

Abstract
Background
5-Fluorouracil (5-FU) is an agent frequently used in the treatment of solid cancers. A deficiency in the enzyme that catabolizes 5-FU leads to severe toxicity. The gene responsible for this enzyme is DPYD, located on chromosome 1q22. The most prevalent alteration described is DPYD*2A, which leads to a splicing defect and thus skipping of the translation of an entire exon. The objectives of this retrospective study were to describe the frequencies of DPYD gene mutations in a Belgian population and to correlate them with the grade of toxicity.
Methods
This was a retrospective, single-center study conducted at the University Hospitals Leuven, by reviewing a database of patients screened for DPYD gene mutations between May 2009 and June 2015 after prolonged grade 3-4 toxicity. Polymerase chain reaction sequencing of exons 2, 6, 10, 11, 13, 18, 19 and 22, and pyrosequencing of exon 14 were performed by an in-house laboratory.
Results
Of the 80 patients screened, 65 were heterozygous or compound heterozygous for DPYD and 3 had a homozygous mutation. The most prevalent mutation in our population was DPYD*9A.
Conclusions
Despite previous reports, in our small retrospective study the most prevalent variation in patients with severe adverse events was DPYD*9A. As this variant has previously been reported to be benign, we suggest that screening for dihydropyrimidine dehydrogenase deficiency should be extended across multiple exons of the DPYD gene.
Keywords: Dihydropyrimidine dehydrogenase, deficiency, 5-fluorouracil, toxicity
Introduction
5-Fluorouracil (5-FU) was first introduced as an anti-cancer agent in the mid-1960s, and ever since it has been the most commonly used agent in the treatment of solid tumors, covering a broad spectrum that includes gastrointestinal, head and neck, gynecological and breast cancers. Fluoropyrimidines were developed as analogs of uracil, one of the 4 bases found in RNA. Once intracellular, 5-FU is converted into its anabolic metabolites: fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP), and fluorouridine triphosphate (FUTP). This interferes with DNA synthesis by FdUMP blockage of thymidylate synthase, the sole pathway responsible for the synthesis of the DNA component thymidylate. In addition, the incorporation of FUTP in RNA synthesis disrupts its normal processing and function [1].
5-FU is associated with adverse side effects, most frequently gastrointestinal toxicity (diarrhea, vomiting, mucositis), plantar-palmar erythrodysesthesia (hand-foot syndrome) and myelosuppression (leukopenia, anemia, and thrombocytopenia). In 1985, Tuchman et al were the first to describe severe toxicity in a 27-year-old woman receiving adjuvant chemotherapy for breast cancer [2]. Because of the high pyrimidinemia and pyrimidinuria, they hypothesized that a genetic defect of pyrimidine-base degradation was the cause of the extreme toxicity. It is now widely recognized that severe 5-FU toxicity, which may be fatal in 1% of severe cases, can be mostly attributed to an inherited deficiency in the enzyme dihydropyrimidine dehydrogenase (DPD) [3].
Only a small portion of infused 5-FU is converted into active metabolites, with approximately 80% being degraded in the liver to 5-fluoro-5,6-dihydrouracil [4]. In the catabolic pathway of pyrimidines and fluoropyrimidine, DPD is the first and a rate-limiting enzyme [1]. The therapeutic window of 5-FU is very narrow, with only a small difference between the required therapeutic dose and the maximum acceptable dose [5]. Cancer patients with low levels of DPD activity are at risk for severe toxic reactions, because the plasma level of the standard administered 5-FU dose exceeds the maximum tolerable dose.
In this manuscript we describe the frequency and type of DPYD mutations found in a population of cancer patients tested after experiencing severe 5-FU related toxicity in a large Belgian University Hospital.
Patients and methods
Patient population
The University Hospitals Leuven is a tertiary academic hospital that administers anti-cancer therapy to around 35,000 patients annually. A prospective database was designed to record patients with prolonged grade III-IV 5-FU toxicity who underwent DPYD mutation screening. Clinical information was extracted from the electronic health records.
