

ABSTRACT
Craniofacial development is regulated through dynamic and complex mechanisms that involve various signaling cascades and gene regulations. Disruption of such regulations can result in craniofacial birth defects. Here, we propose the first developmental stage-specific network approach by integrating two crucial regulators, transcription factors (TFs) and microRNAs (miRNAs), to study their co-regulation during craniofacial development. Specifically, we used TFs, miRNAs and non-TF genes to form feed-forward loops (FFLs) using genomic data covering mouse embryonic days E10.5 to E14.5. We identified key novel regulators (TFs Foxm1, Hif1a, Zbtb16, Myog, Myod1 and Tcf7, and miRNAs miR-340-5p and miR-129-5p) and target genes (Col1a1, Sgms2 and Slc8a3) expression of which changed in a developmental stage-dependent manner. We found that the Wnt-FoxO-Hippo pathway (from E10.5 to E11.5), tissue remodeling (from E12.5 to E13.5) and miR-129-5p-mediated Col1a1 regulation (from E10.5 to E14.5) might play crucial roles in craniofacial development. Enrichment analyses further suggested their functions. Our experiments validated the regulatory roles of miR-340-5p and Foxm1 in the Wnt-FoxO-Hippo subnetwork, as well as the role of miR-129-5p in the miR-129-5p–Col1a1 subnetwork. Thus, our study helps understand the comprehensive regulatory mechanisms for craniofacial development.
KEY WORDS: Craniofacial development, Feed-forward loop, Regulatory network, Transcription factor, MicroRNA
Highlighted Article: Insights into the physical and cellular biology underlying craniofacial development will broaden our understanding of the etiology of orofacial clefts, and guide therapies and prevention strategies for orofacial clefts.
INTRODUCTION
Orofacial clefts, including cleft lip with or without cleft palate (CL/P) and cleft palate only (CPO), are among the most common congenital malformations that severely affect orofacial structure (Watkins et al., 2014). The current treatment for individuals with orofacial clefts requires multiple surgeries and complex interdisciplinary care throughout life, including medical, surgical, speech and behavioral treatments. A comprehensive understanding of the biological processes underlying morphogenesis will facilitate the development of novel interventions and prevention of orofacial clefts.
The etiology of CL/P and CPO is complex and heterogeneous, involving both genetic and environmental factors as well as their interactions. Numerous studies have been conducted for the identification of genetic risk factors for CL/P and CPO, including genome-wide association studies (GWAS), association analyses, animal model studies and direct sequencing of genomes from patients with orofacial clefts. Although a substantial number of genes have been identified in CL/P (Suzuki et al., 2018a,b), the molecular mechanisms in orofacial clefts still remain unclear due to the lack of appropriate approaches for such analyses during craniofacial development.
The development of lip and palate in mice starts with formation of the maxillary processes at embryonic day 9.5 (E9.5), which relies on a tightly wired regulated network that involves the activity of diverse signaling pathways, crucial extracellular matrix molecules and transcription factors (TFs) (Meng et al., 2009). Gene expression is controlled at several levels, including transcriptional (e.g. TFs bind on the promoter regions of genes), post-transcriptional (e.g. miRNAs suppress transcripts) and epigenetic (e.g. DNA methylation and chromatin remodeling) levels. TFs are proteins that activate or repress the expression of genes by binding to the cis-regulatory regions of target DNA at the transcriptional level (Latchman, 1997). MicroRNAs (miRNAs) are a set of small noncoding RNAs, typically 21-22 nucleotides long, that suppress gene expression by binding to miRNA recognition elements (MREs) in the 3′-untranslated regions (3′-UTRs) at the post-transcriptional level (Bartel, 2004). Both TFs and miRNAs play crucial roles in developmental processes such as craniofacial development, and their disruption is linked to craniofacial anomalies such as CL/P (Gajera et al., 2019; Li et al., 2020; Suzuki et al., 2019a,b). Thomason and colleagues used mouse models to demonstrate the cooperation of TF p63 and gene IRF6 in cleft palate (Thomason et al., 2010). In addition, He and coworkers reported the regulation of metadherin (AEG-1; an oncogene) by miRNA-375 in hepatocellular carcinoma xenograft mouse models (He et al., 2012).
It has been demonstrated that TFs can coordinate with miRNAs to co-regulate gene expression (Shalgi et al., 2007; Tsang et al., 2007). The relationship between a TF, a miRNA and a target gene can generate feed-forward loops (FFLs), one of the most commonly occurring network motifs, and then FFLs can form co-regulatory networks, which has proved to be a powerful tool for identifying crucial regulators and interactions (Shalgi et al., 2007; Tsang et al., 2007). This unique co-regulation approach has been applied to various diseases, such as schizophrenia (Guo et al., 2010), cancer (e.g. glioblastoma, prostate cancer, lung cancer, ovarian cancer, colorectal cancer, gastric cancer, and testicular germ cell cancer) (Afshar et al., 2014; Sun et al., 2012) and tuberculosis (Lin et al., 2017). Jiang et al. recognized frequently dysregulated TF-miRNA FFLs across multiple tumor types (Jiang et al., 2016). In addition to these diseases, FFLs have also been applied in lung and craniofacial development. Fibroblast growth factor (FGF) forms an FFL with canonical molecules Wnt2a and Wnt7b in WNT signaling to regulate lung development (Yin et al., 2011). The transcription factor MEF2C is involved in a feed-forward transcriptional circuit with Dlx5/6 and Hand2 and exhibits an unanticipated role in craniofacial development (Verzi et al., 2007). Li et al. showed that the miRNA hsa-mir-27b represses the expression of transcription factor SMAD1 and accordingly activates WNT pathway core gene WNT3A through FFL during lip development (Li et al., 2020). Although various important regulatory motifs for TFs, miRNAs and genes have been identified, most studies have been concerned with only one specific condition (e.g. tumor versus matched control samples) and not with a dynamic process (e.g. time series or developmental stages).
A systematic investigation of the developmental or temporal expression of key regulatory factors and their networks will facilitate our understanding of the molecular basis of control of lip and palate development. In this study, we have, for the first time, developed a novel developmental stage-specific, network-based analytical approach for investigating the co-regulation of TFs and miRNAs in a temporal-specific manner during craniofacial development. We examined how these FFL modules change over embryonic stages, by stage-to-stage comparison, and how this affects craniofacial development. We also experimentally validated the role of crucial miRNAs and TFs in the resultant subnetworks using mouse palatal mesenchymal cells. Our work provides new insights into the physical and cellular biology underlying craniofacial development, broadens understanding of the etiology of orofacial clefts, as well as guides future therapies and prevention strategies for at-risk populations.
RESULTS
Differentially expressed genes, TFs and miRNAs in mouse craniofacial development
In this study, we developed a novel developmental stage-specific approach for studying the expression of genes, TFs and miRNAs, and their co-regulation in a phenotype or disease. Specifically, we utilized gene expression data from the maxillary processes of C57BL6/J mouse embryos (E10.5, E11.5, E12.5, E13.5 and E14.5) available at the FaceBase Portal (Affymetrix Mouse Genome 430 2.0 Array, n=6 per developmental stage, Accession ID FB00000804/GSE67985) and miRNA expression data from the frontonasal prominence of 129S6 mouse embryos (E10.5, E11.5, E12.5, E13.5 and E14.5) available at the FacaBase portal (RNA-Seq, n=2, Accession ID FB00000663.01-FB00000666.01). Our data analysis workflow is illustrated in Fig. 1. Briefly, we downloaded the raw mRNA and miRNA expression data and mapped them to the mouse reference genome assembly GRCm38 (mm10) to obtain mRNA and miRNA expressions. We conducted pairwise comparison between each adjacent embryonic time point to define significantly differentially expressed genes. These molecules, including mRNAs and miRNAs, were used to construct FFLs and then networks. The miRNA mapping results are summarized in Table S1. The average mapping rate was 85.11%. By pairwise comparison, we identified an average of 355 significantly differentially expressed non-TF genes (range 70-470 among all pairwise comparisons), 14.25 TFs (range 1-23) and 17 miRNAs (range 10-34). Hereafter, we refer to genes encoding non-TF proteins as non-TF genes to distinguish them from TF protein-coding genes. The significantly differentially expressed mRNAs and miRNAs were defined as those with an absolute log2 fold change >1 and adjusted P-value <0.05 (Table S2). The P-value was adjusted using the Benjamini–Horchberg procedure (Benjamini and Hochberg, 1995).
