

ABSTRACT
Start is the main decision point in the eukaryotic cell cycle at which cells commit to a new round of cell division. It involves the irreversible activation of a transcriptional programme through the inactivation of Start transcriptional repressors: the retinoblastoma family in mammals, or Whi5 and its recently identified paralogue Whi7 (also known as Srl3) in budding yeast. Here, we provide a comprehensive comparison of Whi5 and Whi7 that reveals significant qualitative differences. Indeed, the expression, subcellular localization and functionality of Whi7 and Whi5 are differentially regulated. Importantly, Whi7 shows specific properties in its association with promoters not shared by Whi5, and for the first time, we demonstrate that Whi7, and not Whi5, can be the main contributor to Start inhibition such as it occurs in the response to cell wall stress. Our results help to improve understanding of the interplay between multiple differentially regulated Start repressors in order to face specific cellular conditions.
KEY WORDS: Cell cycle, Start, Whi7, Whi5, Stress
Highlighted Article: Cells can use the interplay between functionally redundant but differentially regulated cell-cycle repressors in order to confer new repression capabilities and to respond to specific cellular conditions.
INTRODUCTION
Eukaryotic cell division is controlled by a complex network of intertwining regulatory circuits (Morgan, 2007). At the heart of this system is the conserved cyclin dependent kinase (CDK) family (Malumbres, 2014). Distinct CDK complexes are activated in a cell cycle phase-specific order, which allows the phosphorylation of many other proteins that drive cell cycle progression. A hallmark of the cell cycle regulatory system is its extraordinary robustness. This is achieved by the existence of different regulatory mechanisms that affect a particular process, and functionally redundant proteins able to substitute for each other or act in different cellular contexts.
The G1-to-S phase transition (Start) represents a critical point at which cells irreversibly commit to initiate a new cell cycle. The Start regulatory network is well conserved between yeast and mammalian cells (Johnson and Skotheim, 2013). The activation of G1 CDKs initiates a wave of gene expression through the inhibition of specific transcriptional repressors, which provides key cell cycle regulators and effectors of downstream cell cycle events. Positive and negative feedback loops guarantee the coherent activation and inactivation of this transcriptional programme (Bertoli et al., 2013). In the yeast Saccharomyces cerevisiae, activation of Start gene expression is controlled by the CDK Cdc28 associated with the G1 cyclins Cln1, Cln2 and Cln3, and involves the transcription factors SBF (Swi4–Swi6) and MBF (Mbp1–Swi6) (Haase and Wittenberg, 2014). SBF associates with its target promoters in early G1, but its activity is blocked due to the binding of the transcriptional repressor Whi5 (Costanzo et al., 2004; de Bruin et al., 2004), which mediates the recruitment of histone deacetylase activities to promoters (Huang et al., 2009; Takahata et al., 2009; Wang et al., 2009). Expression is initially triggered by the Cdc28–Cln3 kinase in late G1, when it extensively phosphorylates the Whi5 repressor and the SBF and MBF transcriptional factors, activating gene expression (Costanzo et al., 2004; de Bruin et al., 2004; Palumbo et al., 2016; Wagner et al., 2009). This process is coordinated with cell growth, although the molecular mechanism is still a matter of controversy (Aldea et al., 2017; Dorsey et al., 2018; Heldt et al., 2018; Litsios et al., 2019; Moreno et al., 2019; Schmoller et al., 2015; Wang et al., 2009). As a result of this initial activation, Cdc28–Cln1 and Cdc28–Cln2 accumulate and further phosphorylate Whi5 and Swi6, promoting the complete dissociation of the Whi5 repressor. This positive-feedback mechanism strengthens a sharp transcriptional response and makes the Start transition coherent and irreversible (Charvin et al., 2010; Skotheim et al., 2008). Later in the cell cycle, SBF-dependent transcription is silenced by Cdc28–Clb CDKs (Koch et al., 1996), whereas MBF is inactivated in a negative feedback loop by the transcriptional repressor Nrm1, whose gene is regulated by MBF (de Bruin et al., 2006). A similar regulatory circuitry controls Start (referred to as the restriction point) in mammalian cells, where the roles of Cdc28–Cln3, SBF/MBF, Whi5 and Cdc28–Cln1/Cln2 are played by CDK4/6–cyclin D, the E2F–DP transcription factor family, retinoblastoma transcriptional repressors [Rb (also known as RB1), p107 (RBL1) and p130 (RBL2)] and CDK2–cyclin E, respectively (Bertoli et al., 2013; Johnson and Skotheim, 2013).
Although SBF and MBF preferentially regulate a subset of specific genes (cyclins CLN1 and CLN2, and cell wall genes for SBF; cyclins CLB5 and CLB6, and DNA metabolism genes for MBF), there is an important functional redundancy between them: SBF can bind to the MBF binding sites and vice versa, one factor can replace the other in its absence for a significant number of genes, and there are common target genes for SBF and MBF (Bean et al., 2005; Ferrezuelo et al., 2010). In fact, in some genes there is an SBF-to-MBF switch during the G1-S transition (Bastos de Oliveira et al., 2012). The crosstalk between both transcription factors also includes the regulation of the SWI4 gene by MBF (Harris et al., 2013). The functional redundancy between SBF and MBF, as well as the importance of the Start transcriptional programme, are evidenced by the lethality of the swi4 swi6 and swi4 mbp1 double mutations (Koch et al., 1993). Strikingly, mbp1 swi6 cells are viable, which indicates that Swi4 could act alone in the absence of Swi6.
An important aspect of cellular behaviour is the ability to properly respond to changes in environmental conditions. To overcome these stresses, cells process and integrate information from their environment into the cell cycle control network, in particular at Start (Aldea et al., 2017; Ewald, 2018). In fact, multiple signalling pathways impinge on different regulators to promote or prevent passage through Start: molecular mechanisms connect the response to nutrient availability with Swi4 (Amigoni et al., 2015), Whi5 (Talarek et al., 2017), the cyclin-dependent kinase inhibitor Sic1 (Moreno-Torres et al., 2015), the SBF-interacting proteins Msa1 and Msa2 (Miles et al., 2016) or the cyclin Cln3 (Gallego et al., 1997; Menoyo et al., 2013); osmotic stress targets Sic1 (Escote et al., 2004), Whi5 (Gonzalez-Novo et al., 2015) and Cip1 (Chang et al., 2017); and genotoxic stress affects transcriptional factors Nrm1 (Travesa et al., 2012), Swi6 (Sidorova and Breeden, 1997) as well as Cip1 (Zhang et al., 2017). Therefore, multiple regulatory pathways might cooperate in regulating the G1-S transition for cells to cope with environmental changes.
One of the S. cerevisiae signalling pathways involved in the response to environmental conditions is the CWI (cell wall integrity) or PKC (protein kinase C) pathway (Levin, 2011). This pathway responds to a variety of conditions that challenge the cells – such as thermal, osmolarity and pH changes, cell wall perturbing agents and oxidative stress – as well as to endogenous cellular processes such as polarized growth. The main function of the pathway is the maintenance of cell integrity in these particular conditions, which have an impact on the cell surface (cell wall and/or plasma membrane) integrity. This stress is detected by sensor proteins that activate the protein kinase C (Pkc1), which controls the activity of the MAPK Slt2 cascade, which in turn induces the expression of genes important for cell wall remodelling and morphogenesis in order to fortify the cell wall and maintain its integrity (Sanz et al., 2017). This transcriptional response is mostly executed by Rlm1, whose activity is regulated by Slt2-mediated phosphorylation. Slt2 also controls the expression of a small subset of cell wall genes through the regulation of the transcription factor SBF by a non-catalytic mechanism (Baetz et al., 2001; Kim et al., 2008; Truman et al., 2009). Importantly, the CWI pathway has a more general role, because components of the pathway also affect other aspects of yeast cell biology (Heinisch and Rodicio, 2018; Jiménez-Gutierrez et al., 2020).
Direct connections between components of the CWI pathway with the regulation of the cell cycle have been described. As regards the G1-S transition, Slt2 phosphorylates Swi6, blocking its nuclear import in response to cell wall stress (Kim et al., 2010), and Sic1, restraining the progression to S phase (Moreno-Torres et al., 2015, 2017), and Rlm1 delays Start in cells grown on a poor medium (Piccirillo et al., 2017). Additionally, the CWI pathway acts on different targets to negatively (Darieva et al., 2012; Harrison et al., 2001; Negishi et al., 2016; Yano et al., 2013) or positively (Thai et al., 2017) regulate the G2-M transition. All these data highlight the central role of the CWI pathway in the response to a wide variety of environmental challenges. However, we still lack a complete picture of how the pathway affects the cell cycle regulatory system to coordinate cell proliferation with the response to such a diverse set of environmental conditions.
