

ABSTRACT
A lack of biological markers has limited our ability to identify the invasive cells responsible for glioblastoma multiforme (GBM). To become migratory and invasive, cells must downregulate matrix adhesions, which could be a physical marker of invasive potential. We engineered murine astrocytes with common GBM mutations, e.g. Ink4a (Ink) or PTEN deletion and expressing a constitutively active EGF receptor truncation (EGFRvIII), to elucidate their effect on adhesion. While loss of Ink or PTEN did not affect adhesion, counterparts expressing EGFRvIII were significantly less adhesive. EGFRvIII reduced focal adhesion size and number, and these cells – with more labile adhesions – displayed enhanced migration. Regulation appears to depend not on physical receptor association to integrins but, rather, on the activity of the receptor kinase, resulting in transcriptional integrin repression. Interestingly, EGFRvIII intrinsic signals can be propagated by cytokine crosstalk to cells expressing wild-type EGFR, resulting in reduced adhesion and enhanced migration. These data identify potential intrinsic and extrinsic mechanisms that gliomas use to invade surrounding parenchyma.
KEY WORDS: Cancer, Extracellular matrix, Adhesion, Invasion
Summary: Constitutively active and truncated EGFR transcriptionally represses integrins via a SHC–MEK signaling axis, and communicates with neighboring cells through TNFα to promote cooperative invasion.
INTRODUCTION
Although the prevalence of brain tumors is below that of other tumor types, e.g. mammary or prostate (Siegel et al., 2017; American Cancer Society, Cancer Facts and Figures 2015), the mortality rate for brain tumors is much higher (5-year survival rate is 35% for brain tumors vs >90% of mammary or prostate tumors). Patient outcomes are even more drastic for glioblastoma multiforme (GBM), a stage-4 astrocytoma characterized by infiltration into healthy parenchyma, tumor heterogeneity, resistance to apoptosis and genomic instability (Furnari et al., 2007). GBM accounts for ∼45% of all invasive brain cancers and >12,800 new cases annually in the US (Ostrom et al., 2019). Average GBM patient survival is low – 12–15 months (Dunn et al., 2012) – despite the high standard of care, i.e. tumor resection, radiation therapy and treatment with the DNA-alkylating agent temozolomide (Paw et al., 2015; Stupp et al., 2005). Poor prognosis is partly due to the poorly margined, highly invasive phenotype of GBM, recurring anywhere within 1 cm of the original lesion to the other side of the corpus callosum (Cuddapah et al., 2014; Demuth and Berens, 2004), thus, prohibiting a surgical cure. Tumor cell invasion requires conversion away from a proliferative phenotype (Cuddapah et al., 2014; Demuth and Berens, 2004) and is characterized by a significant increase in migration and interaction with, and degradation of, multiple brain ECM proteins (Bellail et al., 2004; Brösicke and Faissner, 2015; Rao, 2003). Despite advances in our understanding of glioma invasion and migration, little is known about the molecular mechanisms that regulate this switch and whether these mechanisms are cell intrinsic, extrinsic or a combination of both.
The presence of multiple invasive mechanisms may be due to significant intratumoral genetic heterogeneity (Kleihues et al., 2002). GBM tumors with the worst prognoses typically have issues regarding one or more of three genes, i.e. deletion of the cell cycle regulator Ink4a/Arf (Holland et al., 1998), deletion of the tumor suppressor PTEN (Verhaak et al., 2010), and amplification and truncation of epidermal growth factor receptor (EGFR) (Gan et al., 2009; Inda et al., 2010). For example, changes in EGFR occur in ∼60% of GBM (Francis et al., 2014; Furnari et al., 2015), and its most common EGFR variant – truncation of exons 2–7, i.e. EGFRvIII – causes constitutive self-phosphorylation, pathway activation (Narita et al., 2002) and reduced apoptosis (Nagane et al., 2001). None of these properties are conferred to cells overexpressing wild-type EGFR (wtEGFR), which cannot drive glioma formation alone (Bachoo et al., 2002; Hesselager and Holland, 2003; Holland et al., 1998). We have previously found that EGFRvIII-positive cells, which are often scattered diffusely within a tumor (Nishikawa et al., 2004), actively communicate with neighboring wtEGFR cells (Bonavia et al., 2011; Inda et al., 2010; Zanca et al., 2017), hinting that inter-clonal communication could illustrate a paradigm for cooperativity of GBM cells. However, the mechanisms that EGFR alterations, individually or collectively, use to drive GBM migration and invasion are less clear.
To ensure dissemination into healthy tissue, cells at the invasive front must detach from the tumor mass, changing adhesion from largely cell–cell to cell–matrix. For epithelial tumors, invasive potential and adhesion strength are inversely correlated (Fuhrmann et al., 2017) owing to altered focal adhesion assembly (Fuhrmann et al., 2014) and turnover (Bijian et al., 2013) allowing cells to move through the tissue effectively. As a result, changes in adhesion of cancer cells to ECM proteins are becoming a more accepted metric for metastatic potential (Reticker-Flynn et al., 2012; Yates et al., 2014). Although this relationship is not clear for GBM, the studies mentioned above suggest that EGFR variants play an intrinsic role in directly binding to and indirectly modifying signaling pathways that affect adhesion. By contrast, wtEGFR cells could cooperatively invade with EGFRvIII cells that recruit and convert them epigenetically. Utilizing a spinning disc assay (Engler et al., 2009; Fuhrmann et al., 2017), which subjects cell populations to radially increasing shear stress, we investigated these possibilities by using an isogenic mouse glioma cell line containing various permutations of Ink4a/Arf deletion (Holland et al., 1998), PTEN deletion (Verhaak et al., 2010), and wtEGFR or EGFRvIII overexpression (Gan et al., 2009; Inda et al., 2010). We found that combinations of Ink4a/Arf deletion, PTEN deletion or EGFR overexpression did not reduce adhesion, but EGFRvIII overexpression did. Given the lower frequency of these cells in heterogeneous tumors (Brennan et al., 2013; Nishikawa et al., 1995), we further found that EGFRvIII-expressing cells produced cytokine signals that, when applied to wtEGFR cells, were able to reduce their adhesion and increase their migration. Together, these data suggest that EGFRvIII creates cell-intrinsic signals that regulate adhesion strength, as well as extrinsic signals that instruct heterogeneous tumor cell populations to invade the surrounding parenchyma.
RESULTS
GBM driver mutations reduce adhesion strength and increase migration via labile adhesions
Tumor recurrence post-resection suggests that some subset of GBM cells have transitioned from a proliferative (Cuddapah et al., 2014; Demuth and Berens, 2004) to an invasive and migratory phenotype (Demuth and Berens, 2004; Paw et al., 2015) by using cell–matrix adhesions. To determine which of the most-common mutations can affect adhesion, we utilized low-passage isogenic murine astrocytes expressing combinations of Ink4A/Arf deletion, PTEN deletion or EGFR alterations, i.e. overexpression of wild-type receptor or a constitutively active truncation mutant (Table S1) (Bachoo et al., 2002). Cell genotypes were confirmed by western blot analysis (Fig. 1A) and then adhesion characterized by spinning disk assay (Boettiger, 2007), i.e. a quantitative population-based assay where cells are detached from a fibronectin-coated coverslip by radially increasing shear stress (Fig. S1). In the absence of cations, many cell lines exhibited similar adhesion strength (Fig. 1B, striped bars); yet, in the presence of cations, adhesion strength was lower only for lines containing EGFRvIII (Fig. 1B, solid bars). This difference might indicate that a significant role for EGFRvIII in modulating cation-dependent astrocyte adhesion. By contrast, epithelial tumor adhesion is reduced in the absence of cations (Fuhrmann et al., 2017).
Fig. 1.

Cation-dependent astrocyte adhesion is reduced by EGFRvIII. (A) Western blot of indicated genotypes for Pten, EGFR and actin. (B) Shear stress at which 50% of the population detaches, i.e. adhesion strength of τ50, is plotted for the indicated genotypes. Cells assayed in buffer with (solid bars) and without (hatched bars) cation-containing medium are shown. *P<0.05 and **P<0.01 by paired Student’s t-test (n=4 per genotype and condition; solid and dashed lines indicate comparisons of cells with and without cation-containing medium, respectively).
