

Abstract
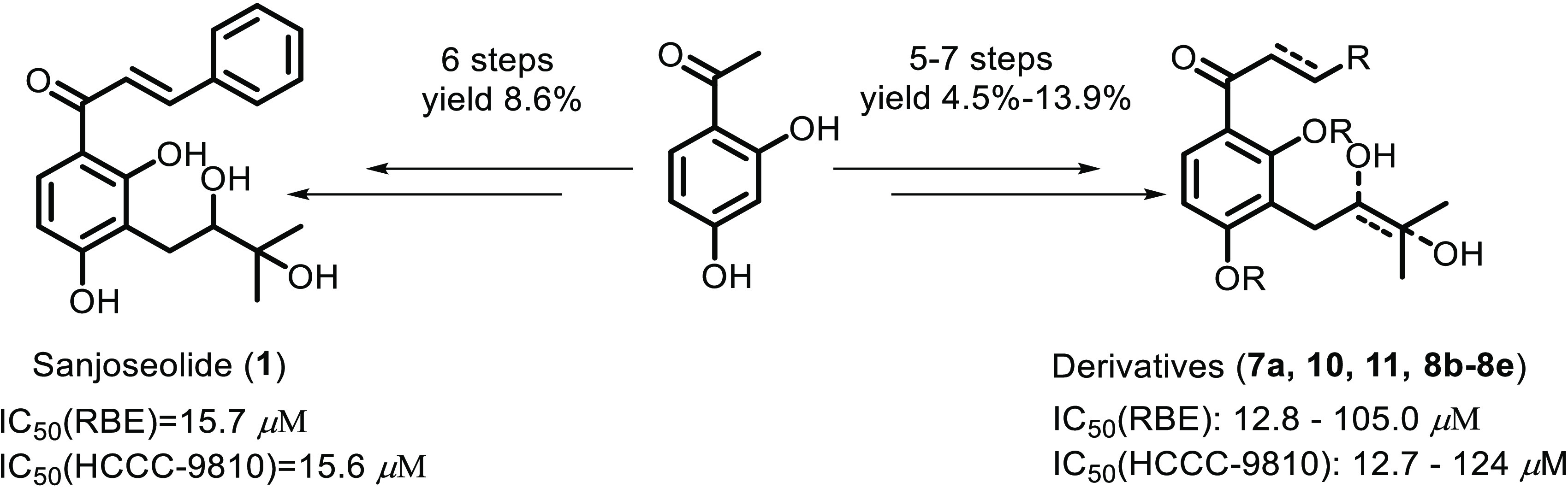
The first total synthesis of sanjoseolide (1), which was originally obtained from Dalea frutescens A, was achieved via an efficient route with a longest linear sequence of six steps from the commercially available 2,4-dihydroxyacetophenone in 8.6% overall yield. Meanwhile, a series of sanjoseolide representative analogues were synthesized and assessed for their antiproliferative potency against cancer cells of different origins. Compound 8e inhibited the survival of all tested cancer cell lines in a dose-dependent manner, the IC50 values of the treatment were about 12.8 μM for human cholangiocarcinoma cell lines RBE and 12.7 μM for human cholangiocarcinoma cell lines HCCC-9810, which was more active than sanjoseolide (1). Analysis of the structure–activity relationships revealed that the presence of a trifluoromethyl group may be beneficial in terms of both RBE and HCCC-9810 inhibition.
Introduction
In the 21st century, cancer is still a challenging health problem. It caused more than 9.6 million deaths all over the world in 2018 and become the second dominating cause of human death worldwide.1 Even though scientists have made many efforts in the past few decades, human mortality rates for numerous types of cancers have not been distinctly reduced. Among them, cholangiocarcinoma (CCA) is the second most common primary liver malignancy, which is highly resistant to available chemotherapeutic agents and confers a 5-year relative survival rate of less than 5%.2 In the past years, a lot of efforts have been made to treat this disease, but outcomes have not been satisfactory. What is worse, CCA is always diagnosed at an advanced stage and patients always lose the chance to have surgery at this stage.3−5 Therefore, people urgently need more effective therapeutic strategies for the treatment of CCA.
Natural products and their derivatives remain a rich source of successful drug leads. Chalcones are precursors in the biosynthesis of flavonoids and occur widely in medicinal plants. It was reported that the hydroxyl and alkoxy groups at different positions on the benzene ring confer antibacterial,6 antifungal,7 antioxidant,8 and antimalarial9 activities to the chalcone molecule. So the scientists persist synthesizing new natural products and their derivatives to find new medicines that act on cancer cells with more effectiveness, specificity, and less toxicity.
