clarified how NOTCH4 interferes with vesicular trafficking: N4ICD and its target gene HES5 access promoter regions of the RAB5A, RAB7A, and RAB11A genes. NOTCH-induced rerouting of endovesicular trafficking required differential effects on these genes; RAB5A and RAB11A were upregulated, whereas RAB7A was repressed. The result was the expansion of early endosomes, diversion of endosomes away from lysosomal degradation, and failure to produce exosomes (Figure 8). It is unlikely that NOTCH4-induced changes in the endocytic pathway are selective for NOX2. Rather, other cargo proteins internalized and passed through endosomes may be similarly altered in subcellular distribution and function.
Figure 8. Scheme of vesicular trafficking system in CD8+ Treg cells.

Healthy Treg cells: Low NOTCH4 signaling induces low expression of RAB5A and RAB11A and high expression of RAB7A, leading to the repression of early and recycling endosomes and expansion of late endosomes. NOX2 is secreted on the surface of exosomes and reaches surrounding cells. GCA Treg cells: High NOTCH4 signaling induces high expression of RAB5A and RAB11A and suppresses expression of RAB7A, rerouting endosomal trafficking. Early and recycling endosomes are favored and late endosomes are disfavored, keeping NOX2 in an intracellular, nonsecretory compartment.
The primary role of NAPDH oxidase is the generation of superoxide to kill pathogens, but also to provide ROS for rapid intracellular redox signaling. ROS release must be highly compartmentalized to avoid oxidative damage. Placing NOX2 onto the plasma membrane guides ROS release to the extracellular milieu. Pathogens internalized in phagosomes are exposed to free radicals by the fusion of intracellular vesicles with the phagosomal membrane (44). Endosomal NADPH oxidase generates ROS for intracellular redox signaling (45). Implicating NOX2 in T cell–T cell communication, specifically in the suppression of CD4+ T cell activation, extends the role of the oxidase as a critical immunoregulator. Functional and nonfunctional CD8+ Treg cells were easily distinguished by the subcellular NOX2 distribution; plasma membrane localization was tightly associated with immunosuppressive ability. Loss of function was linked to sequestering NOX2 in an intracellular storage compartment, consisting of early endosomes and slow recycling endosomes. In macrophages, intracellular NADPH oxidase has been localized to a RAB5+RAB11+ endosomal fraction, and fusion of RAB27+ organelles with the plasma membrane has been implicated in bringing NOX2 to the cell surface (46). NOX2+ exosomes are the core principle through which CD8+ Treg cells mediated their suppressive function (8). Dysfunctional CD8+ Treg cells from patients with GCA failed to build NOX2-containing exosomes. Instead, they accumulated NOX2 in RAB5+ early endosomes and RAB11+ recycling endosomes. NOTCH-induced RAB7 downregulation prevented NOX2 packaging for exosomal expulsion. Defective CD8+ Treg cells essentially assigned NOX2 to a cytoplasmic storage pool, instead of releasing the enzyme into the extracellular environment. CD8+ Treg cells showed a dichotomous distribution in NOX2 surface expression, indicating that cell membrane–located NADPH oxidase is not a universal property of these immunosuppressive cells. NOX2 participates in the formation of a multiprotein complex that ultimately functions as a NADPH oxidase and ROS producer. Intracellular trafficking and cell surface retention of NOX2 may be determined by additional factors that regulate the availability and trafficking of other components of the enzyme complex. Our data mechanistically linked loss of function of GCA CD8+ Tregs cells with failed ROS production, thus involving assembly and activation of the 5-protein complex. Patients with chronic granulomatous disease, caused by the inherited loss of one of the subunits of NADPH oxidase, not only develop severe immunodeficiency, but frequently present with autoimmune and autoinflammatory disease (47, 48). Whether CD8+ Treg cell failure contributes to the burden of autoimmune disease in these patients is unknown.
CD8+ T cells, best known for their ability to kill target cells, rely on the fusion of cytotoxic granules with endosomal elements before these vesicles fuse with the plasma membrane as the carriers of cytotoxic mediators (49). Utilization of the endosomal machinery for granule expulsion thus appears to be fundamentally associated with the CD8+ T cell subset, providing an explanation for the unique mechanism of action of CD8+ Treg cells. Our data predict that compromised production of immunosuppressive exosomal vesicles is not the only defect of GCA CD8+ T cells. Rather, CD8+ T cells in these patients may also have impaired cytotoxic capability.
