

ABSTRACT
Irf6 and Esrp1 are important for palate development across vertebrates. In zebrafish, we found that irf6 regulates the expression of esrp1. We detailed overlapping Irf6 and Esrp1/2 expression in mouse orofacial epithelium. In zebrafish, irf6 and esrp1/2 share expression in periderm, frontonasal ectoderm and oral epithelium. Genetic disruption of irf6 and esrp1/2 in zebrafish resulted in cleft of the anterior neurocranium. The esrp1/2 mutant also developed cleft of the mouth opening. Lineage tracing of cranial neural crest cells revealed that the cleft resulted not from migration defect, but from impaired chondrogenesis. Analysis of aberrant cells within the cleft revealed expression of sox10, col1a1 and irf6, and these cells were adjacent to krt4+ and krt5+ cells. Breeding of mouse Irf6; Esrp1; Esrp2 compound mutants suggested genetic interaction, as the triple homozygote and the Irf6; Esrp1 double homozygote were not observed. Further, Irf6 heterozygosity reduced Esrp1/2 cleft severity. These studies highlight the complementary analysis of Irf6 and Esrp1/2 in mouse and zebrafish, and identify a unique aberrant cell population in zebrafish expressing sox10, col1a1 and irf6. Future work characterizing this cell population will yield additional insight into cleft pathogenesis.
KEY WORDS: IRF6, ESRP1, Craniofacial, Cleft, Development
Summary: Experiments using complementary models indicate that disruption of Irf6 and Esrp1/2 in mouse and zebrafish results in orofacial clefts, with an aberrant mesenchymal/epithelial cell population identified in the cleft of the esrp1/2 zebrafish mutant.
INTRODUCTION
Development of vertebrate craniofacial structures requires coordinated cellular induction, migration, proliferation and differentiation, which allow for the positioning of adjacent epithelial-lined facial processes that ultimately merge (Reid et al., 2011; O'Donoghue et al., 2020; Knight and Schilling, 2006; Jiang et al., 2006; Helms et al., 2005; Dougherty et al., 2012; Creuzet et al., 2005; Cordero et al., 2011; Abramyan and Richman, 2015). Morphogenesis of facial structures such as the midface, lip and palate requires convergence of the medial and lateral nasal prominences and the fusion of the secondary palatal shelves at the midline (Losa et al., 2018; Jiang et al., 2006; Abramyan and Richman, 2015). Failure of these processes to fuse results in orofacial clefts of the lip, primary palate or secondary palate (Gritli-Linde, 2008). Orofacial clefts are among the most common congenital structural anomalies (Yuan et al., 2011; Juriloff and Harris, 2008; Goodwin et al., 2015). From genome-wide association studies carried out over a decade ago to more recent whole-genome sequencing projects of orofacial cleft cohorts, cleft-associated genetic loci continue to be identified, and the transcription factor IRF6 is one of the most commonly associated genes (Zucchero et al., 2004; Marazita, 2012; Yu et al., 2017; Cox et al., 2018). IRF6 disruption is causal for syndromic cleft in Van der Woude and popliteal pterygium syndromes, and associated with non-syndromic orofacial clefts (Zucchero et al., 2004; Leslie et al., 2013; Kondo et al., 2002; Beaty et al., 2016).
Several IRF6 transcriptional targets – such as GRHL3, WDR65 (CFAP57), OVOL1 and KLF4 – have been identified, which are also important for palate development and implicated in human cleft pathogenesis (Rorick et al., 2011; Liu et al., 2016; Kousa and Schutte, 2016; de la Garza et al., 2013). These studies support the premise that investigation of Irf6 and its transcriptional network will identify key genes that regulate palate development. Multiple mouse models have been generated to investigate Irf6 function, including a total Irf6 knockout and substitution of key functional residue Irf6R84C in the DNA-binding domain (Richardson et al., 2006; Ingraham et al., 2006). These Irf6 mutant mouse models exhibited disrupted epithelial terminal differentiation and lack of a functional periderm, leading to pathological adhesions of epithelial embryonic tissues (Richardson et al., 2006; Iwata et al., 2013; Ingraham et al., 2006; Ferretti et al., 2011). The epithelial differentiation and adhesion defects are thought to prevent elevation of the palatal shelves, and ultimately these mice develop a cleft in the secondary palate. Additionally, the midface of these mice were hypoplastic, a phenotype that was attributed to the dysfunctional embryonic epithelium (Richardson et al., 2006; Ingraham et al., 2006).
Epithelial splicing regulatory proteins 1 and 2 (Esrp1, Esrp2) are also important in embryonic epithelial differentiation and palate development (Lee et al., 2020, 2018; Bebee et al., 2015). Esrp2 and its homolog Esrp1 are regulators of RNA splicing that are specifically expressed in the epithelium (Warzecha et al., 2009). Esrp1/2 knockout mice exhibit bilateral cleft of the lip and primary palate, as well as a secondary palate cleft (Bebee et al., 2015). Esrp1/2 are unusual among regulators of RNA splicing in that they are tissue restricted and exhibit dynamic expression during embryogenesis (Burguera et al., 2017; Bebee et al., 2015). The developmental importance of Esrp1/2 is underscored by their conservation across species, from ascidians to zebrafish, Xenopus, mouse and humans (Burguera et al., 2017). Gene variant in ESRP2 was also recently reported in human orofacial cleft cohorts (Cox et al., 2018).
The mouse has been an important experimental model to study craniofacial and palate morphogenesis (Gritli-Linde, 2008). Secondary palate development in the mouse is similar to that in humans, with the analogous stages of vertical outgrowth, elevation, horizontal growth and fusion (Juriloff and Harris, 2008; Gritli-Linde, 2008). Many genes associated with cleft lip and palate (CL/P) in humans, when disrupted in the mouse, result in cleft of the secondary palate without affecting morphogenesis of the primary palate and lip (Van Otterloo et al., 2016; Gritli-Linde, 2008). So while the mouse model can be useful to study the secondary palate, the use of mouse models to study cleft of the lip and primary palate has been less effective as there are remarkably few mouse models in which development of the lip and primary palate are perturbed (Gritli-Linde, 2008). Meanwhile, clinically, CL/P is more common than isolated cleft of the palate only (CPO), and human genetic studies have suggested that the genetics underpinning CL/P and CPO are distinct (Juriloff and Harris, 2008; Gritli-Linde, 2008). The developmental processes of outgrowth of the facial prominences followed by convergence and fusion are thought to be conserved across mammals (Abramyan and Richman, 2015). Therefore, it is hypothesized that differences in mouse versus human phenotypic presentation are caused by spatiotemporal differences in craniofacial development (Gritli-Linde, 2008). In this context, the phenotype of bilateral clefts affecting the lip, primary and secondary palate in the Esrp1/2 mutant mouse is unique among mouse models and is a valuable tool to study lip and palate morphogenesis.
Zebrafish has been favored by embryologists as an animal model to study craniofacial development, owing to its accessibility, transparency and genetic tractability (Schilling and Le Pabic, 2009; Lieschke and Currie, 2007; Kimmel, 1989). Although a secondary palate, which partially or entirely separates the oral and nasal cavities, is reserved to amniotes, the primary palate is appreciably conserved across vertebrates (Abramyan and Richman, 2015). The primary palate establishes the intact upper jaw (Abramyan and Richman, 2015), which in the larval zebrafish consists of the ethmoid plate, also known as the anterior neurocranium (ANC). In all vertebrates, the most anterior cranial neural crest cells (CNCCs), that migrate rostral then turn caudal and ventral to the eye, contribute to the median frontonasal prominence, and a second CNCC stream, that migrates inferior to the eye and into the first pharyngeal arch, generates the paired maxillary prominences (Reid et al., 1986; Swartz et al., 2011; Kimmel, 1989; Dougherty et al., 2012). The ANC of the zebrafish is formed from the convergence of the median element, which is derived from the frontonasal prominence, and paired lateral elements that are derived from the maxillary prominences (Swartz et al., 2011; O'Donoghue et al., 2020; Duncan et al., 2017). Zebrafish homologs of human genes associated with orofacial clefts will disrupt morphology of the ANC, as have been observed for a number of genes, such as capzb, pitx2, pdgfra, smad5, tgfb2, fgf10a and wnt9a (O'Donoghue et al., 2020; Van Otterloo et al., 2016; Duncan et al., 2017).
Here, we carried out detailed gene expression analysis of Irf6, Esrp1 and Esrp2 in mouse and zebrafish in order to understand the comparative morphogenesis of facial structures and periderm between these important vertebrate genetic models. We analyzed and compared the Irf6 and Esrp1/2 mutant phenotypes to elucidate the comparative morphologies and genetic epistasis between these genes. Further, we generated zebrafish irf6 and esrp1/2 zebrafish mutants and examined their requirement in morphology of the stomodeum opening and ANC. Interestingly, we identified an aberrant cell population with epithelial and mesenchymal molecular signatures that localized to the region of the ANC cleft. This work highlights the relative strengths of the mouse and zebrafish models for investigating the morphogenetic mechanisms of orofacial clefts, and contributes new insights into the function of Irf6 and Esrp1/2 during palatogenesis.
RESULTS
irf6 null zebrafish embryos have decreased expression of esrp1
We previously generated a functionally null irf6 zebrafish allele (Li et al., 2017). Using CRISPR/Cas9, an 8 bp deletion in exon 6 of the irf6 coding region resulting in a frameshift and premature stop codon, leading to the ablation of irf6 function. It was observed that embryos lacking maternally expressed irf6 exhibited epiboly arrest and periderm rupture at 4-5 h post-fertilization (hpf) (Li et al., 2017). Utilizing this irf6 null model, we aimed to identify genes that were differentially downregulated in irf6 null versus wild-type (WT) embryos. We performed RNA sequencing (RNA-seq) on WT and maternal/zygotic irf6 null (mz-irf6-8bp/-8bp) embryos at 4.5 hpf, just before embryo rupture at the onset of gastrulation. Differential expression analysis revealed a substantial number of significantly differentially expressed genes (DEGs; n=10,299, adjusted P-value <0.05) (Fig. 1A-C). Full differential expression results are available in Table S1. To visualize changes in this large number of DEGs, a heat map was generated, which illustrates 1377 upregulated and 1799 downregulated genes with an absolute fold change greater than 2 in irf6 null relative to WT (Fig. 1A). The patterns of gene expression among these strongly DEGs were highly reproducible across the two genotypes, and demonstrated relatively similar numbers of downregulated and upregulated genes. When we visualized DEGs using significance values relative to fold change in expression, we found that the most significant and strongest effects on gene expression were biased toward those downregulated in the irf6 null embryos (Fig. 1B). The RNA-seq results revealed significant downregulation of genes previously known to be downregulated with disruptions in irf6 function (Fig. 1B,C). Disruption of irf6 via injecting dominant-negative irf6 mRNA led to downregulation of many periderm-enriched genes [including grhl1, krt5, krt18 (krt18a.1), tfap2a and klf2b] and genes for adhesion molecules (including claudins and cadherins) (de la Garza et al., 2013). Here, we found a similar expression profile in the mz-irf6-8bp/-8bp embryos relative to WT (Fig. 1B,C).
