

Abstract
A series of halo-substituted mixed ester/amide-based analogues 4a-l have been prepared as jack bean urease inhibitor, which showed good to excellent inhibition of enzyme activity. The role of halo-substituted benzoyl moieties and alkyl substituted anilines in urease inhibitory kinetics was also investigated. The alkyl-substituted anilines 1a–b reacted with chloroacetyl chloride to afford intermediates 2a-b, which were then reacted with different halo-substituted benzoic acids 3a–f to prepare the title compounds 4a-l. The chemical structures of final products 4a-l were ascertained by FTIR, 1H NMR, 13C NMR, and mass spectra. The compound 4b showed remarkable activity with IC501.6 ± 0.2 nM, better than the standard thiourea having IC50472.1 ± 135.1 nM. The 2-chloro-substituted phenyl ring on one side of compound 4b and 4-isopropyl-substituted benzene on the other side play an essential role in inhibition of urease activity. Lineweaver–Burk plots (kinetics study) indicated about 4b derivative as a mixed type of inhibitor. The virtual screening performed against urease enzyme (PDBID 4H9M) showed that compounds 4b and 4e have binding energies of −7.8 and −7.9 Kcal/mol, respectively. Based upon our results, it was found that derivative 4b is a highly potent urease inhibitor, better than the standard thiourea.
1. Introduction
Urease (EC.3.5.1.5) is an enzyme of the amidohydrolase and phosphotriesterase family with nickel atoms present in their active binding sites. They catalyze hydrolysis of urea into carbamic acids which then cleaved into carbon dioxide and ammonia (Figure 1) [1–4] which in turn increased the pH. The increased level of urease enzyme is also associated with serious health problems like stomach cancer, peptic ulceration, and pyelonephritis [5–7].
Figure 1.
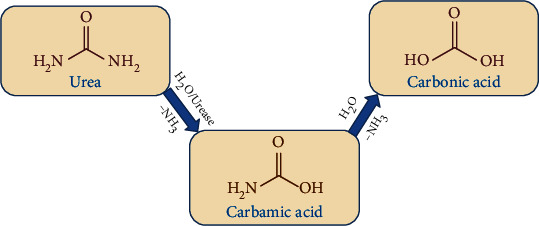
Hydrolysis of urea by urease enzyme.
The bacterial ureases increase the rate of urea hydrolysis which is associated with different biological disorders like gastric cancer, urinary tract infections, liver cirrhosis, liver inflammation, and hepatic coma [8]. The Helicobacter pylori and other bacteria live at lower pH of the stomach by creating a shielding ammonium cloud during colonization [9–14]. It is statistically calculated that nearly half of the world's population suffer due to Helicobacter pylori [15–18]. Some other bacteria, mainly Proteus, Klebsiella, Pseudomonas, and Staphylococcus species, lead to kidney stones, often called infectious stones, such as struvite (magnesium ammonium phosphate) [NH4MgPO4.6H2O)] and carbonate apatite [Ca10(PO4)6CO3] [19, 20]. The congenital problems associated with a well-known urease inhibitor hydroxamic acids limit its clinical utility. The need of new antibiotics is also necessary due to the emergence of resistance in Helicobacter pylori against metronidazole, clarithromycin, and levofloxacin [21, 22].
The already reported urease inhibitors are urea derivatives, hydroxamic acids, heavy metal ions, quinones, polyphenols, and organosulfur compounds. Thiourea and hydroxyurea bind with the active binding site of target enzyme in the same way as urea binds with urease. It has been studied that amides and hydroxyl-substituted acids display antimicrobial activity [23]. It is expected that the mixed ester and amide functionalities may assist in designing more potent urease inhibitors. Our previous work focusses on drug development through enzyme-based assay [24, 25]. Hence, the compounds 4a–l were designed and synthesized having ester amide linkages along with halo-substituted benzoyl moiety for their improved urease inhibitory activity by creating secondary interactions with the protein part of the enzyme.
2. Materials and Methods
2.1. Chemistry
Reagents and chemicals were used as received without further purification. A digital Gallen Kamp (SANYO) apparatus was used to record the melting points. The Perkin Elmer spectrophotometer was used to record FTIR spectra by using the KBr pellet technique and is expressed in centimeters. The 1H NMR (300 MHz) and 13C NMR (75 MHz) (δ-ppm) analyses were done using a Bruker AM-300 spectrometer. The mass spectra were recorded on a GC–MS spectrometer by using the 6890 Network GC system (Agilent Technologies).
2.1.1. Synthesis of Compounds 4a–l
In the first step, 1a (4-isopropyl aniline) and 1b (2-methyl-5-isopropyl aniline) were reacted with chloroacetyl chloride to form 2a and 2b, respectively. In the second step, 2a and 2b were also made to react with different halo-substituted benzoic acids 3a–f to form 4a–l, the final products.
2.1.2. Synthesis of Chloroacetyl Alkylated Aniline Derivative 2a–b
4-Isopropylaniline 1a (0.3 g, 0.01 mol) and 5-isopropyl-2-methylaniline 1b (0.3 g, 0.01 mol) were mixed in a reaction flask; then, triethylamine (0.01 mol) was added in dichloromethane (30 mL). To this reaction mixture, 0.01 mol of chloroacetyl chloride was slowly added with constant stirring for 1 h at 0–5°C. After completion of the reaction, mixture was washed by 1% HCl (2 × 50.0 mL), 1% sodium hydroxide solution (2 × 50.0 mL), and, finally, 1% sodium chloride solution (2 × 50.0 mL) and then dried by magnesium sulfate to get the intermediate products 2a–b. These products were purified by silica gel column chromatography by using n-hexane and ethyl acetate as mobile phase.
2.1.3. Synthesis of Compounds 4a–l
The intermediates 2a–b (0.01 mol) were reacted with 0.01 mol of different halogen-substituted benzoic acids 3a–f in the presence of equimolar amounts of triethylamine and KI in 10 mL DMF; mixture was stirred at 25°C for 8–10 h. The mixture was poured into ice-cold water, and product was extracted in ethyl acetate (3 × 25 mL). The product was washed with 1% HCl (2 × 50.0 mL), 1% NaOH solution (2 × 50.0 mL), and 1% NaCl solution (2 × 50 mL), and then, crude products were purified by silica gel column chromatography (Scheme 1).
2.1.4. 2-((4-Isopropylphenyl)amino)-2-oxoethyl 2-Chlorobenzoate 4a
Yield: 60%; m.p 94–96°C; Rf = 0.38 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3316 (N–H), 2965 (sp3 C–H aromatic), 2850 (C–H saturated), 1742 (C=O aromatic ester), 1685 (C=O amide), 1588 (C=C aromatic), 1237 (C–O ester), 746 (C–Cl); 1H NMR (CDCl3): 7.99 (1H, s, N–H), 7.58 (d, J = 4.2 Hz, 1H), 7.54 (1H, m,), 7.49 (2H, d, J = 8.4 Hz), 7.42 (1H, m), 7.23 (2H, d, J = 8.5 Hz), 4.98 (2H, s, methylene), 2.90 (1H, sep), 1.25 (6H, d, J = 6.9 Hz alkyl); 13C NMR: 164.53 (C=O aromatic ester), 164.32 (C=O amide), 145.79, 134.46, 133.63, 133.17, 132.57, 131.33, 128.90, 127.27, 127.06, 120.18 (Aromatic), 64.20, 33.65, 24.02 (Aliphatic); EI-MS (m/z, %): 331 (M+, 40), 318 (14), 296 (0.6), 258 (1.1), 197 (6), 139 (100), 136 (56), 119(64), 111 (36), 91 (21), 75 (18), 50 (8).
