

Abstract
Macrophages are main effectors of heme metabolism, increasing transiently in the liver during heightened disposal of damaged or senescent RBCs (sRBCs). Macrophages are also essential in defense against microbial threats, but pathological states of heme excess may be immunosuppressive. Herein, we uncovered a mechanism whereby an acute rise in sRBC disposal by macrophages led to an immunosuppressive phenotype after intrapulmonary Klebsiella pneumoniae infection characterized by increased extrapulmonary bacterial proliferation and reduced survival from sepsis in mice. The impaired immunity to K. pneumoniae during heightened sRBC disposal was independent of iron acquisition by bacterial siderophores, in that K. pneumoniae mutants lacking siderophore function recapitulated the findings observed with the WT strain. Rather, sRBC disposal induced a liver transcriptomic profile notable for suppression of Stat1 and IFN-related responses during K. pneumoniae sepsis. Excess heme handling by macrophages recapitulated STAT1 suppression during infection that required synergistic NRF1 and NRF2 activation but was independent of heme oxygenase-1 induction. Whereas iron was dispensable, the porphyrin moiety of heme was sufficient to mediate suppression of STAT1-dependent responses in human and mouse macrophages and promoted liver dissemination of K. pneumoniae in vivo. Thus, cellular heme metabolism dysfunction negatively regulated the STAT1 pathway, with implications in severe infection.
Keywords: Infectious disease, Pulmonology
Keywords: Bacterial infections, Innate immunity, Macrophages

Introduction
Approximately 2 × 1011 RBCs are synthesized each day in the healthy adult human (1, 2). The vast majority of iron required for erythropoiesis is obtained from recycling of iron after ingestion of damaged or senescent RBCs (sRBCs) by the mononuclear phagocyte system within the bone marrow, liver, and spleen (3). When numbers of sRBCs increase acutely or there is increased erythrophagocytosis — such as in hemolytic anemias, transfusion iron overload, anemia of inflammation, or severe infection — macrophages in the liver become the chief cells charged with sRBC disposal and excess heme iron handling (4, 5). Although erythrophagocytosis under homeostatic conditions is largely an immunologically silent event, enhanced sRBC delivery to the macrophage during infection provides a competing stressor that may alter integration of signals when the macrophage is also tasked with elaborating mediators and sensing, engulfing, and destroying invading pathogens. How the macrophage is able to process competing signals, respond, and prioritize function is largely unknown.
Gram-negative extracellular bacteria such as Klebsiella pneumoniae (KP) are common causes of acute lower respiratory tract infection and sepsis worldwide (6, 7). We and others have shown that the initial encounter by macrophages dictates the ability of the host to control KP infection in the lungs (8, 9). Indeed, a major mechanism of KP virulence is the expression of a polysaccharide capsule that enables the bacteria to evade phagocytosis (10) and increases pathogenicity in mice and humans (11–13). KP also secrete a number of siderophores (14, 15) that exhibit higher affinity for ferric iron than transferrin and lactoferrin and could therefore theoretically scavenge iron from these host proteins (16). Enhanced delivery of sRBCs to macrophages may increase pathogen replication by boosting nutritional iron sources (17), and another consequence of sRBC delivery may be through direct alterations of host immune response to infection that are unrelated to iron scavenging by the pathogen. Macrophages in the liver are the primary cells that support rapid clearance of stressed, damaged red cells (4), but remarkably little is known about whether this competing physiological stressor weakens macrophage function during infection, thereby impairing host defense. Moreover, states of intracellular heme iron excess such as hemochromatosis are associated with increased risk of infection (18–20), a finding that has been attributed to iron overload. Growing evidence indicates heme is an important regulator of macrophage function (21–23), however, and invites the possibility that excess heme handling has profound effects on host immunity.
Intracellular heme excess can also occur with repeated RBC transfusions, such as in the case of transfusion iron overload. Red cells that undergo progressive biochemical and biophysical changes during standard storage conditions are collectively termed the red cell storage lesion (24). These stored, damaged RBCs, once transfused, are recognized and rapidly cleared by macrophages. It is noteworthy that randomized controlled trials in anemic patients requiring transfusion have not shown that red cell storage duration significantly affects clinical outcomes (25, 26). Although these trials were not intended to address the issue of excess heme handling, here we utilized RBC transfusion as a model to deliver sRBCs to macrophages in order to examine pathological states of intracellular heme excess during infection. We showed that increased sRBC disposal by macrophages led to defective signaling through STAT1, a master transcription factor involved in antiviral and antibacterial host response (27, 28), which marks the host for an immunosuppressive fate during infection. Intracellular heme excess induced by stressed erythrophagocytosis during infection suppressed STAT1 responses through NRF1 and NRF2 activation, but was independent of heme oxygenase-1 (HO-1) induction. Surprisingly, the porphyrin moiety of heme, but not iron, was sufficient to induce STAT1 suppression. Thus, in pathological states of intracellular heme excess, our findings suggest that the ability to mount optimal STAT1 responses during infection is impaired.
Results
sRBC delivery impairs bacterial control at extrapulmonary sites and worsens survival after KP intrapulmonary infection.
To examine the effect of sRBC disposal on host outcome during acute bacterial infection in vivo, mice initially underwent intratracheal inoculation with KP and were challenged 1 hour later with 200 μL of freshly isolated young RBCs (yRBCs) or sRBCs damaged by extended storage. This volume approximates 1 packed RBC unit in humans (29) and serves as a model of acute increase in erythrophagocytosis (30). Mice challenged with sRBCs after acute intrapulmonary KP infection showed increased mortality compared with mice challenged with yRBCs from the same pool of donor mice blood (Figure 1A). The median survival after KP infection was 9.5 days for the yRBC-challenged group and 2.5 days for the sRBC-challenged group over a 2-week observational period (log-rank Mantel-Cox test, P = 0.005).
Figure 1. sRBC delivery impairs bacterial control at extrapulmonary sites and worsens survival after K. pneumoniae intrapulmonary infection.

(A) KP was instilled intratracheally into C57BL/6 mice and was followed by challenge with young RBCs (yRBCs) or senescent RBCs (sRBCs). n = 10 mice per group. **P = 0.005 by log-rank test. (B) Total BAL cell count/mL. (C) Total BAL PMN count/mL. (D) Total BAL mononuclear cell count/mL. (E) Lung bacterial burden was estimated by CFU count/mL of tissue homogenates. (F) Spleen CFU/mL. (G) Liver CFU/mL. (H) Blood CFU/mL. (B–H) each point indicates individual mice, n = 7–8 mice/group, line indicates the median. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-tailed Mann-Whitney U test.
To determine whether increased mortality observed in mice challenged with sRBC was due to impaired host defense, we evaluated lung bacterial burden and extrapulmonary dissemination. Because initial mortality was observed by 48 hours, we examined mice prior to this time point to minimize survivor bias. At 4 and 24 hours after KP inoculation, airspace leukocyte numbers were evaluated in the bronchoalveolar lavage (BAL) fluid. Total BAL leukocyte, neutrophil, and mononuclear cell counts were decreased in mice challenged with sRBCs at 24 hours (Figure 1, B–D). Although mice challenged with sRBCs showed increased bacterial burden in the lungs compared with mice challenged with yRBCs at 4 hours, the lung bacterial burden by 24 hours was comparable in both groups (Figure 1E). These findings suggest a modest effect in the lungs after sRBC delivery. In contrast, mice challenged with sRBCs showed increased splenic bacterial burden by 4 and 24 hours (Figure 1F). Consistent with the spleen data, liver and blood CFU burdens were increased at 24 hours (Figure 1, G and H). Moreover, the inability to adequately control bacterial replication was accompanied by a systemic inflammatory response: sRBC-challenged mice showed elevated plasma C5a, CXCL10, IFN-γ, IL-1β, IL-6, IL-10, and MBL2 levels at 4 hours (Supplemental Figure 1, A–G; supplemental material available online with this article; https://doi.org/10.1172/JCI137468DS1), with sustained plasma CXCL10, IL-10, and TNF-α elevation at 24 hours (Supplemental Figure 1, B, F, and H). Notably, RBC washing prior to transfusion did not mitigate extrapulmonary bacterial proliferation (Supplemental Figure 2, A and B). Thus, in a KP model of acute bacterial pneumonia, sRBC disposal impaired the ability of mice to control pathogen replication after breach of the lung mucosal barrier, resulting in increased extrapulmonary bacterial proliferation, dysregulated hyperinflammatory systemic response, and worse survival.
Impaired bacterial control observed with sRBC delivery is independent of iron acquisition by bacterial siderophores.
KP utilizes siderophores enterobactin (Ent), glycosylated enterobactin (gly-Ent), and yersiniabactin (Ybt) as virulence mechanisms to scavenge iron in iron-restricted environments such as the lung (14, 15). We utilized an entB ybtS isogenic KP mutant strain lacking the ability to produce all 3 siderophores, Ent, gly-Ent, and Ybt, to assess whether sRBC delivery enhanced the pathogenicity of this KP isolate. We found that sRBC challenge enhanced plasma transferrin-bound iron when compared with mice challenged with yRBCs or untreated mice by electron paramagnetic resonance spectroscopy (Figure 2A). Others have also reported a rise in non–transferrin-bound iron after challenge with damaged RBCs in humans (31) and mice (29). Although in vitro growth of the entB ybtS KP mutant was similar when compared with the WT parent strain (Figure 2B), the entB ybtS KP mutant exhibited impaired proliferation in iron-rich plasma obtained from mice challenged with sRBCs when compared with the WT strain (Figure 2C). In addition, the entB ybtS KP mutant replicated more slowly in the lung than WT KP (ref. 32 and Figure 2D) and showed reduced dissemination to the spleen, liver, and blood (Figure 2, E–G). After challenge with sRBCs, mice infected with the entB ybtS KP mutant showed no difference in BAL total protein and lung bacterial burden when compared with mice challenged with either PBS or yRBCs (Figure 2, H and I). However, challenge with sRBCs resulted in increased extrapulmonary bacterial proliferation of the less virulent entB ybtS KP mutant (Figure 2, J–L). Collectively, these findings suggest that, although bacterial siderophores are key virulence factors that enhance microbial dissemination within the host, sRBC delivery enhances extrapulmonary bacterial proliferation through an alternative mechanism that is independent of pathogen siderophore function.