Mutations in endolysosomal proteins have been associated with a wide range of diseases and phenotypes, in line with their fundamental role in endocytosis, lysosome biogenesis, phagocytosis, and autophagy (50). The composition of the plasma membrane depends upon continuous endocytic uptake — routing of cargo from the early to the late endosome and the lysosome for degradation. Endosomes and autophagosomes sequester their cargo through different mechanisms, with convergence of pathways upon fusion with lysosomes. Alternatively, recycling endosomes carry the cargo back to the cell membrane. Transport of cargo-loaded secretory vesicles to predetermined areas affects cell polarity and maturation, and release of multivesicular bodies shapes the secretory repertoire of the cell (27, 51). Data presented here demonstrated that NOTCH4 signaling introduced a switch from making NOX2 available for exosomal release to keeping it in an intracellular storage compartment. Transcriptome analysis in functional and nonfunctional CD8+ Treg cells identified RAB5A, RAB7A, and RAB11A as differentially expressed. RAB GTPases function as key regulators in intracellular vesical transport, controlling different steps of membrane budding, vesicle transport, tethering, and fusion (20). We demonstrated that all 3 RAB GTPases are NOTCH4 target genes. By transcriptional enhancement of RAB5A and RAB11A, NOTCH4 favored uptake into early endosomes and passage into recycling endosomes, resulting in intracellular entrapment of NADPH oxidase. In parallel, the NOTCH4 target gene HES5 functioned as a RAB7A repressor, paralyzing the passage of cargo into the lysosomal pathway. Our data provided no evidence that NOX2 is degraded in the endolysosomal system. Sequestering of a cell surface protein in an intracellular storage compartment, where it is available for recycling to the cell surface, has been described for the water channel aquaporin in renal tubule epithelium and glucose transporter 4 (GLUT-4). In muscle and adipose tissues, GLUT-4 traffics between the cell surface and exocytic storage vesicles but is partially retained in early and recycling endosomes. Flux of GLUT-4 through the endosomal system is regulated by insulin, and aberrations in the trafficking pattern lead to insulin resistance (52–54).
In summary, we have discovered that exosomal NOX2 is the key mediator of CD8+ Treg–dependent immunosuppression and that Treg fitness is directly related to intracellular vesicular trafficking of NOX2. In large vessel vasculitis, CD8+ Treg cells failed to release NOX2-containing vesicles, leading to unopposed vascular inflammation. Functional failure of antiinflammatory CD8+ Treg cells was a consequence of aberrant signaling through the NOTCH4 receptor. The molecular signature of NOTCH4hiCD8+ Treg cells included upregulation of the GTPases RAB5 and RAB11 and repression of RAB7. Reprogramming of intracellular vesicle trafficking deviated NOX2 toward the recycling endosome, assigning the oxidase to a storage pool. Restraining NOX2-containing vesicles from entering the late endosomal pathway essentially paralyzed exosomal packing, maturation, and release. NOTCH4 knockdown consistently forced NOX2 back onto the plasma membrane and into immunoinhibitory exosomes. Interfering with the NOTCH4 signaling pathway was sufficient to protect arteries from inflammatory attack, opening opportunities for the design of potentially novel immunomodulatory treatments with potential application in autoimmune disease, tissue inflammation, and cancer immunotherapy.
Methods
Patients and healthy individuals.
