

Abstract
The COVID-19 pandemic has spread rapidly and poses an unprecedented threat to the global economy and human health. Broad-spectrum antivirals are currently being administered to treat severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). China's prevention and treatment guidelines suggest the use of an anti-influenza drug, arbidol, for the clinical treatment of COVID-19. Reports indicate that arbidol could neutralize SARS-CoV-2. Monotherapy with arbidol is superior to lopinavir-ritonavir or favipiravir for treating COVID-19. In SARS-CoV-2 infection, arbidol acts by interfering with viral binding to host cells. However, the detailed mechanism by which arbidol induces the inhibition of SARS-CoV-2 is not known. Here, we present atomistic insights into the mechanism underlying membrane fusion inhibition of SARS-CoV-2 by arbidol. Molecular dynamics (MD) simulation-based analyses demonstrate that arbidol binds and stabilizes at the receptor-binding domain (RBD)/ACE2 interface with a high affinity. It forms stronger intermolecular interactions with the RBD than ACE2. Analyses of the detailed decomposition of energy components and binding affinities revealed a substantial increase in the affinity between the RBD and ACE2 in the arbidol-bound RBD/ACE2 complex, suggesting that arbidol generates favorable interactions between them. Based on our MD simulation results, we propose that the binding of arbidol induces structural rigidity in the viral glycoprotein, thus restricting the conformational rearrangements associated with membrane fusion and virus entry. Furthermore, key residues of the RBD and ACE2 that interact with arbidol were identified, opening the door for developing therapeutic strategies and higher-efficacy arbidol derivatives or lead drug candidates.
Keywords: Umifenovir, COVID-19, Membrane fusion, Pandemic, Antiviral, Influenza virus, Receptor-binding domain, Drug repurposing, Protein interaction
Graphical abstract
1. Introduction
We are living through an unprecedented existential crisis. The rapid spread of the new coronavirus disease (COVID-19) across all continents over a short span of a few months has posed a severe global threat. In March 2020, the World Health Organization declared COVID-19 a pandemic, and by early January 2021, COVID-19 had spread to 218 countries and territories, with over 86 million confirmed cases and over 1.8 million deaths. The causative pathogen of COVID-19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), represents a serious threat to human health and the global economy. Shortly after the outbreak, the genome sequence and organization of SARS-CoV-2 were determined (Lu et al., 2020). SARS-CoV-2 is a large, enveloped, single-positive-strand RNA coronavirus (CoV). The envelope-anchored spike glycoprotein (S-protein) binds to the human angiotensin-converting enzyme 2 (ACE2) receptor, facilitating membrane fusion. Upon interaction, the S-protein/ACE2 complex undergoes structural rearrangements that allow for the fusion of viral and cellular membranes and entry into host cells.
The receptor-binding domain (RBD) of the S-protein is the central region that is involved in direct interactions with the ACE2 receptor. Several complex structures of RBD/ACE2 have been elucidated, and the interfacial residues have been identified (Lan et al., 2020; Wrapp et al., 2020). A cryo-EM structure reveals the different conformational states of the RBD, namely, the active open conformation, the semiactive conformation, and the closed state (Wrapp et al., 2020). Structurally, the SARS-CoV-2 spikes consist of three receptor-binding S1 heads (responsible for receptor recognition) and a trimeric membrane fusion S2 stalk (responsible for membrane fusion). S1 is further divided into an N-terminal domain (NTD) and a C-terminal domain (CTD). An extended insert contains the receptor-binding motif of the RBD that contains most of the interfacial residues. The binding affinity of the RBD for ACE2 is the key determinant of SARS-CoV-2 transmissibility (Padhi et al., 2020). Interestingly, the SARS-CoV-2 RBD displays a stronger affinity for ACE2 than the RBDs of other CoVs (Tai et al., 2020), which is attributed to the higher number of residue interaction networks between SARS-CoV-2 and human ACE2 (hACE2). Thus, SARS-CoV-2 encodes epitope features distinct from those of other CoVs in the RBD. Blockade of the S-protein from ACE2 receptor interaction and cell fusion is among the key targets for antiviral development (Zhou et al., 2020). The SARS-CoV-2 virus is not actively mutating; however, with time, it can acquire mutations with fitness advantages and develop resistance strategies (Callaway, 2020; Padhi et al., 2021; Padhi and Tripathi, 2020).
To date, no clinically proven effective antiviral strategy exists for the treatment of COVID-19. Consequently, the management for SARS-CoV-2 is mostly supportive, with the only aim being mortality reduction. With limited scientific evidence, clinicians are treating patients with ritonavir-lopinavir, hydroxychloroquine, azithromycin, corticosteroids, and interleukin-6 inhibitors outside of their approved uses and without study protocols. However, an array of drugs approved for other viral infections are being studied for COVID-19 treatment in hundreds of clinical trials around the globe (Gordon et al., 2020; Guy et al., 2020; Horby et al., 2020; Rajarshi et al., 2020; The, 2020). Recent data have indicated that remdesivir treatment could shorten the time to recovery in adults (Beigel et al., 2020). However, another human trial showed that the drug did not help patients in China with severe COVID-19 (Wang et al., 2020d). Several clinical trials are also underway to evaluate the suitability and efficacy of other antiviral drugs, such as favipiravir, darunavir, ribavirin, galidesivir, oseltamivir, and arbidol, for treating COVID-19 (De Clercq, 2004; Dong et al., 2020; Elfiky, 2020; Koren et al., 2003; Li et al., 2020; Rajter et al., 2020; Runfeng et al., 2020; Sarzi-Puttini et al., 2020; Tu et al., 2020; Velavan and Meyer, 2020; Wang et al., 2020a; Wu and Yang, 2020).
Arbidol, also known as umifenovir and ethyl-6-bromo-4-[(dimethylamino)methyl]-5-hydroxy-1-methyl-2 [(phenylthio)methyl]-indole-3-carboxylate hydrochloride monohydrate, is a small indole derivative. It is a potent broad-spectrum antiviral with proven activity against several enveloped and nonenveloped viruses, including influenza, parainfluenza, coronavirus, polio, Lassa fever, respiratory syncytial virus, adenovirus, Coxsackie B5, and hepatitis B and C (Blaising et al., 2014; Boriskin et al., 2008; Herod et al., 2019; Pecheur et al., 2016). Arbidol has been used for decades in China and Russia for treating influenza and other respiratory viral infections with no major adverse effects (Blaising et al., 2014; Boriskin et al., 2008). It suppresses influenza virus propagation and modulates the expression of inflammatory cytokines in vitro and in vivo (Wang et al., 2017). Currently, arbidol use is recommended in China's prevention and treatment guidelines and is used in the clinical treatment of COVID-19. Arbidol interferes with multiple stages of the virus life cycle by directly targeting viral proteins or virus-associated host factors (Blaising et al., 2014). In the influenza virus, arbidol binds to hemagglutinin (HA), the major cell-surface glycoprotein, and prevents fusion of the viral membrane (Kadam and Wilson, 2017). A recent study revealed that arbidol efficiently inhibits SARS-CoV-2 infection. Arbidol blocks both viral entry (by interfering with viral binding to host cells) and postentry stages (by blocking intracellular vesicle trafficking) (Wang et al., 2020c). However, the mechanism by which arbidol modulates the RBD/ACE2 interaction has not been completely elucidated, thereby obscuring its development as a specific therapeutic for COVID-19. Thus, we aimed to investigate the molecular basis underlying the RBD/ACE2 interaction inhibition using all-atom molecular dynamics (MD) simulations. Our data provide details on why arbidol is effective against SARS-CoV-2. The mechanistic understanding gained from this study will be useful for the design and development of more specific SARS-CoV-2 inhibitors.