Adhesion changes are likely to manifest themselves in migration differences (Beri et al., 2020); so, we next determined whether reduced EGFRvIII cell adhesion improved migration speed and persistence on fibronectin in cation-containing medium. Cells expressing EGFRvIII had a significantly longer pathlength (i.e. the total distance traveled) and migrated significantly faster compared to Ink−/−- and wtEGFR-expressing cells, implying that amplification of wtEGFR does not increase tumor cell migration but that receptor truncation is beneficial (Fig. 2A,B; Fig. S2). However, wtEGFR cells had significantly greater average displacement (i.e. the shortest distance drawn from initial to final position), resulting in more processive motility (i.e. ratio of displacement to total distance traveled) compared to Ink−/− cells, whereas EGFRvIII cells had slightly lower displacement and, thus, less processive migration (Fig. 2C,D; Fig. S2), indicating that they explored a greater area than cells simply with EGFR amplification. Therefore, these data indicate that EGFRvIII cells tend to rapidly investigate a larger portion of their environment – which would be consistent with the invasiveness of GBM.
Fig. 2.

Migratory characteristics scale with adhesion and depend on EGFR. (A–C) Total cell migration distance (A), speed (B), displacement (C) and persistence (D) over 24 h is plotted for the indicated genotypes (n=3). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 assessed by paired Student’s t-test (n=30, 91 and 74 cells for Ink−/−-, wtEGFR- and EGFRvIII-expressing cells, respectively).
The link between diminished cation-dependent adhesion strength and migration further suggests that EGFRvIII, in some manner, affects focal adhesion dynamics. Thus, we investigated focal adhesion structures on fibronectin-coated coverslips by using immunofluorescence, and found significant structural differences between EGFRvIII and wtEGFR cells (Fig. 3A,B). When normalized to cell area, the number of focal adhesions in EGFRvIII cells were reduced by ∼50% and had smaller focal adhesions per cell area (Fig. 3C–E). Moreover, EGFR was enriched in the plasma membrane at focal adhesions for cells expressing EGFRvIII (Fig. 3F), suggesting a direct regulation of integrins by EGFR amplification and truncation, which, in turn, might reduce cell adhesion strength and increase migration speed.
Fig. 3.
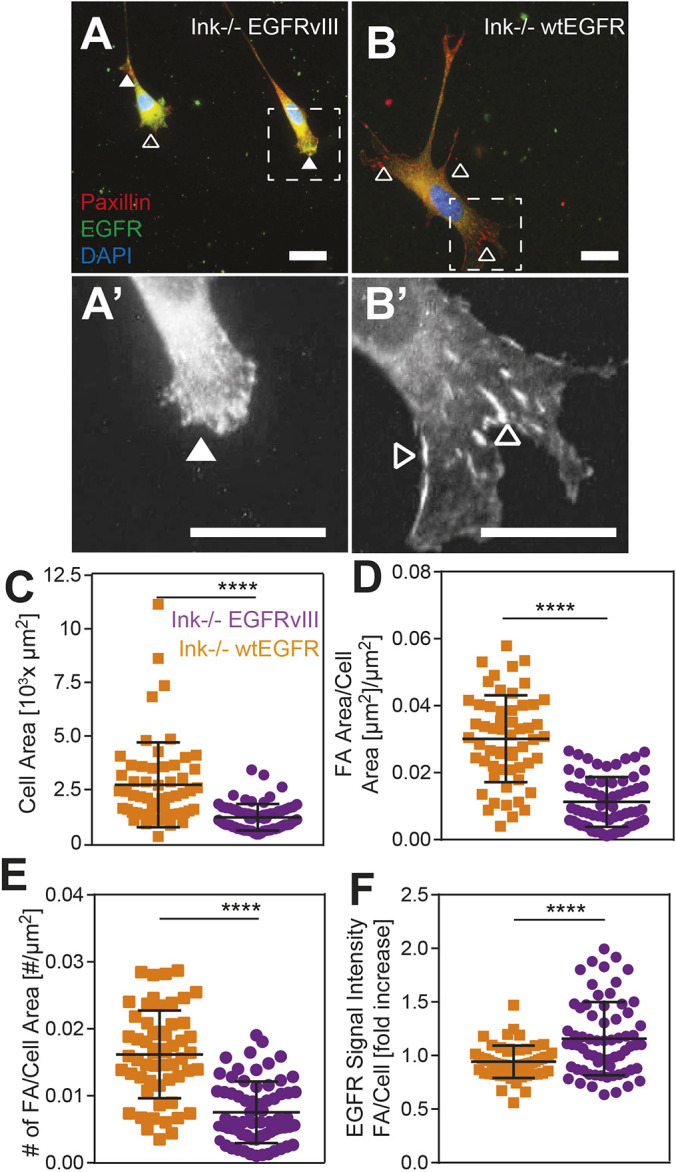
Focal adhesion assembly is suppressed by EGFRvIII. (A–B’) Immunofluorescence images of EGFRvIII (A) and wtEGFR (B) cells stained for paxillin (red) and EGFR (green); the nucleus was stained with DAPI (blue). Areas surrounded by dashed boxes are magnified in A′ and B′. Filled and open arrowheads indicate regions of diffuse and assembled adhesion complexes. Scale bars: 10 μm. (C–F) Cell area (C), area of focal adhesion-to-cell area (D), number of focal adhesions to cell area (E) and EGFR signal intensity within a focal adhesion per average cell intensity (F) are plotted for both EGFRvIII (orange) and wtEGFR (purple) cells, with n=3. ****P<0.0001 was assessed by paired Student’s t-test (n=68 and 57 cells for wtEGFR and EGFRvIII cells, respectively).
Labile focal adhesions are the result of intrinsic kinase-dependent signaling
EGFR appears to be more localized within focal adhesion sites in EGFRvIII-expressing cells compared to cells that overexpress wtEGFR. However, it is unclear whether the constitutively active kinase activity of the receptor (Holland et al., 1998) or its potential interactions with integrin (Liu et al., 2016) create more labile adhesions. To determine whether physical associations between EGFRvIII and integrins reduce adhesion, levels of αV and β3 integrin were analyzed by western blotting. Consistent with immunostaining experiments, both were found to be expressed less in EGFRvIII compared with wtEGFR cells. Yet, immunoprecipitation of αV and β3 integrins in the presence of a membrane-impermeable crosslinker to maximize detection of transient interactions did not detect a direct interaction (Fig. 4A). These data indicate that, despite enhanced colocalization, EGFRvIII does not directly interact with integrins but, rather, may indirectly regulate adhesion through enzymatic activity of its kinase domain and downstream signaling effectors. Western blotting of phosphorylated EGFR, however, did confirm the constitutively active kinase activity of the receptor for EGFRvIII (Fig. 4B), which suggests that adhesion regulation is kinase-dependent. To determine whether this is indeed the case, a kinase-defect mutant was created by substitution of lysine to methionine (K721M) (Honegger et al., 1987) within the catalytic domain of the receptor (Fig. S3A), i.e. an EGFRvIII kinase-dead mutant (EGFRvIIIKD), and validated by FACS sorting (Fig. S3B) and western blotting to confirm equal EGFR protein expression and loss of autophosphorylation for EGFRvIIIKD (Fig. S3C). Although EGFRvIII reduced adhesion strength, the loss of kinase activity in EGFRvIIIKD restored adhesion to native wtEGFR levels (Fig. 4C). Similarly, cell migration speed, which increased with EGFRvIII, was decreased in EGFRvIIIKD cells as well as the distance traveled (Fig. 4D; Fig. S4). These data indicate that the constitutive activation of EGFRvIII, and not its residence on the cell surface (Schmidt et al., 2003) is responsible for altered adhesion.
Fig. 4.