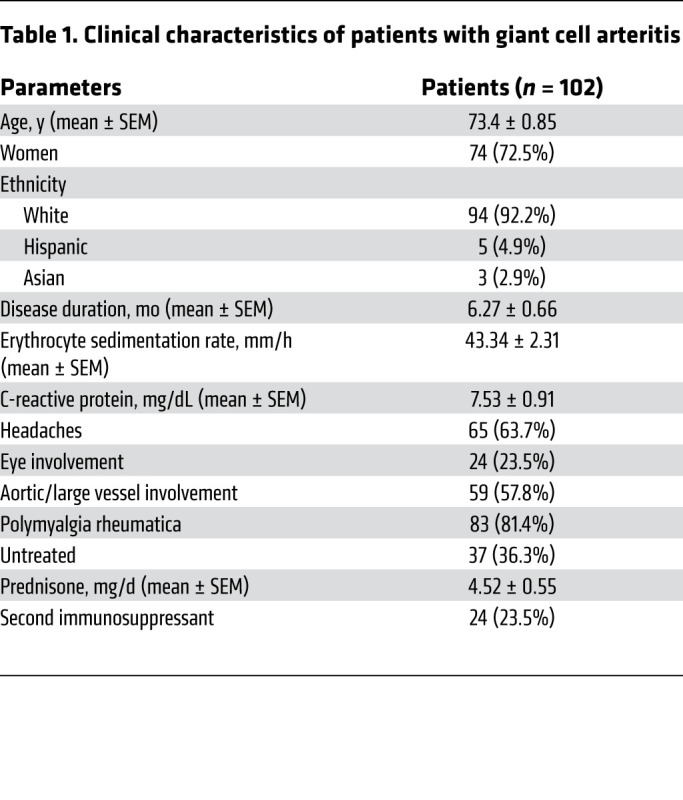
A total of 102 patients with biopsy-positive GCA and 63 age-matched healthy controls were enrolled in this study. Demographic and clinical characteristics of GCA patients are summarized in Table 1. The study was approved by the Stanford University IRB and written informed consent was obtained from each patient and healthy individual as appropriate.
Table 1. Clinical characteristics of patients with giant cell arteritis.
Cell isolation.
PBMCs from GCA patients and healthy controls were isolated by gradient centrifugation with Lymphoprep Medium (Cosmo Bio USA) and cultured in RPMI 1640 (Thermo Fisher Scientific) medium with 10% FBS (Gemini) plus penicillin/streptomycin (Thermo Fisher Scientific). Naive CD4+ T cells were purified from PBMCs using EasySep human naive CD4+ T cell enrichment kits (STEMCELL Technologies, Inc.). Purity of cell populations was consistently greater than 90%.
Reagents and transfections.
All reagents were used according to the manufacturer’s instructions. The human HES5 expression plasmid was purchased from Origene Technology. RAB5DN (13051) and RAB7DN (12662) plasmids were obtained from Addgene. siRNAs targeting human NOTCH4, HES5, and RAB5A were purchased from Santa Cruz Biotechnology, Inc. The N4ICD plasmid was provided by Anthony Capobianco (Molecular Oncology Program, Division of Surgical Oncology, Dewitt Daughtry Family Department of Surgery, Miller School of Medicine, University of Miami, Miami, Florida, USA). Using the T Cell Nucleofector Kit (Lonza, VVPA-1002), 0.5 μg plasmid or 10 pmol siRNA was transfected into 2 × 106 cells.
In vitro generation of CD8+ Treg cells.
CD8+ Treg cells were induced in an in vitro culture system as previously described (8, 55). Specifically, PBMCs (1 × 106 cells/mL) were stimulated with 0.1 ng/mL anti-CD3 antibody (BioLegend, OKT3, 317315) and 5 ng/mL recombinant human IL-15 protein (Sino Biological, GMP-10360-HNCE) for 6 days. CD8+CCR7+ Treg cells were negatively selected with EasySep human naive CD8+ T cell isolation kits (STEMCELL Technologies, Inc.). Over 90% of the purified cells had a CD8+FOXP3+CCR7+ phenotype.
Exosome isolation.
CD8+CCR7+ Treg cells generated in vitro were cultured in RPMI 1640 medium with 10% exosome-free FBS (System Biosciences) and 5 ng/mL recombinant human IL-15 for 24 hours. Exosomes were isolated from the culture medium with the Total Exosome Isolation Reagent (Thermo Fisher Scientific) and quantified using the ExoELISA-ULTRA Complete Kit (System Biosciences).
In vitro T cell suppressor assays.