2. Materials and methods
2.1. System preparation and molecular docking
The cocrystal structure of the SARS-CoV-2 RBD/ACE2 complex was retrieved from the RCSB Protein Data Bank (PDB ID: 6LZG) and used for molecular docking (Fig. 1 A) (Wang et al., 2020b) with arbidol (umifenovir) [PubChem ID: 131,411] (Fig. 1B). The CB-Dock protein-ligand docking method was used for the docking of arbidol with ACE2 and the RBD separately and into the RBD/ACE2 complex. This method automatically identifies the binding sites, calculates the center and size, customizes the docking box size according to the query ligands, and then performs molecular docking with AutoDock Vina (Liu et al., 2020). First, arbidol was docked to ACE2 and the RBD separately and into the RBD/ACE2 complex. It could bind at several positions in all three conditions. However, since arbidol inhibits SARS-CoV-2 membrane attachment to host cells, arbidol was docked into the RBD/ACE2 complex structure. During this process, twenty cavities were generated and used, and molecular docking was performed at each one. The binding modes were analyzed in cerebro, and the docked pose with the highest AutoDock Vina score and cavity size was selected for subsequent experiments. Hereafter, the RBD/ACE2 complex without arbidol will be termed the “apo-RBD/ACE2 complex”, and the RBD/ACE2 complex bound to arbidol will be named the “RBD/ACE2-arbidol complex”.
Fig. 1.

Representation of the RBD/ACE2 complex and arbidol. (A) Ribbon structure of the RBD/ACE2 complex (ACE2: green and RBD: cyan) showing the interface residues of RBD (inset) as yellow sticks, and (B) a 2D structural representation of arbidol.
2.2. Molecular dynamics simulations
The selected docked complex of RBD/ACE2-arbidol was prepared using Schrödinger Maestro (Schrödinger Release, 2016–4: Maestro, Schrödinger, New York) and subsequently used for the MD simulations. As a first step, the topology of arbidol was generated using Automated Topology Builder and Repository, and the resulting complex was subjected to the addition of hydrogen atoms (Koziara et al., 2014; Malde et al., 2011). Next, the complex was solvated in a dodecahedron box with simple point charge water in the center at least 1.0 nm from the box edge (Berendsen et al., 1981). Appropriate counterions were then added to neutralize the system electrostatically. MD simulations for the resulting system were performed using the GROMACS 5.1.2 software package with the gromos96 54A7 forcefield (Huang et al., 2011; Liu et al., 1996; Schmid et al., 2011). The system was subjected to 50,000 energy minimization steps until the energy was stabilized, followed by a heating step from 0 to 300 K in 200 ps (ps) and a constant temperature equilibration for 1000 ps at 300 K. During the equilibration stage, velocity impact was prevented by employing Parrinello-Rahman barostat pressure coupling (Parrinello and Rahman, 1981). After monitoring the convergence of the complex, a 300 ns (ns) production run was performed with periodic boundary conditions in the NPT ensemble, in which a modified Berendsen temperature coupling and a constant pressure of 1 atm were employed (Bussi et al., 2007). During this process, the LINCS algorithm and the particle mesh Ewald method were used to calculate the long-range electrostatic forces (Darden et al., 1993; Hess et al., 1997). During the simulation, the Fourier grid spacing and Coulomb radius were set at 0.16 and 1.4 nm, respectively, and the van der Waals interactions were limited to 1.4 nm. The MD simulated trajectories were saved every 10 ps for energy stabilization. As a series of experiments, to understand the dynamics between the RBD and ACE2 in the absence of arbidol, the simulation for the apo system was performed under similar conditions for 300 ns?
2.3. Analysis of MD simulations
The MD simulated trajectories were analyzed using the gmx rms, gmx rmsf, and gmx gyrate GROMACS utilities to obtain the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (Rg) of each system, respectively. The hydrogen bonds for the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes were computed using the hydrogen bond plug-in of visual molecular dynamics (VMD) (Humphrey et al., 1996).
2.4. Essential dynamics of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes
To understand the dominant and collective modes from the overall dynamics of the MD trajectory for the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes, principal component analysis (PCA) and essential dynamics (ED) were performed using the protein backbones, during which a variance/covariance matrix was constructed by calculating the eigenvectors and eigenvalues and their projections along with the first two principal components (PCs) by ED (Amadei et al., 1993). The movements of the complexes in the essential subspace were identified by projecting the Cartesian trajectory coordinates along the important eigenvectors. The eigenvalues of the first two eigenvectors that were derived from the PCAs of the simulation trajectories were computed to understand the dynamics of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes. The eigenvalues associated with each eigenvector of the complexes were used to calculate the percentage of variability. The GROMACS built-in gmx covar and gmx anaeig modules were used to obtain the eigenvalues and eigenvectors by calculating and diagonalizing the covariance matrix. To study the conformational changes in the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes, the 2D representation of the free energy landscape (FEL) was computed using the gmx sham package from the first two eigenvectors extracted after PCA (Brooks et al., 2001; Mitsutake et al., 2001).
2.5. Binding free energy calculations
To calculate the binding free energies between the RBD-arbidol, ACE2-arbidol, and RBD-ACE2-arbidol complexes, the molecular mechanics/Poisson-Boltzmann surface area (MM-PBSA) methodology employed in the g_mmpbsa tool of GROMACS was used (Homeyer and Gohlke, 2012; Kumari et al., 2014). In MM-PBSA, the binding free energy of the protein and ligand is typically defined as
where ΔGcomplex, ΔGprotein, and ΔGligand represent the total free energies of the protein, the ligand, and the protein-ligand complex, respectively, measured separately in the solvent (Gohlke et al., 2003; Wang and Kollman, 2000). In general, the free energy of the individual entity is represented as
where EMM represents the average molecular mechanical potential energy in the vacuum, Gsolvation denotes the free energy of solvation, and TS represents the entropic contribution to the free energy in a vacuum, where T and S denote the temperature and entropy, respectively. Furthermore, the EMM comprises bonded and nonbonded terms, including the bond angle, torsion, and electrostatic (Eelec) and van der Waals (Evdw) interactions. Last, the solvation free energy and Gsolvation consider both electrostatic and nonelectrostatic (Gpolar and Gnonpolar) components. The binding free energies of the RBD-arbidol, ACE2-arbidol, and RBD-ACE2 complexes were calculated for 200 snapshots obtained from the last 30 ns of the trajectories.