Whi7 (also known as Srl3), Whi5 and Nrm1 constitute a family of proteins characterized by the presence of a GTB (G1/S transcription binding factor) motif (Travesa et al., 2013). WHI7 gene expression is induced in different situations of cellular stress (Garcia et al., 2004; Gasch et al., 2000; Waern and Snyder, 2013), and Whi7 protein is unstable, being degraded by the SCFGrr1 ubiquitin ligase in a Cdc28-dependent and cell cycle-regulated way (Gomar-Alba et al., 2017). Unlike whi5 mutation, whi7 mutation has no effect on cell size distribution in exponentially growing cultures, questioning a role for this protein at the beginning of the cell cycle (Costanzo et al., 2004). However, Whi7 acts as a negative regulator of Start, helping to retain the Cdc28–Cln3 complex at the ER surface (Yahya et al., 2014), and loss of Whi7 has a slight effect on re-entry into the cell cycle after quiescence (Miles et al., 2016). More recently, our group reported that Whi7 acts as a transcriptional repressor of SBF-dependent transcription, acting as a genuine Whi5 paralogue (Gomar-Alba et al., 2017). However, little is known about the mechanisms of action of Whi7 and how they potentially differ from those of Whi5. The different effects of whi5 and whi7 mutations on cell size indicate that Whi5 has a greater relevance than Whi7 during the Start transition. The differences between cellular levels of Whi5 and Whi7, caused by the high instability of Whi7 protein (Gomar-Alba et al., 2017), could contribute to explaining this; however, we wonder whether, in addition to this quantitative difference, Whi7 and Whi5 could also show qualitative differences. In this work, we have carried out a comprehensive comparison of Whi7 and Whi5 Start repressors. The results reveal significant intrinsic differences in the regulation and function of both proteins and a major role for Whi7 in Start control during the response to cell wall stress.
RESULTS
Comparative analysis of Whi7 and Whi5 association with promoters of target genes
We first wondered whether Whi7 and Whi5 could show qualitative differences in their affinity for target genes. The Whi7 and Whi5 repressors are associated with the transcription factor SBF to bind to Start gene promoters (Costanzo et al., 2004; de Bruin et al., 2004; Gomar-Alba et al., 2017). Because this binding is strongly periodic and occurs only at a very specific moment in the cell cycle, we decided to analyse the association of both Whi7 and Whi5 with promoters in the same cells to accurately investigate differences in their binding. The genes analysed included the G1/S cyclin genes CLN2 and CLN1, and genes encoding proteins involved in cell wall and DNA biosynthesis, such as the glucan synthase Fks1, the mannosyltransferase Mnn1, the glucanosyltransferase Gas1 and the ribonucleotide reductase subunit Rnr1. The CLN2, CLN1, FKS1, MNN1 and GAS1 genes are mainly regulated by SBF, whereas regulation of the RNR1 gene depends mainly on MBF (Harris et al., 2013). The results show that both Whi7 and Whi5 were able to bind to the promoters of all genes. However, their relative binding pattern to these genes was different, revealing that Whi5 had a marked preference for the Start cyclin genes (in particular CLN2), and Whi7 for the cell wall genes (FKS1 and MNN1) (Fig. 1). This suggests that Whi7 and Whi5 association with promoters involves some distinct mechanisms.
Fig. 1.
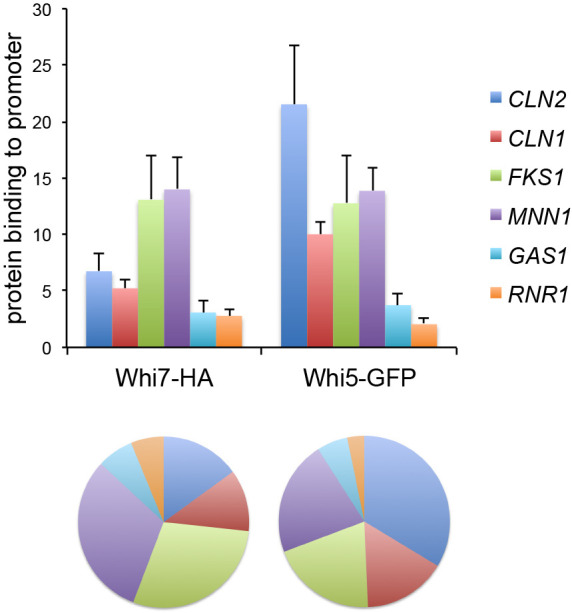
Whi7 and Whi5 show different binding preferences for Start gene promoters. Whi7 binding to CLN2, CLN1, FKS1, MNN1, GAS1 and RNR1 promoters was investigated using ChIP assays in WHI5–GFP (JCY2116) cells transformed with the centromeric pWHI7–HA plasmid. The wild-type W303-1a strain transformed with an empty vector was used as a no-tag control in the assays. Top: graph represents Whi7 and Whi5 binding relative to the no-tag control for each gene. Data are presented as mean±s.d. of three experiments. Bottom: a chart with the relative binding pattern of the analysed genes is shown.
Differential determinants in the binding of Whi7 and Whi5 to promoters
Whi5 binding to SBF and Start gene promoters depends on the integrity of the transcription factor, because the absence of either of its components (Swi4 or Swi6) abolishes the interaction (Costanzo et al., 2004; de Bruin et al., 2004). We previously described that Whi7 binds to the CLN2 promoter mainly through SBF (Gomar-Alba et al., 2017). To obtain further insight into the association between Whi7 and the transcription factor SBF, the binding of Whi7 to the Start genes CLN2, FKS1, MNN1 and RNR1 in swi4 and swi6 mutant strains was investigated. In agreement with the fact that Whi5 interaction with SBF requires the intact transcription factor complex, Whi5 binding to promoters was completely lost in the absence of either Swi4 or Swi6. Strikingly, this was not the case for Whi7. Whi7 was still substantially associated with all genes in the absence of Swi6, although to a lesser extent (Fig. 2A). We envisaged the possibility that Swi4 alone could mediate this binding. Supporting this, deletion of Swi6 reduced, but did not abolish, association of Swi4 with CLN2, FKS1, MNN1 and RNR1 promoters (Fig. 2B). Importantly, co-immunoprecipitation experiments demonstrated that unlike Whi5, Whi7 was able to physically interact in vivo with Swi4 in the absence of Swi6 (Fig. 2C). All these results demonstrate that, whereas Whi5 only interacts with the integral transcriptional factor SBF, Whi7 is also able to associate with Swi4 in the absence of Swi6 in order to bind to Start promoters. On the other hand, Whi7 association was drastically reduced in the absence of Swi4, especially in the case of the CLN2 promoter (Fig. 2A). This result reflects that the Whi7 binding to promoters is strongly dependent on Swi4, either when it is part of the SBF complex with Swi6 or on its own. However, it is important to note that, unlike for Whi5, a residual binding of Whi7 was still observed in the absence of Swi4, mainly to the FKS1 and MNN1 promoters. We wondered whether this residual binding in the absence of Swi4 could be due to MBF. Supporting this, Mbp1 was found to be associated with all the analysed promoters in both a wild-type strain and the swi4 mutant (Fig. 2D). More importantly, inactivation of Mbp1 in the absence of Swi4 caused a reduction in Whi7 binding to FKS1, MNN1 and RNR1 promoters (Fig. 2E). Although no physical interaction between Whi7 and Mbp1 could be detected by co-immunoprecipitation assays, this observation strongly suggests that in the absence of Swi4, MBF mediates Whi7 recruitment to target genes. In short, although both Whi5 and Whi7 share a function as Start transcriptional repressors, their association with target genes involves specific molecular determinants (Fig. 2F).
Fig. 2.

Whi7 and Whi5 present differences in the Start transcription factors required for binding to promoters. (A) Left: Whi7 binding to FKS1, MNN1, CLN2 and RNR1 promoters was investigated using ChIP assays in wild-type (wt; W303-1a), swi4 (JCY167) and swi6 (JCY325) cells transformed with centromeric pWHI7–HA plasmid. W303-1a transformed with an empty vector was used as the no-tag control (no tag). Right: Whi5 binding to the same promoters was investigated using ChIP assays in cultures of the W303-1a (no tag), WHI5–HA (wt; JCY1346), WHI5–HA swi4 (JCY740) and WHI5–HA swi6 (JCY1934) strains. Graphs represent Whi7 and Whi5 binding relative to the no-tag control for each gene. Data are presented as mean±s.d. of at least three experiments. (B) Swi4 binding to CLN2, RNR1, FKS1 and MNN1 promoters was investigated using ChIP assays in cultures of the W303-1a (no tag), SWI4–HA (wt; JCY1956) and SWI4–HA swi6 (JCY1958) strains. Graph represents Swi4 binding relative the no-tag control for each gene. Data are presented as mean±s.d. of three experiments. (C) Top: SWI4–myc (JCY1879) and SWI4–myc swi6 (JCY1884) cells were transformed with the centromeric pWHI7–HA plasmid (WHI7–HA and WHI7–HA swi6, respectively), alongside SWI4–myc cells transformed with a control vector (no tag). Whi7 was immunoprecipitated from crude extracts, and the presence of Whi7 and Swi4–myc in the input, unbound and immunoprecipitated (IP) fractions was determined by western blotting. Bottom: Whi5 was immunoprecipitated from crude extracts from no-tag control SWI4–myc (no tag; JCY1879), SWI4–myc WHI5–HA (JCY2134) and SWI4–myc WHI5–HA swi6 (JCY2135) cells, and the presence of Whi5 and Swi4–myc in the input, unbound and immunoprecipitated fractions was determined by western blotting. Input, 5%; unbound, 5%. (D) Mbp1 binding to CLN2, RNR1, FKS1 and MNN1 promoters was investigated using ChIP assays in cultures of the W303-1a (no tag), MBP1–HA (wt; JCY2084) and MBP1–HA swi4 (JCY2100) strains. Graph represents Mbp1 binding relative to the no-tag control for each gene. Data are presented as mean±s.d. of three experiments. (E) Wild-type (wt; W303-1a), mbp1 (JCY2194), GAL1:SWI4 (JCY2268) and mbp1 GAL1:SWI4 (JCY2304) cells transformed with the centromeric pWHI7–HA plasmid were grown in SGal and incubated overnight in SD medium to repress SWI4 expression. Whi7 binding to FKS1, MNN1 and RNR1 promoters was investigated using ChIP assays. W303-1a transformed with an empty vector was used as the no-tag control. Graph represents Whi7 binding relative to the no-tag control for each gene. Data are presented as mean±s.d. of at least six experiments. (F) Scheme of Whi7 binding to gene promoters of the Start transcriptional programme (see Discussion).