Fig. 1.

esrp1 expression is downregulated in irf6 null zebrafish embryos. (A) Hierarchical clustering of top differentially expressed genes (DEGs) defined by RNA-seq performed on wild-type (WT) versus mz-irf6-8bp/-8bp (irf6−/−) zebrafish embryos at 4-5 hpf. Top DEGs were identified by selecting genes with an adjusted P-value (Benjamini–Hochberg) <0.01 and absolute log2-fold change >2. Data are shown for three biological replicates. Color scale on the bottom left represents relative levels of expression, with yellow showing higher expression levels and blue showing lower expression. (B) Volcano plot from the RNA-seq dataset, showing the distribution of DEGs based on P-values (P) and log2-fold change (Log2 FC). NS, not significant. Previously published irf6-regulated genes are expressed at significantly higher levels in WT relative to mz-irf6−/−, including grhl3, klf17 and wnt11. The newly identified cleft-associated gene esrp1 is also expressed significantly higher in WT relative to irf6−/−. Vertical dashed lines represent the P-value cutoff of 0.01 and the log2-fold change cutoff of 2, respectively. (C) Gene ontology (GO) gene-concept network analysis of RNA-seq data, showing that irf6−/− embryos have perturbations in processes such as transcription factor activity, signal receptor binding and structural molecule activity. Note that many of these genes – such as wnt11, fgf8, tgfb1, krt4 and krt5 – are implicated in ectoderm development and cell specification. Gray nodes show GO terms, colored nodes show individual genes from the RNA-seq dataset, and black lines connect genes to one or more associated GO terms. Colored nodes show relative enrichment (measured by fold change) of genes in WT samples relative to irf6−/− embryos. Maps were generated using the enrichplot package in R. (D) qPCR gene expression analysis for esrp1, showing ∼80% downregulation in mz-irf6-8bp/-8bp embryos compared with WT at 4 hpf, and rescued esrp1 gene overexpression in mz-irf6-8bp/-8bp embryos injected with WT zebrafish irf6 mRNA. n=4. Unpaired Student’s t-test, *P<0.05.
To further understand the molecular pathways and biological functions being affected in the irf6 null embryo we performed gene ontology (GO) analyses on upregulated and downregulated gene sets. Of particular interest were significant changes in genes enriched for functions related to transcription factor activity, signal receptor binding and structural molecule expression (Fig. 1C). When compared with previously published IRF6 siRNA human keratinocyte DEG expression data (Botti et al., 2011), there were major overlaps of genes in molecular pathways responsible for epithelial regulation, including gata3, krt18 and cldn4 (Fig. 1B,C and Figs S1-S3). Many key developmental signaling pathways including Fgf (fgf8a, fgf17 and fgf24) and Wnt (wnt11, dact2, rspo3, frzb, fzd5) pathways were also heavily represented in our dataset as genes downregulated because of irf6 ablation (Fig. 1B,C). Further, a number of genes associated with human orofacial clefts were also downregulated in the irf6 null embryos, including hey1, gata3, wnt11 and fgf8 (Fig. 1B,C).
Interestingly, one of the most downregulated genes was esrp1. esrp1 and its paralog esrp2 are epithelial-restricted RNA splicing regulators. ESRP2 genetic variants in humans are associated with orofacial clefts (Cox et al., 2018), and Esrp1 and Esrp1/2 knockout mice display a bilateral cleft of the lip, primary and secondary palate (Bebee et al., 2015; Lee et al., 2020). To confirm the RNA-seq results, we performed quantitative PCR (qPCR) on mz-irf6-8bp/-8bp and WT embryos at 4-5 hpf. Relative to WT, esrp1 expression in mz-irf6-8bp/-8bp embryos was reduced ∼80%. Additionally, injection of mz-irf6-8bp/-8bp embryos with irf6 mRNA at the one-cell stage rescued esrp1 expression, resulting in an increase in expression to ∼3-fold higher than WT (Fig. 1D). These rescued fish were phenotypically normal, as previously shown (Li et al., 2017).
We tested Esrp1 and Esrp2 mRNA expression in embryonic day (E)11.5 Irf6 mutant mouse embryos (Irf6R84C/R84C) and found expression to be significantly decreased relative to that in littermate WTs (Fig. S4). Additionally, Shh expression was decreased in Irf6R84C/R84C embryos (Fig. S4), consistent with a previous report of decreased Shh expression in Esrp1−/− mice (Lee et al., 2020). These results in zebrafish and mouse suggest that Esrp1 gene expression is dependent on Irf6, either through direct regulation or the requirement of a normal periderm.
irf6, esrp1 and esrp2 are co-expressed in the oral epithelium of zebrafish during craniofacial development
Previous mouse studies have described Irf6 (Knight et al., 2006) and Esrp1/2 (Bebee et al., 2015; Lee et al., 2020; Warzecha et al., 2009) gene expression in oral epithelium during palate development. To determine the gene expression of irf6 and esrp1/2 in the zebrafish during epithelial and craniofacial development, we performed whole-mount in situ hybridization (WISH). Maternal deposition of irf6, esrp1 and esrp2 mRNA was detectable at eight-cell stage (Fig. 2A). The maternal transcripts were also detected in the periderm of the gastrulating embryo, although expression of esrp2 appeared lower than that of irf6 and esrp1 (Fig. 2A). During craniofacial development, WISH demonstrated specific expression of irf6, esrp1 and esrp2 lining the embryonic oral epithelium, and circumscribing surface epithelium concentrated around the developing stomodeum (Fig. 2B,C).
Fig. 2.
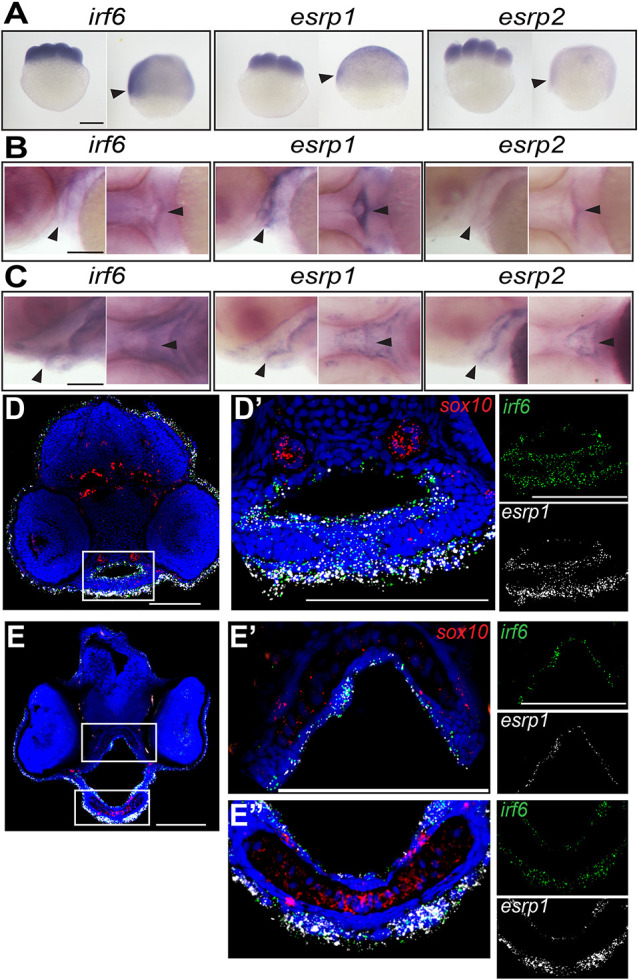
irf6, esrp1 and esrp2 are co-expressed in the oral epithelium of zebrafish embryos. (A-C) Whole-mount in situ hybridization (WISH), showing that irf6, esrp1 and esrp2 maternal deposited transcripts are detected at the eight-cell and shield stage (A; arrowheads indicate periderm), and circumscribe the developing stomodeum and line the oral epithelium of zebrafish embryos at 48 (B) and 72 (C) hpf (arrowheads). All whole-mount embryos are oriented with anterior left and dorsal top. (D-E″) Coronal sections of 48 (D) and 72 (E) hpf embryos analyzed by RNAscope in situ hybridization (ISH), (dorsal top), showing cellular RNA co-expression of irf6 (green) and esrp1 (white) in surface and oral epithelial cells. sox10 (red) staining depicts cartilage elements of the palate. Boxed areas are shown at higher magnification in D′, E′ and E″. Scale bars: 250 μm (A) and 100 μm (B-E″).
To resolve the specific cell populations that express irf6, esrp1 and esrp2, we performed RNAscope in-situ hybridization (ISH) of coronal cryosections taken through the developing mouth and palate at 48 hpf and 72 hpf. We found that irf6 and esrp1 were co-expressed within epithelial cells lining the oral cavity as well as the surface epithelium (Fig. 2D,E). No expression of these genes was detected within the cartilage elements, identified by sox10 expression. Further, we detected irf6 and esrp1 transcripts within the same cells, importantly within cells separating adjacent mesenchymal elements (Fig. 2D′); these cells are likely in the epithelial lineage as esrp1 is an epithelia-specific gene.
