

Abstract
Patient: Female, 62-year-old
Final Diagnosis: Medullary thyroid microcarcinoma
Symptoms: No pain or swelling in her neck • no dysphagia or odynophagia • no changes in voice
Medication: —
Clinical Procedure: Genetic analysis
Specialty: Genetics
Objective:
Unusual clinical course
Background:
RET p.V804M is a known activating oncogenic variant that confers an increased risk for medullary thyroid carcinoma (MTC). Based on age-specific penetrance, the American Thyroid Association (ATA) categorizes this variant as posing moderate risk. Therefore, ATA guidelines endorse prophylactic thyroidectomy for carriers in childhood (by age 5–10 years) or adulthood, or when the serum calcitonin level becomes elevated. The recommendation for thyroidectomy is increasingly controversial due to the recently reported low penetrance of the RET p.V804M variant in a large unbiased ascertainment cohort.
Case Report:
We describe the unexpected identification of this variant in a 62-year-old woman undergoing broad, multigene cancer panel testing for her personal and family history of breast cancer. There was no known family history of MTC. Biochemical screening prompted by the RET p.V804M result revealed a mildly elevated serum calcitonin. Pathology examination of her thyroidectomy specimen revealed multifocal medullary thyroid microcarcinoma; her sibling’s prophylactic thyroidectomy after a RET p.V840M-positive result similarly revealed early-stage MTC.
Conclusions:
This report demonstrates the value of genetic counseling, shared decision-making, cascade testing, and timely thyroidectomy in the management of a patient with an unexpected RET p.V804M result.
MeSH Keywords: Multiple Endocrine Neoplasia Type 2a, Proto-Oncogene Proteins c-ret, Thyroid Neoplasms, Thyroidectomy
Background
Medullary thyroid carcinoma (MTC) is an aggressive malignancy of parafollicular C cells of the thyroid gland connective tissue, resulting in abnormal production of calcitonin hormone. MTC accounts for 5%–8% of all thyroid cancers [1]. While most cases are sporadic, 25% of MTCs occur in patients with familial multiple endocrine neoplasia type 2 (MEN2) syndrome. MEN2 has 3 distinct phenotypes: MEN2A, MEN2B, and familial MTC. MEN2A and familial MTC have an autosomal dominant pattern of inheritance characterized by the presence of heterozygous pathogenic variants in the RET proto-oncogene. The RET gene encodes a transmembrane receptor protein kinase. Oncogenic activating variants in specific RET codons are associated with different MEN2 phenotypes [2,3].
The p.V804M variant is the most common pathogenic variant (PV) in RET associated with MTC [4]. The 2015 revised MTC Clinical Guidelines by the American Thyroid Association (ATA) placed RET p.V804M in the moderate-risk category, with the recommendation of thyroidectomy in heterozygous carriers in childhood (by 5–10 years of age) or adulthood, or when the serum calcitonin level becomes elevated [5]. Estimates of age-specific MTC penetrance vary widely. In a study of 160 subjects with PVs in codon 804 ascertained through family cancer history who underwent pathologic or clinical evaluation, MTC penetrance was reported to be approximately 85% by age 70 [6]. In contrast, a more recent study of MTC in a large, unbiased ascertainment cohort from the Exome Aggregation Consortium (ExAC) dataset (n=51 000) estimated the lifetime penetrance to be only 4% [4]. In that study, penetrance was calculated using conservative, best-baseline estimates (MTC lifetime risk, 1 in 3000; genetic heterogeneity, 20%; allelic heterogeneity, 25%). Based on their results, the authors contended that prophylactic thyroidectomy may be inappropriate for individuals with an incidental RET p.V804M finding and no apparent family history of disease, and they therefore called for urgent review of the ATA guidelines [4].
Herein, we present our experience with and clinical management of a patient with breast cancer who had an unexpected finding of the RET p.V804M variant. The finding led to screening, surgical management, and diagnosis of MTC.
Case Report
A 62-year-old woman with a personal and family history of breast cancer was referred to the Division of Cancer Genetics and Prevention at Dana-Farber Cancer Institute for evaluation. One year prior to her visit, she had been diagnosed with multifocal, ER-positive, PR-positive, and HER-2 negative invasive lobular carcinoma of the right breast. She was treated with curative intent with a unilateral mastectomy and aromatase inhibitor systemic therapy.