Some natural chalcone molecules often contain an isopentenyl structure, which has been shown to have a wide range of biological properties, including anti-inflammatory, antitumor, and antidiabetic activity.10 They are an important source of clinical development drugs.11 In 2016, the natural product sanjoseolide (1), which possessed an isoprenylated chalcone skeleton, was isolated from the extracts of Dalea frutescens A. Gray (Leguminosae). The compound has good inhibitory activity on PC-3 and DU 145 prostate cancer cells, with the IC50 values of 35.0 and 25.5 μM.12 Although the isolation and biological activity of sanjoseolide (1) have been partially studied, its synthesis has not been reported. At the same time, it may be an important lead compound, and modification of the structure has great significance in future drug development.11a Therefore, an efficient synthetic strategy has been developed for the first total synthesis of sanjoseolide (1) and representative analogues using the commercially available 2,4-dihydroxyacetophenone as the starting materials.
Results and Discussion
The retrosynthetic analysis of the synthesis of sanjoseolide (1) is presented in Scheme 1. The target molecule, sanjoseolide (1), could be constructed through the Claisen–Schmidt condensation reaction between the protected acetophenone 6 and benzaldehyde, followed by the deprotection of two phenolic hydroxyl groups. Alcohol 6 could be synthesized from precursor 5 via Sharpless asymmetric dihydroxylation. Compound 5 would be originated from a protected 4, which was synthesized through Stille coupling. Compound 4 is a known compound that can be synthesized according to the literature. Using the same strategy, the derivatives could be synthesized for a structure–activity relationship (SAR) analysis, which can further study the antitumor activity of these compounds for the development of new drugs.
Scheme 1. Retrosynthesis of Sanjoseolide (1).

The syntheses of sanjoseolide (1) and the derivatives are described in Scheme 2. First, 1-(3-iodo-2,4-bis(methoxymethoxy)phenyl)ethan-1-one 4 was synthesized from the commercially available 2,4-dihydroxyacetophenone 2 using the protocol available in the literature.13,14 Next, the Stille coupling reaction of the double protected 2,4-bis-(methoxymethoxy)-3-iodoacetophenone 4 with tributyl(3-methyl-2-buten-1-yl)) stannane in dimethylformamide (DMF) using Pd(PPh3)4 as a catalyst produced product 5 in 42% yield.15 Using the Sharpless asymmetric dihydroxylation reaction, compound 6 was prepared in only 33% ee values.16 Because the natural product 1 was isolated as a racemate by chiral high-performance liquid chromatography (HPLC), we did not try more to improve the enantioselectivity. By the Claisen–Schmidt condensation reaction under alkaline conditions, compound 6 successfully yields chalcone 7a. Finally, the reaction of the bis-MOM-protected chalcone intermediates with hydrochloric acid in methanol and THF at 55 °C provided natural product sanjoseolide (1) in 8.6% yield over six steps.17 In a hydrogen atmosphere, reduction of the sanjoseolide (1) provided derivatives 11 in 8.0% yield over seven steps. The other derivatives (7a, 10, 8b–8e) were synthesized similarly and the overall yields ranged from 4.5 to 13.9%.18 The spectroscopic data of 1 were identical to those of sanjoseolide (1) reported in the literature.12
Scheme 2. Syntheses of Sanjoseolide (1) and Their Derivatives.

Finally, we prepare to evaluate the cytotoxicity of the synthetic compounds in human cancer cell lines, namely, REB and HCCC-9810 (cholangiocarcinoma). Interestingly, among all of the synthesized compounds, 8e and sanjoseolide (1) have a more obvious inhibitory effect on RBE and HCCC-9810. Table 1 shows that the IC50 values with synthetic 8e and sanjoseolide (1) were 12.8 and 15.7 μM for RBE and 12.7 and 15.6 μM for HCCC-9810, respectively. The cytotoxic effects of other compounds were not significant because the IC50 values were higher than 20 μM. Compared to the natural product sanjoseolide (1), it is not difficult to find that adding the trifluoromethyl group in the molecule has a better inhibitory effect on the growth of cancer cells (8e and 1). At the same time, we found that adding the methoxy group on the benzene ring with sanjoseolide (1) showed no significant difference in the HCCC-9810 tumor cell inhibitory activity (8d and 1). When we use the hydrogen reduction of the double bond in α,β-unsaturated ketone, the IC50 values increased notably, meaning that the α,β-unsaturated ketone moiety is believed to be responsible for the biological activities of chalcones (11 and 1). Meanwhile, compared with compounds 10 and 1, the introduction of two hydroxy groups at isoprenyl in the chalcone led to a considerable increase in the activity. Of course, the benzene ring is also crucial (8b, 8c, and 1). Currently, our laboratory is also doing research on the cytotoxicity against other tumor cell lines.
Table 1. IC50 Values (μM) of Sanjoseolide (1) and Their Derivatives against Cholangiocarcinoma Cell Linesa.
| compound | REB | HCCC-9810 |
|---|---|---|
| 7a | 25.7 ± 0.5 | 31.0 ± 1.0 |
| 11 | 59.1 ± 3.8 | 101.1 ± 1.8 |
| 10 | 28.3 ± 1.4 | 29.5 ± 1.4 |
| 8b | 73.2 ± 2.6 | 102.5 ± 4.4 |
| 8c | 105.0 ± 2.8 | 124.0 ± 7.4 |
| 8d | 31.2 ± 1.0 | 20.5 ± 1.6 |
| 8e | 12.8 ± 0.4 | 12.7 ± 0.6 |
| sanjoseolide(1) | 15.7 ± 0.3 | 15.6 ± 1.2 |
| cisplatin | 5.0 ± 0.3 | 1.8 ± 0.11 |
Data presented as a mean ± standard deviation (SD) of three replicate measurements of the same sample.