Irf6, Esrp1 and Esrp2 are co-expressed in murine frontonasal and oral epithelium during palate and lip development
Irf6 expression within the embryonic oral epithelium and surrounding the developing palatal shelves has been well established (Knight et al., 2006; Iwata et al., 2013; Kousa et al., 2017). Esrp1/2 expression was previously shown in the oral epithelium of developing mice (Bebee et al., 2016; Lee et al., 2020; Warzecha et al., 2009). Ablation of Irf6 or Esrp1/2 causes a cleft of the secondary palate, but the disruption of the lip and primary palate phenotypes differ between the Irf6 and Esrp1/2 mutants (Richardson et al., 2006; Ingraham et al., 2006; Bebee et al., 2015; Lee et al., 2020). To determine whether Irf6, Esrp1 and Esrp2 transcripts colocalize during mouse craniofacial development, we performed WISH for each gene at E10.5, as the frontonasal prominences and lambdoidal junction are taking shape at this time point. We found that Irf6, Esrp1 and Esrp2 were expressed similarly, with high levels of expression in areas of craniofacial development (Fig. 3A). The mouse gene expression pattern was similar to that observed in zebrafish, with more concentrated expression to the developing head. Higher-resolution imaging with RNAscope ISH detected Irf6, Esrp1 and Esrp2 transcripts in the periderm and the basal epithelium across all time points examined (Fig. 3B-F). Irf6, Esrp1 and Esrp2 were co-expressed in the surface ectoderm overlying the developing frontonasal prominences (Fig. 3B), a cell population with important signaling and inductive functions (Hu et al., 2003). Further, co-expression included cells at critical fusion points, specifically between the medial and lateral nasal prominences (Fig. 3C) and the palatal shelves (Fig. 3E,F). The co-expression of Irf6 and Esrp1/2 within cells with key roles during epithelial fusion supports the existence of an Irf6-Esrp1/2 regulatory axis during craniofacial morphogenesis.
Fig. 3.

Irf6, Esrp1 and Esrp2 are co-expressed in the oral epithelium of mouse embryos. (A) WISH of E10.5 embryos, showing Irf6, Esrp1 and Esrp2 mRNA expression in the surface epithelium and concentrated within the ectoderm of the frontonasal prominences (arrowheads) and first brachial arch. Oblique and frontal orientation. Scale bars: 500 μm. (B-F′) Sections of E10 (B,B′), E11.5 (C-D′), E13.5 (E,E′) and E15 (F,F′) embryos analyzed by RNAscope ISH, showing mRNA cellular co-expression of Irf6 (green), Esrp1 (red) and Esrp2 (white) in the surface ectoderm (E10), lining the frontonasal and maxillary prominences, including expression in periderm (arrows) (E11.5), and lining the palatal shelves (E13.5, E15). Sagittal (B,B′) and coronal (C-F′) sections; boxed areas are shown at higher magnification in B′, C′, D′, E′ and F′. dapi, 4′,6-diamidino-2-phenylindole; lnp, lateral nasal prominence; mnp, medial nasal prominence; mxp, maxillary prominence; ps, palate shelf; t, tongue; tel, telencephalon. Scale bars: 100 μm.
Interestingly, in addition to Irf6 expression in the epithelium, RNAscope ISH detected Irf6 mRNA expression in the mesenchyme, particularly at E10 and E15.5 (Fig. 3B,E). Expression of Irf6 in this craniofacial mesenchyme has not been reported previously, and high transcript detection with RNAscope ISH might be delineating gene expression not previously observed. Non-epithelial Irf6 expression was detected in CNCCs of the first and second pharyngeal arches at E9 (Fakhouri et al., 2017), and Irf6 is expressed in cells of the developing tongue (Goudy et al., 2013). Further, we previously reported that zebrafish expressing the irf6R84C variant under a sox10 promoter exhibit a partial cleft of the ANC (Dougherty et al., 2013). Together, these results suggest an additional role of Irf6 in craniofacial development beyond its role in epithelial cell differentiation.
Disruption of irf6 during neural crest cell migration results in cleft in zebrafish
Germline mutation of irf6 results in early embryonic lethality as a result of periderm rupture, which precluded evaluation of palate morphogenesis (Sabel et al., 2009; Li et al., 2017). To circumvent embryonic lethality, we employed an optogenetic gene-activation system based on the light-sensitive protein EL222, which serves to induce the expression of genes downstream of the C120 promoter (Motta-Mena et al., 2014). To this end, a dominant-negative form of irf6 consisting of a fusion protein of the irf6 protein-binding domain and the engrailed repressor domain (irf6-ENR) was cloned downstream of the C120 promoter (C120-irf6-ENR; Fig. 4A) (Sabel et al., 2009). When co-injected with VP-16 mRNA, this light-activated irf6-ENR construct enabled us to control the timing of irf6 disruption by exposing the embryos to a 465 nm light source later in embryogenesis (Fig. 4A). Zebrafish embryos injected with the optogenetic system and continuously exposed to blue light from 10 hpf to 96 hpf were able to survive, but developed with a slightly curved body axis and a dysmorphic ventral cartilage phenotype (Fig. 4E), which were not observed in uninjected embryos (Fig. 4B,C) or injected embryos that were raised in the dark (Fig. 4D). Further analysis of the cartilage in these embryos (Fig. 4F-Q) revealed a cleft in the ANC, where a population of cells in the median portion was absent (Fig. 4Q). Moreover, injecting increasing doses of EL222-VP-16 mRNA and/or C120-irf6-DN (dominant negative) plasmid led to a dose-dependent effect on the proportion of zebrafish embryos with a cleft phenotype, which was more pronounced for injected embryos grown in blue light starting at 10 hpf compared with embryos grown in the dark (Fig. S5). Consistent with decreased expression of esrp1 in mz-irf6-8bp/-8bp embryos, disruption of irf6 using this optogenetic system resulted in decreased expression of esrp1 (Fig. S5).
Fig. 4.
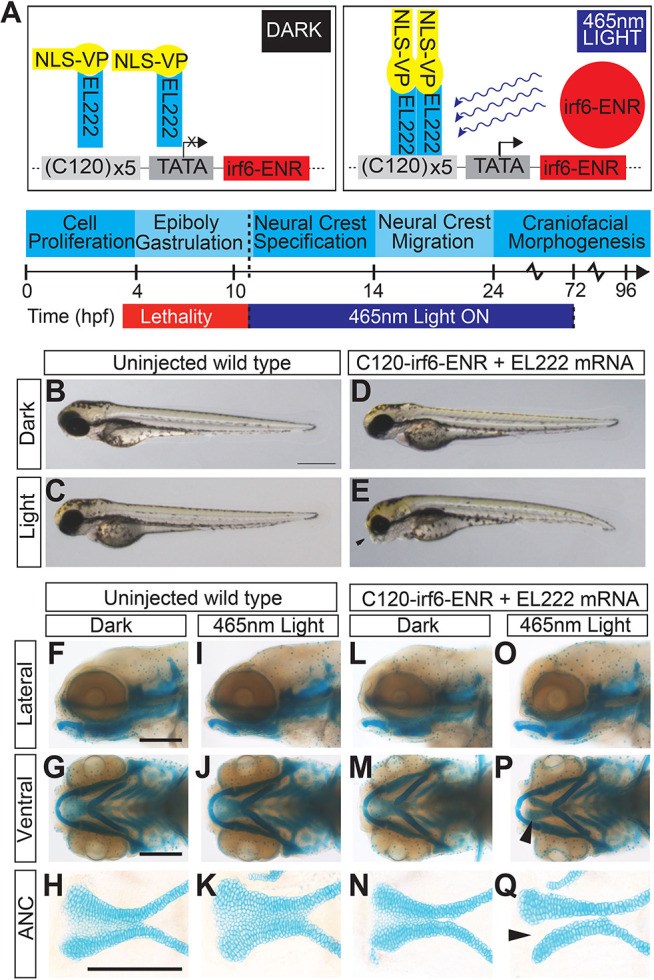
EL222 optogenetic disruption of irf6 circumvents early embryonic lethality and causes a cleft palate phenotype. (A) Schematic of EL222 optogenetic system. VP16-EL222 monomers are inactive under dark conditions. Upon stimulation by 465 nm light, VP16-EL222 dimerizes, drives gene expression downstream of the C120 promoter and induces the expression of a dominant-negative form of irf6 (irf6-ENR). Embryos were exposed to blue light from 10 hpf to 72 hpf to circumvent embryonic lethality in mz-irf6-8bp/-8bp embryos. (B-E) Brightfield microscopy of 72 hpf zebrafish embryos injected with the optogenetic system and grown in the dark (D) or exposed to blue light from 10-72 hpf (E) compared with control injected embryos (B,C). Injected fish exposed to blue light exhibit retrusion of the midface (arrowhead) and a curved body not observed in the other groups. (F-Q) Alcian Blue staining of cartilage and microdissection of the palate of 72 hpf embryos reveals a midface retrusion and cleft phenotype through the medial ethmoid plate (arrowhead in P, Q) in the C120-irf6-ENR-injected embryos grown under blue light (O-Q), which is not seen in control injected embryos (I-K) or injected embryos grown in the dark (L-N). Scale bars: 150 µm.
The compound homozygote of esrp1 and esrp2 exhibits cleft lip and ANC in zebrafish
To investigate the genetic requirement for esrp1 and esrp2 in zebrafish craniofacial development, CRISPR/Cas9 genome editing was utilized to generate esrp1 and esrp2 mutant alleles. Several alleles of esrp1 and esrp2 were obtained, where alleles harboring −4 bp and −14 bp indels that lead to frameshift mutations and early protein truncation were selected for breeding, hereafter referred to as esrp1-4bp/-4bp and esrp2-14bp/-14bp, respectively (Fig. S6). No phenotype was observed in the esrp1-4bp/-4bp embryos, and esrp2-14bp/-14bp fish developed normally (Fig. S6), except that females were infertile, as previously published in independently derived CRISPR alleles of esrp1/2 (Burguera et al., 2017). However, compound homozygote esrp1-4bp/-4bp; esrp2-14bp/-14bp zebrafish exhibit several phenotypes, consistent with previously published mutants (Burguera et al., 2017). The espr1-4bp/-4bp; esrp2-14bp/-14bp embryos also failed to inflate the swim bladder, and the pectoral fins were formed but diminutive, and the margins of the fin appeared dysplastic with irregular morphology. Further, Alcian Blue staining of double knockouts revealed cleft in the ANC, whereas the ventral cartilages, including the Meckel's cartilage, were formed and appeared wild-type (Fig. 5A).
Fig. 5.