To quantify the suppressive function of CD8+ Treg cells, naive CD4+ T cells (5 × 104 cells per 100 μL) were mixed with CD8+ Treg cells (2.5 × 104 cells per 100 μL) and activated with anti-CD3/anti-CD28 beads (Thermo Fisher Scientific, 11132D) at a 1:1 ratio for 15 minutes. CD4+ T cell activation was evaluated by quantifying phosphorylated ZAP70 by flow cytometry. For DAPT treatment, DAPT was added during the CD8+ Treg differentiation process and washed off before mixing the cells with CD4+ T cells. The inhibition efficiency of CD8+ Treg cells was calculated as follows: Inhibition efficiency = (pZAP70%(No Treg) – pZAP70%(Treg)) / pZAP70%(No Treg).
Naive CD4+ T cells were CFSE labeled and incubated with CD8+ Treg cells for 30 minutes. CD8+ Treg cells were then removed and CD4+ T cells were stimulated with T-Activator anti-CD3/anti-CD28 beads for 5 days. Proliferation of CD4+ T cells was determined by flow cytometric measurement of CFSE dilution.
T cell suppressor assay in vivo.
NSG mice (The Jackson Laboratory) were housed in institutional animal facilities under specific pathogen–free conditions and at 8 weeks of age were reconstituted by i.p. injection of healthy PBMCs (10 million/mouse) with or without CD8+ Treg cells (5 million/mouse). Five days later, cell numbers of CD4+ T cells in the spleen and the blood were quantified to assess the suppressive function of CD8+ Treg cells.
Flow cytometry.
For cell surface proteins, cells were fixed with Fix I buffer (BD Phosflow) and stained with antibodies listed in Table 2. For intercellular proteins, cells were fixed with Fix I buffer (BD Phosflow), permeabilized with Perm buffer III (BD Phosflow), and stained with antibodies listed in Table 2. For quantification of IFN-γ– and IL-17–producing CD4+ T cells, cells were treated with 50 ng/mL PMA (Tocris), 500 ng/mL ionomycin (Tocris), and 5 μg/mL Brefeldin A (BioLegend) for 5 hours, followed by fixation, permeabilization, and staining with specific antibodies. LysoTracker staining was performed with LysoTracker Red DND-99 (L7528, ThermoFisher) according to the manufacturer’s instruction. Flow cytometry was performed on an LSRII flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star Inc.).
Table 2. Antibody list.

Real-time PCR.
Total mRNA was extracted with a Direct-zol RNA MiniPrep Kit (Genesee Scientific). cDNA was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Real-time PCR was performed with SYBR Green qPCR Master Mix (Bimake) and β-actin was used as an internal control. All primers are listed in Table 3.
Table 3. Primer list.

Western blot.
Immunoblots were performed using standard protocols as previously described (56). Lysates were mixed with 6× SDS loading buffer and boiled at 95°C for 5 minutes. Lysates were resolved by Mini SDS-PAGE Gel (Bio-Rad) and transferred onto PVDF membranes, blocked in 5% milk or 2.5% BSA at room temperature for 1 hour, and incubated with appropriate primary antibodies at 4°C overnight. Subsequently, membranes were incubated with secondary antibody (room temperature, 1 hour), and proteins were detected using Pico PLUS Chemiluminescence Reaction (Thermo Fisher Scientific); β-actin was used as an internal control. All antibodies are listed in Table 2.
Cell surface protein isolation.
Isolation of cell surface proteins was achieved using Cell Surface Protein Isolation Kits (Abcam). Briefly, CD8+ Treg cells were incubated with sulfo-NHS-SS-biotin for 30 minutes and quenched for 5 minutes. Cells were then lysed and centrifuged at 10,000g for 2 minutes. Supernatant was incubated with streptavidin beads for 1 hour. Streptavidin beads were collected and washed with lysis buffer, followed by incubating with elution buffer for 30 minutes to release the proteins for subsequent analysis.
ChIP.
ChIP assays were performed using ChIP-IT High Sensitivity Kits (Active Motif). Specifically, 5 × 105 CD8+ Tregs were cross-linked with Complete Fix buffer for 30 minutes at room temperature and quenched with Quench buffer for 5 minutes. Cells were resuspended with ChIP Prep buffer and nuclei were extracted with a Dounce homogenizer, resuspended with ChIP buffer, and sonicated into yield chromatin fragments of 300 to 1000 bp. Lysates were incubated with Protein G beads and immunoprecipitated with 8 μg of anti-NOTCH4 (Cell Signaling Technology, 2423) or anti-HES5 antibody (Abcam, ab194111) overnight at 4°C. Proteins were washed and eluted from Protein G beads, followed by incubation with RNase A and proteinase K. DNA was cleaned using a PCR purification kit (Qiagen) and promoter regions of RAB5A, RAB7A, and RAB11A were amplified by qPCR via specific primers. All primer pairs are included in Table 3.