Figure 2. Impaired K.

pneumoniaecontrol at extrapulmonary sites after sRBC delivery is independent of iron acquisition by bacterial siderophores. (A) Plasma transferrin-bound iron from mice 2 hours after challenge with either yRBCs or sRBCs, with unchallenged mice serving as baseline control. Growth curve of entB ybtS isogenic KP mutant and WT parent strain in (B) tryptic soy broth and (C) plasma obtained from mice challenged with sRBCs. (D–G) EntB ybtS isogenic KP mutant and WT KP were instilled intratracheally into C57BL/6 mice (104 CFU inoculum each). Bacterial burden was obtained from homogenates of (D) lung, (E) spleen, (F) liver, and (G) blood as CFU/mL at 24 hours after infection. Each point indicates individual mice, n = 6 mice per group, line indicates median. **P < 0.01 by 2-tailed Mann-Whitney U test. (H–L) EntB ybtS isogenic KP mutant bacteria were instilled into C57BL/6 mice (103 CFU inoculum), followed by challenge with either PBS, yRBCs, or sRBCs. (H) BAL total protein and bacterial burden were obtained from (I) lung, (J) spleen, (K) liver tissue homogenates, and (L) blood as CFU/mL at 24 hours. Each point indicates individual mice, n = 5–7 mice per group, line indicates median. *P < 0.05, **P < 0.01 by Kruskal-Wallis test with Dunn’s multiple comparisons test.
sRBC delivery reveals a unique transcriptomic profile notable for suppression of Stat1 and IFN-related responses harbored in the liver of mice after acute intrapulmonary KP infection.
Prior studies have highlighted the important role of the liver in the innate immune response to acute bacterial pneumonia (33). Moreover, in an initial evaluation of the innate immune transcriptional response using a PCR array to profile 87 antibacterial response genes, minimal differences in gene expression were noted between the lungs of KP-infected mice challenged with yRBCs versus sRBCs (Supplemental Figure 3, A and B). This contrasted with the liver, where a significant reduction in innate immune gene expression was observed in infected sRBC-challenged mice (Supplemental Figure 3, A and C). To comprehensively examine the liver transcriptome during sRBC disposal, we evaluated the gene expression profile by RNA-Seq 24 hours after acute intrapulmonary KP infection in mice challenged with either yRBCs or sRBCs. We observed excellent separation between these 2 groups in terms of global transcriptomic profile and detected 4891 differentially expressed genes (threshold 1.5-fold change, FDR-adjusted P value ≤ 0.05) in livers of KP-infected mice challenged with yRBCs or sRBCs (Supplemental Figure 4A). Although we observed elevated plasma alanine transaminase (ALT) and aspartate transaminase (AST) levels in mice challenged with sRBCs 24 hours after KP infection (Supplemental Figure 4, B and C), we did not observe gross differences in histopathological evaluations in livers of KP-infected mice challenged with yRBCs or sRBCs in the model (Supplemental Figure 4, D–I). Furthermore, others have reported induction of ferroptosis in splenic red pulp macrophages after sRBC delivery (30). Whereas 2 highly oxygenated polyunsaturated phosphatidylethanolamine (PE) species — PE(40:7) and PE(40:8) — were significantly elevated in the liver after sRBC delivery (Supplemental Figure 5, A–C), neither of these species containing 3 oxygens has been previously identified as a proferroptotic predictive biomarker (34). Additionally, we did not observe significant differences in oxidized PE levels in the livers of KP-infected mice challenged with yRBCs or sRBCs for the majority of the molecular species evaluated (Supplemental Figure 5D). Hence, although sRBC delivery causes changes in PE oxidation in the liver, these changes are most likely distinct from execution of the ferroptotic cell death program.
Given impaired immunity in the acute infection model, we focused on altered innate immune gene profiles in the RNA-Seq data (Supplemental Table 1). Although Nfkb-related genes Rela, Relb, Nfkbia, Nfkbib, Nfkbiz, and Stat3 were increased in the KP + sRBC group, the data were notable for suppression of Stat1 (–3.2-fold change, FDR P value = 0) and IFN regulatory factors such as Irf1, Irf2, Irf3, Irf5, and Irf8 (Figure 3A). Suppression of IFN responses was consistent with upregulation of Socs3 (2.76-fold change, FDR P value = 1.5 × 10–12) and Socs1 (7.6-fold change, FDR P value = 0) gene expression (Figure 3A). Notably, heme oxygenase 1 (Hmox1), the enzyme catalyzing the rate-limiting step of heme catabolism, was also increased (3.46-fold change, FDR P value = 0). Reduced Irf1, Irf3, Irf8, and Stat1 gene expression and increased Rela, Socs1, Socs3, Stat3, and Hmox1 gene expression were confirmed by qRT-PCR (Figure 3, B–J).
Figure 3. RNA-Seq of the liver in mice after sRBC delivery reveals a unique transcriptomic profile notable for suppression of Stat1 and IFN-related responses during K. pneumoniae infection.

KP was instilled intratracheally into C57BL/6 mice followed by challenge with either yRBCs or sRBCs. Mice were euthanized at 24 hours. (A) Volcano plot of innate immune genes. Red depicts downregulated genes. Blue depicts upregulated genes. (B–J) qPCR validation of RNA-Seq data. Floating bar plots indicate median and 25% to 75% quartiles. n = 4 mice per group. *P < 0.05 by 2-tailed Mann-Whitney U test. Fold change relative to yRBC + KP. (K) IRF1 immunoblot in livers of KP-infected mice challenged with either yRBCs or sRBCs. Each line indicates individual mice. (L) Relative density of blot depicted in K. n = 4 mice per group, line indicates the median. *P < 0.05 by 2-tailed Mann-Whitney U test.
STAT1 is essential for transducing IFN-α and IFN-γ responses (28) and is required for optimal defense against KP infection (Supplemental Figure 6, A–D, and ref. 35). We evaluated one of its key downstream effectors, IRF1, a transcription factor induced by STAT1 activation, in liver tissue homogenates of KP-infected mice challenged with either yRBCs or sRBCs. KP-infected mice challenged with sRBCs showed impaired IRF1 expression in the liver compared with KP-infected mice challenged with yRBCs (Figure 3, K and L, full uncut gels provided in the online supplemental material), supporting the findings of RNA-Seq. Taken together, these results suggest that delivery of sRBCs to the infected host suppresses a proximal regulator of IFN response and antimicrobial immune defense.
KP infection increases erythrophagocytosis, leading to upregulation of heme iron transcriptional responses and suppression of STAT1.
sRBC disposal occurs through transient accumulation of macrophages in the liver that enable restoration of iron homeostasis (4). Macrophages are also the initial effector cells of host defense in tissue, but how the macrophage processes competing signals and prioritizes function during infection is less known. We challenged RAW 264.7 macrophages with live KP to evaluate the effect on downstream innate immune signaling in the presence or absence of a competing stressor such as sRBCs (generated by heating at 48°C and continuous agitation). Others have previously shown that RBCs stressed by either aging or heating show no differences in clearance from circulation in mouse models (4). We first demonstrated uptake of sRBCs after delivery to macrophages (Figure 4A and Supplemental Figure 7A). Bacterial infection with KP potentiated uptake of sRBCs by macrophages (Figure 4B) in a TLR4-dependent manner (refs. 36, 37, and Figure 4C). However, the increased erythrophagocytosis did not significantly alter macrophage uptake of KP (Figure 4D).
Figure 4. K. pneumoniae enhances erythrophagocytosis, leading to upregulation of heme iron transcriptional responses and suppression of STAT1.

(A) sRBC internalization in RAW cells incubated with vehicle (PBS), sRBCs (50 sRBC:1 Mφ), KP (MOI 10:1), or KP + sRBC for 90 minutes. (B) Quantification of sRBC uptake shown in A. (C) Bone marrow–derived macrophages (BMDMs) obtained from WT and Tlr4–/– mice were challenged with KP + sRBC (10sRBC:1 Mφ) for 2 hours. (B and C) n = 3 technical replicates per group and data are indicative of 2 independent experiments. *P < 0.05, **P < 0.01 by 2-tailed t test. (D) Intracellular CFU/mL in RAW cells that were challenged with KP or KP + sRBC for 90 minutes. (E and F) Heme iron transcriptional genes Hmox1, Scl40a1, and (G–K) Stat1 and STAT1 target genes C3, Cfb, Irf1, and Nos2 evaluated in RAW cells challenged with KP or KP + sRBC for 4 hours. (L–N) STAT1 and IRF1 immunoblots in RAW cells challenged with vehicle (PBS), sRBCs, KP, or KP + sRBC for 4 hours. Blots are indicative of at least 3 independent experiments. (O–Q) CCL5, CXCL10, and TNF-α were measured in cell culture supernatant by ELISA 4 hours after infection. (R–W) Stat1, C3, Cfb, Irf1, Nos2, and Rela in Stat1+/+ and Stat1–/– BMDMs challenged with KP or KP + sRBC for 4 hours. (E–K and R–W) Gene expression was evaluated by qPCR analysis. Fold change is relative to PBS-treated macrophages. Floating bar plots indicate median and 25% to 75% quartiles, n = 3 technical replicates per group. *P < 0.05, ***P < 0.001, ****P < 0.0001 by 2-tailed t test. (X and Y) CXCL10 and TNF-α were measured in cell culture supernatant by ELISA 4 hours after infection. (O–Q, X, and Y) n = 3 technical replicates per group. ****P < 0.0001 by 1-way ANOVA with Tukey’s multiple comparisons test.