Conclusions
In summary, we have designed an efficient and concise route to the first total synthesis of sanjoseolide (1) and representative analogues. Although some of these compounds do not show good activity on cholangiocarcinoma cells, we are also actively exploring the activity on other cancer cells. We firmly believe that this strategy will show its potential application value in future drug development. Further research studies, which include constructing libraries with diverse structures through this synthetic strategy and studying their biological activities, are underway.
Experimental Section
General Experimental Procedures
All solvents were redistilled before the experiment. The chemicals were purchased from Aladdin and had the highest commercial purity. The mixture was separated and purified using 200–300 or 300–400 silica gel and reversed silica gel. Glass instruments were dried before use. Characterizations include NMR, high-resolution electrospray ionization MS, and melting point. 1H NMR spectra were recorded at 500 MHz using a Bruker NMR spectrometer. All split modes are specified as follows: m (multiplet), q (quartet), t (triplet), dd (doublet of doublets), d (doublet), and s (singlet). 13C NMR spectra were recorded at 125 MHz, which was identified by the number and location of the carbon. 2D HMBC, HMQC, 1H–1H COSY, and NOESY are used to determine the absolute configuration of a synthetic compound. Deuterated methanol and chloroform were purchased from CIL and TMS as internal standards. High-resolution electrospray ionization MS was carried out using Thermo Scientific. The melting points were retained on a SGW X-4 precision melting point tester.
Preparation of 1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-bis(methoxymethoxy)-phenyl]ethan-1-one (6)
1-(2,4-Dihydroxy-3-iodophenyl)ethanone (3)
A literature procedure was followed.13 To a solution of 2,4-dihydroxyacetophenone (1.52 g, 10 mmol) in ethanol (8 mL) and water (12 mL) was added iodine (1.142 g, 4.5 mmol) and potassium iodate (428 mg, 2 mmol). The reaction was stirred for 10 h at room temperature. TLC tracks the progress of the reaction. The mixture was diluted with water and extracted with ethyl acetate (3 × 30 mL); the combined organic layers were washed with brine and dried over Na2SO4, filtered, and concentrated under reduced pressure to provide a pale yellow solid. Compound 3 (2.7 g, 98%) was isolated by chromatography on a silica gel column (petroleum ether/ethyl acetate = 2/1) as a off-white solid; 1H NMR (500 MHz, CDCl3) δ 13.76 (s, −OH), δ 7.66 (1H, d, J = 8.8 Hz), δ 6.62 (1H, d, J =8.8 Hz), δ 2.60 (3H, s).
1-[3-Iodo-2,4-bis(methoxymethoxy)phenyl]ethanone (4)
A literature procedure was followed.14 To a stirred solution of compound 3 (278 mg, 1.0 mmol) in dry THF (5 mL) was slowly added anhydrous 70% NaH (137 mg, 4.0 mmol) and chloromethyl methyl ether (483 mg, 6.0 mmol) at 0 °C and the mix stirred for 2 h. TLC tracks the progress of the reaction. After the completion of the reaction, the resulting mixture was quenched with ice water and diluted with ethyl acetate (3 × 20 mL), the organic layer was washed with brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 8/1) to afford compound 4 (330 mg, 90%) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.57 (1H, d, J = 8.7 Hz), δ 6.90 (1H, d, J = 8.6 Hz), δ 5.28 (2H, s), δ 5.05 (2H, s), δ 3.55 (3H, s), δ 3.50 (3H, s), δ 2.59 (3H, s).
2,4-Bis(methoxymethoxy)-3-prenylacetophenone (5)
A literature procedure was followed.15 To a solution of 1-[3-Iodo-2,4-bis(methoxymethoxy)phenyl]ethanone (4) (366 mg, 1.0 mmol) in reagent grade DMF (16 mL) was added Pd(PPh3)4 (58 mg, 0.05 mmol) and prenyltributylstannane (431 mg, 1.2 mmol). The reaction mixture was heated at 100 °C for 40 h under a nitrogen atmosphere. TLC tracks the progress of the reaction. The solution was cooled to room temperature, diluted with ethyl acetate, and filtered through Celite. DMF was removed by vacuum distillation to give a yellow oil. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 16/1) to afford 2,4-bis(methoxymethoxy)-3-prenylacetophenone 5 (129 mg, 42%) as a dark yellow oil. Also, 2,4-bis(methoxymethoxy)acetophenone (103 mg, 42%) was achieved accidentally; 1H NMR (500 MHz, CDCl3) δ 7.46 (1H, d, J = 8.7 Hz), δ 6.88 (1H, d, J = 8.7 Hz), δ 5.21 (2H, s), δ 5.18–5.15 (1H, m), δ 4.94 (2H, s), δ 3.49 (3H, s), δ 3.44 (3H, s), δ 3.43-3.41 (2H, m), δ 2.55 (3H, s), δ 1.76 (3H, d, J = 1.4 Hz), δ 1.65 (3H, d, J = 1.4 Hz).
