

Abstract
Cancer patients with malignant involvement of tumor-draining lymph nodes (TDLNs) and distant metastases have the poorest prognosis. A drug delivery platform that targets the primary tumor, TDLNs, and metastatic niches simultaneously, remains to be developed. Here, we generated a novel monoclonal antibody (MHA112) against peripheral node addressin (PNAd), a family of glycoproteins expressed on high endothelial venules (HEVs), which are present constitutively in the lymph nodes (LNs) and formed ectopically in the tumor stroma. MHA112 was endocytosed by PNAd-expressing cells, where it passed through the lysosomes. MHA112 conjugated antineoplastic drug Paclitaxel (Taxol) (MHA112-Taxol) delivered Taxol effectively to the HEV-containing tumors, TDLNs, and metastatic lesions. MHA112-Taxol treatment significantly reduced primary tumor size as well as metastatic lesions in a number of mouse and human tumor xenografts tested. These data, for the first time, indicate that human metastatic lesions contain HEVs and provide a platform that permits simultaneous targeted delivery of antineoplastic drugs to the three key sites of primary tumor, TDLNs, and metastases.
Graphical Abstract
Introduction
The development of antibody-drug conjugates (ADCs) as efficient targeting agents for cancer therapy has created much excitement. Over 100 ADCs are in preclinical development, more than 60 are in clinical development, and three are Food and Drug Administration (FDA)-approved currently for cancer therapy[1–3]. ADCs consist of monoclonal antibodies (mAbs) connected by a specified linkage to antitumor cytotoxic drugs. ADCs stand in contrast to the traditional methods of cancer therapy (chemotherapy, radiotherapy, antibody immunotherapy, and targeted therapies based on nanoparticles), for which efficacy has been hindered by troubling safety profiles[4]. Indeed, ADCs have been utilized successfully for cancer therapy in recent years[5–8].
High endothelial venule (HEV) is a highly specialized blood vessel found primarily in the lymph nodes (LNs)[9]. Peripheral node addressin (PNAd) is a family of sulfated and fucosylated glycoproteins expressed exclusively on HEVs that is recognized by the monoclonal antibody, MECA79[9, 10]. Interestingly, HEVs also have been incredibly recognized to be formed ectopically within the tumor environment in numerous cancer models[11–14]. However, there is no evidence on generation of HEVs in metastatic lesions.
Remodeling of the stromal compartment of tumor-draining LNs (TDLNs), especially the novel growth of intranodal lymphatic vessels, may accelerate the spread of a tumor to more distant LNs[15, 16]. Metastases to LNs is a well-known poor prognostic factor for many solid malignancies[17, 18]. Cancer cells migrate from the primary tumor to adjacent LNs through invasion of the surrounding lymph vessels, forming an organized colony in the LNs that becomes a source of dissemination (distant metastasis) to other organs[19]. Importantly, TDLNs are major sites for mounting tumor immunity, where antigen-specific immune responses are directed against tumor cells[20]. TDLNs have been found to contain an immunosuppressive environment when metastases are present[21, 22]. TDLNs involvement can, therefore, have an impact on tumor progression in a number of ways.
In addition to the delivery of chemotherapy drugs, targeted delivery of immune checkpoint inhibitors or cancer cell-specific antigens to TDLNs also have the potential to improve cancer therapies[23–25] by eradicating metastatic disease from LNs and increasing tumor immunity, respectively[26]. However, only a small fraction of systemically delivered therapeutics accumulate in the LNs[19, 27, 28]. Several attempts have been undertaken to enhance the drug delivery pharmacokinetics to the TDLNs through administration of payloads directly to lymphatic vessels (not through the intravenous [i.v.] route) or the LNs themselves[23, 29, 30]. However, most of these strategies face significant technical limitations.
In addition to TDLNs involvement, distant metastases account for the majority of cancer-associated deaths[31, 32]. Typically, strategies such as immunotherapy, radiation therapy, surgery, or a combination thereof[33], fail to halt the progression of metastatic cancers. Furthermore, high-dose systemic chemotherapeutic drugs for metastatic cancer can cause significant toxicity and intolerance for the patients[34, 35]. Just a minority of metastasis-specific targets have been exploited therapeutically, so effective prevention as well as suppression of metastatic disease remains an elusive goal[36]. Drugs that reach well-vascularized primary tumors may not accumulate in metastases that are poorly vascularized[26, 37, 38]. Clearly, a targeted therapy platform using a simple intravenous injection that delivers a drug simultaneously to the primary tumor, TDLNs, and distant metastases would represent a major paradigm-shifting approach to improve the outcomes of lethal cancers.
Here, we isolated and characterized a new anti-PNAd monoclonal antibody called MHA112 by immunizing GlcNAc6ST-1,2,4 triple-knockout mouse, a novel PNAd-deficient model with PNAd-expressing CHO cells. MHA112 demonstrated better affinity than MECA-79, an antibody used widely for PNAd binding. Our MHA112 delivery platform permitted the targeted delivery of cytotoxic agents to the triad of crucial sites for effective antineoplastic therapy—the primary tumor, TDLNs, and distant metastases. Notably, we also demonstrated the potential utility of MHA112 as an imaging agent that can enhance the accuracy of cancer staging by increasing the sensitivity of detecting malignant involvement of the LNs. Treatment with MHA112-Taxol suppressed growth of murine breast cancer model, as well as both murine and human pancreatic cancer. Moreover, LN-targeted delivery via MHA112 restored host immunity to tumors and halted the growth of metastatic lesions as well as fibrosis within the TDLNs.
Results
1. Generation, characterization and bio-distribution of MHA112
GlcNAc6ST1,2,4 triple-knockout (TKO) mouse, a new PNAd-deficient model, was generated by interbreeding GlcNAc6ST1,2 double deficient mice[39] and GlcNAc6ST4 single deficient mice. To generate novel antibodies targeting HEVs, we immunized GlcNAc6ST-1,2,4 TKO mice three times with 1×107 PNAd-expressing CHO (CHO-PNAd) cells[40] (Fig. 1A). After single-clone screening, one clone designated as MHA112 bound successfully to CHO-PNAd cells (Fig. S1A) and HEVs in mouse LNs as well as human tonsil (Fig. 1B). The staining results were similar to commercial anti-HEV antibody MECA-79, which was used as a positive control (Fig. S1A). Isotyping ELISA results indicated that MHA112 mAb was a mouse IgM isotype antibody (Fig. S1B). Then, cell-based ELISA was performed to compare the binding affinity of MHA112 to MECA-79. CHO-PNAd antigen was coated onto 96-well plates and incubated either with MHA112 or MECA79. As shown in Fig. 1C, best-fit curves for the binding affinities of MHA112 and MECA-79 were constructed, and the Kd values were calculated as 0.97nM and 1.41nM, respectively. These results indicated that the binding affinity of MHA112 was 30% higher than MECA-79.
Figure 1. Generation, characterization and bio-distribution of MHA112.
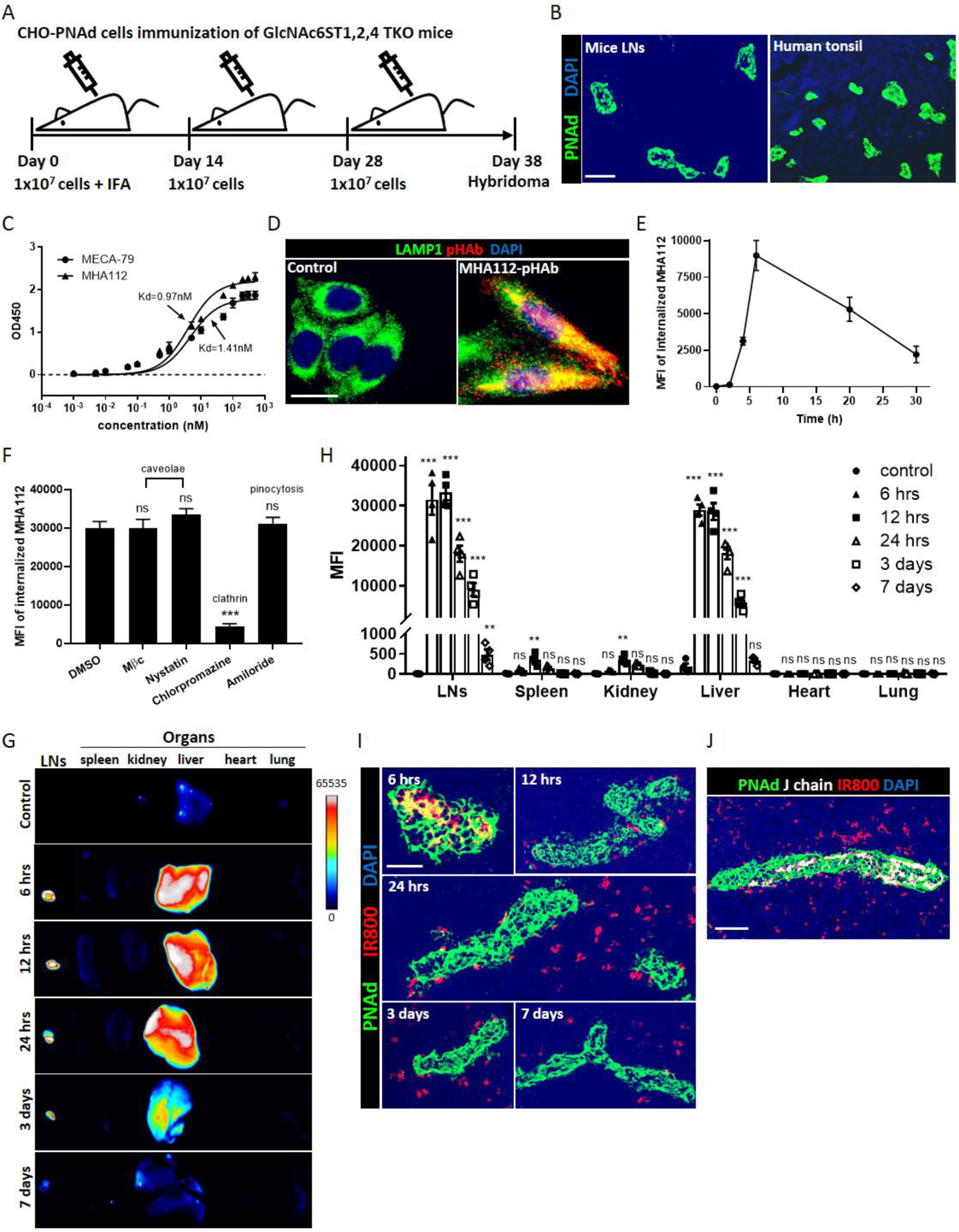
(A) Immunization Schedule. GlcNAc6ST1,2,4 TKO mice were immunized three times at intervals of 14 days (primary immunization with 1 × 107 CHO-PNAd cells and IFA, followed by boosting with 1 × 107 CHO-PNAd cells), and spleens were collected for hybridoma generation at 10 days following the third immunization. (B) Fluorescence micrographs showed HEVs (green) in mice LNs and human tonsil were stained by MHA112. DAPI (blue) was cell nuclei. Scale bar: 50μm. (C) Determination of binding affinity of MECA-79 and MHA112 mAb to CHO-PNAd by cell-based ELISA. Data are representative of three independent experiments (n=3). (D) Fluorescence micrograph showed colocalization of MHA112-pHAb (red) with lysosome marker LAMP1 (green). DAPI (blue) was cell nuclei. Scale bar: 10μm. (E) Flow cytometry MFI showed time-dependent changes in intracellular levels of MHA112 in CHO-PNAd cells. Data are representative of three independent experiments (n=3). (F) Flow cytometry MFIs of intracellular MHA112 were measured in the presence of caveolae-, clathrin- and pinocytosis-pathway inhibitors. Data are expressed as means ± SEM. ***P < 0.001, NS (no significant difference). (G-H) In vitro fluorescence imaging and semiquantitative analysis of whole organs showed the bio-distribution of MHA112-IR800 to LNs and organs at different time points. Data are expressed as means ± SEM. **P < 0.01, ***P < 0.001, NS (no significant difference). (I) Fluorescence micrographs showed the distribution of MHA112-IR800 (red) in the vicinity of HEVs (green) in the LNs at different time points. DAPI (blue) was cell nuclei. Scale bar: 20μm. (J) Fluorescence micrographs showed the distribution of J-chain (MHA112, white) and IR800 (red) in the vicinity of HEVs (green) in the LNs. DAPI (blue) was cell nuclei. Scale bar: 20μm.
One of the key aspects of ADC and drug delivery relies on the internalization of the antibody inside the target cells and transferring to the lysosomes where the linker is cleaved, and the drug of interest is released. Isolating and culturing HEV is a daunting task. We therefore examined whether PNAd expressing CHO cells can internalize the MHA112 antibody. We incubated CHO-PNAd cells with MHA112 that was labeled with pH-sensitive dye pHAb to investigate whether MHA112 can be internalized by target cells. We found that MHA112-pHAb (red) colocalized with the lysosome marker LAMP1 (green), indicating that MHA112 was located in the lysosomes after internalization (Fig. 1D). In the lysosomes, classically the drug is released from ADC following the cleavage of the linker by proteases[41]. To investigate the internalization of MHA112 by the target cells, we performed intracellular staining for flow cytometry at different time points, as described previously[42]. As shown in Fig. 1E, the intracellular levels of MHA112 rose quickly within 4 hr. of incubation, peaked around 6 hr., and slowly decreased through 30 hr. Clathrin- and caveolin-dependent pathways are the major routes for the endocytosis of a wide variety of molecules by endothelial cells [43, 44]. We investigated the identity of the internalization pathway for MHA112 in CHO-PNAd cells. Intracellular staining for flow cytometry showed that MHA112 was internalized readily by CHO-PNAd cells in the control group (DMSO) (Fig. 1F). The MHA 112 signal was not significantly different from the control group in the presence of caveolae-mediated endocytosis inhibitors [methyl-beta-cyclodextrin (MβC) and nystatin] and a pinocytosis inhibitor (amiloride). In contrast, MHA112 endocytosis was decreased dramatically by the clathrin-mediated endocytosis inhibitor chlorpromazine. These data indicated that MHA112 was internalized through a clathrin-dependent route.