Cell-cycle regulation of Whi7 subcellular localization
Whi5 subcellular localization is cell cycle regulated. Specifically, Whi5 is located in the nucleus in the G1 phase, from late mitosis (telophase) until Start activation (Costanzo et al., 2004; Taberner et al., 2009). Here, we analysed Whi7 subcellular localization in parallel with that of Whi5. Given the low amount of Whi7 protein, we first used the pADH1:WHI7–GFP4 and pADH1:WHI5–GFP4 constructs, in which both transcriptional repressors are fused to four copies of the GFP protein and are expressed under the control of the ADH1 promoter. The analysis of asynchronous cultures confirmed that, like Whi5, the localization of Whi7 changes throughout the cell cycle. Whi7, like Whi5, was located in the nucleus of cells in G1 phase and telophase (Fig. 3A). The assay was also performed in α-factor synchronized cultures. The results indicated that Whi7 was nuclear in G1 cells (0–30 min), disappeared from the nucleus after activation of Start coinciding with budding, and only returned to the nucleus at the end of the cell cycle (150 min) (Fig. 3B). Time-lapse analysis of cells expressing Whi5–mCherry and Whi7–GFP further demonstrated that Whi7 localization is regulated throughout the cell cycle with the same localization kinetics as Whi5 (Fig. 3C; Movie 1). However, it is important to note that in all the experimental approaches, whereas Whi5 was totally nuclear during G1 phase, Whi7 never became completely nuclear, but rather was distributed between the nucleus and the cytosol of the cell. Finally, we investigated Whi7 localization at endogenous levels using the mNeonGreen tag. Notably, the results confirmed the Whi7 cell cycle-regulated pattern described above (Fig. 3D; Movie 2). Whi7 is an unstable protein (Gomar-Alba et al., 2017), so we wondered whether the disappearance of nuclear signal could be due to a selective degradation of nuclear Whi7. However, Whi7 stabilization in a grr1 mutant strain did not alter the cell cycle-regulated pattern of Whi7 localization (Fig. S1). This result points to the existence of an active nuclear export mechanism controlling Whi7 subcellular localization in a cell cycle-regulated manner.
Fig. 3.

Whi7 subcellular localization is cell cycle regulated. (A) Exponentially growing cells of the wild-type (W303-1a) strain transformed with plasmids pADH1:WHI7–GFP4 or pADH1:WHI5–GFP4 were analysed by fluorescence microscopy. GFP signal and DIC images are shown. Cells were scored as unbudded, budded with one nucleus (1nl) or budded with two nuclei (2nl), and as cells with fluorescence signal only in the nuclei (N), in the nuclei and the cytoplasm (N+C) or only in the cytoplasm (C). Graphs show mean±s.d. percentage protein localization derived from three (Whi7) or two (Whi5) independent experiments (n>300). (B) Exponentially growing cells of the wild-type (W303-1a) strain transformed with plasmid pADH1:WHI7–GFP4 were blocked in G1 using α-factor. The localization of Whi7 was investigated at the indicated times after the release from the arrest. Budding index (% budded cells) is shown as an indication of cell cycle progression. Graph shows the percentage of cells in which nuclear signal was detected. One of two replicates is shown. (C) Time-lapse analysis by confocal fluorescence microscopy of exponentially growing cells of the WHI5–mCherry MYO1–mCherry (YMM3056) strain transformed with plasmid pADH1:WHI7–GFP4. Disappearance of Myo1 signal (marked with arrowheads) from the neck indicates the end of cytokinesis, referred as to time 0. Approximately 50 cells were analysed with identical results. Dashed lines indicate cell outlines. (D) Exponentially growing cells of the WHI5–mCherry (YMM3055), WHI5–mCherry WHI7–mNeonGreen (YMM5682) and WHI5–mNeonGreen (YMM5680) strains were analysed by confocal fluorescence microscopy. mNeonGreen and mCherry signals are shown. Scale bars: 8 μm (A,D), 4 μm (B), 2 μm (C).
Whi7 is a highly phosphorylated protein. In the case of Whi5, the phosphorylation of specific residues by Cdc28 mediates its nuclear export (Costanzo et al., 2004; Taberner et al., 2009; Wagner et al., 2009). For this reason, the localization of Whi7NP, a version of Whi7 protein with all the putative Cdc28 phosphorylation sites mutated to Ala (Yahya et al., 2014), was investigated. The results demonstrated that the non-phosphorylatable Whi7 was nuclear in all cell cycle phases (Fig. 4; Movie 3). This strongly suggests that Cdc28-dependent phosphorylation of Whi7 is necessary for Whi7 nuclear export, as occurs with Whi5.
Fig. 4.

The non-phosphorylatable Whi7 protein is nuclear in all cell cycle stages. (A) Exponentially growing cells of the wild-type (W303-1a) strain transformed with either plasmid pADH1:WHI7–GFP4 or plasmid pADH1:WHI7NP–GFP4, which expresses a Whi7 protein mutated in all consensus CDK phosphorylation sites, were analysed by fluorescence microscopy. GFP signal and DIC images are shown. All cells showed nuclear fluorescence signal (n>300 cells from three experiments). (B) Time-lapse analysis by confocal fluorescence microscopy of exponentially growing cells of the WHI5–mCherry MYO1–mCherry (YMM3056) strain transformed with plasmid pADH1:WHI7NP–GFP4, as described in Fig. 3C. Scale bars: 8 μm (A), 2 μm (B).
Whi5 nuclear export is driven by karyopherin Msn5 through the recognition of a NES (nuclear export signal) in a phosphorylation-dependent manner (Taberner et al., 2009). Given the results described above, we envisaged the possibility that Msn5 could also mediate the nuclear export of Whi7. However, contrary to what happens with Whi5, inactivation of Msn5 did not significantly change Whi7 localization (Fig. 5A–C; Movie 4). We also tested whether the Whi770–110 fragment, covering the equivalent region to the Msn5-dependent NES of Whi5, has NES activity. Unlike what happens with Whi551–167, Whi770–110 was not able to mediate nuclear export when fused to a constitutive nuclear localization protein NLSSV40–GFP (Fig. S2). Although it cannot be completely ruled out that Msn5 contributes to Whi7 export (nuclear Whi7 was consistently detected in ∼10% of msn5 mutant cells outside G1), these results indicate that, in contrast to export of Whi5, Whi7 nuclear export does not require Msn5. Concerning nuclear import, it is known that Whi5 is imported by the classical nuclear import pathway dependent on the Kap60 (Srp1)–Kap95 karyopherin (Taberner et al., 2009). In contrast, inactivation of Kap95 had no effect on the nuclear accumulation of Whi7NP (Fig. 5D). In short, although nuclear localization of Whi7 changed along the cell cycle similarly to that of Whi5, the two proteins use different pathways to control their nuclear localization.
Fig. 5.

Msn5 and Kap95 karyopherins control Whi5 but not Whi7 subcellular localization. (A) Exponentially growing cells of the msn5 (JCY1018) strain transformed with plasmids pADH1:WHI7–GFP4 or pADH1:WHI5–GFP4 were analysed by fluorescence microscopy, as described in Fig. 3A. Graphs show mean±s.d. percentage protein localization derived from three (Whi7) or two (Whi5) independent experiments (n>300). (B) Whi7 localization was investigated in α-factor synchronized cultures of the msn5 (JCY1018) strain transformed with plasmid pADH1:WHI7–GFP4, as described in Fig. 3B. One of two replicates is shown. (C) Time-lapse analysis by confocal fluorescence microscopy of exponentially growing cells of the WHI5–mCherry MYO1–mCherry msn5 (YMM5382) strain transformed with plasmid pADH1:WHI7–GFP4. as described in Fig. 3C. Arrowheads indicate Myo1–mCherry. Dashed lines indicate cell outlines. (D) Exponentially growing cells of the tetO7:KAP95 strain (JCY970) transformed with plasmid pADH1:WHI7NP–GFP4 or pADH1:WHI5NP–GFP4 were incubated in the presence of 5 μg/ml doxycycline (+ dox) for 12 h to repress KAP95 expression. GFP signal and DIC images are shown. Scale bars: 8 μm (A,D), 4 μm (B), 2 μm (C).
The PKC pathway controls the cellular levels of Whi7, but not those of Whi5
In a preliminary global transcriptomic analysis in a pkc1 mutant strain, we observed decreased expression of the WHI7 gene (I.Q. and J.C.I., unpublished), which was consistent with results from other groups connecting WHI7 gene expression and cell wall stress (Boorsma et al., 2004; Garcia et al., 2004; Lagorce et al., 2003). Because of that, we first investigated Whi7 protein level in a thermosensitive pkc1 mutant strain (pkc1ts). As shown in Fig. 6A, there was an increase in Whi7 levels in the wild-type strain at 37°C that was dependent on Pkc1, because it was not observed in the pkc1 mutant strain at restrictive temperature. The analysis of WHI7 mRNA demonstrated that this Pkc1-dependent induction probably occurs at the transcriptional level (Fig. 6B).
