

Abstract
Lateralized overgrowth (LO), or segmental overgrowth, is defined as an increase in growth of tissue (bone, muscle, connective tissue, vasculature, etc.) in any region of the body. Some overgrowth syndromes, characterized by both generalized and lateralized overgrowth, have been associated with an increased risk of tumor development. This may be due to the underlying genetic and epigenetic defects that lead to disrupted cell growth and proliferation pathways resulting in the overgrowth and tumor phenotypes. This chapter focuses on the four most common syndromes characterized by LO: Beckwith-Wiedemann spectrum (BWSp), PIK3CA-related overgrowth spectrum (PROS), Proteus syndrome (PS), and PTEN hamartoma tumor syndrome (PHTS). These syndromes demonstrate variable risks for tumor development in patients affected by LO, and we provide a comprehensive literature review of all common tumors reported in patients diagnosed with an LO-related disorder. This review summarizes the current data on tumor risk among these disorders and their associated tumor screening guidelines. Furthermore, this chapter highlights the importance of an accurate diagnosis when a patient presents with LO as similar phenotypes are associated with different tumor risks, thereby altering preventative screening protocols.
Keywords: lateralized overgrowth, hemihypertrophy, hemihyperplasia, Beckwith-Wiedemann spectrum, Beckwith-Wiedemann syndrome (BWS), PIK3CA-related overgrowth spectrum (PROS), Proteus syndrome (PS), PTEN hamartoma tumor syndrome
Introduction
Lateralized overgrowth (LO) is defined as any type of segmental overgrowth (1) (Figure 1). The nomenclature was developed to classify patients who were previously described with overgrowth due to both hyperplasia (OMIM 235000), a proliferation of cells, and hypertrophy (OMIM 235000), an increase in cell size. The overgrowth defined by LO is not specific to the type of tissue affected and can include skeletal, muscular, adipose, and/or vascular tissues. Some patients present with isolated LO, in which patients are primarily affected by LO. Overgrowth of organs is not required for the designation of LO, but it can be present and typically occurs in patients with overgrowth syndromes associated with LO.
Figure 1.
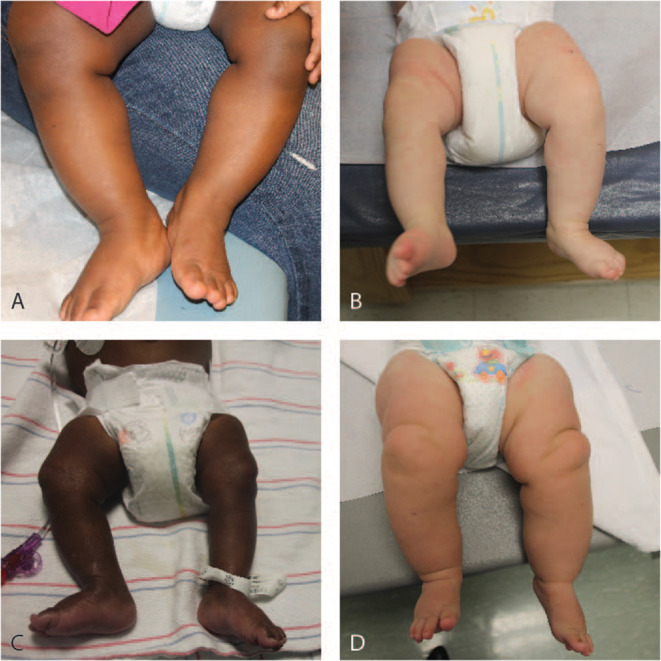
Legs of patients with lateralized overgrowth. (A) 12-month old patient with Beckwith-Wiedemann syndrome. (B) 3-month old patient with Beckwith-Wiedemann syndrome. (C) 9-month old patient with Beckwith-Wiedemann syndrome. (D) 6-month old patient with PIK3CA-related overgrowth spectrum.
Patients with isolated lateralized overgrowth (ILO), those affected by LO but lacking other features and patterns of malformations, dysplasia, and morphologic variants, have been reported to have an increased development of tumors, primarily the embryonal tumors Wilms tumor (WT) and hepatoblastoma (HB) (2, 3), similar to the most common tumor types observed among patients affected by LO and overgrowth disorders (4). A prior study of patients with isolated hemihypertrophy, now referred to as ILO, reported 9 out of 168 developing a tumor (5) and two cases of HB in patients with isolated hemihyperplasia, now also termed ILO (6). Retrospectively, it is likely that many of these patients could be classified with an overgrowth or cancer predisposition syndrome.
There are several genetic and epigenetic syndromes associated with LO and ILO. These molecular changes may influence the tissue type, location of the observed overgrowth, and associated tumor risk in patients. In this chapter, we review the clinical characteristics of the most common genetic and epigenetic syndromes associated with LO. We focus on tumor development and risks associated within each syndrome and summarize current screening recommendations.
Common considerations for all suspected LO-related overgrowth disorder include the underlying molecular cause and appropriateness for tumor surveillance.
Molecular Considerations
The underlying mechanisms for the disorders described are complex and beyond the focus of this review. A brief description of the currently understood mechanisms for each disorder is summarized and includes both genetic and epigenetic mechanisms. One consideration for molecular investigation for these disorders is that some defects can present as mosaic, in which the proportion of normal cells to cells with the molecular change varies in any given tissue, leading to patients with somatic molecular defects. This means that positive molecular detection may only be found in affected tissue(s), whereas blood sample analyses may yield negative results. Other patients affected by LO and overgrowth have the molecular defect change(s) detectable in blood samples (constitutional defects), with some patients affected by changes that are inheritable or considered germline defects.