Transferrin uptake assay.
CD8+ Treg cells were starved in FBS-free medium for 1 hour and incubated with 20 μg/mL FITC-transferrin (Thermo Fisher Scientific, T13342) at 37°C for 10 minutes or indicated time periods. Surface-bound FITC-transferrin was removed with cold acidic buffer (PBS containing 0.5% acetic acid and 500 mM NaCl) for 30 seconds. Cells were washed with cold PBS and fixed with Fix I buffer, followed by detecting the intracellular FITC-transferrin using flow cytometry and/or immunofluorescence staining.
Immunofluorescence.
Cells were fixed with Fix I buffer (BD Phosflow) and permeabilized with Perm buffer III (BD Phosflow), followed by staining with primary antibodies (4°C, overnight) plus secondary antibodies (room temperature, 2 hours). Cell nuclei were stained with DAPI in mounting medium (Sigma-Aldrich). Cells were visualized using the LSM710 confocal microscope (Carl Zeiss) with a Plan-Neofluar ×63/1.3 NA oil objective lens. All primary and secondary antibodies are listed in Table 2.
IHC.
CD3 protein expression in tissue sections was detected using IHC as previously described. Briefly, frozen sections of arterial tissues were stained with anti–human CD3 antibody (Dako, A0452) and developed with anti-rabbit secondary antibody (Invitrogen, A11304). The LSM710 confocal microscope (Carl Zeiss) with a Plan-Neofluar ×40/1.3 NA oil objective lens was used to acquire images.
Human artery–NSG mouse chimeras.
Human artery–NSG mouse chimeras were generated as previously described (3). Briefly, NSG mice (from The Jackson Laboratory) were used at the age of 8 weeks. Human temporal and axillary arteries (diameters of 4–5 mm and 6–9 mm, respectively) were cut into fragments and transplanted subcutaneously into the back. Chimeras were i.p. injected with LPS (10 μg/mouse) on day 7 and reconstituted with PBMCs from GCA patients (10 million/mouse) on day 8. Mice transplanted with the arteries from the same donor were randomly assigned into 2 groups receiving control or genetically manipulated CD8+ T cells. CD8+ Treg cells (5 million/mouse) were coadministered with patient-derived PBMCs on day 8. Arterial grafts were harvested on day 15 and processed for RNA extraction or embedded for IHC analyses. For all animal experiments, the investigators performing the tissue analysis were blinded to the treatment assignment of the mice. All protocols were approved by the Stanford University IACUC.
Statistics.
All data are presented as mean ± SEM. Paired and unpaired Mann-Whitney-Wilcoxon rank tests were used for 2-group comparisons as appropriate. One-way ANOVA was used for comparison of more than 2 groups and post-ANOVA pairwise 2-group comparisons were conducted with Tukey’s method. Data were analyzed with Prism 7 (GraphPad Software), and P values less than 0.05 were considered statistically significant. All data analyses were overseen by the Department of Biomedical Data Science at Stanford University.
Study approval.
The human studies were approved by the Institutional Review Board at Stanford University. The animal protocol was approved by the Animal Care and Use Committee at Stanford University. Written informed consent was obtained from participants before their participation in the study as appropriate.
Author contributions
CMW, KJ, and JJG designed the study and analyzed the data. KJ, ZW, BW, HZ, JQ, and YW performed experiments. GB and KJW provided expertise in case identification. CMW, KJ, and JJG wrote the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the NIH (R01 AR042527, R01 HL117913, R01 AI108906, R01 HL142068, and P01 HL129941 to CMW and R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191 to JJG). KJ received fellowship support from the Govenar Discovery Fund.
Version 1. 09/22/2020
In-Press Preview
Version 2. 01/04/2021
Print issue publication
Funding Statement
KJ received fellowship support from the Govenar Discovery Fund.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(1):e136042.https://doi.org/10.1172/JCI136042.