Fig. 6.

The PKC pathway regulates expression of WHI7 but not WHI5. (A,C) Exponentially growing cells of the WHI7–HA pkc1ts (JCY1730) or WHI5–HA pkc1ts (JCY2164) strains transformed with plasmid pPKC1 or an empty vector were incubated at 37°C for 3 h. Whi7 (A) or Whi5 (C) protein level was analysed by western blotting. Cdc28 is shown as loading control. Graphs show the mean±s.d. protein levels derived from six (Whi7) or two (Whi5) experiments, normalized to Cdc28. (B,D) The level of WHI7 (B) or WHI5 (D) mRNA relative to ACT1 mRNA was analysed by quantitative RT-PCR in the strains described in A and C. Graphs show the mean±s.d. mRNA level derived from three (Whi7) or four (Whi5) experiments. (E) Whi7 protein level was analysed in exponentially growing cells of WHI7–GFP (wt; JCY1746), WHI7–GFP slt2 (slt2; JCY2039) and WHI7–GFP slt2 (JCY2039) transformed with plasmid pSLT2 or an empty vector. Slt2 and Cdc28 are shown as controls. Graph shows the mean±s.d. Whi7 protein level derived from three experiments, normalized to Cdc28. (F) Whi7 protein level was analysed in exponentially growing cells of WHI7–HA (wt; JCY1728) and WHI7–HA rlm1 (JCY2166) strains. Cdc28 is shown as loading control for protein analysis. (G) WHI7 mRNA level relative to ACT1 mRNA level was analysed by quantitative RT-PCR in the same strains described in E and F. Graphs show the mean±s.d. mRNA level derived from four (slt2) or three (rlm1) experiments. (H) Whi7 protein level was analysed in exponentially growing cells of the wild-type (W303-1a; SLT2) and slt2 (JCY2040) strains transformed with plasmid pGAL1:WHI7–HA, alongside wild-type cells transformed with empty vector (no tag). Cdc28 is shown as loading control for protein analysis. (I) Left: Whi5 protein level was analysed in exponentially growing cells of WHI5–HA (wt; JCY1346), WHI5–HA slt2 (JCY2140) and WHI5–HA slt2 (JCY2140) transformed with plasmid pSLT2 or an empty vector. Middle: graph shows mean±s.d. protein levels derived from three experiments, normalized to Cdc28. Right: WHI5 mRNA level relative to ACT1 mRNA level was analysed by quantitative RT-PCR in the WHI5–HA slt2 (JCY2140) strain transformed with plasmid pSLT2 or an empty vector and in the wild-type (wt: JCY1728) and rlm1 mutant (JCY2166) strains. Graphs show the mean±s.d. mRNA level derived from seven (slt2) or three (rlm1) experiments.
We extended the analysis to Whi5. Unlike what happens with Whi7, Whi5 levels were reduced at 37°C in wild-type cells (Fig. 6C). The same result was observed after the inactivation of Pkc1. Levels of WHI5 mRNA were not affected in the wild-type strain at 37°C, whereas they increased approximately twofold in the pkc1 mutant strain (Fig. 6D). This could be an indirect effect, because pkc1 mutant cells tend to accumulate in S phase, when the WHI5 gene has its maximum expression (Pramila et al., 2006). Taken together, these results suggest that Pkc1 activity promotes expression of Whi7 but not expression of its functional paralogue Whi5.
Pkc1 activates the MAP kinase Slt2. It has been previously described that expression of the WHI7 gene is induced by cell wall stress by Slt2 through the transcription factor Rlm1 (Garcia et al., 2004; Sanz et al., 2012). To address whether the role of Slt2 and Rlm1 in the regulation of WHI7 expression can be more general, Whi7 levels in slt2 or rlm1 mutants were investigated in basal conditions. The Whi7 protein levels were severely reduced in the absence of the MAPK Slt2 or the transcription factor Rlm1 (Fig. 6E,F). In addition, WHI7 mRNA levels also decreased drastically in the absence of Slt2 or Rlm1 (Fig. 6G). Inactivation of Slt2 had no effect on Whi7 protein level when the WHI7 gene was ectopically expressed (Fig. 6H). Furthermore, no significant differences in Whi7 protein stability were observed in the slt2 mutant strain (Fig. S3). These results indicate that, in basal conditions, the MAP kinase Slt2 promotes Whi7 expression through the transcriptional factor Rlm1.
Finally, both Whi5 protein and WHI5 mRNA levels in the slt2 and rlm1 mutant strains were investigated. Contrary to what happens with Whi7, neither the Whi5 protein nor the mRNA levels were altered in the absence of Slt2 or Rlm1 (Fig. 6I). These observations indicate that, unlike what it is observed for Whi7, Whi5 cellular levels are completely independent of the MAP kinase Slt2.
Cellular regulation and function of Whi7 and Whi5 under cell wall stress
Heat shock stress induces the activation of the PKC pathway, and above, we describe an increase in the cellular levels of Whi7 in this stress condition. We wondered whether this could reflect a more preponderant role for Whi7 compared with that played in non-stress conditions. To test this possibility, we tested the effect of WHI7 or WHI5 deletion on the phenotype of a cln3 mutant strain at high temperature, in which the Start process is compromised. Growth assays revealed that cln3 mutation was lethal at 38°C and that this lethality was suppressed by either WHI7 or WHI5 deletion (Fig. S4). This result suggests, for the first time, that in particular conditions Whi7 could play a major role, comparable at least to that developed by Whi5, in the control of Start.
To further compare Whi7 and Whi5 cellular function under cell wall stress conditions, the effect of Congo Red, a dye that binds to chitin, generating cell wall stress and thus activating the PKC pathway, was studied. Congo Red induces expression of the WHI7 gene (Garcia et al., 2004). Given this, we tested the effect of Congo Red on the protein levels of Whi7 and Whi5, as well as the effect of this compound on the stability of the Whi7 protein. In line with previous studies of gene expression, Whi7 protein levels increased more than fourfold in the presence of Congo Red (Fig. 7A). The same result was observed with another cell wall stressor, Calcofluor White (Fig. S5A). Translational shut-off assays revealed that Whi7 protein stability was not altered in response to Congo Red (Fig. S6), which supports Whi7 induction being a transcriptional response. No change in Whi7 subcellular localization after cell wall stress was observed (Fig. S5B). Interestingly, in contrast to what occurs with Whi7, Whi5 protein levels were not affected by cell wall stress.
Fig. 7.

Whi7 plays a more important role than Whi5 in Start regulation under cell wall stress conditions. (A) Exponentially growing cells of WHI7–HA (JCY2015) or WHI5–HA (JCY2036) strains were incubated in the presence of 40 μg/ml Congo Red (CR). Whi7 and Whi5 protein level was analysed by western blotting at the indicated times. The Ponceau staining of the membrane is shown as loading control. Graph shows the mean±s.d. derived from three experiments. (B) Tenfold serial dilutions from cultures of wild-type (wt; W303), cln3 (MT244), whi5 cln3 (JCY1875) and whi7 cln3 (JCY1868) strains were spotted onto YPD plates with or without 40 μg/ml Congo Red and incubated at 25°C for 3 d. Images shown are representative of five experiments. (C) Exponentially growing cultures of the cln3 (MT244) strain were split and incubated in the absence or presence of 40 μg/ml Congo Red. Cell number was determined at the indicated times. Graph shows the mean±s.d. derived from three experiments. (D) Cell cycle distribution of cln3 (MT244) cells at 4 h after the addition of Congo Red (+CR). Untreated cells are shown as a control (−CR). Fixed cells were also treated with zymolyase (zym) to digest the cell wall. Cells were scored as unbudded, budded with one nucleus (1nl), budded with two nuclei (2nl) or re-budded. Graph shows the mean±s.d. percentage of cells in each category derived from three experiments. Pictures show DIC images (top) and DAPI staining of DNA (bottom). In the +CR fluorescence images, note the presence of Congo Red fluorescence in the neck associated with a cell separation defect. (E) Cell cycle distribution of cln3 (MT244), whi5 cln3 (JCY1875) and whi7 cln3 (JCY1868) cells at 4 h after the addition of Congo Red. Cells were scored as described in D. Graph shows the mean±s.d. percentage of cells in each category derived from three experiments. Pictures show DIC images (top) and DAPI staining of DNA (bottom). Scale bars: 8 μm.
Next, the effect of Congo Red on cell growth in wild-type, whi7 mutant and whi5 mutant strains was studied. As observed in Fig. S7A, growth of the wild-type strain was impaired in the presence of Congo Red. Microscopic analysis of the cells revealed a significant increase in the percentage of dumbbell cells (cells with large bud and segregated nuclei) in Congo Red-containing medium. Remarkably, digestion with zymolyase led to the disappearance of dumbbell cells and an increase in the percentage of unbudded cells (Fig. S7B). This indicated that dumbbell cells completed mitosis but had a defect in cell separation caused by the Congo Red. The accumulation of cells in the G1 phase suggests a partial blockage of Start in response to cell wall stress. Interestingly, both the whi7 and whi5 mutations alleviated, to a similar extent, the growth defect of wild-type cells in Congo Red medium (Fig. S7A). However, microscopic analysis revealed that Whi7 inactivation suppressed the accumulation of cells in G1 to a greater extent than Whi5 inactivation (Fig. S7B). This points to Whi7 transcriptional repressor as an important mediator of the partial repression of Start caused by Congo Red-induced cell wall stress.