Finally, we tested the LN-targeting capacity and biodistribution of MHA112 in mice by labeling MHA112 with the near-infrared marker IRDye 800CW (MHA112-IR800). First, fluorescence imaging showed that MHA112 conjugation did not impact the fluorescence of the IR800 probe (Fig. S1C). Control mice were injected with an equal amount of free IR800 dye. The mice were euthanized at time points between 6 hr. and 7 days post-injection. Whole-organ fluorescent imaging revealed that MHA112-IR800 accumulated mainly in the LNs and liver with very low signals in other organs (Fig. 1G–H). Images of MHA112-IR800 at different time points revealed significant dynamic changes. At 6 hr., the signals in the LNs and liver were the highest. Early transient accumulation in the liver was likely due to the nonspecific, innate protein degradative function of this organ. By 3 days, the signal of MHA112-IR800 in the liver faded significantly, while the signal in the LNs persisted. Histological examination of the LNs revealed the presence of MHA112-IR800 (red) inside the HEVs (green) at 6 hr. (Fig. 1I). From 12 hr. to 7 days, MHA112-IR800 dispersed from the vicinity of the HEVs (Fig. 1I). We also investigated the identity of the cells that internalized MHA112-IR800. As shown in Fig. S1D, around 40% of the IR800 signal was found in dendritic cells (DCs; CD11c+), while a small portion of IR800 was detected in macrophages (CD11b+) and fibroblastic reticular cells (FRCs; podoplanin [PDPN]+ and ER-TR7+). To assess whether the IR800 signal in the LNs was from cleaved IR800 dye or intact MHA112-IR800 conjugates, we stained the LNs of MHA112-IR800-injected mice with an antibody to the J-chain, a protein component of the IgM antibody, to indicate the location of intact MHA112. As shown in Fig. 1J, the J-chain signal was detected within the HEV cells, and the IR800 signal in the LN interstitium did not colocalize with the J-chain. All these data together indicated that MHA112-conjugates were cleaved inside HEV cells, and the cleaved conjugates were accessible to LN-resident cells.
2. Treatment with MHA112-Taxol suppressed breast tumor LN metastases and tumor growth in vivo
To examine the therapeutic potential of MHA112, we employed a xenograft tumor mouse model derived from the mammary cancer cell line 4T1. First, we established that MHA112-IR800 accumulated in TDLNs by fluorescence imaging to demonstrate its capacity to target LNs following systemic delivery in a mouse breast tumor model (Fig. 2A). We conjugated MHA112 mAbs with Taxol, a chemotherapeutic agent that interferes with the growth and spread of cancer cells. To calculate the conjugation ratio of MHA112 and Taxol, we first conjugated Oregon Green 488-labeled Taxol (Taxol*) to MHA112. Based on the molar extinction coefficients (ε) of MHA112 and Taxol* (ε MHA112: 280 nm = 1.2 × 106 cm−1M−1, ε Taxol*: 500 nm = 4.2 × 104 cm−1M−1), we confirmed that the drug-antibody ratio (DAR) was 3.01±0.23 (Fig. 2B). We also performed a high-performance liquid chromatography (HPLC) assay to evaluate the DAR of MHA112-Taxol. The DAR measured by HPLC was 2.811±0.12 (Fig. S2A), which was consistent with Oregon Green 488-labeled Taxol conjugation. 4T1 mouse mammary tumors cells were implanted in the mammary glands of mice. These mice were injected intravenously (i.v.) with MHA112-Taxol or the equivalent amount of free Taxol (0.5 mg/kg) every other day from days 9 to 31 post-implantation. A control group was injected with the same volume of phosphate buffered saline (PBS). Expansion of both the HEVs and lymphatic vasculature coincide with greater infiltration of the TDLN by the tumor[19]. Lymphatic vessel expansion is also associated with spread of the tumor to adjacent downstream LNs[45]. To examine HEVs and lymphatics expansion and metastatic lesions in the TDLNs, we stained the TDLNs with a tumor marker (pan-cytokeratin) and various vascular. As shown in Fig. 2C–D, the expansion of HEVs and lymphatic vessels in the TDLNs was lower in the MHA112-Taxol group than the free Taxol or control groups. Fewer cells stained positive for pan-cytokeratin in the MHA112-Taxol group, as compared to the other two groups, indicating that the metastatic lesions in the TDLNs were smaller (Fig. 2C–D). Therefore, MHA112-Taxol treatment reduced the metastases of 4T1 mammary tumor cells to TDLNs.
Figure 2. Treatment with MHA112-Taxol suppressed breast tumor LNs metastases and tumor growth in vivo.
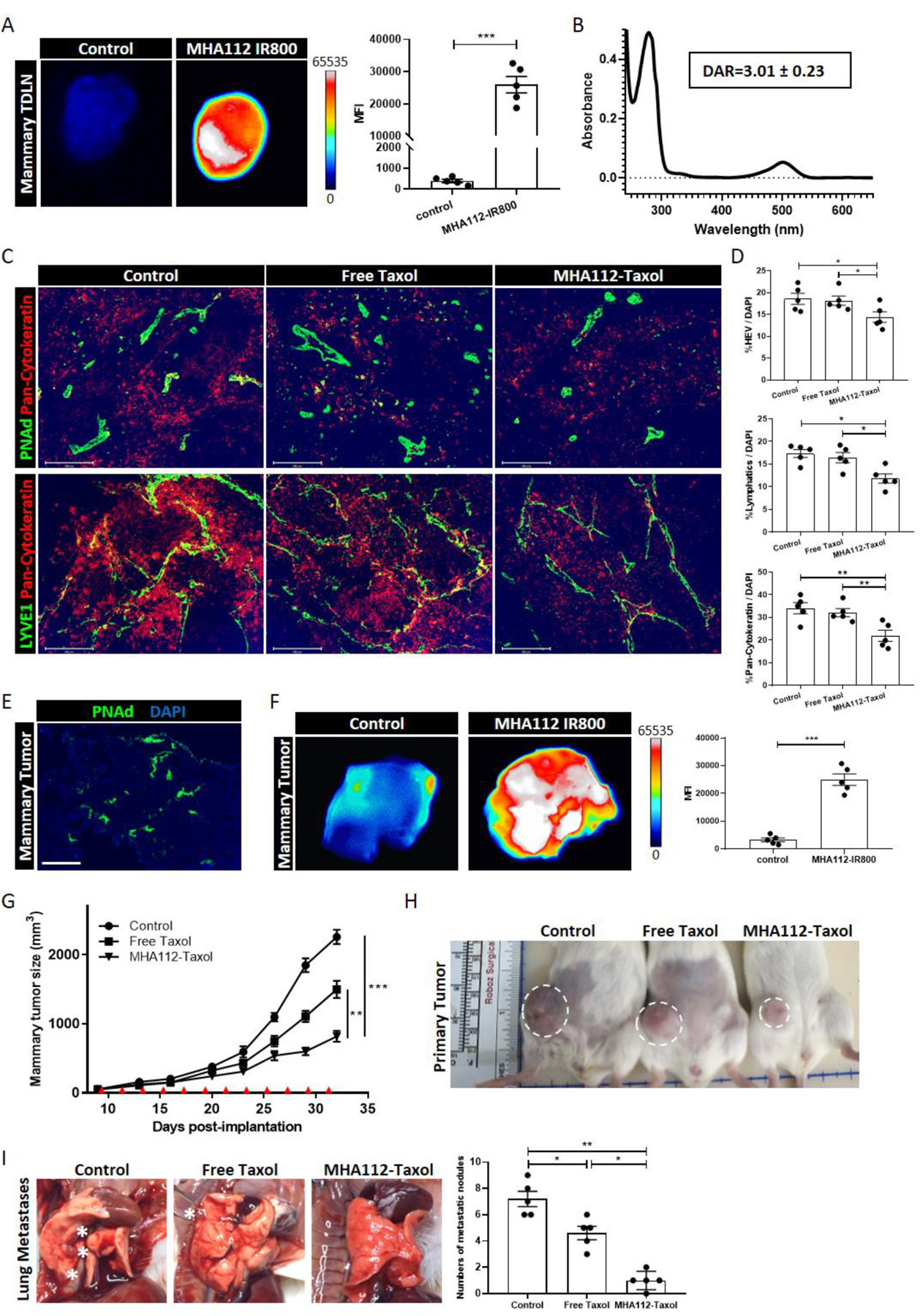
(A) Fluorescence micrographs and semiquantitative analysis showed that MHA112-IR800 mAbs accumulated more robustly in mouse mammary TDLNs at 24 hr. following i.v. injection in comparison to free IR800. ***p < 0.001. (B) The absorption of MHA112-Taxol* (Oregon Green 488-labeled Taxol) at 280nm and 500nm wavelength. Based on the molar extinction coefficients (ε) of MHA112 and Taxol* (ε MHA112: 280 nm = 1.2 × 106 cm−1M−1, ε Taxol*: 500 nm = 4.2 × 104 cm−1M−1), the drug-antibody ratio (DAR) was calculated as 3.01 ± 0.23. (C) Fluorescence micrographs of TDLNs showed less expansion of HEVs (green in upper panel), lymphatic vessels (green in lower panel), as well as fewer metastatic cancer cells (pan-cytokeratin, red) in MHA112-Taxol group than free Taxol and control groups. DAPI (blue) was cell nuclei. Scale bar: 200μm. (D) Quantification data from two independent experiments with five mice/group (n=5) are summarized in bar chart. *p < 0.05, **p < 0.01. (E) Fluorescence micrograph revealed the presence of HEVs (green) in 4T1 tumors. DAPI (blue) was cell nuclei. Scale bar: 100μm. (F) Fluorescence micrographs and semiquantitative analysis indicated that MHA112-IR800 accumulated more robustly in the 4T1 tumors at 24 hr. following iv injection in comparison to free IR800 dye. ***p < 0.001. (G) Tumor growth curve demonstrated significantly slower growth of 4T1 mouse mammary tumors in the BALB/c-WT mice treated with MHA112-Taxol (n=12) than free Taxol and control groups (n=12). Data are expressed as means ± SEM. **P < 0.01, ***P < 0.001. Red triangles indicated the injection timepoints. (H) Representative photographs showed significantly smaller size of 4T1 mouse mammary tumors in the BALB/c-WT mice treated with MHA112-Taxol than free Taxol and control groups. (I) Representative photographs and semiquantitative analysis showed fewer metastatic nodules in the lung in the MHA112-Taxol group. White stars indicated the metastatic lesions. *p < 0.05, **p < 0.01.
HEVs in some tumor tissues have been reported to be formed ectopically[11–14]. First, immunofluorescence staining revealed the presence of HEVs in 4T1 mammary tumor tissue (Fig. 2E). To further investigate whether MHA112 also accumulated in tumor tissue, mice bearing 4T1 mammary tumors were injected with MHA112-IR800 at day 25 post-implantation. MHA112-IR800 signal was significantly stronger than the free IR800 dye signal in the 4T1 tumor tissue at 24 hr. following i.v. injection (Fig. 2F). Tumor growth was suppressed significantly in the mice that received MHA112-Taxol in comparison to the mice that received free Taxol as well as those that received the vehicle (Fig. 2G–H) (**P < 0.01, ***P < 0.001). In addition, metastatic lesions in the lung were less extensive in the mice treated with MHA112-Taxol (Fig. 2I). We also stained the tumor sections of the three groups for HEVs. As shown in Fig. S2B, no significant difference of HEV density (%HEV/DAPI) was found among the three groups. Altogether, these data indicated that MHA112-Taxol suppressed the growth of the primary mammary tumor in vivo.
Next, we evaluated the role of the enhanced permeability and retention (EPR) effect on potential non-specificity in the targeting of the 4T1 tumor and TDLNs by MHA112 conjugates through the assessment of the trafficking of an isotype control antibody. Tumor-bearing C57BL/6 mice were injected with free IR800, isotype control-IR800 and MHA112-IR800 (n=4/group). Tumors, TDLNs, and organs were collected for fluorescence imaging at 24 hr. post-injection. MHA112-IR800 targeted the TDLN and tumor with significantly greater efficacy than the isotype control (Fig. S2C–D). We also compared the half-life (t1/2) of free Taxol, isotype control-Taxol, and MHA112-Taxol. Mice were injected with free Taxol*, IgM isotype control-Taxol*, and MHA112-Taxol*, and the sera were collected at different time points from 0 to 72 hr. As shown in Fig. S2E, t1/2 of isotype-Taxol* and MHA112-Taxol* were 26.72 hr. and 24.80 hr., respectively, while free Taxol* was 1.98 hrs. These data indicated that IgM conjugation increased the circulation of Taxol, but it did not affect its localization to the TDLN and tumor. Therefore, the trafficking of MHA112 to the TDLNs and tumor did not correlate to the circulation time or a passive EPR effect. These data indicated collectively that the targeting efficacy of MHA112-Taxol is related directly to the interaction between MHA112 antibody and its ligand PNAd.