Tumor Risk and Screening
Specific recommendations and implementation of tumor surveillance protocols are determined by the risk of tumor development in a particular syndrome, the uniformity of the tumors that develop (i.e., can they be screened for in a non-invasive manner), and the health care environment in which the screening is occurring (i.e., the threshold of acceptable risk) (4). In some syndromes with an established tumor risk, tumor screening has been demonstrated to detect tumors at an early age. For example, in Beckwith-Wiedemann spectrum (BWSp), patients who underwent ultrasonographic screening had on average earlier tumor stages at diagnosis than those who did not undergo screening (7). Diagnosing tumors in their earlier stages may allow for less invasive treatment and the prevention of possible metastasis.
Here, we review the most common syndromes characterized by LO: BWSp (OMIM 130650), PIK3CA-related overgrowth spectrum (PROS), Proteus syndrome (PS) (OMIM 176920), and PTEN hamartoma tumor syndrome (PHTS). Tumor development in these four syndromes is variable and discussed below.
Beckwith-Wiedemann Spectrum (OMIM 130650)
Overview
BWSp is the most common and well-characterized overgrowth and cancer predisposition disorder and is caused by a variety of molecular defects in the chromosome 11p15 region. The disorder is estimated to affect 1 in 10,340 live births and disproportionately affects patients conceived by assisted reproduction techniques, estimated to affect 1 in 1,100 live births (8, 9). The clinical manifestations and subsequent phenotype of patients with BWSp can be highly variable, leading to the reclassification of the disorder from a syndrome [Beckwith-Wiedemann syndrome (BWS)] to a spectrum [BW spectrum (BWSp)] by an international consensus group (10). The consensus group created a clinical scoring system to guide molecular and clinical diagnosis. They classified features as those classically associated with the disorder (cardinal features) and features associated with the disorder but that can also occur in the general population (suggestive features). This scoring system was implemented to determine if genetic testing is necessary (10). Cardinal features include macroglossia, omphalocele, muscular LO, bilateral WT, hyperinsulinism, adrenal cytomegaly, pancreatic adenomatosis, and placental mesenchymal dysplasia, and suggestive features include macrosomia, facial nevus simplex, polyhydramnios or placentomegaly, ear creases or pits, transient hypoglycemia, embryonal tumors, nephromegaly and/or hepatomegaly, and umbilical hernia or diastasis recti. Each cardinal feature receives two points, and each suggestive feature receives one point. A total clinical score greater or equal to 2 indicates the need for genetic testing for BWSp. A clinical score greater or equal to 4 (typically including at least one cardinal feature) is sufficient for a clinical diagnosis of BWSp even if no molecular defect on chromosome 11p15 is identified. Genetic testing is also recommended for patients with a family history of BWSp caused by a heritable alteration.
Molecular Considerations
BWSp is caused by a variety of genetic and epigenetic alterations in the BWS critical region on chromosome 11p15.5 (10). The BWS critical region contains two imprinted regions, which control the normal regulation of fetal and postnatal growth genes through a process called methylation. The majority of patients are affected by abnormal methylation in the imprinting control 1 (IC1) and/or imprinting control 2 (IC2) regions, with the most common cause being loss of methylation at KCNQ1OT1:TSS DMR (IC2 LOM) (~50% of patients) (10). Other causes of BWSp include paternal uniparental isodisomy of chromosome 11p15 (pUPD11), gain of methylation at H19/IGF2:IG DMR (IC1 GOM), mutations of CDKN1C, and other genetic aberrations including deletions, duplications, and translocations that affect chromosome 11p15 (10).
Tumor Risk in BWSp
The risk for WT, HB, and neuroblastoma in BWSp is well documented (11–18). A patient's tumor risk varies based on the molecular etiology of BWSp. According to the recent international consensus for BWSp, for patients with IC1 GOM, the overall risk of tumor development is 28%, and the risk for WT is 24%. For patients with IC2 LOM, the overall tumor risk is 2.5%. For patients with pUPD11, the overall tumor risk is 16%. The risk for developing a WT is 8%, and the risk for developing a HB is 3.5%. Screening guidelines are constantly evolving based on ongoing research on this topic and are dependent on geographical location and cultural context of clinical practice. The European guidelines include abdominal ultrasounds every 3 months until the age of 7 years for patients with BWSp due to IC1 GOM, pUPD11, CDKN1C mutations, and other chromosome aberrations of the BWS region (10). The United States guidelines developed by the American Association for Cancer Research (AACR) Childhood Predisposition Workshop include abdominal ultrasounds and alpha-fetoprotein (AFP) screening every 3 months until the 4th birthday and renal ultrasounds every 3 months from the 4th to the 7th birthday for all patients with BWSp (4). In addition, patients with CDKN1C mutations, those at the highest risk for developing a neuroblastoma among patients with BWSp, should receive urine vanillylmandelic acid (VMA), homovanillic acid (HVA), and chest X-rays screening every 3 months until the 6th birthday and every 6 months from the 6th to the 10th birthday (10). Patients with BWSp caused by genome-wide paternal isodisomy (GWpUPD) have been reported to have additional tumors and beyond these screening windows. Patients with this molecular subtype should be monitored closely (10, 19).
PIK3CA-Related Overgrowth Spectrum
Overview
The phenotypic variety and overlap of individual syndromes caused by PIK3CA mutations prompted the establishment of the term PROS (20). The specific overgrown tissue observed in patients with PROS is typically adipose or vascular; however, muscular and skeletal overgrowth has also been observed (20). Other common clinical characteristics include epidermal nevus, macrodactyly, hemimegalencephaly (HMEG), seborrheic keratoses, and benign lichenoid keratoses (20). To determine the eligibility for genetic testing, clinical characteristics are divided into two categories: category A, which includes a spectrum of overgrowth, vascular malformations, and epidermal nevus phenotypes, and category B, which includes isolated features, such as lymphatic malformations or macrodactyly. Genetic testing is warranted if a patient presents with two or more features from category A, or one feature from category B, that was/were congenital or developed during early childhood.