Heme accumulation after erythrophagocytosis induces Hmox1 (38) and ferroportin-1 (Slc40a1) transcription (39). Indeed, we observed increased Hmox1 and Slc40a1 gene expression in KP + sRBC–treated macrophages (Figure 4, E and F). As observed in vivo, we noted suppression of Stat1 in KP + sRBC–treated macrophages (Figure 4G). STAT1 is a master transcription factor that regulates transcription of critical immune effectors such as complement component 3 (C3), complement factor b (Cfb), IRF-1 (Irf1), and inducible NO synthase (iNOS or Nos2) in macrophages in response to IFN stimulation (28). After sRBC delivery to KP-infected macrophages, C3, Cfb, Irf1, and Nos2 gene expression was also impaired (Figure 4, H–K).
We next investigated STAT1 protein expression and found that sRBC delivery to macrophages dose-dependently reduced both phosphorylated STAT1 and total STAT1 expression during KP infection (Figure 4L). sRBC delivery to KP-infected macrophages also dose-dependently inhibited IRF1, a downstream target of STAT1 (Figure 4, M–N). The mechanism appeared to be independent of autocrine type I or II IFN receptor signaling, in that Ifngr1–/– and Ifnar1–/– bone marrow–derived macrophages (BMDMs) retained the ability to induce IRF1 during KP infection (Supplemental Figure 7, B and C). Although hepatocytes constitute approximately 80% of liver volume and have been shown to recognize and respond directly to microbial products (40–42), we found that hepatocytes did not regulate the IFN response to KP, and that sRBC delivery had no effect on IRF1 expression in hepatocytes (Supplemental Figure 7D).
To determine whether sRBC-mediated suppression of STAT1 and IRF1 induction resulted in impaired macrophage cytokine responses, we evaluated CCL5, CXCL10, and TNF-α secretion in KP + sRBC–treated macrophages. We observed that sRBC delivery markedly reduced production of the IFN-related cytokines CCL5 and CXCL10 in response to KP (Figure 4, O and P) but did not alter TNF-α secretion (Figure 4Q). Moreover, Stat1–/– BMDMs showed impaired Stat1, C3, Cfb, Irf1, and Nos2 but not Rela transcriptional responses after KP infection, and CXCL10 but not TNF-α protein secretion was STAT1 dependent (Figure 4, R–Y). Taken together, these findings indicated that KP infection heightened erythrophagocytosis, resulting in selective transcriptional responses notable for heme iron metabolism and suppression of STAT1 and its downstream targets that are independent of type I or II IFN receptor signaling.
Heme is the constituent of RBC that mediates suppression of STAT1 during KP infection.
We next examined which component of sRBC mediates suppression of STAT1. Macrophages were challenged with vehicle (PBS), sRBCs, stroma-free sRBC lysates, and hemoglobin-depleted ghost RBCs prepared from an equivalent number of sRBCs with or without KP infection. IRF1 expression was utilized as a STAT1 target readout. Although KP strongly induced IRF1 expression, KP + sRBC exposure suppressed IRF1 expression that was recapitulated by KP + sRBC lysate but not KP + sRBC ghosts (Figure 5A). Hemoglobin is the principal cytoplasmic component of RBCs (43), and purified hemoglobin recapitulated sRBC-mediated IRF1 suppression (Figure 5B). Moreover, depletion of hemoglobin from sRBCs severely limited extrapulmonary dissemination of entB ybtS KP to the spleen and liver (Supplemental Figure 8).
Figure 5. Heme is the constituent of RBCs that mediates STAT1 suppression during K. pneumoniae infection.

IRF1 immunoblots in (A) RAW cells challenged with vehicle (PBS), sRBCs (30 sRBC:1 Mφ), sRBC lysates obtained from equivalent numbers of sRBCs, ghosts obtained from equivalent numbers of sRBCs, KP (MOI 10:1), KP + sRBC, KP + sRBC lysate, or KP + sRBC ghost for 4 hours, and (B) RAW cells challenged with vehicle, purified hemoglobin from an equivalent number of lysed RBCs (Hb), KP, KP + sRBC, or KP + Hb for 4 hours. Blot is indicative of 2 independent experiments. (C) HO-1, IRF1, p-STAT1 (Ser727), and STAT1 in RAW cells challenged with hemin (100 μM) or KP at 0.5, 1, 2, or 4 hours. (D) HO-1, IRF1, p-STAT1 (Ser727), p-STAT1 (Tyr701), and STAT1 in RAW cells challenged with KP and increasing concentrations of hemin for 4 hours. (E) CXCL10 and (F) TNF-α were measured in cell culture supernatant by ELISA 4 hours after infection. (G) HO-1, p-STAT1 (Ser727), and STAT1 in BMDMs challenged with vehicle (PBS), KP, KP + sRBC (50 sRBC:1 Mϕ), or KP + sRBC +hemopexin (HPX, 200 μg/mL) for 4 hours. (H) CXCL10 was measured in cell culture supernatant by ELISA 4 hours after infection. (E, F, and H) n = 3 technical replicates per group. ***P < 0.001, ****P < 0.0001 by 1-way ANOVA with Tukey’s multiple comparisons test.
The heme moiety of hemoglobin has been implicated as an immunomodulating agent and can trigger (44) or dampen (45, 46) the immune response. Although heme and hemin have been shown to directly bind to TLR4 and induce proinflammatory cytokine secretion (47), hemin alone was a weak activator of STAT1 in macrophages (Figure 5C) but potently induced HO-1 (Figure 5, C and D). However, we noted that hemin dose-dependently inhibited KP-induced STAT1 phosphorylation, at both its serine and tyrosine sites, in addition to total STAT1 and IRF1 expression (Figure 5D). Furthermore, CXCL10 — but not TNF-α — was dose-dependently suppressed by hemin in KP-infected macrophages (Figure 5, E and F), indicating that hemin recapitulated sRBC-mediated STAT-1 suppression.
To delineate extracellular versus intracellular heme contribution to STAT1 suppression, RAW macrophages challenged with KP + sRBC were incubated in the presence or absence of hemopexin (HPX). HPX is hydrophilic with no known transcellular transport mechanism and would thus chelate extracellular heme but not heme concentrated intracellularly. As expected, sRBC delivery to KP-infected macrophages potently induced HO-1 and markedly suppressed STAT1 activation and total STAT1 protein expression (Figure 5G). However, extracellular heme scavenging by HPX failed to restore STAT1 (Figure 5G) or STAT1-dependent CXCL10 (Figure 5H) in macrophages challenged with KP + sRBC. Collectively, these findings suggest that intracellular heme resulting from sRBC breakdown is responsible for STAT1 suppression during KP infection.
STAT1 suppression requires NRF1 and NRF2 activation but is independent of HO-1 induction.
Heme binds to the transcriptional repressor BTB (bric-a-brac, tramtrack, broad complex) domain and CNC (cap’n’collar) homolog 1 (BACH1) (48) and induces BACH1 proteasome-dependent degradation (49) that results in derepression of Hmox1 (ref. 50 and Figure 6A). As expected, we observed a reduction in BACH1 expression in KP-infected macrophages after sRBC delivery (Supplemental Figure 9A). Bach1 silencing enhanced Hmox1 (Supplemental Figure 9B) but not Stat1 transcription in response to KP (Supplemental Figure 9B). Moreover, Bach1 silencing induced HO-1 expression but did not alter STAT1 protein expression in KP-infected macrophages (Supplemental Figure 9C). Based on these findings, we concluded that BACH1 did not contribute to STAT1 suppression after KP + sRBC delivery. Furthermore, activation of JAK2, through binding of hepcidin to ferroportin-1 in conditions of iron surplus, may result in STAT3-mediated transcriptional activation of SOCS3 and subsequent dampening of inflammation (51). As we observed in the liver, we showed that sRBC delivery upregulated Stat3 and Socs3 gene expression in KP-infected macrophages (Supplemental Figure 9, D and E). However, dose-dependent inhibition of STAT3 failed to rescue STAT1-dependent IRF1 suppression in KP + sRBC–treated macrophages (Supplemental Figure 9F). Based on these findings, we concluded that neither BACH1 nor STAT3 contributed to STAT1 suppression after KP + sRBC delivery.
Figure 6. STAT1 suppression requires NRF1 and NRF2 activation but is independent of HO-1 induction.

(A) Schematic depicting heme-BACH1-NRF1/2 interaction. Intracellular heme accumulation after erythrophagocytosis induces degradation of BACH1 and stabilization of NRF1 and NRF2, with nuclear translocation of NRF2 resulting in Hmox1 transcription. (B) Hmox1 and (C) Stat1 gene expression in RAW cells transfected with control or Hmox1 siRNA and subsequently challenged with KP or KP + sRBC for 4 hours. (B and C) Fold change relative to uninfected control siRNA-transfected RAW cells. Data are average of 3 independent experiments. (D) NRF2 target gene (Nqo1) expression in RAW cells challenged with KP or KP + sRBC for 4 hours. (E–G) Hmox1, Slc40a1, and Stat1 expression in BMDMs obtained from WT and Nrf2–/– mice challenged with KP or KP+ RBC for 4 hours. Fold change relative to PBS-treated BMDMs. (H) NRF1 target gene (Mt1) expression in RAW cells challenged with KP or KP + sRBC for 4 hours. **P < 0.01, ****P < 0.0001 by 2-tailed t-test (D and H). (I) Nrf1 and (J) Stat1 expression in BMDMs transfected with control siRNA or Nrf1 siRNA and subsequently challenged with KP or KP + hemin (50 μM) for 4 hours. (D–K) n = 3 technical replicates per group, and representative of at least 2 independent experiments. (K) Stat1 expression in BMDMs obtained from WT and Nrf2–/– mice, transfected with control siRNA or Nrf1 siRNA, and subsequently challenged with KP for 4 hours. (I–K) Fold change relative to uninfected control siRNA-transfected BMDMs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA with Tukey’s multiple comparisons test (B, C, E–G, and I–K).