Adhesion strength is modulated by kinase-dependent mechanism(s). (A) Western blots of whole-cell lysate (WCL; left) confirm genotypes and expression patterns of integrins αV and β3. Additional blots (right) for EGFRvIII and wtEGFR show the results of EGFR immunoprecipitation with supernatant (Supp.) and pulldown (IP) lanes for the integrins αV and β3, phosphorylated SHC (pShc) and actin. (B) Western blot for total EGFR (pan-EGFR), phosphorylated EGFR (pEGFR) and actin for wtEGFR (wt), EGFRvIII (vIII) and EGFRvIIIKD (vIIIKD). (C) Adhesion strength, i.e. τ50, plotted for the different genotypes as indicated. *P<0.05 and **P<0.01 assessed by paired Student’s t-test (n=4 for each genotype). (D) Cell speed and total migration distance plotted for the same genotypes as listed in panel C. *P<0.05; ****P<0.0001; *****p<0.00001; assessed by paired Student’s t-test (n=4 biological replicates, analyzing 39, 88, 65 and 93 cells for Ink−/−, wtEGFR, EGFRvIII and EGFRvIIIKD cells, respectively).
Although kinase-dependent, it is uncertain which downstream signaling effectors result in focal adhesion disassembly, thus we assessed adhesion protein expression and phosphorylation status in EGFRvIII cells compared to the Ink−/− and wtEGFR cells. We found that no phosphorylation or protein expression differences in FAK or vinculin but marked decreases in the fibronectin-binding integrins αV, α5 and β1, and increases in paxillin and talin in EGFRvIII cells (Fig. 5A). Consistent with smaller focal adhesions in EGFRvIII cells, these data could indicate that the truncated receptor prevents integrin production but that cells attempt to compensate this by overexpressing adhesion components. To better understand at what point integrins are downregulated, we first assessed transcript levels, and found that EGFRvIII-expressing cells downregulate αV and β1 integrins (Fig. 5B), i.e. those fibronectin-binding integrins that require the most force to rupture (Bharadwaj et al., 2017). Next, because constitutive receptor activation induces a plethora of downstream signaling exclusive to EGFRvIII (Huang et al., 2009), we assessed which kinase-dependent signals could result in adhesion changes by detecting and perturbing these signals. Together with different expression levels of the above mentioned adhesion proteins, we observed that levels of phosphorylation of SHC (here referring to SHC1), JNK (MAPK8) and JUN, SRC, Stat3, and MEK (here referring to MEK1 and MEK2, also known as MAP2K1 and MAP2K2, respectively) in EGFRvIII cells were different compared to wtEGFR or EGFRvIIIKD cells (Fig. 5C). To validate these pathways, we first chose to block phosphorylation of EGFR/SHC and MEK (using the tyrosine kinase inhibitors Erlotinib or Trametinib, respectively), as shown by western blotting (Fig. 5D). Small-molecule inhibition of EGFR with Erlotinib and of MEK with Trametinib decreased signaling by 58% and 56%, respectively. To determine their effect on adhesion, cells were treated with inhibitor for 48 h.
Fig. 5.

Kinase signaling downstream of EGFR transcriptionally silences integrins. (A) Western blots of indicated adhesive proteins expressed in cells expressing EGFRvIII (vIII), wtEGFR (wt) or Ink−/− (−/−). Those that show differential expression with EGFRvIII as being different from the other isogenic cell lines are shown in red. (B) Quantification of transcript (RNA) expression of the indicated integrin genes showing fold decrease of EGFRvIII cells normalized to wtEGFR cells, i.e. truncated and amplified receptor normalized to amplified receptor only. *P<0.05 assessed by one-way ANOVA with Dunn's multiple comparisons test (n=6 biological replicates). (C) Western blots of indicated receptor, adhesive and signaling proteins expressed in wtEGFR, EGFRvIII or EGFRvIIIKD (KDv3) cells. (D) Western blots of indicated proteins expressed in non-treated (-) wtEGFR, EGFRvIII or EGFRvIIIKD cells, as well as in EGFRvIII cells treated with (+) and without (-) Erlotinib or Trametinib. (E) Adhesion strength τ50 plotted for different genotypes as indicated, including those selectively treated with (+) and without (-) Erlotinib or Trametinib (EGFRvIII hatched bars; n=3 biological replicates). *P<0.05, ***P<0.001, ****P<0.0001 assessed by paired Student’s t-test as indicated. (F) RNA levels of integrin α5, αv and β1 expressed in EGFRvIII cells treated with Erlotinib or Trametinib normalized to and shown as fold increase of levels in mock-treated (DMSO) cells. $P<0.1 assessed by one-way ANOVA with Dunn's multiple comparisons test (n=3 biological replicates).
Inhibition of EGFR signaling with Erlotinib increased adhesion strength and, in addition, inhibition of MEK with Trametinib also increased adhesion strength (Fig. 5E), implicating its involvement in adhesion modulation. To determine whether transcriptional repression of the integrins observed in EGFRvIII-expressing cells was reversed by inhibiting either EGFRvIII with Erlotinib or MEK via Trametinib, cells were selectively treated and assayed for transcript levels. Both inhibitors increased transcription of integrin subunits αV, α5 and β1, although Erlotinib was generally more effective (Fig. 5F), perhaps because it directly inhibits EGFR activity. These data support our conclusion that intrinsic signaling can transcriptionally repress integrins to increase EGFRvIII cell migration.
Extrinsic crosstalk modulates the adhesion of receptor-amplified cells
Given that GBMs are exceedingly heterogeneous and can contain cells that exhibit a variety of Ink4a/Arf, PTEN and EGFR genetic alterations, we next sought to determine whether cell extrinsic communication, which has been previously observed for wtEGFR and EGFRvIII (Inda et al., 2010; Zanca et al., 2017), can alter adhesion of cells expressing amplified EGF receptor alone. To determine whether paracrine communication was sufficient, we first treated wtEGFR cells with EGFRvIII-conditioned medium for 24 h to ‘educate’ them prior to shear exposure, thereby reducing their cell adhesion strength near to levels seen in EGFRvIII-expressing cells. However, for Ink−/− and EGFRvIIIKD cells, exposure to conditioned medium did not affect adhesion strength (Fig. 6A). These data indicate that the kinase activity of the receptor is necessary to alter adhesion strength, but do not identify the specific cytokine(s) required to alter cellular adhesion strength. Although cytokines IL6 and LIF have been suggested (Inda et al., 2010) to be involved in the extrinsic alteration of cellular phenotype, we performed a cytokine screen on EGFRvIII- and wtEGFR-conditioned medium to broadly determine the components necessary for paracrine signaling. Through analysis of those cytokines that demonstrated a greater than 2-fold change, we identified a subset of candidates with differential secretion in EGFRvIII-conditioned medium, including TNFα (Fig. 6B). To determine to what extent TNFα signaling affects adhesion strength of wtEGFR-expressing cells, EGFRvIII-conditioned medium was supplemented with either neutralizing antibody against TNFα or with rabbit IgG before treatment of wtEGFR and EGFRvIII cells. Inhibition of TNFα eliminated the decrease in adhesion strength conveyed by EGFRvIII-conditioned medium for wtEGFR cells. In contrast, the presence of rabbit IgG did not reverse the decrease. These data also show that, for EGFRvIII-expressing cells, extrinsic signaling from TNFα does not play an autocrine role in their reduced adhesion because cells remained less adherent independently of TNFα neutralization (Fig. 6C). Similarly, extrinsic signaling to wtEGFR cells from EGFRvIII-conditioned medium did not appear to be mediated by transcriptional silencing integrin because no clear pattern in transcript repression was found (Fig. S5). These data indicate that TNFα, among other cytokines, is necessary for cellular crosstalk and cooperative adhesion modulation of neighboring cells that express wtEFGR. These findings further suggest that EGFRvIII not only modulates adhesion strength intrinsically but that it modulates the adhesion strength of non-EGFRvIII-expressing cells through the secretion of TNFα (Fig. 7).
Fig. 6.