3. LN-targeted delivery of Taxol via MHA112 restored host immunity to tumors and improved the fibrosis of TDLNs
Tumor cells have evolved methods to evade the immune response and suppress immune activation[46, 47], so we hypothesized that a lower spread of 4T1 to TDLNs combined with more effective delivery of Taxol to TDLNs with MHA112 would be associated with more robust anti-tumor immunity. 4T1 tumor cells were implanted in the mammary glands of mice, and these mice received MHA112-Taxol, free Taxol, or PBS. Flow cytometric analysis of TDLNs revealed higher percentages of CD8+CD44hi T cells (activated T cells; MHA112-Taxol 72.7% vs free Taxol 49.6% vs control 50.8%) and CD8+TNFα+ T cells (MHA112-Taxol 56.9% vs free Taxol 40.4% vs control 32.7%), and a lower percentage of CD4+CD25+FoxP3+ regulatory T cells (Tregs) (MHA112-Taxol 28.8% vs free Taxol 38.3% vs control 40.8%) in MHA112-Taxol group than free Taxol and control groups (Fig. 3A–B,). Moreover, the ratio of CD8+TNFα+ T cells to Tregs was significantly higher in the MHA112-Taxol group in comparison to the other two groups, indicating a more robust pro-inflammatory immune response (Fig. 3B). We also examined the immune response in the tumors. As shown in Fig. S3, these results were consistent with TDLNs. The percentages of CD8+TNFα+ and CD8+IFNγ+ T cells were significantly higher, while CD4+CD25+FoxP3+ Tregs was lower in the MHA112-Taxol group in comparison to the other two groups.
Figure 3. LN-targeted delivery of Taxol via MHA112 restored host immunity to tumors and improved the fibrosis of TDLNs.
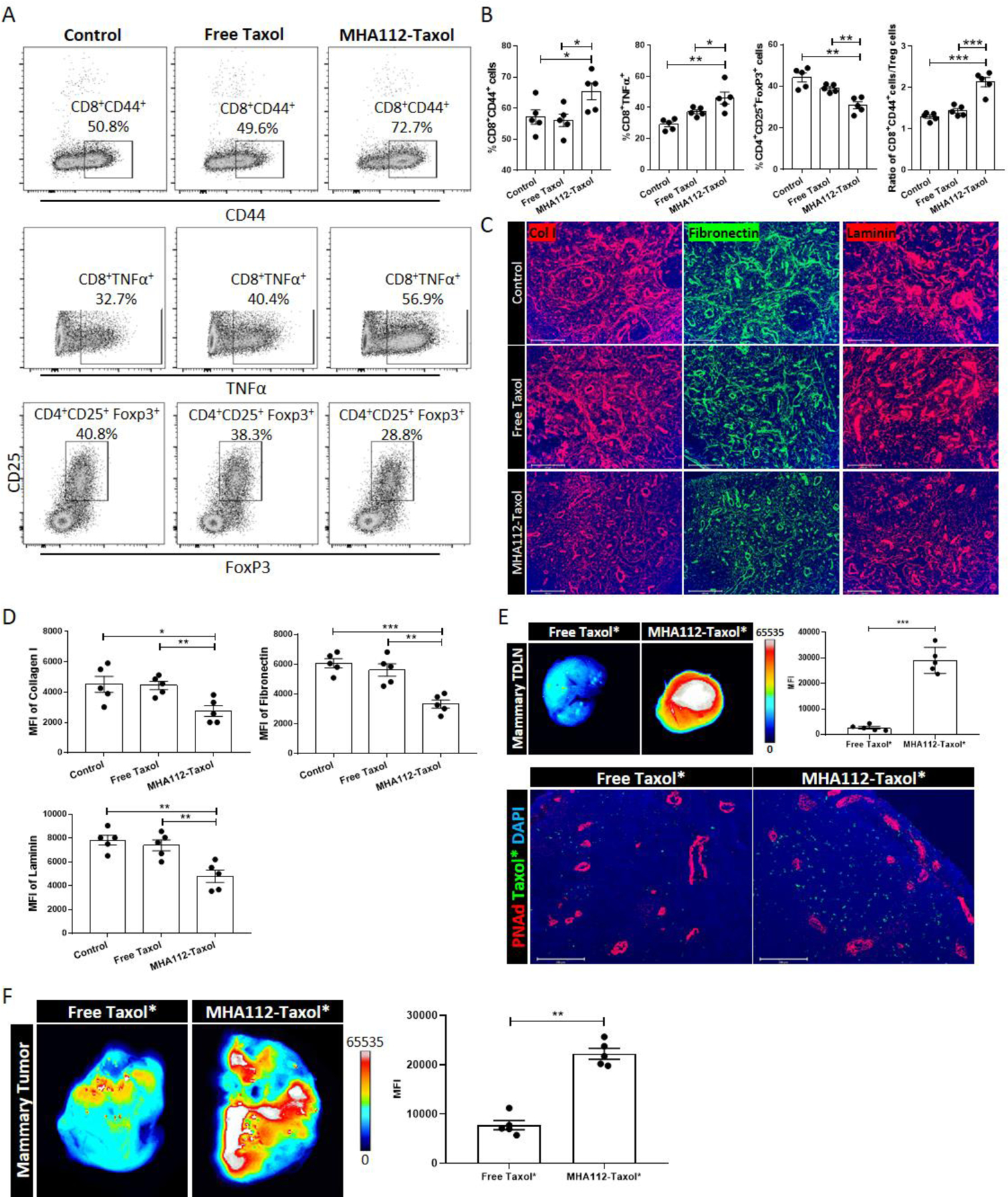
(A-B) Representative flow cytometry plots (A) and analysis (B) revealed higher percentages of CD8+CD44+ T cells and CD8+TNFα+ T cells, and lower percentage of CD4+CD25+FoxP3+ Tregs in TDLNs of MHA112-Taxol than free Taxol and control groups at 25 days post-implantation. All listed populations were gated under CD3+CD45+ cell population. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Fluorescence micrographs of TDLNs revealed that collagen I (red), fibronectin (green), and laminin (red) fibers were significantly sparser in the MHA112-Taxol group than the free Taxol and control groups. Scale bar: 200μm. (D) Quantification data of collagen I, fibronectin, and laminin from two independent experiments with five mice/group (n=5) are summarized in bar chart. *p < 0.05, **p < 0.01, ***P < 0.001. (E) Representative in vitro fluorescence images and semiquantitative analysis revealed that MHA112-Taxol* accumulated in TDLNs, and fluorescence micrographs of TDLNs revealed higher presence of MHA112-Taxol*(green) within the vicinity of HEVs (red), as compared to the free Taxol* group. Scale bar: 200μm. ***P < 0.001. (F) The MFI of tumors was significantly higher in MHA112-Taxol* group, as compared to the free Taxol* group (**p < 0.01). The data were presented as mean ± SEM.
Fibrosis formed within the tumor, referred to as desmoplastic reaction, is associated with poor prognosis[48, 49]. Such phenomenon within the TDLN has received less attention. Examination by fluorescence microscopy of the extracellular matrix (ECM) deposited by stromal cells in the TDLNs revealed that collagen I, fibronectin, and laminin fibers were less extensive in the MHA112-Taxol group (Fig. 3C–D). These data indicated that MHA112-Taxol treatment restored host immunity to the tumors and reduced ECM deposition caused by tumor metastases to the TDLNs.
Next, to assess the efficiency by which MHA112 delivers Taxol to the TLDNs and primary tumors, 4T1 mammary tumor-bearing mice were injected with either MHA112 conjugated to Oregon Green 488-labeled Taxol (Taxol*) or free Taxol* at 25 days post-implantation. At 24 hr. after injection, more MHA112-Taxol* than free Taxol* accumulated in the TDLNs (Fig. 3E, upper panel). Specifically, more MHA112-Taxol* was located specifically in the vicinity of the HEVs, as compared to free Taxol* group (Fig. 3E, lower panel). In addition, the mean fluorescence intensity (MFI) of Taxol* was significantly higher in the MHA112-Taxol* group, as compared to the free Taxol* group as shown in Fig. 3F (**p < 0.01).
4. MHA112 imaging identifies early metastasis to the TDLNs
Pursuant to the evidence that MHA112 mAbs localized to the TDLNs, we tested the capacity of MHA112 as a tool to detect metastases to the TDLNs of mammary cancer in mice. 4T1 tumor-bearing mice were injected with MHA112-IR800 at various time points post-implantation, and one group of naive mice was set aside as the control. Fluorescence imaging at 24 hr. post-implantation revealed no significant difference between MFIs of TDLNs and the control group. However, accumulation of MHA112-IR800 in the TDLNs at 48 hr. and thereafter until 2 weeks post-implantation became significantly higher than in the control group (***P < 0.001) (Fig. 4A). At 24 hr., the expansion of the HEVs in the TDLNs was not significantly different between the two groups (Fig. 4B). However, after 48 hr. post-implantation, HEVs in the TDLNs expanded significantly over time, especially in comparison to those in the naïve LNs (Fig. 4B) (**P < 0.01, ***P < 0.001). This expansion in HEVs over time was substantiated by significantly higher gene expression of the PNAd core proteins (Glycam1, CD34, Emcn [coding endomucin], Cd300lg [nepmucin], and Podxl [podocalyxin-like protein]) and the modifying enzymes Chst2 (carbohydrate [N-acetylglucosamine 6-O] sulfotransferase 2, GlcNAc6ST1), Chst4 (carbohydrate [N-acetylglucosamine 6-O] sulfotransferase 4, GlcNAc6ST2), and Fut7 (fucosyltransferase 7) in the TDLNs at 2 weeks (Fig. 4C). Moreover, immunofluorescence staining of the TDLNs of other cancers (melanoma, glioblastoma, and lung cancer) revealed that the HEVs were also similarly expanded in these mouse models (Fig. 4D). Together, these data demonstrate that MHA112 can be used to detect early malignant invasion of the TDLNs in vivo as well as to evaluate the progress of HEVs expansion in the TDLNs.
Figure 4. MHA112 imaging identifies early metastasis to the TDLNs.
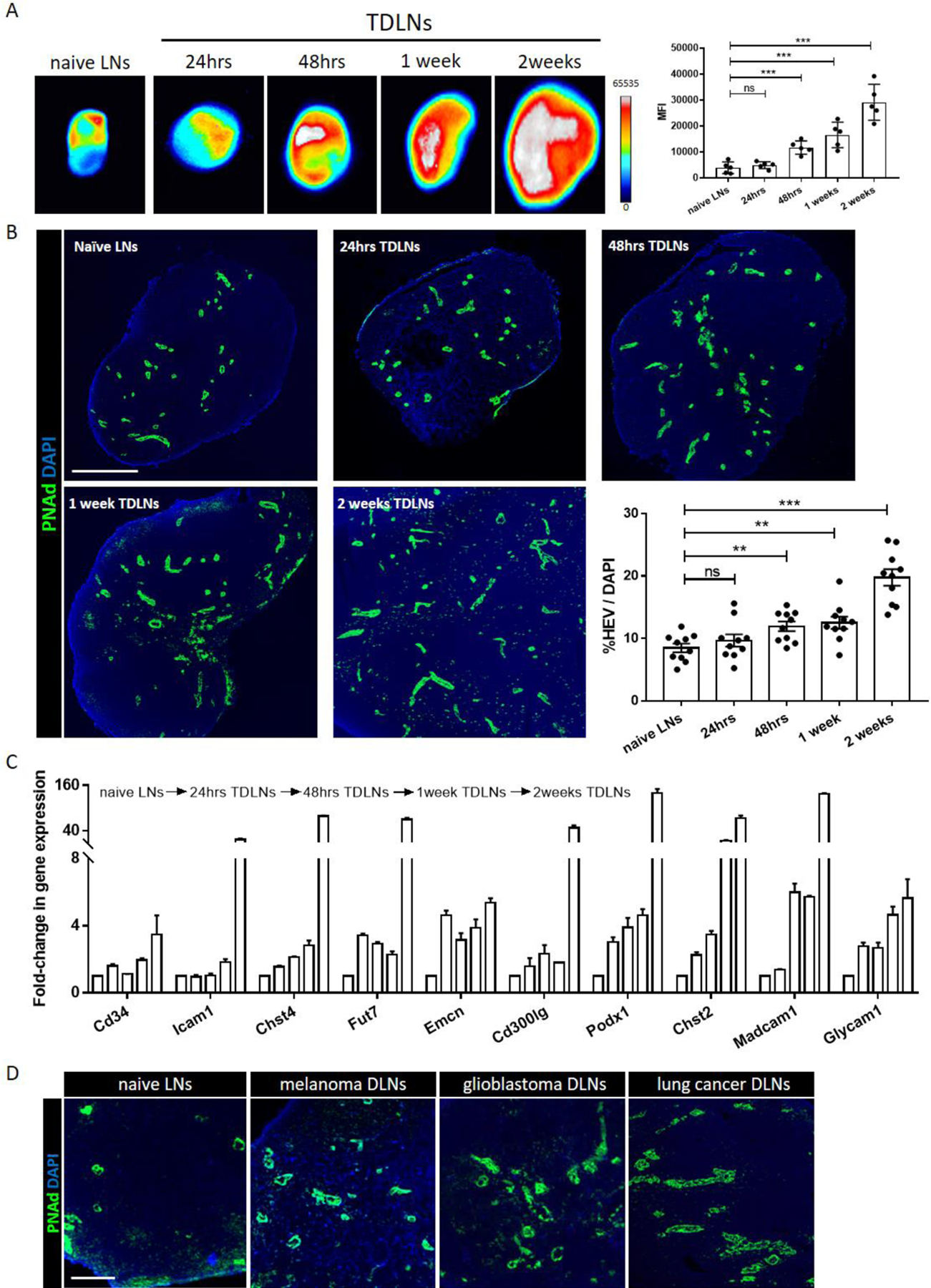
(A) Fluorescence micrographs and semiquantitative analysis of TDLNs in 4T1 tumor-bearing mice showed accumulation of MHA112-IR800 mAbs 24 hr. post-injection at 24 hr., 48 hr., 1 week, and 2 weeks post-tumor implantation in comparison to naïve LNs. ***P < 0.001, NS (no significant difference). (B) Fluorescence micrographs of TDLNs and accompanying semiquantitative analysis display expansion of HEVs (green) at 24 hr., 48 hr., 1 week, and 2 weeks post tumor implantation, as compared with naïve LNs. Scale bar: 500μm. Data were presented as mean ± SEM. **P < 0.01, ***P < 0.001, NS (no significant difference). (C) RT-qPCR analysis demonstrated that HEV-related genes were upregulated in TDLNs. (D) Fluorescence micrograph revealed the expansion of HEVs (green) in the TDLNs from melanoma, glioblastoma, and lung cancer-bearing mice. Scale bar: 100μm.
