

Abstract
Haemochromatosis is defined as systemic iron overload of genetic origin, caused by a reduction in the concentration of the iron regulatory hormone hepcidin, or a reduction in hepcidin-ferroportin binding. Hepcidin regulates the activity of ferroportin, which is the only identified cellular iron exporter. The most common form of haemochromatosis is due to homozygous mutations (specifically, the C282Y mutation) in HFE, which encodes hereditary haemochromatosis protein. Non-HFE forms of haemochromatosis due to mutations in HAMP, HJV or TFR2 are much rarer. Mutations in SLC40A1 (also known as FPN1; encoding ferroportin) that prevent hepcidin-ferroportin binding also cause haemochromatosis. Cellular iron excess in HFE and non-HFE forms of haemochromatosis is caused by increased concentrations of plasma iron, which can lead to the accumulation of iron in parenchymal cells, particularly hepatocytes, pancreatic cells and cardiomyocytes. Diagnosis is noninvasive and includes clinical examination, assessment of plasma iron parameters, imaging and genetic testing. The mainstay therapy is phlebotomy, although iron chelation can be used in some patients. Hepcidin supplementation might be an innovative future approach.
In this Primer, haemochromatosis is defined as systemic iron overload of genetic origin caused by a deficiency of hepcidin, including decreased production or decreased activity of hepcidin-ferroportin binding. Normally, hepcidin restricts iron transport into plasma. The definition of haemochromatosis used herein excludes acquired iron overload, such as overload caused by multiple blood transfusions, dyserythropoiesis or excessive parenteral iron supplementation. In addition, this definition excludes rare genetic disorders that lead to systemic iron excess by mechanisms other than primary hepcidin deficiency. These disorders include the loss-of-function ferroportin disease, atransferrinaemia, aceruloplasminaemia or divalent cation transporter 1 (DMT1; also known as NRAMP2)-related iron overload. Some of these disorders are occasionally considered as part of a wider haemochromatosis spectrum; indeed, ferroportin disease is officially referred to as type 4A haemochromatosis.
In most cases, haemochromatosis is caused by homozygous p.Cys282Tyr (C282Y) mutations in HFE (encoding hereditary haemochromatosis protein, or HFE, which has a role in hepcidin regulation). This mutation is found almost exclusively in white individuals and leads to HFE-associated haemochromatosis (also known as type 1 haemochromatosis). Haemochromatosis can also be caused by mutations in other genes (collectively referred to as non-HFE haemochromatosis), including HAMP (encoding hepcidin), HJV (also known as HFE2; encoding haemojuvelin) and TFR2 (encoding transferrin receptor protein 2) in addition to gain-of-function mutations in SLC40A1 (also known as FPN1, encoding ferroportin; BOX 1). Non-HFE haemochromatosis can affect both white and non-white individuals. Both HFE-associated and non-HFE-associated haemochromatosis lead to hepcidin deficiency, resulting in increased iron release from splenic macrophages and cells of the small intestine into plasma. Increased plasma iron levels lead to increased iron transport into parenchymal cells (particularly hepatocytes, pancreatic cells and cardiomyocytes), leading to predominantly hepatic, pancreatic and cardiac iron overload. The corresponding phenotype is characterized by increased plasma iron and transferrin saturation. Transferrin saturation is the ratio of the number of occupied iron binding sites to the total number of iron binding sites on plasma transferrin; increases in this parameter is critical for the diagnosis of haemochromatosis. In addition, strict interpretation of serum ferritin (the principal iron storage protein) concentration should be used for diagnosis. In general, patients with haemochromatosis are asymptomatic for many years and, in HFE-associated haemochromatosis, they develop symptoms only around 30–40 years of age. When symptoms manifest, they can be general and affect several organ systems (FIG. 1).
Box 1 |. Haemochromatosis subtypes.
|
Denotes juvenile haemochromatosis.
Sometimes corresponds to juvenile haemochromatosis.
Figure 1 |. Symptoms of haemochromatosis.
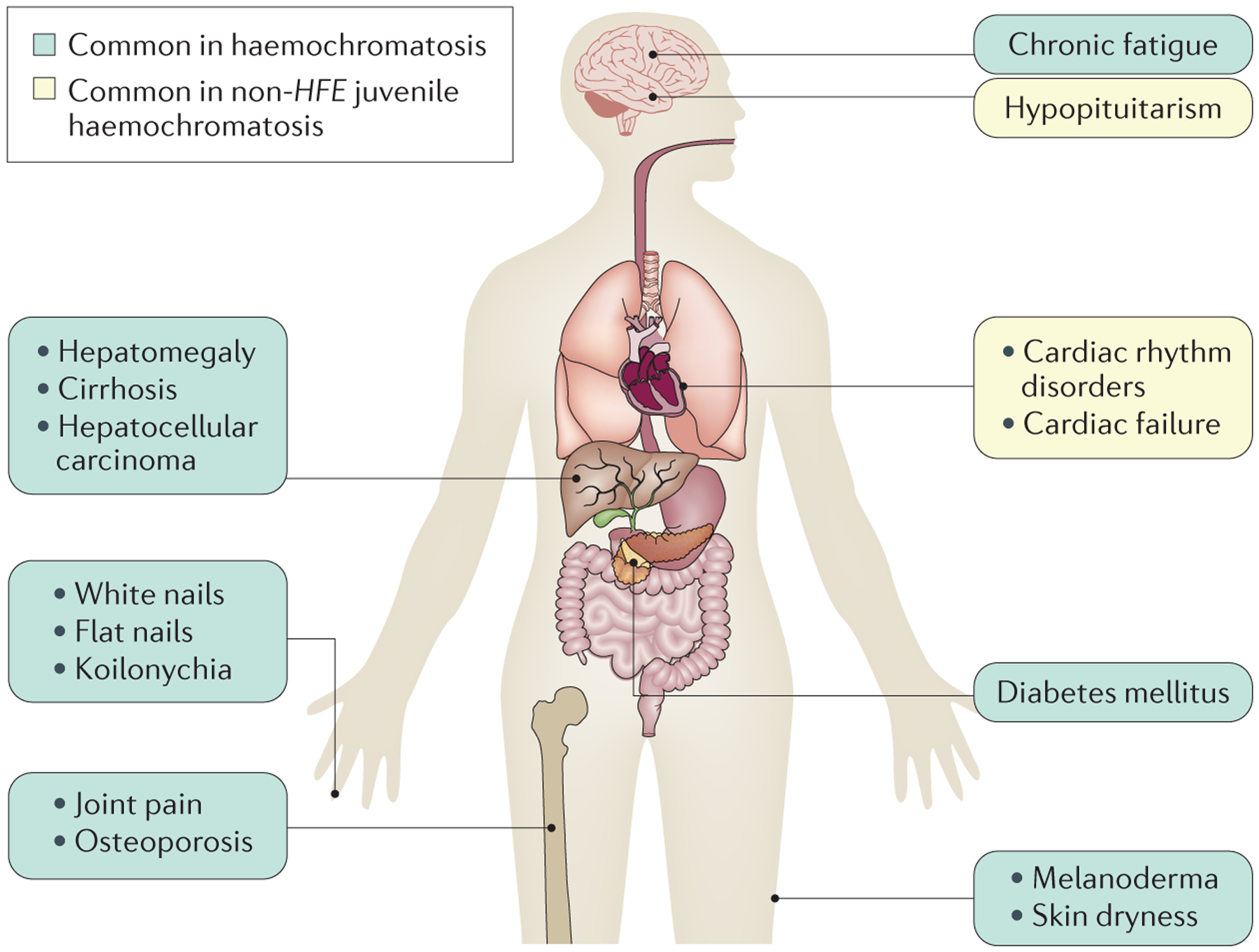
When symptoms develop, chronic fatigue is prominent. In addition, joint pain is frequent and is caused by acute or chronic monoarthritis, oligoarthritis or polyarthritis; arthritis in the second and third metacarpophalangeal joints and the ankles is particularly suggestive of haemochromatosis. Spontaneous fractures (particularly of the vertebrae) can occur owing to early-onset osteoporosis209. Dermatological signs are primarily melanoderma (darkening of the skin) but can also include skin dryness and nail changes, such as white nails, flat nails and koilonychia (that is, abnormally thin nails that curve inwards, also called ‘spoon’ nails). The main hepatic symptom is hepatomegaly. In contrast to most other liver diseases (notably due to alcohol use, viral infection or nonalcoholic fatty liver), cirrhotic livers in patients with haemochromatosis are well functioning, such that neither hepatocellular insufficiency or portal hypertension are usually observed in the absence of hepatotoxic cofactors. This finding explains why patients with haemochromatosis rarely develop complications of liver failure but develop hepatocellular carcinoma. In addition, diabetes mellitus and, more rarely, adrenal insufficiency or hypopituitarism can occur. Cardiac symptoms consist of cardiac rhythm disorders and cardiac failure. Anaemic syndrome is not observed in haemochromatosis; the presence of this symptom together with signs of iron excess suggests congenital atransferrinaemia210, hereditary aceruloplasminaemia125 and divalent cation transporter 1 (DMT1)-related iron overload208. Arthropathies are a frequent complication of haemochromatosis that frequently persists in patients despite otherwise successful iron depletive treatments, suggesting that this manifestation is not directly related to iron excess211. Whether persistent high plasma transferrin saturation levels159 with the presence of labile plasma iron are involved in arthropathies needs further investigation.