esrp1/2 double mutants display a cleft lip and palate. (A) Alcian Blue staining of 4 dpf zebrafish. Representative images of WT, esrp1 CRISPR mutant (esrp1−/−) and esrp1/2 double CRISPR mutant (esrp1−/−; esrp2−/−), as well as esrp1 CRISPR mutant treated with esrp2 morpholino and WT treated with esrp1 and esrp2 morpholino (esrp1 MO, esrp2 MO). Flat-mount images of the anterior neurocranium (ANC) show a cleft (arrowheads) between the median element and lateral element of the ANC when both esrp1 and esrp2 function were disrupted. Lateral images and flat-mount images of the ventral cartilage (VC) show only subtle changes in morphology between WT and esrp1/2−/− zebrafish. (B) Morphant phenotypes observed over a range of esrp1 and esrp2 MO doses. Single esrp2 MO injections in the esrp1−/− background achieves nearly 100% phenotype penetrance, even at very low MO doses. (C) SEM of 5 dpf zebrafish showing discontinuous upper lip (filled arrowheads) in the esrp1/2 double CRISPR mutant as well as absent preoptic cranial neuromasts (open arrowheads) and abnormal keratinocyte morphology. The white arrowhead indicates an aberrant cell mass. (D) Representative images of Alizarin Red/Alcian Blue staining of 9 dpf esrp1/2 double CRISPR mutant zebrafish and WT clutch-mate controls. Esrp1/2 ablation causes abnormal morphology of the mineralizing parasphenoid bone; the bone appears wider and with a cleft (arrowhead). Scale bars: 150 µm (A,D); 100 µm (C).
Inter-cross of esrp1-4bp/-4bp; esrp2wt/-14bp produces predicted Mendelian ratio of 25% esrp1-4bp/-4bp; esrp2-14bp/-14bp embryos for downstream phenotypic analysis, where 75% of the embryos appeared wild type. In order to increase the percentage of embryos that can be utilized for analysis to 100%, we asked whether morpholino (MO) disruption of esrp2 in the esrp1-4bp/-4bp background would consistently yield a cleft ANC phenotype that phenocopied the esrp1-4bp/-4bp; esrp2-14bp/-14bp mutant. We successfully phenocopied the cleft ANC phenotype by co-injecting esrp1 and esrp2 MOs into WT embryos. However, the MO concentrations needed were relatively high, requiring 2-8 ng of each MO to be injected for ∼25-50% of embryos to develop a cleft (Fig. 5A,B). Importantly, when esrp1-4bp/-4bp embryos were injected with esrp2 MO, the cleft ANC phenotype was consistent and observed in nearly 100% of embryos, even when the MO concentration was reduced as low as 0.4 ng (Fig. 5A,B). One explanation for this observation is that transcriptional compensation between esrp1 and esrp2 occurs when each gene is targeted, thereby requiring higher doses of each MO to ablate esrp activity sufficiently (Rossi et al., 2015). But when one of the esrp genes is already disrupted in the homozygous esrp1-4bp/-4bp mutant, the threshold for full esrp loss of function is lower, requiring a much smaller dose of MO to generate the cleft ANC phenotype.
Using scanning electron microscopy (SEM), we observed that the cleft of the upper margin of the stomodeum had invaginated and extended into the cleft of the ANC. Additionally, the keratinocyte morphology of the surface epithelium appeared irregular and round with epithelial blebs in the esrp1-4bp/-4bp; esrp2-14bp/-14bp embryo. By contrast, the WT surface epithelium keratinocytes appeared octagonal or hexagonal without epithelial blebs (Fig. 5C). Alizarin Red staining of the larvae at 9 days post-fertilization (dpf) also revealed a lack of mineralization at the midline of the parasphenoid bone (Fig. 5D), consistent with a cleft of ANC that persisted to the ossification stage and subsequent larval development.
Zebrafish ANC morphogenesis is dependent on epithelial interactions with infiltrating cranial neural crest cells
Formation of the zebrafish ANC involves migration of anteriormost CNCCs to populate the median portion (frontonasal derived), while more posterior CNCCs migrate from each side (maxillary derived). These three discrete embryonic elements fuse to form the ANC. Concurrent with these cellular movements, the CNCCs undergo differentiation to chondrocytes (Reid et al., 1986; Dougherty et al., 2013). We found that the ablation of irf6 (a key periderm/epithelial gene) and esrp1/2 (epithelial-restricted genes) both resulted in a cleft in the ANC, where chondrocytes were absent along the fusion plane between the frontonasal-derived median element and one side of the maxillary-derived lateral element (Fig. 4C and Fig. 5A).
To investigate the absence of these ANC chondrocytes, we performed lineage tracing of CNCCs in esrp1/2-ablated embryos. Previously, we and others identified that the anteriormost CCNC populations at 20 somites migrate to and populate the median (frontonasal) element of the ANC (Reid et al., 1986; Dougherty et al., 2013). Accordingly, we labeled the CNCCs at 20-somite stage through photo-conversion of Kaede under the lineage specificity of the sox10 promoter. CNCCs of WT or esrp1/2 CRISPR mutants or esrp1/2 morphants were photo-converted at 12-15 hpf (Fig. 6A,B). Embryos were imaged at 4 dpf to determine the population of the ANC contributed by photo-converted cells. We found that esrp1/2 ablation did not affect the ability of CNCCs to migrate into the ANC and reached posterior positions without clustering anteriorly (Fig. 6A,B). These results suggest that the cleft of the ANC in the esrp1/2 mutants is not caused by total absence of progenitor cells or a defect in CNCC migration into the ANC. Nevertheless, Alcian Blue staining confirmed that chondrocytes were absent from a cleft in the ANC in the esrp1/2 mutants (Fig. 5A).
Fig. 6.

esrp1/2 null cranial neural crest cells (CNCCs) migrate to the ANC but do not differentiate to chondrocytes. (A) Lineage tracing of WT or esrp1/2 morphant zebrafish embryos using the Tg(sox10:kaede) line, native Kaede fluorescence is shown in green, and photo-converted Kaede is shown in magenta. Sagittal and horizontal views of zebrafish embryos at 19 hpf and 4.5 dpf, respectively. The anteriormost neural crest frontonasal prominence (FNP) progenitors were photoconverted at 19 hpf. At 4.5 dpf, the WT signal tracks to the medial portion of the ANC. Both the esrp1/esrp2 double CRISPR mutants and esrp1/2 morphants exhibit a cleft in the ANC with absence of a portion of sox10+ cells in the medial portion of the ANC, but the labeled CNCCs representing FNP progenitors did reach and populate the entire length of the ANC. (B) Illustrative summary of lineage tracing results showing that photo-converted anteriormost CNCCs contributing to FNP do migrate into the ANC in esrp1/2 mutant embryos, but a cleft forms at the juxtaposition of the FNP-derived median element and the maxillary-derived lateral element. Scale bars: 150 µm.
To investigate the cellular composition of the ANC cleft, we performed RNAscope ISH staining of WT and esrp1/ 2 mutants at 4 dpf. Sections through ANC clefts showed a dense population of cells in the location of the cleft (Fig. 7). In fact, this mass of cells can be localized in the SEM image of the esrp1/2 mutant larvae (Fig. 5C). These cells are col2a1–, consistent with absent Alcian Blue staining. Instead, this aberrant cell population expresses irf6, while krt4 and krt5 staining is restricted to the periphery, consistent with the epithelial lining of the oral cavity (Figs 7 and 8). Coronal and sagittal sectioning through the medial ANC of WT and esrp1/2 mutant embryos confirmed the ectopic expression of irf6 and revealed sox10 expression in these aberrant, Alcian Blue– cells (Fig. 8A,B). Like krt4, the expression of krt5 outlines the oral cavity (Fig. 8B). The expression of sox10 suggests that at least a portion of these cells was CNC derived, whereas krt4 expression indicates an epithelial lineage. The presence of irf6 expression could be indicative of epithelial/periderm cells, or indicative of expression by CNCCs, as has previously been reported (Dougherty et al., 2013; Kousa et al., 2019). Based on these results, we hypothesize that epithelial (and/or periderm) cells associated with frontonasal and maxillary prominence derivatives are defective in the esrp1/esrp2 null mutants, and either disrupt or fail to promote fusion of the median and lateral elements of the ANC, causing a cleft to form (Fig. 10). In this way, this is the first direct evidence of cleft pathogenesis in the zebrafish as a result of epithelial defect, and suggests a model to consider how cleft pathogenesis involving the primary palate is conserved across vertebrates (Iwata et al., 2013; Ingraham et al., 2006; Bebee et al., 2015; Richardson et al., 2006).
Fig. 7.

ANC of esrp1/2 double mutants is populated by undifferentiated cells. Representative z-stacks of RNAscope ISH of coronal sections of esrp1/2 double CRISPR mutants and WT clutch-mate controls at 4 dpf. (A) Sections through ANC anterior to the eyes. col2a1 (red) staining depicts normal morphology of the ANC cartilage elements in WT, while a cleft is apparent in the esrp1/2−/− zebrafish, with dapi (blue)-stained cells between adjacent trabeculae (arrowheads). These col2a1– cells do not express epithelial markers krt4 (cyan) or krt5 (magenta), except around the periphery. (B) Sections posterior to those in A show col2a1– cells continuing inferior to the trabeculae in the esrp1/2 mutant zebrafish, and cells have low expression of irf6 (boxed area). (C) Zoomed image of col2a1– cells from the boxed area in B, showing irf6 expression (green). Dashed lines outline ANC cartilage elements. Scale bars: 50 μm.
Fig. 8.

Aberrant ANC cells of esrp1/2 double mutants express CNCC and epithelial cell markers. Representative z-stacks of RNAscope ISH of coronal sections of esrp1/2 double CRISPR mutants and WT clutch-mate controls at 4 dpf. (A) Sections through the ANC anterior to the eyes. (B) Medial sagittal sections through the ANC (anterior to left). Dashed lines outline the ANC cartilage elements. col1a1 (white) staining depicts perichondrium surrounding the aberrant mass of cells in the esrp1/2 mutant zebrafish, consistent with chondrogenic condensation (leftmost arrowhead). irf6 (green) and sox10 (red) expression is apparent in these cells (indicated by arrowheads in respective columns); dapi is shown in blue. Scale bars: 20 μm.
Fig. 10.

Illustrative summary of results. Ablation of the epithelial-restricted splicing factors esrp1 and esrp2 led to the dysregulation of CNCC integration and differentiation in the medial ANC, causing a cleft between lateral ANC elements. These results suggest that epithelial-specific splice variants of yet to be determined factors are required for directing the juxtaposed mesenchymal-derived cells and promoting normal morphogenesis.