2.1.5. 2-((4-Isopropylphenyl)amino)-2-oxoethyl 3-Chlorobenzoate 4b
Yield: 68%; m.p 96–98°C; Rf = 0.41 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3257 (N–H), 3117 (unsaturated C-H), 2963 (saturated C–H), 1739 (C=O aromatic ester), 1670 (C=O amide), 1537 (C=C aromatic), 1275 (C–O ester), 759 (C–Cl); 1H NMR (CDCl3): 8.18 (1H, s), 8.01 (1H, dd, J1 = 5.4 Hz, J2 = 1.2 Hz), 7.72 (1H, s, N–H), 7.64 (1H, dd, J1 = 7.3 Hz, J2 = 0.9 Hz), 7.49 (2H, d, J = 7.8 Hz), 7.44 (1H, m), 7.23 (2H, d, J = 8.4 Hz), 4.97 (2H, s), 2.91 (1H, sep, J = 6.9 Hz), 1.25 (6H, d, J = 6.9 Hz); 13C NMR: 164.68 (C=O aromatic ester), 164.17 (C=O amide), 145.99, 134.99, 134.18, 133.90, 130.60, 130.09, 129.89, 127.94, 127.05, 120.55 (Aromatic), 63.95, 33.64, 23.98 (Aliphatic); EI-MS (m/z, %): 331 (M+, 40), 318 (10), 258 (1.1) 197 (9), 139 (100), 136 (74), 119 (88), 111 (58), 91 (32), 75 (23), 50 (8).
2.1.6. 2-((4-Isopropylphenyl)amino)-2-oxoethyl 3,4-Dichlorobenzoate 4c
Yield: 65%; m.p 107–108°C; Rf = 0.41 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3279 (N–H stretch), 3066 (unsaturated C–H), 2962 (saturated C–H), 1724 (C=O aromatic ester), 1658 (C=O amide), 1541 (C=C aromatic), 1232 (C–O ester), 782 (C–Cl); 1H NMR (DMSO): 10.11 (1H, s, N–H), 8.16 (1H, s), 7.94 (1H, d, J = 8 Hz), 7.84 (1H, d, J = 8 Hz), 7.46 (2H, d, J = 8 Hz), 7.15 (2H, d, J = 9 Hz), 4.91 (2H, s), 2.83 (1H, sep, J = 8 Hz), 1.14 (6H, d, J = 8 Hz); 13C NMR: 165.17 (C=O aromatic ester), 164.08 (C=O amide), 144.16, 137.05, 136.52, 132.24, 131.78, 130.17, 129.90, 126.95, 119.93, 110.97 (Aromatic), 64.03, 33.30, 24.30 (Aliphatic); EI-MS (m/z, %): 365 (M+, 37), 350 (20), 203 (4), 173 (100), 145 (44), 119 (98), 135 (68), 157 (33), 91 (42), 65 (6).
2.1.7. 2-((4-Isopropylphenyl)amino)-2-oxoethyl 2-Bromobenzoate 4d
Yield: 70%; m.p 89–90°C; Rf = 0.52 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3307 (N–H), 2922 (unsaturated C–H), 2853 (saturated C–H), 1742 (C=O aromatic ester), 1685 (C=O amide), 1537 (C=C aromatic), 1106 (C–O ester), 743 (C–Br); 1H NMR (CDCl3): 8.30 (1H, s, N–H), 7.93 (1H, m), 7.75 (1H, m), 7.52 (2H, d, J = 8.4 Hz), 7.47 (1H, m), 7.45 (1H, m), 7.23 (2H, d, J = 8.5 Hz), 4.98 (2H, s), 2.91 (1H, sep, J = 6.2 Hz), 1.25 (6H, d, J = 9.0 Hz); 13C NMR: 164.79 (C=O ester), 164.51 (C=O amide), 145.81, 134.51, 134.44, 133.54, 132.41, 131.19, 127.76, 127.06, 121.18, 120.26 (Aromatic), 64.27, 33.65, 24.02 (Aliphatic); EI-MS (m/z, %): 375 (M+, 50), 355 (16), 243 (12), 183 (100), 157 (33), 135 (90), 119 (64), 91 (48), 75 (32), 50 (14).
2.1.8. 2-((4-Isopropylphenyl)amino)-2-oxoethyl 3-Bromobenzoate 4e
Yield: 70%; m.p 91–92°C; Rf = 0.50 (C6H14/CH3CO2C2H5 3:1); FTIR ѵmax cm−1: 3256 (N–H), 2964 (Ar–H), 2877 (sp3 C–H), 1739 (C=O aromatic ester), 1669 (C=O amide), 1536 (C=C aromatic), 1250 (C–O ester), 729 (C–Br); 1H NMR (CDCl3): 8.26 (1H, s), 8.07 (1H, dd, J1 = 7.8 Hz, J2 = 1.2 Hz), 7.81 (1H, dd, J1 = 8.1 Hz, J2 = 0.9 Hz), 7.70 (1H, s, N–H), 7.47 (2H, d, J = 8.4 Hz), 7.40 (1H, m), 7.23 (2H, d, J = 8.4 Hz), 4.99 (2H, s), 2.91 (1H, sep, J = 6.9 Hz), 1.25 (6H, d, J = 6.9 Hz); 13C NMR: 165.28 (C=O aromatic ester), 164.60 (C=O amide), 144.13, 136.78, 136.56, 132.25, 13.60, 130.09, 131.89, 131.58, 128.89, 126.95, 122.29, 119.90 (Aromatic), 63.89, 33.30, 24.37 (Aliphatic); EI-MS (m/z, %): 375 (M+, 38), 355 (16), 243 (12), 157 (33), 135 (100), 119 (98), 91 (48), 75 (32), 50 (14).
2.1.9. 2-((4-Isopropylphenyl)amino)-2-oxoethyl 2-Iodobenzoate 4f
Yield: 70%; m.p 91–92°C; Rf = 0.50 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3269 (N–H stretch), 3133 (Ar–H), 2953 (sp3 C–H), 1734 (C=O aromatic ester), 1671 (C=O amide), 1583 (C=C aromatic), 1241 (C–O ester), 741 (C–I); 1H NMR (DMSO): 10.03 (1H, s, N–H), 8.03 (1H, d, J = 8), 7.87 (1H, m), 7.70 (1H, m) 7.54 (1H, m), 7.47 (2H, d, J = 8 Hz), 7.16 (2H, d, J = 8 Hz), 4.88 (2H, s), 2.80 (1H, sep, J = 8 Hz), 1.92 (6H, d, J = 9.0 Hz); 13C NMR: 166.20 (C=O aromatic ester), 165.23 (C=O amide), 144.90, 141.35, 136.61, 134.91, 133.83, 132.02, 131.45, 129.09, 128.73, 126.96 ((Aromatic)), 63.84, 33.30, 24.38 (Aliphatic); EI-MS (m/z, %): 423 (M+, 64), 289 (8), 231 (100), 261 (6), 203 (38), 146 (9), 135 (52), 119 (64), 91 (46), 75 (35), 50 (12).