This Primer focuses on haemochromatosis caused by hepcidin deficiency and addresses the epidemiology, mechanistic and pathophysiological aspects of this disorder and the diagnosis, screening and management of patients. This Primer also discusses the quality of life of patients and summarizes some key remaining issues in the field. Other causes of iron overload aside from hepcidin deficiency are addressed, when relevant.
Epidemiology
The prevalence of the most common mutations in HFE (that is, C282Y and p.His63Asp (H63D)) that can cause haemochromatosis varies among ethnic groups1. The HEIRS study evaluated the prevalence and genetic and environmental determinants, among other factors, of haemochromatosis in a multi-ethnic primary care-based sample of 100,000 adults over a 5-year period in the United States and Canada2. Out of the 99,711 participants who did not have a family member participating in the study, 299 individuals were homozygous for C282Y. The estimated prevalence of C282Y homozygosity was 0.44% in non-Hispanic white individuals, 0.11% in native and indigenous Americans, 0.027% in Hispanic individuals, 0.014% in black individuals, 0.012% in individuals of Pacific Island descent and 0.000039% in Asian individuals3. By contrast, the prevalence of C282Y homozygosity was 1.2% in Ireland4. Moreover, an average prevalence of 0.4% for C282Y homozygosity and 9.2% for C282Y heterozygosity was demonstrated in one review of 27 studies that included 6,302 samples from European countries5. The same study demonstrated a prevalence of 0.5% for C282Y homozygosity and 9% for C282Y heterozygosity in North America. In Asian, Indian subcontinent, African, Middle Eastern and indigenous Australasian populations (including Aboriginal and Vanuatuan Australians and Papuans), C282Y homozygosity was not detected in 3,752 samples, although the frequency of C282Y heterozygosity ranged from 0% to 0.5%. The prevalence of C282Y/H63D compound heterozygosity and H63D homozygosity was 2% (for both) in the European general population; in the Americas, the prevalence of compound heterozygosity was 2.5% and the prevalence of H63D homozygosity was 2.1%5. Other studies reported the highest frequency of C282Y in non-Finnish Europeans (an allele frequency of 5.14%)6. TFR2, HJV and HAMP-related haemochromatosis were rare, with allele frequencies ranging between 0.00007 and 0.0004.
SLC40A1 variants have an allele frequency of 0.0004 in several populations and were most prevalent in African individuals6. Iron overload in individuals in sub-Saharan Africa has been recognized for many years7 and is not uncommon among indigenous South Africans8. This syndrome is referred to as African iron overload, Bantu siderosis or dietary iron overload9. The prevalence of African iron overload is up to 20% in sub-Saharan Africa9,10. In a survey of 505 rural Zimbabweans, iron overload was found almost exclusively in men who consumed traditional beer brewed in steel drums. In drinkers >45 years of age, 21% had high serum ferritin concentration and a transferrin saturation of >70%10.
Penetrance
Despite the high prevalence of C282Y homozygosity, only a minority of individuals will accumulate enough iron to cause organ damage. Given the autosomal recessive inheritance of C282Y, the frequency of C282Y homozygosity is similar in men and women, but the prevalence of clinical manifestations differs. Indeed, in one study, only 28.4% of men and 1.2% of women with C282Y homozygosity satisfied criteria for iron-related disease, consistent with a clinical diagnosis of haemochromatosis11. However, at the start of the study, 81.8% of men and 55.4% of women had increased serum ferritin levels, suggesting that biochemical penetrance is higher than clinical penetrance. Other studies have estimated that the clinical manifestations of haemochromatosis develop in 25–60% of individuals homozygous for C282Y12,13. Accordingly, other factors, such as genetic modifiers, environmental factors or lifestyle factors must modify susceptibility to haemochromatosis in individuals with C282Y homozygosity.
Modifier genes.
A single-nucleotide polymorphism (SNP) in CYBRD1 (rs884409) is a modifier gene for haemochromatosis and has exclusively been found in individuals homozygous for C282Y14, although this polymorphism alone is not a major modifier of the haemochromatosis phenotype15. In addition, the p.D519G polymorphism in GNPAT is associated with a high-iron phenotype in individuals with C282Y homozygosity16. Another study confirmed the association between the p.D519G polymorphism and increased iron stores in individuals homozygous for C282Y after correction for age, iron-related variables and alcohol consumption17. Mutations in other genes might also influence phenotypes in individuals homozygous for C282Y, including HAMP, HJV, TFR2, SLC40A1 and TMPRSS6 (REFS 18–25). In addition, mutations in BMP6 (REFS 26,27) increase iron overload and a polymorphism in PCSK7 is a strong genetic cofactor favouring the development of liver fibrosis in HFE-associated haemochromatosis28. Nutritional factors that could have a major role in the development of fibrosis include excessive alcohol intake29 and diabetes mellitus30, which could increase the effects of oxidative stress.
Other factors.
Iron is absorbed from the diet as haem iron (present in meat and fish) or as non-haem iron (mainly present in vegetables, cereals and other foods, although this type of iron is also found in meat)31,32. Haem iron is absorbed more readily than non-haem iron33 and dietary haem iron content is a significant predictor of iron stores in individuals homozygous for C282Y34,35. Indeed, the amount of dietary haem iron, but not non-haem iron, is positively associated with serum ferritin concentration36,37. However, one study did not demonstrate a significant relationship between serum ferritin concentration and dietary haem iron content, dietary non-haem iron content or supplemental iron use in individuals homozygous for C282Y38.
Heavy alcohol use by individuals with haemochromatosis increases the risk of cirrhosis39. Indeed, 7% of individuals with haemochromatosis caused by C282Y homozygosity who consumed <60 g of alcohol per day had severe fibrosis and/or cirrhosis compared with 61% of patients who consumed excess alcohol (that is, individuals who consume >60 g alcohol per day). Patients with haemochromatosis who consume >60 g alcohol per day are approximately nine-times more likely to develop cirrhosis than those who drink <60 g per day40.
Other environmental factors that could exert a protective effect against haemochromatosis in individuals homozygous for C282Y by decreasing iron absorption or increasing iron loss have been examined. For example, blood donations and physiological blood loss can influence iron stores and, accordingly, the phenotype of haemochromatosis. However, one study demonstrated that routine blood donation does not, on average, decrease the severity of iron overload in patients with haemochromatosis41 and one review concluded that these effects on disease penetrance are small42. The phenotypic expression of haemochromatosis is usually less pronounced in women than men, mainly due to menstruation, pregnancy or breastfeeding. In addition, hormonal factors — namely, testosterone — that can decrease hepcidin expression might contribute to the different phenotypic expression in men and women43.
In an investigation of genetic modifiers of HFE-associated haemochromatosis caused by the H63D mutation, no correlation was found between potential genetic modifiers and phenotype44. However, the H63D mutation together with alcohol, iron and calcium intake affects iron status45. Recessively inherited haemochromatosis includes juvenile haemochromatosis and TFR2-associated haemochromatosis, which have similar patterns of iron loading, albeit with differing severities and age at onset9.
Mechanisms/pathophysiology
Normal iron metabolism
Systemic iron regulation.
The maintenance of iron homeostasis includes the control of both iron concentration and biochemical forms of iron in plasma. Plasma iron is the main source of bioavailable iron for cells. Adequate body iron content and iron distribution require the maintenance of plasma iron concentration within normal limits (12–25 μM)46 and the regulation of transferrin saturation. Normal transferrin saturation is between 20% and 45% and allows adequate iron delivery to cells, thereby avoiding diseases of iron metabolism47. In mammals, iron is bound to transferrin in plasma, forming holotransferrin, which is the main biochemical form of iron in plasma. Transferrin is secreted by hepatocytes and receives up to two atoms of ferric iron (Fe(iii)) through an active process involving ferroxidase enzymes.
The main sources of plasma iron are enterocytes (which absorb iron from the gut lumen as non-haem or haem iron) and macrophages (which acquire iron from erythrophagocytosis; FIG. 2). The transport of iron from enterocytes and macrophages into the plasma occurs through ferroportin48–50, which is highly expressed on the basolateral membrane of enterocytes and the plasma membrane of macrophages and is the only identified iron exporter in mammals. Hepcidin — an iron-regulated peptide that is secreted by hepatocytes — is the main regulator of ferroportin activity51–53. Ferroportin-hepcidin binding induces the internalization, ubiquitination and degradation of ferroportin and, therefore, plasma hepcidin levels strongly affect plasma iron concentration54. Indeed, low hepcidin levels increase the export of iron from enterocytes and macrophages, thereby increasing plasma iron concentration and transferrin saturation. When transferrin saturation increases, non-transferrin bound iron (NTBI) can appear in plasma, which can lead to cell toxicity (see Iron-induced toxicity, below). Accordingly, the control of hepcidin expression levels is a cardinal factor in the control of systemic iron homeostasis.
Figure 2 |. Iron uptake, cycling and distribution within the body.
