

Abstract
Motivation
The novel coronavirus (SARS-CoV-2) currently spreads worldwide, causing the disease COVID-19. The number of infections increases daily, without any approved antiviral therapy. The recently released viral nucleotide sequence enables the identification of therapeutic targets, e.g. by analyzing integrated human-virus metabolic models. Investigations of changed metabolic processes after virus infections and the effect of knock-outs on the host and the virus can reveal new potential targets.
Results
We generated an integrated host–virus genome-scale metabolic model of human alveolar macrophages and SARS-CoV-2. Analyses of stoichiometric and metabolic changes between uninfected and infected host cells using flux balance analysis (FBA) highlighted the different requirements of host and virus. Consequently, alterations in the metabolism can have different effects on host and virus, leading to potential antiviral targets. One of these potential targets is guanylate kinase (GK1). In FBA analyses, the knock-out of the GK1 decreased the growth of the virus to zero, while not affecting the host. As GK1 inhibitors are described in the literature, its potential therapeutic effect for SARS-CoV-2 infections needs to be verified in in-vitro experiments.
Availability and implementation
The computational model is accessible at https://identifiers.org/biomodels.db/MODEL2003020001.
1 Introduction
In December 2019, an outbreak of pneumonia in Wuhan, Hubei province, in China, has aroused the interest of the international community by showing alarming similarities to the outbreaks caused by other -coronaviruses (-CoV) like the Severe Acute Respiratory Syndrome (SARS) virus (Huang et al., 2020; Hui et al., 2020).
The febrile respiratory illness caused by the novel coronavirus (SARS-CoV-2) is thought to have spread as a zoonosis from the Huanan Seafood Wholesale Market, which was as a consequence shut down on January 1, 2020 to prevent further transmission events (Hui et al., 2020).
On January 7, first isolation and subsequent deep-sequencing of SARS-CoV-2 from the human lower respiratory tract samples have made the genetic sequence of the virus available to the public by January 12, 2020, thus allowing for the identification of the virus as a Group 2B -CoV (Huang et al., 2020; Hui et al., 2020). SARS-CoV-2 has 82% sequence similarity with the SARS virus, which has caused an outbreak originating in China in 2002 (Hui et al., 2020; Zhang et al., 2020). The outbreak in 2002 has peaked at a total of 8098 documented cases with a case fatality rate of 9.6% (Huang et al., 2020; Hui et al., 2020). In contrast, the ongoing SARS-CoV-2 has reached, at time of writing, a total of ∼3 million cases worldwide and caused over 200 000 deaths (John Hopkins University, 2020; WHO, 2020). The resemblance and the severe global health threat have initiated a swift and determined implementation of public health measures by the Chinese Authorities (Chen et al., 2020; Hui et al., 2020).
While human-to-human transmissions in a nosocomial setting were the primary route of transmission of the SARS virus outbreak, Chinese horseshoe bats have been identified as putative primary reservoir and source of zoonosis for SARS (Huang et al., 2020; Hui et al., 2020; Paraskevis et al., 2020). Moreover, Himalayan palm civets, raccoon dogs and Chinese ferret badgers from the Guangdong wet markets (live animal retail markets) were identified as intermediate reservoirs of zoonosis (Hui et al., 2020; Perlman and Netland, 2009). In the case of SARS, the switch to the human host was allowed by an adaption of the receptor binding domain of the spike (S) protein, conferring to binding capabilities to the human angiotensin-converting enzyme 2 (ACE2) (Huang et al., 2020; Perlman and Netland, 2009). For the SARS-CoV-2, there was initially no proof of efficient human-to-human transmission, however, the rapid increase in cases and distinct clustering of the disease have made it clear that an efficient transmission route in-between humans exists (Chan et al., 2020; Hui et al., 2020). Moreover, Paraskevis et al. (2020) have suggested, based on full-genome evolutionary analysis, a zoonosis of SARS-CoV-2 from bats to humans. Accordingly, Letko et al. have recently identified ACE2 as the SARS-CoV-2 entry receptor (Letko and Munster, 2020).
As mentioned above, the SARS-CoV-2 belongs to the subfamily of -CoV (Huang et al., 2020; Hui et al., 2020). The members of the family are enveloped, single-stranded RNA (ssRNA) viruses with a positive polarity genome of up to 34 kb (Chen et al., 2020; Huang et al., 2020; Zhang et al., 2020). Replication of the RNA genome is performed via an RNA-dependent RNA polymerase (RdRP) in double-membrane vesicles (DMVs), modified to form a reticulovesicular network (Chen et al., 2020; de Wilde et al., 2015; Perlman and Netland, 2009). In the DMVs, the 16 non-structural proteins (NSPs) are directly expressed as polyproteins pp1a and pp1ab from the (+)RNA genome (Chen et al., 2020). Processing of pp1a/1ab by the viral main protease (Mpro) is essential to form the replication-transcription complex for the subsequent expression of the viral structural proteins (Chen et al., 2020; Perlman and Netland, 2009).
In total, the NSPs constitute two thirds of the genome’s coding capacity (Chen et al., 2020). The rest encodes for structural proteins, such as the spike (S), membrane (M) and envelope (E) proteins, which cover a helical nucleocapsid made up of the nucleocapsid (N) proteins (Chen et al., 2020; Perlman and Netland, 2009). Unlike NSPs, the structural and additional accessory proteins of coronaviruses require synthesis as subgenomic messenger RNAs via discontinuous transcription from (-)RNA templates (Chen et al., 2020).
Clinically, the SARS-CoV-2 seems to be milder than the former SARS virus, although they share the symptoms of febrile illness and pneumonia (Chen et al., 2020; Huang et al., 2020; Hui et al., 2020). It was recently shown that the SARS-CoV-2 does not only infect lower respiratory tract cells, but also upper cells in the pharyngeal region (Chen et al., 2020; Huang et al., 2020). Moreover, many coronaviruses also infect macrophages and subsequently inhibit an interferon-stimulated genes-mediated antiviral response, increasing immune evasion and pathogenicity (Chen et al., 2020; Cheung et al., 2005; Deng et al., 2017; Huang et al., 2020).