2.1.10. 2-((5-Isopropyl-2-methyl phenyl) Amino)-2-oxoethyl 2-Chloro Benzoate 4g
Yield: 65%; m.p 115–116°C; Rf = 0.48 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3256 (N–H), 2990 (unsaturated C-H), 2878 (saturated C–H), 1745 (C=O aromatic ester), 1685 (C=O amide), 1541 (C=C aromatic), 1124 (C–O ester), 747 (C–Cl); 1H NMR (CDCl3): 8.02 (1H, s, N–H), 7.99 (1H, d, J = 8.4 Hz), 7.72 (1H, s), 7.55 (1H, d, J = 4.5 Hz), 7.53 (1H, m), 7.48 (1H, m), 7.15 (1H, d, J = 7.8 Hz), 7.02 (1H, d, J = 6.3 Hz), 5.02 (2H, s), 2.91 (1H, sep, J = 10.2 Hz), 2.24 (3H, s), 1.25 (6H, d, J = 8.5 Hz); 13C NMR: 164.80 (C=O aromatic ester), 164.38 (C=O amide), 147.48, 133.60, 132.24, 131.41, 131.36, 130.91, 130.53, 128.82, 128.80, 127.13, 123.92, 121.67 (Aromatic), 64.34, 33.79, 23.99, 17.39 (Aliphatic); EI-MS (m/z, %): 345 (M+, 64), 206 (8), 176 (9), 148 (54), 139 (100), 111 (26), 75 (11), 50 (2).
2.1.11. 2-((5-Isopropyl-2-ethylphenyl) Amino)-2-oxoethyl 3-Chlorobenzoate 4h
Yield: 76%; m.p 119–120°C; Rf = 0.54 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3258 (N–H), 2957 (unsaturated C–H), 2869 (saturated C–H), 1727 (C=O aromatic ester), 1666 (C=O amide), 1575 (C=C aromatic), 1249 (C–O ester), 756 (C–Cl); 1H NMR (CDCl3): 8.11 (1H, s), 8.03 (1H, d, J = 7.8 Hz), 7.83 (1H, s), 7.75 (1H, s), 7.64 (1H, d, J = 8.4 Hz), 7.48 (1H, m), 7.15 (1H, d, J = 7.8 Hz), 7.03 (1H, d, J = 8.1 Hz), 5.02 (2H, s), 2.92 (1H, sep, J = 6.9 Hz), 2.25 (3H, s), 1.25 (6H, d, J = 6.9 Hz); 13C NMR: 165.64 (C=O aromatic ester), 164.68 (C=O amide), 146.48, 135.76, 133.93, 133.88, 131.72, 131.28, 130.66, 129.94, 129.45, 128.59, 124.04, 123.51 (Aromatic), 63.91, 33.40, 24.32, 17.28 (Aliphatic); EI-MS (m/z, %): 345 (M+, 12), 206 (8), 176 (100), 148 (8), 139 (76), 119 (8), 75 (20), 50 (6).
2.1.12. 2-((5-Isopropyl-2-methyl phenyl) Amino)-2-oxoethyl 2-Bromobenzoate 4i
Yield: 68%; m.p 112–113°C; Rf = 0.43 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3257 (N–H), 2958 (unsaturated C–H), 2870 (saturated C–H), 1741 (C=O aromatic ester), 1666 (C=O amide), 1541 (C=C aromatic), 1242 (C–O ester), 743 (C–Br); 1H NMR (CDCl3): 8.02 (1H, s, N–H), 7.99 (1H, dd, J1 = 6.3 Hz, J2 = 2.7 Hz), 7.75 (1H, dd, J1 = 7.2 Hz, J2 = 2.7 Hz), 7.47 (1H, m), 7.43 (1H, m), 7.15 (1H, d, J = 7.5 Hz), 7.03 (1H, d, J = 8.5 Hz), 5.01 (2H, s), 2.91 (1H, sep, J = 7.5 Hz), 2.24 (3H, s), 1.25 (6H, d, J = 7.2 Hz); 13C NMR: 165.53 (C=O aromatic ester), 164.07 (C=O amide), 146.70, 137.02, 135.73, 132.19, 131.71, 131.59, 130.67, 130.17, 129.96, 124.09, 123.56, 110.94 (Aromatic), 64.05, 33.40, 24.32, 17.81 (Aliphatic); EI-MS (m/z, %): 391 (M+, 35), 216 (16), 241 (6), 148 (96), 105 (24), 76 (23), 50 (10).
2.1.13. 2-((5-Isopropyl-2-methyl phenyl) Amino)-2-oxoethyl 3-Bromobenzoate 4j
Yield: 72%; m.p 116-118°C; Rf = 0.62 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3254 (N–H), 2956 (unsaturated C–H), 2922 (saturated C–H), 1735 (C=O ester), 1667 (C=O amide), 1569 (Ar–C=C), 1224 (C–O ester), 740 (C–Br); 1H NMR (CDCl3): 8.27 (1H, s,), 8.06 (1H, dd, J1 = 7.8 Hz, J2 = 0.9 Hz), 7.83 (1H, dd, J1 = 6.6 Hz, J2 = 0.9 Hz), 7.78 (1H, s), 7.76 (1H, s, N–H), 7.42 (1H, m), 7.15 (1H, d, J = 7.8 Hz), 7.02 (1H, d, J = 7.5 Hz), 5.01 (2H, s), 2.92 (1H, sep, J = 6.9 Hz), 2.25 (3H, s), 1.26 (6H, d, J = 6.9 Hz); 13C NMR: 165.56 (C=O aromatic ester), 165.37 (C=O amide), 146.68, 135.79, 134.59, 134.01, 132.05, 131.70, 130.68, 129.77, 128.29, 123.95, 123.34, 121.14 (Aromatic), 63.86, 33.41, 24.33, 17.85 (Aliphatic); EI-MS (m/z, %): 389 (M+, 26), 216 (16), 241 (6), 149 (100), 148 (96), 105 (24), 76 (23), 50 (10).