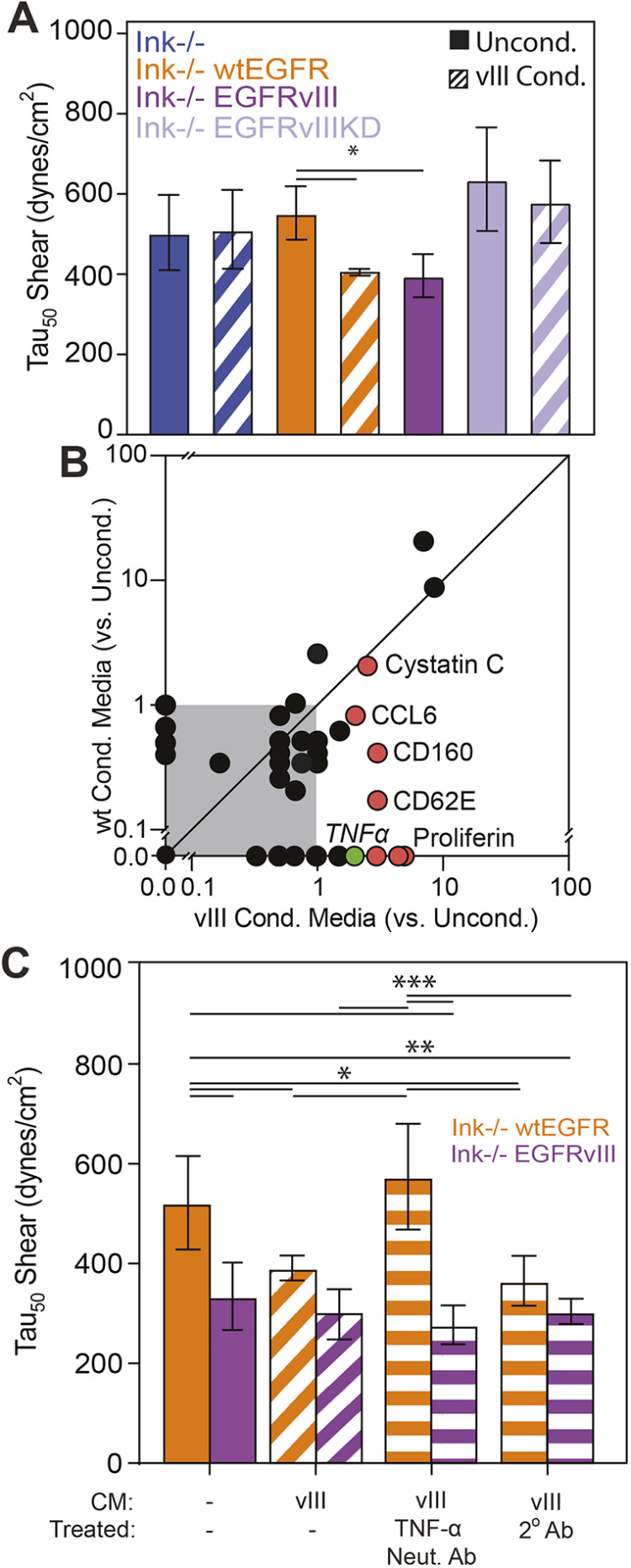
Cell extrinsic cytokines EGFRvIII reduce adhesion of wtEGFR cells. (A) Quantification of adhesion strength for cells of the indicated genotypes in unconditioned (solid bars) or EGFRvIII-conditioned medium (hatched bars; n=3 biological replicates). (B) Cytokine production was measured by microarray and plotted for wtEGFR and EGFRvIII-conditioned medium as normalized to unconditioned medium. Gray area indicates all cytokines measured below a ratio of 1. Unity line, i.e. y=x, is shown to illustrate those cytokines differentially expressed in wtEGFR- or EGFRvIII-conditioned medium. Red data points indicate cytokines (as indicated) overproduced in EGFRvIII-conditioned medium. Green data point indicates TNFα. (C) Adhesion strength τ50 plotted for wtEGFR- or EGFRvIII-expressing cells selectively cultured in their own medium or in EGFRvIII-conditioned medium that had been pre-treated with antibodies as indicated (hatched bars represent cells treated with EGFRvIII-conditioned medium and horizontal-striped bars represent cells treated with EGFRvIII-conditioned medium and an antibody as indicated; n=3 biological replicates). *P<0.05; **P<0.01; ***P<0.001; assessed by paired Student’s t-test for the indicated comparisons.
Fig. 7.
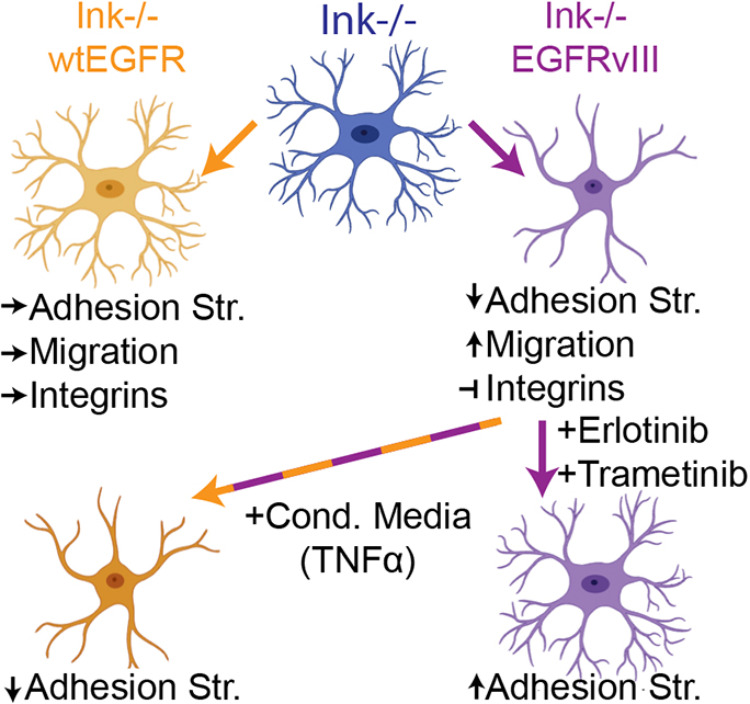
Cell extrinsic cytokines from EGFRvIII reduce adhesion of wtEGFR cells. Schematic of how expression of EGFR or its variant EGFRvIII affect cell functions and integrin expression in the Ink−/− parental line. Indicated drug treatments that affect the EGFR–MEK signaling axis alter adhesion of EGFRvIII cells, whereas cytokines from those cells can effect adhesion of wtEGFR-expressing cells.
DISCUSSION
Tumor heterogeneity and complex cooperative signaling between cells have impaired our ability to dissect the signaling pathways that promote the highly invasive nature of GBM in vivo (Cuddapah et al., 2014; Demuth and Berens, 2004). Although crosstalk between cells certainly does occur in this heterogeneous mass (Bonavia et al., 2011; Inda et al., 2010; Zanca et al., 2017), current in vivo methods to elucidate pathway details in GBM, e.g. laser capture microdissection (Daubon et al., 2019), are limited in throughput and cannot resolve crosstalk between individual cells. However, dissemination cues are not limited to biochemical signals, e.g. as cells detach from the tumor mass to invade, significant adhesive changes must occur that should be ubiquitous for solid tumors. We have previously demonstrated that in mammary epithelial tumors, changes in cell biophysical properties (i.e. decreased cellular adhesion strength) correspond with increased migratory and metastatic potential (Fuhrmann et al., 2017). Such assays also stratify tumor heterogeneity and provide a clearer understanding of the molecular mechanisms responsible for cancer cell migration (Beri et al., 2020). Here, for example, we used a spinning disk shear assay to engineer murine astrocytes, and found that cells expressing the EGFRvIII mutation had decreased adhesion strength relative to cells expressing similar wild-type receptor levels, regardless of additional mutations. Cell adhesion scales inversely with migration and is the result of EGFRvIII kinase-mediated integrin transcriptional repression. Our data demonstrate the utility of biophysical analyses to predict cell migration and show that cellular adhesion strength serves as an effective means to determine which cell populations may affect tumor recurrence.