Genetic interaction of Irf6R84C with Esrp1 and Esrp2
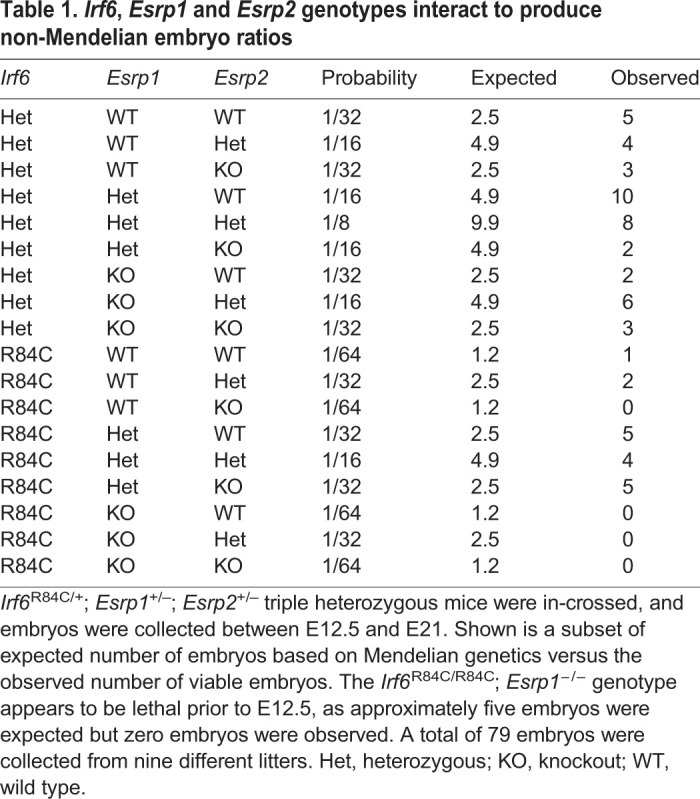
To test the hypothesis that Irf6 and Esrp1/2 genes function in the same developmental pathway, we carried out genetic epistasis analysis and generated Irf6; Esrp1; Esrp2 compound mutants. We hypothesized that if Irf6 and Esrp1/2 genetically interact, then Irf6 and Esrp1 heterozygosity on an Esrp2 null background might result in a cleft phenotype, when Irf6 and Esrp1 heterozygotes do not normally form a cleft. As expected, we observed that Irf6R84C/+; Esrp1+/−; Esrp2+/− mice developed and reproduced normally. To generate Irf6R84C/+; Esrp1+/−; Esrp2−/− embryos, we intercrossed the triple heterozygous mice. We collected a total of 79 embryos from nine litters from E12.5 to E18.5 and tabulated the resulting genotypes (Table 1, Table S2). Based on Mendelian genetics, we expected approximately five Irf6R84C/R84C; Esrp1–/– double homozygous mice. However, these breedings did not produce any Irf6R84C/R84C; Esrp1−/– embryos (Table 1). This result suggested that compound ablation of Irf6 and Esrp1 is more deleterious to development than either genotype alone, and supports a genetic interaction between Irf6 and Esrp1, which could be essential early in development.
Table 1.
Irf6, Esrp1 and Esrp2 genotypes interact to produce non-Mendelian embryo ratios
To test for phenotypic effects in the resulting Irf6; Esrp1; Esrp2 compound mutants, we imaged embryos at E18.5. We did not observe any cleft lip or palate in any genotype collected except for the expected clefts when null for Irf6 (Fig. S7) or Esrp1 (Fig. 9). Irf6R84C/+; Esrp1+/−; Esrp2−/− embryos that we predicted to be susceptible to cleft lip and/or palate were grossly normal (Fig. S7). We noticed some differences in the shape of the palate between heterozygous genotypes and measured the length (from philtrum to first rugae) relative to the width (space between lips). We found that Irf6R84C/+ heterozygotes exhibited a shorter palate than WT (Fig. 9A,B). A shorter snout has previously been reported in Irf6 KO mice (Ingraham et al., 2006). The length/width ratio of Esrp1/2 double heterozygotes was similar to WT, and in the Irf6; Esrp1; Esrp2 triple heterozygote, the shorter palate phenotype of the Irf6 heterozygote was reversed (Fig. 9A,B). Taken together, these breeding and morphologic analyses suggest an overlapping role of Irf6 and Esrp1/2 in regulating midface morphogenesis.
Fig. 9.

Irf6 and Esrp1/2 interact to modify palate phenotypes. Mice compound heterozygous for Irf6R84C, Esrp1 and Esrp2 were generated by breeding Irf6R84C/+ with Esrp1+/−; Esrp2−/− mice. The triple heterozygotes were then inter-crossed and embryos were collected at E18.5. (A) Representative lateral, frontal and oral images of embryos, comparing WT (Irf6+/+; Esrp1+/+; Esrp2+/+), Irf6R84C heterozygote (Het) (Irf6R84C/+; Esrp1+/+; Esrp2+/+), Esrp1/2 double heterozygote (Irf6+/+; Esrp1+/−; Esrp2+/−) and triple heterozygote (Irf6R84C/+; Esrp1+/−; Esrp2+/−). (B) Measurements of palate length (L) relative to width (W). Irf6R84C/+ embryos tend to have a shorter palate compared with WT; however this genotype on an Esrp1+/−; Esrp2+/− background results in significantly increased palate length relative to Irf6R84C/+; Esrp1+/+; Esrp2+/+ (one-way ANOVA, *P<0.05; n=3,5,6,9). (C) Representative frontal and oral images of embryos, comparing Irf6+/+; Esrp1−/−; Esrp2+/− with Irf6R84C/+; Esrp1−/−; Esrp2+/− and Irf6+/+; Esrp1−/−; Esrp2−/− with Irf6R84C/+; Esrp1−/−; Esrp2−/−. Scale bars: 50 μm. (D) Hematoxylin and Eosin staining of coronal sections through the vomeronasal cavity and primary palate of the same embryos. Irf6R84C heterozygosity modifies the Esrp1 knockout (KO) and Esrp1/2 double KO cleft lip and palate such that the cleft space between adjacent elements is narrower (arrowheads; C,D), and, in some cases, we noticed epithelial adhesions that limited the cleft. Scale bars: 100 μm.
As previously reported, Esrp1 null and Esrp1/2 double null embryos displayed bilateral CL/P (Fig. 9C). Interestingly, we noted a modification of this cleft phenotype when Esrp1 and Esrp1/2 null embryos were also heterozygous for Irf6. Irf6R84C/+; Esrp1−/−; Esrp2+/− and Irf6R84C/+; Esrp1−/−; Esrp2−/− embryos had less space or less wide clefts between the lateral lips and maxilla and the midline nasal capsule (Fig. 9C). This decreased space between tissue (or cleft severity) but persistence of a cleft was confirmed in histological sections (Fig. 9D). Further, histological sections showed presumed epithelial adhesions between lateral and medial portions of the nasal cavity in Irf6 heterozygotes, whereas this space was open in Esrp1 null and Esrp1/2 double null embryos (Fig. 9D).
DISCUSSION
Orofacial clefts are a common birth defect, and genome-wide association studies have identified some crucial genes associated with syndromic and non-syndromic cleft. Here, we describe mouse and zebrafish models using genes with known genetic variants in human cleft patients, IRF6 and ESRP1/2. We present evidence to support that Irf6 and Esrp1/2 function in the same regulatory pathway. We observed that mz-irf6-8bp/-8bp zebrafish embryos have significantly decreased expression of esrp1, and this is rescued upon introduction of irf6 mRNA. This finding is consistent with esrp1 being a transcriptional target of irf6, and putative irf6 response elements (Khan et al., 2018) can be found surrounding the esrp1 transcriptional start site. Additionally, RNA-seq identified known irf6 targets, including grhl3 and tfap2a. Direct molecular experiments are needed, however, to test transcriptional regulation of esrp1 by irf6. We found that Irf6 and Esrp1/2 are consistently co-expressed in the embryonic frontonasal ectoderm and oral epithelium associated with the palate, and epithelium of the mouth opening, in both mouse and zebrafish.
In zebrafish, irf6 null embryos ruptured during gastrulation, whereas esrp1/2 null embryos survive to larval stage. However, post-gastrulation ablation of irf6 resulted in a similar cleft morphology of the ANC as the esrp1/2 null. Further analysis of the esrp1/2 null showed that the cleft of the ANC correlated with a cleft in the upper margin of the mouth opening, reminiscent of a human cleft lip. Further, using a neural crest-specific photo-convertible reporter line, we were able to show that migration of CNCCs to the developing ANC occurred but chondrogenesis was impaired.
The early lethality of irf6 null zebrafish initially precluded analysis of the irf6 zygotic requirement in craniofacial development. Here, we utilized an optogenetics strategy to disrupt irf6 function after gastrulation when the embryonic body axis had formed, thereby revealing the zygotic requirement for irf6. Future studies will use the irf6 optogenetic model to study the roles of irf6 during ANC and lip morphogenesis. Interestingly, periderm markers identified in the mouse lambdoidal junction were found to be dysregulated in the irf6 mutant zebrafish model, specifically grhl3, tfap2a and perp. Additionally, gata3, which was identified as a mesenchymal marker at the fusion zone of mice (Li et al., 2019), is dysregulated in the irf6 null zebrafish. This work highlights the utility of complementary studies of palate morphogenesis in zebrafish and mouse models. The zebrafish model affords the transgenic tractability and visualization of CNCC migration, enabling us to determine the cellular mechanism responsible for the cleft ANC. The mouse mutants provide the mammalian anatomic contexts to examine cleft malformation.
Although the esrp1-4bp/-4bp; esrp2-14bp/-14bp zebrafish exhibited consistent cleft lip and cleft ANC phenotype, the infertility of the esrp2-14bp/-14bp fish preclude large-scale experiments to analyze downstream mechanisms of the development of cleft palate. We generated a robust esrp1-4bp/-4bp; esrp2 morphant assay that can be applied in chemical screening experiments and functional testing of human ESRP1/2 gene variants.
In humans, CPO is less common than CL/P (Gritli-Linde, 2008; Van Otterloo et al., 2016; Bush and Jiang, 2012). Although humans and mice share ∼99% of their genes and the early craniofacial development of the mouse embryo closely mirrors that of human (Swartz et al., 2011), there is a striking difference in the manifestation of orofacial cleft defects (Gritli-Linde, 2008). Most often, when a human CL/P-associated gene has been disrupted in mice, a cleft of the palate forms but the lip appears normal. Our current understanding in humans is that CL/P and CPO are different genetic disorders (Gritli-Linde, 2008; Juriloff and Harris, 2008; Dixon et al., 2011). These discrepancies between humans and mouse models hamper understanding of the etiopathogenesis of human CL/P. Here, we characterize the Esrp1/2 null mouse, exhibiting bilateral CL/P, as an important model for studying orofacial cleft etiopathogenesis. Additionally, as we place ESRP1 in the IRF6 gene-regulatory pathway, we hope to better understand how alternative isoforms regulated by ESRP1 may, in turn, be important for palate development.
