

Abstract
Objective:
To determine whether statin exposure is associated with decreased cancer and mortality risk among persons with HIV (PWH) and uninfected persons. Statins appear to have immunomodulatory and anti-inflammatory effects and may reduce cancer risk, particularly among PWH as they experience chronic inflammation and immune activation.
Design:
Propensity score matched cohort of statin-exposed and unexposed patients from 2002–2017 in the Veterans Aging Cohort Study (VACS), a large cohort with cancer registry linkage and detailed pharmacy data.
Methods:
We calculated Cox regression hazard ratios (HRs) and 95% confidence intervals (CI) associated with statin use for all cancers, microbial cancers (associated with bacterial or oncovirus coinfection), non-microbial cancers, and mortality.
Results:
The propensity score-matched sample (N=47,940) included 23,970 statin initiators (31% PWH). Incident cancers were diagnosed in 1,160 PWH and 2,116 uninfected patients. Death was reported in 1,667 (7.0%) statin-exposed, and 2,215 (9.2%) unexposed patients. Statin use was associated with 24% decreased risk of microbial associated cancers (HR 0.76; 95% CI 0.69–0.85), but was not associated with non-microbial cancer risk (HR 1.00; 95% CI 0.92–1.09). Statin use was associated with 33% lower risk of death overall (HR 0.67; 95% CI 0.63–0.72). Results were similar in analyses stratified by HIV status, except for non-Hodgkin lymphoma where statin use was associated with reduced risk (HR 0.56; 95% CI 0.38–0.83) for PWH, but not for uninfected (p-interaction = 0.012).
Conclusions:
In both PWH and uninfected, statin exposure was associated with lower risk of microbial, but not non-microbial cancer incidence, and with decreased mortality.
Keywords: neoplasms, cancer, hypolipidemic agents, HIV
INTRODUCTION
Beyond their lipid-lowering properties, 3-hydroxy-3-methylglutaryl coenzyme (HMG-CoA) reductase inhibitors, commonly known as statins, have multiple benefits. Statins inhibit conversion of HMG-CoA to mevalonic acid, an early and major rate-limiting step of cholesterol biosynthesis. In addition to cholesterol biosynthesis, this pathway also mediates protein prenylation and regulates T cell cycle progression and function including migration, proliferation and cytotoxic effector responses [1, 2]. Further, statins might interfere with leukocyte trafficking and T cell activation through inhibition of the beta2 integrin leukocyte function antigen-1 (LFA-1)/intercellular adhesion molecule (ICAM)-1 interaction [3]. Statins therefore have a variety of anti-inflammatory [4] and immune-modulatory [5] effects and could potentially enhance immune response against invading pathogens and tumor cells [6].
In the general population, the potential association of statin use with cancer risk and mortality has been inconsistent. A Dutch analysis of over 3,000 statin-exposed and 17,000 matched unexposed persons reported statin use was associated with 20% reduction in cancer risk [7]. A Canadian analysis of over 50,000 patients with acute myocardial infarction found that compared to non-statin users, those with a high-dose statin prescription at hospital discharge had 25% lower risk of cancer over the following 7 years [8]. Similarly, U.S. Veterans using statins had 25% lower risk of cancer compared to those using anti-hypertensives in the absence of statins [9]. However, a meta-analysis of 27 studies evaluating the efficacy of statins in reducing cardiovascular disease showed no association with incidence of, or mortality from, cancer [10, 11]. The association of statin exposure with decreased site-specific cancer risk has been observed in some studies [12–16], but not in others [17–20]. A Danish population study showed an association between statin use at the time of cancer diagnosis and reduced risk of both cancer-related and all-cause mortality [21]. Reduced cancer-related mortality was observed for all 13 included cancer types. Inconsistent findings in the general population could be related to differences in those studied including age [14], statin type, dose and duration [7, 8], and methodologies. Finally, lack of accounting for “confounding by indication” is a major concern in most observational studies [22, 23]. We are unaware of any published randomized controlled trials (RCT) specifically designed for statin exposure with cancer endpoints. Meta-analyses of trials designed for other endpoints generally considered all cancers together and found no significant associations between statins and cancer [10, 24].
While associations between statins and cancer risk have been inconsistent in the general population, statin effects may be particularly pronounced among persons with HIV (PWH), due to long-term effects of HIV viral replication and the prevalence of viral and bacterial coinfections known to increase cancer risk. Three small studies of PWH found statin use associated with decreased incidence of AIDS- and non-AIDS-defining cancers [25–27]. Also, statin use has been associated with significantly lower risk of death in a single center US HIV cohort [28], but non-significantly associated with lower mortality in the Danish HIV cohort [29].
The effect of statins on cancer incidence has not been compared among PWH and demographically similar uninfected individuals. Further, analysis of the association of statins with specific cancer types and mortality in PWH has been limited by small sample size and short follow-up time. We used the Veterans Aging Cohort Study (VACS), a large cohort of PWH and demographically-matched uninfected individuals receiving care in the Veterans Health Administration (VA), to examine the effect of statin exposure on the incidence of any cancer, microbial cancers (cancers associated with bacterial or oncovirus infection), non-microbial cancers, specific cancer types, and with all-cause mortality. We used a propensity score matched cohort design to reduce the impact of confounding by indication [30]. We hypothesized that the association of statins with cancer would be strongest among PWH and for microbial cancers.
METHODS
Data source
The VACS is a prospective cohort of all PWH in the VA, the largest integrated healthcare system in the US. Each newly identified PWH is matched to two uninfected Veterans under VA care at that time by age, sex, race/ethnicity, year, and the clinical site where they receive care, as described previously [31]. The full cohort is predominantly male (97%) and about half non-Hispanic black.
Patients have been continuously enrolled each year since 1998 using a validated existing algorithm from the VA national electronic health record system [32]. The VACS database consists of detailed demographics, hospital and outpatient diagnoses (recorded using International Classification of Diseases, Ninth Revision [ICD-9] codes), procedures, laboratory results, and dispensed medications data. Death date was determined from the VA vital status file, and cancer diagnosis information was linked from the VA national cancer registry. The VA Connecticut Healthcare System and Yale University Institutional Review Boards have approved the VACS.
Study population
We identified statin users from October 1, 1998 to September 30, 2015. Statin-exposed persons were defined as newly-initiating statin use (atorvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, and simvastatin) between fiscal year 2002–2015 and having at least two prescription fills within 180 days and clinic visits at the following VA clinics: general internal medicine, cardiology, endocrinology, diabetes, gastroenterology, hypertension, infectious disease, pulmonary, renal/nephrology, geriatrics, women’s clinic, primary care, and hepatology. These clinics were chosen because nearly all statin-exposed patients (97.6%) had a visit to one of these clinics in the year prior to first statin prescription in the VA. Statin regimens used by fewer than 100 patients (pitavastatin, cerivastatin, and nicostatin) were considered rare. Rare statin regimens and patients with statin exposure before 2002 were excluded. We randomly selected one outpatient visit date per calendar year to identify patients who attended one of the listed clinics but did not receive a statin to ensure that unexposed patients came from the same source population and had an equal opportunity to receive a statin prescription.