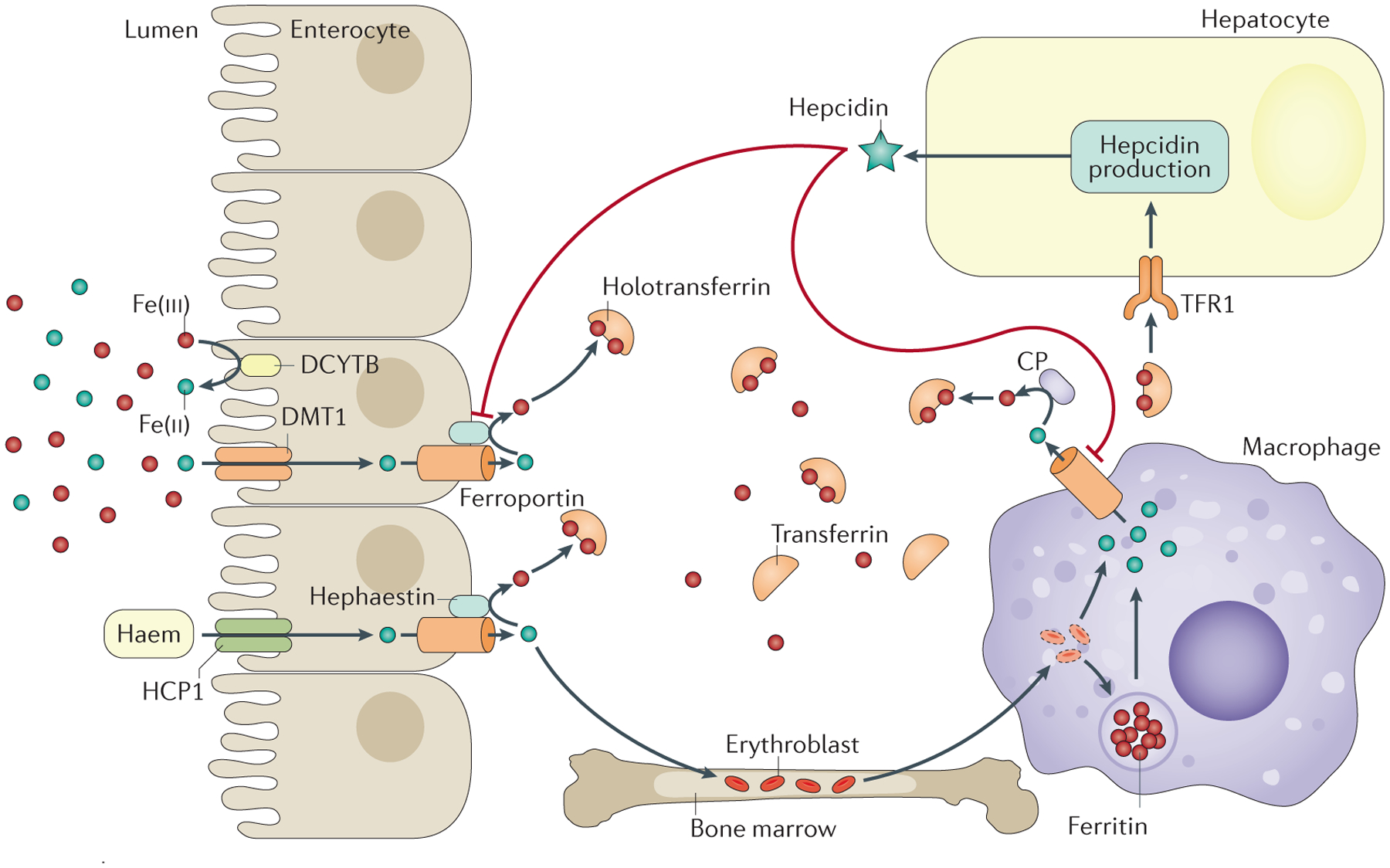
The main sources of plasma iron are enterocytes and macrophages. Non-haem iron is absorbed through divalent cation transporter 1 (DMT1), which is found on the apical surface of enterocytes212 after conversion of iron from its ferric (Fe(iii)) to its ferrous (Fe(ii)) form by a reduction process that can involve the duodenal cytochrome b reductase 1 (CYBDR1; also known as DCYTB) or other enzymes213. The mechanism of haem iron uptake by enterocytes is not fully elucidated, although it likely involves proton-coupled folate transporter (SLC46A1; also known as HCP1). By contrast, macrophages acquire iron from erythrophagocytosis (whereby erythrocytes are degraded and the contained iron is recycled). Iron is released from enterocytes and macrophages into plasma through ferroportin. Iron is then oxidized by ceruloplasmin (CP, which circulates in plasma) and hephaestin (which is anchored to enterocytes) and binds to transferrin (to form holotransferrin)203,214. CP has a role in the control of iron export from reticuloendothelial cells215, and both hephaestin and CP might have a role in cell iron metabolism in other tissues204,205. Holotransferrin delivers iron to every cell type, although erythroblasts (immature erythrocytes) are the main consumers. Holotransferrin levels are also sensed by hepatocytes that take up iron through transferrin receptor 1 (TFR1). In response to high levels of holotransferrin, hepcidin — which induces the degradation of ferroportin — is secreted into plasma to control the iron export. Figure adapted from REF. 216, Macmillan Publishers Limited.
The transcription activity of HAMP is the main regulator of hepcidin expression. Several pathways have been implicated in the regulation of HAMP transcription, including cell signalling in response to alterations in iron stores or transferrin saturation (FIG. 3; reviewed in REF. 47). The signalling pathways controlling HAMP transcription from iron stores involve a complex of proteins localized on the hepatocyte membrane (including HJV and the bone morphogenetic protein receptor (BMPR) that is reactive to bone morphogenetic protein 6 (BMP6) and, to a lesser extent, BMP2 (REFS 55,56). Interestingly, transmembrane pro-tease serine 6 (also known as matriptase-2; encoded by TMPRSS6) cleaves HJV and, therefore, strongly modulates this pathway; patients with TMPRSS6 mutations have very high levels of hepcidin, leading to severe iron deficiency that is resistant to oral iron supplementation (iron-refractory iron deficiency anaemia)57. Accordingly, modulating TMPRSS6 expression by altering HJV expression could modify the phenotype of haemochromatosis in patients, as suggested from data from haemochromatosis mouse models58. In addition, transferrin saturation is speculated to regulate hepcidin expression through HFE59,60 and TFR2 (REFS 61,62), which are expressed on hepatocyte membranes (FIG. 3), although an alternative or complementary mechanism could be interactions between HFE and the BMPR-HJV complex63. Hepcidin expression is downregulated by hypoxia through hypoxia-inducible factor (HIF)64 and platelet-derived growth factor BB65. Erythropoiesis might reduce hepcidin expression through erythroferrone that is excreted by erythroblasts66 and transforming growth/differentiation factor 15 (REF. 67). Chronic inflammation induces hepcidin expression at least partly through IL-6 and signal transducers and activators of transcription 3 (STAT3) signalling68–71 (FIG. 3). Hormones can also modulate hepcidin expression; for example, oestrogens and testosterone increase and decrease hepcidin expression, respectively43,72.
Figure 3 |. Hepcidin regulation.
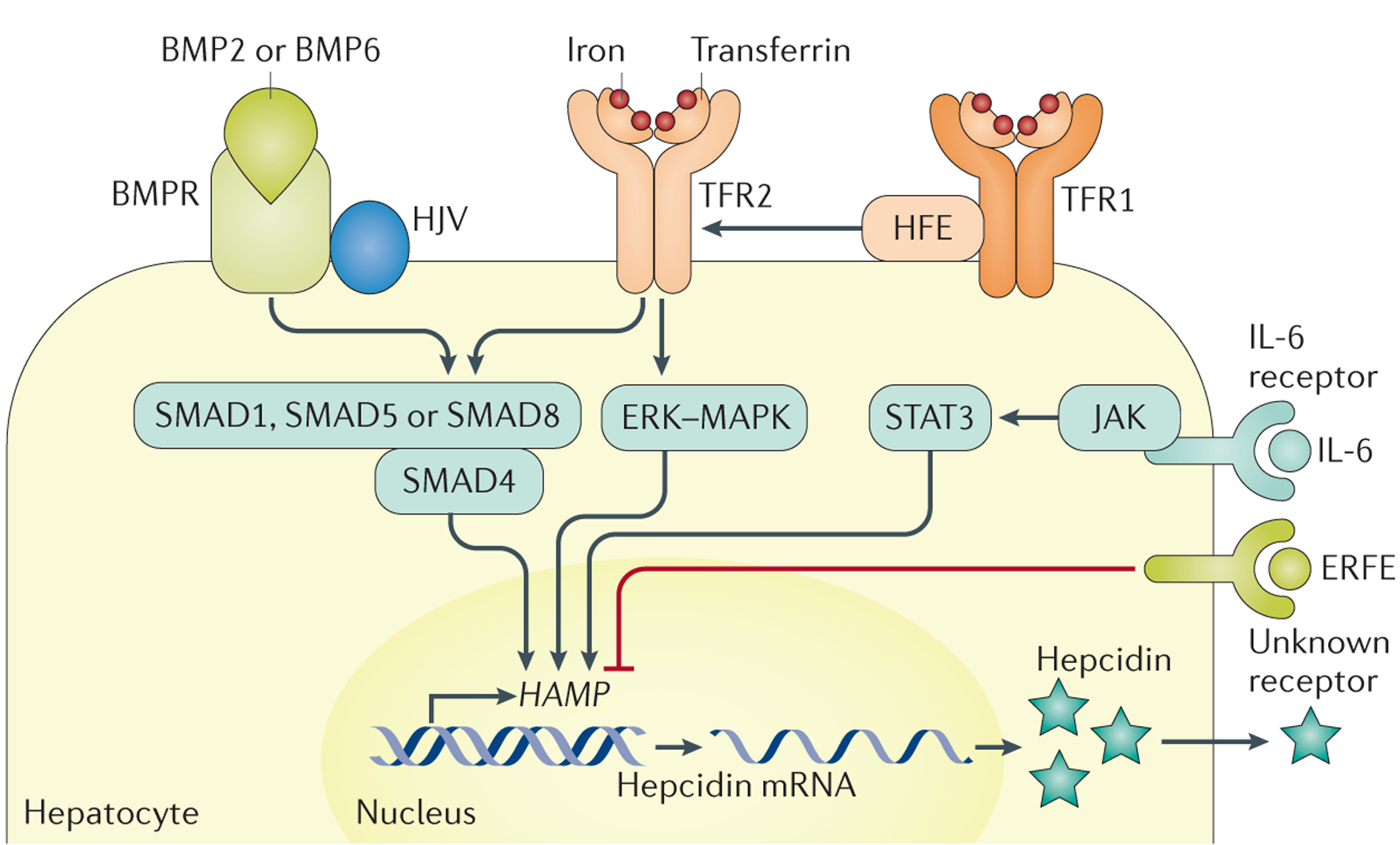
Increased cell iron stores lead to increased bone morphogenetic protein 6 (BMP6) expression in liver cells (sinusoidal cells are probably the major producers217,218, but hepatocytes and stellate cells might also be involved). BMP6 binds to the heterodimeric BMP receptor (BMPR) type 1 and type 2, which are bound to haemojuvelin (HJV)219,220. BMP-BMPR binding leads to BMPR phosphorylation, which leads to the phosphorylation of mothers against decapentaplegic homologue 1 (SMAD1), SMAD5 or SMAD8 (REF. 221). These proteins form a complex with SMAD4, which is transported into the nucleus and interacts with BMP-responsive elements on the HAMP promoter, leading to HAMP transcription and hepcidin expression102,222–224. Transferrin saturation is also involved in hepcidin regulation. This process could involve a shift of the interactions between hereditary haemochromatosis protein (HFE), transferrin receptor 2 (TFR2) and transferrin receptor 1 (TFR1) with increased transferrin saturation190, leading to signalling — potentially through mitogen-activated protein kinase (MAPK) and extracellular-signal-regulated kinase (ERK)225 — to increase HAMP transcription, although this process is not fully understood. The HFE-TFR1-TFR2 complex could also interact with the BMPR-HJV complex63. Other regulators of hepcidin expression include chronic inflammation, which is mediated by IL-6 produced by inflammatory cells and induces Janus kinase (JAK) and signal transducers and activators of transcription 3 (STAT3) activation68–71, in addition to erythroferrone (ERFE), which is produced by erythroblasts and interacts with unknown partners to decrease HAMP transcription. Figure adapted from REF. 216, Macmillan Publishers Limited.