2.1.14. 2-((5-Isopropyl-2-methyl phenyl) Amino)-2-oxoethyl 3,4-Dichlorobenzoate 4k
Yield: 76%; m.p 115–116°C; Rf = 0.38 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3264 (N–H), 2926 (unsaturated C–H), 2850 (saturated C–H), 1741 (C=O aromatic ester), 1667 (C=O amide), 1557 (C=C aromatic), 1240 (C–O ester), 768 (C–Cl); 1H NMR (DMSO): 9.54 (1H, s, N–H), 8.16 (1H, s), 8.04 (1H, d, J = 9), 7.90 (1H, d, J = 8 Hz), 7.52 (1H, d, J = 8 Hz), 7.23 (1H, s), 7.12 (1H, d, J = 8 Hz), 4.95 (2H, s), 2.82 (1H, sep, J = 8 Hz), 2.14 (3H, s), 1.15 (6H, d, J = 4 Hz); 13C NMR: 165.64 (C=O aromatic ester), 164.58 (C=O amide), 146.68, 136.75, 135.76, 132.33, 131.90, 131.50, 130.66, 129.92, 128.94, 124.03, 123.49, 122.26 (Aromatic), 63.91, 33.40, 24.32, 17.28 (Aliphatic); EI-MS (m/z, %): 379 (M+, 29), 231 (8), 206 (9), 173 (100), 149 (88), 119 (7), 135 (68), 157 (33), 91 (7), 65 (5).
2.1.15. 2-((5-Isopropyl-2-methyl phenyl) Amino)-2-oxoethyl 2-Iodobenzoate 4l
Yield: 72%; m.p 99–101°C; Rf = 0.69 (C6H14/CH3CO2C2H5 3:1); FTIR νmax cm−1: 3256 (N–H), 2957 (unsaturated C–H), 2921 (saturated C–H), 1740 (C=O aromatic ester), 1663 (C=O amide), 1569 (C=C aromatic), 1221 (C–O ester), 739 (C–I); 1H NMR (DMSO): 9.54 (1H, s, N–H), 8.03 (1H, d, J = 8 Hz), 7.89 (1H, d, J = 8 Hz), 7.54 (1H, m), 7.30 (1H, m), 7.25 (1H, s), 7.10 (1H, d, J = 8 Hz), 6.96 (1H, d, J = 8 Hz), 4.92 (2H, s), 2.76 (1H, sep, J = 8 Hz), 2.15 (3H, s), 1.14 (6H, d, J = 8 Hz); 13C NMR: 166.12 (C=O aromatic ester), 165.60 (C=O amide), 146.68, 141.37, 135.80, 134.82, 133.84, 131.53, 130.68, 129.78, 128.69, 123.35, 110.96 (Aromatic), 63.58, 33.41, 24.34, 17.98 (Aliphatic); EI-MS (m/z, %): 423 (M+, 47), 289 (5), 261 (2), 231 (100), 203 (29), 176 (10), 149 (46), 105 (23), 76 (31), 50 (8).
2.2. Jack Bean Urease Inhibition Assay
The inhibitory effects on urease activity were done based on the Weatherburn method, using the indophenol scheme by determining the amount of ammonia produced [26]. Briefly, 20 μL of jack bean urease enzyme (0.135 units) and 20 μL of the tested compounds were taken in 50 μL buffer and were incubated for 30 min. Temperature was kept at 37°C in a 96-well plate reader. The buffer solution was made by 0.01 molar K2HPO4/KH2PO4, 1 mM EDTA, 100 mM urea, and 0.01 M LiCl; pH was maintained at 7.0. Then, 50 μL of phenol reagent (1% w/v phenol and 0.005% w/v sodium nitroprusside) was added in 50 μL of alkali reagent in each well. The alkali reagent was prepared by 0.5% w/v NaOH and 0.1% NaOCl. Absorbance was measured at 625 nm using OPTIMax, a tunable microplate reader, after 10 min interval in a triplicate manner. The thiourea was used as a standard jack bean urease inhibitor. The data has been analyzed using software, GraphPad Prism, and statistical analysis (SD) was performed by using SigmaPlot software to get SD values of the bioassay results.
2.3. Molecular Docking Studies
The Protein Data Bank (PDB) was used to retrieve the structure of the jack bean urease enzyme PDBID 4H9M [27]. The University of California, San Francisco (UCSF) Chimera 1.10.1 tool was used to save the crystal structure of the enzyme in its stable conformation [28]. The MolProbity server (http://molprobity.biochem.duke.edu/) was used to calculate the Ramachandran plot values and the stereochemical properties of the urease structure [29] and ProtParam [30]. Hydrophobicity graphs of the targeted protein were generated using Discovery Studio 4.1 client tool (BIOVIA 5005 Wateridge Vista Drive, San Diego, CA 92121, USA) [31]. VANDAR 1.8, the online server, was used to predict protein architecture statistical percentage of receptor protein helices, beta sheets, coils, and turns [32]. The UCSF Chimera 1.10.1 tool was used to save the stable structures of the synthesized compounds (4a–l), which were electronically sketched using the ACD/Chem sketch tool. The molecular docking experiment was performed by the PyRx docking tool. Before going for the docking experiment, the active site of the target protein was analyzed from the PDB and was also compared with literature data [33]. The grid was generated based on binding pocket residues with appropriate coordinate values in XYZ dimensions, respectively. The center values of the grid box were set as center X_ = 1.48, center_Y = −55.22, and center_Z = −26.48. The size values of grid box were adjusted accordingly as X = 66.60, Y = 60.08, and Z = 56.84. Exhaustiveness value = 8 was set as default and was used to maximize the conformational analysis of binding. Each of the synthesized ligands was docked separately against the urease enzyme to predict the binding affinity. The predicted energies were compared on the lowest energies and were used to generate structure–activity relationship. Discovery Studio 2.1.0 was used to depict three-dimensional graphs for all of the docked ligand–protein complexes. The AutoDock tool has been used for determination of inhibition constant (Ki) for the most potent derivative 4b.
3. Results
3.1. Chemistry
The halo-substituted mixed ester/amide derivatives 4a-l were successfully synthesized by following the simple reaction route in good yields. The final products 4a-l were prepared by using our previously developed method with minor modifications [34]. Chloroacetyl chloride was reacted with alkyl-substituted anilines in the presence of dichloromethane as a solvent (Scheme 1). In this reaction, the amino group in compounds 1a–b displaced the chlorine atom attached with carbonyl carbon of the chloroacetyl chloride to give chloroacetyl derivatives 2a–b. According to the FTIR spectrum, compounds 4a–l showed a characteristic peak of amide carbonyl (C=O) at 1640–1685 cm−1, ester (C=O) at 1724–1745 cm−1, and secondary (N–H) stretching at 3250–3307 cm−1. The amide carbonyl absorption appeared at 1670 cm−1 in 2a and 1655 cm−1 in 2b and secondary (N–H) absorption at 3270 cm−1 in 2a and 3257 cm−1 in 2b in the FTIR spectrum, which confirmed the formation of 2a–b. The final products 4a–l were synthesized by nucleophilic substitution of chloroacetyl derivative 2a–b by halo-substituted benzoic acids 4a–f.
3.2. Biological Evaluation
The compounds 4a–l having ester and amide linkages were designed and synthesized as jack bean urease inhibitors. We have already synthesized some heterocyclic derivatives as jack bean urease inhibitors. The iminothiazoline-sulfonamide 1 and 4-aminocoumarine thiourea derivative 2 (Figure 2) showed good activity with IC50 values 58 nM and 6.5 nM, respectively [35, 36]. These derivatives possess iminothiazoline bearing sulfonamide moieties and thiourea bearing coumarin ring system. These derivatives exhibited good urease inhibitory activity especially compound 2 comparable to 4b reported in present studies.