Fig. 1.

Developmental stage-specific TF-miRNA co-regulatory network approach for studying craniofacial development. (A) Flowchart. (B-E) Candidate co-regulation types in FFLs. Arrows denote a directional regulation (either positive or negative role). The symbol ‘––|’ represents a repression role. DE, differentially expressed; DEG, differentially expressed gene; FDR, false discovery rate; FFL, feed-forward loop; miRNA, microRNA; PCC, Pearson's correlation coefficient; TF, transcriptional factor.
FFL assembling and network construction
The curated interaction pairs between nodes (TF-gene, miRNA-gene and TF-miRNA) were obtained from databases (see Materials and Methods for more details). They were further validated by calculating the Pearson's correlation coefficient (PCC) using the expression data to decrease the number of false positives. A sample size of six is sufficient for detecting a correlation coefficient of 0.9 with power of 0.8 (Bujang and Baharum, 2016). Li and colleagues used PCC>0.3 to nominate co-expression (Li et al., 2019). Thus, we only retained significant pairs with a coefficient greater than 0.3 (P<0.05), which resulted in a total of 34,198 significant TF-miRNA pairs. Then, all possible TF-miRNA-gene FFLs were assembled. Specifically, there are four patterns of FFLs with concordant regulation: (1) gene expression is activated by a TF and is repressed by a miRNA, where miRNA may repress the TF (miRNA-FFL, Fig. 1B); (2) vice versa (TF-FFL, Fig. 1C); (3) a TF and a miRNA are mutually repressed (composite-FFL, Fig. 1E); and (4) gene expression is repressed by both a TF and a miRNA, where the TF also activates the miRNA (TF-FFL, Fig. 1D). In all these four regulation patterns, the direction between each pair of TF, miRNA and gene is concordant in biology (e.g. the expression correlation between a miRNA and a gene is required to be negative). For example, if a gene is activated by a TF and repressed by a miRNA, then the regulation type between this TF and miRNA can only be repression. The classification of FFLs depends on the regulation type between TF, gene and miRNA. In a TF-FFL loop, TF is the master regulator and dominates expression of gene and miRNA. In a miRNA-driven FFL loop, the miRNA expression affects both gene and TF. In a composite-based FFL, in which TF and miRNA repress each other, gene expression is mainly affected by the regulator in the FFL with higher expression change (Sun et al., 2012). We obtained a total of 144,466 FFLs.
To evaluate the significance of each FFL, we next conducted a hypergeometric test to assess the probability that each TF-miRNA pair targets the same gene. As a result, 33,988 TF-miRNA pairs in 140,684 FFLs were filtered out by this test. After this step, we obtained a total of 3782 FFLs with 334 unique genes, 157 TFs and 44 miRNAs. The FFLs containing at least two differentially expressed nodes between any two adjacent stages were extracted to construct the developmental stage-specific regulatory network. Considering the data from the five developmental stages in the mouse maxillary processes, we used the FFLs derived from each pair of two consecutive days and constructed four co-regulatory networks: E10.5-E11.5 network, E11.5-E12.5 network, E12.5-E13.5 network and E13.5-E14.5 network (Fig. 2). Table 1 summarizes the node and edge information of these four co-regulatory networks.
Fig. 2.

TF-miRNA co-regulatory network at E10.5-E11.5. (A) Co-regulation networks visualized by Cytoscape, Green triangles denote miRNAs, brown diamonds represent TFs and pink circles denote non-TF protein-coding genes. The area of the node is proportional to the degree in the network. (B) Bubble plots depict the pathways enriched in the regulatory networks. GO biological process terms (circles) and KEGG pathways (triangles) were used for the enrichment analysis. Color scheme reflects the adjusted P-values. The area of a circle or triangle is proportional to the number of enriched genes. (C) Wnt-FoxO-Hippo subnetwork for the expression and regulation changes of E10.5 to E11.5. (D) Putative model of Foxm1 and miR-340-5p regulation in the Wnt-FoxO-Hippo subnetwork. (E) Expression profiles of genes in the Wnt-FoxO-Hippo subnetwork along the developmental stage.
Table 1.
Summary of the four TF-miRNA co-regulatory networks in craniofacial development (E10.5 to E14.5)

TF-miRNA co-regulatory network at E10.5-E11.5
The co-regulatory network at E10.5-E11.5 contained a total of 76 unique nodes (TF, non-TF genes and miRNAs) and 193 edges (the links between two nodes), with 88.2% (67/76) of the nodes showing differential expression (Fig. 2A). Among the 76 unique nodes, there were 58 genes, 9 TFs and 9 miRNAs. Among the 193 edges, 102 were TF-gene pairs, 79 were miRNA-gene pairs and 12 were TF-miRNA pairs (Table 1).
The topological characteristics, such as the degree of nodes and their distribution, were measured to provide an overview of the network. Specifically, we used the degree to measure the number of interactors of a node in the network, regardless of the edge direction. A higher degree implies more important roles in the network (Sun et al., 2012). Overall, the degree distribution of the nodes was strongly right skewed, indicating that the majority of nodes had relatively small degrees, whereas only a small proportion of nodes had high degrees (e.g. hub nodes) (Fig. S1A). This is consistent with previous studies showing that the degree of regulatory networks tended to follow a scale-free distribution (Sun et al., 2018). Specifically, the degree of genes, TFs and miRNAs ranged from 2 to 8, 2 to 29 and 2 to 35, respectively. The average degree of genes, TFs and miRNAs were 3.12, 12.67 and 10.11, respectively (Fig. S1B). Hubs are conservatively defined as nodes with degree more than 7 and lie above the 0.95 quantile of the degree distribution (Yu et al., 2017). Based on this definition, we pinpointed one hub miRNA (miR-340-5p, degree 35) and three hub TFs (Foxm1, degree 29; Hif1a, degree 20; and Zbtb16, degree 19), where miR-340-5p might reduce mesenchymal traits and cell migration, and Foxm1 might be involved in cell proliferation and epithelial-to-mesenchymal transition (EMT) (Kim et al., 2019; Luo et al., 2019). Our results suggest their important regulatory roles in the network (Fig. 2A). The transcription factor Foxm1 might repress the expression of miR-340-5p through the FFL and, accordingly, affect EMT, cell migration and disappearance of medial edge epithelium during lip formation.
The gene set enrichment analysis was performed to categorize the genes and TFs functionally in the network. In total, 17 Gene Ontology (GO) terms and 14 Kyoto Encyclopedia of Genes and Genomes (KEGG) canonical pathways were found to be significantly enriched with the nodes in the co-regulatory network from E10.5 to E11.5 (multiple-testing adjusted P<0.05, Fig. 2B). Interestingly, all of these GO terms were related to craniofacial development: ‘cell-cell adhesion’, ‘trabecula morphogenesis’, ‘ossification’, ‘cartilage development’, ‘tissue development’, ‘epithelial to mesenchymal transition’ and ‘tissue remodeling’. Of note, three important pathways were identified: Wnt signaling pathway (six genes, Col1a1, Egfr, Fzd3, Mapk14, Tcf7, Tnks2, adjusted P=0.040), FoxO signaling pathway (five genes, Egfr, Mapk14, Plk4, Tgfb2, Tgfbr1, adjusted P=0.006), and Hippo signaling pathway (four genes, Fzd3, Tcf7, Tgfb2, Tgfbr, adjusted P=0.040). These three pathways have been well studied in craniofacial and neural crest development.
The subnetwork for these three pathways, obtained by extracting the enriched genes and their first neighbors, is shown in Fig. 2C. It is interesting that Foxm1 and miR-340-5p emerged as crucial regulators. Based on the edge direction, it can be inferred that Foxm1 activates the expression of a list of enriched nodes, including Fzd3, Hif1a, Mapk14, Plk4, Tgfbr1 and Tnks2, whereas miR-340-5p represses their expression. Combining the subnetwork and results from previous studies, we propose a putative model in which Foxm1 and miR-340-5p act as promising contributors and are involved in the pathogenic mechanism of orofacial clefts (Fig. 2D). Downregulated Foxm1 expression and upregulated miR-340-5p expression collectively lead to downregulation of genes in the pathways. The expression plot of these genes along the developmental stage showed a decreasing trend from E10.5 to E11.5 (Fig. 2E). Abnormal regulation of Foxm1 and miR-340-5p could lead to the alteration of gene expression in Wnt, FoxO and Hippo signaling pathways, resulting in orofacial clefts. The subnetwork further indicates that our stage-specific co-regulatory network of miRNAs and TFs can summarize previously published findings at the systems level.