2.6. Interaction network of RBD/ACE2-arbidol and binding affinity calculations
Representative structures of the RBD/ACE2-arbidol complex were extracted from the last 30 ns of the stabilized MD simulated trajectory, and all the intermolecular interactions between arbidol and RBD/ACE2 were computed using Schrödinger Maestro. The binding affinity between the RBD/ACE2 and RBD/ACE2-arbidol complex structures was first computed using PROtein binDIng enerGY prediction (PRODIGY), a predictor of the binding affinity in protein-protein and protein-ligand complexes (Kurkcuoglu et al., 2018; Vangone and Bonvin, 2015; Vangone et al., 2019; Xue et al., 2016). In this method, after the extracted snapshot from MD simulations is supplied with a default temperature of 25 °C, the binding affinity is computed. PPCheck was used to quantify the strength of a protein-protein interface, in which the structures of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes extracted from the MD simulations were used to calculate the interaction energies and total stabilizing energy (Et) (Sukhwal and Sowdhamini, 2013) which was the sum of the energy values for all the nonbonded interactions, including van der Waals interactions, electrostatic interactions, and hydrogen bond interactions. In addition, it also computes the number of short contacts, hydrophobic interactions, van der Waals pairs, salt bridges, potential favorable electrostatic interactions, and potential unfavorable electrostatic interactions at the interface, thus providing comprehensive knowledge on the qualitative and quantitative features of RBD/ACE2 with or without arbidol binding.
2.7. Figures and rendering
The figures were rendered and generated using PyMOL and VMD. Ligand interaction diagrams were generated using Schrödinger Maestro (Schrödinger Release, 2016–4: Maestro, Schrödinger, New York).
3. Results
3.1. Molecular docking of arbidol in the ACE2-RBD complex
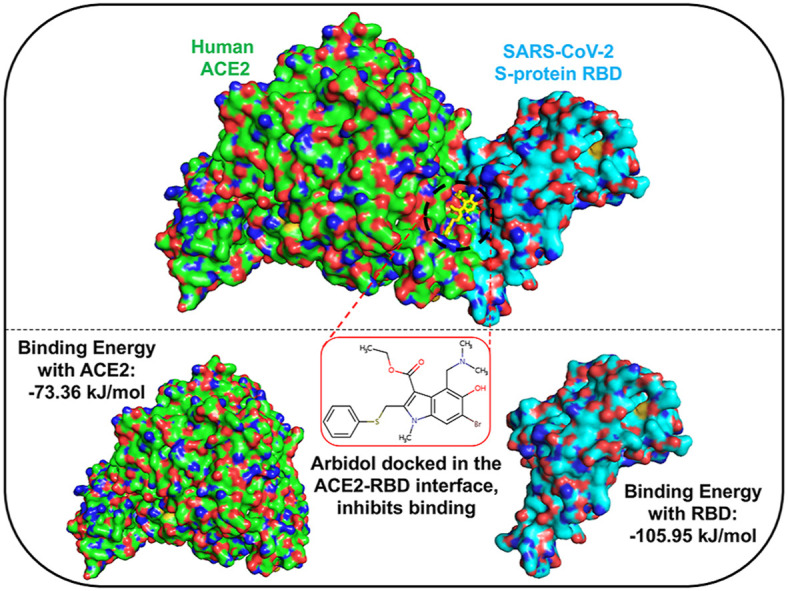
Arbidol inhibits SARS-CoV-2 membrane fusion to host cells, suggesting that it targets S-protein/ACE2 interactions. Since the RBD of the S-protein is involved in ACE2 recognition, arbidol was docked into the RBD/ACE2 complex structure. When arbidol was docked into ACE2 and the RBD separately, it was not docked at the ACE2 and RBD binding interface (Supplementary Fig. 1). Moreover, the largest cavities where arbidol was docked were not at the interface of the RBD side (facing ACE2) or the ACE2 side (facing RBD) (Supplementary Fig. 1). When this drug was docked into the RBD/ACE2 complex, it quickly docked into one of the largest cavities in the RBD/ACE2 interface with a high score [cavity size: 940, center: 176*114*247 (x*y*z) and size: 22*22*22 (x*y*z)]. Arbidol was found to bind and stabilize at the RBD/ACE2 interface with a high binding affinity (AutoDock Vina score of −5.7) (Fig. 2 ). The docked complex was subsequently used to study the detailed structural, dynamic, and binding mechanisms to understand how it targets the RBD/ACE2 interface.
Fig. 2.

Docked pose of arbidol bound the RBD/ACE2-interacting interface. Surface representation of the RBD/ACE2 interface showing the docking site of arbidol (yellow stick) and the docking score after molecular docking simulation.
3.2. Stability of the RBD/ACE2-arbidol complex during MD simulations
The MD simulation trajectories comprising 300 ns of independent simulations for the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes were examined for their structural and dynamic behaviors. The RMSD of the backbone atoms computed over 300 ns revealed that the apo-RBD/ACE2 complex reached stability after approximately 75 ns, whereas the RBD/ACE2-arbidol complex took nearly 125 ns to converge (Fig. 3 A). Beyond 125 ns, both systems were stabilized until the end of the production run and converged overall; however, the RMSD profiles suggested that the last 30 ns were most preferable for further structural and dynamics analyses. The analysis of hydrogen bonds formed for the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes revealed that upon arbidol binding, the RBD/ACE2 complex was able to form more hydrogen bond interactions than the apo-RBD/ACE2 complex (Fig. 3B). While the apo-RBD/ACE2 complex formed an average of 188 hydrogen bond interactions, the RBD/ACE2-arbidol complex formed 247 hydrogen bond interactions on average during the 300 ns simulations (Fig. 3B). This result indicated that the hydrogen bonds probably played an essential role in stabilizing the RBD/ACE2-arbidol complex during the simulation and confer stability to the RBD/ACE2-arbidol complex.
Fig. 3.

MD simulation-derived structural and dynamic parameters from the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes. (A) Plots showing the backbone RMSD profiles of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes over the course of the MD simulations, (B) the total number of hydrogen bonds formed during the 300 ns simulations for both complexes, (C) the per-residue RMSF profiles of ACE2 from both complexes, (D) the per-residue RMSF profiles of the RBD from both complexes over the 300 ns simulations, and (E) the computed radii of gyration by the C⍺ atoms for both complexes as a function of time at 300 K.