Study design
A database audit was conducted, following approval by the human research ethics committee of the University Hospitals Leuven. The Common Terminology Criteria for Adverse Events (CTCAE version 4.0) were used for grading toxicity.
DPYD gene analysis
DPYD gene analysis was undertaken using standard techniques in the University Hospitals Leuven accredited laboratory. In brief, leukocytes were isolated (Buffy Coat technique) and the DNA was extracted from the obtained leukocyte pellets using the BioRobot® EZ1™ (Qiagen®, Valencia, CA, USA). The DNA was then quantified by measuring the optical density using the spectrophotometer NanoDrop ND-1000 (Thermo Fisher Scientific, Wilmington, DE, USA). A genetic analysis was performed by sequencing of exons 2, 6, 10, 13, 18, 19 and 22, and pyrosequencing of exon 14. Polymerase chain reaction (PCR) was used to synthesize a large amount of specific target DNA by successive steps of denaturation, hybridization and elongation. After amplification by PCR, the amplified sequences were subjected to electrophoresis on an agarose gel and visualized using an ultraviolet transilluminator in order to determine which ones could be used for the eventual sequencing. These were further purified to eliminate salts and unnecessary dNTP, and then committed in a cycle sequencing reaction to incorporate marked ddNTP. Finally, these marked products were purified using the BigDye® XTerminator™ Purification Kit and sequenced (Applied Biosystems, Foster City, California, USA). For exon 14, pyrosequencing was used. This is a real-time DNA sequencing technique that detects pyrophosphate released during DNA synthesis as visible light. The technique is used to quickly obtain a short nucleotide sequence of a DNA fragment from a PCR product that has a primer at its 5’ terminus. The obtained sequences were then compared to a reference.
Results
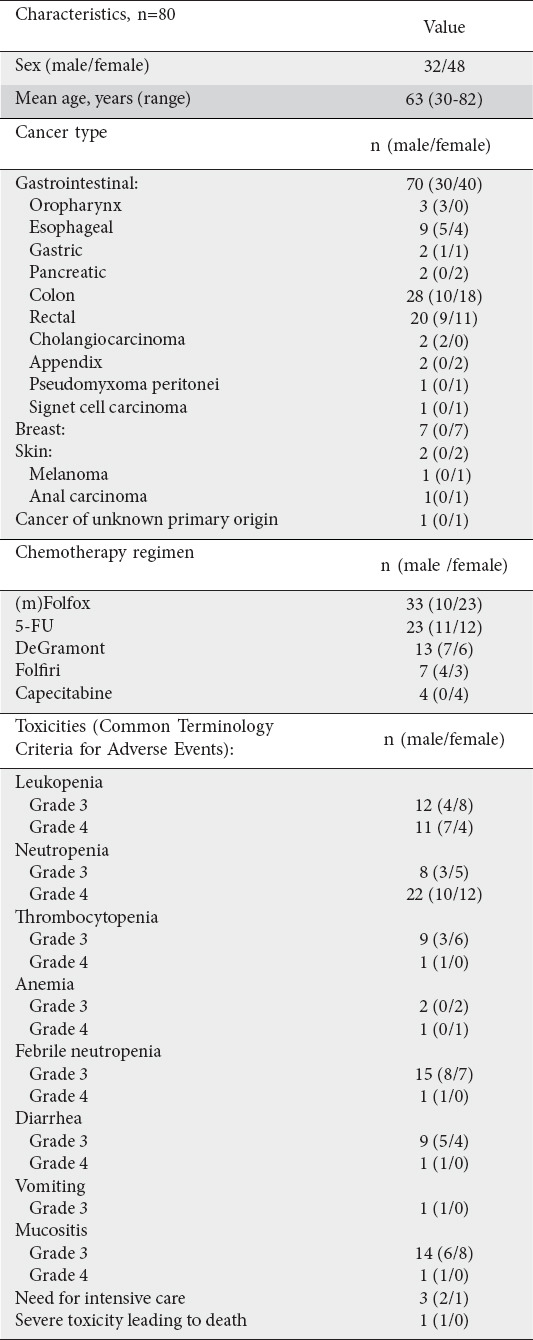
Eighty patients had results of screening for DPYD gene mutations between May 2009 and June 2015, after experiencing severe toxicity whilst receiving 5-FU. Patient characteristics and toxicity are summarized in Table 1. Patients experienced 2 major classes of adverse events: hematological and gastrointestinal.