1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-bis(methoxymethoxy)phenyl]ethan-1-one (6)
The AD-mix-β (700 mg, 140 mg/0.1 mmol) and methane sulfonamide (52 mg, 0.55 mmol) were dissolved in t-BuOH/H2O (1/1) at 0 °C and stirred for 30 min; then, the compound 5 (154 mg, 0.5 mmol) was added to the reaction mixture. The reaction was kept at 0 °C for 48 h and quenched by saturated Na2S2O3. The mixture was extracted with ethyl acetate (3 × 20 mL). The organic extracts were washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 2/1) to give 6 (91 mg, 44%, 33% ee) as a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.57 (1H, d, J = 9.0 Hz), δ 6.96 (1H, d, J = 9.0 Hz), δ 5.26 (2H, s), δ 5.10 (1H, d, J = 6.4 Hz), δ 5.00 (1H, d, J = 6.4 Hz), δ 3.68–3.64 (1H, m), δ 3.59 (3H, s), δ 3.49 (3H, s), δ 3.18 (1H, d, J = 6.5 Hz), δ 3.00–2.91 (2H, m), δ 2.55 (3H, s), δ 1.32 (3H, s), δ 1.29 (3H, s); 13C NMR (125 MHz, CDCl3) δ 198.6, 159.1, 156.6, 130.0, 126.6, 123.0, 109.5, 101.9, 94.4, 78.2, 72.9, 57.8, 56.5, 29.6, 26.7, 25.8, 23.6. The enantiomeric ratio was determined by Daicel Chiralpak AD-H (25 cm), Hexanes/IPA = 75/25, 1 mL/min, λ = 254 nm, tmajor = 14.79 min, tminor = 23.73 min. HRMS(ESI) m/z 365.1577 [M + Na]+ (calcd for C17H26O7Na m/z, 365.1571).
(E)-1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-bis(methoxymethoxy)phenyl]-3-phenylprop-2-en-1-one (7a)
To a solution of 6 (150 mg, 0.44 mmol) and benzaldehyde (0.99 mL, 0.97 mmol) in EtOH was added KOH (45 mg, 0.79 mmol) slowly. The reaction mixture was stirred at room temperature for 24 h, diluted with H2O (10 mL), and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on a silica gel (petroleum ether/ethyl acetate = 16/1) to afford 7a (160 mg, 85%) as a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.62 (1H, d, J = 15.0 Hz), δ 7.57–7.53 (2H, m), δ 7.50 (1H, d, J = 9.0 Hz), δ 7.37–7.35 (3H, m), δ 7.23 (1H, d, J = 5.0 Hz), δ 6.96 (1H, d, J = 5.0 Hz), δ 5.23 (2H, s), δ 4.98 (1H, d, J = 5.0 Hz), δ 4.94 (1H, d, J = 5.0 Hz), δ 3.68–3.64 (1H, m), δ 3.47 (3H, s), δ 3.46 (3H, s), δ 3.11 (1H, d, J = 6.0 Hz), δ 3.00 (1H, dd, J = 2.5 and 13.5 Hz), δ 2.90 (1H, dd, J = 10.5 and 13.5 Hz), δ 2.50 (1H, s), δ 1.30 (3H, s), δ 1.26 (3H, s); 13C NMR (125 MHz, CDCl3) δ 192.0, 158.9, 156.7, 144.3, 134.9, 130.7, 129.9, 129.1, 128.6, 127.5, 126.1, 122.7, 109.9, 101.9, 94.6, 78.4, 73.1, 58.0, 56.6, 26.9, 26.0, 23.8; HRMS(ESI) m/z 453.1891 [M + Na]+ (calcd for C24H30O7Na m/z, 453.1884).
Sanjoseolide (1): To a solution of 7a (111 mg, 0.26 mmol) in MeOH (3 mL) and THF (3 mL) was added HCl dropwise (2.0 mol/L, 1.5 mL). The reaction mixture was stirred at 55 °C for 6 h. The reaction was quenched with water (5 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate = 8/1) to afford the target compound 1 (52 mg, 58%) as a bright yellow powder: mp 160–162 °C; 1H NMR(500 MHz, CDCl3) δ 13.85 (s, −OH), δ 7.88 (1H, d, J = 15.0 Hz), δ 7.76 (1H, d, J = 10.0 Hz), δ 7.66–7.64 (2H, m), δ 7.59 (1H, d, J = 15.0 Hz), δ 7.44–7.41 (3H, m), δ 6.54 (1H, d, J = 10.0 Hz), δ 3.68 (1H, dd, J = 2.0 and 9.5 Hz), δ 3.25 (1H, dd, J = 2.0 and 15.0 Hz), δ 2.53 (1H, dd, J = 9.5 and 15.0 Hz), δ 1.39 (3H, s), δ 1.30 (3H, s); 13C NMR (125 MHz, CDCl3) δ 192.2, 164.3, 163.5, 144.3, 135.0, 130.7, 130.0, 129.1, 128.7, 120.6, 113.8, 113.8, 109.4, 80.6, 73.7, 26.7, 25.3, 22.8; HRMS(ESI) m/z 365.1367 [M + Na]+ (calcd for C20H22O5Na m/z, 365.1359).