3.3. Structural flexibility and compactness of the RBD/ACE2-arbidol complex
The structural flexibilities of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes was evaluated by computing the per-residue RMSF. As expected, the RBD/ACE2-arbidol complex exhibited an overall lower RMSF than the apo-RBD/ACE2 complex (Fig. 3C and D). While the apo-RBD/ACE2 complex showed an average RMSF of 2.49 Å, the RBD/ACE2-arbidol complex showed a lower average RMSF of 1.67 Å during the simulation, corroborating the RMSD and hydrogen bond interaction profiles (Fig. 3C and D). We then investigated how the two molecular systems displayed their compactness during the simulations. For this reason, we computed the Rg for both complexes over the 300 ns period. The Rg profiles revealed that the RBD/ACE2-arbidol complex exhibited a more compact behavior than the apo-RBD/ACE2 complex (Fig. 3E). The average Rg values were 5.29 Å and 3.27 Å for apo-RBD/ACE2 and the RBD/ACE2-arbidol complexes, respectively. Interestingly, the Rg profile of the RBD/ACE2-arbidol complex was more stable than that of the apo-RBD/ACE2 complex due to a higher number of favorable interactions. All these data indicate that the formation of more hydrogen bonds, reduced per-residue fluctuation, and higher compactness in the RBD/ACE2-arbidol complex drive its overall stability and convergence.
3.4. Principal component and free energy analyses of the RBD/ACE2-arbidol complex
PCA is an important technique that can provide insights into the correlation of atomic movements in enzyme and substrate interactions, which originate from the collective motion of atoms controlled by protein secondary structures. Typically, the largest associated eigenvalues define the essential subspace, in which most of the protein dynamics occur. For this purpose, the clusters of stable PCA states for the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes were visualized and analyzed (Supplementary Fig. 2). The trace values calculated from the covariance matrix of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes were 80.76 nm2 and 69.51 nm2, respectively, suggesting that the RBD/ACE2 complex was subject to compaction upon arbidol binding. However, a higher trace value for the apo-RBD/ACE2 complex suggested comparatively high flexibility. Moreover, the majority of the dynamics came from a small number of eigenvectors representing the overall collective motions. The percentage contributions of the first two eigenvectors described more than 80% and 90% of the total dynamics for the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes, respectively. Next, the Gibbs FEL plots were generated from the principal component 1 (PC1) and principal component 2 (PC2) coordinates. In the FEL plots, the ΔG values of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes ranged from 0 to 15.8 kJ/mol (Fig. 4 ). This further demonstrates that the two systems behaved in comparatively different manners during the simulations.
Fig. 4.

Free energy landscapes of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes. The free energy landscapes generated by projecting the principal components PC1 and PC2 of (A) the apo-RBD/ACE2 complex and (B) the RBD/ACE2-arbidol complex from MD simulations at 300 K. The free energies are represented by -kBT ln P(PC1, PC2), with P(PC1, PC2) being the distribution probability calculated using the structures sampled at 300 K. The blue, green, and cyan colors represent the metastable conformations with low-energy states, while the red color signifies the high-energy protein conformations.
3.5. Binding free energy calculations between ACE2-arbidol and RBD-arbidol
To investigate how strongly arbidol binds to ACE2 and the RBD and its associated binding modes, the binding free energies were computed using the MM-PBSA method. To this end, the last 30 ns of the MD simulation trajectories were investigated to obtain the binding affinities and insights into the binding mechanisms of arbidol. Arbidol was found to display a binding affinity of −73.36 kJ/mol for ACE2 (Table 1 ). The detailed decomposition of the energy components revealed that except for the polar solvation energy, the van der Waals energy and SASA energy contributed to the tighter binding of arbidol with ACE2. Computation of the binding affinity of arbidol for the RBD revealed that it exhibited a higher affinity (−105.95 kJ/mol) for the RBD than for ACE2 (Table 1). An energy decomposition profile further revealed energy components similar to those of ACE2; however, the van der Waals and polar solvation energies were found to positively contribute towards tighter binding with the RBD than with ACE2. The electrostatic energy played a comparatively minor role in binding. A molecular interaction obtained from the average structure of the MD simulations supported the binding mechanism of arbidol to the RBD/ACE2 complex (Fig. 5 A). Additionally, the well-superimposed structures of arbidol in the RBD/ACE2 interface obtained from the last 30 ns simulation further confirmed the stability and preferred binding mechanism (Fig. 5B). Only the thio-phenyl group appeared to be slightly flexible in a few snapshots. The detailed binding free energy calculations and a visual assessment led to the conclusion that arbidol has a higher binding affinity towards the RBD, which is most likely due to the establishment of more favorable interactions with the surrounding key residues.
Table 1.
Comparison of the binding free energy and various energy components between Arbidol-ACE2 and Arbidol-RBD from MD simulation trajectory.
| Energy components | Arbidol-ACE2 (kJ/mol) | Arbidol-RBD (kJ/mol) |
|---|---|---|
| Van der Waals energy | −141.42 ± 27.85 | −133.03 ± 12.72 |
| Electrostatic energy | −11.19 ± 4.11 | −10.20 ± 5.58 |
| Polar solvation energy | 96.72 ± 16.98 | 52.30 ± 9.66 |
| SASA energy | −17.48 ± 1.80 | −14.98 ± 1.04 |
| Binding energy | −73.36 ± 16.79 | −105.95 ± 14.25 |
Fig. 5.

Molecular interaction of arbidol with RBD/ACE2. (A) A 3D interaction diagram showing the contacts and interactions of arbidol (yellow stick) with the RBD/ACE2 complex residues during MD simulations. The interacting residues of ACE2 and the RBD are labeled and shown as green and cyan sticks, respectively, and interactions are shown as orange dashes and (B) superimposed structures of arbidol (stick) in the RBD/ACE2 complex (ACE2: green surface and RBD: cyan surface) over the last 30 ns MD simulation for the RBD/ACE2-arbidol complex.
3.6. Key residues governing the binding of arbidol to ACE2 and the RBD
Following the estimation of arbidol binding affinities for ACE2 and the RBD, we identified the critical residues in ACE2 and the RBD that positively and negatively impacted arbidol binding. First, the contribution energies of individual interacting residues on ACE2 revealed that the residues Lys 26, Asp 30, His 34, Val 93, Ala 387, Pro 389, and Phe 390 contributed the most to the ACE2 affinity for arbidol (Fig. 6 A). In addition, arbidol showed an even higher binding affinity for the RBD, in which many residues positively contributed to its tighter binding. An array of residues, such as Arg403, Asp405, Glu406, Gln409, Gly416, Lys417, Ile418, and Tyr505, were found to contribute to the higher binding affinity of the RBD to arbidol (Fig. 6B). Interestingly, they were also found to interact with the RBD/ACE2 interface, which further confirms that arbidol binds and inhibits the interfacial residues of the RBD/ACE2 complex, where it establishes more contacts and interactions with the RBD than with the ACE2 receptor. Furthermore, to understand the intermolecular interactions between arbidol and the RBD/ACE2 complex, representative structures were extracted from the MD simulation trajectory and analyzed. While arbidol exhibited few intermolecular interactions with the residues of ACE2, it exhibited relatively more interactions with the RBD. Overall, the analysis of the RBD/ACE2-arbidol complex interactions provided insights into the vital residue interactions and to the differences between ACE2-arbidol and RBD-arbidol binding and its inhibitory mechanism.