Intracellular regulation of iron metabolism.
Holotransferrin binds to transferrin receptor 1 (TFR1) on the surface of every cell and is internalized by endocytosis73. Thereafter, ferric iron is reduced to ferrous iron by metalloreductase STEAP3 (REF. 74) and is transported into the cytosol by DMT1 (REFS 75,76). Cytosolic iron is used as a cofactor for many proteins and enzymes or is stored in a complex with ferritin. Ferritin is composed of 24 subunits of two different types (H and L) that can accumulate up to 4,500 iron atoms in a biochemically inactive form. The H subunit of ferritin has ferroxidase activity77 that allows the iron ingress as ferric iron and in turn its subsequent deposition within the ferritin protein in a chemically inactive form after a nucleation process78. This mechanism of iron sequestration avoids the oxidative stress that can occur through the Haber-Weiss and Fenton reaction when cytosolic iron concentration increases79.
Both the cellular influx and storage of iron are controlled by the iron responsive element-iron regulatory protein (IRE-IRP) system80. In cellular iron deficiency, the IRPs interact with the IRE nucleotide sequence located in the 3′ untranslated region (UTR) of the TFRC mRNA (encoding TFR1) and the 5′ UTR of ferritin mRNAs. The IRE-IRP interaction stabilizes TFRC mRNA, leading to increased TFR1 expression and increased holotransferrin internalization80. In addition, IRE-IRP interaction reduces ferritin translation and iron storage. Thus, IRP-IRE interaction increases cytoplasmic iron availability. Conversely, when iron accumulates in cells, IRE-IRP interaction is reduced, leading to decreased TFR1 expression and increased ferritin expression80.
Iron-induced toxicity
Cellular iron toxicity due to iron excess leads to the formation of reactive oxygen species, which can cause organ damage. Increased transferrin saturation can lead to the formation of abnormal forms of iron in plasma, such as NTBI81. Thus, NTBI can form when transferrin saturation is >45% (such as in patients with HFE-associated haemochromatosis)82. NTBI is not fully characterized but seems bound to low molecular weight molecules, including citrate and acetate83 or to albumin. In addition, small amounts of labile plasma iron (a highly reactive form of iron) can be detected in the plasma of patients with haemochromatosis when transferrin saturation is >75%84,85. Labile plasma iron, which could be one type of NTBI but does not cover the whole NTBI, is involved in the production of reactive oxygen species through the Haber-Weiss and Fenton reactions86. It is noteworthy that cellular iron excess alters mitochondrial function in the liver87,88.
NTBI has specific kinetics that contribute to cell toxicity. Indeed, uptake of NTBI occurs independently of TFR1 (REF. 89) and can involve zinc transporter ZIP14 (REFS 90,91), which is expressed by hepatocytes, and ZIP8, which is expressed by cardiomyocytes. In addition, L-type calcium channels might have a role in NTBI uptake by cardiomyocytes92, and pharmacological blockade of these channels has been shown to reduce iron accumulation in animal models93. NTBI uptake occurs despite iron overload94, in contrast to transferrin iron uptake that is downregulated through the IRE-IRP system in such situations. Parenchymal cells are prone to take up NTBI, especially hepatocytes and cells of the pancreas and heart95,96, which explains the main complications of haemochromatosis, such as liver fibrosis, cirrhosis, hepatocellular carcinoma, diabetes mellitus and heart failure.
Genetic haemochromatosis
Mutations causing plasma hypohepcidinaemia.
Plasma hypohepcidinaemia is caused by mutations in HAMP or in genes involved in the regulation of hepcidin levels. Mutations in HFE lead to HFE-associated haemochromatosis. HFE is a human leukocyte antigen (HLA) class I molecule that is expressed on cell membranes in association with β2-microglobulin. As previously mentioned, most cases of haemochromatosis involve homozygosity for C282Y59, which, owing to the disruption of a disulfide bond, affects the conformation of HFE and the interaction of this protein with β2-microglobulin60,97 and, consequently, the interaction with other potential binding partners, including TFR2 and/or BMPR-HJV. The phenotypic expression of HFE-associated haemochromatosis is less severe than that of haemochromatosis caused by mutations in HAMP, as hepcidin levels, although not adapted to the iron status, remain detectable98,99.
HAMP mutations cause haemochromatosis type 2B. This form of haemochromatosis is inherited in a recessive manner and is caused by homozygous or compound heterozygous mutations in the HAMP gene coding sequence that are extremely rare but lead to the juvenile form of haemochromatosis. Indeed, the absence of normal hepcidin in plasma leads to a bioclinical phenotype that fully recapitulates haemochromatosis100. Also, mutations in the HAMP promoter, although extremely rare, can affect hepcidin expression101,102. The absence of hepcidin allows constant ferroportin expression independent of the iron status, which allows excessive iron transport into plasma. Increased plasma iron levels increase transferrin saturation and favour the production of plasma NTBI89.
Homozygous mutations in HJV can cause haemochromatosis type 2A (also known as juvenile haemochromatosis), which is closely related to homozygous HAMP mutations25. These mutations, by limiting the activity of the BMPR-HJV-SMAD pathway, impair the upregulation of HAMP transcription in response to cellular iron store increases, therefore underlying the major role of the BMPR-HJV-SMAD pathway in the iron-related regulation of hepcidin expression.
Mutations in TFR2 can also lead to haemochromatosis103,104, corresponding to haemochromatosis type 3. The phenotype is usually intermediate between HFE-associated, HJV-associated and HAMP-related haemochromatosis.
The combination of heterozygous mutations in two different genes involved in iron metabolism has been reported to cause iron overload105–108. This finding suggests that mutations in different genes involved in the control of hepcidin transcription have a cumulative effect on hepcidin level and that additional rare mutations in HFE and in other genes that have a role in iron metabolism should be investigated109. Moreover, every mechanism potentially modulating hepcidin expression in addition to iron gene mutations can modulate the phenotype of patients.
Mutations causing loss of activity of hepcidin.
A loss of hepcidin activity can mainly result from missense mutations in HAMP or SLC40A1. The biological activity of hepcidin requires an interaction with ferroportin on the cell membrane, which involves specific domains in both proteins.
In ferroportin, some amino acids that interact with hepcidin have been identified, and mutations that affect these amino acids cause a loss of ferroportin sensitivity to hepcidin110–114. The identification of these amino acids explains why some mutations in SLC40A1 can lead to an iron overload phenotype that is similar to hepcidin deficiency. This form of haemochromatosis corresponds to a rare form of iron overload caused by some rare mutations in SLC40A1 and should be, in our view, named haemochromatosis type 4 (BOX 1). Other mutations in SLC40A1 lead to ferroportin loss of function, and patients have a different phenotype to haemochromatosis (ferroportin disease).
A domain of hepcidin that mediates the biological effect on ferroportin has been reported115. It is noteworthy that missense mutations in HAMP could cause a decrease of hepcidin activity that could have a role in the development of iron overload owing to non-functionality of hepcidin. Similarly, mutations in the HAMP promoter that strongly decrease hepcidin expression might favour or aggravate iron overload phenotype101,102.
Ferroportin disease.