Syntheses of Derivatives 8b–8e, 10
(E)-1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-dihydroxyphenyl]-3-(thiophen-2-yl)prop-2-en-1-one (8b)
By following the procedure described above for the preparation of 10, the reaction was performed using 6 (80 mg, 0.234 mmol) and thiophene-2-carbaldehyde (44 μL, 0.468 mmol) as the starting material. Purification by column chromatography afforded 8b (36 mg, 45%) as a bright yellow powder; mp 172–173 °C; 1H NMR(500 MHz, CD3OD) δ 7.98 (1H, d, J = 15.0 Hz), δ 7.81 (1H, d, J = 5.0 Hz), δ 7.59 (1H, d, J = 5.0 Hz), δ 7.52 (1H, d, J = 15.0 Hz), δ 7.49 (1H, d, J = 5.0 Hz), δ 7.14 (1H, dd, J = 3.5 and 5.0 Hz), δ 6.48 (1H, d, J = 10.0 Hz), δ 3.62 (1H, dd, J = 2.5 and 10.0 Hz), δ 3.09 (1H, dd, J = 2.5 and 15.0 Hz), δ 2.72 (1H, dd, J =10.0 and 15.0 Hz), δ 1.26 (6H, s); 13C NMR (125 MHz, CD3OD) δ 192.9, 165.4, 164.8, 141.6, 137.5, 133.2, 130.9, 130.5, 129.5, 120.4, 115.1, 114.5, 109.0, 79.9, 74.0, 26.0, 25.6, 25.2; ESIMS m/z 347.09583 [M – H]− (calcd for C18H19O5S m/z, 347.09587).
(E)-1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-dihydroxyphenyl]-3-(furan-2-yl)prop-2-en-1-one (8c)
By following the procedure described above for the preparation of 10, the reaction was performed using 6 (80 mg, 0.234 mmol) and furan-2-carbaldehyde (39 μL, 0.468 mmol) as the starting materials. Purification by column chromatography afforded 8c (22 mg, 28%) as a bright yellow powder; mp 148–150 °C; 1H NMR(500 MHz, CD3OD) δ 7.81 (1H, d, J = 5.0 Hz), δ 7.69 (1H, d, J = 5.0 Hz), δ 7.64 (1H, dd, J = 15.0), 7.57 (1H, d, J = 15.0), δ 6.88 (1H, d, J = 5.0 Hz), δ 6.59 (1H, dd, J = 2.0 and 3.5 Hz), δ 6.48 (1H, d, J = 10.0 Hz), δ 3.62 (1H, dd, J = 2.5 and 10.0 Hz), δ 3.09 (1H, dd, J = 2.5 and 15.0 Hz), δ 2.72 (1H, dd, J = 10.0 and 15.0 Hz), δ 1.26 (6H, d, J = 1.5 Hz); 13C NMR (125 MHz, CD3OD) δ 193.0, 165.4, 164.8, 153.2, 146.8, 131.1, 130.9, 119.2, 117.4, 115.1, 114.5, 113.8, 109.1, 79.9, 74.0, 26.0, 25.6, 25.2; ESIMS m/z 331.11884 [M – H]− (calcd for C18H19O6m/z, 331.11871).
(E)-1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-dihydroxyphenyl]-3-(2,3,4-trimethoxyphenyl)prop-2-en-1-one (8d)
By following the procedure described above for the preparation of 10, the reaction was performed using 6 (158 mg, 0.46 mmol) and 2,3,4-trimethoxybenzaldehyde (181 mg, 0.92 mmol) as the starting materials. Purification by column chromatography afforded 8d (71 mg, 36%) as a bright yellow powder; mp 133–135 °C; 1H NMR(500 MHz, CD3OD) δ 8.05 (1H, d, J = 15.0 Hz), δ 7.85 (1H, d, J = 10.0 Hz), δ 7.74 (1H, dd, J = 1.0 and 15.0 Hz), δ 7.57 (1H, dd, J = 1.0 and 10.0 Hz), δ 6.86 (1H, dd, J = 1.0 and 10.0 Hz), δ 6.48 (1H, d, J = 10.0 Hz), δ 3.93 (3H, s), δ 3.90 (3H, s), δ 3.84 (3H, s), δ 3.63 (1H, dd, J = 2.5 and 10.0 Hz), δ 3.09 (1H, dd, J = 2.5 and 15.0 Hz), δ 2.72 (1H, dd, J = 10.0 and 15.0 Hz), δ 1.27 (3H, s), δ 1.26 (3H, s); 13C NMR (125 MHz, CD3OD) δ 193.8, 165.5, 164.6, 157.5, 155.0, 143.6, 140.0, 131.0, 124.9, 123.0, 120.5, 115.1, 114.7, 109.3, 109.0, 80.0, 74.0, 62.1, 61.3, 56.6, 26.1, 25.6, 25.2; ESIMS m/z 431.17120 [M – H]− (calcd for C23H27O8m/z, 431.17114).