Table 1.
Patients characteristics and toxicities
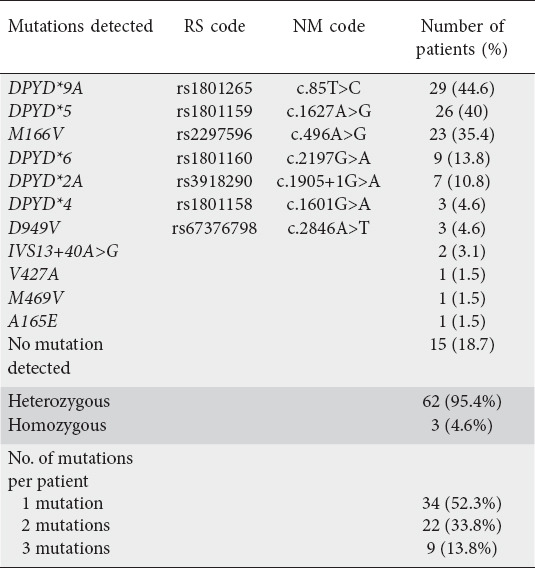
DPYD mutation analysis is presented in Table 2. Sixty-five patients carried one or more mutations, with 105 mutations documented in total. Thirty-four patients had one mutation, while 22 and nine had 2 and 3 mutations respectively. DPYD*9A was the most common, occurring in 45% of the cohort. Almost all mutations were heterozygous (95%); only 3 patients had homozygous mutations. Of these patients, 1 was homozygous for DPYD*5; 1 for M166V with additional compound heterozygosity for DPYD*9A and DPYD*5; and the remaining patient was also homozygous for M166V, with additional concurrent heterozygosity in DPYD*9A and D949V.
Table 2.
Type of DPYD mutations
In addition, we explored the type of mutation in 15 patients with combined grade III-IV hematological toxicity and grade III-IV gastrointestinal toxicity (including mucositis). We found a higher frequency of the DPYD*2A variant (6/15), while the other patients had either no mutation (3/15), DPYD*9A (3/15), DPYD*5 (2/15), or D949V (1/15).
Correlation of toxicity with mutation revealed that, of the 3 patients who required intensive care, 2 were mutation carriers. The patient who subsequently died from neutropenic sepsis had a heterozygous mutation in DPYD*5, whilst the other patient, who recovered, carried a DPYD*2A mutation. Importantly, 15 patients with severe toxicity were not found to have any DPYD mutations; however, since all were receiving combination therapy, toxicity from causes other than DPD cannot be ruled out.
Discussion
The DPYD gene is located on chromosome 1q22 and consists of 23 exons, ranging from 69 bp to 961 bp, with an estimated total size of at least 950 kb [6]. To date, there have been more than 50 genetic alterations described in DPYD, although in most cases either they do not impair enzyme activity or their impact is uncertain. DPYD gene alterations are present in approximately 3-5% of the Caucasian population, with some debate over the effect of sex and ethnicity [7]. The most common mutation (1% of the general population), DPYD*2A (c.1905+1G>A), results in a guanine base being replaced by adenine at the intron boundary of exon 14, resulting in a splicing defect [8,9]. This leads to skipping of an entire exon (165 bp mRNA deletion) and consequently formation of a non-functioning enzyme [6]. In patients with a heterozygous DPYD*2A mutation, the DPD activity is reduced by up to 48%, with a corresponding 50% dose reduction of 5-FU recommended by the Clinical Pharmacogenetics Implementation Consortium Guidelines (CPIC; www.cpicpgx.org) [10]. If this starting dose is well tolerated, increasing the dose can be considered [5]. Similar advice is given for patients with the 2 other genetic variations commonly associated with severe 5-FU toxicity, DPYD*13 and D949V (c.2846A>T). Patients homozygous for any of these 3 mutations are advised against 5-FU use [5]. A recent prospective study also showed evidence to support a suggestion for a 50% dose reduction in heterozygous carriers of the decreased function variants c.1129–5923C>G (rs75017182; HapB3 or its tagging SNP c.1236G>A; rs56038477) [11].