TF-miRNA co-regulatory network at E11.5-E12.5
The regulatory network for E11.5 to E12.5 was comprised of 93 FFLs, which involved 41 non-TF genes, eight TFs and nine miRNAs (Fig. 3A). There were 89.1% (49/55) of nodes showing differential expression during embryonic development (Table 1). Among the 147 edges within the network, 93 were TF-gene pairs, 45 were miRNA-gene pairs and nine were TF-miRNA pairs. The average degrees of genes, TFs and miRNAs were 3.37 (range 2-7), 12.88 (range 2-27), and 9.17 (range 2-33), respectively. Notably, one hub miRNA (miR-129-5p, degree 33) and two hub TFs (Myog, degree 27; Myod1, degree 24) were identified in the network (Fig. 3B). In the literature, the muscle-specific TFs Myog and Myod1 are reported to play a role in the formation of myofibers (Rosero Salazar et al., 2020).
Fig. 3.

TF-miRNA co-regulatory network at E11.5-E12.5. (A) Co-regulation networks visualized by Cytoscape. (B) Bubble plots depict the pathways enriched in the regulatory networks.
Enrichment analysis of the nodes in the network from E11.5 to E12.5 indicated the importance of hormones, as shown by enrichment of GO terms such as ‘response to estradiol’ (five genes, adjusted P=0.064), ‘oxytocin signaling pathway’ (six genes, adjusted P=0.005), ‘synthesis and secretion of aldosterone’ (three genes, adjusted P=0.094) and ‘synthesis of parathyroid hormone’ (three genes, adjusted P=0.094) (Fig. 3B).
TF-miRNA co-regulatory network at E12.5-E13.5
The subsequent co-regulatory network from E12.5 to E13.5 contained a total of 108 edges and 60 unique nodes, with 83.3% (50/60) being differentially expressed nodes (Fig. 4A). The average degrees of miRNAs were the highest (9.17, range 2-22), followed by TFs (6.00, range 2-22) and genes (2.56, range 2-5). Two hub miRNAs (miR-340-5p, degree 22; miR-129-5p, degree 16) and one hub TF (Tcf7, degree 22) were pinpointed (Fig. S1B). Tcf7 is an HMG box protein that associates with molecules in the nucleus to modulate the Wnt signaling pathway (Cadigan and Waterman, 2012).
Fig. 4.

TF-miRNA co-regulatory network at E12.5-E13.5. (A) Co-regulation networks visualized by Cytoscape. (B) Bubble plots depict the pathways enriched in the regulatory networks. (C) Tissue remodeling subnetwork for the change from E12.5 to E13.5.
Twenty GO terms were significantly enriched in the network from E12.5 to E13.5. These GO terms highlighted the importance of bone and tissue development and remodeling (Fig. 4B), including ‘biomineral’ (six genes, adjusted P=0.005) and ‘connective tissue development’ (seven genes, adjusted P=0.006), ‘tissue remodeling’ (six genes, adjusted P=0.006), ‘regulation of osteoblast differentiation’ (six genes, adjusted P=0.006) and ‘regulation of ossification’ (six genes, adjusted P=0.008). Among the KEGG pathways, we observed enrichment in ‘adherens junction’ (four genes, adjusted P=0.029) and ‘ECM-receptor interaction’ (four genes, adjusted P=0.029). The tissue remodeling-related subnetwork was extracted and composed of three motifs (Tcf7–miR-340-5p–Igf1/Pdk4/Spp1; Egr1–miR-129-5p–Cspg4/Tnfsf11; and Nr5a1/Nr6a1/Sp2/Smad3–miR-7b-5p–Car2) (Fig. 4C). For instance, in the motif Tcf7–miR-340-5p–Igf1/Pdk4/Spp1, Tcf7 regulates several important genes for tissue remodeling (Igf1, Pdk4 and Spp1) by binding to the promoter regions of these genes during palatogenesis, and it also regulates them through suppression of miR-340-5p.
TF-miRNA co-regulatory network at E13.5-E14.5
There were no FFLs with two differentially expressed nodes from E13.5 to E14.5. Hence, FFLs with only one node showing differential expression were utilized to construct the network, where the differential expression proportion was 8.7% (6/69) (Table 1). The resultant co-regulatory network had a total of 131 edges and 69 unique nodes (Fig. 5A). Interestingly, the majority of nodes were TFs (71.01%, 49/69), with a small proportion of miRNAs (17.39%, 12/69) and genes (8.70%, 6/69). We found that 45.04% of the edges in the regulatory network (59/131) were TF-gene pairs, 42.75% (56/131) were TF-miRNA pairs and 12.21% (16/131) were miRNA-gene pairs.
Fig. 5.

TF-miRNA co-regulatory network at E13.5-E14.5. (A) Co-regulation networks visualized by Cytoscape. (B) Bubble plots depict the pathways enriched in the regulatory networks.
In contrast to the regulatory networks in the previous stages, the genes showed high connectivity and skewed the node degree distribution (Fig. S1A). Specifically, the average degree of the genes was 12.00 (range 2-30). TFs and miRNAs had relatively low average degrees of 2.39 (range 2-6) and 5.50 (range 2-32), respectively. One hub miRNA (miR-129-5p, degree 32) and three hub genes (Col1a1, degree 30; Sgms2, degree 16; Slc8a3, degree 10) were identified (Fig. 5A).
The co-regulatory network from E13.5 to E14.5 was significantly enriched in GO terms that are closely related to embryonic limb and appendage morphogenesis and development (Fig. 5B): ‘cell fate commitment’ (12 genes, adjusted P=1.0×10−8), ‘embryonic limb morphogenesis’ (nine genes, adjusted P=1.6×10−7), ‘embryonic appendage morphogenesis’ (nine genes, adjusted P=1.6×10−7) and ‘skeletal system development’ (eight genes, adjusted P=1.4×10−6).
Comparisons between stage-specific regulatory networks
When we compared the nodes in these four regulatory networks, interestingly, we found that the overlapped nodes were primarily the hub nodes in the regulatory network at specific stages (Fig. 6), further suggesting the importance of these hub nodes. For example, Col1a1 appeared in the network at every stage and acted as the hub gene in the network from E13.5 to E14.5. The hub miRNA was miR-129-5p in the three networks across four time points from E11.5 to E14.5. Myod1 was the hub TF in the network from E11.5 to E12.5 and also appeared in other networks (E10.5 to E11.5 and E13.5 to E14.5).
Fig. 6.

Comparisons between stage-specific regulatory networks. (A-C) Venn diagrams show the overlap of genes (A), TFs (B) and miRNAs (C) in four craniofacial development-specific networks. The tables on the right summarize the overlapped nodes for the comparison. (D) The miR-129-5p–Col1a1 subnetwork along the developmental stages.
Additionally, all four co-regulatory networks comprised Col1a1, miR-129-5p and miR-466l-5p (Fig. 6). Col1a1 and miR-129-5p constitutionally presented at all stages analyzed and were targeted by several TFs, including Myod1 (E10.5 to E12.5), Myog (E11.5 to E12.5), Erg1 (E11.5 to E13.5) and other TFs (E13.5 to E14.5) (Fig. 6D). These TFs might suppress the expression of miR-129-5p to alleviate its inhibition of Col1a1, which could promote cell migration and proliferation in development. Thus, the subnetwork containing the Col1a1 and miR-129-5p pair might represent an important mechanism for craniofacial development.
Experimental validations
To validate the results from the regulatory network analyses, we performed a series of experiments. We focused on the Wnt-FoxO-Hippo subnetwork as shown in Fig. 2C. We analyzed expression of the genes predicted to be downstream targets of either miR-340-5p or Foxm1 in O9-1 cells, a mouse cranial neural crest cell line. We conducted quantitative RT-PCR (qRT-PCR) analyses for Tgfbr1, Fzd3, Mapk14, Tnks2 and Plk4 after treatment with a miR-340-5p mimic and found that the miR-340-5p mimic significantly downregulated expression of Fzd3, Mapk14, Plk4, Tgfbr1 and Tnks2 (Fig. 7A). In addition, we conducted qRT-PCR analyses for these genes after overexpression of Foxm1 in O9-1 cells. We found that Foxm1 overexpression significantly upregulated expression of Fzd3, Mapk14, Plk4, Tgfbr1 and Tnks2 (Fig. 7B). Furthermore, we evaluated whether Foxm1 overexpression attenuated miR-340-5p expression and found that Foxm1 overexpression significantly downregulated miR-340-5p expression (Fig. 7C). Taken together, our experiments using cultured O9-1 cells validate the Wnt-FoxO-Hippo subnetwork identified through our bioinformatic analyses.