Fig. 6.

Contribution energies of the ACE2 and RBD residues towards arbidol. (A) The per-residue energy contributions of the (A) ACE2 and (B) RBD residues towards arbidol binding as obtained from the MM-PBSA binding affinity calculations.
3.7. Intermolecular interactions and energy components between ACE2 and the RBD upon arbidol binding
Next, we used PRODIGY to compute the binding affinities of the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes extracted from MD simulations. We found that the RBD/ACE2-arbidol complex exhibited a higher binding affinity (ΔG) of −11.5 kcal/mol, whereas the apo-RBD/ACE2 complex exhibited a relatively lower binding affinity (ΔG) of −10.8 kcal/mol. We observed that the higher binding affinity of the RBD/ACE2-arbidol complex was due to increased numbers of intermolecular, charged-polar, charged-apolar, and apolar-apolar contacts. The detailed decomposition of intermolecular contacts governing the difference in binding energies is shown in Table 2 . PPCheck was also used to compute the Et and the types of interactions in the apo-RBD/ACE2 and RBD/ACE2-arbidol complexes based on the MD simulations. The Et for the apo-RBD/ACE2 complex was −113.46 kJ/mol, and it was −171.57 kJ/mol for the RBD/ACE2-arbidol complex (Table 3 ). A closer inspection of interactions and energy components revealed that the hydrogen bonds and van der Waals pairs contributing to the hydrogen bond and van der Waals energies largely governed the differences in the Et values and between the affinity of the RBD/ACE2 complex in its apo-RBD/ACE2 and RBD/ACE2-arbidol complexes (Table 3)..
Table 2.
PRODIGY-derived decomposition of intermolecular contacts for the apo-RBD/ACE2 complex and RBD/ACE2-arbidol complex from the last 30 ns MD simulation trajectories.
| Types of contacts | Apo-RBD/ACE2 complex | Types of contacts | RBD/ACE2-arbidol complex |
|---|---|---|---|
| No. of intermolecular contacts | 59 | No. of intermolecular contacts | 74 |
| No. of charged-charged contacts | 1 | No. of charged-charged contacts | 1 |
| No. of charged-polar contacts | 9 | No. of charged-polar contacts | 13 |
| No. of charged-apolar contacts | 18 | No. of charged-apolar contacts | 23 |
| No. of polar-polar contacts | 4 | No. of polar-polar contacts | 3 |
| No. of apolar-polar contacts | 18 | No. of apolar-polar contacts | 18 |
| No. of apolar-apolar contacts | 9 | No. of apolar-apolar contacts | 16 |
| % of apolar NIS residues | 35.58 | % of apolar NIS residues | 34.94 |
| % of charged NIS residues | 26.76 | % of charged NIS residues | 27.71 |
| Predicted binding affinity (kcal/mol) | −10.8 | Predicted binding affinity (kcal/mol) | −11.5 |
Table 3.
PPCheck-derived energy components for the apo-RBD/ACE2 complex and RBD/ACE2-arbidol complex from the last 30 ns MD simulation trajectories.
| Energy components | Apo-RBD/ACE2 complex | RBD/ACE2-arbidol complex |
|---|---|---|
| Hydrogen bond energy | −7.56 kJ/mol | −8.43 kJ/mol |
| Electrostatic energy | −1.92 kJ/mol | 10.22 kJ/mol |
| Van der Waals energy | −103.99 kJ/mol | −173.35 kJ/mol |
| Total stabilizing energy | −113.46 kJ/mol | −171.57 kJ/mol |
| No. of interface residues | 75 | 74 |
| Normalized energy per residue | −1.51 kJ/mol | −2.32 kJ/mol |
| No. of short contacts | 0 | 2 |
| No. of hydrophobic interactions | 1 | 3 |
| No. of van der Waals pairs | 1915 | 2595 |
| No. of salt bridges | 0 | 0 |
| No. of potential favorable electrostatic interactions | 1 | 0 |
| No. of potential unfavorable electrostatic interactions | 1 | 2 |
4. Discussion
The COVID-19 pandemic heightens the need for preparedness to respond to emerging viral threats rapidly (Mishra and Tripathi, 2021). No therapies have been shown to be clinically effective to date. To assist in efforts to control the rapidly evolving pandemic, drug repurposing is being employed as an effective and fast method. Given the urgency of the situation, scientists must understand and characterize the molecular mechanisms of action for the repurposed drugs being tested for COVID-19 treatment. A computational understanding of the molecular mechanisms underlying unproven anti-COVID-19 drugs may guide scientists towards developing a specific therapeutic against SARS-CoV-2.
Arbidol, a broad-spectrum antiviral drug, is currently being used as a standard treatment option for COVID-19 (Anonymous, 2020; Hulseberg et al., 2019; Pei-Fang, 2020; Pshenichnaya et al., 2019). It shows significant antiviral and anti-inflammatory efficacy both in vitro and in vivo against the influenza virus (Wang et al., 2017). Since the SARS-CoV-2 and influenza viruses exhibit similar disease pathologies, arbidol could exhibit anti-SARS-CoV-2 activity. Arbidol is patented for its medicinal use as an antiviral agent against atypical pneumonia induced by SARS-CoV (http://www.arbidol.org/arbidol-patent-2004-sars-russian.pdf). It has been found to inhibit SARS-CoV-2 effectively at a concentration of 4.11 μM in vitro, suggesting its potential for treating COVID-19 patients (Wang et al., 2020c). Currently, at least four clinical trials are being conducted with arbidol as a single agent for COVID-19 treatment. A few case reports have shown that patients with COVID-19 successfully recovered after receiving arbidol and lopinavir/ritonavir therapy (Lim et al., 2020; Wang et al., 2020e). Arbidol has been shown to be superior to the antiviral favipiravir, which did not improve the clinical recovery rate at day 7 compared to that in the arbidol group (Chen et al., 2020). A recent result of a clinical trial in China indicated that arbidol monotherapy was also superior to lopinavir/ritonavir at treating COVID-19 (Zhu et al., 2020). A study with a small sample size reported that arbidol could not improve the prognosis of COVID-19 patients not in the ICU; however, that study was preliminary and limited by several factors because i) the research involved a single-center and retrospective study, ii) pharyngeal swabs were not collected every day, and iii) the study included only patients with moderate and severe COVID-19, thus prohibiting the confirmation of arbidol efficacy in the treatment of mild and critical patients (Lian et al., 2020). Arbidol binds to the interface of the viral glycoprotein and human cell-surface receptors, thereby blocking membrane fusion and viral entry (Boriskin et al., 2008; Leneva et al., 2009; Pécheur et al., 2007; Teissier et al., 2011). The binding of arbidol is topologically distant from the active site of the ACE2 receptor. In SARS-CoV-2, arbidol acts by interfering with virus binding to host cells (Wang et al., 2020c). A few molecular docking studies on arbidol against the SARS-CoV-2 RBD and human ACE2 complex have revealed the specific residues involved in the interaction; however, the detailed mechanism underlying the process and the associated dynamics are not known (Barik et al., 2020; Vankadari, 2020).