By now, no antiviral treatment for coronaviruses has been proven efficacious in a clinical trial (Chen et al., 2020; Huang et al., 2020). Recently, Zhang et al. (2020) have found -ketoamides to be broad-spectrum inhibitors of coronaviruses by tissue-dependently inhibiting the Mpro of SARS in Vero cells. This inhibition has occurred in a micromolar EC50 range, indicating -ketoamides to be a potential antiviral for the SARS-CoV-2 (Zhang et al., 2020). However, since no therapy or vaccination is available for clinical use, the current standard of care for a SARS-CoV-2 infection is limited to the supportive treatment of symptoms (Chen et al., 2020; Huang et al., 2020).
As no antiviral treatment is currently available for coronaviruses, the identification of potential antiviral targets is of great interest. One possibility of identifying new targets is the analysis of metabolic changes in infected cells. Aller et al. introduced a procedure for integrated human-virus metabolic models to predict host-based antiviral targets against Chikungunya, Dengue and Zika viruses. The analysis of the integrated human-virus models predicted inhibiting effects of constrained host-reactions on virus production. These predictions included already known targets of existing antiviral drugs, demonstrating the applicability of such analysis methods (Aller et al., 2018).
In our study, we integrated and analyzed a human genome-scale metabolic model (GEM) infected with SARS-CoV-2. As it is shown that coronaviruses infect alveolar macrophages (Cheung et al., 2005; Deng et al., 2017; Joel Funk et al., 2012), and Bordbar et al. (2010) already published an extensive metabolic model of human alveolar macrophages, this model was used as a host model. For SARS-CoV-2, a virus biomass objective function (VBOF) was generated according to Aller et al. (2018). Subsequent analysis of the integrated host-virus model revealed potential targets for antiviral therapies.
2 Materials and methods
The methods used in this article are based on the article by Aller et al. (2018). Methods and analyses were adapted and expanded for the coronavirus SARS-CoV-2.
2.1. Generation of VBOF
Analogous to the biomass production or maintenance function in prokaryotic or eukaryotic metabolic models, the VBOF is a pseudo-reaction simulating the production of virus particles. It comprises nucleotides, amino acids and associated energy metabolites required for the production of the virus particles. Due to a lack of knowledge, the stoichiometric information of the virus envelope, and the dynamic information of lipids are not included in the VBOF. Hence, virus entry or exit cannot be modeled. The generation of the VBOF was performed following the seven steps described by Aller et al. (2018).
Step 1: Virus genome and protein information. The recently published virus genome and protein sequence were obtained from the NCBI database (Geer et al., 2010) with the accession number NC_045512.2 in February 2020. Essential for the calculation of the nucleotide count is the classification of the SARS-CoV-2 in the Baltimore Classification System (Baltimore, 1971) that characterizes viruses based on the replication method. Coronaviruses fall into the Group IV classification. These viruses replicate their positive single-stranded RNA (+ssRNA) genomes via a complementary negative ssRNA. The nucleotide counts of the positive strand can be taken from the genome sequence. The nucleotides of the negative strand can be calculated by counting the complementary nucleotides of the positive strand. Both nucleotides are summed up to receive the total nucleotide count.SARS-CoV-2 has a total of 12 proteins, four structural proteins, and 8 NSPs. Structural and NSPs are not expressed equally, and this ratio needs to be considered during the calculation of the VBOF. To this date, no information about the copy number of structural proteins in Coronaviruses is reported.
Step 2: Nucleotide investment. The nucleotide count of the virus genome and its replication intermediate are summed per nucleotide. The genome copy number (Cg) was assumed to be one. According to Aller et al., the moles of nucleotides were converted into grams of nucleotides per mole of the virus. After similar calculations of the amino acids and the calculation of the total molar weight of the virus based on nucleotide and amino acid content, the stoichiometric coefficients of each nucleotide in the VBOF were calculated.
Step 3: Amino acid investment. Analogous to the nucleotide investment, the stoichiometric coefficient of each amino acid was calculated. Instead of the genome copy number, a copy number for structural and NSPs is required. As already mentioned, the copy number of structural proteins is not reported to date. Therefore, we repeated all analyses described in this article for copy numbers of structural proteins Csp=200, 500, 800 and 1200. With the information of the total count of each amino acid, weighted by the copy number of structural proteins, and the inclusion of the respective molar mass, the total viral molar mass was calculated (see also Step 6) to eventually calculate the stoichiometric coefficient for every amino acid.
Steps 4 and 5: Adenosine triphosphate (ATP) requirements, and pyrophosphate liberation. The calculations of the ATP requirements for the polymerization of amino acids and the calculation of pyrophosphate (PPi) liberation from the nucleotide polymerization were performed as described by Aller et al. based on the results of the previous steps. The constants and were chosen, as suggested by Aller et al. is the required amount of four ATP molecules for the polymerization of amino acid monomers. The constant stands for the liberation of one diphosphate molecule per polymerization of nucleotide monomers.
Steps 6 and 7: Total viral molar mass and final construction of the VBOF. The total molar mass of SARS-CoV-2 and the final VBOF were calculated and constructed in accordance with Aller et al.
2.2 Integration of the SARS-CoV-2 virus into iAB-AMØ-1410
Since Coronaviridae infect alveolar macrophages (Cheung et al., 2005; Deng et al., 2017), the cell-specific GEM of human alveolar macrophages iAB-AMØ-1410 was utilized. This GEM was published in 2010 by Bordbar et al. (2010) to give insight into human alveolar macrophages infected with Mycobacterium tuberculosis. The cell-specific GEM was constructed based on the human metabolic reconstruction Recon 1 (Duarte et al., 2007). It consists of 3394 reactions and 2572 metabolites. The model was downloaded in SBML Level 2 Version 4 format (Hucka et al., 2008) and upgraded to SBML Level 3 Version 1 format (Hucka et al., 2018) using libSBML (Bornstein et al., 2008). The VBOF was incorporated into the existing macrophage model, using flux bounds of 0 and 1000 as lower and upper bounds. Additionally, aggregated subsystems were added to the reactions as additional information for further analyses (Aller et al., 2018; Zielinski et al., 2015).