The CPIC recommendations apply to patients with known mutations, usually sought following the toxic event. Guidelines for routinely screening patients prior to 5-FU administration vary between jurisdictions. Some cancer centers have for some time been screening all patients prospectively. The National Comprehensive Cancer Network guidelines for colon cancer state that carriers of certain DPYD variants have an elevated risk for life-threatening toxicity and acknowledge that DPYD genotyping and fluoropyrimidine dose individualization is feasible and cost-effective, while the European Society for Medical Oncology guidelines state that DPD testing before 5-FU administration remains an option but is not routinely recommended [12]. In France, health authorities introduced mandatory screening in 2020, and earlier this year (March 2020) the European Medicines Agency safety committee recommended that all patients should be tested for DPD before starting 5-FU. However, this measure has not yet been implemented in all European countries [13].
There are 2 ways of screening for DPD deficiency: genotyping by sequencing the DPYD gene and phenotyping tests assessing DPD activity, either directly by peripheral blood mononuclear cells enzyme activity or indirectly by measurement of pyrimidine metabolites. Van Staveren et al published an overview of the different possibilities for phenotyping. Phenotyping offers an advantage over genotyping because it takes into account genetic and non-genetic sources of variation in the DPD enzyme activity [14,15]. In general, genotyping is simpler but has a low sensitivity and specificity, while phenotyping is more complex and a routine phenotypic method has not yet been validated and standardized [16].
Although DPYD genotyping reduces overall healthcare costs, clinical uptake of preemptive DPYD genotyping has been slow. In our center, patients have not been routinely tested for DPD activity or DPYD mutations before receiving 5-FU, because of the upfront cost and inconvenience (long waiting time before results are available) [17]. This audit shows that screening after severe toxicities yields a high number of predictive mutations. However, we were unable to determine the total number of patients treated with 5-FU who suffered severe toxicity and were not evaluated for mutations, nor the total number treated who had any or no toxicity during the study period.
An interesting finding was the different mutation profile to that previously described [5,14,17]. The most prevalent mutation in our cohort was DPYD*9A, in just under half of the cases. Although this variant has been associated with fluoropyrimidine toxicity in some series, there are no large confirmatory studies [18]. In our study population, DPYD*2A, reported elsewhere as the most common predictor of 5-FU toxicity, was infrequent.
Broad gene analysis by next-generation sequencing has led to the possibility of evaluating entire genome sequencing with an affordable cost and a short turnaround time. However, the high number of genetic polymorphisms in DPYD makes it difficult to make genotype-phenotype correlations, because not all these SNPs will lead to the same reduction in enzymatic activity [8,16]. Recommendations by groups such as the CPIC have based at least some of their guidelines on in vitro work, with the resulting conclusions that the variants DPYD*4, DPYD*5, DPYD*6 and DPYD*9A confer normal enzyme activity [19]. However, in our study a patient heterozygous for DPYD*5 died from neutropenic fever after his first administration of chemotherapy. Additionally, 6 patients with a single DPYD*9A mutation, 14 with a single DPYD*5 mutation and 4 with a single DPYD*6 mutation were all documented to have severe toxicity. A recent large study of 1254 patients also found a significant association between DPYD*6 and severe 5-FU toxicity [20]. We also recorded 31 patients who had more than one DPYD gene variant. Our study therefore concurs with several others to show that limiting DPD genetic analysis to one or only a few variants may miss clinically relevant predictors of severe 5-FU toxicity [17]. Additionally, the effect of multiple mutations on the enzyme activity and their interaction is still unclear but may be of clinical relevance.