Our analyses of the 300 ns MD simulation trajectories revealed that when arbidol was docked at the RBD-ACE2 complex interface, it stabilized and formed favorable interactions with both the RBD and ACE2. However, arbidol displayed a higher binding affinity for the RBD than for ACE2. The analysis of hydrogen bonds revealed that following arbidol binding, the RBD/ACE2 complex could form more hydrogen bond interactions than the apo-RBD/ACE2 complex. The binding also induced structural rigidity and compaction in the RBD/ACE2-arbidol complex. The compaction was also confirmed by PCAs, in which the RBD/ACE2-arbidol complex formed a more stable cluster. The detailed decomposition of energy components revealed that the binding of arbidol to ACE2 is due to positive contributions from van der Waals, electrostatic, and SASA energies, while the tighter binding of arbidol to the RBD is primarily due to positive contributions from the van der Waals and polar solvation energies. Furthermore, arbidol binding leads to a substantial increase in the binding affinity between RBD and ACE2, indicating that arbidol induces favorable interactions between them. Overall, the data indicated that arbidol binding drives an increase in the structural stability, compactness, and convergence of the RBD/ACE2 complex. In the absence of any arbidol-bound structure of the RBD/ACE2 complex, our data offer the possible hypothesis that arbidol binding to the interface of viral glycoprotein and human receptors induces structural rigidity, leading to the inhibition of conformational changes in the S-protein that are associated with viral entry.
5. Conclusions
The Chinese Center for Disease Control and Prevention recommends arbidol, an anti-influenza drug, for the management and treatment of COVID-19. Arbidol has been found to be superior to other antiviral combinations. In SARS-CoV-2, arbidol acts by interfering with viral binding to host cells. However, the detailed mechanism is not known. To develop effective therapeutics or higher-efficacy arbidol derivatives, there is an ample need to gain insights into its mechanisms of action. With guidance from MD simulations and structure network analysis, we demonstrate that arbidol binds at the RBD/ACE2 interface, establishing stronger intermolecular interactions with the RBD and inducing favorable interactions between them, resulting in an increased affinity between the RBD and ACE2. Arbidol binding may induce structural inflexibility, leading to inhibition of the conformational dynamics required during virus entry. Our study provides a first-hand explanation of why arbidol is effective against SARS-CoV-2. We highlight discrete regions in the interaction interface that could be potentially druggable pockets to destabilize the RBD/ACE2 complex. The mechanistic understanding gained from this study should be useful for the design and development of additional specific SARS-CoV-2 inhibitors.
CRediT authorship contribution statement
Aditya K. Padhi: performed the experiments and generated the data, and, and, and. Aniruddha Seal: and. Javed Masood Khan: and. Maqusood Ahamed: and. Timir Tripathi: conceived the study, participated in its design and coordination, analyzed the data, drafted the, manuscript. All, the authors read and approved the final manuscript.
Declaration of competing interest
The authors declare no conflict of interests.
Acknowledgments
The author (MA) is grateful to the Researchers Supporting Project number (RSP-2020/129), King Saud University, Riyadh, Saudi Arabia. The authors also thank Dr. Kam Y.J. Zhang (Laboratory for Structural Bioinformatics, RIKEN, Yokohama) for his continuous support and valuable suggestions for improving the manuscript. The authors acknowledge RIKEN ACCC for the Hokusai supercomputing resources. AKP acknowledges the Japan Society for the Promotion of Science (JSPS), Govt. of Japan, for the postdoctoral research fellowship. AS thanks the Department of Atomic Energy (DAE), Govt. of India, for the DISHA Fellowship.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejphar.2020.173836.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Anonymous . In: China National Health Commission, China. seventh ed. ed. Cardiology C.S.o., editor. 2020. Chinese clinical guidance for COVID-19 pneumonia diagnosis and treatment. [Google Scholar]
- Barik A., Rai G., Modi G. Molecular docking and binding mode analysis of selected FDA approved drugs against COVID-19 selected key protein targets: an effort towards drug repurposing to identify the combination therapy to combat COVID-19. arXiv. 2020 [Google Scholar]
- Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., Lopez de Castilla D., Finberg R.W., Dierberg K., Tapson V., Hsieh L., Patterson T.F., Paredes R., Sweeney D.A., Short W.R., Touloumi G., Lye D.C., Ohmagari N., Oh M.D., Ruiz-Palacios G.M., Benfield T., Fätkenheuer G., Kortepeter M.G., Atmar R.L., Creech C.B., Lundgren J., Babiker A.G., Pett S., Neaton J.D., Burgess T.H., Bonnett T., Green M., Makowski M., Osinusi A., Nayak S., Lane H.C. Remdesivir for the treatment of covid-19 - final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. In: Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, April 13–16, 1981. Pullman B., editor. Springer Netherlands; Dordrecht: 1981. Interaction models for water in relation to protein hydration; pp. 331–342. [Google Scholar]
- Blaising J., Polyak S.J., Pecheur E.I. Arbidol as a broad-spectrum antiviral: an update. Antivir. Res. 2014;107:84–94. doi: 10.1016/j.antiviral.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriskin Y.S., Leneva I.A., Pecheur E.I., Polyak S.J. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008;15:997–1005. doi: 10.2174/092986708784049658. [DOI] [PubMed] [Google Scholar]
- Brooks C.L., 3rd, Onuchic J.N., Wales D.J. Statistical thermodynamics. Taking a walk on a landscape. Science. 2001;293:612–613. doi: 10.1126/science.1062559. [DOI] [PubMed] [Google Scholar]
- Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126 doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Callaway E. Making sense of coronavirus mutations. Nature. 2020;585:174–177. doi: 10.1038/d41586-020-02544-6. [DOI] [PubMed] [Google Scholar]
- Chen C., Zhang Y., Huang J., Yin P., Cheng Z., Wu J., Chen S., Zhang Y., Chen B., Lu M., Luo Y., Ju L., Zhang J., Wang X. Favipiravir versus arbidol for COVID-19: a randomized clinical trial. medRxiv. 2020 [Google Scholar]
- Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- De Clercq E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L., Hu S., Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug Discov Ther. 2020;14:58–60. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- Elfiky A.A. 2020. Anti-HCV, Nucleotide Inhibitors, Repurposing against COVID-19. Life Sci 117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohlke H., Kiel C., Case D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., Tummino T.A., Huettenhain R., Kaake R.M., Richards A.L., Tutuncuoglu B., Foussard H., Batra J., Haas K., Modak M., Kim M., Haas P., Polacco B.J., Braberg H., Fabius J.M., Eckhardt M., Soucheray M., Bennett M.J., Cakir M., McGregor M.J., Li Q., Meyer B., Roesch F., Vallet T., Mac Kain A., Miorin L., Moreno E., Naing Z.Z.C., Zhou Y., Peng S., Shi Y., Zhang Z., Shen W., Kirby I.T., Melnyk J.E., Chorba J.S., Lou K., Dai S.A., Barrio-Hernandez I., Memon D., Hernandez-Armenta C., Lyu J., Mathy C.J.P., Perica T., Pilla K.B., Ganesan S.J., Saltzberg D.J., Rakesh R., Liu X., Rosenthal S.B., Calviello L., Venkataramanan S., Liboy-Lugo J., Lin Y., Huang X.-P., Liu Y., Wankowicz S.A., Bohn M., Safari M., Ugur F.S., Koh C., Savar N.S., Tran Q.D., Shengjuler D., Fletcher S.J., O'Neal M.C., Cai Y., Chang J.C.J., Broadhurst D.J., Klippsten S., Sharp P.P., Wenzell N.A., Kuzuoglu D., Wang H.-Y., Trenker R., Young J.M., Cavero D.A., Hiatt J., Roth T.L., Rathore U., Subramanian A., Noack J., Hubert M., Stroud R.M., Frankel A.D., Rosenberg O.S., Verba K.A., Agard D.A., Ott M., Emerman M., Jura N., von Zastrow M., Verdin E., Ashworth A., Schwartz O., d'Enfert C., Mukherjee S., Jacobson M., Malik H.S., Fujimori D.G., Ideker T., Craik C.S., Floor S.N., Fraser J.S., Gross J.D., Sali A., Roth B.L., Ruggero D., Taunton J., Kortemme T., Beltrao P., Vignuzzi M., García-Sastre A., Shokat K.M., Shoichet B.K., Krogan N.J. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy R.K., DiPaola R.S., Romanelli F., Dutch R.E. Rapid repurposing of drugs for COVID-19. Science. 2020;368:829. doi: 10.1126/science.abb9332. [DOI] [PubMed] [Google Scholar]
- Herod M.R., Adeyemi O.O., Ward J., Bentley K., Harris M., Stonehouse N.J., Polyak S.J. The broad-spectrum antiviral drug arbidol inhibits foot-and-mouth disease virus genome replication. J. Gen. Virol. 2019;100:1293–1302. doi: 10.1099/jgv.0.001283. [DOI] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H.J., Fraaije J.G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- Homeyer N., Gohlke H. Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Mol Inform. 2012;31:114–122. doi: 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., Prudon B., Green C., Felton T., Chadwick D., Rege K., Fegan C., Chappell L.C., Faust S.N., Jaki T., Jeffery K., Montgomery A., Rowan K., Juszczak E., Baillie J.K., Haynes R., Landray M.J. Dexamethasone in hospitalized patients with covid-19 - preliminary report. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Lin Z., van Gunsteren W.F. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J. Chem. Theor. Comput. 2011;7:1237–1243. doi: 10.1021/ct100747y. [DOI] [PubMed] [Google Scholar]
- Hulseberg C.E., Feneant L., Szymanska-de Wijs K.M., Kessler N.P., Nelson E.A., Shoemaker C.J., Schmaljohn C.S., Polyak S.J., White J.M. Arbidol and other low-molecular-weight drugs that inhibit lassa and ebola viruses. J. Virol. 2019;93 doi: 10.1128/JVI.02185-18. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kadam R.U., Wilson I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. U. S. A. 2017;114:206–214. doi: 10.1073/pnas.1617020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren G., King S., Knowles S., Phillips E. Ribavirin in the treatment of SARS: a new trick for an old drug? CMAJ (Can. Med. Assoc. J.) 2003;168:1289–1292. [PMC free article] [PubMed] [Google Scholar]
- Koziara K.B., Stroet M., Malde A.K., Mark A.E. Testing and validation of the automated topology builder (ATB) version 2.0: prediction of hydration free enthalpies. J. Comput. Aided Mol. Des. 2014;28:221–233. doi: 10.1007/s10822-014-9713-7. [DOI] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Lynn A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Kurkcuoglu Z., Koukos P.I., Citro N., Trellet M.E., Rodrigues J., Moreira I.S., Roel-Touris J., Melquiond A.S.J., Geng C., Schaarschmidt J., Xue L.C., Vangone A., Bonvin A. Performance of HADDOCK and a simple contact-based protein-ligand binding affinity predictor in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018;32:175–185. doi: 10.1007/s10822-017-0049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Leneva I.A., Russell R.J., Boriskin Y.S., Hay A.J. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antivir. Res. 2009;81:132–140. doi: 10.1016/j.antiviral.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Li H., Zhou Y., Zhang M., Wang H., Zhao Q., Liu J. Updated approaches against SARS-CoV-2. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.00483-20. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian N., Xie H., Lin S., Huang J., Zhao J., Lin Q. Umifenovir treatment is not associated with improved outcomes in patients with coronavirus disease 2019: a retrospective study. Clin. Microbiol. Infect. 2020;26:917–921. doi: 10.1016/j.cmi.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J., Jeon S., Shin H.Y., Kim M.J., Seong Y.M., Lee W.J., Choe K.W., Kang Y.M., Lee B., Park S.J. Case of the index patient who caused tertiary transmission of COVID-19 infection in korea: the application of lopinavir/ritonavir for the treatment of COVID-19 Infected pneumonia monitored by quantitative RT-PCR. J. Kor. Med. Sci. 2020;35:e79. doi: 10.3346/jkms.2020.35.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Mark A.E., van Gunsteren W.F. Estimating the relative free energy of different molecular states with respect to a single reference state. J. Phys. Chem. B. 1996;100:9485–9494. [Google Scholar]
- Liu Y., Grimm M., Dai W.T., Hou M.C., Xiao Z.X., Cao Y. CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin. 2020;41:138–144. doi: 10.1038/s41401-019-0228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W.J., Wang D., Xu W., Holmes E.C., Gao G.F., Wu G., Chen W., Shi W., Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malde A.K., Zuo L., Breeze M., Stroet M., Poger D., Nair P.C., Oostenbrink C., Mark A.E. An automated force field topology builder (ATB) and repository: version 1.0. J. Chem. Theor. Comput. 2011;7:4026–4037. doi: 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Mishra S.K., Tripathi T. One year update on the COVID-19 pandemic: where are we now? Acta Trop. 2021;214:105778. doi: 10.1016/j.actatropica.2020.105778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsutake A., Sugita Y., Okamoto Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers. 2001;60:96–123. doi: 10.1002/1097-0282(2001)60:2<96::AID-BIP1007>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Padhi A.K., Kalita P., Zhang K.Y.J., Tripathi T. High throughput designing and mutational mapping of RBD-ACE2 interface guide non-conventional therapeutic strategies for COVID-19. bioRxiv. 2020 [Google Scholar]
- Padhi A.K., Shukla R., Saudagar P., Tripathi T. High-throughput rational design of the remdesivir binding site in the RdRp of SARS-CoV-2: Implications for potential resistance. iScience. 2021 doi: 10.1016/j.isci.2020.101992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A.K., Tripathi T. Can SARS-CoV‐2 accumulate mutations in the S‐protein to increase pathogenicity? ACS Pharmacol Transl Sci. 2020;3:1023–1026. doi: 10.1021/acsptsci.0c00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- Pecheur E.I., Borisevich V., Halfmann P., Morrey J.D., Smee D.F., Prichard M., Mire C.E., Kawaoka Y., Geisbert T.W., Polyak S.J. The synthetic antiviral drug arbidol inhibits globally prevalent pathogenic viruses. J. Virol. 2016;90:3086–3092. doi: 10.1128/JVI.02077-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pécheur E.I., Lavillette D., Alcaras F., Molle J., Boriskin Y.S., Roberts M., Cosset F.L., Polyak S.J. Biochemical mechanism of hepatitis C virus inhibition by the broad-spectrum antiviral arbidol. Biochemistry. 2007;46:6050–6059. doi: 10.1021/bi700181j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei-Fang W. Diagnosis and treatment protocol for novel coronavirus pneumonia (trial version 7) Chin. Med. J. 2020;133:1087–1095. doi: 10.1097/CM9.0000000000000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pshenichnaya N.Y., Bulgakova V.A., Lvov N.I., Poromov A.A., Selkova E.P., Grekova A.I., Shestakova I.V., Maleev V.V., Leneva I.A. Clinical efficacy of umifenovir in influenza and ARVI (study ARBITR) Ter. Arkh. 2019;91:56–63. doi: 10.26442/00403660.2019.03.000127. [DOI] [PubMed] [Google Scholar]
- Rajarshi K., Khan R., Singh M.K., Ranjan T., Ray S., Ray S. Essential functional molecules associated with SARS-CoV-2 infection: potential therapeutic targets for COVID-19. Gene. 2020:145313. doi: 10.1016/j.gene.2020.145313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajter J.C., Sherman M.S., Fatteh N., Vogel F., Sacks J., Rajter J.J. Use of ivermectin is associated with lower mortality in hospitalized patients with coronavirus disease 2019: the ICON study. Chest. 2020 doi: 10.1016/j.chest.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runfeng L., Yunlong H., Jicheng H., Weiqi P., Qinhai M., Yongxia S., Chufang L., Jin Z., Zhenhua J., Haiming J. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2) Pharmacol. Res. 2020;156:104761. doi: 10.1016/j.phrs.2020.104761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzi-Puttini P., Giorgi V., Sirotti S., Marotto D., Ardizzone S., Rizzardini G., Antinori S., Galli M. COVID-19, cytokines and immunosuppression: what can we learn from severe acute respiratory syndrome? Clin. Exp. Rheumatol. 2020;38:337–342. [PubMed] [Google Scholar]
- Schmid N., Eichenberger A.P., Choutko A., Riniker S., Winger M., Mark A.E., van Gunsteren W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Sukhwal A., Sowdhamini R. Oligomerisation status and evolutionary conservation of interfaces of protein structural domain superfamilies. Mol. Biosyst. 2013;9:1652–1661. doi: 10.1039/c3mb25484d. [DOI] [PubMed] [Google Scholar]
- Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020;17:613–620. doi: 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier E., Zandomeneghi G., Loquet A., Lavillette D., Lavergne J.P., Montserret R., Cosset F.L., Böckmann A., Meier B.H., Penin F., Pécheur E.I. Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PloS One. 2011;6 doi: 10.1371/journal.pone.0015874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The WHO.React Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID-19: a meta-analysis. J. Am. Med. Assoc. 2020;324:1330–1341. doi: 10.1001/jama.2020.17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y.F., Chien C.S., Yarmishyn A.A., Lin Y.Y., Luo Y.H., Lin Y.T., Lai W.Y., Yang D.M., Chou S.J., Yang Y.P., Wang M.L., Chiou S.H. A review of SARS-CoV-2 and the ongoing clinical trials. Int. J. Mol. Sci. 2020;21:2657. doi: 10.3390/ijms21072657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A., Bonvin A.M. Contacts-based prediction of binding affinity in protein-protein complexes. Elife. 2015;4 doi: 10.7554/eLife.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A., Schaarschmidt J., Koukos P., Geng C., Citro N., Trellet M.E., Xue L.C., Bonvin A. Large-scale prediction of binding affinity in protein-small ligand complexes: the PRODIGY-LIG web server. Bioinformatics. 2019;35:1585–1587. doi: 10.1093/bioinformatics/bty816. [DOI] [PubMed] [Google Scholar]
- Vankadari N. Arbidol: a potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020;56:105998. doi: 10.1016/j.ijantimicag.2020.105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velavan T.P., Meyer C.G. The COVID‐19 epidemic. Trop. Med. Int. Health. 2020;25:278. doi: 10.1111/tmi.13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.Y., Wang Q., Zhou H., Yan J., Qi J. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894–904. doi: 10.1016/j.cell.2020.03.045. e899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Kollman P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000;303:567–582. doi: 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- Wang X., Cao R., Zhang H., Liu J., Xu M., Hu H., Li Y., Zhao L., Li W., Sun X., Yang X., Shi Z., Deng F., Hu Z.A.-O., Zhong W., Wang M.A.-O. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020;6:28. doi: 10.1038/s41421-020-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Ding Y., Yang C., Li R., Du Q., Hao Y., Li Z., Jiang H., Zhao J., Chen Q., Yang Z., He Z. Inhibition of the infectivity and inflammatory response of influenza virus by Arbidol hydrochloride in vitro and in vivo (mice and ferret) Biomed. Pharmacother. 2017;91:393–401. doi: 10.1016/j.biopha.2017.04.091. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., Fu S., Gao L., Cheng Z., Lu Q., Hu Y., Luo G., Wang K., Lu Y., Li H., Wang S., Ruan S., Yang C., Mei C., Wang Y., Ding D., Wu F., Tang X., Ye X., Ye Y., Liu B., Yang J., Yin W., Wang A., Fan G., Zhou F., Liu Z., Gu X., Xu J., Shang L., Zhang Y., Cao L., Guo T., Wan Y., Qin H., Jiang Y., Jaki T., Hayden F.G., Horby P.W., Cao B., Wang C. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Chen X., Lu Y., Chen F., Zhang W. Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci Trends. 2020;14:64–68. doi: 10.5582/bst.2020.01030. [DOI] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Yang X.O. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect. 2020;53:368–370. doi: 10.1016/j.jmii.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L.C., Rodrigues J.P., Kastritis P.L., Bonvin A.M., Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics. 2016;32:3676–3678. doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Lu Z., Xu T., Chen C., Yang G., Zha T., Lu J., Xue Y. Arbidol monotherapy is superior to lopinavir/ritonavir in treating COVID-19. J. Infect. 2020;81:e21–e23. doi: 10.1016/j.jinf.2020.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.