Fig. 7.

Effect of Foxm1 and miR-340-5p in the Wnt-FoxO-Hippo subnetwork. (A) qRT-PCR for the indicated genes after treatment with control or miR-340-5p mimic in O9-1 cells. (B) qRT-PCR for the indicated genes after Foxm1 overexpression in O9-1 cells. Each gene expression was compared with mock control. (C) qRT-PCR for miR-340-5p after Foxm1 overexpression or for mock control in O9-1 cells. Expression of miR-340-5p was normalized with miR-26a-5p expression, a housekeeping miRNA in the developing palate. *P<0.05, **P<0.01, ***P<0.001.
Next, to validate the miR-129-5p–Col1a1 subnetwork shown in Fig. 6D, we performed qRT-PCR for miR-129-5p and its target genes, which were predicted to regulate each other's expression. The miR-129-5p mimic significantly downregulated Col1a1 expression (Fig. 8A). In addition, overexpression of Crebpb, Myod1, Myog, Smad2, Sp2 or Wt1 significantly upregulated Col1a1 expression (Fig. 8B), whereas overexpression of these TFs downregulated miR-129-5p expression (Fig. 8C). Taken together, the results show that the miR-129-5p–Col1a1 subnetwork identified through our bioinformatic analyses was also validated with our experiments using cultured O9-1 cells.
Fig. 8.

Effect of TFs and miR-129-5p in the miR-129-5p–Col1a1 subnetwork. (A) qRT-PCR for Col1a1 after treatment with control or miR-129-5p mimic in O9-1 cells. (B) qRT-PCR for Col1a1 after overexpression of the indicated TFs in O9-1 cells. Each gene expression was compared with mock control. (C) qRT-PCR for miR-129-5p after overexpression of the indicated TFs in O9-1 cells. Expression of miR-129-5p was normalized with miR-26a-5p expression, a housekeeping miRNA in the developing palate. **P<0.01, ***P<0.001.
DISCUSSION
Precise control of embryonic developmental processes is encoded by a series of tight regulations in cells. Such multifactorial systems have been difficult to decode because of the complex and dynamic interactions between molecules, and also largely because of the lack of the appropriate data and analytical approaches. In this study, we present a novel network-based analytical approach for identification of key regulators during craniofacial development. By coupling stage-specific transcriptional profiling of different molecules (TFs, miRNAs and non-TF genes), we identified and dissected a list of key regulators governing the developmental processes from E10.5 to E14.5. The results not only confirmed previous findings in craniofacial development, but also offered some new insights into the molecular interactions that define morphogenesis and cell fate commitment. This approach can be generally applied to any developmental or disease stage-specific research in order to decode molecular regulation; it can be applied to other organisms, such as humans, and it can be extended to the single cell level.
During mouse craniofacial development from E10.5 to E11.5, the maxillary processes of the first branchial arch start to grow toward the medial and nasal processes, which gradually fuse together to form the upper lip (Suzuki et al., 2016). Consistent with such biological processes, functional enrichment analysis of our regulatory network directly revealed branchial arch growth-related terms, such as ‘trabecula morphogenesis’ and ‘cartilage development’. The pinpointed three important signaling pathways (namely Wnt, FOXOs and Hippo pathways) have been reported as having a strong relation to orofacial clefts. Specifically, the Wnt signaling pathway is considered to be one of the most important pathways and its deficiency results in malformations of the upper lip (Lan et al., 2006; Reynolds et al., 2019). Foxo6, which is expressed particularly in craniofacial tissues, activates Hippo signaling to control the growth of the craniofacial complex (Sun et al., 2018). In this study, the subnetwork of these three pathways (namely the Wnt-FoxO-Hippo subnetwork) was validated with qRT-PCR analyses, which showed that miR-340-5p overexpression significantly downregulated the expression of targeted genes, whereas Foxm1 overexpression upregulated expression of these genes and downregulated miR-340-5p expression. These results suggested the importance of miR-340-5p and Foxm1 in craniofacial development. In addition, there is evidence showing the correlation of the targeted genes (Tgfbr1, Hif1a, Mapk14, Fzd3, Tnks2 and Plk4) with orofacial clefts. For example, Tgfbr1, a transmembrane receptor for TGFβ ligands and ubiquitously expresses in craniofacial epithelium and mesenchyme, plays crucial roles in palate development (Iwata et al., 2011). Although Foxm1 has not been reported as a causative TF for orofacial clefts, several studies imply indirect evidence for the involvement of Foxm1 in palatogenesis. For example, Foxm1 connects with Pitx2, a downstream target of TGFβ signaling, via Lef1 (Iwata et al., 2012; Pelikan et al., 2013). Foxm1 is also involved in EMT, one of three important ways for disappearance of medial edge epithelium, and in cell proliferation and survival in oral squamous cell carcinoma cells (Luo et al., 2019).
Although the association of other two hub TFs, Hif1a and Zbtb16, has not been considered in orofacial cleft and lip and palate development, they may be good candidates for further human and mouse genetic studies. Hif1a is a transcriptional activator that responds to cellular hypoxia and regulates expression of hypoxia-inducible genes such as EPO, VEGF and ENO1 (Minet et al., 2000; Semenza et al., 1997). Although the alteration of expression of Hif1a is involved in congenital birth defects, embryonic development, tumorigenesis and many other diseases (Semenza, 1998; Tong et al., 2018; Yildirim and Kocak, 2018), there is no association between HIF1A polymorphism and orofacial cleft in humans (Küchler et al., 2018). Hypoxia is known as a risk factor for developmental defects, including orofacial cleft in animal models (Fajersztajn and Veras, 2017; Küchler et al., 2018; Millicovsky and Johnston, 1981; Nagaoka et al., 2012). Hif1a knockout mice are embryonic lethal by E11.0, with severe developmental defects such as open neural tube and cardiovascular defects (Iyer et al., 1998). Zbtb16 (also known as PLZF), a transcriptional repressor that works with histone deacetylase, is involved in the maturation of myeloid and organogenesis (Barna et al., 2005; Clotaire et al., 2019; Mao et al., 2017). Zbtb16 knockout mice exhibit defects in the limbs, immune system and spermatogenesis (Barna et al., 2000; Costoya et al., 2004; Kovalovsky et al., 2008). However, it is unknown whether Zbtb16 is associated with craniofacial development or orofacial cleft.
The fusion between the medial nasal and maxillary processes occurs at E11.5 and E12.5. The epithelial seam between these processes disappears and forms the upper lip. Intriguingly, our co-regulatory network at this stage (E11.5-E12.5) suggests a role for focal adhesions, which transmit mechanical force and signals from the extracellular matrix (ECM) to cytosol and regulate migration and/or invasion and cell survival (Mitra et al., 2005; Wozniak et al., 2004). The results also imply the importance of hormones, including estrogen, oxytocin, aldosterone and parathyroid hormone. Estrogen can facilitate the growth and migration of cranial neural crest cells (Furukawa et al., 1997), from which derive the mesenchyme of lip and palatal shelves (Chai et al., 2000). The hub TFs Myog and Myod1 are muscle-specific TFs and belong to the myogenic regulatory factor (MRF) family, which drives the formation of myofibers, a crucial process in orofacial muscle generation (Rosero Salazar et al., 2020; Singh and Dilworth, 2013).
Upper lip formation is complete by E12.5 and secondary palate formation starts from E12.5. A set of palatal shelves elongates from the maxillary processes and grows vertically along with the tongue through E13.5. At E14.0, the palatal shelves are elevated, grow horizontally on the tongue and fuse together at the midline of the oral cavity at E14.5 (Gritli-Linde, 2007; Iwata et al., 2011; Jin et al., 2010; Parada and Chai, 2012). Although tremendous morphological and mechanical changes occur during palate elevation, how mechanical forces are generated and perceived by the cells for tissue remodeling remains poorly understood. Our study sheds light on the process of tissue remodeling by looking into the co-regulatory network during this process. The functional analysis highlights the importance of bone and tissue development and remodeling, which are concordant in biology with the growth, elevation and reorientation of the palate shelves (Bush and Jiang, 2012). The tissue remodeling-related subnetwork suggests the importance of six genes (Car2, Cspg4, Igf1, Pdk4, Spp1 and Tnfsf11) and five regulators (Egr1, Tcf7, miR-7b-5p, miR-129-5p and miR-340-5p) (Fig. 4C). Among them, the hub TF Tcf7 is associated with cleft palate, based on gene-disease association datasets from the curated Comparative Toxicogenomics Database (CTD) (Davis et al., 2019). Expression of Tcf7 is higher in the posterior nasal compartment of the palate than in the oral compartment of the palate (Potter and Potter, 2015).