Reported family history was notable for the proband’s mother having multiple primary cancers, including breast cancer at age 49, colorectal cancer at age 60, and leukemia at age 65 (Figure 1). There was no reported family history of thyroid cancer, calcium disorder, or pheochromocytoma. The patient’s maternal ancestry is English and French, and her paternal ancestry is Czech, with no known Ashkenazi Jewish ancestry or consanguinity. Given her personal and family breast cancer histories, the patient was counseled and consented to genetic testing analyzing 84 cancer susceptibility genes (Invitae, CA, USA). Results were positive for the heterozygous pathogenic variant RET p.V804M (NM_020975.6: c.2410G>A) (ClinVar ID: 37102). The remainder of the genetic testing was unremarkable.
Figure 1.
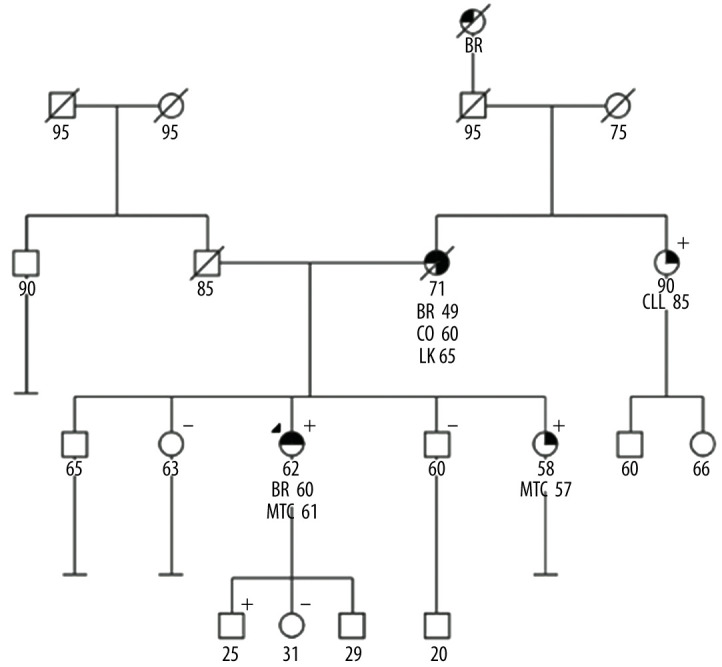
Pedigree of a family with RET p.V804M variant. The pedigree depicts individuals with cancer, cancer types, and RET p.V804M genotype when available. Positive and negative signs denote presence and absence of RET p.V804M, respectively. Circles and squares denote female and male family members, respectively. Arrow indicates proband. Numbers under circles or squares reflect current age or age at death if deceased, and numbers beside diagnosis reflect age at diagnosis. Filled quadrants denote affected conditions. BR – breast cancer; CO – colon cancer; MTC – medullary thyroid cancer; CLL – chronic lymphoid leukemia.
Given the association of RET p.V804M with MTC, the patient was counseled about the associated cancer risks. Both the historical lifetime MTC penetrance estimates of 80–90% and the more recently published attenuated risks of 4% were discussed. The low risks for other tumors, specifically pheochromocytoma and parathyroid adenoma, were reviewed. Risk-reducing thyroidectomy was discussed as the historical cornerstone of management for RET activating variants due to incomplete sensitivity of serum calcitonin markers and aggressiveness of MTC. Additionally, cascade testing for family members was recommended.
The patient underwent thyroid oncology evaluation. She was asymptomatic at the time with no pain or swelling in her neck, no dysphagia or odynophagia, and no changes in voice. Physical examination was normal with no evidence of enlarged thyroid, palpable lymph nodes, or thyroid masses. A thyroid ultrasound and biochemical screening were obtained. The ultrasound results were normal (with no thyroid nodules or abnormal adenopathy) as were plasma metanephrine, parathyroid hormone (PTH), serum calcium, and carcinoembryonic antigen (CEA). Calcitonin (electrochemiluminescence assay by Roche Diagnostics Inc., IN, USA) was mildly elevated at 9.9 pg/mL (normal <7.6 pg/mL). The calcitonin remained elevated 2 weeks later at 10.4 pg/mL.