Whereas zebrafish have historically been an excellent model organism for forward genetic screens, CRISPR/Cas9 gene-editing technology has permitted relatively efficient reverse genetic engineering of zebrafish (Liu et al., 2019). This utility of the zebrafish embryo for studying developmental processes and modeling human cleft-associated genes necessitates further study into their craniofacial morphogenesis. Transplant and lineage-tracing experiments have illuminated the neural crest origin of the zebrafish ANC, and how the frontonasal and paired maxillary cartilage elements converge into a continuous cartilage structure (Reid et al., 1986; Dougherty et al., 2012; Dougherty et al., 2013). We show that IRF6 and ESRP1 are conserved in their requirement for ANC morphogenesis, where disruption results in orofacial cleft in human, mouse and zebrafish. These findings provide evidence of conserved molecular and morphological processes occurring in the merging and fusion of the mouse and zebrafish midface.
We suspect that non-epithelial expression of Irf6 contributes to normal craniofacial morphogenesis and may explain some differences in the Irf6 and Esrp1/2 mutant phenotypes. Future research utilizing tissue-specific knockout of Irf6 will address this hypothesis. We also suspect that the Irf6 phenotype is more severe because Irf6 acts upstream of Esrp1, along with additional targets, and ongoing experiments on the transcriptional activity of Irf6 will be important. Recently, an in-depth analysis of a lineage-specific Esrp1 knockout mouse was completed and found that Esrp1 regulates proliferation of the mesenchyme of the lateral nasal prominences, along with being required for fusion of the medial and lateral nasal prominences (Lee et al., 2020). Ongoing work to identify Esrp1/2 molecular targets and mechanistic studies of these targets will provide new insight into palate morphogenesis.
These studies highlight the utility of complementary mouse and zebrafish models to elucidate mechanisms of orofacial cleft development. Additionally, this work has expanded the scope of Irf6 gene regulation in craniofacial development.
MATERIALS AND METHODS
Animal breeding and gene editing
All animal experiments were performed in accordance with protocols approved by Massachusetts General Hospital Animal Care and Usage Committee. C57Bl/6J (WT) animals were obtained from The Jackson Laboratory. Irf6R84C/+ mice were a gift from Dr Yang Chai (University of Southern California, Los Angeles, USA). Esrp1+/–; Esrp2−/− mice were received from Dr Russ Carstens (University of Pennsylvania, Philadelphia, USA). E0.5 was considered to be 12:00 on the day of the copulatory plug.
Zebrafish (Danio rerio) adults and embryos were maintained in accordance with approved institutional protocols at Massachusetts General Hospital. Embryos were raised at 28.5°C in E3 medium (5.0 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) with 0.0001% Methylene Blue. Embryos were staged according to standardized developmental timepoints by hpf or dpf (Liu et al., 2001). All zebrafish lines used for experimentation were generated from the Tübingen strain.
CRISPR sgRNA target sites were identified by a variety of online CRISPR computational programs such as ZiFiT Targeter Version 4.2 (zifit.partners.org/ZiFiT) (Sander et al., 2007), crispr.mit.edu (https://zlab.bio/guide-design-resources) (Ran et al., 2013) and ChopChop (https://chopchop.cbu.uib.no) (Montague et al., 2014). sgRNAs were designed with the traditional sequence constraint of a 3′ protospacer adjacent motif sequence containing NGG and an additional sequence constraint of a 5′ NG for in vitro RNA synthesis.
The esrp1, esrp2 and irf6 CRISPR sgRNAs were generated by in vitro transcription from an SP6 promoter as described (Gagnon et al., 2014). Lyophilized Cas9 protein (PNA Bio) was resuspended in ddH2O to a stock concentration of 1 µg/µl and stored in single-use aliquots in −80°C and kept for 6 months. One-cell-staged zebrafish embryos were microinjected directly in the cytoplasm with 2 nl of a solution containing 15 ng/µl sgRNA and 100 ng/µl Cas9 protein pre-complexed for 5-10 min at room temperature (RT). A subset of embryos injected with the sgRNA and Cas9 protein mixture was harvested for genomic DNA to confirm the presence of indels, and the rest were grown into adulthood as F0 mosaic fish. F0 adult fish were subsequently outcrossed with WT fish to generate F1 founders with germline transmission of indel alleles. F1 founders were further outcrossed with WT fish to yield a large number of heterozygotes and minimize the presence of off-target edits. Lastly, F2 heterozygotes were in-crossed to generate homozygote embryos for phenotypic analysis.
DNA for genotyping was isolated from either whole 24 hpf embryos or tail fin clips using the HotSHOT method as previously described (Meeker et al., 2007). Genotyping primers flanking the CRISPR sgRNA site were designed using a combination of ChopChop (https://chopchop.cbu.uib.no) and NCBI primer BLAST (ncbi.nlm.nih.gov/tools/primer-blast/). Forward primers were synthesized by Invitrogen with 5′-FAM modifications. Microsatellite sequencing analyses were used to determine indel mutation sizes and frequencies (Massachusetts General Hospital DNA Core), and Sanger sequencing was performed on PCR amplicons of CRISPR sgRNA to confirm the exact sequence changes resulting from CRISPR mutagenesis.
mRNA sequencing and qPCR
Total RNA was isolated from 4 hpf WT and maternal-null irf6−/− embryos by TRIzol and phenol-chloroform ethanol precipitation. Total RNA was quantified with the Nanodrop 2500 and assessed for quality with Bioanalyzer 2100 RNA chips (Agilent). Samples with RNA integrity numbers (RIN) over 9 were selected to proceed with sequencing library preparation. mRNA sequencing (mRNA-seq) libraries were prepared with the NEBNext Ultra RNA library preparation kit with poly(A) mRNA magnetic isolation module (NEB) essentially according to manufacturer protocols. Resulting cDNA libraries were quantified by a Qubit fluorometer and assessed for quality with a Bioanalyzer. The sequencing-ready cDNA libraries were quantified with the NEBNext library quantification kit for Illumina (NEB). mRNA-seq libraries were sequenced with single-end 50 at ≈20 million reads per sample with biological triplicates. Sequencing data are available at Gene Expression Omnibus (GEO; accession number GSE153828).
For qPCR, ∼30 zebrafish embryos per sample were flash frozen in liquid nitrogen. Mouse embryos from E11.5 timed pregnancies were isolated and dissected so that the head portion was flash frozen for RNA isolation and a posterior portion was frozen for genotyping. Samples were homogenized using a rotor-stator homogenizer, and RNA was isolated using an RNeasy Mini Kit (Qiagen). Total mRNA was quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific) and used for cDNA synthesis. qPCR was performed with Taqman probes and reagents (Thermo Fisher Scientific), and expression was normalized to 18s rRNA or TBP expression.
Zebrafish embryo microinjection of mRNA and MOs
Microinjection of mRNA was performed by injecting 2 nl mRNA solution with 0.05% Phenol Red directly into the cytoplasm of one-cell-staged embryos. Lyophilized MOs were resuspended with ddH2O to a stock concentration of 20 ng/µl and stored at RT in aliquots. Individual aliquots were heated to 70°C and briefly vortexed before preparation of the injection mix to ensure full dissolution. Mismatch control MOs were injected under identical conditions to control for potential toxicities. Embryos from all methods of microinjection were examined at 3 hpf to remove unfertilized embryos, which were quantified against the total number of microinjected embryos to ensure that no fertilization defects were observed.
WISH
Embryos were isolated at various time points and fixed in 4% formaldehyde at 4°C for 12-16 h. Subsequently, embryos were washed and stored in methanol. WISH- and digoxigenin (DIG)-labeled riboprobes were synthesized as described (Thisse and Thisse, 2008). Briefly, for riboprobe synthesis, PCR was performed using embryonic cDNA as templates and T7 promoter sequence-linked reverse primers to generate cDNA templates for in vitro transcription. PCR reactions were purified using the NucleoSpin gel and PCR clean-up kit (Machery-Nagel). In vitro transcription was performed using a T7 polymerase (Roche) and DIG labeling mix (Roche). DIG-labeled riboprobes were isolated with ethanol-NaOAc precipitation, resuspended in diethyl pyrocarbonate-treated ddH2O and stored at −20°C. All PCR products were TOPO cloned into pGEM-T Easy vectors (Promega) and sequence verified by Sanger sequencing. WISH colorimetric signal detection was performed using an alkaline phosphatase (AP)-conjugated anti-DIG antibody (Roche) and BM Purple AP substrate (Roche).
RNAscope ISH
Zebrafish and mouse embryos were fixed in 4% formaldehyde, taken through a sucrose gradient and cryo-embedded and sectioned. Probes were designed and purchased from ACD Bio, and hybridization and staining were performed according to the manufacturer's protocol. Stained sections were imaged using a Leica SP8 confocal microscope, where a z-stack was obtained and analyzed on ImageJ for z-stack maximum-intensity projections. In cases where a larger field was imaged, Leica LAS X software was utilized to perform a tile scan and to reconstruct the tiled images.
Skeletal staining and brightfield imaging
Zebrafish embryos were fixed at 96 hpf or 120 hpf in 4% formaldehyde and stored at 4°C overnight, washed with PBS, dehydrated in 50% ethanol, and stained with acid-free Alcian Blue overnight on a rotating platform at RT as described (Thisse and Thisse, 2008). Stained embryos were washed with ddH2O and subsequently bleached (0.8% w/v KOH, 0.1% Tween 20, 0.9% H2O2) until cell pigmentation was no longer present. For double-stained embryos (Alcian Blue and Alizarin Red), embryos were stained with a 0.05% Alizarin Red solution in ddH2O for 30 min on a rotating platform at RT following bleaching with KOH and H2O2. Then, double-stained embryos were placed in three changes of a tissue-clearing solution consisting of 25% glycerol and 0.1% KOH, each for 25 min. Whole and dissected stained embryos were mounted in 3% methylcellulose on a depression slide and imaged using a Nikon Eclipse 80i compound microscope with a Nikon DS Ri1 camera. Z-stacked images were taken to increase the depth of field with NIS Element BR 3.2 software. Stacked images were processed by ImageJ to generate maximum-intensity projection images.
For SEM, 4 dpf embryos were fixed in half-strength Karnovsky fixative. Samples were processed, and images were obtained by CBSET (Lexington, MA, USA). Mouse embryos from Irf6R84C/+; Esrp1+/−; Esrp2+/− crosses were collected into PBS. Tail clips were saved for genotyping and embryos were fixed in 10% formalin for brightfield imaging. After imaging, skulls (excluding the lower jaw) were cryosectioned and sections were stained with Hematoxylin and Eosin.