Of note, the Col1a1 and miR-129-5p pair presented in all four regulatory networks. Furthermore, Col1a1 acts as the hub node in the regulatory network from E13.5 to E14.5 and the networks from E11.5 to E14.5, indicating that it plays crucial roles in craniofacial development. It has been reported that miR-129-5p regulates EMT (Xiao et al., 2015) and is expressed in tubular epithelial cells and colorectal cancer (Li et al., 2015; Lin et al., 2018). Also, miR-129-5p is thought to bind COL1A1 and directly suppress its expression, which results in the inhibition of cell invasion and proliferation in gastric cancer cells (Wang and Yu, 2018). Similarly, we found that miR-340-5p was a key regulator in regulatory networks from E10.5 to E11.5 and E12.5 to E13.5. A previous study has shown that overexpression of miR-340-5p reduces mesenchymal traits in glioblastoma multiforme (Kim et al., 2019). Additionally, Egr1 (early growth response 1), a transcriptional regulator involved in cell proliferation, differentiation and mitosis, is a key regulator in our co-regulatory networks from E11.5 to 13.5. Another study showed that miR-340-5p is involved in the development of several craniofacial tissues in mice (McMahon et al., 1990). Our experiments support the proposal that miR-340-5p plays a crucial role in regulation of the miR-129-5p–Col1a1 subnetwork.
Although our developmental stage-specific network analysis has successfully identified hub regulators for craniofacial development, there are some limitations. First, TF expression revealed by transcriptomic data is limited because TF expression levels can be changed via various processes (transcription, post-translation, translation, post-translational modification, protein transportation, etc.). Therefore, it would be interesting to integrate other sequencing methods, such as chromatin immunoprecipitation followed by sequencing (ChIP-seq), DNase-seq (Song and Crawford, 2010), and ATAC-seq (Buenrostro et al., 2013), to reveal a more comprehensive landscape of TF expression at the molecular level. Second, the interaction pairs between a TF, a gene and a miRNA were obtained from the curation databases based on computational approaches or large-scale experiments. Pearson correlation restrictions were applied to reduce the false positive rate in this study (e.g. PCC>0.3 and P<0.05; see ‘FFL assembling and network construction’ in Results). However, the calculations were based on a limited number of samples, which may result in unstable coefficient and P-values and the existence of false interaction pairs. Third, the in vitro experimental validations were conducted using O9-1 cells, a multipotent cranial neural crest cell line established from E8.5 mouse embryo. However, the FFLs in our study were constructed in tissues at stages E10.5 to E14.5, when the neural crest has been specified into craniofacial mesenchymal progenitors. Thus, the usage of O9-1 cells has some limitations in the conservation of regulatory networks. It is worth noting that, although some false positive results may exist in our analysis, our network approach reveals overall regulatory information at each comparison and can tolerate some false signals (i.e. most of the nodes and edges are true signals, whereas only one or a few non-hub nodes are false signals). Therefore, such a systems biology approach can still reveal major biological signals.
In this study, we have introduced a developmental stage-specific network approach by integrating two common regulators, TF and miRNA, to study their co-regulation during craniofacial development. We applied this approach to currently available data in mice (mouse strain C57BL/6J). The obtained networks can be further used for interpretation of genetic signals from human GWAS and transcriptomic studies, or both. For example, network tools such as dmGWAS and EW_dmGWAS can detect the enrichment of network modules (typically small modules with enriched signals) for the genetic association signals from GWAS and gene expression in humans using a large network (e.g. regulatory network) (Jia et al., 2011a,b; Wang et al., 2015).
MATERIALS AND METHODS
Differentially expressed genes, TFs and miRNAs
The mouse gene expression data of 30 C57BL/6J samples isolated from the maxillary processes at embryonic days 10.5, 11.5, 12.5, 13.5 and 14.5 (E10.5-E14.5) was downloaded from the FaceBase Portal (Affymetrix Mouse Genome 430 2.0 Array, n=6, Accession ID FB00000804/GSE67985). The differentially expressed genes were identified using GEO2R through the comparison between two adjacent time points (E11.5 versus E10.5, E12.5 versus E11.5, E13.5 versus E12.5 and E14.5 versus E13.5).
The miRNA sequencing profiles of ten mouse 129S6 samples across five developmental stages (E10.5-E14.5) were downloaded from the FacaBase portal (RNA-Seq, n=2, Accession ID: FB00000663.01-FB00000666.01). Reads were trimmed by cutadapt (Martin, 2011) (v2.4) and quality control was performed using fastQC (v0.11.8) (Andrews, 2010, http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads were mapped to the mouse genome and expression was calculated using miRDeep2 (v 2.0.1.0) (Friedländer et al., 2012). After post-processing steps, namely filtering low expression miRNAs and quantile normalization, differential gene expression analysis was performed using the DESeq2 (v1.26.0) (Love et al., 2014) package in the R (v.3.6.2) environment.
Regulation between TF and target (gene or miRNA)
The promoter regions, defined as 1000 bp downstream to 200 bp upstream of the transcription start sites, of mouse protein-coding genes as well as precursor miRNAs were obtained from UCSC Table Browser (Kent et al., 2002). TRANSFAC, the largest database of eukaryotic transcription factors, was utilized to obtain the TF motifs (Matys et al., 2006). Because the new version of TRANSFAC (release 2019.03) added the information for human and fruit fly TFs, but not mouse TFs, the old version (release 2016.1) was used for analyses, as described in our previous study (Li et al., 2019). The Find Individual Motif Occurrences (FIMO) program was utilized to map the position weight matrix of TF motifs to the promoter regions (Grant et al., 2011). To control for false-positive predictions, a core score of 1.0 and a matrix score of 0.95 were required. A total of 245,743 TF-gene pairs containing 533 TFs and 1288 genes were retrieved. In addition, a total of 71,137 TF-precursor miRNA interaction pairs were collected with 529 TFs and 490 precursor miRNAs. Then, the precursor miRNAs were mapped to mature miRNAs using the genome coordinates file in the miRBase database (Griffiths-Jones et al., 2008). After mapping, there were 529 TFs that regulate 811 mature miRNAs through 126,924 TF-mature miRNA interaction pairs.
Regulation between miRNA and target (gene or TF)
The miRTarBase is a comprehensive database including experimentally validated miRNA-target gene interactions (Chou et al., 2018). It deposits 959 unique miRNAs and 7280 target genes with a total of 40,681 pairs in mice (release 7.0). In addition to the miRTarBase, computationally predicted miRNA-target gene pairs were collected from PITA (version 6) (Kertesz et al., 2007), TargetScan (release 7.2) (Agarwal et al., 2015) and miRanda (released in August 2010) (Betel et al., 2008) (Table S3). To control false discovery rate, only the experimentally validated miRNA-target pairs were collected in miRTarBase plus the pairs predicted by at least two computational algorithms (PITA, TargetScan and miRanda). In total, 130,608 unique miRNA-target pairs were retrieved, including 1183 miRNAs and 11,958 targets. The PCC was calculated for each miRNA-target pair.
Construction of FFLs
Three-node FFLs were assembled, in which at least two nodes were differentially expressed. A cumulative hypergeometric test was conducted to assess whether TF and miRNA pairs randomly regulate the same target genes, using the following formula:
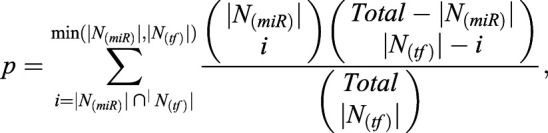 |
where N(miR) denotes the number of genes targeted by a given miRNA, N(tf) represents the number of genes targeted by a given TF and Total is the number of genes targeted by both miRNAs and TFs. The FDR was calculated by the Benjamini–Hochberg method for multiple test correction (Benjamini and Hochberg, 1995). Only the FFLs with significant TF-miRNA pairs were assembled to form the regulatory network.