2.3 Identification of stoichiometric differences
The human alveolar macrophage model iAB-AMØ-1410 included now two pseudo-reactions for the production or maintenance of the virus and hosts biomass, respectively. The stoichiometric coefficients of shared metabolites within these pseudo-reactions can be compared to identify differences in nucleotide or amino acid requirements. First, the individual amino acid and nucleotide stoichiometric coefficients were normalized against the sum of all metabolites present in the respective biomass objective function (BOF), except for the metabolites for energy requirements. Then, the fold-change (FC) of the normalized amino acid or nucleotide was calculated:
| (1) |
with index i over nucleotides or amino acids and k over all biomass precursors (except for energy requirements). Subscripts H and V represent the use of either the host or virus biomass function. Positive values imply higher usage of nucleotides or amino acids in the virus compared to the host, while negative values imply a lower usage.
2.4 Flux balance analysis and flux variability analysis for the comparison of host- and virus-optimized states
The integration of the VBOF into the iAB-AMØ-1410 model paved the way for the analysis of metabolic changes between host and virus-optimized states. To identify an optimal state, an objective function that is optimized needs to be defined. In general, every reaction in the GEM can serve as objective function. However, biologically meaningful objective functions depend on the research question. For analyses of the growth or survival of the studied organism or cell, biomass production or maintenance reactions can be introduced into the model as pseudo-reactions. In industrial settings, the production of a specific metabolite might be of interest, and hence its production reaction can be set as the objective function. The defined objective function is then optimized with flux balance analysis (FBA). FBA is a mathematical approach using linear optimization to analyze the flow of metabolites through a metabolic network while optimizing for an objective function (Orth et al., 2010). This objective function is optimized under a set of constraints. These constraints are, on the one hand, defined by the stoichiometry of the reactions, and, on the other hand, by limitations of reaction fluxes through upper and lower bounds. Hence, not only the biomass production or maintenance reaction can be adapted to a specific organism or cell type, but also the environment, in which it occurs, can be adapted accordingly, e.g. by constraining exchange fluxes. In this work, we optimized the host–virus integrated iAB-AMØ-1410 model for either the host biomass maintenance function or the VBOF using FBA.
Since the solutions calculated by FBA are often not unique, the flux variability analysis (FVA) is a method to identify alternate optimal solutions. In FVA, the maximum and minimum possible flux for each reaction in the network is evaluated while constraining the objective value equal or close to the optimal flux (Orth et al., 2010). As for the FBA, the FVA was conducted for both the host- and virus-optimized states. The results of the FVA were used in the subsequent host-derived enforcement to define the upper and lower bounds.
2.4.1 Copy number analysis
As the copy number of structural proteins in SARS-CoV-2 or coronaviruses, in general, is not reported so far, we evaluated the effect of different Csp values on the growth rate in the virus-optimized state. To do so, we varied the copy number Csp between 1 and 1500. The variation of Csp leads to changing stoichiometric coefficients in the VBOF. Since the VBOF is optimized in the virus-optimized state, changing VBOF reactions can lead to changing optimization results.
2.4.2 Optimization analysis
The host-virus integrated model holds both the host biomass maintenance and the VBOF reaction. First, each biomass reaction was optimized individually using FBA. The objective function is defined by setting the objective coefficient to 1. In general, only one reaction (the objective function) has an objective coefficient of 1. However, one can also set the objective coefficient of several reactions to 1 to optimize for both reactions. Hence, the objective coefficient of both biomass functions from host and virus was set to 1 to optimize for both functions. Last, we used COBRApy’s (Ebrahim et al., 2013) tailored objectives option to weight the influence of the two biomass reactions on the objective function. To do so, we created a linear combination of the two biomass reactions weighted by factors varying between 0 and 1, and not exceeding 1 in total. When the host maintenance function weighted 0.2, the VBOF had a weight of 0.8 in the objective function. For each combination, the model was optimized using FBA, and the flux through the two biomass reactions was reported. This analysis was repeated using the results from the host-derived enforcement analysis by adapting the bounds of the reported reactions (Section 2.5.2).
2.4.3 Metabolic analysis
The metabolic analysis was performed for the host- and virus-optimized states. The fluxes of each reaction in the two optimized states were compared by calculating the FCr:
| (2) |
where the indexation r is over all reactions of the model, and the superscript H indicates flux values from the host and V from the virus-optimized models. The FCs were aggregated into the included subsystems.
2.5 Antiviral target identification
For the identification of potential antiviral targets that preferentially alter the virus growth rate while maintaining the host’s biomass maintenance, each reaction in the model was evaluated using two different approaches.
2.5.1 Reaction knock-out
The first approach is a reaction knock-out that is already implemented in COBRApy. The single_reaction_deletion function subsequently sets both bounds of every reaction to zero and optimizes for the chosen objective function. We ran this function twice, once with the host maintenance reaction as the objective function and once with the VBOF. Possible targets were only reported when the growth of the virus was diminished (<99% of its initial growth rate) and when the growth rate of the virus was below the growth rate of the host-optimized state.
2.5.2 Host-derived enforcement
In the second approach, the effects of changes in flux ranges of the reactions on the VBOF are analyzed, while the metabolic system of the host-optimized state is maintained. To change the flux bounds ensuring maximal biomass maintenance of the host while optimizing for the VBOF, the results of the FVA were utilized. For each reaction, the flux range was defined as described by Aller et al. The corresponding minimum (F–) and maximum (F+) flux values from the FVA of the host (H) and virus (V) optimization and their relation define the minimum () and maximum () flux values. Since the cases suggested by Aller et al. did not cover all reactions, we added the two more conditional arguments cases 3 and 7:
The flux values and are set as upper and lower bounds of the corresponding reactions and the model was optimized for the VBOF. The resulting optimization result was compared to the original VBOF growth rate. When the growth rate with adapted bounds was below the threshold of 80% of the initial growth rate, the reaction was reported as potential antiviral target.