We defined an index date as date of first statin fill or as a randomly chosen clinic date during the same fiscal year for statin-unexposed persons. Follow-up started 180 days following the index date, for both exposed and unexposed persons, to prevent immortal time bias (due to the requirement of two statin fills in 180 days) [33, 34] and ended at the event of interest (cancer diagnosis, death) or the last follow-up date (last patient interaction in the VA) prior to September 30, 2017.
Study outcomes
Study outcomes included incident cancer diagnosis and all-cause mortality. We linked VACS with the VA national cancer registry, a database of cancer cases diagnosed and/or treated at the VA. We mapped International Classification of Diseases for Oncology, third edition (ICD-O-3) [35] topography and morphology codes from these databases to specific cancer types, consistent with Surveillance, Epidemiology, and End Results (SEER) algorithms [36]. We classified cancer types into the following groupings: all cancers, microbial cancers, and non-microbial cancers. Microbial cancers were defined as cancers associated with either known oncoviruses (cancers of the oral cavity and pharynx, stomach, anus, liver, cervix, vagina, vulva, penis, Hodgkin lymphoma, non-Hodgkin lymphoma, and Kaposi sarcoma) or chronic bacterial infection (lung and bronchus), using morphology and detailed topography (Appendix Table 1). For example, squamous cell carcinoma of the anus is a microbial cancer, whereas other morphological types of anal cancer are non-microbial. We also examined risk of specific cancers of interest, with sufficient numbers.
Propensity score model
We used propensity score matching to account for potential confounding by indication. We created separate propensity score models by HIV status, that included known and potential confounders of the association between statin use and cancer. We explored a wide range of variables related to patient demographics, clinical data, laboratory results, hospitalizations, and comorbidities. The final model included calendar year, demographic variables: age, gender, race/ethnicity; clinical variables: comorbid conditions (diabetes, hepatitis C virus [HCV], hepatitis B virus [HBV]), body mass index (BMI), smoking status, anti-hypertensive medication exposure history; laboratory variables: glucose, FIB-4 (calculated from age, aspartate aminotransferase, platelet count, and alanine aminotransferase), hemoglobin, cholesterol (LDL, HDL, and total), triglycerides, blood pressure; facility level prescription patterns, numbers of unique clinic visits in the prior year, and hospitalizations (Appendix Table 2). We used the measurement prior and closest to the index date for all variables. In the PWH propensity score model (c-statistic=0.893), we included laboratory values for HIV viral load and CD4 cell count as well as interactions for LDL cholesterol with HIV viral load and LDL cholesterol with HCV. In the uninfected model (c-statistic=0.901), we included diabetes medication history and an interaction for diabetes diagnosis status with LDL cholesterol.
Matching
We matched statin-exposed to unexposed persons using greedy matching algorithm without replacement [37]. We matched each statin-exposed to one unexposed person within a caliper of 0.20 SD of the logit of propensity score [37]. The final dataset included only matched statin-exposed and unexposed persons. We assessed covariate balance before and after matching. Covariates were considered imbalanced if the standardized difference between statin-exposed and unexposed was >0.1 [38].
Outcome analysis
We used Cox proportional hazards regression models to estimate hazard ratios (HRs) and 95% confidence intervals (CI) associated with statin use for all cancers, cancer groups, individual cancer types, and mortality. We ran three sets of models, first including all patients and then stratified by HIV status. We examined whether the association between statins and cancer varied by HIV status in a model with all patients, adjusting for HIV, and noted if there was a significant HIV and statin interaction.
We calculated standardized differences with Stata version 14.2 (StataCorp LLC, College Station, Texas). All other analyses were conducted using SAS version 9.4 (SAS Institute, Inc. Cary, North Carolina).
We conducted sensitivity analyses examining the microbial cancer group definition by calculating the HR estimates for the microbial and non-microbial cancers with and without lung cancer. We also calculated HR estimates by statin type at initiation (Simvastatin versus all others). We used the Benjamini-Hochberg method for multiple-comparison corrections [39].
RESULTS
Among VACS participants, there were 12,153 PWH and 34,561 uninfected statin initiators during the study period (Table 1, Appendix Figure 1). There were 27,876 PWH and 46,642 uninfected patients without a statin prescription fill in the VA health system among patients alive in the cohort during the study follow-up period. Statin-exposed patients were older (mean age 54.0 years for PWH, 53.1 years for uninfected) than patients without a statin prescription (mean age 49.0 years for PWH, 48.4 years for uninfected).
Table 1.