HO-1 is the rate-limiting enzyme in heme catabolism, yielding ferrous iron, carbon monoxide, and biliverdin. These biologically active end products, carbon monoxide and biliverdin, have been implicated as antiinflammatory (52–54). To determine whether sRBC-mediated STAT1 suppression in KP-infected macrophages is due to increased HO-1 induction, we silenced Hmox1 in KP-infected macrophages challenged with sRBCs. We achieved, on average, approximately 60% Hmox1 knockdown in KP-infected macrophages with or without sRBC delivery (Figure 6B). Hmox1 silencing resulted in further suppression rather than an increase of Stat1 in KP + sRBC–treated macrophages (Figure 6C). This suggests that HO-1 induction does not mediate STAT1 suppression after sRBC delivery and may instead be vital for augmenting STAT1 expression during KP infection.
Heme evokes nuclear translocation of nuclear factor erythroid 2–related factor 2 (NFE2L2, NRF2) (55), and activation of NRF2 is required for heme iron transcriptional response after heme stimulation in macrophages (refs. 39, 55, and Figure 6A). In addition, NRF2 has been shown to regulate the innate immune response to microbial stimuli (56) and can inhibit proinflammatory cytokine gene transcription (57) or repress STING-dependent IFN response (58) — highlighting a potential link between heme iron transcriptional response and the innate immune response. We found that sRBC delivery activated NRF2 as assessed by upregulation of the NRF2 target gene NAD(P)H quinone oxidoreductase 1 (Nqo1) (59) in KP + sRBC–treated macrophages (Figure 6D). To determine whether NRF2 activation mediated the STAT1 suppression seen in macrophages challenged with KP + sRBC, we examined WT and Nrf2–/– BMDMs. Deletion of NRF2 resulted in diminished heme iron transcriptional response, as assessed by reduced Hmox1 and Slc40a1 expression in Nrf2–/– macrophages challenged with KP + sRBC (Figure 6, E and F). Furthermore, deletion of NRF2 prevented Stat1 suppression in KP + sRBC–treated macrophages but did not fully restore Stat1 induction (Figure 6G). Moreover, although treatment with a known NRF2 activator possessing antioxidant activity, isothiocyanate sulforaphane (SFN) (60), phenocopied the effect of KP + sRBC–treated macrophages (Supplemental Figure 10, A–E), suppression of STAT1 target gene Nos2 (Supplemental Figure 10F) and IRF1 protein (Supplemental Figure 10G) expression persisted even in the absence of NRF2 (Supplemental Figure 10G) — suggesting that NRF2 is necessary but not sufficient for sRBC-mediated STAT1 suppression.
Because NFE2–related factor 1 (NFE2L1, NRF1) binds to similar cis-regulatory regions of antioxidant response elements as NRF2 (61), we sought to determine whether Nrf1 knockdown further boosted Stat1 expression observed in Nrf2–/– macrophages challenged with KP + sRBC. We observed that sRBC delivery activated NRF1 target gene metallothionein 1 (Mt1) (62) in KP + sRBC–treated macrophages (Figure 6H). Given that heme recapitulated sRBCs’ effect on macrophages during KP infection, we utilized hemin in macrophages transfected with Nrf1 siRNA. We achieved greater than 80% Nrf1 knockdown in macrophages with or without KP + hemin (Figure 6I). Nrf1 silencing reversed hemin-mediated Stat1 suppression in KP-infected macrophages (Figure 6J). Moreover, Nrf2–/– macrophages showed higher Stat1 transcript levels compared with WT macrophages during KP infection, but Nrf1 knockdown further boosted Stat1 transcript levels in Nrf2–/– macrophages (Figure 6K), suggesting synergism between NRF1 and NRF2 in control of Stat1 even in the absence of hemin. Collectively, these data showed that although sRBC delivery induced HO-1 and activated NRF1 and NRF2 target genes, STAT1 suppression did not require HO-1 but was mediated, in part, by NRF1 and NRF2.
The porphyrin moiety of heme is necessary and sufficient for NRF1/NRF2 activation and STAT1 suppression.
Iron liberated from heme by HO-1 may weaken immunity either by direct iron provision to the pathogen or through dysregulated iron recycling in the host cell. To investigate the contribution of iron to heme-mediated STAT1 suppression during KP infection, we utilized iron chelators deferasirox (DFX) and deferoxamine (DFO). DFX is lipophilic and can bind intracellular ferric iron in a 2:1 binding ratio (63, 64). DFO, on the other hand, is highly hydrophilic with no known transcellular transport mechanism and binds extracellular ferric iron in a 1:1 binding ratio (65). Neither chelation of host intracellular iron by DFX nor chelation of extracellular iron available to KP by DFO restored STAT1 activation or total STAT1 expression in KP-infected macrophages challenged with hemin (Figure 7A). STAT1-dependent CXCL10 secretion in macrophages challenged with KP + hemin was also not rescued by introduction of iron chelators (Figure 7B). Moreover, transfusion of iron dextran that approximated iron contained in 1 unit of packed RBC did not recapitulate enhanced extrapulmonary proliferation observed with sRBC delivery in KP-infected mice (Supplemental Figure 11, A–D) or entB ybtS KP mutant–infected mice (Supplemental Figure 11, E–H), indicating that enhanced iron delivery was not the major mechanism for increased extrapulmonary dissemination observed with sRBC delivery.
Figure 7. The porphyrin moiety of heme is necessary and sufficient for NRF1/NRF2 activation and STAT1 suppression.

(A) STAT1 immunoblot in BMDMs challenged with KP (MOI 10:1), KP + hemin (25 μM), KP + hemin + DFX (300 μM), or KP + hemin + DFO (300 μM) for 4 hours. All groups contained vehicle (DMSO, ~1%). Blot is representative of 2 independent experiments. (B) CXCL10 was measured in cell culture supernatant by ELISA 4 hours after infection. (C) Chemical structures of hemin (iron protoporphyrin IX, FePPIX), cobalt protoporphyrin IX (CoPPIX), and protoporphyrin IX (PPIX). (D) Mt1 and (E) Nqo1 expression, (F) Hmox1, and (G) Stat1 gene expression, (H) CXCL10 secretion in BMDMs challenged with KP, KP + FePPIX (50 μM), KP + CoPPIX (50 μM), or KP + PPIX (50 μM) for 4 hours. (D–G) Fold change relative to PBS-treated BMDMs. (B and F–H) n = 3 technical replicates per group. **P < 0.01, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA with Tukey’s multiple comparisons test. (I) BACH1, HO-1, p-STAT1, and total STAT1 immunoblot in BMDMs challenged with KP and increasing concentrations of PPIX. (J) IRF1 immunoblot in human monocyte–derived macrophages (HMDMs) challenged with vehicle (PBS), KP, KP + FePPIX (25 μM), or KP + PPIX (25 μM) for 4 hours. (K) CXCL10 and (L) TNF-α secretion from HMDMs in J. n = 3 donors. ****P < 0.0001 by 2-way ANOVA with Tukey’s multiple comparisons test. KP was instilled intratracheally into C57BL/6 mice and followed by i.p. challenge with vehicle (2.5% DMSO) or CoPPIX (5 mg/kg, 25 mg/kg) 1 hour after KP instillation. Bacterial burden was obtained from (M) lung, (N) spleen, (O) liver tissue homogenates, and (P) blood of mice 24 hours after KP infection and reported as CFU/mL. Each point indicates individual mice, n = 10–11 mice per group combined from 2 independent studies. Line indicates median. *P < 0.05, **P < 0.01 by Kruskal-Wallis test with Dunn’s multiple comparisons test.
Given these findings, we tested whether a metal within the porphyrin ring was necessary for NRF1/2 activation and STAT1 suppression observed in macrophages challenged with KP + hemin. We utilized hemin (iron protoporphyrin IX, FePPIX); cobalt-substituted protoporphyrin IX (CoPPIX); and protoporphyrin IX (PPIX), which lacks a metal ion at its center (Figure 7C). The metalloporphyrins, FePPIX and CoPPIX, activated NRF1 and NRF2 target genes, as assessed by Mt1 and Nqo1 induction, respectively (Figure 7, D and E), and induced Hmox1 in KP-infected macrophages (Figure 7F). PPIX also activated NRF1 and NRF2 target genes (Figure 7, D and E) but did not induce Hmox1 in KP-infected macrophages (Figure 7F). Interestingly, the presence of a metal ion within the porphyrin macrocycle appeared dispensable because all 3 protoporphyrins — FePPIX, CoPPIX, and PPIX — suppressed Stat1 in KP-infected macrophages (Figure 7G). STAT1-dependent CXCL10 secretion was also impaired by all 3 protoporphyrins (Figure 7H). PPIX dose-dependently degraded BACH1 but, unlike hemin, did not induce HO-1 protein expression in KP-infected macrophages (Figure 7I). As observed with hemin, PPIX dose-dependently suppressed STAT1 activation and total STAT1 protein expression in KP-infected macrophages (Figure 7I). Moreover, FePPIX and PPIX suppressed IRF1 (Figure 7J) and CXCL10 (Figure 7K) but not TNF-α secretion (Figure 7L) in KP-infected human monocyte-derived macrophages, suggesting that response was conserved in human macrophages. Collectively, these findings indicate that the presence of iron within the porphyrin ring was not essential for STAT1 suppression during KP infection.