Figure 2.
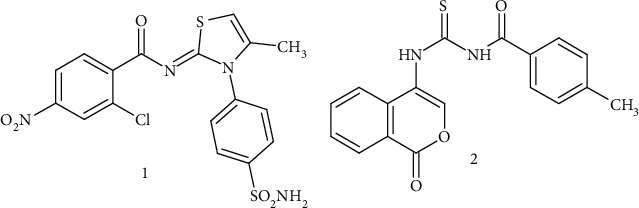
Structures of already reported urease inhibitors.
In the present work, halo-substituted benzoic acid moiety was attached to create secondary interactions with amino acid residues of urease enzyme, and we obtained excellent activity compared to standard thiourea. The derivatives 4a–l exhibited good to excellent urease inhibitory activity, compared to thiourea used as positive control. The bioassay results and mean standard errors n = 3 are presented in Table 1.
Table 1.
Urease inhibition (IC50) values of synthesized derivatives 4a–l.
| Compound | Urease (jack bean) inhibition IC50 ± SD (nM) |
|---|---|
| 4a | 57.1 ± 1.9 |
| 4b | 1.6 ± 0.2 |
| 4c | 18.2 ± 0.6 |
| 4d | 3.8 ± 0.2 |
| 4e | 62.7 ± 2.1 |
| 4f | 31.2 ± 1.1 |
| 4g | 3.8 ± 0.2 |
| 4h | 4.5 ± 0.2 |
| 4i | 21.1 ± 0.7 |
| 4j | 2.1 ± 0.1 |
| 4k | 50.1 ± 1.7 |
| 4l | 11.5 ± 3.9 |
| Thiourea | 472.1 ± 135.1 |
3.3. Kinetic Study
The derivative 4b was selected on the basis of its high activity for the determination of its kinetic mechanism on jack bean urease. The enzyme inhibition EI and enzyme-substrate inhibition ESI constants for compound 4b have been determined. The inhibitor concentration used in the kinetic experiments was 0.00, 0.0008, and 0.0016 μM while concentration of substrate was 0.0, 0.1, 0.2, 0.3, and 0.4 mM. The kinetic mechanism was determined by plotting of 1/V versus 1/[S] in the presence of inhibitor concentrations which gave a series of straight lines as shown in Figure 3. The results revealed intersection of lines in the second quadrant. The kinetic results showed that maximum velocity (Vmax) decreased with increasing Michaelis constant (Km) as a result of increasing concentration of 4b. The EI dissociation constant (Ki) and ESI dissociation constant (Ki′) for compound 4b have also been determined from Lineweaver-Burk plots. The stronger binding of derivative 4b with enzyme has been assured by a lower value of Ki than Ki′ which also confirmed the mixed-type behaviour with a Ki value of 0.0007 μM and a Ki′ value of 0.0018 μM, respectively (Table 2).
Figure 3.
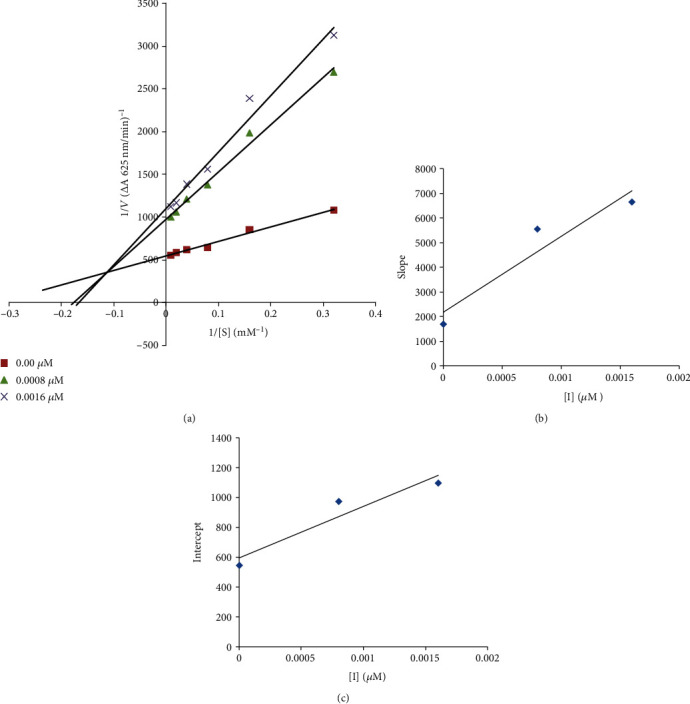
Enzyme inhibitory kinetic mechanism of the most potent derivative 4b by Lineweaver–Burk plots. 1/Vmax: reciprocal of maximum velocity; 1/[S]: reciprocal of substrate concentration.
Table 2.
Kinetic parameters of the jack bean urease for urea activity in the presence of different concentrations of compound 4b.
| Code | Dose (μM) | 1/Vmax (ΔA/min) | K m (mM) | Inhibition type | K i (μM) | K i′ (μM) |
|---|---|---|---|---|---|---|
| 4b | 0.00 | 0.001796 | 3.076 | Mixed inhibition | 0.0007 | 0.0018 |
| 0.0008 | 0.00099 | 5.263 | ||||
| 0.0016 | 0.00088 | 5.555 |
3.4. Molecular Docking Study
In order to show best conformations, derivatives (4a–l) were docked with target enzyme (PDBID 4H9M). The resulting complexes were observed based on the minimum energy values (kJmol−1) and pattern of bonding (hydrophobic and hydrophilic). Docking results justified good binding energies presented in Figure 4 with standard error—2.5 Kcal/mol. The binding energies showed the best conformations of the synthesized inhibitors in active binding sites of enzymatic protein. The derivatives 4e and 4b showed docking energies (−7.90 and 7.80 kJmol−1, respectively). Most of the docked molecules had closely related docking energies due to similar ester/amide functionalities.
Figure 4.
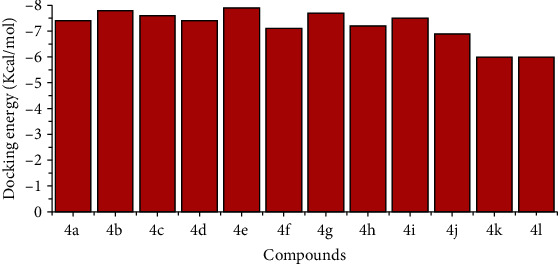
The docking energy values of synthesized compounds 4a-l.
3.4.1. Binding Pocket Analysis of Urease-Docked Complexes
Based on the in vitro and in silico results, the 4b-docked complex was further analyzed to learn about the structure–activity relationship (SAR) based on interactions. Docking energy values showed that docked molecules were bound in the active binding site of enzyme. The docking complexes of the most potent derivative 4b are shown in Figure 5.
Figure 5.