3 Results
We developed a GEM of a human macrophage infected with the SARS-CoV-2. To integrate the virus into the human macrophage, a virus biomass reaction representing the virus particle production was added to the model. This virus biomass reaction was generated based on the nucleotide and amino acid sequence.
3.1 Stoichiometric differences
The human alveolar macrophage biomass maintenance function is comprised of several macromolecules, including amino acids, deoxyribonucleic acid (DNA) and RNA nucleotides, components for the energy requirements and others, such as fatty acids or phospholipids. In contrast, the VBOF only consists of amino acids, RNA nucleotides and the components for the energy requirements. The stoichiometric coefficients of amino acids and nucleotides are compared by calculating the FC. The result is visualized in Figure 1. The stoichiometric coefficients of L-asparagine (N), L-phenylalanine (F), L-threonine (T) and L-tyrosine (Y) are increased. In contrast, the coefficients of L-glutamate (E), L-histidine (H) and L-methionine (M) are reduced. These findings might indicate an up- or down-regulation of the respective metabolic pathways.
Fig. 1.

FC differences in amino acid and nucleotides usage. The stoichiometric coefficients of the alveolar macrophage biomass maintenance function and the VBOF are compared using Equation (1). The left panel displays the FC of all 20 proteinogenic amino acids. The one letter code of the amino acids is used for labeling the x-axis. The right panel displays the FC of the four RNA nucleotides. The one letter code of the RNA nucleotides is used for labeling the x-axis
3.2 Influence of the copy number of the structural proteins
The calculations for obtaining a virus VBOF include the parameter for the copy number of the structural proteins (Csp). The copy number of structural proteins for some viruses, such as Alpha- and Flaviviruses, is known and ranges from 180 for Flaviviruses (Mukhopadhyay et al., 2005) to 240 for Alphaviruses (Strauss and Strauss, 1994). Coronaviruses have four major structural proteins: the envelope (E) protein, membrane (M) protein, nucleocapsid (N) protein and the spike (S) protein (Masters, 2006; Mortola and Roy, 2004; Wang et al., 2017). However, the copy number of those structural proteins is not reported so far. To evaluate the influence of the copy number on the modeling results, we varied the parameter Csp between 1 and 1500. As shown in Figure 2, the optimization result of the VBOF depends on the copy number. The growth rate increases for copy numbers ranging from 1 to 58 and decreases for higher copy numbers. For very high copy numbers of structural proteins, the growth rate seems to reach a steady state.
Fig. 2.
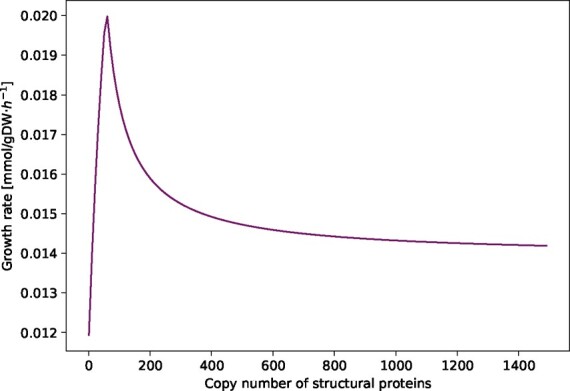
Influence of on the growth rate. The copy number of structural proteins Csp in coronaviruses is not yet reported in literature. Hence, its influence on the growth rate in the virus-optimized state is evaluated by varying Csp between 1 and 1500. For each copy number, the integrated host-virus model was optimized for the VBOF. For Csp values between 58 and 1500, the growth rate decreases
Coronaviruses have a diameter of ∼80–120 nm (Guy et al., 2000), while Alpha- and Flaviviruses have only a diameter of ∼40–80 nm and 30–50 nm, respectively (Fraenkel-Conrat and Wagner, 1974). However, not only the virus size is larger, but also the size of its structural proteins. Since no value for the structure protein copy number in coronaviruses was available, we continued our analyses with a Csp value of 500 and verified the results with Csp values of 200, 800 and 1200.
3.3 Optimization of host and virus metabolism
Potential changes in the human alveolar macrophage metabolism during virus infection were first examined by analyzing the following two scenarios: (i) the host cell is not infected and the metabolic system is optimized for the already included maintenance biomass reaction (host optimization). This scenario reflects the normal physiological state of human alveolar macrophages; (ii) the host cell is infected by the virus and is optimized solely for the production of the virus particles (virus optimization). The host optimization results in a biomass maintenance flux of 0.0267 mmol/(gDW⋅h), while the virus optimization returns a flux of 0.0147 mmol/(gDW⋅h). When defining both the host maintenance and the virus biomass function as objective functions by assigning both an objective coefficient of one simultaneously, the model only optimizes for the host maintenance reaction while the flux through the VBOF is zero. Last, the optimization result of host and virus metabolism was compared using a new constraint. With varying percentages of the host maintenance reaction and the VBOF on the objective expression, the effect on the respective biomass maintenance or growth function was investigated. The model does not predict an equilibrium state, where both the host maintenance reaction and the VBOF are active. As displayed in Figure 3, the switch between the maintenance of the host metabolism and the growth of the virus is at 65% of VBOF contribution to the objective expression. This switch in biomass production is rather insensitive to the structural proteins’ copy number. For Csp=200, the switch occurs at 63% virus BOF contribution, and for Csp=800 and Csp=1200 at 66%.
Fig. 3.