Although the intrinsic role of EGFRvIII in astrocyte adhesion has not been examined previously, the effects of EGFRvIII on cell behavior have been studied in other contexts. Generally, EGFR kinase-dependent autophosphorylation activates downstream growth and cell survival pathways (Guo et al., 2015), and cytoskeletal modifications can cause EGFR and its ErbB2 family members to localize to adhesions and signal via the signaling axis between Ras, MEK and ERK1/2 (MAPK3/1) (Singhai et al., 2014). For cancer cell adhesion, migration and invasion of surrounding parenchyma, however, little is known about the impact of amplified levels or receptor truncation of EGFR. EGFRvIII does modulate adhesion, as shown in ovarian cancer cells (Gan et al., 2013; Ning et al., 2005), but its effects on adhesion in glioblastoma are uncertain. Given that ∼60% of GBM exhibit alterations concerning EGFR (Francis et al., 2014; Furnari et al., 2015), there seems to be a significant gap in our understanding of how biophysical changes in astrocytes affect disease progression. Adhesion data in our study here identify a unique role for EGFRvIII in transcriptional silencing of fibronectin-binding integrins via kinase signaling, which results in less stable adhesions capable to enhance migration. The reliance on kinase-dependent signaling is consistent with the GBM literature (Narita et al., 2002) but different to epidermoid carcinomas, in which EGFRvIII appears to directly associate with α2β1 integrins (Yu et al., 2000). So, although there is a precedent for interactions elsewhere, we believe our data establish a new function for the EGFRvIII variant in glioma.
Whereas intrinsic signaling can modulate a cell subtype, glioma invasion is rarely the result of a single subtype and more likely to be the result of cooperative signals. EGFR-expressing astrocytes are not tumorigenic upon intracranial injection unless hyperphysiological levels of EGF ligand are infused into the injection site (Bachoo et al., 2002). Amplified wild-type receptor can also respond to EGFRvIII-secreted cytokines to cooperatively invade into healthy parenchyma (Inda et al., 2010; Zanca et al., 2017). We found that these extrinsic signals may modulate adhesion of wild-type receptor counterparts to cooperatively invade but may use extrinsic mechanisms that do not involve transcriptional silencing of integrins. A variety of cytokines, such as IL6, LIF, IL8 and TNFα, are differentially expressed in EGFRvIII cells. TNFα has been linked to more-aggressive and migratory phenotypes in other tumors (Wu and Zhou, 2010), and our analysis indicated that TNFα is not only differentially expressed, but seems to be a necessary component in modulating the adhesion strength of cells overexpressing wtEGFR. Our data highlight the importance of cell-extrinsic factors to modulate adhesion and, thus, migration, and provide insight into possible cooperative behaviors that lead to larger, collective migration of tumor cells.
GBM is a heterogeneous disease, and our data suggest that additional attention should be paid to how certain receptor variants modulate adhesion and subsequent migration intrinsically, as well as how they recruit other cells to cooperatively migrate and locally invade adjacent parenchyma. Similar to that in epithelial tumor cells (Beri et al., 2020), GBM cell adhesion strength is inversely correlated with migration and, thus, adhesion may serve as a physical means to stratify tumors and an alternative to genetic markers. The most-aggressive gene variants may dominate their behavior, e.g. EGFRvIII, but the plasticity we observed in adhesion strength of wtEGFR cells when exposed to conditioned medium suggests that stratification by genetics alone only explains a part of tumor outcomes; cell state plasticity has recently been shown to give rise to tumor diversity (Dirkse et al., 2019). So, it is conceivable that cytokine crosstalk drives phenotype and possible epigenomic changes that reduce adhesion strength throughout a population, as observed here. Overall, our data are supportive of a shift to more-collective migration that has recently been thought to be a significant mode in which tumors invade in vivo (Alieva et al., 2019).
MATERIALS AND METHODS
Cell lines, media and mutagenesis
Mouse astrocytes, mAstr-Ink4a/Arf−/−, mAstr-Ink4a/Arf−/−-wtEGFR, mAstr-Ink4a/Arf−/−-EGFRvIII, were obtained and cultured as previously described (Bachoo et al., 2002; Inda et al., 2010; Wykosky et al., 2015). Briefly, cells were cultured in high-glucose Dulbecco's modified Eagle medium (DMEM), supplemented with L-glutamine4 (ThermoFisher Scientific, Prod#: 11965), 10% fetal bovine serum (Gemini Bio-Products, Prod#: 900-208), 1% Penicillin/Streptomycin (10,000 μ/ml, ThermoFisher Scientific, Prod#: 15140122), and 1% L-glutamine (200 mM, ThermoFisher Scientific Prod#: 25030081). All cells were cultured at 37°C in a humidified incubator containing 5% CO2, and all medium was sterile filtered. Cells were grown on tissue-culture-treated polystyrene (TCPS) substrates unless otherwise indicated. Cells were passaged every 2–3 days, depending on confluency, using 0.25% trypsin (ThermoFisher Scientific Prod#: 25200056), neutralized in medium and resuspended in fresh complete medium at dilutions appropriate for each cell line. For conditioned medium, EGFRvIII cells were seeded overnight, with medium subsequently being replaced with fresh medium for 24 h. Conditioned medium was collected, filtered using a 0.22 µm steriflip (MilliporeSigma Prod#: SCGP00525) and used immediately or frozen at −80°C in specific assays described below.
To briefly describe the generation of mAstr-Ink4a/Arf−/−-EGFRvIIIKD cells, EGFRvIII in pBABE-puro (Zanca et al., 2017) was altered by site-directed mutagenesis to generate lysine 721 to methionine (K721M) substitution (Honegger et al., 1987) within the kinase domain of the receptor (Fig. S3A). To produce retrovirus, HEK 293T cells were plated and adhered for ∼18 h before transfection by Lipofectamine 2000 (ThermoFisher Scientific Prod#: 11668019) together with retrovirus packaging construct pCL10A1 and the kinase-dead receptor construct pBABE-puro EGFRvIII kinase-dead. Retroviral supernatant was harvested up to 48 h post transfection, filtered and used to transduce mAstr-Ink4a/Arf−/−. Following overnight incubation, transduced cells were selected with 2 μg/ml puromycin for 4 days. To ensure equivalent EGFRvIIIKD and EGFRvIII expression, cells were subjected to fluorescence-activated cell sorting (Fig. S3B). Cells were grown to ∼80% confluency under selection of puromycin (5 μg/ml, ThermoFisher Scientific Prod#: A1113802), and detached with Verscene (ThermoFisher Scientific Prod#: 15040066). Cells were counted, pelleted and resuspended in flow-cytometry buffer (FACS buffer, 1×DPBS, 2% BSA, 1 mM EDTA pH 7.4) at a concentration of 1×107 cells/ml. After resuspension, DH8.3 anti EGFRvIII (Novus Biologicals Prod#: NBP2-50599) primary antibody was added to the resuspended cells and incubated on ice for 1 h. Cells were then washed, labeled at 1:100 with goat anti-mouse IgG H&L Alexa Fluor 488 (Abcam Prod#: ab150113) and washed again. Propidium iodide (1 mg/ml; ThermoFisher Scientific Prod#: P1304MP) was added at 1:1000 to differentiate between live and dead cells prior to sorting. Sorting parameters were set using non-EGFRvIII-expressing and EGFvIII-expressing cells, and kinase dead cells were sorted to equivalent expression levels of cell surface receptor by using a SH800S Cell Sorter (Sony Biotechnology).