Heterozygous mutations in SLC40A1 that lead to ferroportin loss of function might lead to systemic iron overload, termed ferroportin disease. In our view, ferroportin disease does not correspond to haemochromatosis despite the current classification as haemochromatosis type 4A116,117. Mutations in SLC40A1 can be found all along the coding sequence of the gene118. The deficiency in iron export that occurs in ferroportin disease might result from a defect of intracellular ferroportin trafficking to the plasma membrane and/or loss of the iron-export function of ferroportin113,119. In ferroportin disease, iron is sequestered in macrophages. Although macrophages deliver ~19–20 mg of iron per day to plasma, plasma iron concentration and transferrin saturation are rarely decreased in patients with ferroportin disease. Moreover, accumulation of iron in hepatocytes can occur, and increased transferrin saturation might be observed in the late stage of disease. Such findings suggest119 that macrophages are severely affected by ferroportin disease owing to the important volume of iron that they provide to plasma, whereas digestive iron absorption could be less affected, with the remaining normal gene being sufficient to ensure iron trafficking.
Diagnosis, screening and prevention
In general, diagnosis of haemochromatosis involves a sequential noninvasive strategy that combines clinical, imaging and biological data120 (FIG. 4).
Figure 4 |. Diagnostic chart for systemic iron overload of genetic origin.
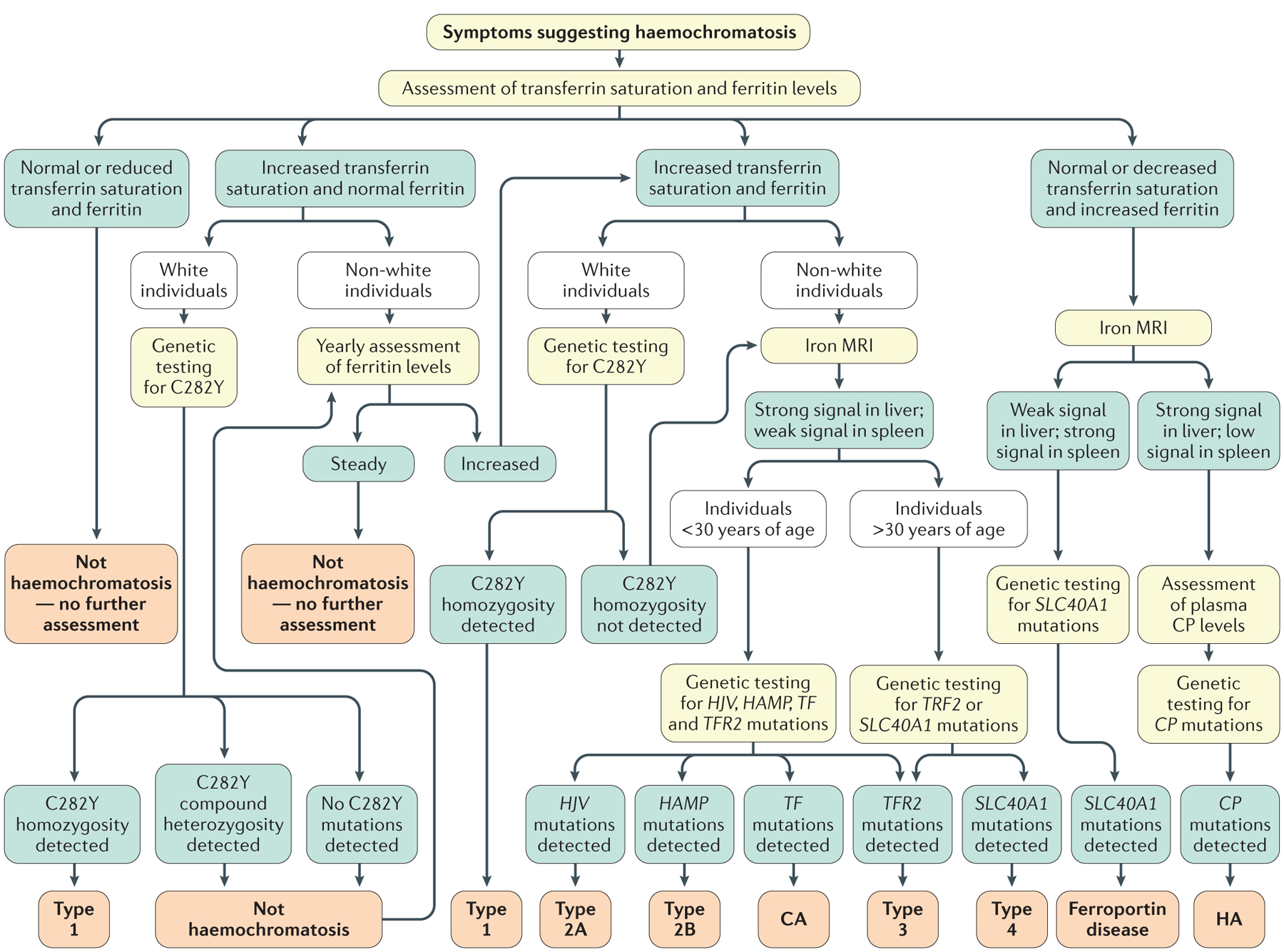
This algorithm excludes the diagnosis of acquired iron overload owing to blood transfusions, dyserythropoiesis or parenteral iron supplementation. Several factors should be considered as part of the initial diagnostic workup for haemochromatosis, such as patient ethnicity (as HFE-associated haemochromatosis is observed almost exclusively in white individuals) and sex (as the phenotypic expression of haemochromatosis is usually less pronounced in women). In addition, patient age should be taken into consideration, as HFE-associated (type 1) or TFR2-associated (type 3) haemochromatosis is generally observed in individuals >30 years of age, whereas clinical expression in younger individuals is typical of HJV-related (type 2A) or HAMP-related (type 2B) haemochromatosis. The diagnostic approach should also consider that non-HFE haemochromatosis diseases are rare, in contrast to HFE-associated haemochromatosis6. Type 4 refers to haemochromatosis caused by gain-of-function mutations in SLC40A1, previously named haemochromatosis type 4B. CA, congenital atransferrinaemia; CP, ceruloplasmin; HA, hereditary aceruloplasminaemia.
Clinical features
Haemochromatosis can be asymptomatic for several years. Indeed, the absence of symptoms can persist until adolescence or young adulthood in most forms of non-HFE haemochromatosis and up to 30–40 years of age in men and 40–50 years of age in women with HFE-associated disease. In general, symptoms are miscellaneous, variable and frequently of mundane nature (FIG. 1), which explains the frequent and damaging diagnostic delay121–123. Clinically, multiple, diversely associated symptoms should suggest the possibility of iron overload.
Biological factors.
Several biological parameters, including increased serum ferritin levels and elevated transferrin saturation can indicate the presence of haemochromatosis.
Increased transferrin saturation (>45%) is the earliest biochemical sign observed in all haemochromatosis sub-types. However, mechanisms aside from iron overload can cause increased transferrin saturation, such as other causes of increased plasma iron (for example, haemolysis and cytolysis) or decreased plasma transferrin concentration (for example, by acquired hepatocellular failure, poor nutrition, proteinuria or genetic alterations)124. Moreover, normal or even low transferrin saturation can be present despite overt body iron overload, such as in individuals with ferroportin disease117 or in hereditary aceruloplasminaemia125 (BOX 2).
Box 2 |. Other genetic disorders leading to systemic iron load.
Hereditary aceruloplasminaemia is caused by homozygous mutations in CP (encoding ceruloplasmin). This disorder is characterized by very low levels of plasma iron and transferrin saturation, which causes anaemia, but patients have systemic iron excess. One explanation is the loss of the ferroxidase activity of ceruloplasmin203, limiting iron release from macrophages into plasma. However, in patients, the spleen is not overloaded with iron, and the liver has iron deposition in hepatocytes, suggesting that the precise mechanisms governing organ iron distribution are not yet understood125. The potential effect of anaemia in favouring iron excess through the downregulation of hepcidin expression has been proposed. Whether compensatory ferroxidase activity by hephaestin has a role in ensuring iron export from macrophages despite low ceruloplasmin-related ferroxidase activity should be considered204,205.
Mutations in TF (encoding transferrin) lead to the development of anaemia owing to the absence of transferrin-bound iron available for erythroid cells. These mutations lead to very low plasma transferrin levels, which favour the appearance of non-transferrin bound iron (NTBI). Indeed, NTBI is the predominant form of plasma iron in these individuals. In addition, hepcidin expression is decreased owing to anaemia, leading to subsequent improved iron absorption, which, together with plasma NTBI, contributes to the development of iron overload in the liver, pancreas and heart206.
Mutations in DMT1 leading to DMT1-related iron overload affect both iron absorption (owing to the role of DMT1 in non-haem iron uptake by enterocytes) and intracellular iron trafficking (owing to the role of DMT1 in iron transfer from endocytic vesicles towards the cytosol, particularly in erythroblasts). Thus, anaemia is one of the major consequences of DMT1 mutations and presents early in life. It is noteworthy that visceral iron overload is found in parallel, suggesting that other mechanisms are involved in the disease, including the potential role of increased digestive iron absorption by an adaptive mechanism and the presence of NTBI207,208.
Whereas low ferritin levels always correspond to iron deficiency, increased ferritin levels (≥300 μg per litre in men and ≥200 μg per litre in women) need rigorous interpretation before they are assigned to iron overload. Thus, four main mechanisms that can account for increased ferritin levels independent of substantial iron overload should be ruled out: metabolic syndrome (which is the most frequent cause), alcoholism, inflammation and marked cytolysis. Despite these limitations, increased ferritin levels are critical for the diagnosis of haemochromatosis.