5. Treatment with MHA112-Taxol prolonged survival of mice in a metastatic breast cancer model
A platform for targeted drug delivery to metastatic lesions remains to be developed. To mimic a liver metastases model, 1.0 × 105 4T1 cells were injected directly into the portal vein. First, we confirmed the formation of HEVs in the resulting 4T1 liver masses by immunofluorescence staining (Fig. 5A). These lesions in the liver contained HEVs that arose from blood vasculature, as indicated by positive co-staining with von Willebrand factor (vWF) (Fig. 5A). These data are the first report on the HEVs expression in mouse mammary tumor metastatic lesion.
Figure 5. Treatment with MHA112-Taxol prolonged survival of mice in a metastatic breast cancer model.
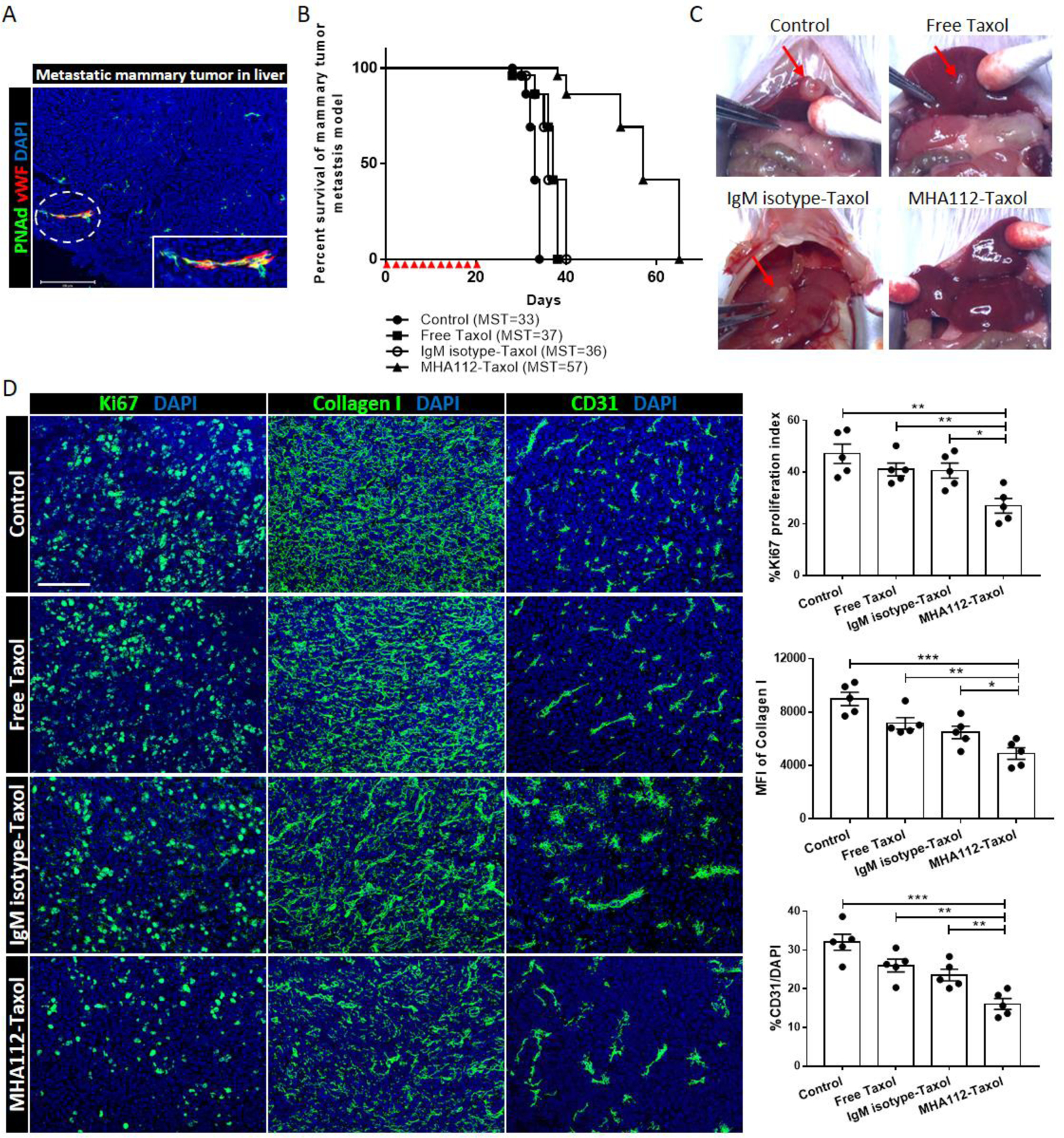
(A) Fluorescence micrograph of 4T1 tumor in the liver revealed overlapping of vWF+ blood endothelial cells (red) with HEVs (green). Scale bar: 200μm. (B) Survival curve of metastatic 4T1 breast tumor mouse model showed significantly longer survival of MHA112-Taxol-treated mice (n=5, MST=57) than the untreated control, free Taxol and IgM isotype-Taxol-treated groups (n=5, MST=33, 37, 36, respectively). Red triangles indicated the injection timepoints. (C) Representative photographs of 4T1 tumors in mouse livers showed significantly smaller size in the MHA112-Taxol group (n=5) than the other three groups (n=5). (D) Fluorescence micrographs of metastatic tumor lesions showed lower tumor cell proliferation (Ki67) and ECM accumulation (collagen I), and less expanded vasculature (CD31) in the MHA112-Taxol group than the other groups. Scale bar: 100μm. Quantification data from two independent experiments with five mice/group (n=5) are summarized in bar chart. *p < 0.05, **p < 0.01, ***p < 0.001.
We also conjugated Taxol to an IgM isotype control to exclude the nonspecific effect of protein degradation of IgM by the liver. Treatments were administered every other day during the first 20 days following injection of the 4T1 cells. The mice that received MHA112-Taxol survived significantly longer (mean survival time [day]; MST=57) than those that received no treatment, free Taxol, and IgM isotype-Taxol (MST=33, MST=37, MST=36, respectively) (Fig. 5B). Photographs of the livers captured at 20 days following injection indicated that tumors were present in the livers of the untreated, free Taxol, and IgM isotype-Taxol groups, but no macroscopic tumor was observed in the MHA112-Taxol group (Fig. 5C). Another set of identically treated mice was sacrificed 32 days following injection of 4T1 cells, and the tumors in their livers were collected for immunofluorescence staining. As shown in Fig. 5D, immunofluorescence staining of the proliferation marker Ki67 in the 4T1 lesions of the four groups indicated that the proliferation of the cancer cells was lower in the MHA112-Taxol group in comparison to the other groups (Fig. 5D). Moreover, deposition of collagen I fibers and expansion of the vasculature were also lower in the MHA112-Taxol group (Fig. 5D). These data indicated that treatment with MHA112-Taxol was effective in reducing the growth of metastatic breast cancer and prolonging survival in these mice.
6. Treatment with MHA112-Taxol suppressed human and murine pancreatic tumor progression in vivo
Pancreatic cancer has some of the highest mortality rate of all major cancers[50]. After confirming the inhibitory effects of MHA112-Taxol treatment on breast tumors model, we tested its efficacy in suppressing the growth of human and murine pancreatic tumors in in vivo mouse models to increase its robustness in treating lethal tumors and demonstrate its clinical translatability. Panc02 murine pancreatic tumor cells were implanted directly in the pancreas. Treatments were administered every other day during the first 20 days post-implantation. All mice were euthanized at 20 days post-implantation to assess the size of the primary tumor and degree of liver metastases. The pancreatic tumors were significantly smaller in the MHA112-Taxol group than the untreated and free Taxol groups (Fig. 6A–B). We also examined metastatic lesions in the liver, which were smaller in size and significantly reduced in abundance in the MHA112-Taxol group (Fig. 6C–D). Immunofluorescence staining of the primary tumor in the pancreas confirmed the presence of HEVs, which co-stained with the blood vasculature marker vWF (Fig. 6E).
Figure 6. Treatment with MHA112-Taxol suppressed human and murine pancreatic tumor progression in vivo.
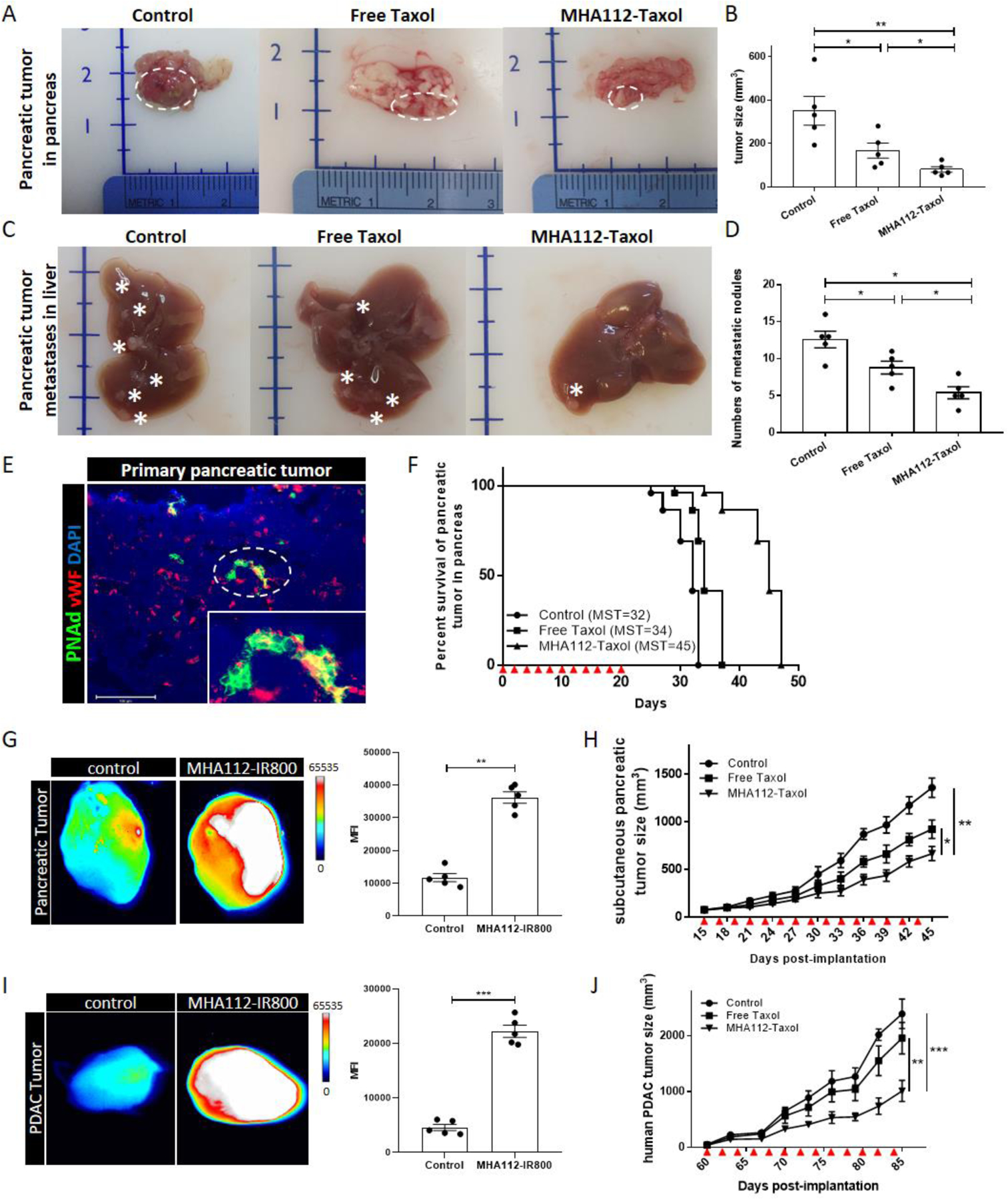
(A-B) Representative photographs (A) and analysis (B) of panc02 pancreatic cancer tumor size in mice revealed significantly smaller size in mice treated with MHA112-Taxol (n=5) than control and free Taxol groups (n=5). Data were presented as mean ± SEM. *P < 0.05, **P < 0.01. (C-D) Representative photographs (C) and analysis (D) of metastatic lesions of panc02 in liver showed significantly fewer metastatic nodules in MHA112-Taxol groups. Stars indicated metastatic lesions. Data were presented as mean ± SEM. *P < 0.05. (E) Fluorescence micrograph of primary panc02 tumor in mice revealed the presence of HEVs (green) that co-stain with blood endothelial marker vWF (red). Scale bar: 200 μm. (F) Survival curve of mice implanted with panc02 tumors in the pancreas showed significantly longer survival of those treated with MHA112-Taxol (n=5, MST=45) in comparison to control and free Taxol (n=5, MST=32, MST=34, respectively). Red triangles indicated injection timepoints. (G) Fluorescence micrographs and semiquantitative analysis showed that MHA112-IR800 accumulated more robustly in the pancreatic tumor 24 hr. following iv injection, as compared with free IR800 dye. **P < 0.01. (H) Tumor growth curve demonstrated slower growth of Panc02 mouse pancreas tumors implanted subcutaneously in the C57BL/6-WT mice treated with MHA112-Taxol (n=10) than control and free Taxol groups (n=10). Data were presented as mean ± SEM. *P < 0.05, **P < 0.01. Red triangles indicated the injection timepoints. (I) Fluorescence micrographs and semiquantitative analysis showed that MHA112-IR800 accumulated more robustly in PDAC tumor 24 hr. following i.v. injection, as compared with free IR800 dye. ***P < 0.001. (J) Tumor growth curve showed that the size of PDAC human pancreatic tumors injected subcutaneously in NSG mice treated with free Taxol and MHA112-Taxol (n=6) were significantly smaller than the control group treated with PBS (n=6). Data were presented as mean ± SEM. **P < 0.01, ***P < 0.001. Red triangles indicated the injection timepoints.