Cell adhesion-strength assay
In the spinning disk device (Boettiger, 2007), cells were seeded onto 25 mm glass coverslips that had been coated with 10 μg/ml human fibronectin (isolated from serum5) for 60 min and blocked with medium (10% FBS) for 60 min at room temperature; all adhesion-strength and cellular-migration assays were performed on fibronectin-coated coverslips unless otherwise noted. Cells were seeded at a density 10,000 cells/cm2 to minimize cell-cell contact. Cells were attached to coverslips for a minimum of 12 h using cation-containing medium. After 12 h cells were mounted on a custom-built spinning-disk device and submerged into temperature-controlled phosphate-buffered saline (PBS) spinning buffer with (PBS+MgCa; Corning Prod#: 20-030-CV) or without cations (PBS; Corning Prod#: 20-031-CV), supplemented with 4.5 g/l dextrose warmed to 37°C. Cells were exposed to a range of fluid sheer – depending on rotational speed – for 5 min. Once spun, cells were fixed with 3.7% formaldehyde. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:2500, ThermoFisher Scientific Prod#: D1306) and mounted on slides with Fluoromount-G (Southern Biotech, cat # 0100-01). Slides were imaged using a CSU-X1 confocal scanner unit (Yokogawa), QuantEM:512SC camera (Photometrics, Tucson, AZ), and MS-2000-WK multi-axis stage controller (Applied Scientific Instrumentation) on a Nikon (Melville, NY) Ti-S microscope. Metamorph 7.6 software. A custom-written MATLAB (Mathworks) program was used to stitch together 1500 individual images of nuclei and to quantify average cell adhesion, i.e. τ50. The latter is defined as the shear stress at which 50% of the initial cell population is removed by fluid sheer stress (Fuhrmann et al., 2017), and was calculated using Eqn 1:
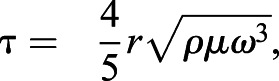 |
(1) |
where r is the radial position from the center of the disk, ρ is the buffer density, μ is the buffer viscosity, and ω is the rotational velocity6. To compare cell adhesion characteristics, cellular adhesion strength was analyzed in at least triplicates and compared using paired Student's t-test. All code associated with image analysis can be obtained at Englerlab.
Co-immunoprecipitation protocol
EGFR was covalently crosslinked to other proteins, isolated from cell lysate and analyzed for integrin association. Briefly, cells were washed and treated with either 2 mM DTSSP or DSP (ThermoFisher Scientific Prod#: 21578, 22585, respectively) for 60 min to covalently crosslink interacting proteins; Tris-buffered saline (TBS) was added to neutralize remaining crosslinker. Cells were then removed from the dish using a cell scraper (Corning, Prod#: 3010) and pelleted. After TBS rinse, cells were resuspended in 200 μl of non-denaturing lysis buffer (20 mM Tris HCL pH 8, 137 mM NaCl, 0.5% Trition X-100, 2 mM EDTA) to lyse cells without destroying protein conformation. Lysates were vortexed and centrifuged to pellet debris. Supernatants were collected to determine protein concentration in a BCA Assay (ThermoFisher Scientific Prod#: 23225). Next, protein G Dynabeads™ (ThermoFisher Scientific Prod#: 10007D) were functionalized with mouse Anti-EGF Receptor Clone 13 antibody (BD Biosciences Prod#: 610017) with or without BS3 crosslinker to covalently bind the antibody to the bead. To immunoprecipitate EGFR, samples containing 37 μg of protein were added to functionalized beads, pipetted to resuspend and incubated overnight at 4°C to allow antigen binding. Samples were magnetically pelleted, and the supernatants decanted and saved for parallel analysis. Proteins were eluted from the beads using denaturing gel loading buffer/dye, 50 mM DTT DTT, 1× Laemmli Sample Buffer (Biorad Prod#:1610747) and filled up to 30 μl with mRIPA [50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1% Triton, 10% glycerol, 25 mM sodium deoxycholate, 0.1% SDS, Roche Complete Protease Inhibitor (SigmaAldrich, Prod#: 11697498001) and PhosSTOP (Sigma-Aldrich, Prod#: 4906845001)]. Samples were heated for 5 min at 95°C, IP solution was separated from beads and western blotting performed for all samples to determine associations between EGFR and integrins.
Immunofluorescence staining
Cells that had been fixed in 3.7% formaldehyde in solution A (1× DPBS with 0.5 mM MgCl2 and 1 mM CaCl2) for 15 min were washed and stained with Deep Red CellMask (ThermoFisher Scientific Prod#: C10046) to label cell membranes. Cells were washed again, permeabilized, blocked (0.3 M glycine, 10% goat serum, 15 BSA, 0.1% BSA) for 1 h at room temp, and stained 1:500 with mouse primary anti-EGFR antibody (BD Biosciences Prod#: 610017) and rabbit anti-paxillin (abcam Prod#: ab32084) at 4°C. Cells were washed and incubated at 1:1000 with secondary antibodies goat anti-rabbit Alexa Fluor 568 (ThermoFisher Scientific Prod#: A11011) and goat anti-mouse Alexa Fluor 488 (ThermoFisher Scientific Prod#: A11001) at room temperature. Cells were then washed, incubated with a 1:2000 dilution of Hoechst 33342 dye for staining (ThermoFisher Scientific Prod#: H3570) at room temperature, and mounted with Fluoromount-G (Southern Biotech, Prod#: 0100-01). Samples were imaged by using the 60× objective on a Nikon Eclipse TI fluorescent microscope with a CSU-X1 confocal scanner unit (Yokogawa), QuantEM:512SC camera (Photometrics, Tucson, AZ), MS-2000-WK multi-axis stage controller (Applied Scientific Instrumentation), and controlled by Metamorph 7.6 (Molecular Devices). A custom-written ImageJ script was then used to quantify cell area and FA number and size. All FA metrics were computed across the entire cell to avoid regional biases.
Migration assays
12-well glass bottom plates with high performance #1.5 cover glass (Cellvis Prod#: P12-1.5H-N) were coated with 1.5 ml of fibronectin in Dulbecco's phosphate-buffered saline (DPBS) at a concentration of 10 μg/cm2. Plates were incubated at room temperature for 1 h to allow for fibronectin adsorption and then blocked with 10% FBS for 1 h at room temperature. Cells were then seeded at a density of 300 cells/cm2 and allowed to adhere for 4 h prior to live-cell imaging for 12 h. Cells were imaged with a Nikon Eclipse Ti-S microscope equipped with a motorized stage (MS-2000-WK multi-axis stage controller (Applied Scientific Instrumentation), as well as a temperature, humidity and CO2-controlled live cell chamber (Pathology Devices Inc., LiveCell). Cells were imaged at 10× in brightfield at multiple positions every 15 min. Cell migration data were analyzed using a combination of Image J (NIH, Bethesda, MD) or a custom MATLAB script (https://github.com/byeoman-eng/CellTracking, MathWorks, Natick, MA) to determine cellular migration characteristics. Average instantaneous cell speed (computed frame-to-frame), path length, displacement and persistence were calculated, plotted in rose plots for each individual cell, and grouped in scatter plots.
Western blotting
Proteins were isolated by collecting cells from an 80% confluent plate using a cell scraper (Corning, Prod#: 3010) and pelleted. Cells were resuspended in mRIPA buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1% Triton, 10% glycerol, 25 mM sodium deoxycholate, 0.1% SDS) containing Roche Complete Protease Inhibitor (Sigma-Aldrich, Prod#: 11697498001) and PhosSTOP (Sigma-Aldrich, Prod#: 4906845001). Lysate concentrations were determined using a Pierce BCA Protein Assay kit (ThermoFisher Scientific, Prod#: 23225). 12 µg of protein from each sample was combined with 50 mM DTT, 1× Laemmli Sample Buffer (BioRad Prod#: 1610747) and heated to 95°C for 5 min to denature the lysates. Protein mixtures were electrophoretically separated on reducing and denaturing gradient Bis-Tris gels (ThermoFisher Scientific, Prod#: NW04120BOX). Proteins were transferred to nitrocellulose membrane using the iBlot 1 semi-dry transfer system (ThermoFisher Scientific, Prod#: IB1001) and membrane cassettes (ThermoFisher Scientific, Prod#: IB301001). Membranes were incubated with 5% Seablock blocking buffer (ThermoFisher Scientific Prod#: 37527) in Tris-buffered saline supplemented with Tween (TBS-T, 150 mM NaCl, 15 mM Tris-HCl, 20 mM Tris Base, 0.1% Tween) and probed with primary antibodies. Membranes were washed, incubated with goat anti-rabbit Alexa Fluor 790 (ThermoFisher Scientific, Prod#: A11374) and goat anti-mouse Alexa Fluor 680 (ThermoFisher Scientific, Prod#: A10038) secondary antibodies. Membranes were washed and imaged utilizing the Odyssey CLx Imaging System and ImageStudio (Licor) software. Immunoblots were normalized to loading control proteins to ensure accurate loading of protein samples.