Baseline characteristics among statin-exposed and unexposed persons in the pre-matched and propensity score-matched patients and standardized differences in the propensity-score-matched patients
| All patients (pre-matched) | Propensity score matched | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PWH | Uninfected | PWH | Uninfected | ||||||||||||||||
| Statin-exposed | Unexposed | Statin-exposed | Unexposed | Statin-exposed | Unexposed | Statin-exposed | Unexposed | ||||||||||||
| N=12,153 | N=27,876 | N=34,561 | N=46,642 | N=7,335 | N=7,335 | Sth | N=16,635 | N=16,635 | Sth | ||||||||||
| N | % | N | % | N | % | N | % | N | % | N | % | diff | N | % | N | % | diff | ||
| Age | Mean +/−st dev (years) | 54.0 | 9.4 | 49.0 | 11.3 | 53.1 | 9.2 | 48.4 | 12.3 | 53.8 | 9.5 | 53.1 | 9.4 | −0.08 | 53.2 | 9.8 | 52.2 | 9.9 | −0.10 |
| Race/ethnicity | Non-Hispanic white | 5,467 | 45.0 | 10,319 | 37.0 | 13,967 | 40.4 | 18,164 | 38.9 | 3,114 | 42.5 | 3,080 | 42.0 | 0.02 | 6,705 | 40.3 | 6,625 | 39.8 | 0.02 |
| Non-Hispanic black | 5,369 | 44.2 | 14,017 | 50.3 | 16,343 | 47.3 | 22,353 | 47.9 | 3,419 | 46.6 | 3,460 | 47.2 | 7,932 | 47.7 | 7,979 | 48.0 | |||
| Hispanic | 949 | 7.8 | 2,260 | 8.1 | 3,086 | 8.9 | 3,806 | 8.2 | 580 | 7.9 | 562 | 7.7 | 1,459 | 8.8 | 1,446 | 8.7 | |||
| Other/unknown | 368 | 3.0 | 1,279 | 4.6 | 1,165 | 3.4 | 2,319 | 5.0 | 222 | 3.0 | 233 | 3.2 | 539 | 3.2 | 585 | 3.5 | |||
| Sex | Female | 327 | 2.7 | 855 | 3.1 | 876 | 2.5 | 1,738 | 3.7 | 216 | 2.9 | 234 | 3.2 | 0.01 | 478 | 2.9 | 484 | 2.9 | <0.01 |
| Male | 11,826 | 97.3 | 27,020 | 96.9 | 33,685 | 97.5 | 44,904 | 96.3 | 7,119 | 97.1 | 7,101 | 96.8 | 16,157 | 97.1 | 16,151 | 97.1 | |||
| Hepatitis C* | HCV negative | 8,991 | 74.0 | 16,815 | 60.3 | 25,948 | 75.1 | 29,099 | 62.4 | 5,158 | 70.3 | 5,133 | 70.0 | 0.03 | 11,997 | 72.1 | 11,765 | 70.7 | 0.04 |
| Chronic HCV | 2,122 | 17.5 | 7,665 | 27.5 | 3,281 | 9.5 | 6,981 | 15.0 | 1,547 | 21.1 | 1,576 | 21.5 | 2,019 | 12.1 | 2,059 | 12.4 | |||
| HCV exposure | 735 | 6.0 | 2,000 | 7.2 | 1,160 | 3.4 | 1,660 | 3.6 | 464 | 6.3 | 434 | 5.9 | 591 | 3.6 | 606 | 3.6 | |||
| Never tested in the VA | 305 | 2.5 | 1,395 | 5.0 | 4,172 | 12.1 | 8,902 | 19.1 | 166 | 2.3 | 192 | 2.6 | 2,028 | 12.2 | 2,205 | 13.3 | |||
| Hepatitis B* | HBV negative | 10,280 | 84.6 | 22,162 | 79.5 | 18,737 | 54.2 | 23,876 | 51.2 | 6,177 | 84.2 | 6,143 | 83.7 | 0.03 | 9,146 | 55.0 | 8,978 | 54.0 | 0.02 |
| HBV positive | 424 | 3.5 | 1,031 | 3.7 | 134 | 0.4 | 209 | 0.4 | 284 | 3.9 | 292 | 4.0 | 72 | 0.4 | 81 | 0.5 | |||
| HBV acute resolved | 140 | 1.2 | 349 | 1.3 | 78 | 0.2 | 116 | 0.2 | 94 | 1.3 | 83 | 1.1 | 43 | 0.3 | 43 | 0.3 | |||
| Unconfirmed HBV | 81 | 0.7 | 324 | 1.2 | 53 | 0.2 | 91 | 0.2 | 49 | 0.7 | 60 | 0.8 | 20 | 0.1 | 24 | 0.1 | |||
| Never tested in the VA | 1,228 | 10.1 | 4,009 | 14.4 | 15,559 | 45.0 | 22,350 | 47.9 | 731 | 10.0 | 757 | 10.3 | 7,354 | 44.2 | 7,509 | 45.1 | |||
| BMI | Under/normal weight (<30) | 8,668 | 71.3 | 21,122 | 75.8 | 16,596 | 48.0 | 26,783 | 57.4 | 5,348 | 72.9 | 5,419 | 73.9 | 0.03 | 8,848 | 53.2 | 9,198 | 55.3 | 0.05 |
| Overweight (30–34.9) | 2,152 | 17.7 | 2,914 | 10.5 | 9,964 | 28.8 | 8,948 | 19.2 | 1,283 | 17.5 | 1,264 | 17.2 | 4,449 | 26.7 | 4,372 | 26.3 | |||
| Obese (≥ 35) | 1,027 | 8.5 | 1,122 | 4.0 | 7,003 | 20.3 | 4,901 | 10.5 | 562 | 7.7 | 513 | 7.0 | 2,892 | 17.4 | 2,623 | 15.8 | |||
| Unknown | 306 | 2.5 | 2,717 | 9.7 | 998 | 2.9 | 6,010 | 12.9 | 142 | 1.9 | 139 | 1.9 | 446 | 2.7 | 442 | 2.7 | |||
| Smoking | Non-smoker | 3,583 | 29.5 | 7,261 | 26.0 | 10,194 | 29.5 | 13,751 | 29.5 | 2,068 | 28.2 | 2,045 | 27.9 | 0.05 | 4,905 | 29.5 | 4,831 | 29.0 | 0.06 |
| Current | 6,031 | 49.6 | 15,724 | 56.4 | 16,746 | 48.5 | 24,207 | 51.9 | 3,826 | 52.2 | 3,945 | 53.8 | 8,312 | 50.0 | 8,524 | 51.2 | |||
| Former | 2,385 | 19.6 | 3,654 | 13.1 | 7,267 | 21.0 | 6,700 | 14.4 | 1,352 | 18.4 | 1,236 | 16.9 | 3,245 | 19.5 | 3,017 | 18.1 | |||
| Unknown | 154 | 1.3 | 1,236 | 4.4 | 354 | 1.0 | 1,984 | 4.3 | 89 | 1.2 | 109 | 1.5 | 173 | 1.0 | 263 | 1.6 | |||
| Diabetes | No | 9,509 | 78.2 | 25,804 | 92.6 | 24,281 | 70.3 | 42,879 | 91.9 | 5,920 | 80.7 | 6,085 | 83.0 | 0.06 | 13,283 | 79.8 | 13,846 | 83.2 | 0.09 |
| Yes | 2,644 | 21.8 | 2,071 | 7.4 | 10,280 | 29.7 | 3,763 | 8.1 | 1,415 | 19.3 | 1,250 | 17.0 | 3,352 | 20.2 | 2,789 | 16.8 | |||
| Year of | 2002–2003 | 1,852 | 15.2 | 5,172 | 18.6 | 6,339 | 18.3 | 7,230 | 15.5 | 818 | 11.2 | 818 | 11.2 | <0.01 | 2,094 | 12.6 | 2,094 | 12.6 | <0.01 |
| Index visit | 2004–2006 | 2,905 | 23.9 | 5,203 | 18.7 | 10,482 | 30.3 | 9,228 | 19.8 | 1,506 | 20.5 | 1,506 | 20.5 | 4,068 | 24.5 | 4,068 | 24.5 | ||
| 2007–2009 | 2,861 | 23.5 | 4,556 | 16.3 | 8,495 | 24.6 | 9,093 | 19.5 | 1,641 | 22.4 | 1,641 | 22.4 | 4,119 | 24.8 | 4,119 | 24.8 | |||
| 2010–2012 | 2,554 | 21.0 | 5,268 | 18.9 | 5,658 | 16.4 | 11,108 | 23.8 | 1,713 | 23.4 | 1,713 | 23.4 | 3,492 | 21.0 | 3,492 | 21.0 | |||
| 2013–2015 | 1,981 | 16.3 | 7,676 | 27.5 | 3,587 | 10.4 | 46,642 | 100.0 | 1,657 | 22.6 | 1,657 | 22.6 | 2,862 | 17.2 | 2,862 | 17.2 | |||
| HIV-RNA | ≤ 400 | 7,343 | 60.4 | 11,764 | 42.2 | 4,577 | 62.4 | 4,432 | 60.4 | 0.05 | |||||||||
| >400 | 1,536 | 12.6 | 6,926 | 24.8 | 1,054 | 14.4 | 1,057 | 14.4 | |||||||||||
| Unknown | 3,274 | 26.9 | 9,185 | 33.0 | 1,704 | 23.2 | 1,846 | 25.2 | |||||||||||
| CD4 | ≥500 | 4,317 | 35.5 | 7,182 | 25.8 | 2,754 | 37.5 | 2,564 | 35.0 | 0.06 | |||||||||
| 350–499 | 2,006 | 16.5 | 3,811 | 13.7 | 1,263 | 17.2 | 1,243 | 16.9 | |||||||||||
| 200–349 | 1,658 | 13.6 | 3,726 | 13.4 | 1,025 | 14.0 | 1,051 | 14.3 | |||||||||||
| 0–199 | 851 | 7.0 | 3,862 | 13.9 | 557 | 7.6 | 605 | 8.2 | |||||||||||
| Unknown | 3,321 | 27.3 | 9,294 | 33.3 | 1,736 | 23.7 | 1,872 | 25.5 | |||||||||||
Abbreviations: Std diff = standardized difference, HCV = hepatitis C virus, HBV = hepatitis B virus, BMI = body mass index
Definitions: HCV negative, negative HCV antibody test result(s) only; Chronic HCV, positive HCV RNA test; HCV exposure, positive HCV antibody test, but negative or unknown HCV RNA test; Never tested in the VA, no HCV laboratory test results available from the VA (it is possible that some of these patients were tested for HCV outside the VA)
HBV negative, negative HBV surface antigen test result(s) only; HBV positive, at least two positive HBV surface antigen tests over 6 months apart; HBV acute resolved, positive HBV surface antigen test followed by only negative test results; Unconfirmed HBV, one positive HBV surface antigen test not confirmed with additional testing; Never tested/unknown, no HBV laboratory test results available.
In the unmatched sample, the median propensity score among statin-exposed patients was 0.24 for PWH and 0.38 for uninfected patients, and among patients not exposed to statins was 0.015 for PWH and 0.021 for uninfected patients (Appendix Figure 2). After matching, the median propensity score was 0.13 for PWH and 0.06 for uninfected for both statin-exposed and unexposed patients. All covariate standardized differences were less than 0.1 indicating no imbalance between exposed and unexposed (Table 1). Statin exposed patients who did not have a propensity score match were excluded from the analysis. Most baseline characteristics were similar between the propensity score matched and unmatched statin exposed patients (Appendix Table 3). Both PWH and uninfected unmatched patients were less likely to have hepatitis C, diabetes, and index visit during later years compared to propensity score matched patients.
The propensity score-matched sample (N=47,940) included 23,970 statin initiators (7,335 PWH and 16,635 uninfected) and 23,970 statin-unexposed patients (Table 1). Median follow-up time was 5.7 (IQR: 3.0–9.0) years for PWH and 7.1 (IQR: 3.8–10.4) years for uninfected patients. Mean age was 52–53 years old for the propensity score matched patients. Simvastatin was the most commonly prescribed statin, representing 63.5% of all first statin prescriptions. 70.8% of statin-exposed patients took simvastatin, followed by atorvastatin (54.3%), pravastatin (33.5%), rosuvastatin (13.7%), lovastatin (6.7%), and fluvastatin (5.5%) during the entire follow-up period, including regimen changes. Median duration of statin use was 3.0 years (interquartile range [IQR]: 1.2–5.8 years) overall. Incident cancers were diagnosed in 1,160 PWH (22.8 cancers/1,000 person-years) and 2,116 uninfected patients (17.4 cancers/1,000 person-years). The most common cancer types were lung and prostate cancer. Death was reported in 1,667 (7.0%) statin-exposed and 2,215 (9.2%) unexposed persons.
Overall, statin use was associated with 11% reduced risk of any cancer (HR 0.89; 95% CI 0.83–0.95) and 24% decreased risk of microbial cancers (HR 0.76; 95% CI 0.59–0.85) (Figure 1). Statin use was not associated with non-microbial cancers (HR 1.00; 95% CI 0.92–1.09). Statin use was also associated with lower risk of death (HR 0.67; 95% CI 0.63–0.72). The association between statin use and reduced cancer risk for both PWH and uninfected patients was strongest for hepatocellular carcinoma (HR 0.54; 95% CI 0.42–0.69) and HPV-associated squamous cell carcinomas of the oral cavity and pharynx (HR 0.60; 95% CI 0.40–0.90). Results were similar in analyses stratified by HIV, with a few exceptions. For PWH, statin use was associated with reduced non-Hodgkin lymphoma risk (HR 0.56; 95% CI 0.38–0.83); but not for uninfected patients (p for interaction = 0.012). Also, there was reduced risk of lung and bronchus cancers associated with statin use in the uninfected group (HR 0.82; 95% CI 0.67–0.99) and PWH group (HR 0.93; 95% CI 0.73–1.20); however, the confidence interval was wider for PWH and the finding was not significant. Among PWH, statin use was associated with 51% reduced Kaposi sarcoma risk (HR 0.49; 95% CI 0.26–0.92). There were no Kaposi sarcoma cases among uninfected patients.
Figure 1.