Next, another set of three similarly treated groups of mice were designated for assessment of survival. As shown in Fig. 6F, mice that received MHA112-Taxol survived longer (MST=45) than those that were received PBS (control) or free Taxol (MST=32 and MST=34, respectively). Then, Panc02 cells were implanted subcutaneously to assess directly the effect of MHA112-Taxol on the progression of pancreatic tumor growth. First, we assessed the MHA112 accumulation in mouse pancreatic tumors by fluorescence imaging in Fig. 6G. Mice received the designated treatments every other day from 15 to 44 days following tumor implantation, and the tumors grew more slowly in the mice treated with MHA112-Taxol than those treated with free Taxol or PBS (Fig. 6H).
Finally, we conducted a similar experiment in a patient-derived xenograft tumor of a surgically resected pancreatic ductal adenocarcinoma (PDAC). PDAC tumors were implanted into humanized NOD scid gamma (NSG) mice, which lack LNs. The mice were randomized on the basis of tumor size. Similar to mouse pancreatic tumor, MHA112 also highly accumulated in human PDAC tumor in Fig. 6I. Fig. 6J showed that the PDAC tumors grew more slowly in the mice treated with MHA112-Taxol (equivalent to 0.5 mg/kg of free Taxol) than those treated with free Taxol or PBS. Together, these findings demonstrated that treatment of human and mice pancreatic cancer with MHA112-Taxol was significantly more effective than treatment with free Taxol.
7. HEV expansion of human TDLNs and presence in human primary tumors and metastatic lesions
To further investigate the use of MHA112 for human cancer therapy, we performed immunohistochemical staining on human TDLNs and human tumor tissues (pancreatic cancer, ovarian cancer, prostate cancer, and gastric cancer). We found that HEV expanded in all human TDLNs tested (Fig. 7A) and that human primary tumor tissues also contained HEVs (Fig. 7B). Consistent with mouse mammary tumor, human primary breast tumor and lung metastatic lesion also had HEV expression (Fig. 7C). In our previous study, we had shown that the PDAC primary tumor contains HEVs[51]. Here, PDAC cells were injected directly into the portal vein of NSG mice (Fig. 7D) to examine if the metastatic lesion of PDAC also has HEVs. In Fig. 7E, The PDAC lesion in the liver showed HEVs structures. Moreover, MHA112-IR800 localized to the vicinity of the HEVs in the PDAC metastatic tumor (Fig. 7E). In Fig. 7F, pancreatic metastatic tumor in human duodenum also showed HEVs, which was the first time reported in metastatic lesions of human cancer. These data indicated that the expansion of HEVs in human TDLNs and the presence of HEVs in various human tumors underlies the potential of MHA112 for translation as both a diagnostic and therapeutic agent to human studies.
Figure 7. HEV expansion of human TDLNs and presence in human primary tumors and metastatic lesions.
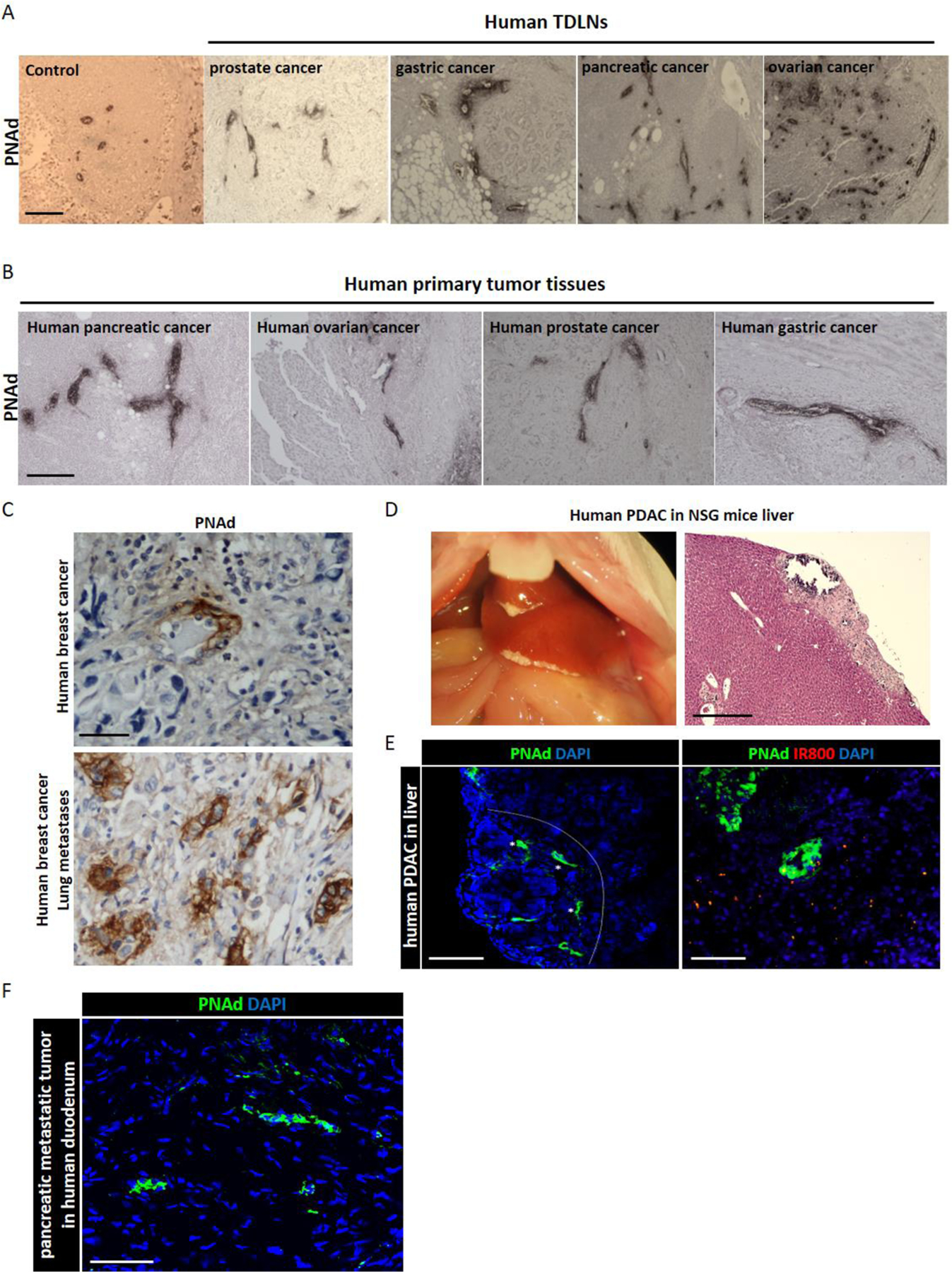
(A) Light micrographs of immunohistochemical staining of TDLNs of various human cancers revealed the expansion of HEVs in pancreatic cancer, ovarian cancer, prostate cancer, and gastric cancer. Scale bar: 200μm. (B) Light micrographs of immunohistochemical staining of primary tumors of various human cancers revealed the presence of HEVs in pancreatic cancer, ovarian cancer, prostate cancer, and gastric cancer. Scale bar: 200μm. (C) Light micrographs of immunohistochemical staining of human primary breast cancer and lung metastases lesion revealed the presence of HEVs. Scale bar: 50μm. (D) Representative photograph and light micrograph of H&E staining of PDAC tumor in liver of NSG mouse. Scale bar: 500μm. (E) Fluorescence micrographs of metastatic PDAC lesions in liver reveal MHA112-IR800 trafficking (red, scale bar: 100μm) to HEVs (green, scale bar: 200μm). (E) Fluorescence micrograph of human pancreatic metastatic lesion in the duodenum revealed the presence of HEVs (green). Scale bar: 100μm.
Discussion
In this study, we generated a new antibody called MHA112 that targets PNAd expressed by HEV. MHA112 was isolated from a novel PNAd-deficient mouse model that had been immunized with PNAd-expressing CHO cells. MHA112 mAbs showed 30% higher binding affinity compared to MECA-79 and cross activity with human HEVs. The effect of ADCs relies on the internalization of targeting antibody into endosomes that subsequently mature and fuse with lysosomes[52, 53]. In the lysosomes, the drug is released via cleavage of the linker by specific proteases or by the degradation of the ADCs. Free drug released from ADCs can cross the plasma membrane to access the extracellular milieu and kill neighbor cells by a process called the bystander effect[54–56]. While isolating HEV endothelia cells and culture are extremely difficult, using PNAd-expressing CHO, we showed that, MHA112 was internalized by target cells and localized in lysosomes in vitro. In vivo study, we showed that MHA112 has strong LN-targeting activity through HEVs binding. The cleaved conjugates from MHA112 can pass through HEVs endothelial cells into LNs interstitium evidenced by LN residence DCs and FRCs uptake. Clathrin- and caveolin-dependent pathways are the major routes by which endothelial cells internalize a wide variety of molecules[43, 44]. Our data showed that MHA112 was internalized via clathrin-dependent endocytosis in the target cells.
Metastasis to the TDLN is a critical prognostic parameter for patients with solid tumors[19]. Indeed, patients with cancer spreading to the LNs have a worse prognosis than those without nodal disease[19, 57]. These metastatic foci in the LNs are extremely difficult to treat, as a small fraction of systemically delivered drugs accumulates in LNs[27], even with high-dose systemic chemotherapeutic drugs, which can also cause significant toxicity and intolerance for patients[34, 35]. Here, we sought to understand whether MHA112-assisted delivery would improve the trafficking of Taxol to TDLNs. Our data showed that Taxol accumulated more robustly in the TDLNs following conjugation to MHA112, as compared with administration of free Taxol. Concentrating Taxol within the LNs can target rapidly growing metastatic cancer cells and kill them directly. Spread of cancer cells from TDLNs is an important mechanism for the formation of distant metastasis and tumor immunity[19, 58, 59]. In addition, an increase in fibrosis within the tumor microenvironment has been shown to occur via activation of cancer-associated fibroblasts (CAFs), promoting tumor progression[60, 61]. The impact of fibrosis in TDLNs is not as clear. TDLN fibrosis could contribute to poor chemotherapy drug penetration[62–64]. Interestingly, we noted a significant amount of fibrosis in the LNs, contributing potentially lesser to poor penetration of systemically administered Taxol in our experiments. Notably, this fibrosis was significantly less extensive in the mice that were treated with MHA112-Taxol. A nanoparticle formulation of albumin-bound Taxol (Abraxane, or nab-paclitaxel) was found previously to interfere with the function of CAFs in a rat model of intrahepatic cholangiocarcinoma[65] as well as in human pancreatic cancer patients[66], and it also was observed to disrupt the migration of CAFs in vitro[67]. A modified nanoparticle formulation of Taxol also inhibited the proliferation of CAFs in a murine pancreatic cancer model[68]. Some cleaved drugs from MHA112 were internalized by FRCs in TDLNs in our study, and we have demonstrated the importance of FRCs to LN fibrosis in previous studies[69–71]. However, whether inhibition by Taxol of ECM secretion by FRCs leads to the reduced fibrosis we observed in the TDLNs of mice treated with MHA112-Taxol is an important question that requires further investigation. Previous studies have examined the link between the formation of tertiary lymphoid organs (TLOs) and cancer outcomes[72]. We are interested in conducting future studies to quantify TLOs as well as HEVs, and examining their association with the progression of slow-growing tumors in mice.