Several parameters can indicate organ-specific increased iron levels, for example, mild hypertransaminasaemia (less than three-times the upper normal limit) can indicate chronic hepatic iron excess, and hyperglycaemia can reflect pancreatic iron excess.
Imaging findings.
Radiological findings, such as chondrocalcinosis of the wrists or knees or subchondral arthropathy that can be detected using X-ray can suggest haemochromatosis. In addition, low T2-weighted signal on liver MRI can suggest haemochromatosis. Indeed, whenever available, iron excess should be confirmed by iron MRI. One of the most convenient techniques for iron MRI is based on the signal intensity ratio approach126–128, which enables the visualization and quantification of hepatic, splenic and pancreatic iron excess by assessing T2-weighted hyposignal, without requiring specific 1.5 Tesla MRI equipment and using free online software (FIG. 5). When using 3 Tesla MRI, which is becoming more widespread, the R2* method provides a more precise quantification of slight or moderate iron overload than signal intensity ratio, whereas signal intensity ratio is more accurate for high iron overload129. The use of R2* measurements in both the liver and the pancreas have been proposed to predict the total amount of iron stores in patients with haemochromatosis130. In addition, iron MRI can be used to differentiate haemochromatosis owing to hepcidin deficiency from ferroportin disease (FIG. 5). CT has a very poor sensitivity for visualizing iron excess, and ultrasonography cannot detect iron overload.
Figure 5 |. MRI findings in haemochromatosis owing to hepcidin deficiency and ferroportin disease.
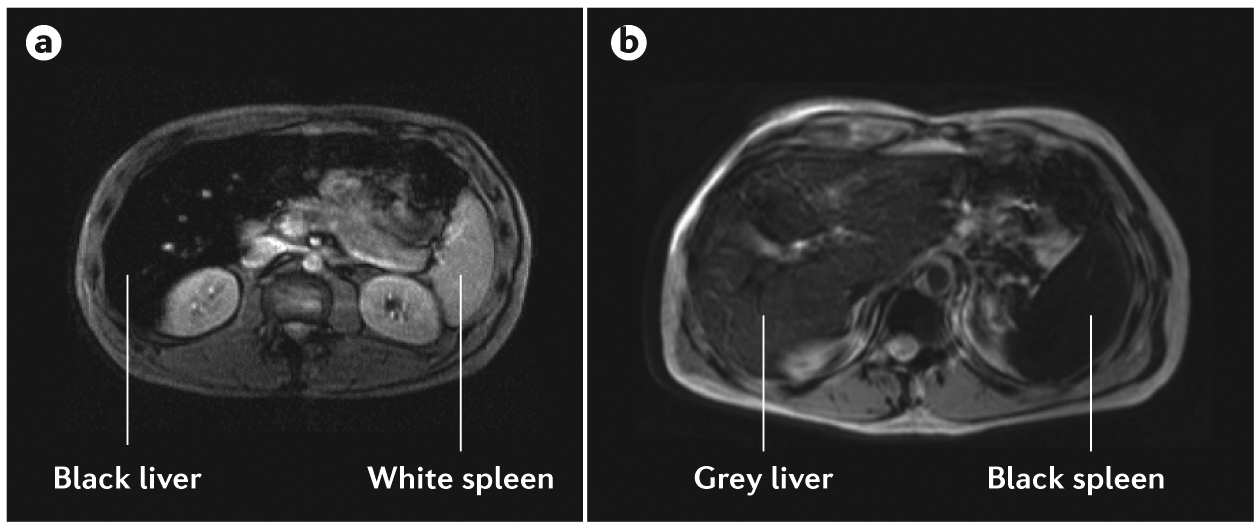
The signal intensity ratio technique with T2-weighted MRI can be used to differentiate patients with haemochromatosis and those with ferroportin disease. a | In patients with hepcidin-deficient haemochromatosis, ‘black’ liver corresponds to a highly iron-overloaded liver, and ‘white’ spleen corresponds to the absence of iron overload. The appearance of the liver and spleen is similar in patients with types 1, 2A, 2B, 3 and 4 haemochromatosis. b | In ferroportin disease, the spleen appears black (highly iron-overloaded) and the liver appears grey (moderately iron-overloaded) or black (highly iron-overloaded) on T2-weighted MRI47.
Liver biopsy.
Liver biopsy is no longer necessary for asserting and quantifying iron excess and cellular iron distribution, and has been largely replaced by the combined evaluation of biological and imaging findings. However, liver biopsy can be important for assessing hepatic complications of haemochromatosis, such as fibrosis131.
Confirming genetic iron overload.
Confirming genetic iron overload requires excluding causes of acquired iron overload. Four main types of disorder should be excluded: haematological disorders, disorders due to excessive iron supplementation, metabolic syndrome and chronic alcoholism. In terms of haematological disorders, iron excess can be caused by transfusional iron overload132 and/or dyserythropoiesis133,134, such as in congenital or acquired chronic anaemias (mostly thalassaemia and myelodysplastic syndromes), as well as hereditary spherocytosis, which might be related to dyserythropoiesis135. In addition, excessive parenteral iron supplementation can lead to iron overload, especially in the setting of chronic renal failure with haemodialysis136. Dysmetabolic iron overload syndrome likely has a multifactorial pathogenesis and can lead to mild iron excess, which is sometimes observed in association with one or several metabolic alterations (increased body mass index, increased blood pressure, hyperlipidaemia, hyperglycaemia, hyperuricaemia or hepatic steatosis). Dysmetabolic iron overload syndrome is associated with increased ferritin levels and mild hepatic iron overload; the key differential parameter from haemochromatosis is that plasma transferrin saturation is not increased137. Finally, chronic alcoholism can cause moderate hepatic iron excess, possibly owing to alcohol-related hepcidin deficiency138.
Identifying the genetic cause of the disease is easy for HFE-associated haemochromatosis139. For example, this disorder is almost always found in white individuals by identifying C282Y homozygosity, which can be carried out by numerous laboratories. As mentioned previously, simple C282Y heterozygosity cannot cause iron excess and C282Y/H63D compound heterozygosity can cause some plasma transferrin saturation increase but not substantially increased ferritin levels or considerable organ iron overload. At most, compound heterozygosity might lead to mild iron overload in situations such as chronic alcoholism or metabolic syndrome. Notably, rare HFE variants are more prevalent than variants in other genes involved in iron metabolism in individuals with suggestive hepcidin deficiency-related haemochromatosis but without C282Y homozygosity109,140. Thus, the detection of C282Y heterozygosity in a patient with considerable systemic iron overload should lead to the search for other genetic and/or acquired causes of iron excess.
The genetic confirmation of non-HFE haemochromatosis requires testing carried out by specialized laboratories after the appropriate selection of required genetic tests by an expert reference centre. High-throughput techniques (especially next-generation sequencing) offer powerful diagnostic tools, although the interpretation of these tests is more demanding than rigorous clinical characterization and selection.
Haemochromatosis severity
After the diagnosis is confirmed, the severity of haemochromatosis should be assessed by determining the amount of body iron excess and the extent of organ damage. A five-grade classification can be used for haemochromatosis related to hepcidin deficiency, which is similar to the classification initially proposed for HFE-associated haemochromatosis141 (FIG. 6). This classification is based on the presence or absence of increased plasma transferrin saturation, increased plasma ferritin, affected quality of life and signs jeopardizing prognosis. For example, plasma ferritin levels >1,000 μg per litre at diagnosis have been reported to correspond to an increased risk of death in HFE-associated haemochromatosis142. This classification does not apply for the loss-of-function ferroportin disease143.
Figure 6 |. Proposed phenotypic classification of haemochromatosis related to hepcidin deficiency.