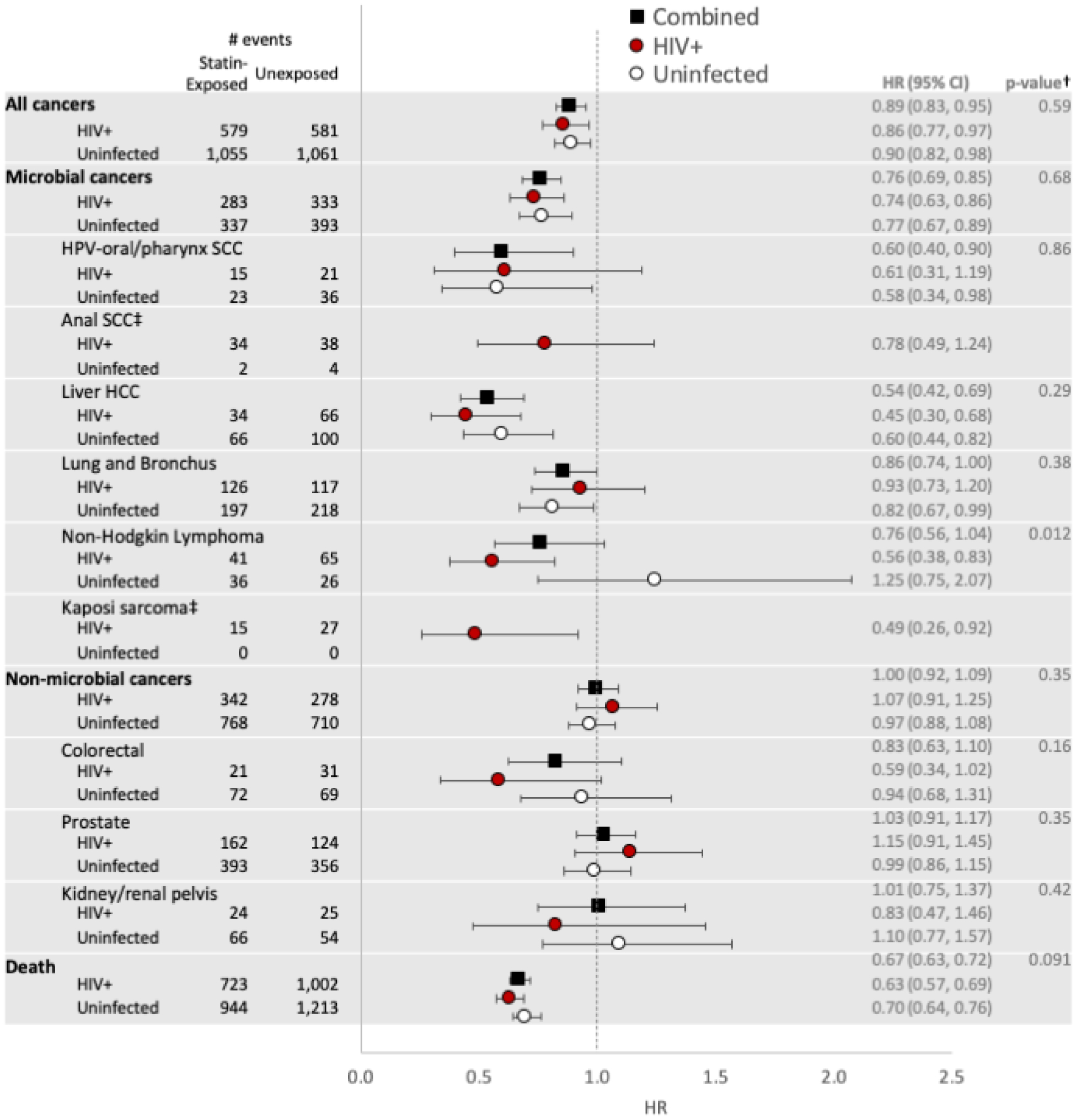
Propensity score-matched hazard ratios (statin-exposed versus unexposed) for cancer groups*, specific cancer types, and mortality
* Microbial cancers include: human papillomavirus (HPV)-related oral cavity and pharynx squamous cell carcinoma (SCC), anal SCC, hepatocellular carcinoma (HCC); cancers of the stomach, lung, cervix, vulva, vagina, and penis; Hodgkin lymphoma, non-Hodgkin lymphoma, and Kaposi sarcoma
† P-value for HIV*statin interaction in combined model with HIV and uninfected patients
‡ Results presented for PWH only because there were only 6 anal squamous cell carcinoma and 0 Kaposi sarcoma cases among uninfected.
In a sensitivity analysis removing lung cancer from the microbial cancer category (Appendix Table 4). This led to minimally stronger association with statin exposure (0.76 vs 0.74). For non-microbial cancers the association with statin exposure remained close to 1. Simvastatin was the dominant initial statin type prescribed through 2012 (Appendix Figure 3). We therefore compared results for patients who initiated Simvastatin versus the other statin types. The hazard ratio patterns were similar with the original analysis except where there were few events, resulting in wide confidence intervals (oral cavity/pharynx and anal cancers, Appendix Figure 4).
DISCUSSION
In this large cohort of PWH and demographically similar uninfected patients, statin exposure was associated with 11% lower risk of any cancer compared to propensity score matched unexposed patients. The strongest associations were for microbial cancers: liver and oral/pharyngeal cancers for both PWH and uninfected, non-Hodgkin lymphoma and Kaposi sarcoma among PWH, and lung cancer among uninfected patients. The decreased risk was generally similar among PWH and uninfected patients. When cancers were grouped, statin exposure was associated with decreased cancer risk among microbial (24% reduced risk) but not among non-microbial cancers. This finding suggests that statins may specifically interfere with the pathogenesis of microbial cancers which are more common among PWH.
Microbial co-infection, chronic inflammation, and immune dysfunction are potent environmental stimuli for oncogenesis. The prevalence of co-infection with HCV, HBV, Epstein Barr virus, cytomegalovirus, etc., is higher among PWH [40–42]. The incidence of AIDS-defining [43–47] and non-AIDS-defining malignancies [43–45, 47–53] is higher among PWH than in the general population, accounting for behavioral risk factors and excess cancer risk remaining after long-term viral suppression [54]. Persistent inflammation and immune dysfunction in HIV patients – even in the context of long-term suppressive antiretroviral therapy (ART) [55, 56] – has been associated with increased risk of non-AIDS complications including cancer [57–59].
Intriguingly, statins have both antimicrobial and anti-inflammatory effects. Statins have in vitro antiviral activity against human cytomegalovirus [60], dengue virus [61, 62], and HIV-1 [63], and statin use was associated with reduced risk of virologic rebound in PWH on suppressive ART [64]. Also, statins may differ in their effect(s) on inflammation and immune activation [65], and as a result, have different effects on cancer risk. Thus, our finding that statin exposure is associated with decreased risk of microbial cancers has biologic plausbility.
Previous studies have suggested a possible dose-response relationship, with longer duration and higher doses of statin use being associated with lower risk of cancer. In the Dutch study, the effect of statin was observed only with longer duration of statin use (more than 4 years) [7], while in the Canadian study, compared to statin-unexposed persons, risk of cancer was lower among high-dose statin-exposed persons (HR: 0.75; 95% CI: 0.60 – 0.95) and marginally lower among low-dose statin-exposed persons (HR: 0.89; 95% CI: 0.75 – 1.07). This could explain, in part, the inconsistent findings of published studies, as most did not account for duration of statin exposure or adherence.