Contributor Information
Ke Jin, Email: kjin88@stanford.edu.
Zhenke Wen, Email: zkwen@suda.edu.cn.
Bowen Wu, Email: bowenwu@stanford.edu.
Hui Zhang, Email: huizh@stanford.edu.
Jingtao Qiu, Email: jtqiu@stanford.edu.
Yanan Wang, Email: yananw@stanford.edu.
Kenneth J. Warrington, Email: warrington.kenneth@mayo.edu.
Cornelia M. Weyand, Email: cweyand@stanford.edu.
References
- 1.Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9(12):731–740. doi: 10.1038/nrrheum.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piggott K, Biousse V, Newman NJ, Goronzy JJ, Weyand CM. Vascular damage in giant cell arteritis. Autoimmunity. 2009;42(7):596–604. doi: 10.1080/08916930903002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen Z, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. 2017;9(399):eaal3322. doi: 10.1126/scitranslmed.aal3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe R, et al. MMP (matrix metalloprotease)-9-producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res. 2018;123(6):700–715. doi: 10.1161/CIRCRESAHA.118.313206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A. 2017;114(6):E970–E979. doi: 10.1073/pnas.1616848114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Watanabe R, Berry GJ, Nadler SG, Goronzy JJ, Weyand CM. CD28 signaling controls metabolic fitness of pathogenic T cells in medium and large vessel vasculitis. J Am Coll Cardiol. 2019;73(14):1811–1823. doi: 10.1016/j.jacc.2019.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation. 2018;137(18):1934–1948. doi: 10.1161/CIRCULATIONAHA.117.030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen Z, et al. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. 2016;126(5):1953–1967. doi: 10.1172/JCI84181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aster JC, Pear WS, Blacklow SC. The varied roles of Notch in cancer. Annu Rev Pathol. 2017;12:245–275. doi: 10.1146/annurev-pathol-052016-100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah DK, Zúñiga-Pflücker JC. An overview of the intrathymic intricacies of T cell development. J Immunol. 2014;192(9):4017–4023. doi: 10.4049/jimmunol.1302259. [DOI] [PubMed] [Google Scholar]
- 12.Radtke F, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10(5):547–558. doi: 10.1016/S1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 13.Ting HA, Schaller MA, de Almeida Nagata DE, Rasky AJ, Maillard IP, Lukacs NW. Notch ligand delta-like 4 promotes regulatory T Cell identity in pulmonary viral infection. J Immunol. 2017;198(4):1492–1502. doi: 10.4049/jimmunol.1601654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piggott K, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123(3):309–318. doi: 10.1161/CIRCULATIONAHA.110.936203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 16.Falcone EL, et al. Colitis susceptibility in p47(phox-/-) mice is mediated by the microbiome. Microbiome. 2016;4:13. doi: 10.1186/s40168-016-0159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savina A, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126(1):205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 18.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12(1):5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ejlerskov P, et al. NADPH oxidase is internalized by clathrin-coated pits and localizes to a Rab27A/B GTPase-regulated secretory compartment in activated macrophages. J Biol Chem. 2012;287(7):4835–4852. doi: 10.1074/jbc.M111.293696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 21.Wandinger-Ness A, Zerial M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol. 2014;6(11):a022616. doi: 10.1101/cshperspect.a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol. 2010;25(1):99–112. doi: 10.14670/hh-25.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu W, Fang F, Ding J, Wu C. Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease. Traffic. 2018;19(4):253–262. doi: 10.1111/tra.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerra F, Bucci C. Multiple roles of the small GTPase Rab7. Cells. 2016;5(3):E34. doi: 10.3390/cells5030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. doi: 10.1007/s00018-017-2595-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H, Wu C. Charcot Marie Tooth 2B peripheral sensory neuropathy: how Rab7 mutations impact NGF signaling? Int J Mol Sci. 2017;18(2):E324. doi: 10.3390/ijms18020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10(9):597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guichard A, Nizet V, Bier E. RAB11-mediated trafficking in host-pathogen interactions. Nat Rev Microbiol. 