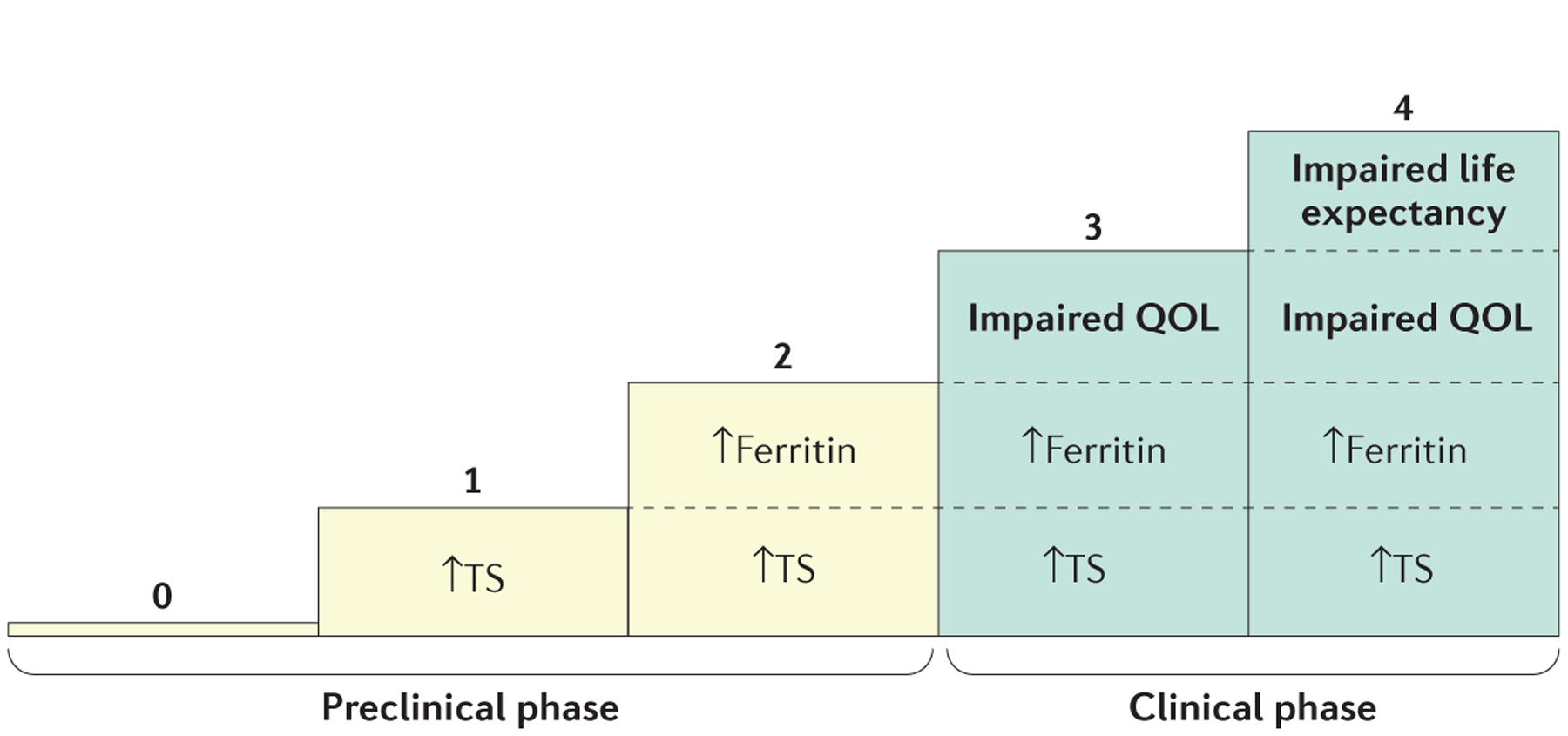
Increased ferritin: ≥300 μg per litre for men and ≥200 μg per litre for women; increased transferrin saturation (TS): >45% (often 60–100%). QOL, quality of life. Adapted with permission from REF. 141, Elsevier.
Screening and prevention
HFE-associated haemochromatosis is theoretically the archetype of genetic diseases, justifying active screening and prevention for several reasons. For example, this disorder is one of the most prevalent genetic diseases, diagnosis is minimally invasive and the disease can be treated with phlebotomy, which is efficient, simple and inexpensive. As soon as an individual has been diagnosed with haemochromatosis, they must be considered as a proband, and cascade family screening should be initiated.
The methodology of screening for haemochromatosis is based on genetic testing. For all forms of haemochromatosis, the absence of the proband’s mutation in their relative means that the relative is free of any risk of that form of haemochromatosis. For individuals with recessive subtypes of haemochromatosis (that is, types 1, 2 and 3), the presence of simple heterozygosity means they do not have a risk of developing the disease but risk transmission to their offspring, whereas for individuals with dominant subtypes of haemochromatosis (such as haemochromatosis type 4), heterozygosity means risk of developing the disease. Siblings of the proband are the preferred target for screening after a family member has been diagnosed with recessive forms of haemochromatosis. However, in individuals with HFE-associated haemochromatosis, the high prevalence of the C282Y mutation among white individuals justifies the screening of offspring (although this screening is usually deferred until the offspring reach adulthood). Despite the efficiency of cascade screening and the fact that this method can determine the pathogenicity of a given genetic variant, this screening strategy is too rarely adopted, likely owing to the practical difficulties in reaching scattered relatives and in collating family data. Specific centres devoted to family screening are the solution but remain quite rare.
Growing evidence highlights the importance of conducting population screening for HFE-associated haemochromatosis144,145. The HEIRS study did not recommend conducting HFE genotyping before phenotyping146; thus, initially phenotyping individuals might be the preferred option. In practice, plasma transferrin saturation could be checked once in young adults and once again, for example, 5 years after menopause, which could form the basis of population screening and allow every individual with increased transferrin saturation to undergo testing for C282Y and, if warranted, treatment for haemochromatosis.
Management
Iron removal
Phlebotomy.
Phlebotomy remains the mainstay of treatment for HFE-associated haemochromatosis. For non-HFE-associated haemochromatosis that is related to hepcidin deficiency, phlebotomy is the preferred treatment147, but adjunctive oral chelation can be used in the most severe cases (for example, individuals with juvenile haemochromatosis). Phlebotomies are also efficient for treatment of patients with loss-of-function ferroportin disease but should be carried out on a less intensive schedule given the risk of anaemia owing to poor iron recycling in these patients. Many physicians and patients insist that high serum ferritin levels equal iron overload and should be treated by phlebotomy. However, as previously mentioned, patients without C282Y homozygosity might not always have iron overload.
The goal of phlebotomy therapy is to remove excess iron to prevent any further tissue damage. For ethical reasons, phlebotomy therapy has not been investigated in randomized clinical trials, which has hindered our understanding of the natural history of untreated disease148,149. Although some individuals, albeit rarely, have suggested phlebotomy therapy has no supporting evidence150, most experts believe that this form of iron depletion can improve chronic fatigue and cardiac function, stabilize liver disease, reverse hepatic fibrosis and reduce skin pigmentation in patients with haemochromatosis147. However, joint symptoms respond poorly to phlebotomy therapy and can worsen151. The effectiveness of phlebotomy is good provided it has been started before the development of cirrhosis. Adverse effects of phlebotomy occur in 37–50% of patients152 and include phlebitis, malaise and fatigue. If adverse effects occur, hot compress to the skin, oral fluid replacement and increasing the interval between treatments can be considered.
For phlebotomy induction therapy, patients usually attend an ambulatory care facility where the treatment is performed by a nurse. Typically, a volume of 400–500 ml of blood is removed over 15–30 minutes with the patient in the reclining position. This process is carried out on a weekly basis until the serum ferritin level is ~50 μg per litre. Haemoglobin levels are also assessed, and the phlebotomy schedule is modified if haemoglobin levels are <11 g per decilitre (for example, to 400–500 ml of blood removal every other week)147. The concomitant oral administration of liquid (such as a salty sport beverage) of an equivalent volume to the volume of blood removed by phlebotomy is important for maintaining plasma volume during the procedure. The duration of induction therapy depends on the severity of iron overload and can range from months to years.
After the induction phase, maintenance phlebotomies are carried out to maintain serum ferritin levels at ~50 μg per litre. If phlebotomy is continued until serum ferritin levels are <20 μg per litre, iron deficiency or iron reaccumulation can occur153. Serum hepcidin levels can decrease with phlebotomy therapy154. Maintenance phlebotomies are carried out on average two to four times per year, and this depends on the rate of iron reaccumulation, which is variable between patients155,156. Checking serum ferritin levels 3–6 months after induction therapy has ended can be useful to estimate the rate of iron reaccumulation. Assessing serum ferritin levels can be carried out monthly during induction phlebotomy and weekly when the serum ferritin has decreased to 100 μg per litre. Once serum ferritin is 50 μg per litre, assessment of serum ferritin levels annually or at the time of each maintenance phlebotomy can be considered. Although the evidence supporting the use of maintenance therapy is lacking, many patients appreciate this therapy, particularly if they can be voluntary blood donors157,158.
Despite successful iron depletion, transferrin saturation will remain increased in several patients. One report suggested that maintaining normal transferrin saturation might improve symptoms to a greater degree than lowering ferritin but not transferrin saturation159. However, transferrin saturation might not decrease until the patient is almost iron deficient, so maintaining appropriate ferritin levels and transferrin saturation might be difficult.
Some patients have been treated by erythrocytapheresis160, but this technique is more expensive and less available than phlebotomy. One study in Australia demonstrated an improvement in a fatigue scale in patients (with ferritin levels of 300–1,000 μg per litre) receiving erythrocytapheresis and who had iron reduction compared with patients receiving sham pheresis, suggesting the potential benefit of blood removal even in mild iron overload161,162.
Chelation therapy.
Chelation therapy with deferoxamine is not recommended for any subtype of haemochromatosis. Deferoxamine is expensive, inefficient and cumbersome and can lead to adverse effects, such as blurred vision, dizziness, tinnitus, flushing, skin rash and diarrhoea163.
Other therapies and dietary factors
Patients with haemochromatosis are advised to avoid oral iron therapy and alcohol abuse, but there are no proven dietary restrictions38. Patient support groups have been discouraged by the iron fortification of foods, although most of this iron is an inexpensive form with poor bio-availability. Iron fortification of foods has been stopped in Sweden, and a reduction in the mean serum ferritin levels has been demonstrated in the general population164. Tea consumption165 and the use of proton pump inhibitors have been shown to decrease intestinal iron absorption166. Indeed, one trial demonstrated a reduction in annual phlebotomy requirements by 1.33 units per year in individuals with C282Y homozygosity who receive daily proton pump inhibitor therapy166. Patients with haemochromatosis should avoid consuming raw shellfish, especially in subtropical areas, as patients have an increased risk of Vibrio spp. infections167.
Liver transplantation
Despite the high prevalence of haemochromatosis, it remains an uncommon indication for liver transplantation. For example, in Brisbane, Australia, 0.6% of liver transplants were carried out for haemochromatosis168. The main indication for liver transplantation in haemochromatosis is hepatocellular carcinoma168. Many individuals diagnosed with haemochromatosis and who received liver transplantation owing to decompensated cirrhosis are more likely to have had iron overload secondary to cirrhosis from other causes169. Pretransplant phlebotomy might improve cardiac function and is recommended if tolerated. The lack of recurrence of iron overload together with normalization of plasma hepcidin levels after liver transplantation have provided the clinical proof that hepcidin is central to the pathogenesis of haemochromatosis170.