(E)-1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-dihydroxyphenyl]-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (8e)
By following the procedure described above for the preparation of 10, the reaction was performed using 6 (110 mg, 0.32 mmol) and 4-(trifluoromethyl)benzaldehyde (88 μL, 0.64 mmol) as the starting materials. Purification by column chromatography afforded 8e (39 mg, 29%) as a bright yellow powder; mp 188–189 °C; 1H NMR(500 MHz, CD3OD) δ 7.97–7.93 (4H, m), δ 7.86 (1H, d, J = 15.5 Hz), δ 7.73 (2H, d, J = 10.0 Hz), δ 6.51 (1H, d, J = 10.0 Hz), δ 3.63 (1H, dd, J = 2.5 and 10.0 Hz), δ 3.09 (1H, dd, J = 2.5 and 14.0 Hz), δ 2.73 (1H, dd, J = 10.0 and 14.0 Hz), δ 1.27 (3H, s), δ 1.26 (3H, s); 13C NMR (125 MHz, CD3OD) δ 193.1, 165.7, 165.2, 142.8, 140.2, 131.4, 130.1, 128.4, 126.8–126.9(q, −CF3), 124.8, 115.2, 114.6, 109.2, 79.9, 74.0, 26.0, 25.6, 25.2; ESIMS m/z 409.12695 [M – H]− (calcd for C21H20O5F3m/z, 409.12683).
(E)-1-[2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl]-3-phenylprop-2-en-1-one (10)
To a solution of 5 (166 mg, 0.54 mmol) and benzaldehyde (0.11 mL, 1.08 mmol) in EtOH was added KOH (54 mg, 0.97 mmol) slowly. The reaction mixture was stirred at room temperature for 24 h, diluted with H2O (10 mL), and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude mixture was directly used for the next reaction. Then, the crude mixture was dissolved in MeOH (3 mL)/THF (3 mL) and HCl was added dropwise (2.0 mol/L, 1.5 mL). The reaction mixture was stirred at 55 °C for 6 h, and the reaction was quenched with water (5 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on a silica gel (petroleum ether/ethyl acetate = 2/1); then, the mixture was repurified using reversed-phase silica gel (MeOH/H2O = 4/1) to afford target compound 10 (36 mg, 16%) as a bright yellow powder; mp 162–162.5 °C; 1H NMR(500 MHz, CD3OD) δ 7.86 (1H, d, J = 5.0 Hz), δ 7.82 (2H, d, J = 5.0 Hz), δ 7.75-7.74 (2H, m), δ 7.46–7.40 (3H, m), δ 6.45 (1H, d, J = 5.0 Hz), δ 5.26-5.22 (1H, m), δ 3.33 (2H, d, J = 10.0 Hz), δ 1.79 (3H, d, J = 1.5 Hz), δ 1.66 (3H, d, J = 1.5 Hz); 13C NMR (125 MHz, CD3OD) δ 193.4, 165.3, 164.4, 144.7, 136.5, 131.9, 131.5, 130.7, 130.1, 129.7, 123.6, 122.1, 116.7, 114.4, 108.6, 26.0, 22.5, 17.9; ESIMS m/z 307.13408 [M – H]− (calcd for C20H19O3m/z, 307.13397).
1-[3-(2,3-Dihydroxy-3-methylbutyl)-2,4-dihydroxyphenyl]-3-phenylpropan-1-one (11)
To a solution of sanjoseolide (1) (15 mg, 0.044 mmol) in ethyl acetate (2 mL) was added Pd/C in a hydrogen atmosphere. The progress of the reaction was monitored by TLC. When the starting material disappeared, the mixture was filtered through Celite. The organic layer was concentrated in vacuo to give compound 11 (15 mg, 99%, white powder) without any purification; mp 123–123.5 °C; 1H NMR(500 MHz, CDCl3) δ 13.21 (s, −OH), δ 8.99 (s, −OH), δ 7.57 (1H, d, J = 10.0 Hz), δ 7.32–7.29 (2H, m), δ 7.25–7.20 (3H, m), δ 6.47 (1H, d, J = 10.0 Hz), δ 3.65 (1H, dd, J = 1.5 and 9.5 Hz), δ 3.51 (s, −OH), δ 3.26–3.19 (3H, m), δ 3.06–3.03 (2H, m), δ 2.50 (1H, dd, J = 9.5 and 15.0 Hz), δ 1.37 (3H, s), δ 1.29 (3H, s); 13C NMR (125 MHz, CDCl3) δ 203.9, 163.3, 162.9, 141.1, 130.2, 128.7, 128.5, 126.4, 113.6, 113.1, 109.3, 80.6, 73.6, 39.8, 30.6, 26.7, 25.2, 22.7; ESIMS m/z 343.15509 [M – H]− (calcd for C20H23O5m/z, 343.15510).