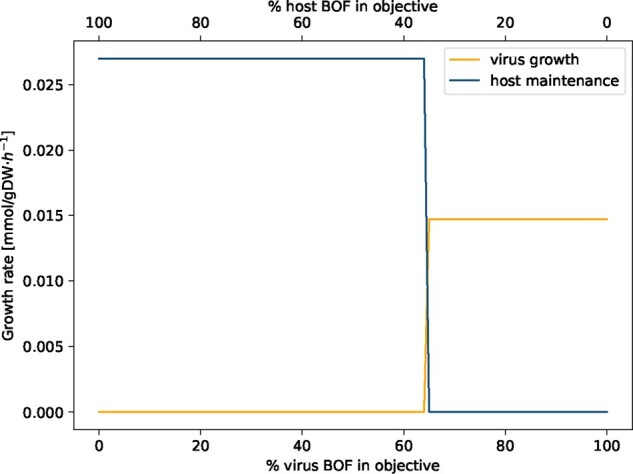
Linear combination of weighted biomass functions. The objective function was defined as linear combination of the host maintenance and VBOF by weighting the proportion of the respective biomass functions between 0 and 1. The sum of the two weights always sums up to 1. At 65% of virus BOF contribution to the objective expression, a switch between host maintenance and virus production occurs
3.4 Metabolic changes in alveolar macrophages after virus infection
The flux distribution for the host- and the virus-optimized states were compared using FBA and FC calculations. For 256 reactions (7.8% of all model reactions), a FC was calculated. As expected from the stoichiometric analysis, reactions related to amino acid and nucleotide metabolism were altered. The main portion of changed reactions consists of transport reactions. However, also several reactions from other subsystems, such as the steroid, fatty acid synthesis and central metabolism, are altered. An overview of altered reactions concerning their FC is given in Figure 4. One needs to keep in mind that the FC can only be calculated if neither of the reaction fluxes in the host- and virus-optimized state was zero. Hence, we calculated the absolute change for reactions that have either in the host- or the virus-optimized state a flux of zero. A total of 97 reactions (2.9% of all model reactions) either turned on a previously turned off reaction or vice versa. For example, in the virus-optimized state, the virus turned off 14 reactions related to lipid metabolism.
Fig. 4.

Altered reactions in virus-optimized state sorted by subsystems. The integrated virus–host model was optimized for the host maintenance and VBOF using FBA. The FC was calculated using Equation (2) and the reactions with FCs were grouped into the aggregated subsystems. Many altered reactions belong to the transport subsystem, or amino acid and nucleotide metabolism
3.5 Identification of metabolic targets for antiviral strategies
FBA and FVA can be used to identify metabolic targets for antiviral strategies by comparing the host- and virus-optimized states after alterations in metabolic pathways.
3.5.1 Knock-out of reactions reveals first promising metabolic target for antiviral strategies
The results from the stoichiometric differences and metabolic changes in alveolar macrophages after virus infections with coronavirus SARS-CoV-2 indicate alternative host- and viral-optimal states. The diverging flux distribution in the two states provides an opportunity for the identification of potential antiviral targets. To identify potential antiviral targets that limit virus production, we implemented two different analysis methods: (i) reaction knock-outs and (ii) host-derived enforcement. The reaction knock-out revealed exactly one reaction (over all tested copy numbers of structural proteins), whose knock-out reduces the flux of the virus biomass to zero, while maintaining the host biomass maintenance at 100%: The guanylate kinase (GK1) reaction that converts ATP and guanosine monophosphate (GMP) to adenosine diphosphate and guanosine diphosphate (GDP):
| (8) |
3.5.2 Host-derived enforcement substantiates the metabolic target and reveals further points of action
This reaction is also observed as a potential target in the second analysis method, the host-derived enforcement. In this approach, the reaction fluxes are constrained to ranges derived from FVA. With flux ranges outside of the optimal state of the virus, the virus production is perturbed while the host maintenance is not affected. For structural protein copy numbers between 500 and 1200, we identified four possible targets, including the GK1 that reduced the virus growth flux to below a threshold of 80% of its initial value. Further potential targets concern the availability of the amino acids L-isoleucine (I), and L-lysine (K), either via the alteration of exchange reactions, or, in case of L-isoleucine, also via the L-isoleucine transporter. In contrast to GK1, where the flux through the reaction is down-regulated (or in case of the reaction knock-outs completely knocked out), the alterations of the mentioned exchange, and transport reactions go into the other direction: the uptake of the amino acids is enabled or even enforced, leading to the host’s maintenance while decreasing the growth of the virus to 50% of its initial growth. The enforcement of exchange reactions can be reached by a sufficient supply of the respective amino acid.
If the host has enough L-lysine in its environment, the maintenance of the host cell is ensured, while the growth of the virus is diminished. We repeated the analysis that evaluates the effect on the growth rates based on the percental involvement of the two biomass functions within the setting of L-lysine excess. Figure 5 illustrates the virus inhibiting effect of L-lysine availability in the environment. Even when the objective function is solely defined by the VBOF (% of virus BOF in objective =100%), the growth rate of the host still exceeds the growth rate of the virus.
Fig. 5.
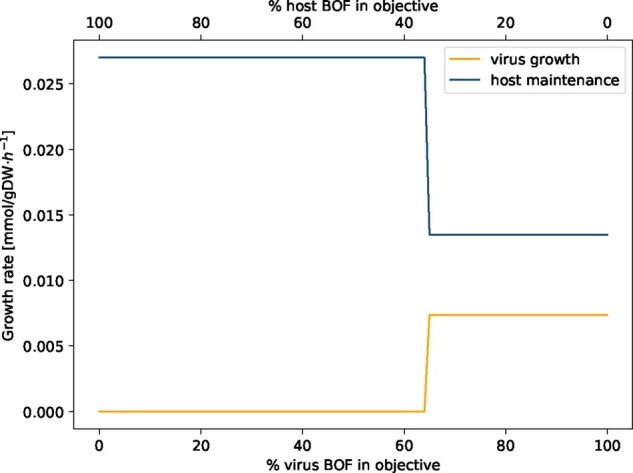
Linear combination of weighted biomass function with adapted boundaries for L-lysine exchange. The objective function was defined as linear combination of the host maintenance and VBOF by weighting the proportion of the respective biomass functions between 0 and 1. The sum of the two weights always sums up to 1. The upper and lower bounds of the L-lysine exchange were adapted based on the host-derived enforcement results (see Section 2.5.2). Even when the objective function is solely defined by the VBOF (% of virus BOF in objective =100%), the growth rate of the host still exceeds the growth rate of the virus
Analogous to L-lysine, the availability of L-isoleucine and L-tyrosine has a positive effect on the growth of the host while decreasing the growth rate of the virus by 50%. The last potential metabolic target for antiviral strategies using the host-derived enforcement approach concerns one transport reaction of L-isoleucine (ILEtec). Enforcing a minimal import of L-isoleucine via this transport has the same effect as the other three targets and results in a diminution of 50% of the virus growth. All targets obtained from the host-derived enforcement for the structural protein copy numbers Csp=500, 800 and 1200 are summarized in Table 1.