Lastly, we tested whether porphyrin lacking iron recapitulated extrapulmonary bacterial proliferation seen with sRBC delivery in KP-infected mice. Mice were intratracheally instilled with KP and challenged an hour later with vehicle, 5 mg/kg CoPPIX, or 25 mg/kg CoPPIX. No differences in bacterial burden in the lung (Figure 7M) or spleen (Figure 7N) were observed among the 3 groups 24 hours after KP instillation. Notably, we observed increased extrapulmonary bacterial proliferation in the liver, the primary organ that supports rapid RBC removal during RBC damage (4), in KP-infected mice challenged with 25 mg/kg CoPPIX (Figure 7O). Although there was a trend toward dose-dependent increase in blood bacterial burden of KP-infected mice challenged with CoPPIX, this did not achieve statistical significance (Figure 7P). Taken together, these findings reinforce the notion that iron was dispensable for STAT1 suppression and extrapulmonary bacterial proliferation observed with sRBC delivery to KP-infected mice.
Discussion
Excess heme handling by macrophages occurs as a consequence of several pathologies where there is increased delivery of damaged RBC precipitating “on-demand” mononuclear phagocytic uptake — including hemoglobinopathies (66, 67), RBC membrane disorders (68), severe infections (45, 69–71), and transfusion iron overload (22, 23). Although comparisons of fresh versus old red cell transfusion did not show differences in clinical outcomes in anemic patients requiring transfusion (25, 26), we utilized the RBC transfusion model here to deliver sRBCs to macrophages and examine the consequences of intracellular heme excess in the face of severe infection. We demonstrated that excess sRBC ingestion disrupted STAT1 signaling during severe KP infection. We further showed that sRBC delivery impaired bacterial control at extrapulmonary sites and was associated with increased systemic inflammatory cytokine response and worsened survival during pneumonia-induced sepsis. Impaired KP control observed with stressed erythrophagocytosis was independent of iron acquisition by bacterial siderophores and supports the notion that excess heme handling by macrophages exerts immune-suppressive effects.
Others have shown that erythrophagocytosis is a conserved innate immune response to microbial products such as LPS (36). This is consistent with our findings that TLR4 stimulation drove macrophages to erythrophagocytosis. It is possible that IFN-γ produced in response to TLR stimulation can act directly on macrophages to provoke red cell uptake (72), and we observed increased systemic IFN-γ levels in KP-infected mice early after sRBC challenge. However, stressed erythrophagocytosis can be IFN-γ independent (36, 37). Regardless of the mechanism driving erythrophagocytosis, we observed consistent suppression of STAT1 and its downstream effectors. STAT1 is a critical transcription factor because STAT1 deficiency renders mice (28) and humans (27) unresponsive to type I or II IFNs and impairs the host’s ability to clear pathogenic microbes and viruses. We identified heme, and specifically the porphyrin macrocycle, as the constituent of RBCs that suppresses STAT1 during KP infection. Although heme has been heralded as an amplifier of inflammation (47), excess heme handling by the macrophage can exert immune-suppressive effects through mechanisms that remain to be fully elucidated (45, 73). Our work highlighted a mechanism by which excess heme handling mediated STAT1 suppression during infection and worsened host immunity.
Under homeostasis, CNC basic leucine zipper (bZIP) transcription factors, NRF1 and NRF2, are constitutively targeted for proteasomal degradation (61, 74). Undue heme exposure dysregulates proteasome activity, resulting in cellular protein accumulation (75). We demonstrated that heme exposure evokes NRF1 and NRF2 activity, as evidenced by increased Mt1 and Nqo1, respectively, in infected macrophages after sRBC delivery or heme or PPIX challenge. NRF2 has been shown to suppress innate immune responses to viral (58) and microbial triggers (56, 57). Here, we uncovered an additional role for NRF2 in tempering macrophage Stat1 expression during KP infection. In contrast to NRF2, relatively little is known about NRF1 and its role in immune regulation; targeted disruption of Nrf1 results in murine embryonic lethality due to impaired fetal liver erythropoiesis (76). Although NRF1 and NRF2 appear to have distinct gene targets (62), NRF1 and NRF2 bind to overlapping antioxidant response element (ARE) sequences as obligate dimers with small musculoaponeurotic fibrosarcoma proteins (77). Indeed, in contrast to late embryonic lethality (16.5–18.5 days) observed in Nrf1 mutants (76), compound deficiency in both Nrf1 and Nrf2 leads to early lethality between embryonic days 9 and 10 (78), indicating shared NRF1 and NRF2 functionality. In this study, we demonstrated that NRF1 and NRF2 synergistically tempered Stat1 during KP infection. The mechanism by which NRF1 and NRF2 regulate Stat1 remains unclear. Although Stat1 does not appear to contain any known ARE sequences in its promoter region, others have shown that NRF2 can bind and inhibit inflammatory cytokine gene expression in an ARE-independent manner (57). Thus, NRF1 and NRF2 may directly bind to and block Stat1 transcription or may induce a yet to be discovered mechanism that represses Stat1 during KP infection.
Owing to the potent pro-oxidant activity of iron and prior studies implicating iron overload in disruption of IFN signaling (79, 80), it came as a surprise that iron appeared dispensable for NRF1 and NRF2 activation and subsequent STAT1 suppression. Our findings indicated that CoPPIX and PPIX lacking a metal ion recapitulated heme-mediated NRF1 and NRF2 activation in macrophages during KP infection. Limitations of our study include examining early STAT1 and related responses mainly at a single time point, and the use of a mouse macrophage cell line or primary BMDMs. Key findings of PPIX suppressing downstream STAT1 targets such as IRF1 and CXCL10, however, were reproduced in human primary monocyte-derived macrophages. Furthermore, CoPPIX — which, unlike FePPIX, does not yield iron that is of benefit to the pathogen — enhanced bacterial proliferation in the liver during KP infection, affirming that provision of a readily available source of iron was not the primary mechanism for enhanced KP pathogenicity after sRBC delivery.
A critical barrier in the field of sepsis immunology is understanding why some individuals acquire an immunosuppressive phenotype after critical illness. We showed here that imposing an additional physiological stressor targeting the mononuclear phagocyte system during infection resulted in an immunosuppressive phenotype due to excess intracellular heme. Cellular heme levels are tightly controlled (81) and perturbations in heme homeostasis may underlie the pathophysiology of many life-threatening hematological disorders affecting millions of people worldwide. We showed that heme, because of its porphyrin ring, activated NRF1/NRF2 target genes and impaired macrophage STAT1 expression during severe infection. These findings demonstrated that the detrimental effects of excess heme extend beyond its propensity as an alarmin (82) and established a link between cellular heme metabolism and regulation of innate immune signaling, with important implications for infection and inflammation.
Methods
Animals.
C57BL/6J, (stock 000664), Ifnar1–/– (stock 32045), and Ifngr1–/– (stock 003288) mice were obtained from The Jackson Laboratory. Tlr4–/– (stock 029015), Nrf2–/– (stock 017009), and Stat1–/+ (stock 012606) mice were also obtained from The Jackson Laboratory, and respective breeding colonies were established at our facilities. Resulting progeny (Tlr4–/–, Nrf2–/–, Stat1–/–, and Stat1+/+) were utilized in select experiments. All mice were fed the same chow within the same room of the vivarium for at least 4 weeks prior to experimentation. For in vivo studies, we used age-matched, sex-matched 8- to 12-week-old male and female mice. Mice were randomly assigned, and a skilled technician who was blinded to the biological hypothesis performed inoculations and organ harvest. All animals were housed and maintained in specific pathogen–free environments.
Human subjects.
Peripheral whole blood (30 mL) was obtained from healthy adult volunteers. Ethnicities and sex were identified by self-reporting. Male and female subjects were included. After collection, monocytes were isolated from whole blood as previously described (83) and utilized in the in vitro study in a deidentified manner.
Bacterial strains.
KP strain 43816 serotype 2 from American Type Culture Collection (ATCC) was utilized for the bacterial pneumonia studies. entB ybtS mutant KP lacking production of siderophores Ent, gly-Ent, and Ybt and its parent WT strain have been previously described (32, 84). The method of bacterial growth and suspension for inoculation has also been previously reported (12).
Cell culture.
RAW 264.7 cells (murine macrophage cell line) were obtained from ATCC. RAW cells were cultured in DMEM supplemented with 10% newborn calf serum in a humidified incubator at 5% CO2 at 37°C. Bone marrow cells were isolated from adult male and female mice and cultured in DMEM supplemented with 20% FBS, 30% L929-conditioned medium, and 1% penicillin-streptomycin for 6 to 7 days in a humidified incubator at 5% CO2 at 37°C to allow for differentiation into macrophages as previously described (85).
Mouse RBC preparation.
C57BL/6J mice were euthanized and blood obtained aseptically via cardiac puncture using citrate dextrose phosphate anticoagulant. For in vivo studies, mouse RBCs were stressed by extended storage (11–13 days). yRBCs indicate freshly isolated RBCs or RBCs stored for only 1 day. The method of RBC storage using citrate phosphate dextrose adenine (CPDA-1) solution has been previously reported (29). Briefly, whole blood was collected from donor mice, pooled, and leukoreduced through a neonatal high-efficiency leukocyte reduction filter (Purecell Neo, Pall Corporation). The pooled blood was centrifuged at 400g for 15 minutes, and the volume reduced to a final hemoglobin level ranging from 17.0 to 17.5 g/dL. The final CPDA-1 concentration in the red cell concentrate was 14% and stored in the dark at 4°C for up to 13 days. For mouse red cells, 14 days corresponds to the limits of storage duration (29, 86). On the day of transfusion, yRBCs and sRBCs were washed with sterile PBS at room temperature and subsequently spun at 800g for 10 minutes. Supernatant was discarded and RBCs were resuspended in sterile PBS to attain hematocrit of 60%.
For in vitro studies, leukoreduced RBCs were stressed by heating at 48°C with continuous agitation at 300 rpm for 20 minutes (4). For RBC ghost preparation, leukoreduced stressed RBCs were centrifuged at 800g for 10 minutes, washed 3 times with cold PBS, and lysed with 20 mOsm phosphate buffer. Lysed RBCs were centrifuged at 30,000g for 30 minutes at 4°C and RBC ghost pellets were washed 3 times with 20 mOsm phosphate buffer. RBC ghosts were reconstituted in a volume of PBS equivalent to the stressed RBC transfusate (87).