The patient underwent surgical evaluation and, after careful consideration of the surgical risks and benefits, proceeded with thyroidectomy. She had an uncomplicated total thyroidectomy with no evidence of extrathyroidal invasion. There were no grossly abnormal lymph nodes. The left upper and bilateral lower parathyroid glands were preserved on their normal blood supplies. The right upper parathyroid gland was devascularized during dissection and was reimplanted into the right sternocleidomastoid muscle. The bilateral recurrent laryngeal nerves were dissected. They were left intact and with normal electromyography signals with stimulation of the ipsilateral vagus nerve at the outset and completion of all dissection.
Pathology examination of the specimen identified 4 foci of medullary thyroid microcarcinomas (largest measuring 0.2 cm) in the right mid-upper and left mid-upper lobes (Figure 2). No lymphovascular invasion and no extrathyroidal extension were identified. Background thyroid tissue showed C-cell hyperplasia, highlighted by calcitonin stains (Figure 2).
Figure 2.

Histologic findings in the thyroidectomy specimen. (A) An example of a medullary microcarcinoma (H&E stain) at ×100 magnification. (B) C-cell hyperplasia (H&E stain) at ×400 magnification. (C) Calcitonin stain highlights the C cells at ×400 magnification. H&E, hematoxylin and eosin.
The patient’s course was complicated by postoperative hypocalcemia with hypoparathyroidism (calcium of 8.2 mg/dL with PTH of 5 pg/mL). She was weaned off calcitriol, and she is maintained on calcium citrate 950 mg (200 mg elemental) 3 times a day and levothyroxine 100 μg daily. Follow-up laboratory results were normal as follows: calcitonin undetectable, calcium 9.2 mg/dL, PTH 14 pg/mL, and thyrotropin 1.94 μIU/mL.
One of the patient’s sisters (age 57) tested positive for the RET p.V804M variant and was found to have elevated calcitonin. She likewise proceeded with thyroidectomy, and pathology also revealed multifocal MTC. As per ATA guidelines, both the patient and her sister will be followed annually with physical evaluation of the neck and thyroid bed, ultrasound of the neck, and blood tests (free thyroxine, thyrotropin, calcitonin, and CEA). In addition, they will also need to undergo annual surveillance for pheochromocytoma with plasma metanephrines and primary hyperparathyroidism. The patient’s maternal aunt (age 90) and an adult child (age 25) were subsequently also found to have the RET p.V804M allele.
Discussion
RET is a proto-oncogene that maps on 10q11.21 and encodes a transmembrane receptor tyrosine protein kinase. Activating variants render the protein oncogenic as the key alteration in both sporadic and inherited MTC.
The p.V804M variant is the most frequent pathogenic variant in the RET gene associated with MTC [4]. The age-related penetrance and expressivity of RET p.V804M is well documented, with clear association with later age of MTC onset [7,8]. There are also reports of kindreds, including large consanguineous families, in which only individuals homozygous for the RET p.V804M variant, and not the heterozygous carriers, presented clinically with MTC [9,10]. Other studies have reported C-cell hyperplasia as the sole finding in thyroidectomy specimens of RET p.V804M carriers [8,11–13]. Notably, in hereditary MTC families, C-cell hyperplasia precedes MTC [14]. The variable penetrance and expressivity of RET p.V804M suggest a possible role for unlinked genetic modifiers, epigenetic changes, or environmental factors related to MTC.
Data from retrospective analysis of 140 cases of MTC have shown that the majority of MTC cases (56%) were diagnosed at stage IV, while only 19% of patient were diagnosed at stage I, 15% at stage II, and 10% in stage III [15]. The 10-year survival rates for patients with stages I, II, III, and IV MTC are reported to be 100%, 93%, 71%, and 21%, respectively [5,16]. A recent Danish nationwide retrospective study of MTC patients (diagnosed with hereditary MTC by screening, without regional metastases, or with stage I-III disease) had similar survival to the general population. However, the presence of distant metastasis resulted in a worse outcome [17]. With late tumor stage being a reliable predictor of survival in MTC patients [18], the efforts to identify at-risk patients and manage risk remain important.
Risk stratification of MEN2 is by the RET genotype [4,5,7]. The 2015 revised ATA guidelines on diagnosis and clinical management of MTC categorizes RET PVs as “moderate,” “high,” or “highest” levels for risk associated with MTC [5]. A retrospective cohort study using an institutional MEN2 registry reported that patients with moderate- and high-risk germline RET PVs had similar time to distant metastatic disease after MTC diagnosis [19]. This finding led the authors to propose RET PV classification by disease age of onset, rather than by risk level (high vs. moderate). The current ATA guidelines report that RET p.V804M is associated with moderate risk, with recommendations for thyroidectomy in carriers in childhood (by 5 or 10 years old) or adulthood or when serum calcitonin level becomes elevated [5].
Measurement of serum calcitonin has been used as an important tumor marker in patients with MTC. However, consensus on its role in the evaluation of a thyroid nodule remains elusive [20]. Studies have reported negative calcitonin in nonsecretory MTC [21], indicating that normal serum calcitonin does not exclude the presence of MTC [22,23]. In a study evaluating 30 family members spanning 3 generations of a Turkish family, 17 members were positive for RET p.V804M. Of the 7 with an MTC diagnosis, only 3 had abnormally elevated calcitonin levels [24]. Further, hypercalcitoninemia has also been reported to be associated with C-cell hyperplasia and not MTC [25,26], and false-positive calcitonin results have reportedly led to unnecessary thyroidectomies [27].
Based on the estimated penetrance of RET p.V804M from the ExAC dataset, the allele contribution of RET p.V804M in MTC has recently been questioned. In that report, unbiased by ascertainment, the penetrance of this allele was estimated to be 4% [4]. Given the low estimated penetrance of RET p.V804M in conjunction with the potential expansion of incidental findings of this variant in nonpenetrant families due to large-scale sequencing efforts, Rich et a.l [6] call for reconsideration of the ATA recommendations for prophylactic thyroidectomy. RET is one of the 59 genes recommended for reporting as an incidental finding in clinical exome or genome sequencing by the American College of Medical Genetics and Genomics [28].
Our group and others have previously reported incidental findings in which the majority of pathogenic or presumed pathogenic cancer variants in various studies were unexpectedly discovered in genes not directly related to the patient’s primary indication [29–31]. Indeed, in this era of large-scale genome sequencing, incidental or unexpected findings involving cancer genes will only increase. Reexamination of classic phenotypes and penetrance across the cancer predisposition syndrome spectrum is required and is occurring for genes including TP53 [32] and CDH1 [33,34].
Conclusions
We have described the early diagnosis of medullary thyroid microcarcinomas through the unexpected finding of the RET p.V804M oncogenic variant precipitated by a patient’s breast cancer and extensive family cancer history. Cascade testing resulted in the early diagnosis of multifocal MTC in the patient’s sister. Management of other relatives found to share this risk allele will likely be informed by the presence of MTC in the proband and her sister.
As the epidemiology of various cancer predisposition syndromes is re-evaluated and guidelines for management reconsidered, shared decision-making with the patient is critical. In this case, patient-specific factors were important in the management of this unexpected cancer predisposition.
Patient-specific factors were good overall health and excellent breast cancer prognosis. Variant-specific factors included the attenuated penetrance in individuals not ascertained through MTC families. Disease-specific factors included sensitivity and specificity of biochemical surveillance. The patient preferred surgical management due to low risk tolerance secondary to prior malignancy and tolerability of the health consequences of surgical intervention.
This report highlights the value of shared decision-making and cascade genetic testing in the clinical management of a patient who was unexpectedly revealed to be a carrier of the RET p.V804M variant.
Acknowledgments
We would like to thank the patient and her family members for permitting us to anonymously share the clinical details in this case report.
Footnotes
Conflict of interest
None.