Network analyses
Cytoscape (v3.7.2) was used to visualize the network (Shannon et al., 2003). The topological analyses were conducted using the NetworkAnalyzer (release 2.7) built in the Cytoscape (Assenov et al., 2007). WebGestalt was utilized to conduct the functional enrichment analysis of the genes and TFs residing in the regulatory network, using annotations from GO and KEGG pathway databases (Liao et al., 2019). The nominal P-values were corrected by the Bonferroni method (Bonferroni, 1936). Only gene sets having more than 5 and less than 300 genes were retained (Jia et al., 2011a,b; Jia and Zhao, 2012). The enriched categories were identified based on the adjusted P-value threshold of 0.1.
Experimental validations by qRT-PCR
O9-1 cells (SCC049, Sigma-Aldrich) were cultured in a conditioning medium obtained from STO cells (a mouse embryonic fibroblast cell line; CRL-1503, ATCC), as previously described (Suzuki et al., 2019b). O9-1 cells were plated onto a 35 mm dish at a density of 15,000 cells/dish. After 24 h, the cells were treated with an miR mimic or plasmid DNA. For the miR mimic, we used miR-129-5p, miR-340-5p or control (mirVana, Thermo Fisher Scientific) with Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific), according to the manufacturer's protocol (3 pmol of mimic in 6 µl of transfection reagent in 2 ml DMEM per dish). pcDNA3.1(-) mouse C/EBP beta (LAP) was a gift from Peter Johnson (Addgene plasmid #12557). pcDNA-Egr1 was a gift from Eileen Adamson (Addgene plasmid #11729). CMV-myc-tagged MyoD was a gift from Andrew Lassar (Addgene plasmid #8399). RGS-6xHis-pcDNA3.1(-) was a gift from Adam Antebi (Addgene plasmid #52534). pCMV-HA (New MCS) was a gift from Christopher A. Walsh (Addgene plasmid #32530). For plasmid DNA, pcDNA-C/EBP beta (Addgene #12557), pcDNA-Egr1 (Addgene #11729), pCMV-Foxm1 (Genomics online #ABIN3808691), pCMV-Myod1 (Addgene #8399), pCMV-Myog (Origene #MR227400), pCMV-Smad2 (Genomics online #ABIN3809501), pCMV-Sp2 (Genomics online #ABIN3826429), pCMV-Wt1 (Origene #MR225851), pcDNA (Addgene #52534) or pCMV (Addgene #32530) were transfected with Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific), according to the manufacturer's protocol (1 µg plasmid DNA in 3.75 µl of Lipofectamine 3000 reagent and 2 µl of P3000 reagent in 2 ml DMEM per dish). After 24 h, total RNA isolated from O9-1 cells (n=6 per group) was extracted with the QIAshredder and miRNeasy Mini Kit (Qiagen), according to the manufacturer's instructions.
Each sample was reverse-transcribed using iScript Reverse Transcription Supermix for qRT-PCR (Bio-Rad) and then cDNA was amplified with iTaq Universal SYBR Green Supermix (Bio-Rad) using the following PCR primers: Gapdh (NM_008084), 5′-AACTTTGGCATTTGGAAGG-3′ and 5′-ACACATTGGGGGTAGGAACA-3′; Col1a1 (NM_007472), 5′-GAAGATGTAGGAGTCGAGGGAC-3′ and 5′-CCTTGGAAACCTTGTGGACC-3′; Hif1a (NM_001313919), 5′- GGTGCTGATTTGTGAACCCATTCC-3′ and 5′- GCCCAAAAGTTCTTCCGGCTC-3′; Mapk14 (NM_011951), 5′- GTGTTCACACCCGCAAGGTC-3′ and 5′- ACGATGTTGTTCAGGTCCGC-3′; Tgfbr1 (NM_009370), 5′-GGCCGGGCCACAAACA-3′ and 5′-CTGAAAAAGGTCCTGTAGTTGGG-3′; Tnks2 (NM_001163635), 5′- AAGGTGAACAGCCGCGAC-3′ and 5′- CATCATCACGCGCTTGCAC-3′; Plk4 (NM_011495), 5′-AGACCGGCGGGAATTTTTCA-3′ and 5′-TAAAGTCCTCGATCCTCTCCCC-3′. The expression of each gene was normalized to GAPDH. Expression of miRNA was measured with probes for miR-129-5p (mmu480913_mir), miR-340-5p (mmu481072_mir), and miR-26a-5p (477995_mir) using the Taqman Fast Advanced Master Mix and Taqman Advanced miRNA cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer's instructions.
A P-value less than 0.05 in two-tailed Student's t-tests or the post-hoc Tukey-Kramer's test was considered to be statistically significant. Data are represented as mean±s.d. in the graphs.
Supplementary Material
Acknowledgements
We thank the members of Bioinformatics and Systems Medicine Laboratory (BSML) for valuable discussion, and the FaceBase investigators shared the data.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: Z.Z.; Methodology: F.Y., P.J., Z.Z.; Validation: H.Y., J.I.; Formal analysis: F.Y.; Data curation: F.Y.; Writing - original draft: F.Y.; Writing - review & editing: P.J., A.S., J.I., Z.Z.; Visualization: F.Y.; Supervision: Z.Z.; Project administration: Z.Z.; Funding acquisition: P.J., J.I., Z.Z.
Funding
This work was partially supported by National Institutes of Health grants (R01LM012806, R03DE027393 and R03DE028103 to Z.Z.; R03DE027711 to P.J.; and R01DE029818, R03DE026208, R03DE026509 and R03DE028340 to J.I.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at https://dev.biologists.org/lookup/doi/10.1242/dev.192948.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.192948.reviewer-comments.pdf
References
- Afshar A. S., Xu J. and Goutsias J. (2014). Integrative identification of deregulated miRNA/TF-mediated gene regulatory loops and networks in prostate cancer. PLoS ONE 9, e100806 10.1371/journal.pone.0100806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V., Bell G. W., Nam J. W. and Bartel D. P. (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenov Y., Ramírez F., Schelhorn S.-E., Lengauer T. and Albrecht M. (2007). Computing topological parameters of biological networks. Bioinformatics 24, 282-284. 10.1093/bioinformatics/btm554 [DOI] [PubMed] [Google Scholar]
- Barna M., Hawe N., Niswander L. and Pandolfi P. P. (2000). Plzf regulates limb and axial skeletal patterning. Nat. Genet. 25, 166-172. 10.1038/76014 [DOI] [PubMed] [Google Scholar]
- Barna M., Pandolfi P. P. and Niswander L. (2005). Gli3 and Plzf cooperate in proximal limb patterning at early stages of limb development. Nature 436, 277-281. 10.1038/nature03801 [DOI] [PubMed] [Google Scholar]
- Bartel D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281-297. 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- Benjamini Y. and Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodological) 57, 289-300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Betel D., Wilson M., Gabow A., Marks D. S. and Sander C. (2008). The microRNA.org resource: targets and expression. Nucleic Acids Res. 36, D149-D153. 10.1093/nar/gkm995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonferroni C. (1936). Teoria statistica delle classi e calcolo delle probabilita. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commericiali di Firenze 8, 3-62. [Google Scholar]
- Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y. and Greenleaf W. J. (2013). Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics. Nat. Methods 10, 1213 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujang M. A. and Baharum N. (2016). Sample size guideline for correlation analysis. WJSSR 3 10.22158/wjssr.v3n1p37 [DOI] [Google Scholar]
- Bush J. O. and Jiang R. (2012). Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231-243. 10.1242/dev.067082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan K. M. and Waterman M. L. (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harbor Perspect. Biol. 4, a007906 10.1101/cshperspect.a007906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y., Jiang X., Ito Y., Bringas P. Jr, Han J., Rowitch D. H., Soriano P., McMahon A. P. and Sucov H. M. (2000). Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671-1679. [DOI] [PubMed] [Google Scholar]
- Chou C.-H., Shrestha S., Yang C.-D., Chang N.-W., Lin Y.-L., Liao K.-W., Huang W.-C., Sun T.-H., Tu S.-J., Lee W.-H. et al. (2018). miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 46, D296-D302. 10.1093/nar/gkx1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clotaire D. Z. J., Wei Y., Yu X., Ousman T. and Hua J. (2019). Functions of promyelocytic leukaemia zinc finger (Plzf) in male germline stem cell development and differentiation. Reprod. Fertil. Dev. 31, 1315-1320. 10.1071/RD18252 [DOI] [PubMed] [Google Scholar]
- Costoya J. A., Hobbs R. M., Barna M., Cattoretti G., Manova K., Sukhwani M., Orwig K. E., Wolgemuth D. J. and Pandolfi P. P. (2004). Essential role of Plzf in maintenance of spermatogonial stem cells. Nat. Genet. 36, 653-659. 10.1038/ng1367 [DOI] [PubMed] [Google Scholar]
- Davis A. P., Grondin C. J., Johnson R. J., Sciaky D., McMorran R., Wiegers J., Wiegers T. C. and Mattingly C. J. (2019). The comparative toxicogenomics database: update 2019. Nucleic Acids Res. 47, D948-D954. 10.1093/nar/gky868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajersztajn L. and Veras M. M. (2017). Hypoxia: from placental development to fetal programming. Birth Defects Res 109, 1377-1385. 10.1002/bdr2.1142 [DOI] [PubMed] [Google Scholar]
- Friedländer M. R., Mackowiak S. D., Li N., Chen W. and Rajewsky N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37-52. 10.1093/nar/gkr688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H., Natsume N., Niimi T., Ueno Y., Miura S. and Kawai T. (1997). Experimental studies of cleft lip and palate, report no. 106: Preventive effect of estradiol on A/J mice. Int. J. Oral Maxillofac. Surg. 26, 247 10.1016/S0901-5027(97)81572-X [DOI] [Google Scholar]
- Gajera M., Desai N., Suzuki A., Li A., Zhang M., Jun G., Jia P., Zhao Z. and Iwata J. (2019). MicroRNA-655-3p and microRNA-497-5p inhibit cell proliferation in cultured human lip cells through the regulation of genes related to human cleft lip. BMC Med. Genomics 12, 70 10.1186/s12920-019-0535-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant C. E., Bailey T. L. and Noble W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017-1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Saini H. K., van Dongen S. and Enright A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154-D158. 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A. (2007). Molecular control of secondary palate development. Dev. Biol. 301, 309-326. 10.1016/j.ydbio.2006.07.042 [DOI] [PubMed] [Google Scholar]
- Guo A.-Y., Sun J., Jia P. and Zhao Z. (2010). A novel microRNA and transcription factor mediated regulatory network in schizophrenia. BMC Syst. Biol. 4, 10 10.1186/1752-0509-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.-X., Chang Y., Meng F.-Y., Wang M.-Y., Xie Q.-H., Tang F., Li P.-Y., Song Y.-H. and Lin J.-S. (2012). MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene 31, 3357-3369. 10.1038/onc.2011.500 [DOI] [PubMed] [Google Scholar]
- Iwata J., Parada C. and Chai Y. (2011). The mechanism of TGF-beta signaling during palate development. Oral Dis. 17, 733-744. 10.1111/j.1601-0825.2011.01806.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J., Tung L., Urata M., Hacia J. G., Pelikan R., Suzuki A., Ramenzoni L., Chaudhry O., Parada C., Sanchez-Lara P. A. et al. (2012). Fibroblast growth factor 9 (FGF9)-pituitary homeobox 2 (PITX2) pathway mediates transforming growth factor β (TGFβ) signaling to regulate cell proliferation in palatal mesenchyme during mouse palatogenesis. J. Biol. Chem. 287, 2353-2363. 10.1074/jbc.M111.280974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer N. V., Leung S. W. and Semenza G. L. (1998). The human hypoxia-inducible factor 1alpha gene: HIF1A structure and evolutionary conservation. Genomics 52, 159-165. 10.1006/geno.1998.5416 [DOI] [PubMed] [Google Scholar]
- Jia P. and Zhao Z. (2012). Personalized pathway enrichment map of putative cancer genes from next generation sequencing data. PLoS ONE 7, e37595 10.1371/journal.pone.0037595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P., Zheng S., Long J., Zheng W. and Zhao Z. (2011a). dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 27, 95-102. 10.1093/bioinformatics/btq615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P., Wang L., Meltzer H. Y. and Zhao Z. (2011b). Pathway-based analysis of GWAS datasets: effective but caution required. Int. J. Neuropsychopharmacol. 14, 567-572. 10.1017/S1461145710001446 [DOI] [PubMed] [Google Scholar]
- Jiang W., Mitra R., Lin C.-C., Wang Q., Cheng F. and Zhao Z. (2016). Systematic dissection of dysregulated transcription factor–mirna feed-forward loops across tumor types. Brief. Bioinform. 17, 996-1008. 10.1093/bib/bbv107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.-Z., Tan M., Warner D. R., Darling D. S., Higashi Y., Gridley T. and Ding J. (2010). Mesenchymal cell remodeling during mouse secondary palate reorientation. Dev. Dyn. 239, 2110-2117. 10.1002/dvdy.22339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M. and Haussler D. (2002). The human genome browser at UCSC. Genome Res. 12, 996-1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M., Iovino N., Unnerstall U., Gaul U. and Segal E. (2007). The role of site accessibility in microRNA target recognition. Nat. Genet. 39, 1278-1284. 10.1038/ng2135 [DOI] [PubMed] [Google Scholar]
- Kim S., Choi J. Y., Seok H. J., Park M.-J., Chung H. Y. and Bae I. H. (2019). miR-340-5p suppresses aggressiveness in glioblastoma multiforme by targeting Bcl-w and Sox2. Mol. Ther. Nucleic Acids 7, 245-255. 10.1016/j.omtn.2019.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalovsky D., Uche O. U., Eladad S., Hobbs R. M., Yi W., Alonzo E., Chua K., Eidson M., Kim H.-J., Im J. S. et al. (2008). The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat. Immunol. 9, 1055-1064. 10.1038/ni.1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küchler E. C., Silva L. A. D., Nelson-Filho P., Saboia T. M., Rentschler A. M., Granjeiro J. M., Oliveira D., Tannure P. N., Silva R. A. D., Antunes L. S. et al. (2018). Assessing the association between hypoxia during craniofacial development and oral clefts. J. Appl. Oral. Sci. 26, e20170234 10.1590/1678-7757-2017-0234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y., Ryan R. C., Zhang Z., Bullard S. A., Bush J. O., Maltby K. M., Lidral A. C. and Jiang R. (2006). Expression of Wnt9b and activation of canonical Wnt signaling during midfacial morphogenesis in mice. Dev. Dyn. 235, 1448-1454. 10.1002/dvdy.20723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman D. S. (1997). Transcription factors: an overview. Int. J. Biochem. Cell Biol. 29, 1305-1312. 10.1016/S1357-2725(97)00085-X [DOI] [PubMed] [Google Scholar]
- Li Y., An H., Pang J., Huang L., Li J. and Liu L. (2015). MicroRNA profiling identifies miR-129-5p as a regulator of EMT in tubular epithelial cells. Int. J. Clin. Exp. Med. 8, 20610-20616. [PMC free article] [PubMed] [Google Scholar]
- Li A., Qin G., Suzuki A., Gajera M., Iwata J., Jia P. and Zhao Z. (2019). Network-based identification of critical regulators as putative drivers of human cleft lip. BMC Med. Genomics 12, 16 10.1186/s12920-018-0458-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A., Jia P., Mallik S., Fei R., Yoshioka H., Suzuki A., Iwata J. and Zhao Z. (2020). Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA-TF-gene network approach in humans and mice. Brief. Bioinform. 21, 1465-1478. 10.1093/bib/bbz082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Wang J., Jaehnig E. J., Shi Z. and Zhang B. (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199-W205. 10.1093/nar/gkz401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Shi Z., Yu Z. and He Z. (2018). LncRNA HIF1A-AS2 positively affects the progression and EMT formation of colorectal cancer through regulating miR-129-5p and DNMT3A. Biomed. Pharmacother. 98, 433-439. 10.1016/j.biopha.2017.12.058 [DOI] [PubMed] [Google Scholar]
- Lin Y., Duan Z., Xu F., Zhang J., Shulgina M. V. and Li F. (2017). Construction and analysis of the transcription factor-microRNA co-regulatory network response to Mycobacterium tuberculosis: a view from the blood. Am. J. Transl. Res. 9, 1962-1976. [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y. D., Ding X., Du H. M., Wu Y. N., Li H. Q., Wu H. M. and Zhang X. M. (2019). FOXM1 is a novel predictor of recurrence in patients with oral squamous cell carcinoma associated with an increase in epithelial-mesenchymal transition. Mol. Med. Report. 19, 4101-4108. 10.3892/mmr.2019.10094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao A.-P., Ishizuka I. E., Kasal D. N., Mandal M. and Bendelac A. (2017). A shared Runx1-bound Zbtb16 enhancer directs innate and innate-like lymphoid lineage development. Nat. Commun. 8, 863 10.1038/s41467-017-00882-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10-12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Matys V., Kel-Margoulis O. V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. et al. (2006). TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108-D110. 10.1093/nar/gkj143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A. P., Champion J. E., McMahon J. A. and Sukhatme V. P. (1990). Developmental expression of the putative transcription factor Egr-1 suggests that Egr-1 and c-fos are coregulated in some tissues. Development 108, 281-287. [DOI] [PubMed] [Google Scholar]