Table 1.
Host-derived enforcement of reactions reduced the growth of the virus
| Reaction ID | Growth host (mmol/gDW⋅h-1) | Growth virus(mmol/gDW⋅h-1) | Regulation |
|---|---|---|---|
| GK1 | 0.027 | 0.00736 | Down |
| EX_ile__L(e) | 0.027 | 0.00736 | Up |
| EX_lys__L(e) | 0.027 | 0.00736 | Up |
| ILEtec | 0.027 | 0.00736 | Up |
Note: The host-derived enforcement analysis (see Section 2.5.2) disclosed reactions reducing the virus growth (initial growth rate 0.0147 mmol/gDW⋅h-1), while maintaining the host’s growth rate at 100%.
The analysis of the host-derived enforcement for Csp=200 showed among Table 1 listed 29 reactions as potential antiviral targets that decrease the virus growth below the threshold of 80%. While the four already known targets again reduced the growth rate by 50%, the growth reduction of the other targets varies between 70% and 80% of the initial value. Of the 25 new targets, almost half (12 reactions) are associated with nucleotide metabolism. Only four further reactions are affecting the amino acid metabolism and transport mechanisms, respectively. The other reactions affected are part of the central metabolism and miscellaneous reactions.
3.6 Existing drugs can target the predicted reactions
For the identified potential targets for antiviral therapies, we have searched for existing drugs or compounds.
3.6.1 Direct inhibition of GK1
As GK1, also known as guanosine monophosphate kinase, was found to be an essential factor for viral growth in this study, the inhibition of the enzyme may be a feasible target in SARS-CoV-2 therapy. The enzyme catalyzes the reversible turnover of GMP or deoxyguanosine monophosphate to GDP or deoxyguanosine diphosphate by binding and transferring a phosphoryl from ATP to GMP (Hible et al., 2006; Khan et al., 2019). GK1 has a highly conserved structure, with a core domain, a GMP-binding domain, a lid domain with catalytic residues, as well as an ATP binding (P)-loop (Hible et al., 2006; Khan et al., 2019). Through activation of ganciclovir in herpes virus treatment and 6-thioguanine or 6-mercaptopurine activation in tumor treatment, GK1 plays an essential role in diverse treatment strategies (Hible et al., 2006; Khan et al., 2019).
Based on the results of this study, we propose a direct inhibitor of GK1 to harbor the potential of SARS-CoV-2 inhibition. Interestingly, such inhibitors have already been described in the context of antiviral therapies (Hible et al., 2006; Jain et al., 2016; Khan et al., 2019; Navé et al., 1992). For instance, Hible et al. have demonstrated P1-(5’-adenosyl) P5-(5’-guanosyl) pentaphosphate (Ap5G) to be a potent, bi-substrate inhibitor of GK1 (Hible et al., 2006; Jain et al., 2016). Although Hible et al. have only shown the high potency of the inhibition in Escherichia coli, Khan et al. have demonstrated the binding of Ap5G to introduce a complete closure of the human GK1, indicating inaccessibility of the substrates upon inhibitor binding (Hible et al., 2006; Jain et al., 2016; Khan et al., 2019).
Potent GK1 inhibitors have also been presented by Navé et al. (1992). Accordingly, 9-phosphonoalkyl derivates, such as 9-(6-phosphonohexyl)guanine and 9-(6,6-difluoro-6-phosphonohexyl)guanine, impede GK1 activity via competitive inhibition of GMP and non-competitive inhibition of ATP (Navé et al., 1992).
3.6.2 Acyclonucleotide analogues require GK1 activation
In subsequent work, Navé et al. (1995) have tested the antiviral activity of 9-phosphonopentenyl derivatives of guanine operating as acyclonucleotide analogues. Acyclonucleotide analogs are pro-drugs, which require the activation by GK1 to form nucleoside triphosphate analogues (Navé et al., 1995). These are known to inhibit viral DNA polymerases via chain termination in diverse herpes virus and retrovirus infections (Navé et al., 1995). In accordance with this, the authors have identified vinyl phosphonates (E)-9-(5-Phosphonopent-4-enyl)guanine and (E)-9-[3-(hydroxymethyl)-5-phosphonopent-4-enyl]guanine to be inhibitors of the human immunodeficiency virus 1 (HIV-1) and human cytomegalovirus (Navé et al., 1995).
As this study has found increased flux through GK1 in SARS-CoV-2 infected cells, a process of pro-drug activation relying on GK1 may allow for increased activation of terminating nucleoside triphosphate analogs in infected cells compared to healthy cells. However, coronaviruses are +ssRNA viruses replicated by an RdRP, unlike most viruses targeted by available acyclonucleotide analogs, such as ganciclovir and acyclovir (Chen et al., 2020; Navé et al., 1995; Navé et al., 1992). Moreover, coronaviruses lack a viral kinase required for activation of these acyclonucleotide analogs (Chen et al., 2020; Hible et al., 2006). Despite these limitations, some market-available analogs, such as cidofovir, brincidofovir or favipiravir, do not require a viral kinase activation, and have shown in vitro activity against other RNA viruses (Ebola virus) and retroviruses (HIV-1) (Arias et al., 2014; Dunning et al., 2016; Garcia et al., 1998; McCarthy et al., 2016; Navé et al., 1995). Thus, they may be drug candidates worth an exploration in the face of the findings in this study and the current SARS-CoV-2 outbreak.
4 Discussion
As proposed by Aller et al. (2018), computational approaches combining FBA and FVA to recover new metabolic antiviral targets are useful, especially in cases of new and emerging viruses, such as the SARS-CoV-2. In this study, we presented a host-virus integrated GEM using the human alveolar macrophage model iAB-AMØ-1410 as host cells and SARS-CoV-2 as virus. We identified potential targets for antiviral therapies using reaction knock-outs and host-derived enforcement approaches and analyzing their metabolic effects on host- and virus-optimized states by optimizing either for the host maintenance or the VBOF.