Optogenetic expression of irf6 in zebrafish
Genes irf6, irf6-ENR, irf6R84C and mCherry were isolated by PCR from various templates and inserted into the pGL4.23-(C120×5)-TATA vector with In-Fusion cloning (Clontech) according to the manufacturer’s instructions using a 1:2 vector-to-insert ratio to generate optogenetic response plasmids. The constructs were transformed in Stellar chemically competent cells (Clontech), and colonies were screened by PCR, restriction digests, Sanger sequencing and whole-plasmid sequencing to verify the sequence identities and accuracy of the constructs. Light-sensitive response protein VP16-EL222 was subcloned into pCS2+8 and in vitro transcribed from the SP6 promoter as described above to generate capped mRNA for embryo microinjections. The optogenetics injection mix consisted of 25 ng/µl EL222 and 10 ng/µl pGL4.23 response plasmid with 0.05% Phenol Red. Each embryo was microinjected with 2 nl of the optogenetics injection mix directly in the cytoplasm at the one-cell stage, immediately wrapped in aluminum foil, and placed into a dark incubator. Unfertilized and abnormal embryos were removed at 3 hpf in a dark room with limited exposure to ambient light. Injected embryos were divided into two groups (dark and light) at the desired developmental stage in E3 medium without Methylene Blue and placed under 465 nm blue light (LED panel, HQRP) at 0.3 mW/cm2 (measured by a PM100D digital power meter with an SV120VC photodiode power sensor, ThorLabs) with constant illumination. Control embryo containers were wrapped in aluminum foil.
Lineage tracing
Embryos originating from an espr1-4bp/-4bp;esrp2+/-14bp in-cross were injected with 8 ng esrp1 MO and 4 ng esrp2 MO at the one-cell stage, or uninjected WT embryos, all in a Tg(sox10:kaede) background, were grown until 20 somites, oriented for imaging in the sagittal position, and encased in 1% low-melt agarose. Using the 405 nm UV laser and ROI setting in a Leica SP8 confocal microscope, the anteriormost portion of neural crest cells that contribute to the FNP were unilaterally photoconverted, keeping the alternate side as an internal control, as previously described (Dougherty et al., 2012). Photoconverted embryos were carefully micro-dissected out of the agar and grown in E3 medium at 28.5°C until 4 dpf and imaged again to track the photoconverted cells. Maximum projections of the photoconverted half of the embryo, or the planes consisting of the palate, in 14 hpf or 4 dpf embryos, respectively, were generated using Fiji/ImageJ.
Statistics
Statistical analyses were performed using Prism Software (GraphPad). An unpaired Student's t-test or one-way ANOVA with multiple comparisons was used as indicated. A P-value <0.05 was considered significant. Graphs represent the mean±s.e.m. or individual values (dots). In all experiments, n represents biological replicates.
Supplementary Material
Acknowledgement
We ae grateful to Jessica Bethoney for excellent management of our aquatics facility. We thank Irimia Manuel for willingness to share esrp1/2 zebrafish mutant alleles (did not survive shipment, not used in this study) and Yang Chai for sharing the Irf6R84C mouse mutant allele.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.H.C., C.M.T., E.B.L., R.P.C., E.C.L.; Methodology: S.H.C., C.M.T., E.B.L., J.C., E.C.L.; Formal analysis: J.C.; Investigation: S.H.C., C.M.T., E.B.L., K.K., N.M., S.A.H., N.A.; Resources: R.P.C., E.C.L.; Data curation: S.H.C., C.M.T., E.B.L., K.K., J.C.; Writing - original draft: S.H.C., C.M.T., E.B.L., E.C.L.; Writing - review & editing: S.H.C., C.M.T., J.C., R.P.C., E.C.L.; Visualization: S.H.C., K.K.; Supervision: E.C.L.; Funding acquisition: E.C.L.
Funding
The Esrp1 and Esrp2 mouse alleles were generated by R.P.C. with funding from the National Institutes of Health (R01DE024749). This work was supported by grants to E.C.L. from the National Institutes of Health (R01DE027983), the Shriners Hospitals for Children and Massachusetts General Hospital (Laurie and Mason Tenaglia MGH Research Scholar Award). Deposited in PMC for immediate release.
Data availability
Sequencing data are available at GEO under accession number GSE153828.
Supplementary information
Supplementary information available online at https://dev.biologists.org/lookup/doi/10.1242/dev.194498.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.194498.reviewer-comments.pdf
References
- Abramyan J. and Richman J. M. (2015). Recent insights into the morphological diversity in the amniote primary and secondary palates. Dev. Dyn. 244, 1457-1468. 10.1002/dvdy.24338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty T. H., Marazita M. L. and Leslie E. J. (2016). Genetic factors influencing risk to orofacial clefts: today's challenges and tomorrow's opportunities. F1000Res 5, 2800 10.12688/f1000research.9503.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee T. W., Park J. W., Sheridan K. I., Warzecha C. C., Cieply B. W., Rohacek A. M., Xing Y. and Carstens R. P. (2015). The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 4, e08954 10.7554/eLife.08954.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee T. W., Sims-Lucas S., Park J. W., Bushnell D., Cieply B., Xing Y., Bates C. M. and Carstens R. P. (2016). Ablation of the epithelial-specific splicing factor Esrp1 results in ureteric branching defects and reduced nephron number. Dev. Dyn. 245, 991-1000. 10.1002/dvdy.24431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botti E., Spallone G., Moretti F., Marinari B., Pinetti V., Galanti S., De Meo P. D., De Nicola F., Ganci F., Castrignano T. et al. (2011). Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc. Natl. Acad. Sci. USA 108, 13710-13715. 10.1073/pnas.1110931108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguera D., Marquez Y., Racioppi C., Permanyer J., Torres-Mendez A., Esposito R., Albuixech-Crespo B., Fanlo L., D'agostino Y., Gohr A. et al. (2017). Evolutionary recruitment of flexible Esrp-dependent splicing programs into diverse embryonic morphogenetic processes. Nat. Commun. 8, 1799 10.1038/s41467-017-01961-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush J. O. and Jiang R. (2012). Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231-243. 10.1242/dev.067082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero D. R., Brugmann S., Chu Y., Bajpai R., Jame M. and Helms J. A. (2011). Cranial neural crest cells on the move: their roles in craniofacial development. Am. J. Med. Genet. A 155A, 270-279. 10.1002/ajmg.a.33702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox L. L., Cox T. C., Moreno Uribe L. M., Zhu Y., Richter C. T., Nidey N., Standley J. M., Deng M., Blue E., Chong J. X. et al. (2018). Mutations in the epithelial cadherin-p120-catenin complex cause mendelian non-syndromic cleft lip with or without cleft palate. Am. J. Hum. Genet. 102, 1143-1157. 10.1016/j.ajhg.2018.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creuzet S., Couly G. and Le Douarin N. M. (2005). Patterning the neural crest derivatives during development of the vertebrate head: insights from avian studies. J. Anat. 207, 447-459. 10.1111/j.1469-7580.2005.00485.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Garza D. E., Schleiffarth G., Dunnwald J. R., Mankad M., Weirather A., Bonde J. L., Butcher G., Mansour S., Kousa T. A., Fukazawa Y. A. et al. (2013). Interferon regulatory factor 6 promotes differentiation of the periderm by activating expression of Grainyhead-like 3. J. Invest. Dermatol. 133, 68-77. 10.1038/jid.2012.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H. and Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167-178. 10.1038/nrg2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty M., Kamel G., Shubinets V., Hickey G., Grimaldi M. and Liao E. C. (2012). Embryonic fate map of first pharyngeal arch structures in the sox10: kaede zebrafish transgenic model. J. Craniofac. Surg. 23, 1333-1337. 10.1097/SCS.0b013e318260f20b [DOI] [PubMed] [Google Scholar]
- Dougherty M., Kamel G., Grimaldi M., Gfrerer L., Shubinets V., Ethier R., Hickey G., Cornell R. A. and Liao E. C. (2013). Distinct requirements for wnt9a and irf6 in extension and integration mechanisms during zebrafish palate morphogenesis. Development 140, 76-81. 10.1242/dev.080473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan K. M., Mukherjee K., Cornell R. A. and Liao E. C. (2017). Zebrafish models of orofacial clefts. Dev. Dyn. 246, 897-914. 10.1002/dvdy.24566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhouri W. D., Metwalli K., Naji A., Bakhiet S., Quispe-Salcedo A., Nitschke L., Kousa Y. A. and Schutte B. C. (2017). Intercellular genetic interaction between Irf6 and Twist1 during craniofacial development. Sci. Rep. 7, 7129 10.1038/s41598-017-06310-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti E., Li B., Zewdu R., Wells V., Hebert J. M., Karner C., Anderson M. J., Williams T., Dixon J., Dixon M. J. et al. (2011). A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell 21, 627-641. 10.1016/j.devcel.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon J. A., Valen E., Thyme S. B., Huang P., Akhmetova L., Pauli A., Montague T. G., Zimmerman S., Richter C. and Schier A. F. (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 9, e98186 10.1371/journal.pone.0098186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin A. F., Kim R., Bush J. O. and Klein O. D. (2015). From bench to bedside and back: improving diagnosis and treatment of craniofacial malformations utilizing animal models. Curr. Top. Dev. Biol. 115, 459-492. 10.1016/bs.ctdb.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudy S., Angel P., Jacobs B., Hill C., Mainini V., Smith A. L., Kousa Y. A., Caprioli R., Prince L. S., Baldwin S. et al. (2013). Cell-autonomous and non-cell-autonomous roles for IRF6 during development of the tongue. PLoS ONE 8, e56270 10.1371/journal.pone.0056270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A. (2008). The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Curr. Top. Dev. Biol. 84, 37-138. 10.1016/S0070-2153(08)00602-9 [DOI] [PubMed] [Google Scholar]
- Helms J. A., Cordero D. and Tapadia M. D. (2005). New insights into craniofacial morphogenesis. Development 132, 851-861. 10.1242/dev.01705 [DOI] [PubMed] [Google Scholar]
- Hu D., Marcucio R. S. and Helms J. A. (2003). A zone of frontonasal ectoderm regulates patterning and growth in the face. Development 130, 1749-1758. 10.1242/dev.00397 [DOI] [PubMed] [Google Scholar]