A significant limitation of this study was that there is no information regarding patients with severe 5-FU toxicity who were not tested, nor information about the mutation rate in patients in the whole population that received 5-FU. Furthermore, our study did not evaluate the effect of reactive versus preemptive DPYD genotyping. A recent study of Deenen et al, in which upfront genotyping of DPYD*2A was performed in patients who were to receive fluoropyrimidine-based chemotherapy, showed that upfront screening for this mutation, and subsequently a 50% dose reduction of 5-FU, was associated with a reduced incidence of grade ≥3 toxicity [17]. They also observed a shorter duration of the adverse events in comparison to carriers of DPYD gene alterations who received a full dose. Besides the effect of upfront genotyping of DPYD*2A on the duration and severity of toxicity, they compared the costs of upfront screening and dose reduction with those patients who received full-dose therapy despite DPD deficiency. They concluded that upfront genotyping was cost-saving, because it reduced the severity and duration of toxicity and led to fewer and shorter hospitalization periods. The mortality rate was also reduced by upfront genotyping. Nevertheless, routine prospective DPD screening is only slowly being adopted.
In conclusion, this single-center study provides evidence that, in patients with severe toxicity to 5-FU, DPD testing should be broader across the DPYD gene; therefore, we favor whole exon sequencing. The effect of the spectrum of DYDP gene variants on enzyme activity remains an important clinical question that needs to be resolved and these variants should be included in pharmacokinetic analysis to better understand their effect. An interesting finding was the high prevalence of DPYD*9A in our patient population, who had already suffered severe toxicity. Reporting of these severe toxicity events and their genomic correlations remains important to inform prescribing practice for one of the world’s most commonly used chemotherapy agents.
Summary Box.
What is already known:
Severe 5-fluorouracil (5-FU) toxicity can be attributed to an inherited deficiency in the enzyme dihydropyrimidine dehydrogenase (DPD)
The 4 most relevant DPYD variants are DPYD*2A, DPYD*13, D949V and c.1236G>A
There are 2 ways of screening for DPD deficiency: genotyping by sequencing the DPYD gene and phenotyping tests assessing DPD activity
What the new findings are:
In this retrospective study, in our population of patients who suffered severe toxicity when treated with 5-FU based chemotherapy, we found a high prevalence of DPYD*9A
Limiting DPD genetic analysis to only a few variants may miss clinically relevant predictors of severe 5-FU toxicity
The effect of multiple mutations on the enzyme activity and their interaction is still unclear
Biography
University Hospitals Leuven, Belgium; Monash University and Monash Health, Melbourne, Victoria, Australia; University of Pisa, Italy; University Hospital Antwerp, Edegem, Belgium
Footnotes
Conflict of Interest: None
References
- 1.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil:mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 2.Tuchman M, Stoeckeler JS, Kiang DT, O'Dea RF, Ramnaraine ML, Mirkin BL. Familial pyrimidinemia and pyrimidinuria associated with severe fluorouracil toxicity. N Engl J Med. 1985;313:245–249. doi: 10.1056/NEJM198507253130407. [DOI] [PubMed] [Google Scholar]
- 3.Fidai SS, Sharma AE, Johnson DN, Segal JP, Lastra RR. Dihydropyrimidine dehydrogenase deficiency as a cause of fatal 5-fluorouracil toxicity. Autops Case Rep. 2018;8:e2018049. doi: 10.4322/acr.2018.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heggie GD, Sommadossi JP, Cross DS, Huster WJ, Diasio RB. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma urine, and bile. Cancer Res. 1987;47:2203–2206. [PubMed] [Google Scholar]