However, the VBOF constructed in this study only considers amino acids, nucleotides and energy requirements. It does not consider or include virus-host cell recognition, viral entry or the lipid envelope production or release (Timm and Yin, 2012). Especially the metabolic process of lipid envelope production of viruses can give further insight into potential targets for antiviral therapies. First studies with other coronaviruses, such as the human coronavirus 229E, suggest elevated and perturbed glycerophospholipids and fatty acids production rates in infected cells (Yan et al., 2019). Yan et al. (2019) suggest the lipid metabolism regulation as a potential druggable target for coronavirus infections. Further information about the lipid metabolism of coronaviruses can enable the integration of lipids into the VBOF. Analyses with the adapted VBOF could highlight additional potential targets for antiviral therapies.
As their coding capacity is limited, coronaviruses strategically regulate host immune response, cell cycle, signaling and metabolism to create a favorable environment for viral replication (de Wilde et al., 2015; Dirmeier et al., 2020; Perlman and Netland, 2009). Accordingly, the viroids depend on cellular enzymes for the formation of progeny, which makes host cell resources a potential target to limit virion production (de Wilde et al., 2015; Dirmeier et al., 2020). The viral hijacking of the cellular metabolic pathways, such as glycolysis, nucleotide and lipid biosynthesis, may shift the environment of the virus to a proliferation promoting environment (Thaker et al., 2019). In this study, we have also demonstrated that SARS-CoV-2 interferes with the host cell organisms, more precisely, the purine biosynthesis pathway to provide for the production of its biomass and, thus, replication.
In our study, we used a GEM. This type of model enables the analysis of metabolic changes under certain constraints. However, only the metabolic changes can be investigated. Further network reconstructions, such as dynamic signaling, regulatory or kinetic network models, can give further insight into changes in signal transduction, regulatory processes or kinetic properties (Bernhard, 2011) of virus infections. Ravindran et al. (2019), e.g. analyzed the effect of HIV-1 and hepatitis C virus infections using a large human signaling network. They demonstrated how the infecting virus could bring the dynamically organized host system into its control. Tan et al. developed a mathematical model describing the virus-induced interferon (IFN) signaling process. Dynamic analysis and numerical simulations led to the suggestion that a balance between viral replication and IFN-induced regulation is responsible for the dynamic behavior of virus-triggered signaling and also for antiviral responses (Tan et al., 2012). Dynamic modeling of infections with coronaviruses, especially with SARS-CoV-2, could broaden the understanding of its effects on the host and give further insight into potential targets for antiviral therapies.
In this study, we used the already published iAB-AMØ-1410 GEM of human alveolar macrophages. This model does currently not include any genes or annotations. It is built upon the first human reconstruction Recon 1 (Duarte et al., 2007). By now, the human reconstruction Recon3D is available with more than 10 000 reactions, 2000 genes and almost 6000 metabolites (Brunk et al., 2018), including numerous annotations. Following the protocol of Bordbar et al. (2010), a new model of the human alveolar macrophage could be generated based on the newer version of the human reconstruction, Recon3D. The newly curated model could then be used to verify the findings from this study and to identify further targets.
As coronaviruses are reported to infect human alveolar macrophages, and a model for these cells was available, we integrated SARS-CoV-2 into this model. Nevertheless, SARS-CoV-2 is reported also to infect upper and lower respiratory tract cells, including pharyngeal regions (Chen et al., 2020; Huang et al., 2020). GEMs for bronchial epithelial cells and airway epithelial cells (AEC) are already available. Wang et al. reconstructed 126 human tissue-specific GEMs using the metabolic Context-specificity Assessed by Deterministic Reaction Evaluation algorithm. Those models are also built upon Recon 1. Furthermore, the models include fewer numbers of reactions (1242 and 1296, respectively) and lack a biomass maintenance function. Following the protocol for generati9.5 ng high-quality genome-scale metabolic reconstructions by Thiele and Palsson, meaningful models of human bronchial and AEC could be generated (Thiele and Palsson, 2010). However, generating a biomass maintenance function requires much data. Tools, such as BOFdat, can be beneficial for the generation of an appropriate BOF (Lachance et al., 2019). These models can then be used to verify the potential antiviral targets that were found in alveolar macrophages.
The integrated host–virus model suggested the supplementation of L-isoleucine and L-lysine as a potential target for antiviral therapies, as well as the inhibition of the GK1. Since compounds that directly inhibit GK1 are already known, their evaluation and verification in cell culture experiments are required for fast responses to the current spread of SARS-CoV-2 worldwide.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy—EXC 2124—390838134 and was supported by the German Center for Infection Research.
Conflict of Interest: none declared.
Data availability
The computational model is accessible at https://identifiers.org/biomodels.db/MODEL2003020001.
Contributor Information
Alina Renz, Computational Systems Biology of Infections and Antimicrobial-Resistant Pathogens, Institute for Bioinformatics and Medical Informatics (IBMI); Department of Computer Science, University of Tübingen, Tübingen 72076, Germany.
Lina Widerspick, Computational Systems Biology of Infections and Antimicrobial-Resistant Pathogens, Institute for Bioinformatics and Medical Informatics (IBMI).
Andreas Dräger, Computational Systems Biology of Infections and Antimicrobial-Resistant Pathogens, Institute for Bioinformatics and Medical Informatics (IBMI); Department of Computer Science, University of Tübingen, Tübingen 72076, Germany; German Center for Infection Research (DZIF), partner site Tübingen, Germany, .