We found that statin exposure was associated with 33% lower risk of all-cause mortality. Although we did not examine cause of death, it is possible that some of the mortality reduction was cancer-related mortality. However, the magnitude of mortality benefit suggests that it might not be entirely mediated through reduced cancer risk or cancer-related mortality. Beyond risk of cancer incidence, statins have been shown to be associated with decreased cancer mortality. In the Danish analysis, statin use was associated with reduced cancer mortality among those with cancer diagnoses, despite lack of association with cancer incidence [29]. Also, results from a small HIV cohort that showed statin exposure associated with lower risk of death, the majority of deaths were cancer-related [28].
Our findings have important clinical implications as microbial malignancies are a leading cause of mortality in the aging population, and cancer-related deaths are increasing in proportion in many HIV cohorts [66, 67]. Rates of malignancies continue to be significantly higher among PWH [54], thus further improvement in HIV survival will likely require biomedical interventions such as statins, in addition to cancer prevention and screening strategies.
Strengths of our study include use of a large national cohort of PWH in the modern ART era and demographically similar uninfected persons followed over a 16-year period, with linked cancer registry data with low rates of misclassification and longitudinal pharmacy dispensing records. This allowed for sufficient cancer and death events to accrue to examine the relationship between statin exposure and both cancer risk and mortality. Further, we used propensity score matching which allowed us to control for confounding by indication, which is a significant hurdle in pharmacoepidemiological studies using real-world data [22, 30]; however, there is always potential for residual and unmeasured confounding. Propensity score matching allows the use of an observational cohort to emulate a randomized controlled trial (RCT) by 1) calculating the propensity score to establish the strength of the indication (criteria that would have been used for inclusion in an RCT) and 2) matching on the propensity score to balance treatment arms by potential confounders, both known and unknown. RCTs often exclude older and sicker patients; however, our study population and results are more generalizable due to a wider array of patients than typically recruited in an RCT.
Limitations of our study include a predominantly male (97%) population, so it is unclear if our findings are generalizable to women. Cancers have long latency periods therefore, longer follow-up may be needed to see the full effects of statins in cancer prevention. Nonetheless, we did see signal in this study spanning 16 years. We also did not examine cumulative exposure to statins. We had a large number of statistical tests; however, the 13 cancer types and groups were selected from a priori hypotheses. Using the Benjamini-Hochberg method with a false discovery rate threshold of 25%, our findings remain significant (for any cancer, microbial cancers, oral cavity and pharynx cancer, hepatocellular carcinoma, lung cancer, Kaposi sarcoma). Non-Hodgkin lymphoma would also meet the threshold for significance. Finally, we did not determine specific causes of mortality and therefore cannot determine whether the associations of statins with decreased cancer risk and decreased mortality are related. Cancer incidence data was obtained from the VA national registry, therefore cancers diagnosed and treated outside the VA system are unlikely to have been ascertained. However, as patients treated with statins in VA care are more likely to have been engaged in primary care within the VA (and thereby diagnosed with cancer within the VA), this would bias the statin arm towards more cancer diagnoses, thereby strengthening the associations noted in our findings. We were only able to propensity score match 60% of PWH and 48% of uninfected statin users, thus our findings may not apply to all statin users. However, this is similar to what happens in randomized trials that apply inclusion and exclusion criteria.
In conclusion, we observed that statin use was associated with at least 10% lower risk of cancer in PWH and uninfected patients, and an even greater (>30%) decreased risk of all-cause mortality. Statin exposure was associated with lower risk of microbial, but not non-microbial, cancer. These findings were largely consistent between PWH and uninfected patients. Prospective, randomized studies, like the REPRIEVE trial, which is examining the efficacy of statins for the primary prevention of major adverse cardiovascular events in PWH with low to moderate traditional risk [68] may be able to assess the effect of specific statins on chronic inflammation/immune activation and HIV persistence. However, REPRIEVE’s main study endpoint is not cancer, therefore, we encourage future research to examine the reproducibility of our findings in both clinical trials and observational cohorts.
Supplementary Material
Funding:
This work was supported by the US Veterans Health Administration and by grants from the National Institute on Alcohol Abuse and Alcoholism (U01-AA020790, U24-AA020794, U01-AA013566, U01-AA026224) and National Cancer Institute (R01-CA210806, R01-CA206465) of the National Institutes of Health.
Contributor Information
Roger J. BEDIMO, Veterans Affairs North Texas Healthcare System, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Lesley S. PARK, Stanford University School of Medicine, Palo Alto, CA, USA.
Fatma SHEBL, Massachusetts General Hospital, Harvard Medical School, Boston MA, USA.
Keith SIGEL, Icahn School of Medicine at Mt. Sinai, New York, NY, USA.
Christopher T. RENTSCH, London School of Hygiene and Tropical Medicine, London, UK.
Kristina CROTHERS, VA Puget Sound Health Care System, University of Washington School of Medicine, Seattle, WA, USA.
Maria C. RODRIGUEZ-BARRADAS, Michael E. DeBakey Veterans Affairs Medical Center, Baylor College of Medicine, Houston, TX, USA.
Matthew Bidwell GOETZ, Veterans Affairs Greater Los Angeles Healthcare System, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA.
Adeel A. BUTT, VA Pittsburgh Healthcare System, Pittsburgh, PA, USA; Weill Cornell Medical College, New York, NY, USA and Doha, Qatar.
Sheldon T. BROWN, James J. Peters Veterans Affairs Medical Center, Bronx, NY, USA; Icahn School of Medicine at Mt. Sinai, New York, NY, USA.
Cynthia GIBERT, Washington DC Veterans Affairs Medical Center, George Washington University School of Medicine and Health Sciences, Washington, DC, USA.