Antibody arrays of phosphorylated kinase
Medium was analyzed using the Proteome Profiler Mouse XL Cytokine Array (R&D Systems, cat # ARY028). Briefly, membranes were blocked for 1 h using array buffer and the medium was combined with array buffer overnight at 4°C while rocking. Membranes were washed, incubated with the antibody cocktail diluted for 1 h, washed, incubated with streptavidin-HRP for 30 min and finally treated with chemiluminescent reagent mix. Membranes were exposed to film and imaged. Pixel quantification was performed in ImageJ and normalized to positive and loading controls. Conditioned medium was normalized to unconditioned medium.
Quantitative PCR
Cells were grown to 80% confluency, RNA was extracted with Trizol-chloroform (ThermoFisher Scientific, Prod#: 15596) and concentration measured via nanodrop 2000 (ThermoFisher Scientific, Prod#: ND-2000). 2 µg RNA was reverse transcribed using Super Script III Reverse Transcriptase (Thermo Scientific, Prod#: 18080093). Quantitative PCR was performed using the iQ SYBR Green Supermix (Bio-Rad Laboratories, Prod#: 1708880) using a 7900HT Fast Real-Time PCR System (ThermoFisher Scientific, Prod#: 4329001) green (45 cycles, 95°C for 15 s, followed by 60°C for 1 min) with primer sets described in Table S2. Data were analyzed by calculating quantities of RNA based on a standard curve generated from a fibronectin plasmid.
Small-molecule treatment
To understand whether small molecule inhibition of EGFRvIII as well as its downstream pathways was able to modulate cellular adhesion, cells were exposed to select inhibitors prior to spinning. In brief, 25 mm glass coverslips were functionalized with fibronectin as previously described. Cells were seeded at a density of 2000 cells/cm2 and allowed to adhere overnight in medium without inhibitor. Once adhered, cells were incubated with either 10 μM Erlotinib (LC Laboratories, Prod#: T-8123) to inhibit constitutive EGFR activation or with 15 nM Trametinib (LC Laboratories, Prod#: T-4007) to inhibit downstream MEK activation for 48 h prior to spinning. Cells were compared to DMSO-treated controls then spun, fixed, stained, imaged and analyzed as previously described. To confirm the effect of the inhibitors on pathway activation, cells were seeded in parallel for western blot analysis and to spinning disk assays. Similar to the cell adhesion-strength assay, cells were seeded into 10 cm tissue culture dishes at a density of 10,000 cells/cm2, allowed to adhere overnight before being exposed to their respective inhibitors (Erlotinib or Trametinib), and incubated for 48 h prior to collection and analysis as previously described.
Statistics
All experiments were performed with at least three biological replicates with the number of technical replicates (n) indicated. Bar graphs show the mean±s.d. Box and whisker graphs are represented as the median and extend to the 25% and 75% quartiles. Statistical differences among two groups were calculated with paired Student’s t-tests or other statistical hypothesis tests as indicated. Statistical analyses were performed using Graphpad Prism software, with the threshold for significance level set at P<0.05 or as indicated.
Supplementary Material
Acknowledgements
The authors thank undergraduate assistants Mana Abbasinik (UCSD), Tristian Saucedo (UCSD), Bryanna Harris (Auburn University), and Shao Chi Chen (National Tsing Hua University) for technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.J.E., F.F.; Methodology: A.B., A.J.E., F.F.; Software: A.B.; Validation: A.B.; Formal analysis: A.B., P.B., A.D.P., B.Y., J.K.P.; Investigation: A.B., M.E., P.B., A.D.P., B.Y., J.K.P.; Resources: A.J.E., F.F.; Writing - original draft: A.B., A.J.E., F.F.; Writing - review & editing: A.B., A.J.E., F.F.; Supervision: A.J.E., F.F.; Project administration: A.J.E., F.F.; Funding acquisition: A.J.E., F.F.
Funding
Funding was provided by direct support from the Ludwig Institute for Cancer Research, National Institutes of Health (grant numbers: R01CA206880 and R21CA217735 to A.J.E., R01NS080939 to F.F., and R01NS116802 to A.J.E. and F.F.), the National Science Foundation (grant number: CMMI-1763139 to A.J.E.) and the National Science Foundation Graduate Research Fellowship program (to A.B.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.247189.supplemental
References
- Alieva M., Leidgens V., Riemenschneider M. J., Klein C. A., Hau P. and van Rheenen J. (2019). Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep. 9, 2054 10.1038/s41598-019-38625-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo R. M., Maher E. A., Ligon K. L., Sharpless N. E., Chan S. S., You M. J., Tang Y., DeFrances J., Stover E., Weissleder R. et al. (2002). Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269-277. 10.1016/S1535-6108(02)00046-6 [DOI] [PubMed] [Google Scholar]
- Bellail A. C., Hunter S. B., Brat D. J., Tan C. and Van Meir E. G. (2004). Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 36, 1046-1069. 10.1016/j.biocel.2004.01.013 [DOI] [PubMed] [Google Scholar]
- Beri P., Popravko A., Yeoman B., Kumar A., Chen K., Hodzic E., Chiang A., Banisadr A., Placone J. K., Carter H. et al. (2020). Cell adhesiveness serves as a biophysical marker for metastatic potential. Cancer Res. 80, 901-911. 10.1158/0008-5472.CAN-19-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj M., Strohmeyer N., Colo G. P., Helenius J., Beerenwinkel N., Schiller H. B., Fässler R. and Müller D. J. (2017). αV-class integrins exert dual roles on α5β1 integrins to strengthen adhesion to fibronectin. Nat. Commun. 8, 14348 10.1038/ncomms14348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijian K., Lougheed C., Su J., Xu B., Yu H., Wu J. H., Riccio K. and Alaoui-Jamali M. A. (2013). Targeting focal adhesion turnover in invasive breast cancer cells by the purine derivative reversine. Br. J. Cancer 109, 2810-2818. 10.1038/bjc.2013.675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger D. (2007). Quantitative measurements of integrin-mediated adhesion to extracellular matrix. Methods Enzymol. 426, 1-25. 10.1016/S0076-6879(07)26001-X [DOI] [PubMed] [Google Scholar]
- Bonavia R., Inda M.-D.-M., Cavenee W. K. and Furnari F. B. (2011). Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 71, 4055-4060. 10.1158/0008-5472.CAN-11-0153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C. W., Verhaak R. G. W., McKenna A., Campos B., Noushmehr H., Salama S. R., Zheng S., Chakravarty D., Sanborn J. Z., Berman S. H. et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462-477. 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brösicke N. and Faissner A. (2015). Role of tenascins in the ECM of gliomas. Cell Adhes. Migr. 9, 131-140. 10.1080/19336918.2014.1000071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah V. A., Robel S., Watkins S. and Sontheimer H. (2014). A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 15, 455-465. 10.1038/nrn3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubon T., Guyon J., Raymond A.-A., Dartigues B., Rudewicz J., Ezzoukhry Z., Dupuy J.-W., Herbert J. M. J., Saltel F., Bjerkvig R. et al. (2019). The invasive proteome of glioblastoma revealed by laser-capture microdissection. Neuro-Oncol. Adv. 1, vdz029 10.1093/noajnl/vdz029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuth T. and Berens M. E. (2004). Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 70, 217-228. 10.1007/s11060-004-2751-6 [DOI] [PubMed] [Google Scholar]
- Dirkse A., Golebiewska A., Buder T., Nazarov P. V., Muller A., Poovathingal S., Brons N. H. C., Leite S., Sauvageot N., Sarkisjan D. et al. (2019). Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 10, 1787 10.1038/s41467-019-09853-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn G. P., Rinne M. L., Wykosky J., Genovese G., Quayle S. N., Dunn I. F., Agarwalla P. K., Chheda M. G., Campos B., Wang A. et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 26, 756-784. 10.1101/gad.187922.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler A. J., Chan M., Boettiger D. and Schwarzbauer J. E. (2009). A novel mode of cell detachment from fibrillar fibronectin matrix under shear. J. Cell Sci. 122, 1647-1653. 10.1242/jcs.040824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis J. M., Zhang C.-Z., Maire C. L., Jung J., Manzo V. E., Adalsteinsson V. A., Homer H., Haidar S., Blumenstiel B., Pedamallu C. S. et al. (2014). EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Disc. 4, 956-971. 10.1158/2159-8290.CD-13-0879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann A., Li J., Chien S. and Engler A. J. (2014). Cation type specific cell remodeling regulates attachment strength. PLoS ONE 9, e102424 10.1371/journal.pone.0102424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann A., Banisadr A., Beri P., Tlsty T. D. and Engler A. J. (2017). Metastatic state of cancer cells may be indicated by adhesion strength. Biophys. J. 112, 736-745. 10.1016/j.bpj.2016.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C. et al. (2007). Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 21, 2683-2710. 10.1101/gad.1596707 [DOI] [PubMed] [Google Scholar]
- Furnari F. B., Cloughesy T. F., Cavenee W. K. and Mischel P. S. (2015). Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 15, 302-310. 10.1038/nrc3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H. K., Kaye A. H. and Luwor R. B. (2009). The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 16, 748-754. 10.1016/j.jocn.2008.12.005 [DOI] [PubMed] [Google Scholar]
- Gan H. K., Cvrljevic A. N. and Johns T. G. (2013). The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 280, 5350-5370. 10.1111/febs.12393 [DOI] [PubMed] [Google Scholar]
- Guo G., Gong K., Wohlfeld B., Hatanpaa K. J., Zhao D. and Habib A. A. (2015). Ligand-Independent EGFR Signaling. Cancer Res. 75, 3436-3441. 10.1158/0008-5472.CAN-15-0989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselager G. and Holland E. C. (2003). Using mice to decipher the molecular genetics of brain tumors. Neurosurgery 53, 685-694, discussion 695 10.1227/01.neu.0000081304.57547.b5 [DOI] [PubMed] [Google Scholar]
- Holland E. C., Hively W. P., DePinho R. A. and Varmus H. E. (1998). A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 12, 3675-3685. 10.1101/gad.12.23.3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger A. M., Dull T. J., Felder S., Van Obberghen E., Bellot F., Szapary D., Schmidt A., Ullrich A. and Schlessinger J. (1987). Point mutation at the ATP binding site of EGF receptor abolishes protein-tyrosine kinase activity and alters cellular routing. Cell 51, 199-209. 10.1016/0092-8674(87)90147-4 [DOI] [PubMed] [Google Scholar]
- Huang P. H., Xu A. M. and White F. M. (2009). Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2, re6 10.1126/scisignal.287re6 [DOI] [PubMed] [Google Scholar]
- Inda M.-D.- M., Bonavia R., Mukasa A., Narita Y., Sah D. W. Y., Vandenberg S., Brennan C., Johns T. G., Bachoo R., Hadwiger P. et al. (2010). Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24, 1731-1745. 10.1101/gad.1890510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleihues P., Louis D. N., Scheithauer B. W., Rorke L. B., Reifenberger G., Burger P. C. and Cavenee W. K. (2002). The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 61, 215-225; discussion 226-9 10.1093/jnen/61.3.215 [DOI] [PubMed] [Google Scholar]
- Liu Z., Han L., Dong Y., Tan Y., Li Y., Zhao M., Xie H., Ju H., Wang H., Zhao Y. et al. (2016). EGFRvIII/integrin β3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget 7, 4680-4694. 10.18632/oncotarget.6730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagane M., Lin H., Cavenee W. K. and Huang H.-J. S. (2001). Aberrant receptor signaling in human malignant gliomas: mechanisms and therapeutic implications. Cancer Lett. 162(Suppl), S17-S21. 10.1016/S0304-3835(00)00648-0 [DOI] [PubMed] [Google Scholar]
- Narita Y., Nagane M., Mishima K., Huang H.-J. S., Furnari F. B. and Cavenee W. K. (2002). Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 62, 6764-6769. [PubMed] [Google Scholar]
- Ning Y., Zeineldin R., Liu Y., Rosenberg M., Stack M. S. and Hudson L. G. (2005). Down-regulation of integrin α2 surface expression by mutant epidermal growth factor receptor (EGFRvIII) induces aberrant cell spreading and focal adhesion formation. Cancer Res. 65, 9280-9286. 10.1158/0008-5472.CAN-05-0407 [DOI] [PubMed] [Google Scholar]
- Nishikawa R., Furnari F. B., Lin H., Arap W., Berger M. S., Cavenee W. K. and Su Huang H. J. (1995). Loss of P16INK4 expression is frequent in high grade gliomas. Cancer Res. 55, 1941-1945. [PubMed] [Google Scholar]
- Nishikawa R., Sugiyama T., Narita Y., Furnari F., Cavenee W. K. and Matsutani M. (2004). Immunohistochemical analysis of the mutant epidermal growth factor, ΔEGFR, in glioblastoma. Brain Tumor Pathol. 21, 53-56. 10.1007/BF02484510 [DOI] [PubMed] [Google Scholar]
- Ostrom Q. T., Cioffi G., Gittleman H., Patil N., Waite K., Kruchko C. and Barnholtz-Sloan J. S. (2019). CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united states in 2012-2016. Neuro Oncol. 21, v1-v100. 10.1093/neuonc/noz150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paw I., Carpenter R. C., Watabe K., Debinski W. and Lo H.-W. (2015). Mechanisms regulating glioma invasion. Cancer Lett. 362, 1-7. 10.1016/j.canlet.2015.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao J. S. (2003). Molecular mechanisms of glioma invasiveness: the role of proteases. Nat. Rev. Cancer 3, 489-501. 10.1038/nrc1121 [DOI] [PubMed] [Google Scholar]
- Reticker-Flynn N. E., Malta D. F. B., Winslow M. M., Lamar J. M., Xu M. J., Underhill G. H., Hynes R. O., Jacks T. E. and Bhatia S. N. (2012). A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat. Commun. 3, 1122 10.1038/ncomms2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. H. H., Furnari F. B., Cavenee W. K. and Bogler O. (2003). Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc. Natl. Acad. Sci. USA 100, 6505-6510. 10.1073/pnas.1031790100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R. L., Miller K. D. and Jemal A. (2017). Cancer statistics, 2017. CA Cancer J. Clin. 67, 7-30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- Singhai A., Wakefield D. L., Bryant K. L., Hammes S. R., Holowka D. and Baird B. (2014). Spatially defined EGF receptor activation reveals an F-actin-dependent phospho-Erk signaling complex. Biophys. J. 107, 2639-2651. 10.1016/j.bpj.2014.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J. B., Belanger K., Brandes A. A., Marosi C., Bogdahn U. et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987-996. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- Verhaak R. G. W., Hoadley K. A., Purdom E., Wang V., Qi Y., Wilkerson M. D., Miller C. R., Ding L., Golub T., Mesirov J. P. et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98-110. 10.1016/j.ccr.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. and Zhou B. P. (2010). TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 102, 639-644. 10.1038/sj.bjc.6605530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J., Hu J., Gomez G. G., Taylor T., Villa G. R., Pizzo D., VandenBerg S. R., Thorne A. H., Chen C. C., Mischel P. S. et al. (2015). A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 75, 394-404. 10.1158/0008-5472.CAN-14-2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates C. M., McGettrick H. M., Nash G. B. and Rainger G. E. (2014). Adhesion of tumor cells to matrices and endothelium. In Metastasis Research Protocols (ed. Dwek M., Schumacher U. and Brooks S. A.), pp. 57-75. New York, NY: Springer New York. [DOI] [PubMed] [Google Scholar]
- Yu X., Miyamoto S. and Mekada E. (2000). Integrin alpha 2 beta 1-dependent EGF receptor activation at cell-cell contact sites. J. Cell Sci. 113, 2139-2147. [DOI] [PubMed] [Google Scholar]
- Zanca C., Villa G. R., Benitez J. A., Thorne A. H., Koga T., D'Antonio M., Ikegami S., Ma J., Boyer A. D., Banisadr A. et al. (2017). Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes Dev. 31, 1212-1227. 10.1101/gad.300079.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.