References
- Aller S. et al. (2018) Integrated human-virus metabolic stoichiometric modelling predicts host-based antiviral targets against Chikungunya, Dengue and Zika viruses. J. R. Soc. Interface, 15, 20180125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias A. et al. (2014) Favipiravir elicits antiviral mutagenesis during virus replication in vivo. Elife, 3, e03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore D. (1971) Expression of animal virus genomes. Bacteriol. Rev., 35, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard Ø.P. (2011) Systems Biology: Simulation of Dynamic Network States. Cambridge University Press, Cambridge. [Google Scholar]
- Bordbar A. et al. (2010) Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol., 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein B.J. et al. (2008) LibSBML: an API library for SBML. Bioinformatics, 24, 880–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk E. et al. (2018) Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol., 36, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F. et al. (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet, 395, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol., 92, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C.Y. et al. (2005) Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol., 79, 7819–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H. et al. (2015) A kinome-wide small interfering RNA screen identifies proviral and antiviral host factors in severe acute respiratory syndrome coronavirus replication, including double-stranded RNA-activated protein kinase and early secretory pathway proteins. J. Virol., 89, 8318–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X. et al. (2017) Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA, 114, E4251–E4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirmeier S. et al. (2020) Host factor prioritization for pan-viral genetic perturbation screens using random intercept models and network propagation. PLoS Comput. Biol., 16, e1007587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte N.C. et al. (2007) Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA, 104, 1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning J. et al. ; for the RAPIDE-BCV trial team. (2016) Experimental treatment of Ebola virus disease with brincidofovir. PLoS One, 11, e0162199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahim A. et al. (2013) COBRApy: COnstraints-based reconstruction and analysis for Python. BMC Syst. Biol., 7, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraenkel-Conrat H., Wagner R.R. (1974) Comprehensive Virology: Descriptive Catalogue of Viruses. Springer, New York. [Google Scholar]
- Garcia C.R. et al. (1998) Cidofovir in the treatment of cytomegalovirus (CMV) retinitis. Ocul. Immunol. Inflamm., 6, 195–203. [DOI] [PubMed] [Google Scholar]
- Geer L.Y. et al. (2010) The NCBI BioSystems database. Nucleic Acids Res., 38, D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J.S. et al. (2000) Characterization of a coronavirus isolated from a diarrheic foal. J. Clin. Microbiol., 38, 4523–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hible G. et al. (2006) Crystal structures of GMP kinase in complex with ganciclovir monophosphate and Ap5G. Biochimie, 88, 1157–1164. [DOI] [PubMed] [Google Scholar]
- Huang C. et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet, 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucka M. et al. (2008) Systems biology markup language (SBML) Level 2: structures and facilities for model definitions. Nat. Preced., 58. 10.1038/npre.2008.2715.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucka M. et al. (2018) Systems biology markup language (SBML) level 3 version 1 core. J. Integr. Bioinform., 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui D.S. et al. (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - the latest 2019 novel coronavirus outbreak in Wuhan. China. Int. J. Infect. Dis., 91, 264–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R. et al. (2016) Insights into open/closed conformations of the catalytically active human guanylate kinase as investigated by small-angle X-ray scattering. Eur. Biophys. J., 45, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel Funk C. et al. (2012) Infection of human alveolar macrophages by human coronavirus strain 229E. J. Gen. Virol., 93, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John Hopkins University. (2020) COVID-19 Map - John Hopkins Coronavirus Resource Center. https://coronavirus.jhu.edu/map.html (29 April 2020, date last accessed).
- Khan N. et al. (2019) Solution structure and functional investigation of human guanylate kinase reveals allosteric networking and a crucial role for the enzyme in cancer. J. Biol. Chem., 294, 11920–11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J.-C. et al. (2019) BOFdat: generating biomass objective function stoichiometric coefficients from experimental data. PLoS Comput. Biol., 15, e1006971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letko M., Munster V. (2020) Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol., 5, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. (2006) The molecular biology of coronaviruses. Adv. Virus Res., 66, 193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S.D. et al. (2016) A rapid screening assay identifies monotherapy with interferon- and combination therapies with nucleoside analogs as effective inhibitors of Ebola virus. PLoS Negl. Trop. Dis., 10, e0004364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortola E., Roy P. (2004) Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett., 576, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S. et al. (2005) A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol., 3, 13–22. [DOI] [PubMed] [Google Scholar]
- Navé J.F. et al. (1992) 9-(Phosphonoalkyl)guanine derivatives as substrates or inhibitors of guanylate kinase. Arch. Biochem. Biophys., 295, 253–257. [DOI] [PubMed] [Google Scholar]
- Navé J.F. et al. (1995) Synthesis, antiviral activity and enzymatic phosphorylation of 9-phosphonopentenyl derivatives of guanine. Antiviral Res., 27, 301–316. [DOI] [PubMed] [Google Scholar]
- Orth J.D. et al. (2010) What is flux balance analysis? Nat. Biotechnol., 28, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevis D. et al. (2020) Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol., 79, 104212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Netland J. (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol., 7, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran V. et al. (2019) Network controllability analysis of intracellular signalling reveals viruses are actively controlling molecular systems. Sci. Rep., 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss J.H., Strauss E.G. (1994) The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev., 58, 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J. et al. (2012) Modeling and dynamical analysis of virus-triggered innate immune signaling pathways. PLoS One, 7, e48114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker S.K. et al. (2019) Viral hijacking of cellular metabolism. BMC Biol., 17, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I., Palsson B. (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc., 5, 93–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm A., Yin J. (2012) Kinetics of virus production from single cells. Virology, 424, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. et al. (2017) MERS-CoV virus-like particles produced in insect cells induce specific humoural and cellular imminity in rhesus macaques. Oncotarget, 8, 12686–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2020) Coronavirus disease (COVID-19) pandemic. https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (29 April 2020, date last accessed).
- Yan B. et al. (2019) Characterization of the lipidomic profile of human coronavirus-infected cells: implications for lipid metabolism remodeling upon coronavirus replication. Viruses, 11, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. (2020) Alpha-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication Structure-based design, synthesis, and activity assessment. J. Med. Chem., 63, 4562–4578. [DOI] [PubMed] [Google Scholar]
- Zielinski D.C. et al. (2015) Pharmacogenomic and clinical data link non-pharmacokinetic metabolic dysregulation to drug side effect pathogenesis. Nat. Commun., 6, 7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The computational model is accessible at https://identifiers.org/biomodels.db/MODEL2003020001.