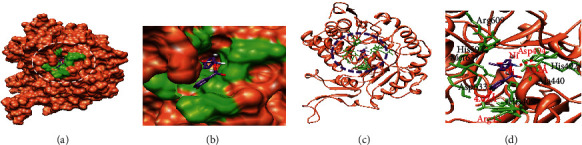
Binding interactions of compound 4b with the active binding site of urease PDBID 4H9M generated using Discovery Studio. (a–c) Show the three-dimensional docking of derivative 4b in a binding pocket. (d) Shows the two-dimensional ligand-protein interactions. The legend inset represents the type of interaction between the ligand atoms and the amino acid residues of the protein.
4. Discussion
The structures of final products 4a-l were ascertained by FTIR spectroscopy, 1H, 13C NMR, and mass spectrometry techniques. The 1H NMR spectra exhibited singlet of methylene (–CH2–) protons at 4.7–4.9 ppm which confirmed the presence of a methylene bridge in the synthesized compounds while peak of ester carbonyl (C=O) at 164.4–164.9 ppm in the 13C NMR spectra confirmed successful synthesis of compounds 4a–l. The mass spectral data of compounds 4a–l showed molecular ion peaks according to their molecular masses with base peaks due to cleavage of ester linkages and loss of alkoxy radical (RO) and base peaks of halo benzoyl cation X–Ph–CO+. The final products, 4d, 4e, 4i, and 4j, containing bromine showed M and M+2 peaks with a 1.1 : 1.0 ratio according to natural abundance of bromine isotopes 79Br and 81Br in 50.5%/49.5%. The 4a, 4b, 4c, 4g, 4h, and 4k containing chlorine atom showed M and M+2 peaks with a ratio of 3 : 1 according to their natural isotopic abundance 35Cl and 37Cl in 75%/25%.
The derivatives with an isopropyl-substituted phenyl ring on one side and a halogen-substituted phenyl ring on the other side showed good activity, especially compound 4b, which exhibited the most potent inhibition with IC50 1.6 nM (Figure 6). The effects on urease inhibitory activity of the chloro, bromo, iodo, and alkyl groups present on the benzene ring were also evaluated. The type and position of the functional groups at acyl core and aniline phenyl ring are the determining factors of urease inhibitory activities. The derivatives 4a and 4b possess the same functional groups -Cl but on a different position of the phenyl ring, thus having different activities. The derivative 4b with 3-chloro at acyl core and 4-isopropyl group at 4-position of aniline phenyl ring is more potent than 4a, which possesses 2-chloro at acyl core but the same aniline substitution pattern. Similarly, derivatives 4d and 4e have the same halogen -Br but on different positions of the benzoyl ring thus having different activities as 2-bromosubstituted derivative 4d is more active than 3-bromosubstituted analogue 4e. Both compounds 4d and 4e possess the same hydrophobic isopropyl substitution at 4-position of the aniline moiety. The presence of alkyl substitution at ortho- and meta-position of aniline also affects the urease inhibitory activity. It is evident from compounds 4g and 4h which possess the same -Cl at a different position of acyl core but having comparable activity. This is due to the presence of ortho- and meta-substituted aniline moiety.
Figure 6.
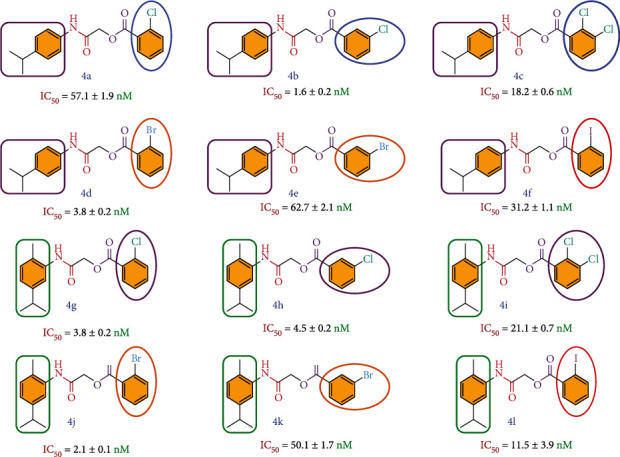
Structure–activity relationship with highlighted functional groups in 4a–l.
The derivatives 4j and 4k having the same bromofunctionalities at the ortho- and meta-positions of the benzoic acid moiety and with similar aniline substitution exhibit clearly different enzyme inhibitory activities. The derivative 4j is 25 times more active than the derivative 4k. The iodosubstituted derivatives in general possess intermediate enzyme inhibitory activity. We may infer that halogen-substituted regioisomers along with alkyl-substituted aniline showed remarkable differences in the inhibitory activity, as in the case of ortho- and meta-chlorosubstituted derivatives 4a and 4b with para-substituted aniline. The bromosubstituted regioisomers 4d and 4e with para-substituted aniline also possess different enzyme inhibitory activities. It has been evident from the results that the derivative 4b is more active as compared to the previously reported heterocyclic derivative 1. The urease inhibitory activity results revealed that halo-substituted benzoic acid moiety, along with ester amide linkages, played an important role in the enzyme inhibitory activity. Different hydrophilic and hydrophobic groups were also introduced at variable positions of the phenyl ring to check their role in the inhibitory activity of jack bean urease. Alkyl substitution on aniline also influences the inhibitory activity, and in most of the cases, increasing the number of alkyl chains also increased the inhibitory activity. This indicates that the presence of hydrophobic groups also affects the inhibitory activity, showing some sort of nonpolar interaction. It also confirmed that the substitution pattern of -Cl at acyl core is not only the determining factor for the activity but the presence of alkyl substitution at aniline also play very important role in urease inhibition. Though both -Cl and -Br are present in the same group and are electron withdrawing inductively, -Cl is more electron withdrawing as compared to -Br which may be the reason of difference in urease inhibitory activity in compounds 4b and 4e. Another important factor which may affect the activity is the size of these atoms. -Br is larger in size compared to -Cl which may affect the enzyme inhibition. The enzyme inhibitory kinetic results revealed that inhibitor 4b inhibits urease enzyme by the mixed type of inhibition.
The computational molecular docking results showed that docked complex of compound 4b interacts with target protein by two hydrogen bonds. The benzyl group directly interacted with Arg439 by hydrogen bonding at a bond distance of 4.02 Å. Phenyl rings with halo substitution also showed π-interactions with Arg439. The carbonyl group in 4b also forms hydrogen bond with Asp494 at 3.02 Å. The literature supports our docking results due to the presence of such functionalities [37–43]. The compound 4b binds with the target enzyme with binding interactions which indicated that the functional groups bind with the amino acid residues present in the active binding site. There are also some interactions between functional groups of 4b and amino acids which are present in the remote sites of the enzyme. This may suggest the allosteric binding of compound 4b with the target enzyme. The inhibition constant (Ki) value determined computationally by using the AutoDock tool was 0.916 μM while the Ki value determined by in vitro studies was 0.0007 μM. Based on the docking and bioassay results, it was found that 4b may act as a lead structure to discover clinical jack bean urease inhibitor.