References:
- 1.Pacini F, Castagna MG, Cipri C, Schlumberger M. Medullary thyroid carcinoma. Clin Oncol (R Coll Radiol) 2010;22(6):475–85. doi: 10.1016/j.clon.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Wagner SM, Zhu S, Nicolescu AC, Mulligan LM. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2. Clinics (Sao Paulo) 2012;67(Suppl. 1):77–84. doi: 10.6061/clinics/2012(Sup01)14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276(19):1575–79. [PubMed] [Google Scholar]
- 4.Loveday C, Josephs K, Chubb D, et al. p.Val804Met, the most frequent pathogenic mutation in RET, confers a very low lifetime risk of medullary thyroid cancer. J Clin Endocrinol Metab. 2018;103(11):4275–82. doi: 10.1210/jc.2017-02529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wells SA, Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567–610. doi: 10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rich TA, Feng L, Busaidy N, et al. Prevalence by age and predictors of medullary thyroid cancer in patients with lower risk germline RET proto-onco-gene mutations. Thyroid. 2014;24(7):1096–106. doi: 10.1089/thy.2013.0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee S, Zakalik D. RET codon 804 mutations in multiple endocrine neoplasia 2: Genotype-phenotype correlations and implications in clinical management. Clin Genet. 2011;79(1):1–16. doi: 10.1111/j.1399-0004.2010.01453.x. [DOI] [PubMed] [Google Scholar]
- 8.Feldman GL, Edmonds MW, Ainsworth PJ, et al. Variable expressivity of familial medullary thyroid carcinoma (FMTC) due to a RET V804M (GTG-->ATG) mutation. Surgery. 2000;128(1):93–98. doi: 10.1067/msy.2000.107103. [DOI] [PubMed] [Google Scholar]
- 9.Lecube A, Hernandez C, Oriola J, et al. V804M RET mutation and familial medullary thyroid carcinoma: Report of a large family with expression of the disease only in the homozygous gene carriers. Surgery. 2002;131(5):509–14. doi: 10.1067/msy.2002.123006. [DOI] [PubMed] [Google Scholar]
- 10.Lesueur F, Cebrian A, Cranston A, et al. Germline homozygous mutations at codon 804 in the RET protooncogene in medullary thyroid carcinoma/multiple endocrine neoplasia type 2A patients. J Clin Endocrinol Metab. 2005;90(6):3454–57. doi: 10.1210/jc.2004-1622. [DOI] [PubMed] [Google Scholar]
- 11.Learoyd DL, Gosnell J, Elston MS, et al. Experience of prophylactic thyroidectomy in multiple endocrine neoplasia type 2A kindreds with RET codon 804 mutations. Clin Endocrinol (Oxf) 2005;63(6):636–41. doi: 10.1111/j.1365-2265.2005.02394.x. [DOI] [PubMed] [Google Scholar]
- 12.Karrasch T, Herbst SM, Hehr U, et al. How to assess the clinical relevance of novel RET missense variants in the absence of functional studies? Eur Thyroid J. 2016;5(1):73–77. doi: 10.1159/000443730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frohnauer MK, Decker RA. Update on the MEN 2A c804 RET mutation: is prophylactic thyroidectomy indicated? Surgery. 2000;128(6):1052–58. doi: 10.1067/msy.2000.11/6/111080. [DOI] [PubMed] [Google Scholar]
- 14.Wolfe HJ, Melvin KE, Cervi-Skinner SJ, et al. C-cell hyperplasia preceding medullary thyroid carcinoma. N Engl J Med. 1973;289(9):437–41. doi: 10.1056/NEJM197308302890901. [DOI] [PubMed] [Google Scholar]
- 15.Simoes-Pereira J, Bugalho MJ, Limbert E, Leite V. Retrospective analysis of 140 cases of medullary thyroid carcinoma followed-up in a single institution. Oncol Lett. 2016;11(6):3870–74. doi: 10.3892/ol.2016.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Modigliani E, Cohen R, Campos JM, et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: Results in 899 patients. The GETC Study Group. Groupe d’etude des tumeurs a calcitonine. Clin Endocrinol (Oxf) 1998;48(3):265–73. doi: 10.1046/j.1365-2265.1998.00392.x. [DOI] [PubMed] [Google Scholar]
- 17.Mathiesen JS, Kroustrup JP, Vestergaard P, et al. Survival and long-term biochemical cure in medullary thyroid carcinoma in Denmark 1997–2014: A nationwide study. Thyroid. 2019;29(3):368–77. doi: 10.1089/thy.2018.0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rios A, Rodriguez JM, Febrero B, et al. [Prognostic value of clinical, histo-pathological and immunohistochemical features in medullary thyroid cancer.] Med Clin (Barc) 2012;139(7):277–83. doi: 10.1016/j.medcli.2011.07.021. [in Spanish] Erratum in: Med Clin (Barc). 2014; 142(5): 233 [in Spanish] [DOI] [PubMed] [Google Scholar]
- 19.Voss RK, Feng L, Lee JE, et al. Medullary thyroid carcinoma in MEN2A: ATA moderate- or high-risk RET mutations do not predict disease aggressiveness. J Clin Endocrinol Metab. 2017;102(8):2807–13. doi: 10.1210/jc.2017-00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baetu M, Dobrescu R. Novel markers for early diagnosis and prognostic classification in medullary thyroid carcinoma. Acta Endocrinol (Buchar) 2017;13(4):519–22. doi: 10.4183/aeb.2017.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gambardella C, Offi C, Patrone R, et al. Calcitonin negative medullary thyroid carcinoma: A challenging diagnosis or a medical dilemma? BMC Endocr Disord. 2019;19(Suppl. 1):45. doi: 10.1186/s12902-019-0367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy M, Chen H, Sippel RS. Current understanding and management of medullary thyroid cancer. Oncologist. 2013;18(10):1093–100. doi: 10.1634/theoncologist.2013-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trimboli P, Giovanella L. Serum calcitonin negative medullary thyroid carcinoma: A systematic review of the literature. Clin Chem Lab Med. 2015;53(10):1507–14. doi: 10.1515/cclm-2015-0058. [DOI] [PubMed] [Google Scholar]
- 24.Basaran MN, Tuna MM, Karakilic E, et al. Characterization of V804M-mutated RET proto-oncogene associated with familial medullary thyroid cancer, report of the largest Turkish family. J Endocrinol Invest. 2015;38(5):541–46. doi: 10.1007/s40618-014-0224-0. [DOI] [PubMed] [Google Scholar]
- 25.Allelein S, Ehlers M, Morneau C, et al. Measurement of basal serum calcitonin for the diagnosis of medullary thyroid cancer. Horm Metab Res. 2018;50(1):23–28. doi: 10.1055/s-0043-122237. [DOI] [PubMed] [Google Scholar]
- 26.Kakita-Kobayashi M, Ueda Y, Tanase-Nakao K, et al. A case of C-cell hyperplasia in an asymptomatic V804M Ret mutation carrier: Can the calcium infusion test predict C-cell hyperplasia? AACE Clin Case Rep. 2015;1(2):e92–95. [Google Scholar]
- 27.Toledo SP, Lourenco DM, Jr, Santos MA, et al. Hypercalcitoninemia is not pathognomonic of medullary thyroid carcinoma. Clinics (Sao Paulo) 2009;64(7):699–706. doi: 10.1590/S1807-59322009000700015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 29.Ghazani AA, Oliver NM, St Pierre JP, et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet Med. 2017;19(7):787–95. doi: 10.1038/gim.2016.191. [DOI] [PubMed] [Google Scholar]
- 30.Aguirre AJ, Nowak JA, Camarda ND, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 2018;8(9):1096–111. doi: 10.1158/2159-8290.CD-18-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs. guideline-based germline testing. JAMA. 2017;318(9):825–35. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rana HQ, Gelman R, LaDuca H, et al. Differences in TP53 mutation carrier phenotypes emerge from panel-based testing. J Natl Cancer Inst. 2018;110(8):863–70. doi: 10.1093/jnci/djy001. [DOI] [PubMed] [Google Scholar]
- 33.Roberts ME, Ranola JMO, Marshall ML, et al. Comparison of CDH1 penetrance estimates in clinically ascertained families vs. families ascertained for multiple gastric cancers. JAMA Oncol. 2019;5(9):1325–31. doi: 10.1001/jamaoncol.2019.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xicola RM, Li S, Rodriguez N, et al. Clinical features and cancer risk in families with pathogenic CDH1 variants irrespective of clinical criteria. J Med Genet. 2019;56(12):838–43. doi: 10.1136/jmedgenet-2019-105991. [DOI] [PubMed] [Google Scholar]