Experimental bacterial pneumonia model.
Bacteria were harvested, washed, and resuspended in PBS just prior to use. Mice were anesthetized with isoflurane and 1 × 103 to 1 × 104 CFU of KP in a total volume of 100 μL was administered intratracheally under direct visualization using a sterile 200 μL pipet with filtered tip positioned just above the vocal cords. One hour after KP inoculation, 200 μL of sterile PBS, washed young RBCs (1 day old) or stored RBCs (11 to 13 days old) were transfused via retro-orbital vein.
In vitro KP stimulation.
RAW cells or BMDMs were seeded at a density of 1 × 106 cells per well of a 6-well tissue culture plate or at a density of 5 × 105 cells per well of a 12-well tissue culture plate in DMEM containing 10% neonatal calf serum 24 hours prior to stimulation. Media were replaced, and opsonized live KP serotype 2 (MOI 10:1, log phase) was introduced. KP was opsonized by incubating with 20% neonatal calf serum for 30 minutes at 4°C. Where indicated, leukoreduced sRBCs, hemin, CoPPIX, or PPIX were introduced concurrently. Leukoreduced sRBCs were resuspended in sterile PBS. In studies where hemin was added, hemin, CoPPIX, and PPIX were first dissolved in 100% DMSO at a concentration of 26 mg/mL (~40 mM) (8). Stock solution was then diluted in warmed DMEM to achieve a final concentration of 100 μM (final concentration of DMSO administered to cell = 0.25%). At the indicated time (usually 4 hours after KP infection), media were collected, spun at 10,000g for 10 minutes at 4°C, and cytokine release was evaluated in the resulting supernatant. Macrophages were washed with PBS and incubated in 800 μL to 1 mL of RBC lysis buffer (eBioscience, Invitrogen, 00-4333) for 30 to 60 seconds at room temperature with continuous swirling to lyse unengulfed RBCs. Macrophages were washed again with PBS and lysed to examine gene and protein expression via qRT-PCR and Western blot, respectively.
BAL fluid collection.
A closed container system was used to euthanize animals at prespecified times using isoflurane. A laparotomy was immediately performed, and mice were exsanguinated via the inferior vena cava using citrate as the anticoagulant. The trachea was cannulated using a 20-gauge catheter secured with silk suture. The left hilum was identified, secured with a second silk suture, and the left lung was removed for immediate tissue homogenization. BAL was performed by instilling 0.6 mL 0.9% normal saline containing 0.6 mM EDTA into the right lung, followed by three 0.5-mL washes. Total cell counts in BAL were determined using a hemocytometer. Cytospins were prepared from BAL fluid and stained with Diff-Quick (Siemens Healthcare Diagnosis Inc.). Differential cell counts were determined by counting a total of 200 cells per slide.
Measurements of lung, spleen, liver, and blood bacterial burden.
The left lung, spleen, and right medial lobe of the liver were removed after euthanasia at predetermined time points. For enumerating bacterial CFU in the lung, spleen, and liver, tissue was homogenized in 1 mL of sterile ddH2O. Bacterial CFU was also measured from whole blood obtained from the inferior vena cava as described above. Next, 10 μL of tissue homogenates or whole blood in triplicates were plated by 10-fold serial dilution on tryptic soy agar plates. Bacterial plates were counted after an overnight incubation at 37°C and recorded as CFU/mL.
Measurement of plasma cytokines.
Plasma samples from mice were diluted to 1:5 for measurement of cytokines in ELISA plates, and the values in pg/mL or ng/mL were calculated based on dilution. ELISA DuoSet antibodies for measuring TNF-α, IL-6, IL-10, C5a, MBL2, IL-1β, CXCL10, and IFN-γ were obtained from R&D Systems.
Liver histology.
The median lobe of the liver was fixed in 2% paraformaldehyde for 4 hours. Subsequently, lobes were embedded in paraffin and processed for sectioning and H&E staining. Whole-section images of the median lobes at ×100 original magnification were obtained using a TissueFAXS PLUS system (Tissuegnostics). Using the overview images, the total numbers of inflammatory foci and necrotic regions in each liver section were assessed by an investigator blinded to experimental groups.
Measurement of transferrin-bound iron.
Transferrin-bound iron was measured by electron paramagnetic spectroscopy. Plasma (~400 μL) was transferred to quartz tubes (Wilmad) for freezing in liquid nitrogen. Measurements of samples were carried out at 5–8 K using an EMX 10/12 spectrometer (Bruker Biospin Corp.) cooled by liquid helium and operated at microwave power of 0.1 mW, microwave frequency 9.387 GHz, modulation amplitude 15 G, with a sweep width of 1500 G, center field of 1250 G, sweep time of 42 seconds, and time constant of 81.92 milliseconds. Transferrin-bound iron concentrations in samples were obtained by fitting (least squares fit) to basis spectra of a standard of known concentration.
RNA-Seq.
Livers were obtained from KP-infected mice challenged with yRBCs or sRBCs 24 hours after KP infection. RNA was purified from liver tissue using RNeasy Plus Universal Mini kit (catalog 73407) according to the manufacturer’s instructions (Qiagen) and the concentration of isolated RNA was determined by NanoDrop. Purified RNA was sequenced using NextSeq 500 System (Illumina) at high output and paired-end read (2 × 150 cycles) by the Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh. Sequencing data were analyzed with CLC Genomics Workbench (Qiagen). Briefly, the sequencing data quality was assessed, and low-quality reads (Phred quality score < 20) and adaptor sequences were trimmed for downstream analysis. RNA-Seq data were mapped against mouse genomic sequence, gene sequence, and mRNA sequence. Expression difference between treatment groups was considered significant when absolute fold change was greater than or equal to 1.5, maximum group mean was greater than or equal to 1, and the FDR P value was less than 0.05.
Quantitative PCR.
Tissues were frozen in cold QIAzol lysis reagent and homogenized using a handheld homogenizer. After centrifugation at 12,000g for 10 minutes, the supernatants were used for total RNA extraction using RNeasy Plus Universal Mini kit. For in vitro studies, cells were lysed in QIAzol lysis reagent and RNA was also extracted using RNeasy Plus Universal Mini kit. RNA samples were reverse transcribed into cDNA using SuperScript III Reverse Transcriptase or MultiScribe Reverse Transcriptase (Moloney murine leukemia virus reverse transcriptase, Invitrogen). qPCR was performed according to the manufacturer’s protocol (Applied Biosystems) by incubating cDNA samples with specified probes and primers of interest and TaqMan Universal PCR Master Mix II and measuring PCR amplification using the 7900HT Real-Time PCR System. Probes and primers for Hmox1 (Mm00516004_m1), Rela (NF-κB subunit p65; Mm00501346_m1), Socs1 (Mm01342740_g1), Socs3 (Mm00545913_s1), Stat3 (Mm01219775_m1), Stat1 (Mm01257286_m1), Nos2 (Mm00440502_m1), Irf1 (Mm01288580_m1), Irf3 (Mm00516784_m1), Irf8 (Mm00492567_m1), Slc40a1 (Mm01254822_m1), C3 (Mm01232779_m1), Cfb (Mm00433909_m1), Gapdh (Mm99999915_g1), and 18S (Hs99999901_s1) were commercially available at Applied Biosystems. Gene expression was analyzed by the ΔΔCt method with 18S rRNA or Gapdh as the endogenous control, and the average ΔCt of unstimulated WT controls served as the calibrator.
Immunoblot.
Macrophages were lysed with Pierce IP lysis buffer (Thermo Fisher Scientific) with cOmplete Mini Protease Inhibitor and Phosphatase Inhibitor (Roche). After brief sonication on ice, samples were centrifuged at 10,000g at 4°C for 10 minutes and supernatants were saved for Western blotting. Next, 5–30 μg protein was loaded per well onto a NuPAGE 4% to 12% Bis-Tris Gel (Life Technologies). After protein transfer, membranes were incubated with 1:1000 dilution of specific antibodies against phospho-STAT1 (Ser727, 9177), phospho-STAT1 (Tyr701, 9167), STAT1 (14994), and IRF1 (8478) obtained from Cell Signaling Technology. Membranes were incubated with 1:2000 dilution of antibody against β-actin (4970) obtained from Cell Signaling Technology or 1:1000 dilution of antibody against α-tubulin (ab4074) obtained from Abcam as loading control. Antibodies against BACH1 (HRP conjugated, sc-271211) and HO-1 (HRP conjugated, sc-390991) were obtained from Santa Cruz Biotechnology and utilized at 1:1000 dilutions.
Purification of mouse hemoglobin.
The process for purification of hemoglobin from RBCs has been previously described (88). Briefly, the initial sample was loaded in a chelating sepharose fast flow resin charged with Zn2+ and equilibrated in 20 mM Tris, 500 mM NaCl, pH 8.3. The column was washed with 20 mM Tris, 500 mM NaCl, pH 8.3 (flow-through sample 1), 200 mM Tris, pH 8.3 (flow-through sample 2), 20 mM Tris, pH 8.3, and then washed with 20 mM Tris, 15 mM EDTA, pH 8.3 (Hb after IMAC small sample). The sample was concentrated and washed with PBS using a 50-kDa filter (final estimated EDTA concentration < 22 nM). Calculated hemoglobin concentration was 4.7 mM (93.5% oxyHb, 6.5% metHb).
Phagocytosis assays.
For sRBC uptake, macrophages were seeded at a density of 1 × 106 cells per well of a 6-well tissue culture plate in DMEM supplemented with 10% neonatal calf serum 24 hours prior to assay. Macrophages were then incubated with vehicle (PBS), sRBCs (50:1), KP (10:1), or KP + sRBC for 90 minutes or 2 hours. After incubation, macrophages were washed with PBS, unengulfed sRBCs were lysed, and macrophages were washed again with PBS. Macrophages were then incubated briefly (<5 minutes) in 0.5% trypsin-EDTA at 37°C to allow cell detachment prior to cytospin. Suspended cells were immobilized onto glass slides by centrifugation at 450 RPM for 3 minutes and slides were stained using Diff-Quik. Phagocytic index was determined as previously described (89):
(Equation 1)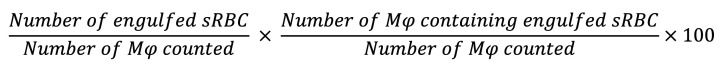
For KP uptake, macrophages were seeded at a density of 2.5 × 105 cells per well of a 24-well tissue culture plate 24 hours prior to assay and subsequently incubated with KP (MOI 10:1) or KP + sRBC for 90 minutes or 2 hours. After incubation, phagocytes were incubated in HBSS containing 100 μg/mL gentamicin for 1 hour to kill extracellular attached bacteria and subsequently lysed with 100 μL HBSS containing 0.1% Triton X-100, and then lysates were plated on tryptic soy agar to reveal intracellular CFU/mL.
siRNA transfection.
Macrophages were plated at a density of 1 × 105 or 2 × 105 cells per well of a 12-well tissue culture plate 24 hours prior to transfection. On the day of transfection, cells were washed with RPMI 1640, incubated in RPMI containing 2% FBS, and transfected with 50 to 240 nM of siRNA using HiPerfect transfection reagent (Qiagen) according to the manufacturer’s instructions. siRNAs were purchased from Dharmacon Inc. (now Horizon Discovery Ltd.). A negative pool of 4 control siRNAs was utilized as control. Experiments were conducted 24 to 48 hours after transfection, and transcription efficiency was determined by qRT-PCR and immunoblot.
Data accessibility.
Data files for RNA-Seq reported in this paper have been deposited in NCBI’s Gene Expression Omnibus database (GEO GSE144902).
Statistics.
Results are reported as the median unless otherwise indicated. Log-rank test was performed to generate the Kaplan-Meier survival curve. For in vivo comparisons between 2 groups, a nonparametric Mann-Whitney U test was undertaken. For in vivo comparisons of multiple groups, Kruskal-Wallis with Dunn’s multiple comparisons test was undertaken. For in vitro comparisons between 2 groups, a 2-tailed t test was undertaken. For in vitro comparisons of multiple groups, a 1-way ANOVA with Tukey’s multiple comparisons test was undertaken. For comparisons between groups over time, a 2-way ANOVA was undertaken. GraphPad Prism software versions 5.0 and 6.0 were used for statistical analysis. A P value less than 0.05 was considered significant.
Study approval.
Studies were conducted in accordance with and approval of the IACUC and IRB (no. 0410173) at the University of Pittsburgh. All subjects underwent venipuncture after providing informed written consent. All subjects provided written informed consent prior to inclusion in the study.
Author contributions
TFO, ZX, MAB, EAH, CS, YT, VEK, and JSL were responsible for designing the research studies. TFO, TS, SGF, ZX, HP, RG, YX, DO, JT, AP, SB, MAB, EAH, CS, YT, and VEK conducted the experiments, analyzed the data, and interpreted the data. RKM, PR, AR, MRR, WM, DBK, and MTG provided resources and important intellectual content to the manuscript. TFO, SGF, DVT, and JSL wrote the manuscript. JSL provided supervision. TFO, RKM, PR, DBK, MTG, and JSL acquired funding.
Supplementary Material
Acknowledgments
We would like to thank Sally Wenzel, Grant Bullock, Jennifer Bomberger, and Saumendra Sarkar for their critique and guidance. We would also like to thank Samit Ghosh for providing additional Nrf2–/– mice and Mei Hulver for animal caretaking. This work was supported by American Heart Association predoctoral fellowship award (to TFO); NIH grants R01 HL136143, R01 HL142084, HL142084-02S1, and K24 HL143285 (to JSL); NIH grant R01 HL098032 (to DBK and MTG); the Vascular Medicine Institute; the Hemophilia Center of Western Pennsylvania; Institute for Transfusion Medicine (to JSL); and NIH grant P01 HL114453 (to RKM, PR, and JSL).
Version 1. 09/17/2020
In-Press Preview
Version 2. 01/04/2021
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(1):e137468.https://doi.org/10.1172/JCI137468.
Contributor Information
Tolani F. Olonisakin, Email: olonisakin.tolani@medstudent.pitt.edu.
Shekina Gonzalez-Ferrer, Email: shg103@pitt.edu.
Zeyu Xiong, Email: xiongz@upmc.edu.
Hernán F. Peñaloza, Email: penalozahf@upmc.edu.
Rick van der Geest, Email: RIV13@pitt.edu.
Yuting Xiong, Email: yux27@pitt.edu.
David O. Osei-Hwedieh, Email: doo8@pitt.edu.
Matthew R. Rosengart, Email: rosengartmr@upmc.edu.
Wendy M. Mars, Email: wmars@pitt.edu.
Daria Van Tyne, Email: vantyne@pitt.edu.
Andreas Perlegas, Email: perlegas@wfu.edu.
Samuel Brashears, Email: sam.brashears@gmail.com.
Daniel B. Kim-Shapiro, Email: shapiro@wfu.edu.
Mark T. Gladwin, Email: gladwinmt@upmc.edu.
Michael A. Bachman, Email: mikebach@med.umich.edu.
Eldad A. Hod, Email: eh2217@cumc.columbia.edu.
Claudette St. Croix, Email: claudette.stcroix@pitt.edu.
Yulia Y. Tyurina, Email: yyt1@pitt.edu.
Valerian E. Kagan, Email: kagan@pitt.edu.
Rama K. Mallampalli, Email: rama.mallampalli2@osumc.edu.
Anuradha Ray, Email: raya@pitt.edu.
Prabir Ray, Email: rayp@pitt.edu.
Janet S. Lee, Email: leejs3@upmc.edu.
References
- 1.Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun. 2012;4(5-6):446–453. doi: 10.1159/000336423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higgins JM, Mahadevan L. Physiological and pathological population dynamics of circulating human red blood cells. Proc Natl Acad Sci U S A. 2010;107(47):20587–20592. doi: 10.1073/pnas.1012747107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. 2017;168(3):344–361. doi: 10.1016/j.cell.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Theurl I, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. 2016;22(8):945–951. doi: 10.1038/nm.4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nairz M, Theurl I, Swirski FK, Weiss G. “Pumping iron”—how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017;469(3–4):397–418. doi: 10.1007/s00424-017-1944-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellani G, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 7.Vincent JL, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302(21):2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 8.Broug-Holub E, et al. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect Immun. 1997;65(4):1139–1146. doi: 10.1128/IAI.65.4.1139-1146.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olonisakin TF, et al. CD36 provides host protection against Klebsiella pneumoniae intrapulmonary infection by enhancing lipopolysaccharide responsiveness and macrophage phagocytosis. J Infect Dis. 2016;214(12):1865–1875. doi: 10.1093/infdis/jiw451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domenico P, Salo RJ, Cross AS, Cunha BA. Polysaccharide capsule-mediated resistance to opsonophagocytosis in Klebsiella pneumoniae. Infect Immun. 1994;62(10):4495–4499. doi: 10.1128/IAI.62.10.4495-4499.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siu LK, Yeh KM, Lin JC, Fung CP, Chang FY. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis. 2012;12(11):881–887. doi: 10.1016/S1473-3099(12)70205-0. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, et al. Thrombospondin-1 restrains neutrophil granule serine protease function and regulates the innate immune response during Klebsiella pneumoniae infection. Mucosal Immunol. 2015;8(4):896–905. doi: 10.1038/mi.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis AJ, Lee JS, Rosengart MR. Translational sepsis research: spanning the divide. Crit Care Med. 2018;46(9):1497–1505. doi: 10.1097/CCM.0000000000003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bachman MA, Miller VL, Weiser JN. Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS Pathog. 2009;5(10):e1000622. doi: 10.1371/journal.ppat.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachman MA, et al. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect Immun. 2011;79(8):3309–3316. doi: 10.1128/IAI.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wooldridge KG, Williams PH. Iron uptake mechanisms of pathogenic bacteria. FEMS Microbiol Rev. 1993;12(4):325–348. doi: 10.1111/j.1574-6976.1993.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 17.Michels KR, et al. Hepcidin-mediated iron sequestration protects against bacterial dissemination during pneumonia. JCI Insight. 2017;2(6):e92002. doi: 10.1172/jci.insight.92002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan FA, Fisher MA, Khakoo RA. Association of hemochromatosis with infectious diseases: expanding spectrum. Int J Infect Dis. 2007;11(6):482–487. doi: 10.1016/j.ijid.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Vadillo M, Corbella X, Pac V, Fernandez-Viladrich P, Pujol R. Multiple liver abscesses due to Yersinia enterocolitica discloses primary hemochromatosis: three cases reports and review. Clin Infect Dis. 1994;18(6):938–941. doi: 10.1093/clinids/18.6.938. [DOI] [PubMed] [Google Scholar]
- 20.Corke PJ, McLean AS, Stewart D, Adams S. Overwhelming gram negative septic shock in haemochromatosis. Anaesth Intensive Care. 1995;23(3):346–349. doi: 10.1177/0310057X9502300313. [DOI] [PubMed] [Google Scholar]
- 21.Soares MP, Hamza I. Macrophages and iron metabolism. Immunity. 2016;44(3):492–504. doi: 10.1016/j.immuni.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin L, et al. Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318(5857):1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 23.Kayama H, et al. Heme ameliorates dextran sodium sulfate-induced colitis through providing intestinal macrophages with noninflammatory profiles. Proc Natl Acad Sci U S A. 2018;115(33):8418–8423. doi: 10.1073/pnas.1808426115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim-Shapiro DB, Lee J, Gladwin MT. Storage lesion: role of red blood cell breakdown. Transfusion. 2011;51(4):844–851. doi: 10.1111/j.1537-2995.2011.03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heddle NM, et al. Effect of short-term vs. long-term blood storage on mortality after transfusion. N Engl J Med. 2016;375(20):1937–1945. doi: 10.1056/NEJMoa1609014. [DOI] [PubMed] [Google Scholar]
- 26.Lacroix J, et al. Age of transfused blood in critically ill adults. N Engl J Med. 2015;372(15):1410–1418. doi: 10.1056/NEJMoa1500704. [DOI] [PubMed] [Google Scholar]