5. Conclusion
The jack bean urease inhibitors 4a–l, having excellent urease inhibitory potential than the standard thiourea, were described in the present work. A series of compounds, 4a–l, having ester/amide functionalities, were synthesized in order to check their inhibitory potential against jack bean urease. Simple synthetic routes were adopted to synthesize the desired compounds in good yield. The inhibitory activity results showed that compound 4b, having a chloro group at the meta-position of acyl core and para-alkyl-substituted aniline, showed excellent inhibitory potential against urease enzyme with an IC50 of 1.6 ± 0.2 nM, much better than the standard thiourea with an IC50 of 472.1 ± 135.1 nM. The presence of a 2-chlorosubstituted phenyl ring and a 4-isopropyl-substituted aniline on the other side, in the case of compound 4b, played a vital role in urease inhibitory activity. The bioassay results confirmed that the substitution pattern of -Cl at the acyl core is not only the determining factor for activity but alkyl substitution at aniline also play very important role in urease inhibition. In the kinetic studies, a mixed-type inhibition mechanism was reflected in the Lineweaver–Burk plots for compound 4b. The molecular docking studies showed that the predicted binding affinities of the synthesized compounds are excellent, especially in the case of compounds 4e and 4b, having energies of −7.9 and − 7.8 Kcal/mol, respectively. It can be concluded from our results that compound 4b is a highly potent urease inhibitor.
Scheme 1.
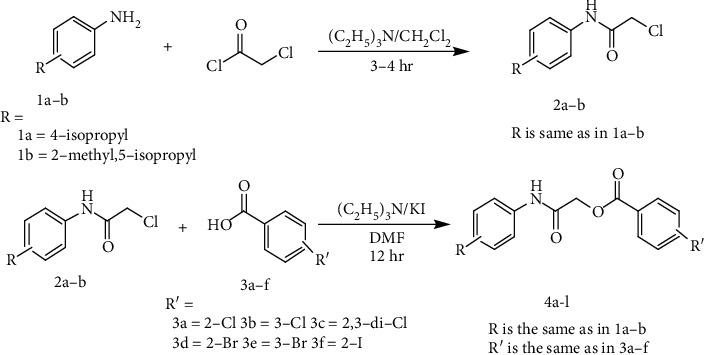
Synthesis of compounds 4a–l.
Acknowledgments
The authors highly acknowledge Dr. Muhammad Hassham Hassan Bin Asad (KFU Russia; CUI Pakistan) for publishing this work.
Contributor Information
Zaman Ashraf, Email: mzchem@yahoo.com.
Muhammad Hassham Hassan Bin Asad, Email: hasshamasad@yahoo.com.
Data Availability
Date could be provided upon request from Dr. Zaman Ashraf.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Eichhorn G. L. In: Advances in Inorganic Biochemistry. Marzilli L. G., editor. Vol. 6. Elsevier; 1984. [Google Scholar]
- 2.Holm L., Sander C. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins: Structure, Function, and Bioinformatics. 1997;28(1):72–82. doi: 10.1002/(SICI)1097-0134(199705)28:1<72::AID-PROT7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 3.Krajewska B. Ureases I. Functional, catalytic and kinetic properties: a review. Journal of Molecular Catalysis B: Enzymatic. 2009;59(1-3):9–21. doi: 10.1016/j.molcatb.2009.01.003. [DOI] [Google Scholar]
- 4.Mazzei L., Musiani F., Ciurli S. The Biological Chemistry of Nickel. Vol. 10. Royal society of chemistry; 2017. CHAPTER 5. Urease; pp. 60–97. [DOI] [Google Scholar]
- 5.Mobley H., Hausinger R. Microbial ureases: significance, regulation, and molecular characterization. Microbiology and Molecular Biology Reviews. 1989;53(1):85–108. doi: 10.1128/mr.53.1.85-108.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krajewska B., van Eldik R., Brindell M. Temperature-and pressure-dependent stopped-flow kinetic studies of jack bean urease. Implications for the catalytic mechanism. JBIC Journal of Biological Inorganic Chemistry. 2012;17(7):1123–1134. doi: 10.1007/s00775-012-0926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taha M., Ismail N. H., Imran S., Wadood A., Rahim F., Riaz M. Synthesis of potent urease inhibitors based on disulfide scaffold and their molecular docking studies. Bioorganic & Medicinal Chemistry. 2015;23(22):7211–7218. doi: 10.1016/j.bmc.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Kosikowska P., Berlicki Ł. Urease inhibitors as potential drugs for gastric and urinary tract infections: a patent review. Expert Opinion on Therapeutic Patents. 2011;21(6):945–957. doi: 10.1517/13543776.2011.574615. [DOI] [PubMed] [Google Scholar]
- 9.Walser M., Bodenlos L. J. Urea metabolism in man. The Journal of Clinical Investigation. 1959;38(9):1617–1626. doi: 10.1172/JCI103940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mobley H., Island M. D., Hausinger R. P. Molecular biology of microbial ureases. Microbiology and Molecular Biology Reviews. 1995;59(3):451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunn B., Cohen H., Blaser M. J. Helicobacter pylori. Clinical Microbiology Reviews. 1997;10:720–741. doi: 10.1128/cmr.10.4.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smoot D. T. How Does _Helicobacter pylori_ Cause Mucosal Damage? Direct Mechanisms. Gastroenterology. 1997;113(6):S31–S34. doi: 10.1016/S0016-5085(97)80008-X. [DOI] [PubMed] [Google Scholar]
- 13.Chen G., Fournier R. L., Varanasi S., Mahama-Relue P. A. Helicobacter pylori survival in gastric mucosa by generation of a pH gradient. Biophysical Journal. 1997;73(2):1081–1088. doi: 10.1016/S0006-3495(97)78140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sachs G., Weeks D. L., Melchers K., Scott D. R. The gastric biology ofHelicobacter pylori. Annual Review of Physiology. 2003;65(1):349–369. doi: 10.1146/annurev.physiol.65.092101.142156. [DOI] [PubMed] [Google Scholar]
- 15.Xiao Z.-P., Peng Z.-Y., Dong J.-J., et al. Synthesis, molecular docking and kinetic properties of β-hydroxy-β- phenylpropionyl-hydroxamic acids as _Helicobacter pylori_ urease inhibitors. European Journal of Medicinal Chemistry. 2013;68:212–221. doi: 10.1016/j.ejmech.2013.07.047. [DOI] [PubMed] [Google Scholar]
- 16.Gioacchini P., Nastri A., Marzadori C., Giovannini C., Vittori Antisari L., Gessa C. Influence of urease and nitrification inhibitors on N losses from soils fertilized with urea. Biology and Fertility of Soils. 2002;36(2):129–135. doi: 10.1007/s00374-002-0521-1. [DOI] [Google Scholar]
- 17.Chung C. S., Chiang T. H., Lee Y. C. A systematic approach for the diagnosis and treatment of idiopathic peptic ulcers. The Korean Journal of Internal Medicine. 2015;30(5):p. 559. doi: 10.3904/kjim.2015.30.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siregar G. A., Parwati I., Achmad T. H., Syukriani Y. F. Risk factors of gastric premalignant lesion in gastritis patients. Sains Malaysiana. 2018;47(8):1811–1818. doi: 10.17576/jsm-2018-4708-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flannigan R., Choy W. H., Chew B., Lange D. Renal struvite stones-pathogenesis, microbiology, and management strategies. Nature reviews Urology. 2014;11(6):p. 333. doi: 10.1038/nrurol.2014.99. [DOI] [PubMed] [Google Scholar]