2014;12(9):624–634. doi: 10.1038/nrmicro3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhuin T, Roy JK. Rab11 in disease progression. Int J Mol Cell Med. 2015;4(1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Prashar A, Schnettger L, Bernard EM, Gutierrez MG. Rab GTPases in immunity and inflammation. Front Cell Infect Microbiol. 2017;7:435. doi: 10.3389/fcimb.2017.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weyand CM, Younge BR, Goronzy JJ. IFN-γ and IL-17: the two faces of T-cell pathology in giant cell arteritis. Curr Opin Rheumatol. 2011;23(1):43–49. doi: 10.1097/BOR.0b013e32833ee946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.To EE, et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat Commun. 2017;8(1):69. doi: 10.1038/s41467-017-00057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casbon AJ, Allen LA, Dunn KW, Dinauer MC. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J Immunol. 2009;182(4):2325–2339. doi: 10.4049/jimmunol.0803476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14(10):1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13(9):654–666. doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66(10):1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21(20):2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 40.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res. 2007;100(11):1556–1568. doi: 10.1161/01.RES.0000266408.42939.e4. [DOI] [PubMed] [Google Scholar]
- 41.Meester JAN, Verstraeten A, Alaerts M, Schepers D, Van Laer L, Loeys BL. Overlapping but distinct roles for NOTCH receptors in human cardiovascular disease. Clin Genet. 2019;95(1):85–94. doi: 10.1111/cge.13382. [DOI] [PubMed] [Google Scholar]
- 42.Shayevitz C, Cohen OS, Faraone SV, Glatt SJ. A re-review of the association between the NOTCH4 locus and schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(5):477–483. doi: 10.1002/ajmg.b.32050. [DOI] [PubMed] [Google Scholar]
- 43.McCarter AC, Wang Q, Chiang M. Notch in leukemia. Adv Exp Med Biol. 2018;1066:355–394. doi: 10.1007/978-3-319-89512-3_18. [DOI] [PubMed] [Google Scholar]
- 44.DeLeo FR, Allen LA, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163(12):6732–6740. [PubMed] [Google Scholar]
- 45.Mumbengegwi DR, Li Q, Li C, Bear CE, Engelhardt JF. Evidence for a superoxide permeability pathway in endosomal membranes. Mol Cell Biol. 2008;28(11):3700–3712. doi: 10.1128/MCB.02038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jancic C, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol. 2007;9(4):367–378. doi: 10.1038/ncb1552. [DOI] [PubMed] [Google Scholar]
- 47.Thomas DC. How the phagocyte NADPH oxidase regulates innate immunity. Free Radic Biol Med. 2018;125:44–52. doi: 10.1016/j.freeradbiomed.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 48.Henrickson SE, Jongco AM, Thomsen KF, Garabedian EK, Thomsen IP. Noninfectious manifestations and complications of chronic granulomatous disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S18–S24. doi: 10.1093/jpids/piy014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ménager MM, et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol. 2007;8(3):257–267. doi: 10.1038/ni1431. [DOI] [PubMed] [Google Scholar]
- 50.van der Beek J, Jonker C, van der Welle R, Liv N, Klumperman J. CORVET, CHEVI and HOPS - multisubunit tethers of the endo-lysosomal system in health and disease. J Cell Sci. 2019;132(10):jcs189134. doi: 10.1242/jcs.189134. [DOI] [PubMed] [Google Scholar]
- 51.Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5(4):317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- 52.Satoh T. Molecular mechanisms for the regulation of insulin-stimulated glucose uptake by small guanosine triphosphatases in skeletal muscle and adipocytes. Int J Mol Sci. 2014;15(10):18677–18692. doi: 10.3390/ijms151018677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foley K, Boguslavsky S, Klip A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry. 2011;50(15):3048–3061. doi: 10.1021/bi2000356. [DOI] [PubMed] [Google Scholar]
- 54.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3(4):267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki M, et al. CD8+CD45RA+CCR7+FOXP3+ T cells with immunosuppressive properties: a novel subset of inducible human regulatory T cells. J Immunol. 2012;189(5):2118–2130. doi: 10.4049/jimmunol.1200122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wen Z, et al. N-myristoyltransferase deficiency impairs activation of kinase AMPK and promotes synovial tissue inflammation. Nat Immunol. 2019;20(3):313–325. doi: 10.1038/s41590-018-0296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.