LNs are extremely compartmentalized organs where naïve T cells home via HEVs to interact with potential antigens presented by LN-resident DCs[70, 73]. Mounting tumor immunity within TDLNs can not only suppress the tumor burden within the TDLNs, but also reduce spreading to distant peripheral organs[16, 74–76]. In this study, we found that localization of Taxol inside the LNs was associated with augmentation of the cytotoxic immune response, as demonstrated by increases in the populations of activated and TNFα-secreting CD8+ T cells and inhibition of the anti-inflammatory immune response, as evidenced by a decrease in the population of Tregs. Prior studies have shown that chemokines secreted by the tumor cells within the TDLNs promote recruitment of Tregs, creating a vicious cycle of immunosuppression[73, 77–80] and constituting one mechanism by which a high density of Tregs is correlated with poor prognosis for many cancers[81–83]. Increasing the destruction of the cancer cells within the TDLNs via MHA112-assisted drug delivery may permit recovery of their stromal compartments and the mounting of an effective anticancer immune response. The activity of DCs has been identified as fundamental to the effectiveness of the anticancer immune response[84, 85] as well as immune checkpoint inhibitor therapy[86]. The previous studies have demonstrated that in vitro treatment of DCs with Taxol boosts their capacity for CD8+ T cell proliferation[87], an effect similar to the results we observed in our in vivo model. Other studies showed that Taxol promoted the differentiation of myeloid-derived suppressor cells to DCs[88] and upregulated the expression of MHC class II by DCs[89]. Maier et al. showed recently that DCs that internalize tumor-associated antigens adopt an immunosuppressive phenotype marked by expression of programmed death-ligand 1, but this phenotype can be reversed by blockade of IL-4 signaling[90]. The accumulation of cleaved drugs from MHA112 within DC population could increase their allergenicity mounting a stronger anti-tumor immunity.
Altogether, our study highlights the importance of delivering therapeutics to LNs. LNs/lymphatic delivery approach has been the subject of several past studies, but many challenges have hindered its success. A vast majority of the current methods for targeting LNs rely on injection of the payloads into the surrounding skin and passive transport through the lymphatics[91]. Numerous factors, including the size of the injected particles or drugs, determine whether they extravasate via capillaries or lymphatics[91, 92]. Some have attempted to target mediastinal lymph nodes by injecting the payloads into the peritoneum of animals and relying on passage through the abdominal lymphatics[93]. Others have injected payloads directly into the LNs[94, 95]. Some of these routes pursued, such as direct injection or lymphatic access, may not be practical for widespread use. Furthermore, alterations of interstitial fluid pressure and lymph angiogenesis at the tumor site may interfere with the delivery of the payload. In addition, the TDLNs of many visceral or thoracic tumors are not accessible through injection into the skin. These factors underline the utility of payload delivery to the TDLNs via systemic injection. Our delivery route provides access to all LNs, since all contain HEVs. However, the TDLNs will receive a higher amount of the payload, as they contain HEVs that are more expanded. Therefore, our platform has the remarkable capacity to amplify the accumulation of the payload in TDLNs indiscriminately, downstream LNs, and other LNs throughout the body that have been infiltrated by the cancer and contain expanded HEVs. Whereas we used antibody-coated nanoparticles previously as vehicles for the therapeutic agents in this HEV-targeting platform[96], here we conjugated the therapeutic agent directly to the antibody, thereby increasing its translatability to clinic. Indeed, MHA112 permits the conjugation of a wide range of drugs other than chemotherapeutic agents, including the delivery of immune checkpoint inhibitors or antifibrotic agents, thereby boosting their anticancer efficacy. Attempts have also been made to increase HEV formation[97, 98]. Therefore, combining a strategy to increase HEVs with the delivery of chemotherapeutic drugs may form a synergistic route for future cancer therapy.
The formation of HEVs at the primary tumor site has been noted in previous studies. HEVs can be formed in the stromal compartment of solid tumors, including melanomas, breast cancer, ovarian cancer, lung cancer, and PDAC[11–14]. Here, we report that HEVs are formed in primary murine breast tumors. Importantly, we also confirmed the presence of HEVs in various human cancers, such as PDAC and its metastatic lesions, as well as human pancreatic cancer, ovarian cancer, prostate cancer, and gastric cancer. To add more rigor and translation, the human PDAC model was utilized in NSG mice to confirm direct tumor targeting by MHA112, due to a deficiency of LNs in these mice. Tumor growth was suppressed markedly by treatment of these mice with MHA112-Taxol. The survival curve following treatment of these mice showed that MHA112-Taxol prolonged survival significantly in comparison to treatment with free Taxol or IgM isotype-Taxol. In addition, tumor proliferation, tumor-associated ECM deposition, and expansion of the vasculature were also inhibited significantly by treatment with MHA112-Taxol. Notably, PNAd molecules are conserved amongst all mammals, which also emphasizes the clinical translatability of our MHA112-based delivery platform.
In addition to invasion of TDLNs by cancer, distant metastasis constitutes a major challenge to the efficacy of cancer treatment. Distant metastasis is responsible for more than 90% of cancer associated death, as these lesions are extremely difficult to treat[31, 99, 100]. Typically, effective treatment of metastatic cancer requires systemic therapy to reach cancer cells throughout the body. Increasing the dosage of drugs in order to achieve therapeutic concentrations within metastatic masses results in toxicity and is not tolerated often by patients[24]. Therefore, increasing drug penetration and accumulation specifically at metastatic sites is a new strategy to minimize systemic toxicity[101]. Here, we have reported for the first time the formation of HEVs in metastatic lesions in both mouse and preclinical human cancer models. Future studies are required to understand the mechanism by which HEVs form in metastatic masses, perhaps from the proliferation of circulating progenitor cells or the effect of molecules secreted by cancer cells that transform the stromal compartment of the surrounding tissue to support the creation of HEVs. Interestingly, our data showed that treatment with MHA112-Taxol was effective in reducing the growth of metastatic lesions comprised of both breast and pancreatic cancer.
Furthermore, MHA112 permits the detection of the primary tumor, TDLNs, and distant metastases. Detection of the cancer cells in the TDLNs as well as distant metastases is extremely important to accurate staging[102, 103]. Since healthy peripheral organs do not have HEVs, the identification of metastatic masses in peripheral tissues by MHA112 conjugated to a tracer, due to the de novo presence of HEVs can alter the treatment course dramatically for these patients from the time points of proper staging to metastasis removal. Due to the presence of HEVs in nascent primary tumor and metastatic sites, our HEV-targeting strategy could also strengthen the sensitivity of early tumor detection. Thus, conjugation of a radiographic marker to MHA112 would afford a significant opportunity for future molecular imaging.
Most ADCs are designed directly against specific antigens on tumor cells, so they are limited to select groups of antigen-positive patients. Antigen loss or antigen-low escape may constitute large obstacles to treatment success in solid malignancies, which display high heterogeneity in target antigen expression[2, 104, 105]. MHA112 permits superior delivery of Taxol to the key sites of the primary tumor, TDLNs, and metastatic lesions. Poor delivery kinetics of chemotherapy drugs to TDLNs, tumor, and metastatic deposits has constituted a key obstacle to effective direct killing of tumor cells by chemotherapy drugs[27, 28]. Taxol kills tumor cells through induction of apoptosis[106]. Accordingly, our data indicate that cancer cells experience a much higher rate of death. Furthermore, our mechanistic data indicate that the anti-tumor immune response in the TDLN was enhanced. Notably, the LN stroma exhibited a less fibrogenic or desmoplastic phenotype as well. This normalization of the LN stroma should support the mounting antitumor immune response. Overall, our innovative HEV-targeted platform provides a novel approach for simultaneous delivery of a payload to three different important sites—primary tumor, metastatic LNs, and metastatic lesions in distant organs—for effective cancer therapy, which constitutes a major urgent clinical need.
Materials and Methods
Mice
All animal experiments and methods were performed in accordance with the relevant guidelines and regulations approved by the Institutional Animal Care and Use Committee of Brigham and Women’s Hospital in Boston, MA (protocol number: 2016N000167/04977). C57BL/6J (WT) (#000664), BALB/c (WT) (#000651) mice, and NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NSG) (#005557) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and used at 8–10 weeks of age. GlcNAc6ST1,2,4 TKO mice were generated by crossbreeding GlcNAc6ST1,2,4 triple heterozygous mice that were produced by interbreeding GlcNAc6ST1/GlcNAc6ST2 doubly deficient mice[39] and GlcNAc6ST4 single deficient mice[107].
Cell lines and cell culture
CHO-PNAd cells were generated as described previously[40]. 4T1 mouse breast cancer cells were purchased from American Type Culture Collection (VA, USA). Panc02 mouse pancreatic cancer cells were provided by Dr. Claudia Gravekamp, Albert Einstein College of Medicine (New York, NY). CHO-PNAd cells were cultured in α-MEM with 10% FBS and 1% penicillin/streptomycin (pen/strep). 4T1 cells were cultured in RPMI-1640 medium with 10% FBS and 1% pen/strep. Panc02 cells were cultured in McCoy’s medium with 10% FBS, glutamine (2 mM), non-essential amino acids, sodium pyruvate (1 mM), HEPES (10 mM), and pen/strep (100 U/ml). Cells were maintained in a 37°C incubator at 5% CO2.
Immunization and hybridoma generation
GlcNAc6ST-1,2,4 TKO mice were immunized with CHO-PNAd cells every other week for 6 weeks. In brief, cell pellet preparation containing 107 cells was emulsified in Incomplete Freund’s Adjuvant (IFA) and administered by intraperitoneal (IP) injection. Mice were bled after each boost, and ELISA was used to monitor anti-HEV antibody titers in the serum. The spleens were collected, and cells were released by gentle pressure applied to the capsule of the organ, which was placed between two frosty glass slides. Splenocytes and Myeloma SP2/0 cell line were mixed in a 1:1 ratio, and the hybridoma were generated by electrical cell fusion. The cells were suspended in 15% FBS-RPMI medium (containing 10% BM-Condimed, PS, 2ME, 1x HAT) at the concentration of 1–2 × 105 total cells/ml and seeded in 96-well plates. ELISA against CHO-PNAd was performed. Hybridoma medium alone and secondary antibody served as the negative controls. Immune polyclonal serum and anti-HEV antibody served as the positive controls. Antibodies were plated directly from culture supernatants. Cells from the positive wells were subcloned by limiting dilution to obtain monoclonal lines.
ELISA
CHO-PNAd (2×104) cells were coated onto 96-well plates and incubated overnight at 37°C. After five washes with PBS buffer, the plates were fixed with 4% paraformaldehyde and blocked with 3% BSA. The antibody dilutions starting at 1ng/ml to 300μg/ml were then added to the wells. After five washes in PBST, secondary antibodies, HRP–conjugated goat anti-mouse (31430, Invitrogen) and anti-rat (31470, Invitrogen) were added at a 1:10,000 dilution and incubated for 1 h at room temperature. Binding was detected with the addition of TMB substrate (N301, Thermo Scientific), and the reaction was stopped by adding TMB Stop Solution (N600, Thermo Scientific). Absorbance signals were read at 450 nm. Isotyping ELISA was performed using Rapid ELISA Mouse mAb Isotyping Kit (37503, Thermo Scientific).
Antibody internalization assay
MHA112 antibody was labeled with pHAb Reactive Dyes (G9841, Promega) and incubated with CHO-PNAd cells for 2 hr. at 37°C. After five washes with PBS buffer, CHO-PNAd cells were stained with Lysosomal Staining Kit (ab112137, Abcam). CHO-PNAd cells were fixed with 4% paraformaldehyde, and DAPI (VECTASHIELD, Vector Laboratories) was used to counterstain the cell nuclei. The cells were visualized using an EVOS™ FL Auto 2 Imaging System (Thermo Fisher Scientific).
Immunofluorescence staining
8-μm tissue sections were cut by cryo-sectioning and stained with conjugated or purified antibodies. Purified antibodies were detected using secondary antibodies. The antibodies included MECA-79 (sc-19602, SCBT), anti-CD11c (117301, BioLegend), anti-CD11b (101202, BioLegend), anti-LYVE1 (ab14917, Abcam), anti-αSMA (19245S, CST), anti-CD31 (14-0311-82, Invitrogen), anti-Collagen I (ab34710, Abcam), anti-Collagen IV (NBP1-91258, Novus), anti-Laminin (ab11575, Abcam), anti-pan-cytokeratin (AE1/AE3, sc-81714, SCBT). DAPI (VECTASHIELD, Vector Laboratories) was used to counterstain the cell nuclei. The stained tissue sections were visualized using an EVOS™ FL Auto 2 Imaging System (Thermo Fisher Scientific). Quantification was performed on 2–3 sections from at least 3 separate mice using image analysis software Celleste (Invitrogen) and ImageJ (NCBI, 1.8.0_112).
In vivo biodistribution studies of antibody.
MHA112 antibody was labeled with IRDye 800CW Protein Labeling Kits (928–38040, LI-COR). C57BL/6 mice were used for biodistribution studies. 100 μg of MHA112-IR800 was administered iv via retro-orbital injection. Trafficking of fluorescent MHA112-IR800 was detected using a UVP iBox Explorer Imaging Microscope, equipped with a 750-to-780-nm excitation filter and an 800-nm long-pass emission filter. LNs and organs were collected and analyzed for the evaluation of the biodistribution of MHA112.