References
- 1.Thurnher M, Gruenbacher G. T lymphocyte regulation by mevalonate metabolism. Sci Signal 2015; 8(370):re4. [DOI] [PubMed] [Google Scholar]
- 2.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 2008; 134(1):97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med 2001; 7(6):687–692. [DOI] [PubMed] [Google Scholar]
- 4.Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov 2005; 4(12):977–987. [DOI] [PubMed] [Google Scholar]
- 5.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med 2000; 6(12):1399–1402. [DOI] [PubMed] [Google Scholar]
- 6.Gruenbacher G, Thurnher M. Mevalonate metabolism governs cancer immune surveillance. Oncoimmunology 2017; 6(10):e1342917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graaf MR, Beiderbeck AB, Egberts AC, Richel DJ, Guchelaar HJ. The risk of cancer in users of statins. J Clin Oncol 2004; 22(12):2388–2394. [DOI] [PubMed] [Google Scholar]
- 8.Karp I, Behlouli H, Lelorier J, Pilote L. Statins and cancer risk. Am J Med 2008; 121(4):302–309. [DOI] [PubMed] [Google Scholar]
- 9.Farwell WR, Scranton RE, Lawler EV, Lew RA, Brophy MT, Fiore LD, et al. The association between statins and cancer incidence in a veterans population. J Natl Cancer Inst 2008; 100(2):134–139. [DOI] [PubMed] [Google Scholar]
- 10.Cholesterol Treatment Trialists C, Emberson JR, Kearney PM, Blackwell L, Newman C, Reith C, et al. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS One 2012; 7(1):e29849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cholesterol Treatment Trialists C, Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380(9841):581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi M, Zheng H, Nie B, Gong W, Cui X. Statin use and risk of liver cancer: an update meta-analysis. BMJ Open 2014; 4(9):e005399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, et al. Statins and the risk of colorectal cancer. N Engl J Med 2005; 352(21):2184–2192. [DOI] [PubMed] [Google Scholar]
- 14.Khurana V, Bejjanki HR, Caldito G, Owens MW. Statins reduce the risk of lung cancer in humans: a large case-control study of US veterans. Chest 2007; 131(5):1282–1288. [DOI] [PubMed] [Google Scholar]
- 15.Simon TG, Bonilla H, Yan P, Chung RT, Butt AA. Atorvastatin and fluvastatin are associated with dose-dependent reductions in cirrhosis and hepatocellular carcinoma, among patients with hepatitis C virus: Results from ERCHIVES. Hepatology 2016; 64(1):47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Facciorusso A, Abd El Aziz MA, Singh S, Pusceddu S, Milione M, Giacomelli L, et al. Statin Use Decreases the Incidence of Hepatocellular Carcinoma: An Updated Meta-Analysis. Cancers (Basel) 2020; 12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lytras T, Nikolopoulos G, Bonovas S. Statins and the risk of colorectal cancer: an updated systematic review and meta-analysis of 40 studies. World J Gastroenterol 2014; 20(7):1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang XL, Liu M, Qian J, Zheng JH, Zhang XP, Guo CC, et al. Statin use and risk of kidney cancer: a meta-analysis of observational studies and randomized trials. Br J Clin Pharmacol 2014; 77(3):458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan M, Song X, Zhang G, Peng A, Li X, Li M, et al. Statins and the risk of lung cancer: a meta-analysis. PLoS One 2013; 8(2):e57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang XL, Geng J, Zhang XP, Peng B, Che JP, Yan Y, et al. Statin use and risk of bladder cancer: a meta-analysis. Cancer Causes Control 2013; 24(4):769–776. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med 2012; 367(19):1792–1802. [DOI] [PubMed] [Google Scholar]
- 22.Kyriacou DN, Lewis RJ. Confounding by Indication in Clinical Research. JAMA 2016; 316(17):1818–1819. [DOI] [PubMed] [Google Scholar]
- 23.Psaty BM, Koepsell TD, Lin D, Weiss NS, Siscovick DS, Rosendaal FR, et al. Assessment and control for confounding by indication in observational studies. J Am Geriatr Soc 1999; 47(6):749–754. [DOI] [PubMed] [Google Scholar]
- 24.Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statins and cancer risk: a meta-analysis. JAMA 2006; 295(1):74–80. [DOI] [PubMed] [Google Scholar]
- 25.Chao C, Xu L, Abrams DI, Towner WJ, Horberg MA, Leyden WA, et al. HMG-CoA reductase inhibitors (statins) use and risk of non-Hodgkin lymphoma in HIV-positive persons. AIDS 2011; 25(14):1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overton ET, Kitch D, Benson CA, Hunt PW, Stein JH, Smurzynski M, et al. Effect of statin therapy in reducing the risk of serious non-AIDS-defining events and nonaccidental death. Clin Infect Dis 2013; 56(10):1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galli L, Spagnuolo V, Poli A, Salpietro S, Gianotti N, Cossarini F, et al. Use of statins and risk of AIDS-defining and non-AIDS-defining malignancies among HIV-1 infected patients on antiretroviral therapy. AIDS 2014; 28(16):2407–2415. [DOI] [PubMed] [Google Scholar]
- 28.Moore RD, Bartlett JG, Gallant JE. Association between use of HMG CoA reductase inhibitors and mortality in HIV-infected patients. PLoS One 2011; 6(7):e21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasmussen LD, Kronborg G, Larsen CS, Pedersen C, Gerstoft J, Obel N. Statin therapy and mortality in HIV-infected individuals; a Danish nationwide population-based cohort study. PLoS One 2013; 8(3):e52828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenbaum PR, Rubin DB. The Central Role of the Propensity Score in Observational Studies for Causal Effects. Biometrika 1983; 70(1):41–55. [Google Scholar]
- 31.Fultz SL, Skanderson M, Mole LA, Gandhi N, Bryant K, Crystal S, et al. Development and verification of a “virtual” cohort using the national VA health information system. Med Care 2006; 44(8):S25–S30. [DOI] [PubMed] [Google Scholar]
- 32.Fultz SL, Skanderson M, Mole LA, Gandhi N, Bryant K, Crystal S, et al. Development and verification of a “virtual” cohort using the National VA Health Information System. Med Care 2006; 448 Suppl 2):S25–30. [DOI] [PubMed] [Google Scholar]
- 33.Suissa S Immortal time bias in observational studies of drug effects. Pharmacoepidemiol Drug Saf 2007; 16(3):241–249. [DOI] [PubMed] [Google Scholar]
- 34.Suissa S Immortal time bias in pharmaco-epidemiology. Am J Epidemiol 2008; 167(4):492–499. [DOI] [PubMed] [Google Scholar]
- 35.Fritz APC, Jack A, Shanmugarathnam K, Sobin L, Parkin DM, Whelan S (editors). International Classification of Diseases for Oncology (ICD-O) Geneva: World Health Organization; 2000. [Google Scholar]
- 36.Surveillance, Epidemiology, and End Results Program. Site Recode ICD-O-3/WHO 2008 Definition. In. Bethesda, MD: Surveillance Research Program, National Cancer Institute; 2008. [Google Scholar]
- 37.Austin PC. A comparison of 12 algorithms for matching on the propensity score. Stat Med 2014; 33(6):1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austin PC. Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity-score matched samples. Stat Med 2009; 28(25):3083–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 1995; 57(1):289–300. [Google Scholar]