Hepcidin therapies
As low hepcidin levels or hepcidin activity have been implicated in the pathogenesis of haemochromatosis, hepcidin analogues have been proposed as a therapy171. Early reports suggest that this approach is better suited for maintenance therapy as these drugs do not decrease iron storage in the liver172. These treatments will likely be parenteral and expensive, which will be difficult to compete with phlebotomy therapy.
Quality of life
Several studies have demonstrated a negative effect of haemochromatosis on health-related quality of life (HRQOL). Elevated iron stores, particularly transferrin saturation and serum ferritin levels and the presence of comorbidities, contribute to reduced HRQOL.
The Short Form 36 (SF-36) is the most commonly used instrument to assess HRQOL for patients with haemochromatosis173. The SF-36 consists of eight domains that provide physical component and mental component scores that range between 0 and 100 (higher scores indicate a higher HRQOL). In the United States, the population normative score is 50 (with a s.d. of 10)174. Individuals with increased transferrin saturation (≥45% for females and ≥50% for males) have a worse HRQOL assessed using the SF-36 than participants with normal transferrin saturation175. Both the physical and mental component scores were significantly lower in individuals with increased transferrin saturation175. Although not examined in this study, the association between higher iron stores and lower HRQOL might be due to increased fatigue and lethargy176,177. The effect of haemochromatosis-related comorbidities on QOL has also been evaluated using the SF-36 (REF. 178). Interestingly, arthritis was associated with a lower overall HRQOL than cirrhosis or diabetes mellitus in patients with completed phlebotomy treatment (with serum ferritin levels <50 μg per litre)178. In addition, one study reported a physical component score of 30.5 (which is 2 s.d. lower than the mean values in the United States) and a mental component score of 77.7 (which is almost 3 s.d. above the normal values in the United States) in patients with painful osteo-arthritis of the hind foot174,179. Patients with a recent diagnosis of haemochromatosis who were deemed clinically affected by the disorder had a significantly lower mean physical component score and a trend towards a lower mental component score180.
Some studies have used HRQOL utility instruments, such as the EuroQol-5D (EQ-5D) and the Assessment of Quality of Life-4D (AQOL-4D), to assess HRQOL in patients with haemochromatosis. These instruments estimate utility values, which enable the estimation of how both quantity and quality of life are affected by a disease, health state and/or intervention181. Utility is measured on a scale of 0 to 1, of which 0 represents death and 1 represents optimal health. Based on self-reported symptoms, a cross-sectional study demonstrated significantly lower mean utility values in patients with either elevated iron levels and early symptoms (0.60) or elevated iron levels and organ damage (0.50), compared with asymptomatic individuals (0.81) and the general population (0.81)177. Further, a negative correlation between the number of haemochromatosis-related symptoms and/or comorbidities and utility was reported.
Only one randomized clinical trial has assessed HRQOL and/or utility in patients receiving different treatments. However, no significant differences were observed in HRQOL in individuals receiving erythrocytapheresis or phlebotomy when assessed using either the SF-36 or the EQ-5D182.
Outlook
Although iron deficiency remains a major health problem worldwide, the clinical effect of hereditary conditions associated with iron overload is now increasingly recognized. Haemochromatosis is the paradigmatic clinical condition associated with systemic iron overload. Whereas haemochromatosis was originally considered an unusual autopsy finding probably owing to excessive alcohol intake, the condition is now recognized as a unique syndromic entity owing to the ground-breaking discovery that a lack of hepcidin causes this disorder.
Evolutionary perspectives
Iron is a vital micronutrient for humans and pathogens, and fighting pathogens (and avoiding starvation) has most likely represented the main challenge in human evolution183,184. Accordingly, it is interesting that hepcidin, an antimicrobial peptide that evolved to withhold iron from blood during pathogen invasion, and a protein used by hepcidin to ‘sense’ blood iron (that is, HFE) are centrally involved in the regulation of body iron homeostasis and in the pathogenesis of haemochromatosis. Yet we still do not understand its profound meaning from an evolutionary perspective. What do the spreading and strong evolutionary pressure of the C282Y polymorphism in white populations have to do with innate immunity and the battle against pathogens (for example, does HFE also have an immunological role? Does the macrophage-deprived environment of haemochromatosis have a role in the response to intracellular pathogens?) and, more generally, does this potential selective advantage still have some relevance in terms of physical performance? These questions remain to be clarified184,185. In addition, an ‘iron-thrifty’ phenotype might have evolved in humans to store energy and accumulate iron reserves184. Although this might have been evolutionarily advantageous to survive long periods of famine, this might be detrimental for contemporary lifestyles and dietary habits that favour iron accumulation. In individuals with chronic and degenerative diseases, a genetic predisposition to a slight excess of pro-oxidant iron in blood owing to variants of HFE or other iron-loading genes could contribute to and amplify the underlying pathophysiological event, which could impact the course and outcome of chronic diseases. This process has been suggested in studies in diabetes186.
Mechanisms of disease
Although the hepcidin-centred pathogenetic basis of haemochromatosis has been largely elucidated, some details are still under investigation, such as the interaction and putative cooperation of different haemochromatosis-associated proteins (such as HFE and TFR2) in hepcidin signalling and the role of BMPs in haemochromatosis187–191. In addition, one concern is that available animal models have been useful to explain iron overload in haemochromatosis, but they do not explain the resulting multiorgan disease. Several general questions still await answers, such as if a role exists for HFE outside the hepatocyte-hepcidin-centred model, for example, in crypt enterocytes (where the HFE-TFR1 complex was originally identified in humans192) or in macrophages (where hepcidin-independent HFE activity has been hypothesized)193–196.
Genetics
In principle, the lack of any gene that limits iron transfer into the blood could cause haemochromatosis. In this context, mutations causing the loss of the iron-retention capability of enterocytes could cause unrestricted iron transfer into blood, leading to haemochromatosis. Indeed, deletion of Fth (encoding H ferritin) causes a haemochromatosis phenotype in mice197, and an unconfirmed report identifying haemochromatosis owing to mutations in SRD5A2 (encoding H ferritin) in humans suggests this is a possibility198.
Other open questions in haemochromatosis relate to the penetrance of HFE-associated haemochromatosis. As previously mentioned, the clinical expression of haemochromatosis varies substantially between patients and follows a gradient ranging from the least penetrant form (HFE and C282Y homozygosity) to the fully penetrant forms (owing to HJV or HAMP mutations)199. Although environmental and physiological factors are important for the clinical expressions of HFE-associated haemochromatosis, genetic factors might also have a role. For example, polymorphic variants in a number of predictable and apparently iron-unrelated genes that potentially affect the phenotype of haemochromatosis patients have been reported16,26,200–202. However, to reach definite conclusions about these variants, the major haemochromatosis referral centres should collaborate to design prospective next-generation sequencing studies in large cohorts of patients and controls.
Diagnosis and management
Although a substantial advancement in the diagnosis and optimal management of the main clinical manifestations of haemochromatosis has been achieved over the past few decades, several areas require further study. For example, the pathophysiology of haemochromatosis arthropathy is still elusive, and this symptom has an unpredictable clinical onset and complex management and is often frustrating for patients and doctors; further study of the pathogenetic basis of haemochromatosis arthropathy is required.
As in the case of diabetes, we can now envisage new and more-effective approaches for the diagnosis and treatment of haemochromatosis199. For example, the measurement of serum hepcidin levels is considered a useful add-on in the diagnostic workup of individuals with suspected haemochromatosis, and, in the near future, hepcidin-sensitivity tests might be available to aid diagnosis of haemochromatosis owing to hepcidin resistance, such as ferroportin-related haemochromatosis. As to the treatment, there is no doubt that phlebotomy is a safe and effective treatment for haemochromatosis. However, severe cases are still cumbersome and difficult to treat, and a hormonal hepcidin-based replacement or phlebotomy-adjuvant therapy might be invaluable in these patients.
Acknowledgements
B.d.G. has received a grant from Haemochromatosis Australia to conduct haemochromatosis research. C.E.M. was supported in part by grant 5R24 DK09984603 from the National Institute of Health and Digestive and Kidney Diseases. The authors thank B. Skikne (University of Kansas Medical Center) for his contributions to the section on Epidemiology.
Footnotes
Competing interests
P.B. has received lecture fees from Novartis and consulting fees from Novartis and La Jolla Pharmaceutical Company. A.P. has received lecture fees from Novartis, and consulting fees from Novartis, La Jolla Pharmaceutical Company and Mitsubishi Tanabe Pharma Corporation. O.L. has received a research grant from Novartis. All other authors declare no competing interests.
References
- 1.Merryweather-Clarke AT, Pointon JJ, Jouanolle AM, Rochette J & Robson KJ Geography of HFE C282Y and H63D mutations. Genet. Test 4, 183–198 (2000). [DOI] [PubMed] [Google Scholar]
- 2.McLaren CE et al. Hemochromatosis and Iron Overload Screening (HEIRS) study design for an evaluation of 100,000 primary care-based adults. Am. J. Med. Sci 325, 53–62 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Adams PC et al. Hemochromatosis and iron-overload screening in a racially diverse population. N. Engl. J. Med 352, 1769–1778 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Kirk L et al. Haemochromatosis gene frequency in a control and diabetic Irish population. Ir. J. Med. Sci 178, 39–42 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Hanson EH, Imperatore G & Burke W HFE gene and hereditary hemochromatosis: a HuGE review. Hum. Genome Epidemiol. