

Abstract
Ribonucleotide reductases (RNRs) are a diverse family of enzymes that are alone capable of generating 2′-deoxynucleotides de novo and are thus critical in DNA biosynthesis and repair. The nucleotide reduction reaction in all RNRs requires the generation of a transient active site thiyl radical, and in class I RNRs this process involves a long-range radical transfer between two subunits, α and β. Due to the transient subunit association, an atomic resolution structure of an active α2β2 RNR complex has been elusive. Here we use a doubly-substituted β2, E52Q/(2,3,5)-trifluorotyrosine122-β2 to trap wildtype-α2 in long-lived α2β2 complex. We report the structure of this complex by cryo-electron microscopy to 3.6-Å resolution, allowing for structural visualization of a 32-Å-long radical transfer pathway that affords RNR activity.
One Sentence Summary:
A structure of the long-range radical transfer pathway within a ribonucleotide reductase.
Ribonucleotide reductases (RNRs) convert ribonucleotides into deoxyribonucleotides and generate the deoxyribonucleotide pools needed for DNA biosynthesis and repair (Fig. 1A, S1). Due to these activities, RNRs are drug targets, with several FDA-approved RNR inhibitors employed clinically in the treatment of various forms of cancer (1). For all RNR classes, catalysis entails formation of a protein-based thiyl radical species in the active site (2). In the case of the prototypic class Ia RNR from Escherichia coli, the generation of this thiyl radical species occurs on the α2 subunit (C439α•) and involves a long-range radical transfer from the β2 subunit’s diferric-tyrosyl radical cofactor (Y122β•) (Fig. 1B (3). The distance that radical species must travel has been estimated to be an incredibly long 35–40 Å, based both on pulsed electron-electron double resonance (PELDOR) spectroscopy (4) and on a docking model of an α2β2 complex that was based on shape complementary of the separate α2 and β2 crystal structures (Fig. 1C, S2A) (5). Importantly, this radical transfer, which is proposed to occur through a series of reversible proton-coupled electron transfer (PCET) steps, does not involve typical electron-transfer cofactors, but instead requires a series of conserved aromatic amino acids (2, 3, 5, 6) (Fig. 1B). Key residue Y356β has never been structurally visualized; this residue is located on the C-terminal β-tail (residues 341β−375β), which is disordered in all β2 structures. The goal of this study is to provide a structure of the active α2β2 state of an RNR that has the critical β-tail ordered and the intact PCET pathway visualized.
Fig. 1: RNR catalysis requires formation of a transient thiyl radical species.
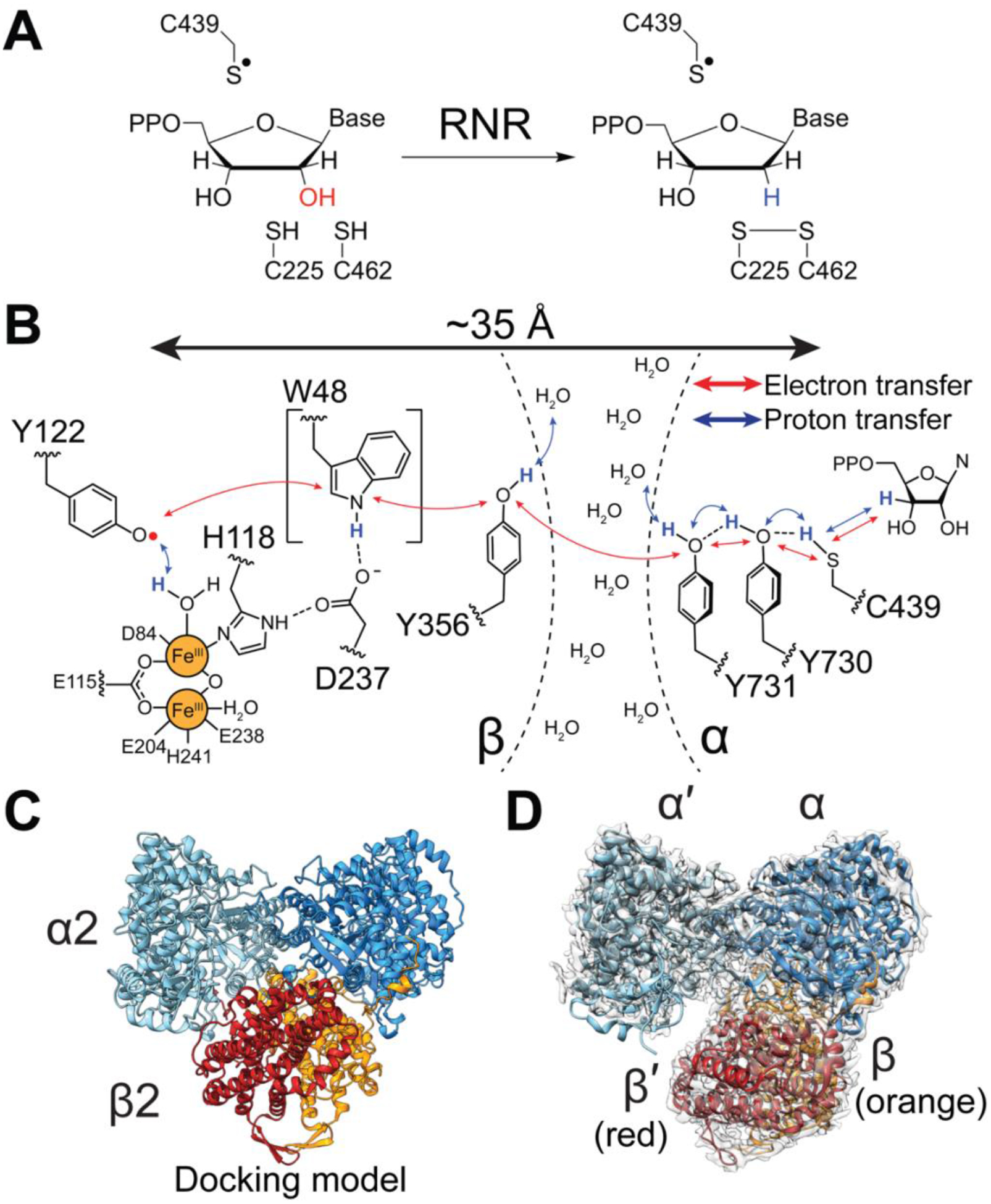
A) Radical-based reduction of ribonucleoside diphosphates by class Ia RNR. Reducing equivalents are provided by a pair of active site cysteines that are oxidized concomitant with product formation. The resulting disulfide is re-reduced via disulfide exchange with NADPH as the ultimate source of electrons (27). B) Proposed proton-coupled electron transfer pathway that results in the transient oxidation of C439-α by Y122•-β. The direct involvement of W48 in the pathway is uncertain. C) Symmetric “docking model” of 260 kDa α2β2 complex that was based on shape complementarity of the individual α2 and β2 crystal structures (5). α2 is shown in cyan/blue and β2 is shown in red/orange. D) 3.6-Å resolution cryo-EM map of an asymmetric, active RNR trapped using a doubly-substituted E52Q/(2,3,5)-trifluorotyrosine122(F3Y122)-β2 with wildtype-α2 in the presence of substrate GDP and specificity effector TTP. Model colors are as in Fig. 1C with cryo-EM density shown in transparent grey.
There are two main reasons why obtaining such a structure has eluded researchers. First, the α2β2 interaction that affords thiyl radical generation is transient, with a koff for β2 from α2 of greater than 60 per sec (7). The PCET pathway is intact during the radical transfer process; however, following the return of the radical to β2, the PCET pathway disassembles. Second, the active α2β2 state is in equilibrium with an inactive α4β4 state (8). The shifting of this equilibrium between active and inactive states is responsible for regulating RNR activity in response to deoxyribonucleotide levels in E. coli (Fig. S2B) (8). For these two reasons, the structure of the active form of RNR has been elusive, requiring some means of trapping the active state to slow subunit dissociation and limit oligomeric state change.
Here we succeed in trapping the active state of RNR by using a previously studied doubly-substituted E52Q/(2,3,5)-trifluorotyrosine122(F3Y122)-β2 (9) with wildtype-α2 in the presence of substrate GDP and specificity effector TTP (Fig. 1D). The overall resolution is 3.6 Å, with local resolution ranging from ~3.3 Å within the core of the protein complex to 5.5 Å on the α2 surface (Fig. S3, Table S1). Regions of highest resolution show clear density for side-chains (Fig. S4). Importantly, we further demonstrate that this RNR construct is active: it is capable of forming product (1 equivalent of dGDP per dimer), and it forms a pathway radical (Y356•) in a kinetically competent manner (Fig. S5). This structure addresses key questions about RNR and about biological PCET, including how PCET residues are arranged with respect to each other and to possible proton acceptors/donors; the molecular basis for the conversion of a disordered stretch of 20 amino acids into an ordered PCET pathway; why the PCET pathway disassembles after every round of turnover; the factors that trigger pathway disassembly and assembly; and to what degree the structure of an active RNR state looks like the docking model.
The first striking observation from our structure is that unlike the E. coli RNR symmetric docking model (Fig. 1C), the cryo-EM structure of the α2β2 complex is asymmetric (Fig. 1D). The α2-β2 interface is formed primarily between one α monomer and β2, and minimal interaction with the other α monomer (hereafter referred to as α′) (Fig. 1D). This positioning of subunits precludes the simultaneous formation of two radical translocation pathways, which is consistent with prior evidence for half-sites reactivity such as the observation that one equivalent of the cancer drug gemcitabine diphosphate inactivates both α monomers (10) We now see that only one α subunit can turnover at a time. Additionally, this finding of asymmetry is consistent with previous structural snapshots of class Ib RNRs (11, 12), the first of which is a structure that is best-described as β2 dangling off of one α monomer (Fig. S2C) (11).
We are able to visualize all residues of the elusive β-tail, including Y356 and the ~20 residues within the ~35 residue-long β-tail (residues 341–375) that have been disordered in previous structures (Fig. 2A). Consistent with complex asymmetry, the tail of the other β monomer (hereafter referred to as β′) is only resolved up to residue 341 (of 375). The ordered β-tail extends far into the active site cavity in α, coming into direct van der Waals contact with substrate before making a sharp turn and extending back toward the β subunit, and finally looping around to a groove on the side of α (Fig. 2A, S6). This observation immediately allows us to answer the question of why the PCET pathway disassembles after every round of turnover: the β-tail completely sequesters the active site such that product dissociation would necessitate β departure (Fig. 2B, S7).
Fig. 2: Fully visualized β-tail extends deep into the α active site.
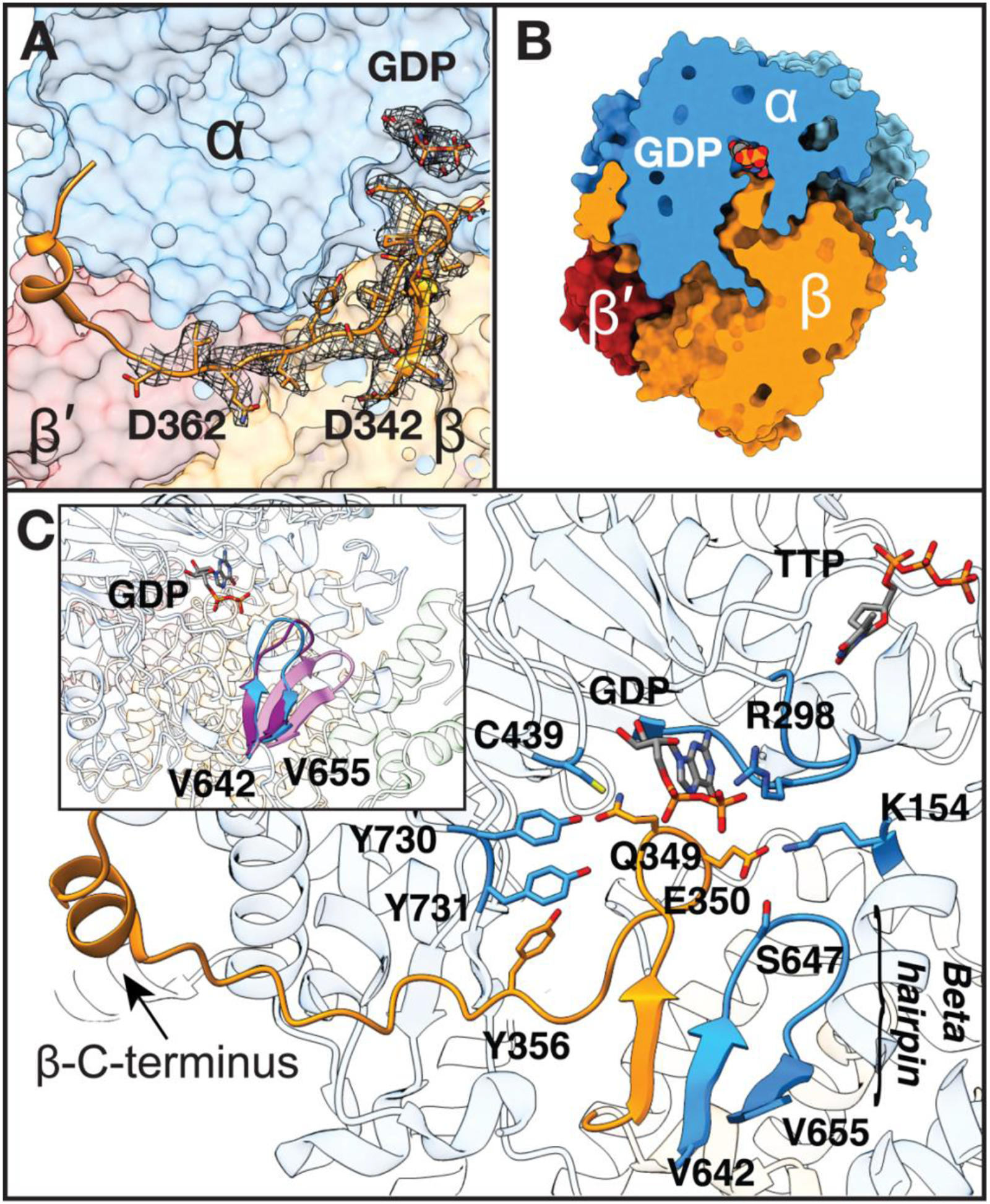
A) Missing β residues 342–362 modeled into cryo-EM density (grey mesh, threshold set to 0.0249). B) Slice through space filling representation showing tight packing between subunits which blocks solvent accessibility. C) β-C-terminus extends up to substrate-binding site in α. Regions of α that contact β-tail are displayed. Substrate GDP and effector TTP are shown in sticks, separated by the specificity loop, labeled at position R298. Inset: β-hairpin of α shifts 5-Å toward the active site upon substrate and effector binding and the ordering of the specificity loop. Three α2 structures were aligned to highlight movement of this β-hairpin: substrate-free α2 (pink, PDB 2R1R (14)), substrate/effector-bound α2 from inhibited α4β4 (purple, PDB 5CNV (13)), and substrate-bound α2 from our structure of active α2β2 (blue).