- 20.Ul-Haq Z., Ashraf S., Al-Majid A., Barakat A. 3D-QSAR studies on barbituric acid derivatives as urease inhibitors and the effect of charges on the quality of a model. International journal of molecular Sciences. 2016;17(5):p. 657. doi: 10.3390/ijms17050657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanafiah A., Binmaeil H., Raja Ali R. A., Mohamed Rose I., Lopes B. S. Molecular characterization and prevalence of antibiotic resistance in Helicobacter pylori isolates in Kuala Lumpur, Malaysia. Infection and Drug Resistance. 2019;12:3051–3061. doi: 10.2147/IDR.S219069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berlicki Ł., Bochno M., Grabowiecka A., Białas A., Kosikowska P., Kafarski P. N-substituted aminomethanephosphonic and aminomethane-P-methylphosphinic acids as inhibitors of ureases. Amino Acids. 2012;42(5):1937–1945. doi: 10.1007/s00726-011-0920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rafiee-Moghaddam R., Salimon J., Haron M. J., et al. Application of methyl fatty hydroxamic acids based on Jatropha curcas seed oil and their metal complexes as antimicrobial agents. Digest Journal of Nanomaterials & Biostructures. 2014;9(1) [Google Scholar]
- 24.Iqbal Z., Iqbal A., Ashraf Z., Latif M., Hassan M., Nadeem H. Synthesis and docking studies of N-(5-(alkylthio)-1, 3, 4-oxadiazol-2-yl) methyl benzamide analogues as potential alkaline phosphatase inhibitors. Drug Development Research. 2019;80:646–654. doi: 10.1002/ddr.21542. [DOI] [PubMed] [Google Scholar]
- 25.Ashraf Z., Rafiq M., Nadeem H., et al. Carvacrol derivatives as mushroom tyrosinase inhibitors; synthesis, kinetics mechanism and molecular docking studies. PLoS One. 2017;12(5, article e0178069) doi: 10.1371/journal.pone.0178069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weatherburn M. Phenol-hypochlorite reaction for determination of ammonia. Analytical Chemistry. 2002;39(8):971–974. [Google Scholar]
- 27.Hanif M., Kanwal F., Rafiq M., et al. Symmetrical heterocyclic cage skeleton: synthesis, urease inhibition activity, kinetic mechanistic insight, and molecular docking analyses. Molecules. 2019;24(2):p. 312. doi: 10.3390/molecules24020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen E. F., Goddard T. D., Huang C. C., et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Chen V. B., Arendall W. B., Headd J. J., et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography. 2010;66(1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasteiger E., Hoogland C., Gattiker A., Wilkins M. R., Appel R. D., Bairoch A. Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook. 2005. pp. 571–607.
- 31.Willard L., Ranjan A., Zhang H., et al. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Research. 2003;31(13):3316–3319. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dallakyan S., Olson A. J. Small-molecule library screening by docking with PyRx. Chemical biology. 2015. pp. 243–250. [DOI] [PubMed]
- 33.Channar P., Saeed A., Albericio F., et al. Sulfonamide-linked ciprofloxacin, sulfadiazine and amantadine derivatives as a novel class of inhibitors of jack bean urease; synthesis, kinetic mechanism and molecular docking. Molecules. 2017;22(8):p. 1352. doi: 10.3390/molecules22081352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iqbal Z., Ashraf Z., Hassan M., Abbas Q., Jabeen E. Substituted phenyl[(5-benzyl-1,3,4-oxadiazol-2-yl)sulfanyl]acetates/acetamides as alkaline phosphatase inhibitors: Synthesis, computational studies, enzyme inhibitory kinetics and DNA binding studies. Bioorganic Chemistry. 2019;90, article 103108 doi: 10.1016/j.bioorg.2019.103108. [DOI] [PubMed] [Google Scholar]
- 35.Saeed A., Mahmood S., Rafiq M., Ashraf Z., Jabeen F., Seo S. Y. Iminothiazoline-sulfonamide hybrids as jack bean urease inhibitors; synthesis, kinetic mechanism and computational molecular modeling. Chemical Biology and Drug Design. 2016;87(3):434–443. doi: 10.1111/cbdd.12675. [DOI] [PubMed] [Google Scholar]
- 36.Fattah T. A., Saeed A., Ashraf Z., et al. 4-Aminocoumarin based aroylthioureas as potential jack bean urease inhibitors; synthesis, enzyme inhibitory kinetics and docking studies. Medicinal Chemistry. 2020;16(2):229–243. doi: 10.2174/1573406415666190715164834. [DOI] [PubMed] [Google Scholar]
- 37.Dige N. C., Mahajan P. G., Raza H., et al. Ultrasound mediated efficient synthesis of new 4-oxoquinazolin-3(4 _H_ )-yl furan-2-carboxamides as potent tyrosinase inhibitors: Mechanistic approach through chemoinformatics and molecular docking studies. Bioorganic Chemistry. 2019;92 doi: 10.1016/j.bioorg.2019.103201. [DOI] [PubMed] [Google Scholar]
- 38.Jadhav P. B., Yadav A. R., Gore M. G. Concept of drug likeness in pharmaceutical research. International journal of pharma and bio sciences. 2015;6:142–154. [Google Scholar]
- 39.Hameed A., Khan K. M., Zehra S. T., et al. Synthesis, biological evaluation and molecular docking of _N_ -phenyl thiosemicarbazones as urease inhibitors. Bioorganic Chemistry. 2015;61:51–57. doi: 10.1016/j.bioorg.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Abdul Fattah T., Saeed A., Channar P. A., et al. Synthesis, enzyme inhibitory kinetics, and computational studies of novel 1-(2-(4-isobutylphenyl) propanoyl)-3-arylthioureas as jack bean urease inhibitors. Chemical Biology and Drug Design. 2018;91(2):434–447. doi: 10.1111/cbdd.13090. [DOI] [PubMed] [Google Scholar]
- 41.Saeed A., ur-Rehman S., Channar P., et al. Jack bean urease inhibitors, and antioxidant activity based on palmitic acid derived 1-acyl-3- Arylthioureas: synthesis, kinetic mechanism and molecular docking studies. Drug Research. 2017;67(10):596–605. doi: 10.1055/s-0043-113832. [DOI] [PubMed] [Google Scholar]
- 42.Saeed A., Rehman S.-u., Channar P. A., et al. Long chain 1-acyl-3-arylthioureas as jack bean urease inhibitors, synthesis, kinetic mechanism and molecular docking studies. Journal of the Taiwan Institute of Chemical Engineers. 2017;77:54–63. doi: 10.1016/j.jtice.2017.04.044. [DOI] [Google Scholar]
- 43.Saeed A., Mahesar P. A., Channar P. A., et al. Hybrid pharmacophoric approach in the design and synthesis of coumarin linked pyrazolinyl as urease inhibitors, kinetic mechanism and molecular docking. Chemistry and Biodiversity. 2017;14(8, article e1700035) doi: 10.1002/cbdv.201700035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Date could be provided upon request from Dr. Zaman Ashraf.