- 27.Dupuis S, et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33(3):388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 28.Meraz MA, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84(3):431–442. doi: 10.1016/S0092-8674(00)81288-X. [DOI] [PubMed] [Google Scholar]
- 29.Hod EA, et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood. 2010;115(21):4284–4292. doi: 10.1182/blood-2009-10-245001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Youssef LA, et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood. 2018;131(23):2581–2593. doi: 10.1182/blood-2017-12-822619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hod EA, et al. Transfusion of human volunteers with older, stored red blood cells produces extravascular hemolysis and circulating non-transferrin-bound iron. Blood. 2011;118(25):6675–6682. doi: 10.1182/blood-2011-08-371849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holden VI, Breen P, Houle S, Dozois CM, Bachman MA. Klebsiella pneumoniae siderophores induce inflammation, bacterial dissemination, and HIF-1α stabilization during pneumonia. MBio. 2016;7(5):e01397-16. doi: 10.1128/mBio.01397-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quinton LJ, et al. Hepatocyte-specific mutation of both NF-κB RelA and STAT3 abrogates the acute phase response in mice. J Clin Invest. 2012;122(5):1758–1763. doi: 10.1172/JCI59408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kagan VE, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poe SL, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 2013;6(1):189–199. doi: 10.1038/mi.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald EM, Pilonieta MC, Nick HJ, Detweiler CS. Bacterial stimulation of toll-like receptor 4 drives macrophages to hemophagocytose. Infect Immun. 2016;84(1):47–55. doi: 10.1128/IAI.01149-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canna SW, et al. Interferon-γ mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. 2013;65(7):1764–1775. doi: 10.1002/art.37958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alam J, Shibahara S, Smith A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J Biol Chem. 1989;264(11):6371–6375. [PubMed] [Google Scholar]
- 39.Marro S, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position –7007 of the FPN1 promoter. Haematologica. 2010;95(8):1261–1268. doi: 10.3324/haematol.2009.020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vodovotz Y, et al. The hepatocyte as a microbial product-responsive cell. J Endotoxin Res. 2001;7(5):365–373. [PubMed] [Google Scholar]
- 41.Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol. 2016;13(3):301–315. doi: 10.1038/cmi.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crispe IN. Hepatocytes as immunological agents. J Immunol. 2016;196(1):17–21. doi: 10.4049/jimmunol.1501668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beutler E. Composition of the erythrocyte. In: Williams Hematology. 8th ed. McGraw-Hill; 2010:chapter 30. [Google Scholar]
- 44.Graça-Souza AV, Arruda MAB, de Freitas MS, Barja-Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood. 2002;99(11):4160–4165. doi: 10.1182/blood.V99.11.4160. [DOI] [PubMed] [Google Scholar]
- 45.Martins R, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. 2016;17(12):1361–1372. doi: 10.1038/ni.3590. [DOI] [PubMed] [Google Scholar]
- 46.Cunnington AJ, de Souza JB, Walther M, Riley EM. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat Med. 2011;18(1):120–127. doi: 10.1038/nm.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Figueiredo RT, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282(28):20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 48.Ogawa K, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20(11):2835–2843. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zenke-Kawasaki Y, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27(19):6962–6971. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun J, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21(19):5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Domenico I, et al. Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. J Clin Invest. 2010;120(7):2395–2405. doi: 10.1172/JCI42011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alcaraz MJ, Fernández P, Guillén MI. Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr Pharm Des. 2003;9(30):2541–2551. doi: 10.2174/1381612033453749. [DOI] [PubMed] [Google Scholar]
- 53.Otterbein LE, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6(4):422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 54.Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80(12):1895–1903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 55.Boyle JJ, et al. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2011;31(11):2685–2691. doi: 10.1161/ATVBAHA.111.225813. [DOI] [PubMed] [Google Scholar]
- 56.Thimmulappa RK, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi EH, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olagnier D, et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun. 2018;9(1):3506. doi: 10.1038/s41467-018-05861-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93(25):14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chorley BN, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40(15):7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang K, Huang R, Fujihira H, Suzuki T, Yan N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J Exp Med. 2018;215(10):2600–2616. doi: 10.1084/jem.20180783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohtsuji M, et al. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283(48):33554–33562. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. National Center for Biotechnology Information. Deferasirox, CID = 214348. NIH website. https://pubchem.ncbi.nlm.nih.gov/compound/Deferasirox Accessed October 27, 2020.
- 64.Glickstein H, et al. Action of chelators in iron-loaded cardiac cells: accessibility to intracellular labile iron and functional consequences. Blood. 2006;108(9):3195–3203. doi: 10.1182/blood-2006-05-020867. [DOI] [PubMed] [Google Scholar]
- 65.Liddell JR, et al. Lipophilic adamantyl- or deferasirox-based conjugates of desferrioxamine B have enhanced neuroprotective capacity: implications for Parkinson disease. Free Radic Biol Med. 2013;60:147–156. doi: 10.1016/j.freeradbiomed.2013.01.027. [DOI] [PubMed] [Google Scholar]
- 66.Williamson D. The unstable haemoglobins. Blood Rev. 1993;7(3):146–163. doi: 10.1016/0268-960X(93)90002-L. [DOI] [PubMed] [Google Scholar]
- 67.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311–323. doi: 10.1016/S0140-6736(17)30193-9. [DOI] [PubMed] [Google Scholar]
- 68. doi: 10.1182/asheducation-2005.1.13. Gallagher PG. Red cell membrane disorders. Hematology Am Soc Hematol Educ Program. 2005;13–18. https://doi.org/10.1182/asheducation-2005.1.13. [DOI] [PubMed]
- 69.Larsen R, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med. 2010;2(51):51ra71. doi: 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- 70.Baskurt OK, Gelmont D, Meiselman HJ. Red blood cell deformability in sepsis. Am J Respir Crit Care Med. 1998;157(2):421–427. doi: 10.1164/ajrccm.157.2.9611103. [DOI] [PubMed] [Google Scholar]
- 71.Piagnerelli M, et al. Alterations of red blood cell shape and sialic acid membrane content in septic patients. Crit Care Med. 2003;31(8):2156–2162. doi: 10.1097/01.CCM.0000079608.00875.14. [DOI] [PubMed] [Google Scholar]
- 72.Zoller EE, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med. 2011;208(6):1203–1214. doi: 10.1084/jem.20102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martins R, Knapp S. Heme and hemolysis in innate immunity: adding insult to injury. Curr Opin Immunol. 2018;50:14–20. doi: 10.1016/j.coi.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 74.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 75.Vallelian F, et al. Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. 2015;22(4):597–611. doi: 10.1038/cdd.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan JY, et al. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998;17(6):1779–1787. doi: 10.1093/emboj/17.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biswas M, Chan JY. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol Appl Pharmacol. 2010;244(1):16–20. doi: 10.1016/j.taap.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leung L, Kwong M, Hou S, Lee C, Chan JY. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J Biol Chem. 2003;278(48):48021–48029. doi: 10.1074/jbc.M308439200. [DOI] [PubMed] [Google Scholar]
- 79.Oexle H, et al. Pathways for the regulation of interferon-γ-inducible genes by iron in human monocytic cells. J Leukoc Biol. 2003;74(2):287–294. doi: 10.1189/jlb.0802420. [DOI] [PubMed] [Google Scholar]
- 80.Dlaska M, Weiss G. Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J Immunol. 1999;162(10):6171–6177. [PubMed] [Google Scholar]
- 81.Ponka P. Cell biology of heme. Am J Med Sci. 1999;318(4):241–256. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 82.Soares MP, Bozza MT. Red alert: labile heme is an alarmin. Curr Opin Immunol. 2016;38:94–100. doi: 10.1016/j.coi.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 83.Pinilla-Vera M, et al. Full spectrum of LPS activation in alveolar macrophages of healthy volunteers by whole transcriptomic profiling. PLoS One. 2016;11(7):e0159329. doi: 10.1371/journal.pone.0159329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bachman MA, Lenio S, Schmidt L, Oyler JE, Weiser JN. Interaction of lipocalin 2, transferrin, and siderophores determines the replicative niche of Klebsiella pneumoniae during pneumonia. MBio. 2012;3(6):e00224-11. doi: 10.1128/mBio.00224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pineda-Torra I, Gage M, de Juan A, Pello OM. Isolation, culture, and polarization of murine bone marrow-derived and peritoneal macrophages. Methods Mol Biol. 2015;1339:101–109. doi: 10.1007/978-1-4939-2929-0_6. [DOI] [PubMed] [Google Scholar]
- 86.et al. Sickle cell trait increases red blood cell storage hemolysis and post-transfusion clearance in mice. EBioMedicine. 2016;11:239–248. doi: 10.1016/j.ebiom.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dodge JT, Mitchell C, Hanahan DJ. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch Biochem Biophys. 1963;100:119–130. doi: 10.1016/0003-9861(63)90042-0. [DOI] [PubMed] [Google Scholar]
- 88.Elmer J, Harris D, Palmer AF. Purification of hemoglobin from red blood cells using tangential flow filtration and immobilized metal ion affinity chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(2):131–138. doi: 10.1016/j.jchromb.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sano H, et al. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112(3):389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data files for RNA-Seq reported in this paper have been deposited in NCBI’s Gene Expression Omnibus database (GEO GSE144902).