Cell Culture
The human hepatocellular carcinoma cell lines RBE and HCCC-9810 were purchased from the Shanghai Cell Bank. The cells are seeded in a cell culture dish containing a certain amount of complete medium, placed in a 37 °C, 5% CO2, saturated in humidity incubator, and the fresh medium is replaced every 24 or 48 h depending on the cell line.
Cell Proliferation Assay
SRB assay was used to detect cell proliferation, and the cell density was determined based on the amount of protein detected. The cells in the logarithmic growth phase were digested with trypsin to adjust the cell density to (3–7) × 104 cells/mL in a fresh medium. Then, they were inoculated in a 96-well plate, 100 μL per well, and cultured in an incubator at 37.0 °C, 5.0% CO2, and saturated humidity overnight so that the cells adhered to the wall. The drug was diluted to twice the detection concentration in the culture medium to ensure that the DMSO content in the drug diluent remained the same, then 100 μL of the drug diluent was pipetted into a 96-well plate, and continued to culture for 72 h. The cell culture medium was discarded and 100 μL of a prechilled 10% (w/v) trichloroacetic acid (TCA) was added gently and left at 4 °C for at least 1 h. The TCA fixing solution was removed and washed five times with slowly flowing water, the remaining water was removed with an absorbing paper, 100 μL of SRB staining solution was added gently, and the mixture incubated at room temperature for 15–30 min. The SRB staining solution was removed and washed five times with 1% glacial acetic acid solution to remove the unbound dye. The mixture was dried at room temperature, 100 μL of 10 mM Tris (pH = 10.0) solution was added, and the OD570 nm was detected with a microplate reader after the dye had completely dissolved.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (NFSC No. 21967015), the Science and Technology Major Project of Gansu Province (18ZD2NA005), the Young Scholars Foundation of Lanzhou Jiaotong University (2017006), and the Natural Science Foundation for Young Scholars of Gansu Province (20JR5RA384).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05546.
1H and 13C NMR spectra of 1–11; growth inhibition rate curve; and HPLC spectra of compound 6 (PDF)
Author Contributions
† T.T. and Z.Z. contributed equally to this work and should be considered co–first authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Bray F.; Ferlay J.; Soerjomataram I.; Siegel R. L.; Torre L. A.; Jemal A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2018, 68, 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Yan L. N. Perihilar cholangiocarcinoma: Current therapy. World J. Gastrointest. Pathophysiol. 2014, 5, 344–354. 10.4291/wjgp.v5.i3.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaib Y. H.; Davila J. A.; Henderson L.; McGlynn K. A.; El-Serag H. B. Endoscopic and surgical therapy for intrahepatic cholangiocarcinoma in the United States a population-based study. J. Clin. Gastroenterol. 2007, 41, 911–917. 10.1097/MCG.0b013e31802f3132. [DOI] [PubMed] [Google Scholar]
- Ustundag Y.; Bayraktar Y.; Poon R. T. Cholangiocarcinoma: a compact review of the literature. World J. Gastroenterol. 2008, 14, 6458–6466. 10.3748/wjg.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. A.; Davidson B. R.; Goldin R. D.; Heaton N.; Karani J.; Pereira S. P.; Rosenberg W. M. C.; Tait P.; Taylor-Robinson S. D.; Thillainayagam A. V.; Thomas H. C.; Wasan H. Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut. 2012, 61, 1657–1669. 10.1136/gutjnl-2011-301748. [DOI] [PubMed] [Google Scholar]
- Liu X. L.; Xu Y. J.; Go M. L. Functionalized chalcones with basic functionalities have antibacterial activity against drug sensitive Staphylococcus aureus. Eur. J. Med. Chem. 2008, 43, 1681–1687. 10.1016/j.ejmech.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Lahtchev K. L.; Batovska D. I.; Parushev S. P.; Ubiyvovk V. M.; Sibirny A. A. Antifungal activity of chalcones: a mechanistic study using various yeast strains. Eur. J. Med. Chem. 2008, 43, 2220–2228. 10.1016/j.ejmech.2007.12.027. [DOI] [PubMed] [Google Scholar]
- Detsi A.; Majdalani M.; Kontogiorgis C. A.; Hadjipavlou-Litina D.; Kefalas P. Natural and synthetic 2′-hydroxy-chalcones and aurones: Synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg. Med. Chem. 2009, 17, 8073–8085. 10.1016/j.bmc.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Yadav N.; Dixit S. K.; Bhattacharya A.; Mishra L. C.; Sharma M.; Awasthi S. K.; Bhasin V. K. Antimalarial activity of newly synthesized chalcone derivatives in vitro. Chem. Biol. Drug Des. 2012, 80, 340–347. 10.1111/j.1747-0285.2012.01383.x. [DOI] [PubMed] [Google Scholar]
- a Zhang L. B.; Lei C.; Gao L. X.; Li J. Y.; Li J.; Hou A. J. Isoprenylated flavonoids with PTP1B inhibition from macaranga denticulate. Nat. Prod. Bioprospect. 2016, 6, 25–30. 10.1007/s13659-015-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu Z. G.; Tang L. G.; Zou P.; Zhang Y. L.; Wang Z.; Fang Q. L.; Jiang L. L.; Chen G. Z.; Xu Z.; Zhang H. J.; Liang G. Synthesis and biological evaluation of allylated and prenylated mono-carbonyl analogs of curcumin as anti-inflammatory agents. Eur. J. Med. Chem. 2014, 74, 671–682. 10.1016/j.ejmech.2013.10.061. [DOI] [PubMed] [Google Scholar]; c Zhang Y. F.; Zhang P.; Cheng Y. Y. Structural characterization of isoprenylated flavonoids from Kushen by electrospray ionization multistage tandem mass spectrometry. J. Mass Spectrom. 2008, 43, 1421–1431. 10.1002/jms.1423. [DOI] [PubMed] [Google Scholar]
- a Das M.; Manna K. Chalcone scaffold in anticancer armamentarium: a molecular insight. J. Toxicol. 2016, 2016, 1–14. 10.1155/2016/8606410. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wright C. W. Recent developments in research on terrestrial plants used for the treatment of malaria. Nat. Prod. Rep. 2010, 27, 961–968. 10.1039/c002196m. [DOI] [PubMed] [Google Scholar]; c Babu M. A.; Shakya N.; Prathipati P.; Kaskhedikar S. G.; Saxena A. K. Development of 3D-QSAR models for 5-lipoxygenase antagonists: chalconesy. Bioorg. Med. Chem. 2002, 10, 4035–4041. 10.1016/S0968-0896(02)00313-9. [DOI] [PubMed] [Google Scholar]; d Yin B. T.; Yan C. Y.; Peng X. M.; Zhang S. L.; Rasheed S.; Geng R. X.; Zhou C. H. Synthesis and biological evaluation of α-triazolyl chalcones as a new type of potential antimicrobial agents and their interaction with calf thymus DNA and human serum albumin. Eur. J. Med. Chem. 2014, 71, 148–159. 10.1016/j.ejmech.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Shaffer C. V.; Cai S. X.; Peng J. N.; Robles A. J.; Hartley R. M.; Powell D. R.; Du L.; Cichewicz R. H.; Mooberry S. L. Texas native plants yield compounds with cytotoxic activities against prostate cancer cells. J. Nat. Prod. 2016, 79, 531–540. 10.1021/acs.jnatprod.5b00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. M.; Yan Z. H.; Lei Y. N.; Sheng K.; Yao Q. W.; Lu K.; Yu P. Concise synthesis of prenylated and geranylated chalcone natural products by regiospecific iodination and Suzuki coupling reactions. Tetrahedron Lett. 2014, 55, 897–899. 10.1016/j.tetlet.2013.12.044. [DOI] [Google Scholar]
- Wang H. M.; Zhang L.; Liu J.; Yang Z. L.; Zhao H. Y.; Yang Y.; Shen D.; Lu K.; Fan Z. C.; Yao Q. W.; Zhang Y. M.; Teng Y. O.; Peng Y. Synthesis and anti-cancer activity evaluation of novel prenylated and geranylated chalcone natural products and their analogs. Eur. J. Med. Chem. 2015, 92, 439–448. 10.1016/j.ejmech.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Grealis J. P.; Müller-Bunz H.; Ortin Y.; Casey M.; McGlinchey M. J. Synthesis of isobavachalcone and some organometallic derivatives. Eur. J. Org. Chem. 2013, 2013, 332–347. 10.1002/ejoc.201201063. [DOI] [Google Scholar]
- Jiang H.; Hamada Y. Highly enantioselective synthesis of angelmarin. Org. Biomol. Chem. 2009, 7, 4173–4176. 10.1039/b913400j. [DOI] [PubMed] [Google Scholar]
- Fang B.; Xiao Z. X.; Qiu Y. D.; Shu S.; Chen X. X.; Chen X. J.; Zhuang F.; Zhao Y. J.; Liang G.; Liu Z. G. Synthesis and anti-inflammatory evaluation of (R)-, (S)-, and (±)-Sanjuanolide isolated from Dalea frutescens. J. Nat. Prod. 2019, 82, 748–755. 10.1021/acs.jnatprod.8b00596. [DOI] [PubMed] [Google Scholar]
- Zhai J.; Fu L.; Li Y. Y.; Zhao R.; Wang R.; Deng H. K.; Liu H. L.; Kong L.; Chen Z. W.; Sang F. Synthesis and biological activities evaluation of sanjuanolide and its analogues. Bioorg. Med. Chem. Lett. 2019, 29, 326–328. 10.1016/j.bmcl.2018.11.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.