Tumor implantation
Mice were anesthetized with isoflurane, and tumor cell lines (4T1and Panc02) were gently injected subcutaneously in the mammary glands or flanks of mice. 1 × 105 cells were injected per mouse for the 4T1 tumor model. 2×106 cells were injected per mouse for the Panc02 tumor model. 1×104 4T1 cells were used for portal vein injection. 2 × 106 Panc02 cells were used for pancreas implantation. Human PDAC tumors for research purposes were collected at the University of Massachusetts Medical School under informed consent. The specimens were completely anonymous, had no direct identifiers, and no codes or indirect identifiers that linked back to the subjects. The human PDAC tumor was cut into 3–5 mm3 pieces with a razor blade on a sterilized petri dish. A small incision was made in the skin on the lower back of NSG mice, and the PDAC tumor was implanted subcutaneously. The tumor growth was monitored three times per week by digital caliper (Fisherbrand™ Traceable™ Digital Calipers).
Flow cytometry
Flow cytometric analysis of TDLNs was performed, and each leukocyte population was quantified. The TDLNs were placed onto a 70-μm cell strainer (BD Falcon), attached to a 50-ml conical tube. The TDLNs was mashed in sterile Dulbecco’s Phosphate-Buffered Saline (DPBS) through the strainer using the plunger end of a syringe. The single-cell suspension was centrifuged at 340 g for 5 minutes. The pellet was resuspended in complete RPMI 1640 at 1 × 107 cells/ml. Cells were plated in 96-well round-bottom plates (Corning, NY) for intracellular cytokine staining and 96-well flat-bottom plates (Corning, NY) for cell-surface and intracellular transcription factor staining. The cell samples that underwent intracellular cytokine staining were incubated first with 100 ng/ml PMA, 1 μg/ml ionomycin (Sigma-Aldrich), and GolgiStop™ protein transport inhibitor (BD Bioscience) at 37°C for 4 hr. All samples were washed with DPBS prior to incubation with Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) diluted 1:1000 in DPBS for 30 min at 4°C. Then, the cells were washed with FACS buffer (DPBS + 2% fetal bovine serum +1 mM EDTA + 0.1% sodium azide) and incubated for 30 min at 4°C with the following cell-surface antibodies: PB anti-CD4 (100428, Biolegend), BV510 anti-CD8 (100752, Biolegend), APC anti-CD44 (103012, Biolegend), PE/Cy7 anti-CD62L (104418, Biolegend), PE anti-CD25 (558642, BD Pharmingen), BV510 anti-CD45 (103138, Biolegend). All the cell-surface antibodies were diluted 1:300 in FACS buffer. The cells were permeabilized using the eBioscience Intracellular Fixation and Permeabilization Buffer Set (Thermo Fisher Scientific) for 30 min at 4°C. Then, they were incubated with the following intracellular antibodies: PerCP/Cy5.5 anti-FoxP3 (45-5773-82, Invitrogen), APC anti-IFNγ (505810, Biolegend), FITC anti-TNFα (506304, Biolegend). All of the intracellular antibodies were diluted 1:300 in the eBioscience Permeabilization Buffer (1x) (Thermo Fisher Scientific). Cells were washed once with Permeabilization Buffer and fixed in FACS buffer containing 1% formalin. Flow cytometry was performed using a BD FACSCanto™ II flow cytometer (BD Biosciences). Analysis of flow cytometry results was performed via FlowJo software (FlowJo LLC, Ashland, OR).
RT-PCR assay
RNA was isolated with Quick-RNA MiniPrep kit (Zymo Research, Irvine, CA, USA), and the first strand of cDNA was synthesized using 2μg of RNA and High-Capacity Reverse Transcriptase (Invitrogen). RT-PCR was performed with SYBR Green PCR reagents. RNA levels were normalized to the level of GAPDH and calculated as delta-delta threshold cycle (ΔΔCT). Primers used for RT-PCR are listed as follows: GAPDH-F:AGCCACATCGCTCAGACAC, GAPDH-R:GCCCAATACGACCAAATCC; Fut7-F:AGCTGGAGGAGCAACATTCAT, Fut7-R:GGATGGTGAGTGTGGACTGAG; Chst2-F:CCGCTCGGGATGAAGGTATTT, Chst2-R:CCACTTGTAGTCCAAGAGGTTGA; Chst4-F:GGGTTCCCAGGTCATCGTTG, Chst4-R:CCGAAAAGCTGTCCCACAAAA; Glycam1-F:GTCCTGCTATTTGTCAGTCTTGC, Glycam1-R: CCTGGGCCTCTTGATTCTCTG; Icam1-F:GTGATGCTCAGGTATCCATCCA, Icam1-R:CACAGTTCTCAAAGCACAGCG; Madcam1-F:CCTGGCCCTAGTACCCTACC, Madcam1-R:CCGTACAGAGAGGATACTGCTG; Cd34-F:GGTAGCTCTCTGCCTGATGAG, Cd34-R:TGGTAGGAACTGATGGGGATATT; Emcn-F:AATACCAGGCATCGTGTCAGT, Emcn-R:CTGATTCTCAGTCTTGTTCTGGG; Cd300lg-F:AAAGCCCCTGTATTCACCGAG, Cd300lg-R: CCTGCATGAGGAGAGGTCG; Podxl-F:GCCACCAAAGTGCCACAAC, Podxl-R:CGGCATAGATGGAGATTGGGTT. All RT-PCR reactions were performed in triplicate.
Antibody and Taxol conjugation
Glutaric anhydride (100 mg, Sigma-Aldrich) and Taxol (33 mg, LC laboratories) were prepared in a 4 mL vial, dried under high vacuum for 24 hr. and dissolved in 1 mL of pyridine. The solution was stirred at room temperature under Ar atmosphere for 2 hr. The reaction was quenched by removal of solvent under high vacuum for 2 hr. 2’-Glutaryl taxol was purified by a reversed phase HPLC (Phenomenex Luna 5 μm C18 250 × 10.0 mm, flow rate 2 mL/min, UV 250 nm detection) with a gradient solvent system (15% to 75% ACN/H2O with 0.1% formic acid for 40 min). 2’-glutaryl taxol (0.2 mg) dissolved in DMSO (Thermo Scientific Fisher) was activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, 0.4 mg, Sigma-Aldrich) and Sulfo-NHS (N-hydroxysulfosuccinimide) (1.1 mg, Thermo Scientific Fisher) for 15 min at room temperature in MES buffer (pH 6.0, Thermo Scientific Fisher) (final solution; ~1 mL in 10% DMSO). The EDC was quenched by 2-mercaptoethanol (1.4 μL, Sigma-Aldrich) for 10 min. Immediately, the pH of solution was increased by NaHCO3 (0.1 M, Sigma-Aldrich) to ~8. MHA112 dissolved in PBS (pH 7.4, Corning) was mixed with the activated 2’-glutaryl Taxol at room temperature for 2 hr. (1:20 molar ratio of MHA112 to Taxol, final solution; 10% DMSO). Dialysis was performed twice by a centrifugal filter (Amicon®, 10 kD MWCO, Sigma-Aldrich) at 10,000 rpm for 15 min. to remove the free taxol. The solution was purified further by a desalting column (Zeba™, 7 kD MWCO, Thermo Scientific Fisher).
Determination of drug to antibody ratio (DAR) for MHA112-Taxol
Reverse-phase HPLC (RP-HPLC) was used to determine the DAR of antibody-drug conjugate. MHA112-taxol conjugates (1 mg/mL) were incubated with 40mM DL-dithiothreitol (Sigma-Aldrich, USA) for 1 hour at 37°C to reduce the interchain disulfide bond. RP-HPLC was performed on both intact and reduced conjugates using the PLRP-S column (5μm, 1000Å, 2.1× 50 mm from Agilent) coupled to Agilent 1260 Infinity II system. The samples were analyzed at 0.8 mL/min using 0.1 % TFA in a water-acetonitrile gradient maintained at 70°C. The reduced conjugate was resolved into a light and heavy chain fragments compared to the intact form. The peak area was quantified from the RP-HPLC chromatogram for the reduced MHA112-Taxol, which comprises of unconjugated light chain (L0) and heavy chain (H0) and conjugated light chain (L1) and heavy chains (H1). The experiment was done in triplicates and DAR was determined from the peak area of the reduced MHA112-Taxol conjugate using the standard equation.
Flow Cytometry for Antibody and Antibody-Drug Conjugate Internalization
CHO-PNAd cells (1 × 106 cells/ml) were incubated with 2μg/ml MHA112 for 30 min on ice, rinsed with ice-cold PBS, and then incubated for 30min. The cells were rinsed with cold PBS, resuspended in growth media, and incubated at 37°C. The samples were harvested at various times and processed for flow cytometry. To detect internalized antibody, the cells were washed with cold PBS, incubated with proteinase K (1μg/ml for 10 min at 37°C), washed to remove cell surface-bound antibody, and incubated with the FITC anti-mouse IgM antibody (ab150121, Abcam). The cells were assessed by flow cytometry using a BD FACSCanto™ II flow cytometer (BD Biosciences). Analysis of flow cytometry results was performed via FlowJo software (FlowJo LLC, Ashland, OR). For inhibitors assay, CHO-PNAd cells were preincubated with inhibitors (20μM methyl-β-cyclodextrin, 3mM amiloride, 10μg/ml chlorpromazine, 10μg/ml nystatin; Sigma) for 30 min at 4°C prior to a 3hrs incubation with MHA112 at 37°C. The cells were processed as described above for internalized antibody.
Statistical Analysis
All experiments were repeated at least three times, each done in triplicate. The statistical significance between two groups was determined by unpaired Student’s t test, whereas comparisons between multiple groups were carried out by a repeated-measures two-way analysis of variance (ANOVA) with Tukey’s test, an ordinary one-way ANOVA with Tukey’s test or Dunnett’s test, using GraphPad Prism 7 software (GraphPad Software, Inc., CA). A probability value of *P < 0.05 was considered to be significant.
Supplementary Material
Highlights.
A novel monoclonal antibody (MHA112) against peripheral node addressin (PNAd) on high endothelial venules (HEVs)
Simultaneous targeting to the three key sites of primary tumor, tumor-draining lymph nodes (TDLNs) and metastatic lesions by MHA112 conjugated antineoplastic drug
Tumor metastatic lesions contain HEVs
Acknowledgments:
This work was supported in part by the National Institute of Allergy and Infectious Diseases and National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH) under award numbers R01HL145813 (R.A.) and P01AI153003 (R.A., J.B.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: All authors declare that they have no competing interests. R.A holds equity in NanoTomer, Inc. a biotech company that has licensed certain patent applications from Mass General Brigham generated by R.A.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability:
All data generated or analyzed for this study are available from the corresponding author upon reasonable request.