Whi7 represses transcription of the G1/S genes in an SBF- and Swi4-dependent manner. For this reason, we investigated the requirement of Swi4 and Swi6 for Whi7 function in response to CR. It is known that swi4 and swi6 mutants are hypersensitive to Congo Red (Garcia et al., 2004). Growth assays showed that deletion of WHI7 or WHI5 did not suppress the lethality of the swi4 and swi6 mutations in the presence of Congo Red (Fig. S8). This result strongly suggests that Whi7 function in response to Congo Red is dependent on Swi4 and Swi6 activity.
Finally, we studied the role of Whi7 and Whi5 during cell wall stress in a cln3 mutant background, in which the Start process is compromised. Note that the cln3 mutation was lethal in the presence of Congo Red (Fig. 7B). Analysis of exponentially growing cultures revealed a first-cycle arrest of cln3 cells after the addition of Congo Red to the medium (Fig. 7C). Practically all of the cln3 mutant cells were dumbbell cells (Fig. 7D). Zymolyase treatment indicated that these cells were in fact in G1 phase. All these data reflect that the lethality of cln3 mutation in the presence of Congo Red is due to a robust arrest in G1, before Start is executed. Importantly, the lethality of the cln3 mutation in the presence of Congo Red was completely abolished by the deletion of WHI7. In contrast, deletion of WHI5 had a very slight effect on growth (Fig. 7B). The same result was observed in the presence of Calcofluor White (Fig. S5C). In addition, microscopic analysis of cln3 whi7 cells in the presence of Congo Red confirmed that the absence of Whi7 overrides the G1 arrest of the cln3 mutant, as revealed by the increase in the percentage of re-budded cells. However, and consistent with what was observed in the growth assay, loss of Whi5 only allowed a small percentage of cells to re-bud (Fig. 7E). In conclusion, all these observations demonstrate that the cln3 mutant suffers a cell cycle arrest in G1 phase, mediated by the Start transcriptional repressor Whi7 and not by Whi5. Thus, whereas Whi5 is the main Start inhibitor during normal growth, Whi7 takes over this role specifically during cell wall stress.
DISCUSSION
In a previous work, our group characterized that the Whi7 protein, which is related at sequence level to the transcriptional repressor Whi5 (the functional paralogue of the Rb proteins in mammalian cells) and functions as a transcriptional repressor of the Start transcriptional programme, demonstrating the existence of functional redundancy between Whi7 and Whi5 (Gomar-Alba et al., 2017). However, this does not necessarily mean that both proteins perform exactly the same function or are regulated in the same way. In fact, Whi5 is more critical than Whi7 for proper execution of the Start transition in normal conditions, as deduced from the fact that Whi5, but not Whi7, inactivation leads to a decrease in cell size distribution in exponentially growing cultures, and WHI5, but not WHI7, overexpression results in a severe increase in cell size. In this work, the differences between Whi7 and Whi5 have been analysed in detail, with the aim of establishing the determinants of functional specificity.
Two possible, non-mutually exclusive scenarios can be considered in order to explain functional differences between redundant proteins that collaborate in a cellular process: a quantitative model, which points to differences in protein level as the origin of functional specificity, and a qualitative model, which proposes the existence of differential intrinsic characteristics. Comparative analysis of cellular levels reveals that Whi5 is in excess over Whi7 (Gomar-Alba et al., 2017). This could explain some functional differences, such as the fact that Whi5 is the repressor that plays the most important role in the control of Start in normal conditions. We have previously reported that WHI7 overexpression rescues the cell size defect of a whi5 mutant (Gomar-Alba et al., 2017), which could support this idea. However, it is necessary to consider that this overexpression was accomplished using the very strong GAL1 promoter. On the other hand, WHI7 overexpression does not lead to its increased binding to the CLN2 promoter (Gomar-Alba et al., 2017). These observations could reflect that the functional specificity of these proteins is not only due to quantitative differences but also to the existence of some differential intrinsic characteristics.
A detailed analysis of Whi7 and Whi5 promoter association demonstrated that Whi7 shares with Whi5 the association with the promoters of all analysed genes: Start cyclin genes (CLN2 and CLN1), cell wall genes (FKS1, MNN1 and GAS1) and DNA metabolism genes (RNR1). Importantly, ChIP experiments in which both Whi7 and Whi5 were immunoprecipitated from the same cells showed a distinct binding pattern to these promoters. Whereas Whi5 had the strongest preference for G1 cyclin genes, Whi7 preference was greater for cell wall biosynthesis genes. The fact that Whi7 and Whi5 were distributed differently among the promoters is an important result that reveals the existence of differential determinants in their promoter association.
The analysis of Whi7 and Whi5 association with promoters in several mutant strains reflects a fundamental difference in their function. Consistently with previous results, Whi5 is associated with promoters through the intact SBF heterodimer transcriptional factor, because no association was detected in either a swi6 or a swi4 mutant. However, in the case of Whi7, binding to promoters was still observed in the absence of Swi6. Different observations support that this association is mediated by Swi4 alone. It has been described that monomeric Swi4 is the unique Start transcription factor active in swi6 mutant cells (Adames et al., 2015; Nasmyth and Dirick, 1991; Wijnen et al., 2002). In fact, we detected Swi4 associated with the same promoters as Whi7 in swi6 mutant cells. Remarkably, we observed stronger Whi7 binding in swi6 cells in the case of the FKS1 gene, a gene for which the binding of Swi4 was barely affected by the inactivation of Swi6. In addition, Whi7, but not Whi5, was capable of physically interacting in vivo with Swi4 in the absence of Swi6. These results reveal a key difference between Whi7 and Whi5 as transcriptional repressors of Start, because, unlike Whi5, Whi7 represses the expression of G1/S genes, not only through SBF, but also through monomeric Swi4 (Fig. 2F). Considering that monomeric Swi4 can function as a transcriptional activator that evades Whi5-mediated regulation, this specific characteristic of Whi7 may be relevant for cellular physiology.
We noted that some residual binding of Whi7 to promoters was still observed in the swi4 mutant. Although Whi7 binding to promoters in the wild-type strain did not depend on Mbp1, the fact that, in the absence of Swi4, inactivation of Mbp1 reduced promoter binding strongly suggests that Whi7 could also associate with promoters through the MBF transcriptional factor. Consistent with this, Mbp1 was able to bind to the same promoters as Whi7 in the absence of Swi4. Remarkably again, the strongest binding of Whi7 in the absence of Swi4 was observed for the case of the FKS1 gene, which among the genes we examined showed the greatest association with Mbp1 in the absence of Swi4. However, we could not confirm a physical interaction between Whi7 and Mbp1 in co-immunoprecipitation assays. From our results on the effect of swi4 and mbp1 mutations on Whi7 promoter association, it can be expected that the interaction between Whi7 and MBF might be weak and secondary, which could explain the failure to detect it. A known repressor of MBF is Nrm1, the third member of the family of GTB-motif G1/S transcriptional repressors (Travesa et al., 2013). Unlike Whi7 and Whi5, Nrm1 mediates repression of MBF after Start execution, once it has been transcribed by MBF itself (de Bruin et al., 2006). The possibility that Whi7 can repress MBF before Start brings into light an additional mechanism to control this transcriptional programme (Fig. 2F). As commented above for the role of Whi7 on monomeric Swi4, this new mechanism could have biological relevance under certain conditions, for example when it is necessary to block the MBF factor before Start.
Spatial regulation is a common mechanism in the control of transcription factors. It is known that Whi5 shuttles between the nucleus and cytoplasm throughout the cell cycle, being located in the nucleus from mitotic exit until the G1-S transition and accumulating in the cytoplasm during the rest of the cell cycle phases (Costanzo et al., 2004; Taberner et al., 2009). In this study, we have described that the subcellular localization of Whi7 is also cell cycle-regulated and follows the same dynamics as Whi5, that is, Whi7 is nuclear only in the G1 phase until Start. However, there is an important difference; Whi5 totally accumulates inside the nucleus in G1, whereas Whi7 is distributed between the nucleus and the cytosol. Consistent with this cytosolic localization, Whi7 plays a function in G1 bound to the ER membrane (Yahya et al., 2014). The determinants responsible for Whi5 spatial regulation are known. Whi5 nuclear export is dependent on the karyopherin Msn5 and the phosphorylation by Cdc28 of specific residues in a nuclear export signal, and nuclear import is mediated by the classical nuclear import pathway (Taberner et al., 2009). We have identified that Whi7 nuclear export is also controlled by Cdc28-dependent phosphorylation. However, in contrast to Whi5, Whi7 nuclear export did not require the Msn5 karyopherin and Whi7 nuclear import did not require the classical nuclear import pathway (Kap60–Kap95 karyopherin). Thus, Whi7 and Whi5 differ in the way their subcellular location is controlled. This difference could provide additional ways to differentially regulate both repressors under specific conditions.
Previous work has found a relationship between the CWI pathway and expression of the WHI7 gene. Global studies have shown an induction of the WHI7 gene in different conditions that affect cell wall integrity (Boorsma et al., 2004; Garcia et al., 2004; Lagorce et al., 2003). Moreover, the expression of WHI7 is induced by the presence of Congo Red in a Slt2-dependent manner (Sanz et al., 2012). In this work, we gained further insight into the characterization of WHI7 gene regulation by the PKC pathway. In particular, we found that Slt2, through the transcriptional factor Rlm1, is responsible for the expression of the WHI7 gene under basal conditions. It should be remarked that Whi7 expression is practically abolished in the slt2 and rlm1 mutants. This points to a very close functional connection between the MAPK Slt2 and the Start transcriptional repressor Whi7, with Whi7 function totally dependent on Slt2. Conversely, Slt2 is activated by distinct stimuli, and these results point to Whi7 as a mediator of the cellular response to these stimuli. Slt2, through Rlm1, regulates the expression of cell wall genes (Sanz et al., 2017). On the other hand, a connection between Slt2 and the components of SBF, Swi4 and Swi6, has been previously described (Baetz et al., 2001; Kim et al., 2008; Truman et al., 2009), and SBF is involved in the expression of cell wall genes (Igual et al., 1996). All these considerations suggest that the function of Whi7 as a repressor of Start could be more relevant under certain conditions, such as cell wall stress, in which there is a higher activity of the PKC pathway. Our results provide support for this hypothesis: WHI7 deletion suppressed the sensitivity and G1 arrest of the cln3 mutant in response to elevated temperatures and to compounds that stress the cell wall. The fact that Whi7 inactivation did not suppress the hypersensitivity of swi4 and swi6 mutants to Congo Red indicates that Whi7 acts under cell wall stress conditions through SBF. Interestingly, in contrast to what was observed with Whi7, the PKC pathway did not affect the cellular levels of Whi5, establishing a new key difference between both transcriptional repressors. Consistent with this, WHI7 deletion had a greater effect than WHI5 deletion on cell growth at high temperature or in the presence of Congo Red or Calcofluor White. Overall, our results indicate that in response to cell wall stress, it is Whi7 and not Whi5 transcriptional repressor that plays a major role as a brake on cell cycle entry at Start. Whi7 induction in the presence of cell wall stress could contribute to this, but the increase in Whi7 cellular content seems quite modest in relation to Whi5 protein level to support its major role. Therefore, how this functional switch could operate is still an open question.
Functional redundancy between proteins is a common trait in cell cycle regulation. One good example is the existence of several transcriptional repressors at the heart of Start control: Rb, p107 and p130 in mammalian cells, and Whi5 and Whi7 in S. cerevisiae cells. However, despite the functional redundancy that exists between the Start transcriptional repressors, we have described in this work that Whi7 and Whi5 greatly differ both in their regulation and in their functionality (Fig. 8). The differential characteristics and regulation of Whi7 compared to Whi5 could contribute to a more robust control of Start under specific circumstances. On one hand, the intrinsic different requirements for Whi7 binding to promoters confers new repression capability of Start genes beyond Whi5-mediated repression, in particular as regards the regulation of the Swi4 transcription factor, which could activate G1/S genes on its own, and MBF before Start. On the other hand, the differential regulation of Whi7 and Whi5 provides alternative ways to affect Start repression depending on internal or external cues. This allows yeast cells to rely on the interplay between distinct Start transcriptional repressors to adapt to different specific cellular conditions: Whi5 stands out as the main transcriptional repressor under normal conditions, whereas the function of Whi7 becomes more relevant in stress conditions, such as cell wall stress. The existence of multiple repressor proteins for the Start transcriptional programme must certainly represent an evolutionary advantage for the cell.
Fig. 8.

Comparison between Whi7 and Whi5. Differences between the budding yeast Start repressors Whi5 and Whi7 regarding protein level, determinants in binding to promoters, control of subcellular localization, regulation of gene expression and response to stress condition are listed.
MATERIALS AND METHODS
Strains, plasmids and growth conditions
Yeast strains used in this work are shown in Table S1. Centromeric plasmid pWHI7-HA expressing a 3×HA-tagged Whi7 protein under the control of the endogenous promoter is a gift from Dr M. Aldea, Molecular Biology Institute of Barcelona (IBMB), Spain (Yahya et al., 2014). pADH1:WHI7–GFP4, pADH1:WHI7NP–GFP4, pADH1:WHI5–GFP4 and pADH1:WHI5NP–GFP4 contain the ADH1 promoter, the coding region of WHI7 and WHI5 and their respective mutations in canonical CDK phosphorylation sites fused to four copies of GFP and the ADH1 terminator cloned in the YCplac33 vector (Gietz and Sugino, 1988). The WHI7NP and WHI5NP coding region were obtained from plasmid pWHI7NP–HA (Yahya et al., 2014) and pGAL1:WHI512Ala–13myc (Wagner et al., 2009). pADH1:NLS–WHI770–110–GFP4 and pADH1:NLS–WHI551–167–GFP4 contain the ADH1 promoter, a fragment coding for the nuclear localization sequence from SV40 and either a WHI7 gene fragment coding for amino acids 70–110 or a WHI5 gene fragment coding for amino acids 51–167 fused to four copies of GFP and the ADH1 terminator in YCplac33 vector. pPKC1 and pSLT2 consist of the PKC1 or SLT2 gene cloned in YCplac33.
Cells were maintained in exponential growth conditions (below 107 cells ml−1) at 25°C in standard YPD medium (1% yeast extract, 2% peptone and 2% glucose) or synthetic complete medium lacking the appropriate compound for selection (Formedium Ltd) supplemented with 2% glucose (SD) or 2% galactose (SGal). Where indicated, cells were incubated in the presence of 40 μg ml−1 Congo Red, 10 μg ml−1 Calcofluor White or transferred to 37°C for 3 h. For cell cycle synchronization, MATa cells were arrested in G1 with 5 μg/ml of α-factor for 3 h and then released into fresh growth medium.
Fluorescence microscopy
GFP, mCherry and mNeonGreen tagged proteins were analysed in living cells grown on synthetic complete medium. Conventional fluorescence microscopy was carried out with an Axioskop 2 microscope (Zeiss). The images were captured with an AxioCam MRm camera (Zeiss) and AxioVision v4.7 software (Zeiss). Where indicated, localization of proteins was monitored by visual inspection.
Confocal fluorescence microscopy was carried out on a confocal spinning-disk microsope (Nikon Ti) equipped with an HCX plan APO 100× objective and a Photometrics Prime 95B camera. Four to eight z-sections, 0.35 μm apart, were collected every 90 or 120 s at 30°C. Images in the figures and movies are 2D maximum projections of the z-stacks. Images were processed using ImageJ (http://rsb.info.nih.gov/ij/). For time-lapse microscopy, exponentially growing cells (below 107 cells ml−1) were grown in synthetic complete medium and plated on concanavalin A-coated (Sigma-Aldrich) Lab-Tek chambers (Thermo Fisher Scientific). Temperature during the images acquisition was controlled with a Tokai Hit Stage Top Incubator.
For DNA staining, cells were fixed for 5 min by addition of 70% ethanol and resuspended in 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole). Where indicated, the cell wall was digested in fixed cells using 0.5 mg/ml zymolyase 20T (SEIKAGAKU CORPORATION) in phosphate spheroplasting buffer (1.2 M sorbitol, 0.1% β-mercaptoethanol and 50 mM KH2PO4, pH 7.0) at 30°C for 30 min.
Miscellaneous
Chromatin immunoprecipitation analysis (ChIP), gene expression analysis by RT-PCR, protein co-immunoprecipitation assays and western blot analysis were performed as previously described (Gomar-Alba et al., 2017). In ChIP assays, Dynabeads Protein G magnetic beads were incubated with HA-probe (F-7) antibody (Santa Cruz Biotechnology; SC-7392) or monoclonal anti-GFP (Roche Diagnostics; 11814460001). The primary antibodies used in the western blot analysis were monoclonal anti-HA peroxidase 3F10 antibody (Roche Diagnostics; 12013819001) diluted 1:5000, monoclonal anti-GFP (Roche Diagnostics; 11814460001) diluted 1:5000 and monoclonal anti Cdc2 p34 (PSTAIRE; Santa Cruz Biotechnology; SC-53) diluted 1:2000. Blots were developed using anti-mouse IgG and anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies (1:20,000; Thermo Fisher Scientific; 170-6516 and 31460, respectively) and Supersignal West Femto Maximum Sensitivity substrate (Thermo Fisher Scientific). Bands were quantified using an ImageQuant LAS 4000mini Biomolecular Imager (GE Healthcare).
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: E.M., M.G.-A., M.C.B., M.M., I.Q., J.C.I.; Validation: E.M., M.G.-A., I.Q., J.C.I.; Formal analysis: E.M., M.G.-A., I.Q., J.C.I.; Investigation: E.M., M.G.-A., M.C.B., I.Q., J.C.I.; Resources: M.M.; Writing - original draft: M.G.-A., I.Q., J.C.I.; Writing - review & editing: E.M., M.G.-A., M.C.B., M.M., I.Q., J.C.I.; Visualization: E.M., M.G.-A., J.C.I.; Supervision: M.C.B., M.M., I.Q., J.C.I.; Project administration: J.C.I.; Funding acquisition: J.C.I.
Funding
This work was supported by Ministerio de Economía y Competitividad (MINECO), Spain, and co-financed by the European Regional Development Fund from the European Union (grant number BFU2017-88692), the Generalitat Valenciana (grant number GVPROMETEO2016-123) and by grant ANR-10-LABX-0030-INRT, which is a French State fund managed by the Agence Nationale de la Recherche under the Programme Investissements d'Avenir ANR-10-IDEX-0002-02 to the IGBMC. E.M. and M.G.-A. were recipients of a Predoctoral Fellowship and Postdoctoral Fellowship, respectively, from the Generalitat Valenciana. Deposited in PMC for immediate release.
Supplementary information
Supplementary information available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.251413.supplemental
Peer review history
The peer review history is available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.251413.reviewer-comments.pdf
References
- Adames N. R., Schuck P. L., Chen K. C., Murali T. M., Tyson J. J. and Peccoud J. (2015). Experimental testing of a new integrated model of the budding yeast Start transition. Mol. Biol. Cell 26, 3966-3984. 10.1091/mbc.E15-06-0358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldea M., Jenkins K. and Csikász-Nagy A. (2017). Growth rate as a direct regulator of the Start network to set cell size. Front. Cell Dev. Biol. 5, 57 10.3389/fcell.2017.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amigoni L., Colombo S., Belotti F., Alberghina L. and Martegani E. (2015). The transcription factor Swi4 is target for PKA regulation of cell size at the G1 to S transition in Saccharomyces cerevisiae. Cell Cycle 14, 2429-2438. 10.1080/15384101.2015.1055997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetz K., Moffat J., Haynes J., Chang M. and Andrews B. (2001). Transcriptional coregulation by the cell integrity mitogen-activated protein kinase Slt2 and the cell cycle regulator Swi4. Mol. Cell Biol. 21, 6515-6528. 10.1128/MCB.21.19.6515-6528.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira F. M., Harris M. R., Brazauskas P., de Bruin R. A. M. and Smolka M. B. (2012). Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 31, 1798-1810. 10.1038/emboj.2012.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean J. M., Siggia E. D. and Cross F. R. (2005). High functional overlap between MluI cell-cycle box binding factor and Swi4/6 cell-cycle box binding factor in the G1/S transcriptional program in Saccharomyces cerevisiae. Genetics 171, 49-61. 10.1534/genetics.105.044560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C., Skotheim J. M. and de Bruin R. A. M. (2013). Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518-528. 10.1038/nrm3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorsma A., de Nobel H., ter Riet B., Bargmann B., Brul S., Hellingwerf K. J. and Klis F. M. (2004). Characterization of the transcriptional response to cell wall stress in Saccharomyces cerevisiae. Yeast 21, 413-427. 10.1002/yea.1109 [DOI] [PubMed] [Google Scholar]
- Chang Y.-L., Tseng S.-F., Huang Y.-C., Shen Z.-J., Hsu P.-H., Hsieh M.-H., Yang C.-W., Tognetti S., Canal B., Subirana L. et al. (2017). Yeast Cip1 is activated by environmental stress to inhibit Cdk1-G1 cyclins via Mcm1 and Msn2/4. Nat. Commun. 8, 56 10.1038/s41467-017-00080-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charvin G., Oikonomou C., Siggia E. D. and Cross F. R. (2010). Origin of irreversibility of cell cycle Start in budding yeast. PLoS Biol. 8, e1000284 10.1371/journal.pbio.1000284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., Nishikawa J. L., Tang X., Millman J. S., Schub O., Breitkreuz K., Dewar D., Rupes I., Andrews B., Tyers M. et al. (2004). CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117, 899-913. 10.1016/j.cell.2004.05.024 [DOI] [PubMed] [Google Scholar]
- Darieva Z., Han N., Warwood S., Doris K. S., Morgan B. A. and Sharrocks A. D. (2012). Protein kinase C regulates late cell cycle-dependent gene expression. Mol. Cell Biol. 32, 4651-4661. 10.1128/MCB.06000-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin R. A. M., McDonald W. H., Kalashnikova T. I., Yates J. III and Wittenberg C. (2004). Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117, 887-898. 10.1016/j.cell.2004.05.025 [DOI] [PubMed] [Google Scholar]
- de Bruin R. A. M., Kalashnikova T. I., Chahwan C., McDonald W. H., Wohlschlegel J., Yates J. III, Russell P. and Wittenberg C. (2006). Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol. Cell 23, 483-496. 10.1016/j.molcel.2006.06.025 [DOI] [PubMed] [Google Scholar]
- Dorsey S., Tollis S., Cheng J., Black L., Notley S., Tyers M. and Royer C. A. (2018). G1/S transcription factor copy number is a growth-dependent determinant of cell cycle commitment in yeast. Cell Syst. 6, 539-554.e11. 10.1016/j.cels.2018.04.012 [DOI] [PubMed] [Google Scholar]
- Escoté X., Zapater M., Clotet J. and Posas F. (2004). Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1. Nat. Cell Biol. 6, 997-1002. 10.1038/ncb1174 [DOI] [PubMed] [Google Scholar]
- Ewald J. C. (2018). How yeast coordinates metabolism, growth and division. Curr. Opin. Microbiol. 45, 1-7. 10.1016/j.mib.2017.12.012 [DOI] [PubMed] [Google Scholar]
- Ferrezuelo F., Colomina N., Futcher B. and Aldea M. (2010). The transcriptional network activated by Cln3 cyclin at the G1-to-S transition of the yeast cell cycle. Genome Biol. 11, R67 10.1186/gb-2010-11-6-r67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego C., Gari E., Colomina N., Herrero E. and Aldea M. (1997). The Cln3 cyclin is down-regulated by translational repression and degradation during the G1 arrest caused by nitrogen deprivation in budding yeast. EMBO J. 16, 7196-7206. 10.1093/emboj/16.23.7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García R., Bermejo C., Grau C., Pérez R., Rodríguez-Peña J. M., Francois J., Nombela C. and Arroyo J. (2004). The global transcriptional response to transient cell wall damage in Saccharomyces cerevisiae and its regulation by the cell integrity signaling pathway. J. Biol. Chem. 279, 15183-15195. 10.1074/jbc.M312954200 [DOI] [PubMed] [Google Scholar]
- Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D. and Brown P. O. (2000). Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241-4257. 10.1091/mbc.11.12.4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D. and Sugino A. (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527-534. 10.1016/0378-1119(88)90185-0 [DOI] [PubMed] [Google Scholar]
- Gomar-Alba M., Méndez E., Quilis I., Bañó M. C. and Igual J. C. (2017). Whi7 is an unstable cell-cycle repressor of the Start transcriptional program. Nat. Commun. 8, 329 10.1038/s41467-017-00374-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Novo A., Jiménez J., Clotet J., Nadal-Ribelles M., Cavero S., de Nadal E. and Posas F. (2015). Hog1 targets Whi5 and Msa1 transcription factors to downregulate cyclin expression upon stress. Mol. Cell Biol. 35, 1606-1618. 10.1128/MCB.01279-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase S. B. and Wittenberg C. (2014). Topology and control of the cell-cycle-regulated transcriptional circuitry. Genetics 196, 65-90. 10.1534/genetics.113.152595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. R., Lee D., Farmer S., Lowndes N. F. and de Bruin R. A. M. (2013). Binding specificity of the G1/S transcriptional regulators in budding yeast. PLoS ONE 8, e61059 10.1371/journal.pone.0061059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J. C., Bardes E. S. G., Ohya Y. and Lew D. J. (2001). A role for the Pkc1p/Mpk1p kinase cascade in the morphogenesis checkpoint. Nat. Cell Biol. 3, 417-420. 10.1038/35070104 [DOI] [PubMed] [Google Scholar]
- Heinisch J. J. and Rodicio R. (2018). Protein kinase C in fungi-more than just cell wall integrity. FEMS Microbiol. Rev. 42, fux051 10.1093/femsre/fux051 [DOI] [PubMed] [Google Scholar]
- Heldt F. S., Lunstone R., Tyson J. J. and Novák B. (2018). Dilution and titration of cell-cycle regulators may control cell size in budding yeast. PLoS Comput. Biol. 14, e1006548 10.1371/journal.pcbi.1006548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D., Kaluarachchi S., van Dyk D., Friesen H., Sopko R., Ye W., Bastajian N., Moffat J., Sassi H., Costanzo M. et al. (2009). Dual regulation by pairs of cyclin-dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Biol. 7, e1000188 10.1371/journal.pbio.1000188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igual J. C., Johnson A. L. and Johnston L. H. (1996). Coordinated regulation of gene expression by the cell cycle transcription factor Swi4 and the protein kinase C MAP kinase pathway for yeast cell integrity. EMBO J. 15, 5001-5013. 10.1002/j.1460-2075.1996.tb00880.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Gutiérrez E., Alegría-Carrasco E., Sellers-Moya A., Molina M. and Martín H. (2020). Not just the wall: the other ways to turn the yeast CWI pathway on. Int. Microbiol. 23, 107-119. 10.1007/s10123-019-00092-2 [DOI] [PubMed] [Google Scholar]
- Johnson A. and Skotheim J. M. (2013). Start and the restriction point. Curr. Opin. Cell Biol. 25, 717-723. 10.1016/j.ceb.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.-Y., Truman A. W. and Levin D. E. (2008). Yeast Mpk1 mitogen-activated protein kinase activates transcription through Swi4/Swi6 by a noncatalytic mechanism that requires upstream signal. Mol. Cell Biol. 28, 2579-2589. 10.1128/MCB.01795-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.-Y., Truman A. W., Caesar S., Schlenstedt G. and Levin D. E. (2010). Yeast Mpk1 cell wall integrity mitogen-activated protein kinase regulates nucleocytoplasmic shuttling of the Swi6 transcriptional regulator. Mol. Biol. Cell 21, 1609-1619. 10.1091/mbc.e09-11-0923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C., Moll T., Neuberg M., Ahorn H. and Nasmyth K. (1993). A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science 261, 1551-1557. 10.1126/science.8372350 [DOI] [PubMed] [Google Scholar]
- Koch C., Schleiffer A., Ammerer G. and Nasmyth K. (1996). Switching transcription on and off during the yeast cell cycle: Cln/Cdc28 kinases activate bound transcription factor SBF (Swi4/Swi6) at Start, whereas Clb/Cdc28 kinases displace it from the promoter in G2. Genes Dev. 10, 129-141. 10.1101/gad.10.2.129 [DOI] [PubMed] [Google Scholar]
- Lagorce A., Hauser N. C., Labourdette D., Rodriguez C., Martin-Yken H., Arroyo J., Hoheisel J. D. and François J. (2003). Genome-wide analysis of the response to cell wall mutations in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 278, 20345-20357. 10.1074/jbc.M211604200 [DOI] [PubMed] [Google Scholar]
- Levin D. E. (2011). Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics 189, 1145-1175. 10.1534/genetics.111.128264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litsios A., Huberts D. H. E. W., Terpstra H. M., Guerra P., Schmidt A., Buczak K., Papagiannakis A., Rovetta M., Hekelaar J., Hubmann G. et al. (2019). Differential scaling between G1 protein production and cell size dynamics promotes commitment to the cell division cycle in budding yeast. Nat. Cell Biol. 21, 1382-1392. 10.1038/s41556-019-0413-3 [DOI] [PubMed] [Google Scholar]
- Malumbres M. (2014). Cyclin-dependent kinases. Genome Biol. 15, 122 10.1186/gb4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menoyo S., Ricco N., Bru S., Hernandez-Ortega S., Escote X., Aldea M. and Clotet J. (2013). Phosphate-activated cyclin-dependent kinase stabilizes G1 cyclin to trigger cell cycle entry. Mol. Cell Biol. 33, 1273-1284. 10.1128/MCB.01556-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles S., Croxford M. W., Abeysinghe A. P. and Breeden L. L. (2016). Msa1 and Msa2 modulate G1-specific transcription to promote G1 arrest and the transition to quiescence in budding yeast. PLoS Genet. 12, e1006088 10.1371/journal.pgen.1006088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno D. F., Parisi E., Yahya G., Vaggi F., Csikász-Nagy A. and Aldea M. (2019). Competition in the chaperone-client network subordinates cell-cycle entry to growth and stress. Life Sci. Alliance 2, e201800277 10.26508/lsa.201800277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Torres M., Jaquenoud M. and De Virgilio C. (2015). TORC1 controls G1-S cell cycle transition in yeast via Mpk1 and the greatwall kinase pathway. Nat. Commun. 6, 8256 10.1038/ncomms9256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Torres M., Jaquenoud M., Péli-Gulli M.-P., Nicastro R. and De Virgilio C. (2017). TORC1 coordinates the conversion of Sic1 from a target to an inhibitor of cyclin-CDK-Cks1. Cell Discov. 3, 17012 10.1038/celldisc.2017.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D. O. (2007). The cell-cycle control system. In The Cell Cycle: Principles of Control, pp. 28-54, New Science Press. [Google Scholar]
- Nasmyth K. and Dirick L. (1991). The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell 66, 995-1013. 10.1016/0092-8674(91)90444-4 [DOI] [PubMed] [Google Scholar]
- Negishi T., Veis J., Hollenstein D., Sekiya M., Ammerer G. and Ohya Y. (2016). The Late S-Phase transcription factor Hcm1 is regulated through phosphorylation by the cell wall integrity checkpoint. Mol. Cell Biol. 36, 941-953. 10.1128/MCB.00952-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo P., Vanoni M., Cusimano V., Busti S., Marano F., Manes C. and Alberghina L. (2016). Whi5 phosphorylation embedded in the G1/S network dynamically controls critical cell size and cell fate. Nat. Commun. 7, 11372 10.1038/ncomms11372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo S., Neog D., Spade D., Van Horn J. D., Tiede-Lewis L. A. M., Dallas S. L., Kapros T. and Honigberg S. M. (2017). Shrinking daughters: Rlm1-dependent G1/S checkpoint maintains Saccharomyces cerevisiae daughter cell size and viability. Genetics 206, 1923-1938. 10.1534/genetics.117.204206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramila T., Wu W., Miles S., Noble W. S. and Breeden L. L. (2006). The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev. 20, 2266-2278. 10.1101/gad.1450606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A. B., García R., Rodríguez-Peña J. M., Díez-Muñiz S., Nombela C., Peterson C. L. and Arroyo J. (2012). Chromatin remodeling by the SWI/SNF complex is essential for transcription mediated by the yeast cell wall integrity MAPK pathway. Mol. Biol. Cell 23, 2805-2817. 10.1091/mbc.e12-04-0278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A. B., García R., Rodríguez-Peña J. M. and Arroyo J. (2017). The CWI pathway: regulation of the transcriptional adaptive response to cell wall stress in yeast. J. Fungi 4, 1 10.3390/jof4010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoller K. M., Turner J. J., Kõivomägi M. and Skotheim J. M. (2015). Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature 526, 268-272. 10.1038/nature14908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova J. M. and Breeden L. L. (1997). Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae. Genes Dev. 11, 3032-3045. 10.1101/gad.11.22.3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim J. M., Di Talia S., Siggia E. D. and Cross F. R. (2008). Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454, 291-296. 10.1038/nature07118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner F. J., Quilis I. and Igual J. C. (2009). Spatial regulation of the Start repressor Whi5. Cell Cycle 8, 3010-3018. 10.4161/cc.8.18.9621 [DOI] [PubMed] [Google Scholar]
- Takahata S., Yu Y. and Stillman D. J. (2009). The E2F functional analogue SBF recruits the Rpd3(L) HDAC, via Whi5 and Stb1, and the FACT chromatin reorganizer, to yeast G1 cyclin promoters. EMBO J. 28, 3378-3389. 10.1038/emboj.2009.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talarek N., Gueydon E. and Schwob E. (2017). Homeostatic control of START through negative feedback between Cln3-Cdk1 and Rim15/Greatwall kinase in budding yeast. eLife 6, e26233 10.7554/eLife.26233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai V., Dephoure N., Weiss A., Ferguson J., Leitao R., Gygi S. P. and Kellogg D. R. (2017). Protein kinase C controls binding of Igo/ENSA proteins to protein phosphatase 2A in budding yeast. J. Biol. Chem. 292, 4925-4941. 10.1074/jbc.M116.753004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travesa A., Kuo D., de Bruin R. A. M., Kalashnikova T. I., Guaderrama M., Thai K., Aslanian A., Smolka M. B., Yates J. R. III, Ideker T. et al. (2012). DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 31, 1811-1822. 10.1038/emboj.2012.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travesa A., Kalashnikova T. I., de Bruin R. A. M., Cass S. R., Chahwan C., Lee D. E., Lowndes N. F. and Wittenberg C. (2013). Repression of G1/S transcription is mediated via interaction of the GTB motifs of Nrm1 and Whi5 with Swi6. Mol. Cell Biol. 33, 1476-1486. 10.1128/MCB.01333-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman A. W., Kim K.-Y. and Levin D. E. (2009). Mechanism of Mpk1 mitogen-activated protein kinase binding to the Swi4 transcription factor and its regulation by a novel caffeine-induced phosphorylation. Mol. Cell Biol. 29, 6449-6461. 10.1128/MCB.00794-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waern K. and Snyder M. (2013). Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 3, 343-352. 10.1534/g3.112.003640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M. V., Smolka M. B., de Bruin R. A. M., Zhou H., Wittenberg C. and Dowdy S. F. (2009). Whi5 regulation by site specific CDK-phosphorylation in Saccharomyces cerevisiae. PLoS ONE 4, e4300 10.1371/journal.pone.0004300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Carey L. B., Cai Y., Wijnen H. and Futcher B. (2009). Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PLoS Biol. 7, e1000189 10.1371/journal.pbio.1000189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnen H., Landman A. and Futcher B. (2002). The G1 cyclin Cln3 promotes cell cycle entry via the transcription factor Swi6. Mol. Cell Biol. 22, 4402-4418. 10.1128/MCB.22.12.4402-4418.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahya G., Parisi E., Flores A., Gallego C. and Aldea M. (2014). A Whi7-anchored loop controls the G1 Cdk-cyclin complex at Start. Mol. Cell 53, 115-126. 10.1016/j.molcel.2013.11.015 [DOI] [PubMed] [Google Scholar]
- Yano K., Uesono Y., Yoshida S., Kikuchi A., Kashiwazaki J., Mabuchi I. and Kikuchi Y. (2013). Mih1/Cdc25 is negatively regulated by Pkc1 in Saccharomyces cerevisiae. Genes Cells 18, 425-441. 10.1111/gtc.12047 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Ren P., Vashisht A. A., Wohlschlegel J. A., Quintana D. G. and Zeng F. (2017). Cdk1-interacting protein Cip1 is regulated by the S phase checkpoint in response to genotoxic stress. Genes Cells 22, 850-860. 10.1111/gtc.12518 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.