- 40.Park LS, Hernandez-Ramirez RU, Silverberg MJ, Crothers K, Dubrow R. Prevalence of non-HIV cancer risk factors in persons living with HIV/AIDS: a meta-analysis. AIDS 2016; 30(2):273–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gianella S, Letendre S. Cytomegalovirus and HIV: A Dangerous Pas de Deux. J Infect Dis 2016; 214 Suppl 2:S67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Compston LI, Li C, Sarkodie F, Owusu-Ofori S, Opare-Sem O, Allain JP. Prevalence of persistent and latent viruses in untreated patients infected with HIV-1 from Ghana, West Africa. J Med Virol 2009; 81(11):1860–1868. [DOI] [PubMed] [Google Scholar]
- 43.Park LS, Tate JP, Sigel K, Rimland D, Crothers K, Gibert C, et al. Time trends in cancer incidence in persons living with HIV/AIDS in the antiretroviral therapy era: 1997–2012. AIDS 2016; 30(11):1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverberg MJ, Chao C, Leyden WA, Xu L, Tang B, Horberg MA, et al. HIV infection and the risk of cancers with and without a known infectious cause. AIDS 2009; 23(17):2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franceschi S, Lise M, Clifford GM, Rickenbach M, Levi F, Maspoli M, et al. Changing patterns of cancer incidence in the early- and late-HAART periods: the Swiss HIV Cohort Study. Br J Cancer 2010; 103(3):416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hleyhel M, Belot A, Bouvier AM, Tattevin P, Pacanowski J, Genet P, et al. Risk of AIDS-defining cancers among HIV-1-infected patients in France between 1992 and 2009: results from the FHDH-ANRS CO4 cohort. Clin Infect Dis 2013; 57(11):1638–1647. [DOI] [PubMed] [Google Scholar]
- 47.Silverberg MJ, Lau B, Achenbach CJ, Jing Y, Althoff KN, D’Souza G, et al. Cumulative incidence of cancer among persons with HIV in North America: a cohort study. Ann Intern Med 2015; 163(7):507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park LS, Tate JP, Rodriguez-Barradas MC, Rimland D, Goetz MB, Gibert C, et al. Cancer incidence in HIV-infected versus uninfected Veterans: comparison of cancer registry and ICD-9 code diagnoses. Journal of AIDS & clinical research 2014; 5(7):1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calabresi A, Ferraresi A, Festa A, Scarcella C, Donato F, Vassallo F, et al. Incidence of AIDS-defining cancers and virus-related and non-virus-related non-AIDS-defining cancers among HIV-infected patients compared with the general population in a large health district of Northern Italy, 1999–2009. HIV medicine 2013; 14(8):481–490. [DOI] [PubMed] [Google Scholar]
- 50.Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Current opinion in oncology 2012; 24(5):506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shiels MS, Cole SR, Kirk GD, Poole C. A meta-analysis of the incidence of non-AIDS cancers in HIV-infected individuals. J Acquir Immune Defic Syndr 2009; 52(5):611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simard EP, Pfeiffer RM, Engels EA. Spectrum of cancer risk late after AIDS onset in the United States. Arch Intern Med 2010; 170(15):1337–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bedimo RJ, McGinnis KA, Dunlap M, Rodriguez-Barradas MC, Justice AC. Incidence of non-AIDS-defining malignancies in HIV-infected versus noninfected patients in the HAART era: impact of immunosuppression. J Acquir Immune Defic Syndr 2009; 52(2):203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park LS, Tate JP, Sigel K, Brown ST, Crothers K, Gibert C, et al. Association of Viral Suppression With Lower AIDS-Defining and Non-AIDS-Defining Cancer Incidence in HIV-Infected Veterans: A Prospective Cohort Study. Ann Intern Med 2018; 169(2):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kristoffersen US, Kofoed K, Kronborg G, Giger AK, Kjaer A, Lebech AM. Reduction in circulating markers of endothelial dysfunction in HIV-infected patients during antiretroviral therapy. HIV medicine 2009; 10(2):79–87. [DOI] [PubMed] [Google Scholar]
- 56.Ross AC, Armentrout R, O’Riordan MA, Storer N, Rizk N, Harrill D, et al. Endothelial activation markers are linked to HIV status and are independent of antiretroviral therapy and lipoatrophy. J Acquir Immune Defic Syndr 2008; 49(5):499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borges AH, Silverberg MJ, Wentworth D, Grulich AE, Fatkenheuer G, Mitsuyasu R, et al. Predicting risk of cancer during HIV infection: the role of inflammatory and coagulation biomarkers. AIDS 2013; 27(9):1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grund B, Baker JV, Deeks SG, Wolfson J, Wentworth D, Cozzi-Lepri A, et al. Relevance of Interleukin-6 and D-Dimer for Serious Non-AIDS Morbidity and Death among HIV-Positive Adults on Suppressive Antiretroviral Therapy. PLoS One 2016; 11(5):e0155100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis 2014; 210(8):1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ponroy N, Taveira A, Mueller NJ, Millard AL. Statins demonstrate a broad anti-cytomegalovirus activity in vitro in ganciclovir-susceptible and resistant strains. J Med Virol 2015; 87(1):141–153. [DOI] [PubMed] [Google Scholar]
- 61.Rothwell C, Lebreton A, Young Ng C, Lim JY, Liu W, Vasudevan S, et al. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 2009; 389(1–2):8–19. [DOI] [PubMed] [Google Scholar]
- 62.Martinez-Gutierrez M, Castellanos JE, Gallego-Gomez JC. Statins reduce dengue virus production via decreased virion assembly. Intervirology 2011; 54(4):202–216. [DOI] [PubMed] [Google Scholar]
- 63.Maziere JC, Landureau JC, Giral P, Auclair M, Fall L, Lachgar A, et al. Lovastatin inhibits HIV-1 expression in H9 human T lymphocytes cultured in cholesterol-poor medium. Biomedicine & Pharmacotherapy 1994; 48(2):63–67. [DOI] [PubMed] [Google Scholar]
- 64.Drechsler H, Ayers C, Cutrell J, Maalouf N, Tebas P, Bedimo R. Current use of statins reduces risk of HIV rebound on suppressive HAART. PLoS One 2017; 12(3):e0172175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Overton ET, Sterrett S, Westfall AO, Kahan SM, Burkholder G, Zajac AJ, et al. Effects of atorvastatin and pravastatin on immune activation and T-cell function in antiretroviral therapy-suppressed HIV-1-infected patients. AIDS 2014; 28(17):2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith CJ, Ryom L, Weber R, Morlat P, Pradier C, Reiss P, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet 2014; 384(9939):241–248. [DOI] [PubMed] [Google Scholar]
- 67.Vandenhende MA, Roussillon C, Henard S, Morlat P, Oksenhendler E, Aumaitre H, et al. Cancer-Related Causes of Death among HIV-Infected Patients in France in 2010: Evolution since 2000. PLoS One 2015; 10(6):e0129550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grinspoon SK, Fitch KV, Overton ET, Fichtenbaum CJ, Zanni MV, Aberg JA, et al. Rationale and design of the Randomized Trial to Prevent Vascular Events in HIV (REPRIEVE). Am Heart J 2019; 212:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.