References
- [1].Kim EG, Kim KM, Biomol Ther (Seoul), 23 (2015) 493–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Collins DM, Bossenmaier B, Kollmorgen G, Niederfellner G, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beck A, Goetsch L, Dumontet C, Corvaia N, Nat Rev Drug Discov, 16 (2017) 315–337. [DOI] [PubMed] [Google Scholar]
- [4].Nejadmoghaddam MR, Minai-Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, Zarnani AH, Avicenna J Med Biotechnol, 11 (2019) 3–23. [PMC free article] [PubMed] [Google Scholar]
- [5].N Engl J Med, 378 (2018) 878.29490175 [Google Scholar]
- [6].Oostra DR, Macrae ER, Breast Cancer (Dove Med Press), 6 (2014) 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yurkiewicz IR, Muffly L, Liedtke M, Drug Des Devel Ther, 12 (2018) 2293–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gbadamosi M, Meshinchi S, Lamba JK, Future Oncol, 14 (2018) 3199–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bahmani B, Vohra I, Kamaly N, Abdi R, Curr Opin Organ Transplant, 23 (2018) 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hemmerich S, Butcher EC, Rosen SD, J Exp Med, 180 (1994) 2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee SY, Chao-Nan Q, Seng OA, Peiyi C, Bernice WH, Swe MS, Chii WJ, Jacqueline HS, Chee SK, J Transl Med, 10 (2012) 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K, Br J Cancer, 112 (2015) 1782–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hiraoka N, Ino Y, Yamazaki-Itoh R, Front Immunol, 7 (2016) 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Colbeck EJ, Ager A, Gallimore A, Jones GW, Front Immunol, 8 (2017) 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alitalo K, Nat Med, 17 (2011) 1371–1380. [DOI] [PubMed] [Google Scholar]
- [16].Ji RC, Int J Mol Sci, 18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gil Z, Carlson DL, Boyle JO, Kraus DH, Shah JP, Shaha AR, Singh B, Wong RJ, Patel SG, Cancer, 115 (2009) 5700–5710. [DOI] [PubMed] [Google Scholar]
- [18].Kowalski LP, Bagietto R, Lara JR, Santos RL, Silva JF Jr., Magrin J, Head Neck, 22 (2000) 207–214. [DOI] [PubMed] [Google Scholar]
- [19].Jones D, Pereira ER, Padera TP, Front. Oncol, 8 (2018) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thomas SN, Rohner NA, Edwards EE, Annu Rev Biomed Eng, 18 (2016) 207–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gerlini G, Urso C, Mariotti G, Di Gennaro P, Palli D, Brandani P, Salvadori A, Pimpinelli N, Reali UM, Borgognoni L, Clin Immunol, 125 (2007) 184–193. [DOI] [PubMed] [Google Scholar]
- [22].Battaglia A, Buzzonetti A, Baranello C, Ferrandina G, Martinelli E, Fanfani F, Scambia G, Fattorossi A, Cancer Immunol Immunother, 58 (2009) 1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Trevaskis NL, Kaminskas LM, Porter CJ, Nat Rev Drug Discov, 14 (2015) 781–803. [DOI] [PubMed] [Google Scholar]
- [24].Milling L, Zhang Y, Irvine DJ, Adv Drug Deliv Rev, 114 (2017) 79–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Batty CJ, Tiet P, Bachelder EM, Ainslie KM, Pharm Nanotechnol, 6 (2018) 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Meijer EFJ, Blatter C, Chen IX, Bouta E, Jones D, Pereira ER, Jung K, Vakoc BJ, Baish JW, Padera TP, Microcirculation, 24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen J, Wang L, Yao Q, Ling R, Li K, Wang H, Breast Cancer Res, 6 (2004) R474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oladipo AO, Oluwafemi OS, Songca SP, Sukhbaatar A, Mori S, Okajima J, Komiya A, Maruyama S, Kodama T, Sci Rep, 7 (2017) 45459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Maisel K, Sasso MS, Potin L, Swartz MA, Adv Drug Deliv Rev, 114 (2017) 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rosenblum D, Joshi N, Tao W, Karp JM, Peer D, Nat Commun, 9 (2018) 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Seyfried TN, Huysentruyt LC, Crit Rev Oncog, 18 (2013) 43–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lambert AW, Pattabiraman DR, Weinberg RA, Cell, 168 (2017) 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tang C, Wang X, Soh H, Seyedin S, Cortez MA, Krishnan S, Massarelli E, Hong D, Naing A, Diab A, Gomez D, Ye H, Heymach J, Komaki R, Allison JP, Sharma P, Welsh JW, Cancer Immunol Res, 2 (2014) 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zavyalova MV, Denisov EV, Tashireva LA, Savelieva OE, Kaigorodova EV, Krakhmal NV, Perelmuter VM, Biochemistry (Mosc), 84 (2019) 762–772. [DOI] [PubMed] [Google Scholar]
- [35].Ko YD, Chirurg, 90 (2019) 997–1002. [DOI] [PubMed] [Google Scholar]
- [36].Fontebasso Y, Dubinett SM, Crit Rev Oncog, 20 (2015) 449–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gu W, Wu C, Chen J, Xiao Y, Int J Nanomedicine, 8 (2013) 2305–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].D’Antonio C, Passaro A, Gori B, Del Signore E, Migliorino MR, Ricciardi S, Fulvi A, de Marinis F, Ther Adv Med Oncol, 6 (2014) 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Uchimura K, Gauguet JM, Singer MS, Tsay D, Kannagi R, Muramatsu T, von Andrian UH, Rosen SD, Nat Immunol, 6 (2005) 1105–1113. [DOI] [PubMed] [Google Scholar]
- [40].Kobayashi M, Mitoma J, Hoshino H, Yu SY, Shimojo Y, Suzawa K, Khoo KH, Fukuda M, Nakayama J, J Pathol, 224 (2011) 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dal Corso A, Cazzamalli S, Gebleux R, Mattarella M, Neri D, Bioconjug. Chem, 28 (2017) 1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sutherland MS, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, Lewis TS, Meyer DL, Zabinski RF, Doronina SO, Senter PD, Law CL, Wahl AF, J. Biol. Chem, 281 (2006) 10540–10547. [DOI] [PubMed] [Google Scholar]
- [43].Sahay G, Alakhova DY, Kabanov AV, J Control Release, 145 (2010) 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mayor S, Pagano RE, Nat Rev Mol Cell Biol, 8 (2007) 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Harrell MI, Iritani BM, Ruddell A, Am. J. Pathol, 170 (2007) 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gonzalez H, Hagerling C, Werb Z, Genes Dev, 32 (2018) 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Messerschmidt JL, Prendergast GC, Messerschmidt GL, Oncologist, 21 (2016) 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zippi M, De Toma G, Minervini G, Cassieri C, Pica R, Colarusso D, Stock S, Crispino P, Saudi J Gastroenterol, 23 (2017) 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rybinski B, Franco-Barraza J, Cukierman E, Physiol Genomics, 46 (2014) 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rawla P, Sunkara T, Gaduputi V, World J Oncol, 10 (2019) 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bahmani B, Uehara M, Ordikhani F, Li X, Jiang L, Banouni N, Ichimura T, Kasinath V, Eskandari SK, Annabi N, Bromberg JS, Shultz LD, Greiner DL, Abdi R, EBioMedicine, 38 (2018) 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ritchie M, Tchistiakova L, Scott N, MAbs, 5 (2013) 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Knewtson KE, Perera C, Hymel D, Gao Z, Lee MM, Peterson BR, ACS Omega, 4 (2019) 12955–12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, Leece BA, Chittenden T, Blattler WA, Goldmacher VS, Cancer Res, 66 (2006) 3214–3221. [DOI] [PubMed] [Google Scholar]
- [55].Kovtun YV, Goldmacher VS, Cancer Lett, 255 (2007) 232–240. [DOI] [PubMed] [Google Scholar]
- [56].Sammet B, Steinkuhler C, Sewald N, Pharm Pat Anal, 1 (2012) 65–73. [DOI] [PubMed] [Google Scholar]
- [57].Wang LY, Ganly I, Future Oncol, 12 (2016) 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Swartz MA, Lund AW, Nat Rev Cancer, 12 (2012) 210–219. [DOI] [PubMed] [Google Scholar]
- [59].Fransen MF, Arens R, Melief CJ, Int J Cancer, 132 (2013) 1971–1976. [DOI] [PubMed] [Google Scholar]
- [60].Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, Yin R, J Hematol Oncol, 12 (2019) 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Liu T, Zhou L, Li D, Andl T, Zhang Y, Front Cell Dev Biol, 7 (2019) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pritchard EM, Dyer MA, Guy RK, Mini Rev Med Chem, 16 (2016) 430–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Han S, Huang K, Gu Z, Wu J, Nanoscale, 12 (2020) 413–436. [DOI] [PubMed] [Google Scholar]
- [64].Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ, Clin Cancer Res, 8 (2002) 878–884. [PubMed] [Google Scholar]
- [65].Chang PM, Cheng CT, Wu RC, Chung YH, Chiang KC, Yeh TS, Liu CY, Chen MH, Yeh CN, Oncol Lett, 16 (2018) 566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Miyashita T, Tajima H, Makino I, Okazaki M, Yamaguchi T, Ohbatake Y, Nakanuma S, Hayashi H, Takamura H, Ninomiya I, Fushida S, Kishimoto K, Harmon JW, Ohta T, Anticancer Res, 38 (2018) 337–343. [DOI] [PubMed] [Google Scholar]
- [67].Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y, Shimada M, Cancer Sci, 109 (2018) 2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ernsting MJ, Hoang B, Lohse I, Undzys E, Cao P, Do T, Gill B, Pintilie M, Hedley D, Li SD, J Control Release, 206 (2015) 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Maarouf OH, Uehara M, Kasinath V, Solhjou Z, Banouni N, Bahmani B, Jiang L, Yilmam OA, Guleria I, Lovitch SB, Grogan JL, Fiorina P, Sage PT, Bromberg JS, McGrath MM, Abdi R, JCI Insight, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Saxena V, Li L, Paluskievicz C, Kasinath V, Bean A, Abdi R, Jewell CM, Bromberg JS, Immunol Rev, 292 (2019) 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li X, Bean A, Uehara M, Banouni N, Ben Nasr M, Kasinath V, Jiang L, Fiorina P, Abdi R, Sci Rep, 9 (2019) 9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lin L, Hu X, Zhang H, Hu H, Front. Immunol, 10 (2019) 1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS, Front Immunol, 10 (2019) 2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lechner MG, Russell SM, Bass RS, Epstein AL, Immunotherapy, 3 (2011) 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chatterjee G, Pai T, Hardiman T, Avery-Kiejda K, Scott RJ, Spencer J, Pinder SE, Grigoriadis A, Breast Cancer Res, 20 (2018) 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Joo JH, Kim YS, Nam JH, Medicine (Baltimore), 97 (2018) e11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chaudhary B, Elkord E, Vaccines (Basel), 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F, Guo C, Xiang B, Zhou M, Ma J, Huang X, Wu X, Li Y, Li GY, Zeng ZY, Xiong W, Mol Cancer, 17 (2018) 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Verma A, Mathur R, Farooque A, Kaul V, Gupta S, Dwarakanath BS, Cancer Manag Res, 11 (2019) 10731–10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Togashi Y, Shitara K, Nishikawa H, Nat Rev Clin Oncol, 16 (2019) 356–371. [DOI] [PubMed] [Google Scholar]
- [81].Shang B, Liu Y, Jiang SJ, Liu Y, Sci Rep, 5 (2015) 15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhang X, Kelaria S, Kerstetter J, Wang J, J Gastrointest Oncol, 6 (2015) 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tanaka A, Sakaguchi S, Cell Res, 27 (2017) 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sanchez-Paulete AR, Teijeira A, Quetglas JI, Rodriguez-Ruiz ME, Sanchez-Arraez A, Labiano S, Etxeberria I, Azpilikueta A, Bolanos E, Ballesteros-Briones MC, Casares N, Quezada SA, Berraondo P, Sancho D, Smerdou C, Melero I, Cancer Res, 78 (2018) 6643–6654. [DOI] [PubMed] [Google Scholar]
- [85].Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, Krummel MF, Cancer Cell, 26 (2014) 938. [DOI] [PubMed] [Google Scholar]
- [86].Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, Freeman GJ, Warren SE, Ong S, Browning E, Twitty CG, Pierce RH, Le MH, Algazi AP, Daud AI, Pai SI, Zippelius A, Weissleder R, Pittet MJ, Immunity, 49 (2018) 1148–1161 e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pfannenstiel LW, Lam SS, Emens LA, Jaffee EM, Armstrong TD, Cell Immunol, 263 (2010) 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR, J Immunotoxicol, 9 (2012) 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].John J, Ismail M, Riley C, Askham J, Morgan R, Melcher A, Pandha H, BMC Immunol, 11 (2010) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, Merad M, Nature, (2020).
- [91].Xie Y, Bagby TR, Cohen MS, Forrest ML, Expert Opin Drug Deliv, 6 (2009) 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Oussoren C, Storm G, Adv Drug Deliv Rev, 50 (2001) 143–156. [DOI] [PubMed] [Google Scholar]
- [93].Maincent P, Thouvenot P, Amicabile C, Hoffman M, Kreuter J, Couvreur P, Devissaguet JP, Pharm Res, 9 (1992) 1534–1539. [DOI] [PubMed] [Google Scholar]
- [94].Fujii H, Horie S, Takeda K, Mori S, Kodama T, J Biophotonics, 11 (2018) e201700401. [DOI] [PubMed] [Google Scholar]
- [95].Kato S, Shirai Y, Sakamoto M, Mori S, Kodama T, Sci Rep, 9 (2019) 13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bahmani B, Uehara M, Jiang L, Ordikhani F, Banouni N, Ichimura T, Solhjou Z, Furtmuller GJ, Brandacher G, Alvarez D, von Andrian UH, Uchimura K, Xu Q, Vohra I, Yilmam OA, Haik Y, Azzi J, Kasinath V, Bromberg JS, McGrath MM, Abdi R, J Clin Invest, 128 (2018) 4770–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, Feyen K, Tawney J, Hanahan D, Michael IP, Bergers G, Sci Transl Med, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Fankhauser M, Broggi MAS, Potin L, Bordry N, Jeanbart L, Lund AW, Da Costa E, Hauert S, Rincon-Restrepo M, Tremblay C, Cabello E, Homicsko K, Michielin O, Hanahan D, Speiser DE, Swartz MA, Sci Transl Med, 9 (2017). [DOI] [PubMed] [Google Scholar]
- [99].Bayraktar S, Rocha-Lima CM, Mt Sinai J Med, 77 (2010) 606–619. [DOI] [PubMed] [Google Scholar]
- [100].Dillekas H, Rogers MS, Straume O, Cancer Med, 8 (2019) 5574–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhang L, Su H, Liu Y, Pang N, Li J, Qi XR, J Control Release, 294 (2019) 1–16. [DOI] [PubMed] [Google Scholar]
- [102].Xia Y, Zhang B, Zhang H, Li W, Wang KP, Shen H, J Thorac Dis, 7 (2015) S231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zhou X, Ou X, Yang Y, Xu T, Shen C, Ding J, Hu C, J Oncol, 2018 (2018) 9172585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Majzner RG, Mackall CL, Cancer Discov, 8 (2018) 1219–1226. [DOI] [PubMed] [Google Scholar]
- [105].Salinas RD, Durgin JS, O’Rourke DM, CNS Drugs, 34 (2020) 127–145. [DOI] [PubMed] [Google Scholar]
- [106].Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, Keyomarsi K, Yarden Y, Seger R, Oncogene, 20 (2001) 147–155. [DOI] [PubMed] [Google Scholar]
- [107].Narentuya, Takeda-Uchimura Y, Foyez T, Zhang Z, Akama TO, Yagi H, Kato K, Komatsu Y, Kadomatsu K, Uchimura K, Sci Rep, 9 (2019) 4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed for this study are available from the corresponding author upon reasonable request.