- Meng L., Bian Z., Torensma R. and Von den Hoff J. W. (2009). Biological mechanisms in palatogenesis and cleft palate. J. Dent. Res. 88, 22-33. 10.1177/0022034508327868 [DOI] [PubMed] [Google Scholar]
- Millicovsky G. and Johnston M. C. (1981). Hyperoxia and hypoxia in pregnancy: simple experimental manipulation alters the incidence of cleft lip and palate in CL/Fr mice. Proc. Natl. Acad. Sci. USA 78, 5722-5723. 10.1073/pnas.78.9.5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minet E., Michel G., Remacle J. and Michiels C. (2000). Role of HIF-1 as a transcription factor involved in embryonic development, cancer progression and apoptosis (review). Int. J. Mol. Med. 5, 253-259. 10.3892/ijmm.5.3.253 [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A. and Schlaepfer D. D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56-68. 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- Nagaoka R., Okuhara S., Sato Y., Amagasa T. and Iseki S. (2012). Effects of embryonic hypoxia on lip formation. Birth Defects Res. A Clin. Mol. Teratol 94, 215-222. 10.1002/bdra.23000 [DOI] [PubMed] [Google Scholar]
- Parada C. and Chai Y. (2012). Roles of BMP signaling pathway in lip and palate development. Front. Oral Biol. 16, 60-70. 10.1159/000337617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelikan R. C., Iwata J., Suzuki A., Chai Y. and Hacia J. G. (2013). Identification of candidate downstream targets of TGFbeta signaling during palate development by genome-wide transcript profiling. J. Cell. Biochem. 114, 796-807. 10.1002/jcb.24417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter A. S. and Potter S. S. (2015). Molecular anatomy of palate development. PLoS ONE 10, e0132662 10.1371/journal.pone.0132662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds K., Kumari P., Sepulveda Rincon L., Gu R., Ji Y., Kumar S. and Zhou C. J. (2019). Wnt signaling in orofacial clefts: crosstalk, pathogenesis and models. Dis. Model. Mech. 12, dmm037051 10.1242/dmm.037051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosero Salazar D. H., Carvajal Monroy P. L., Wagener F. and Von den Hoff J. W. (2020). Orofacial muscles: embryonic development and regeneration after injury. J. Dent. Res. 99, 125-132. 10.1177/0022034519883673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. (1998). Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 8, 588-594. 10.1016/S0959-437X(98)80016-6 [DOI] [PubMed] [Google Scholar]
- Semenza G. L., Agani F., Booth G., Forsythe J., Iyer N., Jiang B.-H., Leung S., Roe R., Wiener C. and Yu A. (1997). Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 51, 553-555. 10.1038/ki.1997.77 [DOI] [PubMed] [Google Scholar]
- Shalgi R., Lieber D., Oren M. and Pilpel Y. (2007). Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 3, e131 10.1371/journal.pcbi.0030131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B. and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498-2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. and Dilworth F. J. (2013). Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 280, 3991-4003. 10.1111/febs.12188 [DOI] [PubMed] [Google Scholar]
- Song L. and Crawford G. E. (2010). DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2, pdb-prot5384 10.1101/pdb.prot5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., da Fontoura C. S. G., Moreno M., Holton N. E., Sweat M., Sweat Y., Lee M. K., Arbon J., Bidlack F. B., Thedens D. R. et al. (2018). FoxO6 regulates Hippo signaling and growth of the craniofacial complex. PLoS Genet. 14, e1007675 10.1371/journal.pgen.1007675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Gong X., Purow B. and Zhao Z. (2012). Uncovering MicroRNA and transcription factor mediated regulatory networks in glioblastoma. PLoS Comput. Biol. 8, e1002488 10.1371/journal.pcbi.1002488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Sangani D. R., Ansari A. and Iwata J. (2016). Molecular mechanisms of midfacial developmental defects. Dev. Dyn. 245, 276-293. 10.1002/dvdy.24368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Abdallah N., Gajera M., Jun G., Jia P., Zhao Z. and Iwata J. (2018a). Genes and microRNAs associated with mouse cleft palate: a systematic review and bioinformatics analysis. Mech. Dev. 150, 21-27. 10.1016/j.mod.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Jun G., Abdallah N., Gajera M. and Iwata J. (2018b). Gene datasets associated with mouse cleft palate. Data Brief 18, 655-673. 10.1016/j.dib.2018.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Li A., Gajera M., Abdallah N., Zhang M., Zhao Z. and Iwata J. (2019a). MicroRNA-374a,-4680, and-133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC Med. Genomics 12, 93 10.1186/s12920-019-0546-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Yoshioka H., Summakia D., Desai N. G., Jun G., Jia P., Loose D. S., Ogata K., Gajera M. V., Zhao Z. et al. (2019b). MicroRNA-124-3p suppresses mouse lip mesenchymal cell proliferation through the regulation of genes associated with cleft lip in the mouse. BMC Genomics 20, 852 10.1186/s12864-019-6238-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W. W., Tong G. H. and Liu Y. (2018). Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 53, 469-476. 10.3892/ijo.2018.4417 [DOI] [PubMed] [Google Scholar]
- Tsang J., Zhu J. and van Oudenaarden A. (2007). MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell 26, 753-767. 10.1016/j.molcel.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason H. A., Zhou H., Kouwenhoven E. N., Dotto G.-P., Restivo G., Nguyen B.-C., Little H., Dixon M. J., van Bokhoven H. and Dixon J. (2010). Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 120, 1561-1569. 10.1172/JCI40266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi M. P., Agarwal P., Brown C., McCulley D. J., Schwarz J. J. and Black B. L. (2007). The transcription factor MEF2C is required for craniofacial development. Dev. Cell 12, 645-652. 10.1016/j.devcel.2007.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Yu H., Zhao Z. and Jia P. (2015). EW_dmGWAS: Edge-Weighted dense module search for genome-wide association studies and gene expression profiles. Bioinformatics 31, 2591-2594. 10.1093/bioinformatics/btv150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. and Yu J. (2018). MiR-129-5p suppresses gastric cancer cell invasion and proliferation by inhibiting COL1A1. Biochem. Cell Biol. 96, 19-25. 10.1139/bcb-2016-0254 [DOI] [PubMed] [Google Scholar]
- Watkins S. E., Meyer R. E., Strauss R. P. and Aylsworth A. S. (2014). Classification, epidemiology, and genetics of orofacial clefts. Clin. Plast. Surg. 41, 149-163. 10.1016/j.cps.2013.12.003 [DOI] [PubMed] [Google Scholar]
- Wozniak M. A., Modzelewska K., Kwong L. and Keely P. J. (2004). Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 1692, 103-119. 10.1016/j.bbamcr.2004.04.007 [DOI] [PubMed] [Google Scholar]
- Xiao L., Zhou X., Liu F., Hu C., Zhu X., Luo Y., Wang M., Xu X., Yang S., Kanwar Y. S. et al. (2015). MicroRNA-129-5p modulates epithelial-to-mesenchymal transition by targeting SIP1 and SOX4 during peritoneal dialysis. Lab. Invest. 95, 817-832. 10.1038/labinvest.2015.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim I. H. and Kocak N. (2018). Studying the C1772T polymorphism of Hif-1alpha and TGF-beta3 IVS5+104 A/G polymorphism in children with congenital non-syndromic neural tube defects and their mothers. Cell. Mol. Biol. (Noisy-Le-Grand) 64, 90-93. 10.14715/cmb/2017.64.15.15 [DOI] [PubMed] [Google Scholar]
- Yin Y., Wang F. and Ornitz D. M. (2011). Mesothelial-and epithelial-derived FGF9 have distinct functions in the regulation of lung development. Development 138, 3169-3177. 10.1242/dev.065110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D., Lim J., Wang X., Liang F. and Xiao G. (2017). Enhanced construction of gene regulatory networks using hub gene information. BMC Bioinformatics 18, 186 10.1186/s12859-017-1576-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.