Additionally, this structure suggests how the binding of substrate/effector pairs could facilitate β-tail binding and PCET pathway assembly (See Fig. S1)substrate/effector pair explanation): the β-tail binding pocket simply does not exist as a pocket in the absence of substrate. Substrate itself forms the top of the pocket (Fig. 2B,C) and the right-side of the pocket is formed by a β-hairpin from the α subunit that is known to swing-in toward the active site when substrate-effector pairs are bound (13, 14) (Fig. 2C). Although we do not fully understand why this β-hairpin is swung-in in the presence of cognate substrate-effector pairs, our E. coli RNR structures bound with all four of these pairs (CDP/dATP, UDP/dATP, GDP/TTP, and ADP/dGTP) (13) show this swung-in position of the β-hairpin whereas substrate-free or effector-free structures do not. In this cryo-EM structure, we observe the same recognition mode for GDP/TTP as was described before (13) (Fig. S8), and again find a swung-in β-hairpin (Fig. 2C). Residues 643α−647α of this hairpin run parallel to residues 343–346 of the β subunit, forming a mini β-sheet, and securing β in position for radical transfer (Fig. 2C, S9).
Further stabilization of the β-tail in its α binding pocket is derived from side chains of β-residues Q349β and E350β (Fig. 2C, S9, Table S2). Q349β contacts PCET residue Y730α and E350β contacts K154α near the effector binding site and S647α of the β-hairpin. E350β is known to play a critical role in catalysis (15, 16) and in PCET gating (17), and we can now propose why this is the case. E350β appears to act as a latch, stabilizing the β-tail and thus the PCET pathway when substrate-effector pairs are bound and the active site is ready for catalysis. Notably, the β-hairpin is also present in class II RNR and shifts inward upon adenosylcobalamin binding to secure this cofactor in position for thiyl radical formation (18). Its conservation in class I RNRs was thought to be a vestige, but now appears to be playing an analogous role but with a different radical-cofactor. In both cases, this β-hairpin secures the cofactor for thiyl radical generation, which for class II RNR involves positioning adenosylcobalamin for a ~3.5-Å direct radical transfer to Cys, and for class Ia RNR involves positioning a PCET pathway for a ~35-Å remote radical transfer.
The full PCET pathway is structurally resolved in our structure, revealing the position of Y356β relative to the other key residues along the PCET pathway (Fig. 3A), and showing that the radical-transfer distance is, in fact, extraordinarily long. The total distance from the hydroxyl group of F3Y122 in β to the thiyl group of C439 in α is 32.4 Å, close to the estimated 35–40 Å (4, 5). Several features of this pathway immediately stand out, including the position of W48β directly in between Y122β and Y356β (Fig. 3A). W48β’s role in PCET (if any) has been enigmatic, since substitution of this residue inactivates RNR at a step prior to PCET, i.e. cofactor formation (19). Now seeing W48β’s location, positioned at the edge of the β subunit just before Y356β, prompts us to propose that this residue plays a role in gating PCET to prevent radical loss when the β-tail is not ready to receive a radical species. Such a role will be hard to test experimentally, but could be probed computationally.
Fig. 3: The interface region of the α2β2 complex.