- 5.Caudle KE, Thorn CF, Klein TE, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin Pharmacol Ther. 2013;94:640–645. doi: 10.1038/clpt.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei X, Elizondo G, Sapone A, et al. Characterization of the human dihydropyrimidine dehydrogenase gene. Genomics. 1998;51:391–400. doi: 10.1006/geno.1998.5379. [DOI] [PubMed] [Google Scholar]
- 7.Van Kuilenburg ABP, Meijer J, Tanck MWT, et al. Phenotypic and clinical implications of variants in the dihydropyrimidine dehydrogenase gene. Biochim Biophys Acta. 2016;1862:754–762. doi: 10.1016/j.bbadis.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Ciccolini J, Gross E, Dahan L, Lacarelle B, Mercier C. Routine dihydropyrimidine dehydrogenase testing for anticipating 5-fluorouracil-related severe toxicities: Hype or hope? Clin Colorectal Cancer. 2010;9:224–228. doi: 10.3816/CCC.2010.n.033. [DOI] [PubMed] [Google Scholar]
- 9.Saif MW, Ezzeldin H, Vance K, Sellers S, Diasio RB. DPYD*2A mutation: the most common mutation associated with DPD deficiency. Cancer Chemother Pharmacol. 2007;60:503–507. doi: 10.1007/s00280-006-0392-5. [DOI] [PubMed] [Google Scholar]
- 10.Van Kuilenburg ABP, Meinsma R, Zoetekouw L, Van Gennip AH. Increased risk of grade IV neutropenia after administration of 5-fluorouracil due to a dihydropyrimidine dehydrogenase deficiency:High prevalence of the IVS14+1G>A mutation. Int J Cancer. 2002;101:253–258. doi: 10.1002/ijc.10599. [DOI] [PubMed] [Google Scholar]
- 11.Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer:a prospective safety analysis. Lancet Oncol. 2018;19:1459–1467. doi: 10.1016/S1470-2045(18)30686-7. [DOI] [PubMed] [Google Scholar]
- 12.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 13.Barin-Le Guellec C, Lafay-Chebassier C, Ingrand I, et al. Toxicities associated with chemotherapy regimens containing a fluoropyrimidine:A real-life evaluation in France. Eur J Cancer. 2020;124:37–46. doi: 10.1016/j.ejca.2019.09.028. [DOI] [PubMed] [Google Scholar]
- 14.van Staveren MC, Guchelaar HJ, van Kuilenburg AB, Gelderblom H, Maring JG. Evaluation of predictive tests for screening for dihydropyrimidine dehydrogenase deficiency. Pharmacogenomics J. 2013;13:389–395. doi: 10.1038/tpj.2013.25. [DOI] [PubMed] [Google Scholar]
- 15.Thomas F, Hennebelle I, Delmas C, et al. Genotyping of a family with a novel deleterious DPYD mutation supports the pretherapeutic screening of DPD deficiency with dihydrouracil/uracil ratio. Clin Pharmacol Ther. 2016;99:235–242. doi: 10.1002/cpt.210. [DOI] [PubMed] [Google Scholar]
- 16.Henricks LM, Lunenburg CA, Meulendijks D, et al. Translating DPYD genotype into DPD phenotype:using the DPYD gene activity score. Pharmacogenomics. 2015;16:1277–1286. doi: 10.2217/pgs.15.70. [DOI] [PubMed] [Google Scholar]
- 17.Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy:a safety and cost analysis. J Clin Oncol. 2016;34:227–234. doi: 10.1200/JCO.2015.63.1325. [DOI] [PubMed] [Google Scholar]
- 18.Gentile G, Botticelli A, Lionetto L, et al. Genotype-phenotype correlations in 5-fluorouracil metabolism:a candidate DPYD haplotype to improve toxicity prediction. Pharmacogenomics J. 2016;16:320–325. doi: 10.1038/tpj.2015.56. [DOI] [PubMed] [Google Scholar]
- 19.Annotation of CPIC Guideline for fluorouracil and DPYD. [Accessed 30 September 2020]. Available from: https://www.pharmgkb.org/guidelineAnnotation/PA166122686 .
- 20.Del Re M, Cinieri S, Michelucci A, et al. DPYD*6 plays an important role in fluoropyrimidine toxicity in addition to DPYD*2A and c. 2846A>T:a comprehensive analysis in 1254 patients. Pharmacogenomics J. 2019;19:556–563. doi: 10.1038/s41397-019-0077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]