A diagnosis of PROS is confirmed with a pathogenic variant found in the PIK3CA gene; however, if a mutation is not detected, the patient retains a clinical diagnosis of PROS if the clinical criteria are met (20, 21). In patients affected by clinical diagnoses of PROS, it is likely that the negative genetic result(s) observed are due to the somatic and therefore mosaic nature of the PIK3CA mutation leading to the phenotype, which may be difficult to detect from a single sample (such as blood).
Molecular Considerations
PIK3CA is a protein coding gene for p110α that is the α subunit of a collection of catalytic subunits for phosphatidylinositol 3-kinase (PI3K) (22). This protein is important for regulating signals for cell proliferation and survival. Mutations in PIK3CA have been identified as the driver for many cancers in asymptomatic patients (those without phenotypes related to PIK3CA abnormalities), with common cancer types including breast (>30%), endometrial (>30%), bladder (>20%), colorectal carcinoma (>17%), and head and neck squamous cell carcinoma (>15%) (23).
PIK3CA mutations have also been identified in patients with the following syndromes: fibroadipose overgrowth (FAO) (24), congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal (CLOVES) syndrome (25, 26), megalencephaly-capillary malformation (MCAP) syndrome (27), Klippel-Trenaunay syndrome (KTS) (26, 28), and HMEG (29). Typically, PIK3CA mutations occur post-fertilization (somatic mutations), but there have been germline PIK3CA mutations reported (30, 31). Allelic heterogeneity in PROS (and other overgrowth disorders) and the overlap of common variants in the genes responsible may influence cancer predisposition, but further study is required.
Tumor Risk in PROS
Tumor risk and surveillance for patients with PROS is currently debated. Gripp et al. suggested similar screening guidelines for patients affected by PROS to the guidelines for patients affected by ILO or BWS, which includes abdominal ultrasounds until the 7th birthday (32). Peterman et al. suggest sonographic screening for patients with CLOVES, MCAP, and diffuse capillary malformations only if LO is present (33, 34). To our knowledge, there have been 12 patients with PROS reported with malignant or potentially malignant renal findings [including WT, nephrogenic rests (NR), and indeterminate WT/NR findings]. NR and nephroblastomatosis (NBL) are capable of transforming into WT, but they are not tumors themselves (35, 36). Among the PROS patients with renal findings, eight patients with findings reported had a molecularly confirmed PROS diagnosis: four reported with WT development, two with reported indeterminate WT/NR findings, and two with NR (26, 28, 32, 37–40). Four additional patients with renal findings and without molecular PROS confirmation have also been reported (41–44). Postema et al. estimated the tumor risk between 1 and 2%, suggesting that under European standards, screening is not warranted (39); however, at that risk level by US guidelines, screening would be warranted (4).
As the focus of this review is to discuss common syndromes associated with LO and tumor screening guidelines, determining the true WT risk in PROS is beyond the scope of this chapter. A meta-analysis of PROS patients and WT development is currently being performed and will be reported separately in the literature once completed. Based on the current literature, the risk depends on how the reported cases are classified (for example, true WT vs. those with indeterminant malignant potential, such as NBL and NR). The tumor risk in the PROS population appears to be slightly less than what Postema et al. reported (~1–2%), and therefore it is unclear whether screening is warranted. The AACR tumor screening guidelines suggest screening when the risk of developing cancer is 1% or greater (4). It is suspected that the total patients with PIK3CA mutations currently classified may be higher than reports suggest (due to difficult detection of low levels of mosaicism). If this is true, the number of patients affected by PROS with tumors and the associated tumor risk for this disorder are likely well below the 1% threshold to warrant screening. Additionally, through our experience and discussions with colleagues, we are aware of many unreported patients with molecularly confirmed PROS who have not developed a WT or NR. We suspect that it is likely that the overall risk falls below 1%, indicating that screening is not warranted. It is also possible there are more patients with PROS and NR that have not been reported, as the NR did not progress to NBL or WT requiring treatment. There is a clear need for further publication of known cases and collaboration among institutions, so the denominator of patients with PROS can be further adjusted to understand true WT risk in this population. In terms of current recommendations, tumor screening should be performed at the discretion of the provider based on the genetic change and clinical features of the PROS presentation, as well as the family perspective.
In addition to WT and NR, there are four case reports of patients with PROS who developed other cancers including leukemia, vestibular schwannoma, retinoblastoma, and a meningioma (45–47); however, these do not suggest a specific predisposition or warrant surveillance.
Additional Considerations
Studies on cell-free DNA of urine of patients with PROS found PIK3CA mutations in urine samples of patients who developed renal abnormalities, but not in patients with PROS who did not have a history of kidney irregularities (48, 49). As a result, it has been suggested that urine may be useful in detecting PIK3CA mutations, and those patients with positive results in urine may represent an increased risk for WT development (48). There may be other specific circumstances that could increase tumor risk, such as known PIK3CA-related changes in proximity to the kidneys or patients affected by specific germline or somatic mutations, but further study using larger cohorts is needed to better understand mechanisms and individual risk.
Proteus Syndrome (OMIM 176920)
Overview
PS is caused by postzygotic de novo activating mutations in AKT1 (50). Clinical features of the syndrome include asymmetric skeletal growth, connective tissue nevi, epidermal nevi, vascular malformations, and dysregulated adipose tissue (lipomas, lipohyperplasia, fatty overgrowth, and partial lipohyperplasia) (51). Overlapping disorders, such as CLOVES, under the umbrella of PROS prompted the creation of a new diagnostic scoring system for PS (52). Five points are attributed for cerebriform connective tissue nevus, disproportionate overgrowth, and organ/visceral overgrowth. Two points are attributed for bullae or cysts of the lungs, dysregulated adipose tissue, linear verrucous epidermal nevus, vascular malformations, deep vein thrombosis/pulmonary embolism, and certain facial features, such as dolichocephaly and a low nasal bridge. Single points are attributed for specific tumors including genital cystadenomas, parotid monomorphic adenoma, and meningiomas (52). Points are subtracted for features, such as substantial prenatal extracranial overgrowth and ballooning overgrowth (52).
A diagnosis is confirmed if a patient has a score of 15 or more regardless of the presence of an AKT1 variant. A patient with 10 or more points with an identified mosaic AKT1 variant is considered to have PS. Those with scores between 2 and 9 points with an AKT1 variant are considered to have AKT1-related overgrowth spectrum (AROS) (52).
Molecular Considerations
The AKT1 gene located on chromosome 14q.32.33 is involved in the mTOR pathway that is responsible for regulating cell proliferation and survival (50). Patients with PS have a somatic, activating mutation in this gene that causes the observed abnormal growth. This mutation is not found in blood cells, and therefore a biopsy of the affected skin or tissue is required for a molecular diagnosis.
Tumor Risk in PS
There are currently no tumor screening guidelines for patients with PS. However, a variety of benign and malignant tumors have been reported. Common neoplasms in patients with PS include lipomas, hamartomas, and vascular malformations (53). There have been multiple reports of patients with PS who developed genital cysts as well as meningiomas (52–55). Other case reports of benign tumors include an optic nerve tumor, pinealoma, monomorphic parotid adenoma, intraductal papilloma, goiter, leiomyomas, papillary adenoma of appendix testis, papillary adenoma of kidney, and epibulbar tumor (53, 54, 56–58). Malignant tumors in patients with PS have also been reported. They include papillary thyroid carcinoma, mesothelioma of tunica vaginalis and peritoneal surface, intraductal carcinoma of the breast, endometrial cancer, ovarian carcinoma, and paratesticular ovarian-type papillary serous carcinoma (53, 54, 59–64).
Early mortality in patients with PS is high yet does not appear to be related to the development of cancer (65), as pulmonary embolisms, postoperative embolisms, and pneumonia are responsible for mortality in 20% of patients with PS (51). It is possible that tumor risk is higher in this population, especially benign tumors, but due to the high mortality, an increased tumor risk is not observed.
PTEN Hamartoma Tumor Syndrome
Overview
PHTS is the umbrella term for genetic syndromes caused by germline PTEN mutations. Common clinical features of pediatric patients with PHTS include macrocephaly, hamartomas, lipomas, cardiac defects, and autism (66). LO is due to adipose and vascular anomalies. Major and minor criteria were implemented to aid in diagnosis. Major criteria include the presence of macrocephaly, macular pigmentation of the glans penis, and multiple mucocutaneous lesions, and minor criteria include autism, lipomas, and vascular malformations (66, 67).
Molecular Considerations
PTEN is a tumor suppressor gene on chromosome 10q23 and is also involved in the mTOR signaling pathway (68). Germline mutations of PTEN cause PHTS and have been identified in patients with Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome (69). There have also been case reports of patients with an initial clinical diagnosis of PS, but a PTEN mutation was identified, leading to the term Proteus-like syndrome (70–72).
Tumor Risk in PHTS
The tumor risk in patients with PHTS is well-documented although the syndrome is not typically associated with early childhood cancer risks. Tumors tend to develop in females more than males. The cumulative cancer risk by age 50 for females is 81% and for males is 48% (73).
Malignant tumors commonly observed in patients with PHTS include breast (25–50%), thyroid (3–17%), endometrium (9–27%), melanoma (1–6%), renal (4–16%), and colorectal cancers (3–13%) (73–82). Lhermitte-Duclos disease (LDD) also known as gangliocytoma of the cerebellum is common to develop late in life in patients with germline PTEN mutations (83, 84). Common benign tumors including hamartomas and lipomas can develop in patients at any age and require attention (evaluation and work-up) because of secondary complications that can arise.
There is no international consensus for tumor screening protocols in PHTS. In pediatric patients with PHTS, annual thyroid ultrasounds for thyroid cancer surveillance are recommended although the age to initiate surveillance is debated. The National Comprehensive Cancer Network (NCCN) guidelines for pediatric patients with PHTS include annual thyroid ultrasounds at the time of diagnosis, but Schultz et al. suggest starting ultrasounds at age 7 since the youngest reported case of thyroid cancer in a patient with PHTS was 7 years old (85, 86). In adult patients, colorectal screening beginning at age 40 is recommended (87), and the NCCN guidelines outline additional cancer surveillance recommendations in adults with PHTS.
Discussion
Narrowing the differential diagnosis and attaining confirmatory molecular testing results are critical for patient care management related to LO (Figure 2). The most common disorders and syndromes leading to LO have many overlapping clinical characteristics, making genetic testing useful for determining the underlying mechanism for the observed phenotype. For instance, PHTS is caused by a germline mutation (i.e., the genetic defect is present in every cell of the body), whereas PROS is mostly due to somatic alterations of the PIK3CA gene, leading to a mosaic distribution of the genetic defect throughout the body (i.e., some positive and negative cells). It is suspected that certain regions of the body are more likely to develop tumors if that region contains the genetic defect. If the genetic defect is widespread as it is in germline mutations and constitutional defects, it is logical that the tumor risks may be higher; however, further research is needed to explore this hypothesis.
Figure 2.

Differential diagnosis flowchart of lateralized overgrowth.
From this review, it is evident that there are drastic differences in tumor risks for patients with syndromic LO, some of which warrant childhood tumor surveillance programs and others that do not seem to contribute an increased tumor risk as part of the phenotype (Table 1). It is therefore of utmost importance to correctly diagnose these patients, so they can receive proper screenings and care. Patients with ILO due to increased muscle bulk but without an identifiable genetic cause are now included under the BWSp umbrella and should undergo routine screening like other patients with BWS (17). Given that the guidelines are still being developed for PROS, a discussion with the family about the risk is recommended. In LO disorders with increased tumor risks, the effectiveness of tumor screening goes beyond diagnosing tumors at earlier stages. One study found that parents of patients with elevated tumor risks prefer screening because when educated about their child's risk, it reduced their worry and psychological stress (88).
Table 1.
Summary of tumor risks in genetic and epigenetic syndromes with lateralized overgrowth.
| Genetic cause | Type of overgrowth | Malignant tumors | Tumor risk | Childhood surveillance recommendation(s) | |
|---|---|---|---|---|---|
| PROS | PIK3CA mutations* | Adipose, vascular | Wilms tumor | ~1% | None (to be determined) |
| BWSp | Genetic and epigenetic alterations on chromosome 11p15.5 | Muscular | Wilms tumor Hepatoblastoma (Neuroblastoma) |
0.2–24% 0–3.5% 0.5–4.2% |
Abdominal ultrasound and AFP screening every 3 months until the 4th birthday and renal ultrasounds from the 4th until the 7th birthday |
| PS | AKT1 mutations | Skeletal, adipose, vascular | None | Unknown | None |
| PHTS | PTEN mutations | Adipose, vascular | Breast Thyroid Endometrium Melanoma Kidney Colorectal |
25–50% 3–17% 9–27% 1–6% 4–16% 3–13% |
Annual thyroid ultrasounds beginning at the time of diagnosis |
Majority are somatic mutations, but there have been case reports of patients with germline PIK3CA mutations.
Overall, syndromes involving LO are heterogenous both within a given syndrome and between syndromes. As a result, tumor risk across the spectrum of LO disorders varies greatly due to the underlying cause of the syndrome, as well as personal tumor risk due to specific abnormalities present. Therefore, following diagnostic criteria to diagnosis, each patient will aid in assessing his/her individualized tumor risk and screening program.
Author Contributions
JG performed the literature search and drafted the manuscript and figures. KD helped conceptualize the project, assisted with literature search, and edited the manuscript. JK conceptualized, organized, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank patients and their families for participating in the many studies compiled to present this review. We thank Christina Gonzalez-Gandolfi for helping put together Figure 1. We thank our funding foundation for supporting this work.
Footnotes
Funding. Alex's Lemonade Stand Foundation for Childhood Cancer provided support for this work.
References
- 1.Kalish JM, Biesecker LG, Brioude F, Deardorff MA, Di Cesare-Merlone A, Druley T, et al. Nomenclature and definition in asymmetric regional body overgrowth. Am J Med Genet A. (2017) 173:1735–8. 10.1002/ajmg.a.38266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hennekam RC, Biesecker LG, Allanson JE, Hall JG, Opitz JM, Temple IK, et al. Elements of morphology: general terms for congenital anomalies. Am J Med Genet A. (2013) 161A:2726–33. 10.1002/ajmg.a.36249 [DOI] [PubMed] [Google Scholar]
- 3.Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet. (2005) 137C:53–71. 10.1002/ajmg.c.30064 [DOI] [PubMed] [Google Scholar]
- 4.Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to wilms tumors and hepatoblastoma. Clin Cancer Res. (2017) 23:e115–22. 10.1158/1078-0432.CCR-17-0710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoyme HE, Seaver LH, Jones KL, Procopio F, Crooks W, Feingold M, et al. Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet. (1998) 79:274–8. [DOI] [PubMed] [Google Scholar]
- 6.Clericuzio CL, Chen E, McNeil DE, O'Connor T, Zackai EH, Medne L, et al. Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr. (2003) 143:270–2. 10.1067/S0022-3476(03)00306-8 [DOI] [PubMed] [Google Scholar]
- 7.Mussa A, Duffy KA, Carli D, Griff JR, Fagiano R, Kupa J, et al. The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J Cancer Res Clin Oncol. (2019) 145:3115–23. 10.1007/s00432-019-03038-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mussa A, Molinatto C, Cerrato F, Palumbo O, Carella M, Baldassarre G, et al. Assisted reproductive techniques and risk of Beckwith-Wiedemann syndrome. Pediatrics. (2017) 140:e20164311. 10.1542/peds.2016-4311 [DOI] [PubMed] [Google Scholar]
- 9.Mussa A, Russo S, De Crescenzo A, Freschi A, Calzari L, Maitz S, et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet. (2016) 24:183–90. 10.1038/ejhg.2015.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. (2018) 14:229–49. 10.1038/nrendo.2017.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. (2001) 10:2989–3000. 10.1093/hmg/10.26.2989 [DOI] [PubMed] [Google Scholar]
- 12.Gaston V, Le Bouc Y, Soupre V, Burglen L, Donadieu J, Oro H, et al. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur J Hum Genet. (2001) 9:409–18. 10.1038/sj.ejhg.5200649 [DOI] [PubMed] [Google Scholar]
- 13.Bliek J, Gicquel C, Maas S, Gaston V, Le Bouc Y, Mannens M. Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J Pediatr. (2004) 145:796–9. 10.1016/j.jpeds.2004.08.007 [DOI] [PubMed] [Google Scholar]
- 14.Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, et al. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics. (2014) 6:11. 10.1186/1868-7083-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mussa A, Molinatto C, Baldassarre G, Riberi E, Russo S, Larizza L, et al. Cancer risk in Beckwith-Wiedemann syndrome: a systematic review and meta-analysis outlining a novel (Epi)genotype specific histotype targeted screening protocol. J Pediatr. (2016) 176:142–9.e1. 10.1016/j.jpeds.2016.05.038 [DOI] [PubMed] [Google Scholar]
- 16.Maas SM, Vansenne F, Kadouch DJ, Ibrahim A, Bliek J, Hopman S, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A. (2016) 170:2248–60. 10.1002/ajmg.a.37801 [DOI] [PubMed] [Google Scholar]
- 17.Duffy KA, Cielo CM, Cohen JL, Gonzalez-Gandolfi CX, Griff JR, Hathaway ER, et al. Characterization of the Beckwith-Wiedemann spectrum: diagnosis and management. Am J Med Genet C Semin Med Genet. (2019) 181:693–708. 10.1002/ajmg.c.31740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brioude F, Lacoste A, Netchine I, Vazquez MP, Auber F, Audry G, et al. Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr. (2013) 80:457–65. 10.1159/000355544 [DOI] [PubMed] [Google Scholar]
- 19.Kalish JM, Conlin LK, Mostoufi-Moab S, Wilkens AB, Mulchandani S, Zelley K, et al. Bilateral pheochromocytomas, hemihyperplasia, and subtle somatic mosaicism: the importance of detecting low-level uniparental disomy. Am J Med Genet A. (2013) 161A:993–1001. 10.1002/ajmg.a.35831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keppler-Noreuil KM, Rios JJ, Parker VE, Semple RK, Lindhurst MJ, Sapp JC, et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. (2015) 167A:287–95. 10.1002/ajmg.a.36836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirzaa G, Conway R, Graham JM, Jr, Dobyns WB. PIK3CA-related segmental overgrowth. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle, WA: University of Washington; (2013). [Google Scholar]
- 22.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. (2002) 2:489–501. 10.1038/nrc839 [DOI] [PubMed] [Google Scholar]
- 23.Madsen RR, Vanhaesebroeck B, Semple RK. Cancer-Associated PIK3CA mutations in overgrowth disorders. Trends Mol Med. (2018) 24:856–70. 10.1016/j.molmed.2018.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindhurst MJ, Parker VE, Payne F, Sapp JC, Rudge S, Harris J, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. (2012) 44:928–33. 10.1038/ng.2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. (2007) 143A:2944–58. 10.1002/ajmg.a.32023 [DOI] [PubMed] [Google Scholar]
- 26.Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. (2012) 90:1108–15. 10.1016/j.ajhg.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. (2012) 44:934–40. 10.1038/ng.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovee JV, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. (2015) 166:1048–54.e1–5. 10.1016/j.jpeds.2014.12.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. (2012) 44:941–5. 10.1038/ng.2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet. (2013) 92:76–80. 10.1016/j.ajhg.2012.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirzaa G, Timms AE, Conti V, Boyle EA, Girisha KM, Martin B, et al. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight. (2016) 1:e87623. 10.1172/jci.insight.87623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gripp KW, Baker L, Kandula V, Conard K, Scavina M, Napoli JA, et al. Nephroblastomatosis or Wilms tumor in a fourth patient with a somatic PIK3CA mutation. Am J Med Genet A. (2016) 170:2559–69. 10.1002/ajmg.a.37758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterman CM, Fevurly RD, Alomari AI, Trenor CC. Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr Blood Cancer. (2017) 64:e26684. 10.1002/pbc.26684 [DOI] [PubMed] [Google Scholar]
- 34.Peterman CM, Vadeboncoeur S, Mulliken JB, Fishman SJ, Liang MG. Wilms tumor screening in diffuse capillary malformation with overgrowth and macrocephaly-capillary malformation: a retrospective study. J Am Acad Dermatol. (2017) 77:874–8. 10.1016/j.jaad.2017.06.014 [DOI] [PubMed] [Google Scholar]
- 35.Bove KE, McAdams AJ. The nephroblastomatosis complex and its relationship to Wilms' tumor: a clinicopathologic treatise. Perspect Pediatr Pathol. (1976) 3:185–223. [PubMed] [Google Scholar]
- 36.Machin GA. Persistent renal blastema (nephroblastomatosis) as a frequent precursor of Wilms' tumor; a pathological and clinical review. Part 2. Significance of nephroblastomatosis in the genesis of Wilms' tumor. Am J Pediatr Hematol Oncol. (1980) 2:253–61. [PubMed] [Google Scholar]
- 37.Hucthagowder V, Shenoy A, Corliss M, Vigh-Conrad KA, Storer C, Grange DK, et al. Utility of clinical high-depth next generation sequencing for somatic variant detection in the PIK3CA-related overgrowth spectrum. Clin Genet. (2017) 91:79–85. 10.1111/cge.12819 [DOI] [PubMed] [Google Scholar]
- 38.Kuentz P, St-Onge J, Duffourd Y, Courcet JB, Carmignac V, Jouan T, et al. Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genet Med. (2017) 19:989–97. 10.1038/gim.2016.220 [DOI] [PubMed] [Google Scholar]
- 39.Postema FAM, Hopman SMJ, Deardorff MA, Merks JHM, Hennekam RC. Correspondence to Gripp et al. nephroblastomatosis or Wilms tumor in a fourth patient with a somatic PIK3CA mutation. Am J Med Genet A. (2017) 173:2293–5. 10.1002/ajmg.a.38290 [DOI] [PubMed] [Google Scholar]
- 40.Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VE, Blumhorst C, Darling T, et al. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A. (2014) 164A:1713–33. 10.1002/ajmg.a.36552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mankad VN, Gray GFJr, Miller DR. Bilateral nephroblastomatosis and Klippel Trenaunay syndrome. Cancer. (1974) 33:1462–7. [DOI] [PubMed] [Google Scholar]
- 42.Ehrich JH, Ostertag H, Flatz S, Kamran D. Bilateral Wilms's tumour in Klippel-Trenaunay syndrome. Arch Dis Child. (1979) 54:405. 10.1136/adc.54.5.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lapunzina P, Gairi A, Delicado A, Mori MA, Torres ML, Goma A, et al. Macrocephaly-cutis marmorata telangiectatica congenita: report of six new patients and a review. Am J Med Genet A. (2004) 130A:45–51. 10.1002/ajmg.a.30235 [DOI] [PubMed] [Google Scholar]
- 44.Wright DR, Frieden IJ, Orlow SJ, Shin HT, Chamlin S, Schaffer JV, et al. The misnomer “macrocephaly-cutis marmorata telangiectatica congenita syndrome”: report of 12 new cases and support for revising the name to macrocephaly-capillary malformations. Arch Dermatol. (2009) 145:287–93. 10.1001/archdermatol.2008.545 [DOI] [PubMed] [Google Scholar]
- 45.Moore CA, Toriello HV, Abuelo DN, Bull MJ, Curry CJ, Hall BD, et al. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet. (1997) 70:67–73. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz IV, Felix TM, Riegel M, Schuler-Faccini L. Atypical macrocephaly-cutis marmorata telangiectatica congenita with retinoblastoma. Clin Dysmorphol. (2002) 11:199–202. 10.1097/00019605-200207000-00010 [DOI] [PubMed] [Google Scholar]
- 47.Mills JR, Moyer AM, Kipp BR, Poplawski AB, Messiaen LM, Babovic-Vuksanovic D. Unilateral vestibular schwannoma and meningiomas in a patient with PIK3CA-related segmental overgrowth: co-occurrence of mosaicism for 2 rare disorders. Clin Genet. (2018) 93:187–90. 10.1111/cge.13099 [DOI] [PubMed] [Google Scholar]
- 48.Biderman Waberski M, Lindhurst M, Keppler-Noreuil KM, Sapp JC, Baker L, Gripp KW, et al. Urine cell-free DNA is a biomarker for nephroblastomatosis or Wilms tumor in PIK3CA-related overgrowth spectrum (PROS). Genet Med. (2018) 20:1077–81. 10.1038/gim.2017.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel ME, Konczyk DJ, Yeung KS, Murillo R, Vivero MP, Hall AM, et al. Causal somatic mutations in urine DNA from persons with the CLOVES subgroup of the PIK3CA-related overgrowth spectrum. Clin Genet. (2018) 93:1075–80. 10.1111/cge.13195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. (2011) 365:611–9. 10.1056/NEJMoa1104017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen MM, Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. (2005) 137C:38–52. 10.1002/ajmg.c.30063 [DOI] [PubMed] [Google Scholar]
- 52.Sapp JC, Buser A, Burton-Akright J, Keppler-Noreuil KM, Biesecker LG. A dyadic genotype-phenotype approach to diagnostic criteria for Proteus syndrome. Am J Med Genet C Semin Med Genet. (2019) 181:565–70. 10.1002/ajmg.c.31744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen MM, Jr. Proteus syndrome: clinical evidence for somatic mosaicism and selective review. Am J Med Genet. (1993) 47:645–52. 10.1002/ajmg.1320470514 [DOI] [PubMed] [Google Scholar]
- 54.Gordon PL, Wilroy RS, Lasater OE, Cohen MM, Jr. Neoplasms in Proteus syndrome. Am J Med Genet. (1995) 57:74–8. 10.1002/ajmg.1320570117 [DOI] [PubMed] [Google Scholar]
- 55.Keppler-Noreuil KM, Baker EH, Sapp JC, Lindhurst MJ, Biesecker LG. Somatic AKT1 mutations cause meningiomas colocalizing with a characteristic pattern of cranial hyperostosis. Am J Med Genet A. (2016) 170:2605–10. 10.1002/ajmg.a.37737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishimura G, Kozlowski K. Proteus syndrome (report of three cases). Australas Radiol. (1990) 34:47–52. 10.1111/j.1440-1673.1990.tb02807.x [DOI] [PubMed] [Google Scholar]
- 57.Zachariou Z, Krug M, Benz G, Daum R. Proteus syndrome associated with a sacrococcygeal teratoma; a rare combination. Eur J Pediatr Surg. (1996) 6:249–51. 10.1055/s-2008-1066522 [DOI] [PubMed] [Google Scholar]
- 58.Trivedi D, Lee SY, Brundler MA, Parulekar MV. Fibrous tumor of the superior oblique tendon in Proteus syndrome. J AAPOS. (2013) 17:420–2. 10.1016/j.jaapos.2013.03.019 [DOI] [PubMed] [Google Scholar]
- 59.Hornstein L, Bove KE, Towbin RB. Linear nevi, hemihypertrophy, connective tissue hamartomas, and unusual neoplasms in children. J Pediatr. (1987) 110:404–8. 10.1016/S0022-3476(87)80502-4 [DOI] [PubMed] [Google Scholar]
- 60.Doucet ME, Bloomhardt HM, Moroz K, Lindhurst MJ, Biesecker LG. Lack of mutation-histopathology correlation in a patient with Proteus syndrome. Am J Med Genet A. (2016) 170:1422–32. 10.1002/ajmg.a.37612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iqbal J, He G, Biesecker LG, Rosen P, Duray PH, Schwartzentruber D, et al. Morphological characterization of the breast in Proteus syndrome complicated by ductal carcinoma in situ. Ann Clin Lab Sci. (2006) 36:469–74. [PubMed] [Google Scholar]
- 62.Leoni C, Gullo G, Resta N, Fagotti A, Onesimo R, Schwartz B, et al. First evidence of a therapeutic effect of miransertib in a teenager with Proteus syndrome and ovarian carcinoma. Am J Med Genet A. (2019) 179:1319–24. 10.1002/ajmg.a.61160 [DOI] [PubMed] [Google Scholar]