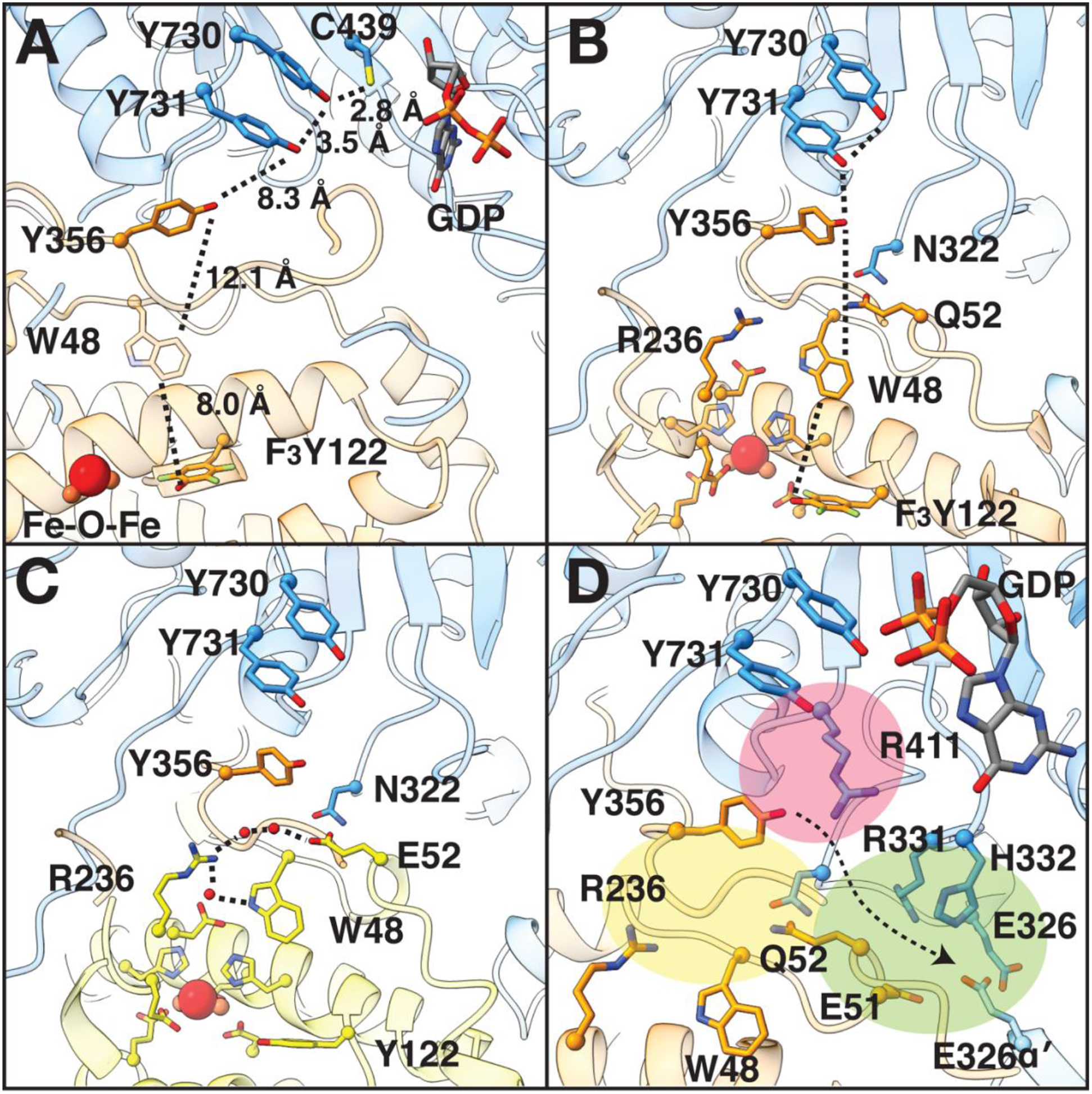
A) Key residues that are involved in PCET, including Y356, are shown with distances between them in angstroms. W48 is shown in partial transparency due to uncertainty regarding its direct participation in PCET. Colors are as in Fig. 1C. B) Positioning of Q52 of β with respect to PCET pathway (dashed lines). The Q52 side-chain is 2.5 Å from the side-chain of N322, 6.9 Å from the side-chain hydroxyl of PCET residue Y356, and 5.2 Å from the C4 position of the indole ring of W48 in β. C) Superimposition of higher-resolution structure of β (yellow, PDB 5CI4 (32)) onto the cryo-EM structure (Fig. S10) shows that E52β connects the diiron site to α residue N322 via a hydrogen-bonding network (dashed lines) that involves several waters (red spheres). D) Regions surrounding E/Q52 are very polar. See text for description of colored regions. Arrow indicates direction towards bulk solvent.
Also noteworthy are the differences in distances between PCET residues in β and those in α (Fig. 3A). PCET residues in α are 2.8–3.5 Å apart and side-chains of Y730α and Y731α are stacked, consistent with colinear transfer of an electron and a proton as suggested experimentally (20, 21). In contrast, PCET residues in β are 8.0–12.1 Å apart and the side-chain of Y356β is not stacked with the other PCET residues. These latter distances are consistent with orthogonal electron and proton transfer (22), as well as with electron-transfer distances observed between cofactors within other enzymes (23).
Despite the fact that PCET requires proton donors and acceptors, key PCET residue Y356β does not make direct contact with any side-chains that could act as proton donor/acceptor. Y356β is however, surrounded by polar residues that could make through-water contacts (Fig. 3). Water molecules have been implicated in orthogonal proton transfer based on spectroscopic studies (24–26), including the proton transfer to Y122β by a water molecule on Fe1, which has been proposed to initiate forward PCET (22) (Fig. 1B). Polar residues along the PCET pathway of note include E52β (Q52β in this structure), which sits at the nexus of three polar-residue regions (colored yellow, pink and green in Fig 3D). In particular, Q52β is located directly at the α2-β2 interface where it contacts α residue N322 (Fig. 3B), which in turn is adjacent to Y356β and R411α. The latter residue has been shown to affect the conformational flexibility of PCET residue Y731α (26) (Fig. 3D-pink). Additionally, E52β was previously shown to be part of a protracted hydrogen-bonding network that extends all the way into the core of the β2 subunit to the diiron cluster (9) (Fig. 3C, 3D-yellow, S10), and finally this structure shows that Q52β is next to a loop containing E326 from both α and α′ in a region of the structure that leads to bulk solvent (Fig. 3D-green). Whereas α makes the vast majority of interactions with β, α′ does help form this part of the α-β interface. Overall, we speculate that E52β’s essential function (9) is related to the proton transfer component of PCET, and we posit that residues and water molecules in the yellow and pink highlighted regions will be involved in proton transfer, with the residues in the green region providing the route for proton release to solvent.
Thus far, we have focused on α-β interaction, but the α′ structure is also worthy of comment. From the biochemical analyses of these RNR constructs ((9), Fig S5), we know that the α2β2 becomes trapped after a single turnover event, i.e. after one α subunit generates product. Consistent with these biochemistry results, α′ appears to be the subunit that underwent turnover, whereas α appears to be the one that is in the pre-turnover state (Fig 4). Briefly, the active site of α (Fig. 4A, S11A) has clear density for a nucleoside diphosphate, and no evidence of a disulfide between C225 and C462, the two redox-active cysteines that become oxidized concomitant with turnover (27). In contrast, α′ (Fig. 4B, S11B) has no observable density in the active site corresponding to substrate/product, and the C225/C462 pair has density suggestive of a disulfide, indicating that nucleotide reduction and dithiol oxidation has occurred.
Fig. 4:
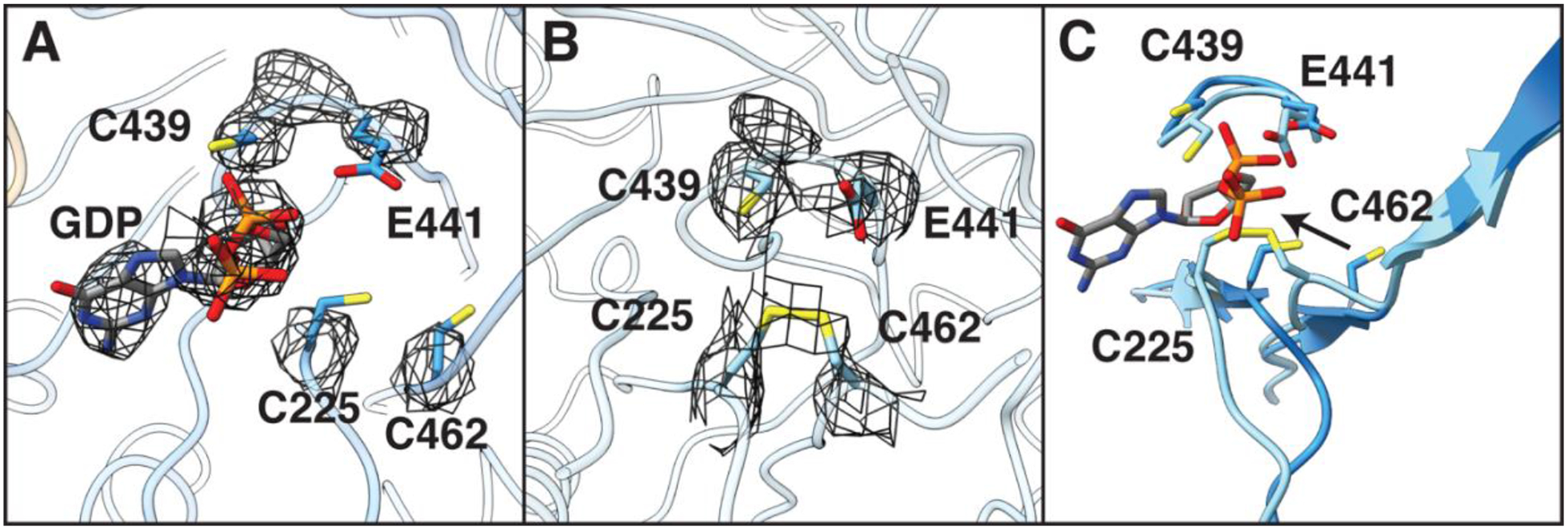
The active sites of α and α′ are structurally different. A) In α, density (shown in mesh) indicates the presence of substrate GDP and the absence of a disulfide between C225 and C462. Catalytic residues C439 and E441 are shown for context. B) In α′, density for GDP is absent and density is present indicative of a disulfide. C) A ~3 Å shift of Cα of both C225 and C462 toward C439 and E441 within the active site of α′ is consistent with a disulfide between C225-C462, suggesting a post-turnover state. Arrow represents the directionality of this movement between α and α′. Density threshold for cryo-EM map in A) and B) were set to same level (0.0384).
The oxidation of the C225/C462 cysteine pair is accompanied by a ~3 Å shift of Cα of both C225 and C462 towards the thiyl radical-forming C439 (Fig. 4C). This movement mirrors that observed in previous α2 crystal structures with reduced (13) and oxidized (14) cysteine residues (Fig. S11C, D). Notably, the disulfide generated by catalysis physically occludes the active site (Fig. 4B), likely facilitating product release. However, our structural data show that product cannot leave when the PCET pathway is intact as its exit is blocked (Fig. 2B). Thus, a possible molecular mechanism for PCET pathway dissolution involves disulfide formation ‘pushing’ on product, and product ‘pushing’ on β, causing β to swing from α′ to α (Fig. S12). The displacement of β from α′ also appears necessary for the re-reduction of the active site disulfide, which is completely buried when the PCET pathway is intact. We observe that α′ still has its specificity effector bound, indicating that product loss is not necessarily coupled with that of the substrate-specificity effector.
These structural views of α and α′ provide insight into both PCET pathway assembly and disassembly, respectively, suggesting how substrate binding could serve to create the PCET pathway pocket, and how turnover (cysteine oxidation) could send a molecular push enabling pathway disassembly. The elegance of this molecular design has not escaped our attention, with one binding event/catalytic step triggering the next, guiding this enzyme through a series of elaborate conformational rearrangements that produce the requisite deoxynucleotide levels for DNA biosynthesis and repair.
The structure of the α2β2 active complex presented here is the culmination of decades of biochemical and structural studies (3, 7–9, 28–31) (Fig. S13) and provides a powerful example of the utility of unnatural amino acids for trapping transient protein-protein complexes. It also provides an illustration of the benefit of cryo-EM for studying transient enzyme complexes. Use of the doubly-substituted β2 slowed complex dissociation to the minute time scale of cryo-EM grid generation, but not to the hour-day time scale that crystallization requires. Without the resolution revolution that the cryo-EM field is experiencing, this work would not have been possible. These resulting structural data will serve as a launching point towards answering other long sought-after questions about both biological PCET and RNR.
Supplementary Material
Acknowledgements:
We thank Dr. E.J. Brignole for his invaluable advice in preparing our samples for cryo-EM data collection and subsequent data processing, A. Kim for performing the activity assays and radical quantifications, and Q. Lin for providing the E52Q/F3Y122-β2 protein that was essential for the success of this study. Cryo-EM data was collected at the UMass Medical School Worcester Cryo-EM Facility with the help of Dr. Kangkang Song and Dr. Chen Xu.
Funding: This work was supported by the National Institutes of Health grants R35 GM126982 (to C.L.D), R01 GM29595 (to J.S.), and F32 GM123596 (A.T.T.), and by a David H. Koch Graduate Fellowship to G.K. CLD is an HHMI Investigator.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: Atomic coordinates and cryo-EM map of the reported structure have been deposited to the Protein Data Bank (accession code 6W4X) and Electron Microscopy Data Bank (accession code EMD-21540), respectively.
Supplementary Materials:
Materials and Methods
Figures S1-S14
Tables S1-S4
References and Notes:
- 1.Aye Y, Li M, Long MJ, Weiss RS, Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34, 2011–2021 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Licht S, Gerfen GJ, Stubbe J, Thiyl radicals in ribonucleotide reductases. Science 271, 477–481 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Minnihan EC, Nocera DG, Stubbe J, Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc Chem Res 46, 2524–2535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyedsayamdost MR, Chan CT, Mugnaini V, Stubbe J, Bennati M, PELDOR spectroscopy with DOPA-beta2 and NH2Y-alpha2s: distance measurements between residues involved in the radical propagation pathway of E. coli ribonucleotide reductase. J Am Chem Soc 129, 15748–15749 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Uhlin U, Eklund H, Structure of ribonucleotide reductase protein R1. Nature 370, 533–539 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Nordlund P, Sjoberg BM, Eklund H, Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 345, 593–598 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Minnihan EC et al. , Generation of a stable, aminotyrosyl radical-induced alpha2beta2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc Natl Acad Sci U S A 110, 3835–3840 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ando N et al. , Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc Natl Acad Sci U S A 108, 21046–21051 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Q et al. , Glutamate 52-beta at the alpha/beta subunit interface of Escherichia coli class Ia ribonucleotide reductase is essential for conformational gating of radical transfer. J Biol Chem 292, 9229–9239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Artin E et al. , Insight into the mechanism of inactivation of ribonucleotide reductase by gemcitabine 5′-diphosphate in the presence or absence of reductant. Biochemistry 48, 11622–11629 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uppsten M, Farnegardh M, Domkin V, Uhlin U, The first holocomplex structure of ribonucleotide reductase gives new insight into its mechanism of action. J Mol Biol 359, 365–377 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Thomas WC et al. , Convergent allostery in ribonucleotide reductase. Nat Commun 10, 2653 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimanyi CM, Chen PY, Kang G, Funk MA, Drennan CL, Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. Elife 5, e07141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eriksson M et al. , Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure 5, 1077–1092 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Climent I, Sjoberg BM, Huang CY, Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2. Effects on catalytic activity and subunit interaction. Biochemistry 31, 4801–4807 (1992). [DOI] [PubMed] [Google Scholar]
- 16.Ravichandran K et al. , Glutamate 350 Plays an Essential Role in Conformational Gating of Long-Range Radical Transport in Escherichia coli Class Ia Ribonucleotide Reductase. Biochemistry 56, 856–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greene BL, Stubbe J, Nocera DG, Photochemical Rescue of a Conformationally Inactivated Ribonucleotide Reductase. J Am Chem Soc 140, 15744–15752 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sintchak MD, Arjara G, Kellogg BA, Stubbe J, Drennan CL, The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat Struct Biol 9, 293–300 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Bollinger JM et al. , Mechanism of Assembly of the Tyrosyl Radical-Diiron(III) Cofactorof E. coli Ribonucleotide Reductase. 3. Kinetics of the Limiting Fe2+ Reaction by Optical, EPR, and Moessbauer Spectroscopies. J Am Chem Soc 116, 8024–8032 (1994). [Google Scholar]
- 20.Argirevic T, Riplinger C, Stubbe J, Neese F, Bennati M, ENDOR spectroscopy and DFT calculations: evidence for the hydrogen-bond network within alpha2 in the PCET of E. coli ribonucleotide reductase. J Am Chem Soc 134, 17661–17670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nick TU et al. , Hydrogen bond network between amino acid radical intermediates on the proton-coupled electron transfer pathway of E. coli alpha2 ribonucleotide reductase. J Am Chem Soc 137, 289–298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Worsdorfer B et al. , Function of the diiron cluster of Escherichia coli class Ia ribonucleotide reductase in proton-coupled electron transfer. J Am Chem Soc 135, 8585–8593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reece SY, Nocera DG, Proton-coupled electron transfer in biology: results from synergistic studies in natural and model systems. Annu Rev Biochem 78, 673–699 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nick TU, Ravichandran KR, Stubbe J, Kasanmascheff M, Bennati M, Spectroscopic Evidence for a H Bond Network at Y356 Located at the Subunit Interface of Active E. coli Ribonucleotide Reductase. Biochemistry 56, 3647–3656 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Greene BL, Taguchi AT, Stubbe J, Nocera DG, Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J Am Chem Soc 139, 16657–16665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasanmascheff M, Lee W, Nick TU, Stubbe J, Bennati M, Radical transfer in E coli ribonucleotide reductase: a NH2Y731/R411A-alpha mutant unmasks a new conformation of the pathway residue 731. Chem Sci 7, 2170–2178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao SS et al. , A model for the role of multiple cysteine residues involved in ribonucleotide reduction: amazing and still confusing. Biochemistry 31, 9733–9743 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Yokoyama K, Uhlin U, Stubbe J, A hot oxidant, 3-NO2Y122 radical, unmasks conformational gating in ribonucleotide reductase. J Am Chem Soc 132, 15368–15379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salowe SP, Ator MA, Stubbe J, Products of the inactivation of ribonucleoside diphosphate reductase from Escherichia coli with 2’-azido-2’-deoxyuridine 5’-diphosphate. Biochemistry 26, 3408–3416 (1987). [DOI] [PubMed] [Google Scholar]
- 30.Ravichandran KR, Minnihan EC, Wei Y, Nocera DG, Stubbe J, Reverse Electron Transfer Completes the Catalytic Cycle in a 2,3,5-Trifluorotyrosine-Substituted Ribonucleotide Reductase. J Am Chem Soc 137, 14387–14395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennati M et al. , EPR distance measurements support a model for long-range radical initiation in E. coli ribonucleotide reductase. J Am Chem Soc 127, 15014–15015 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Oyala PH et al. , Biophysical Characterization of Fluorotyrosine Probes Site-Specifically Incorporated into Enzymes: E. coli Ribonucleotide Reductase As an Example. J Am Chem Soc 138, 7951–7964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge J, Yu G, Ator MA, Stubbe J, Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry 42, 10071–10083 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Zheng SQ et al. , MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penczek PA et al. , CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy 140, 9–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moriya T et al. , High-resolution Single Particle Analysis from Electron Cryo-microscopy Images Using SPHIRE. J Vis Exp, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bepler T et al. , Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Res Comput Mol Biol 10812, 245–247 (2018). [PMC free article] [PubMed] [Google Scholar]
- 38.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kucukelbir A, Sigworth FJ, Tagare HD, Quantifying the local resolution of cryo-EM density maps. Nat Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goddard TD et al. , UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Afonine PV et al. , Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams CJ et al. , MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barad BA et al. , EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morin A et al. , Collaboration gets the most out of software. Elife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown NC, Reichard P, Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J Mol Biol 46, 39–55 (1969). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.