- Ingraham C. R., Kinoshita A., Kondo S., Yang B., Sajan S., Trout K. J., Malik M. I., Dunnwald M., Goudy S. L., Lovett M. et al. (2006). Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335-1340. 10.1038/ng1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J., Suzuki A., Pelikan R. C., Ho T. V., Sanchez-Lara P. A., Urata M., Dixon M. J. and Chai Y. (2013). Smad4-Irf6 genetic interaction and TGFbeta-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development 140, 1220-1230. 10.1242/dev.089615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R., Bush J. O. and Lidral A. C. (2006). Development of the upper lip: morphogenetic and molecular mechanisms. Dev. Dyn. 235, 1152-1166. 10.1002/dvdy.20646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff D. M. and Harris M. J. (2008). Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol 82, 63-77. 10.1002/bdra.20430 [DOI] [PubMed] [Google Scholar]
- Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J. A. V. AN., Der Lee R., Bessy A., Cheneby J., Kulkarni S. R., Tan G. et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D1284 10.1093/nar/gkx1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B. (1989). Genetics and early development of zebrafish. Trends Genet. 5, 283-288. 10.1016/0168-9525(89)90103-0 [DOI] [PubMed] [Google Scholar]
- Knight R. D. and Schilling T. F. (2006). Cranial neural crest and development of the head skeleton. Adv. Exp. Med. Biol. 589, 120-133. 10.1007/978-0-387-46954-6_7 [DOI] [PubMed] [Google Scholar]
- Knight A. S., Schutte B. C., Jiang R. and Dixon M. J. (2006). Developmental expression analysis of the mouse and chick orthologues of IRF6: the gene mutated in Van der Woude syndrome. Dev. Dyn. 235, 1441-1447. 10.1002/dvdy.20598 [DOI] [PubMed] [Google Scholar]
- Kondo S., Schutte B. C., Richardson R. J., Bjork B. C., Knight A. S., Watanabe Y., Howard E., De Lima R. L., Daack-Hirsch S., Sander A. et al. (2002). Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285-289. 10.1038/ng985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousa Y. A. and Schutte B. C. (2016). Toward an orofacial gene regulatory network. Dev. Dyn. 245, 220-232. 10.1002/dvdy.24341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousa Y. A., Moussa D. and Schutte B. C. (2017). IRF6 expression in basal epithelium partially rescues Irf6 knockout mice. Dev. Dyn. 246, 670-681. 10.1002/dvdy.24537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousa Y. A., Zhu H., Fakhouri W. D., Lei Y., Kinoshita A., Roushangar R. R., Patel N. K., Agopian A. J., Yang W., Leslie E. J. et al. (2019). The TFAP2A-IRF6-GRHL3 genetic pathway is conserved in neurulation. Hum. Mol. Genet. 28, 1726-1737. 10.1093/hmg/ddz010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Cieply B., Yang Y., Peart N., Glaser C., Chan P. and Carstens R. P. (2018). Esrp1-regulated splicing of Arhgef11 isoforms is required for epithelial tight junction integrity. Cell Rep. 25, 2417-2430.e5. 10.1016/j.celrep.2018.10.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Sears M. J., Zhang Z., Li H., Salhab I., Krebs P., Xing Y., Nah H. D., Williams T. and Carstens R. P. (2020). Cleft lip and cleft palate in Esrp1 knockout mice is associated with alterations in epithelial-mesenchymal crosstalk. Development 147, dev187369 10.1242/dev.187369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E. J., Mancuso J. L., Schutte B. C., Cooper M. E., Durda K. M., L'heureux J., Zucchero T. M., Marazita M. L. and Murray J. C. (2013). Search for genetic modifiers of IRF6 and genotype-phenotype correlations in Van der Woude and popliteal pterygium syndromes. Am. J. Med. Genet. A 161A, 2535-2544. 10.1002/ajmg.a.36133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E. B., Truong D., Hallett S. A., Mukherjee K., Schutte B. C. and Liao E. C. (2017). Rapid functional analysis of computationally complex rare human IRF6 gene variants using a novel zebrafish model. PLoS Genet. 13, e1007009 10.1371/journal.pgen.1007009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Jones K. L., Hooper J. E. and Williams T. (2019). The molecular anatomy of mammalian upper lip and primary palate fusion at single cell resolution. Development 146, dev174888 10.1242/dev.174888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieschke G. J. and Currie P. D. (2007). Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367. 10.1038/nrg2091 [DOI] [PubMed] [Google Scholar]
- Liu W. S., Pesold C., Rodriguez M. A., Carboni G., Auta J., Lacor P., Larson J., Condie B. G., Guidotti A. and Costa E. (2001). Down-regulation of dendritic spine and glutamic acid decarboxylase 67 expressions in the reelin haploinsufficient heterozygous reeler mouse. Proc. Natl. Acad. Sci. USA 98, 3477-3482. 10.1073/pnas.051614698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Leslie E. J., Jia Z., Smith T., Eshete M., Butali A., Dunnwald M., Murray J. and Cornell R. A. (2016). Irf6 directly regulates Klf17 in zebrafish periderm and Klf4 in murine oral epithelium, and dominant-negative KLF4 variants are present in patients with cleft lip and palate. Hum. Mol. Genet. 25, 766-776. 10.1093/hmg/ddv614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Petree C., Requena T., Varshney P. and Varshney G. K. (2019). Expanding the CRISPR toolbox in zebrafish for studying development and disease. Front. Cell Dev. Biol. 7, 13 10.3389/fcell.2019.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losa M., Risolino M., Li B., Hart J., Quintana L., Grishina I., Yang H., Choi I. F., Lewicki P., Khan S. et al. (2018). Face morphogenesis is promoted by Pbx-dependent EMT via regulation of Snail1 during frontonasal prominence fusion. Development 145, dev157628 10.1242/dev.157628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita M. L. (2012). The evolution of human genetic studies of cleft lip and cleft palate. Annu. Rev. Genomics Hum. Genet. 13, 263-283. 10.1146/annurev-genom-090711-163729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker N. D., Hutchinson S. A., Ho L. and Trede N. S. (2007). Method for isolation of PCR-ready genomic DNA from zebrafish tissues. BioTechniques 43, 610, 612,, 614 10.2144/000112619 [DOI] [PubMed] [Google Scholar]
- Montague T. G., Cruz J. M., Gagnon J. A., Church G. M. and Valen E. (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401-W407. 10.1093/nar/gku410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta-Mena L. B., Reade A., Mallory M. J., Glantz S., Weiner O. D., Lynch K. W. and Gardner K. H. (2014). An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol. 10, 196-202. 10.1038/nchembio.1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'donoghue S., Green T., Ross J. L., Hallmayer J., Lin X., Jo B., Huffman L. C., Hong D. S. and Reiss A. L. (2020). Brain development in school-age and adolescent girls: effects of turner syndrome, estrogen therapy, and genomic imprinting. Biol. Psychiatry 87, 113-122. 10.1016/j.biopsych.2019.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A. and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid J. S., Carmichael A. F., Sri-Pathmanathan R. and Douglas S. (1986). Ectodermal dysplasia: case with an unusual combination of dental features. J. Oral. Med. 41, 259-261. [PubMed] [Google Scholar]
- Reid B. S., Yang H., Melvin V. S., Taketo M. M. and Williams T. (2011). Ectodermal Wnt/beta-catenin signaling shapes the mouse face. Dev. Biol. 349, 261-269. 10.1016/j.ydbio.2010.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R. J., Dixon J., Malhotra S., Hardman M. J., Knowles L., Boot-Handford R. P., Shore P., Whitmarsh A. and Dixon M. J. (2006). Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 38, 1329-1334. 10.1038/ng1894 [DOI] [PubMed] [Google Scholar]
- Rorick N. K., Kinoshita A., Weirather J. L., Peyrard-Janvid M., De Lima R. L., Dunnwald M., Shanske A. L., Moretti-Ferreira D., Koillinen H., Kere J. et al. (2011). Genomic strategy identifies a missense mutation in WD-repeat domain 65 (WDR65) in an individual with Van der Woude syndrome. Am. J. Med. Genet. A 155A, 1314-1321. 10.1002/ajmg.a.33980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A., Kontarakis Z., Gerri C., Nolte H., Hölper S., Kruger M. and Stainier D. Y. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230-233. 10.1038/nature14580 [DOI] [PubMed] [Google Scholar]
- Sabel J. L., d'Alençon C., O'brien E. K., Van Otterloo E., Lutz K., Cuykendall T. N., Schutte B. C., Houston D. W. and Cornell R. A. (2009). Maternal interferon regulatory factor 6 is required for the differentiation of primary superficial epithelia in danio and Xenopus embryos. Dev. Biol. 325, 249-262. 10.1016/j.ydbio.2008.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander J. D., Zaback P., Joung J. K., Voytas D. F. and Dobbs D. (2007). Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res. 35, W599-W605. 10.1093/nar/gkm349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T. F. and Le PABIC P. (2009). Fishing for the signals that pattern the face. J. Biol. 8, 101 10.1186/jbiol205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz M. E., Sheehan-Rooney K., Dixon M. J. and Eberhart J. K. (2011). Examination of a palatogenic gene program in zebrafish. Dev. Dyn. 240, 2204-2220. 10.1002/dvdy.22713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C. and Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69. 10.1038/nprot.2007.514 [DOI] [PubMed] [Google Scholar]
- Van Otterloo E., Williams T. and Artinger K. B. (2016). The old and new face of craniofacial research: How animal models inform human craniofacial genetic and clinical data. Dev. Biol. 415, 171-187. 10.1016/j.ydbio.2016.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha C. C., Sato T. K., Nabet B., Hogenesch J. B. and Carstens R. P. (2009). ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33, 591-601. 10.1016/j.molcel.2009.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Zuo X., He M., Gao J., Fu Y., Qin C., Meng L., Wang W., Song Y., Cheng Y. et al. (2017). Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 8, 14364 10.1038/ncomms14364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q., Blanton S. H. and Hecht J. T. (2011). Genetic causes of nonsyndromic cleft lip with or without cleft palate. Adv. Otorhinolaryngol. 70, 107-113. 10.1159/000322486 [DOI] [PubMed] [Google Scholar]
- Zucchero T. M., Cooper M. E., Maher B. S., Daack-Hirsch S., Nepomuceno B., Ribeiro L., Caprau D., Christensen K., Suzuki Y., Machida J. et al. (2004). Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N. Engl. J. Med. 351, 769-780. 10.1056/NEJMoa032909 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.