- 63.Sadaf A, Nguyen A, Meyers AB, Nguyen CK, Wehry MA, Berkow R. Patient with Proteus syndrome and paratesticular ovarian-type papillary serous carcinoma. Pediatr Blood Cancer. (2018) 65:e27099. 10.1002/pbc.27099 [DOI] [PubMed] [Google Scholar]
- 64.Virgone C, Alaggio R, Dall'Igna P, Buffa P, Tonegatti L, Ferrari A, et al. Epithelial tumors of the ovary in children and teenagers: a prospective study from the Italian TREP project. J Pediatr Adolesc Gynecol. (2015) 28:441–6. 10.1016/j.jpag.2014.12.010 [DOI] [PubMed] [Google Scholar]
- 65.Sapp JC, Hu L, Zhao J, Gruber A, Schwartz B, Ferrari D, et al. Quantifying survival in patients with Proteus syndrome. Genet Med. (2017) 19:1376–79. 10.1038/gim.2017.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macken WL, Tischkowitz M, Lachlan KL. PTEN Hamartoma tumor syndrome in childhood: a review of the clinical literature. Am J Med Genet C Semin Med Genet. (2019) 181:591–610. 10.1002/ajmg.c.31743 [DOI] [PubMed] [Google Scholar]
- 67.Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet. (2011) 48:505–12. 10.1136/jmg.2011.088807 [DOI] [PubMed] [Google Scholar]
- 68.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. (1997) 275:1943–7. 10.1126/science.275.5308.1943 [DOI] [PubMed] [Google Scholar]
- 69.Eng C. PTEN: one gene, many syndromes. Hum Mutat. (2003) 22:183–98. 10.1002/humu.10257 [DOI] [PubMed] [Google Scholar]
- 70.Zhou X, Hampel H, Thiele H, Gorlin RJ, Hennekam RC, Parisi M, et al. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet. (2001) 358:210–1. 10.1016/S0140-6736(01)05412-5 [DOI] [PubMed] [Google Scholar]
- 71.Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. (2000) 9:765–8. 10.1093/hmg/9.5.765 [DOI] [PubMed] [Google Scholar]
- 72.Smith JM, Kirk EP, Theodosopoulos G, Marshall GM, Walker J, Rogers M, et al. Germline mutation of the tumour suppressor PTEN in Proteus syndrome. J Med Genet. (2002) 39:937–40. 10.1136/jmg.39.12.937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. (2013) 50:255–63. 10.1136/jmedgenet-2012-101339 [DOI] [PubMed] [Google Scholar]
- 74.Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns. (2009) 18:13–27. 10.1007/s10897-008-9187-7 [DOI] [PubMed] [Google Scholar]
- 75.Riegert-Johnson DL, Gleeson FC, Roberts M, Tholen K, Youngborg L, Bullock M, et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract. (2010) 8:6. 10.1186/1897-4287-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. (2010) 139:1927–33. 10.1053/j.gastro.2010.06.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. (2012) 18:400–7. 10.1158/1078-0432.CCR-11-2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Milas M, Mester J, Metzger R, Shin J, Mitchell J, Berber E. Should patients with Cowden syndrome undergo prophylactic thyroidectomy? Surgery. (2012) 152:1201–10. 10.1016/j.surg.2012.08.055 [DOI] [PubMed] [Google Scholar]
- 79.Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. (2013) 163C:114–21. 10.1002/ajmg.c.31364 [DOI] [PubMed] [Google Scholar]
- 80.Shuch B, Ricketts CJ, Vocke CD, Komiya T, Middelton LA, Kauffman EC. Germline PTEN mutation Cowden syndrome: an underappreciated form of hereditary kidney cancer. J Urol. (2013) 190:1990–8. 10.1016/j.juro.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nieuwenhuis MH, Kets CM, Murphy-Ryan M, Yntema HG, Evans DG, Colas C, et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer. (2014) 13:57–63. 10.1007/s10689-013-9674-3 [DOI] [PubMed] [Google Scholar]
- 82.Yakubov E, Ghoochani A, Buslei R, Buchfelder M, Eyupoglu IY, Savaskan N. Hidden association of Cowden syndrome, PTEN mutation and meningioma frequency. Oncoscience. (2016) 3:149–55. 10.18632/oncoscience.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Padberg GW, Schot JD, Vielvoye GJ, Bots GT, de Beer FC. Lhermitte-Duclos disease and Cowden disease: a single phakomatosis. Ann Neurol. (1991) 29:517–23. 10.1002/ana.410290511 [DOI] [PubMed] [Google Scholar]
- 84.Jiang T, Wang J, Du J, Luo S, Liu R, Xie J. Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum) and Cowden syndrome: clinical experience from a single institution with long-term follow-up. World Neurosurg. (2017) 104:398–406. 10.1016/j.wneu.2017.04.147 [DOI] [PubMed] [Google Scholar]
- 85.Schultz KAP, Rednam SP, Kamihara J, Doros L, Achatz MI, Wasserman JD. PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res. (2017) 23:e76–82. 10.1158/1078-0432.CCR-17-0629 [DOI] [PubMed] [Google Scholar]
- 86.Smith JR, Marqusee E, Webb S, Nose V, Fishman SJ, Shamberger RC, et al. Thyroid nodules and cancer in children with PTEN hamartoma tumor syndrome. J Clin Endocrinol Metab. (2011) 96:34–7. 10.1210/jc.2010-1315 [DOI] [PubMed] [Google Scholar]
- 87.Nieuwenhuis MH, Kets CM, Murphy-Ryan M, Colas C, Moller P, Hes FJ, et al. Is colorectal surveillance indicated in patients with PTEN mutations? Colorectal Dis. (2012) 14:e562–6. 10.1111/j.1463-1318.2012.03121.x [DOI] [PubMed] [Google Scholar]
- 88.Duffy KA, Grand KL, Zelley K, Kalish JM. Tumor screening in Beckwith-Wiedemann syndrome: parental perspectives. J Genet Couns. (2018) 27:844–53. 10.1007/s10897-017-0182-8 [DOI] [PMC free article] [PubMed] [Google Scholar]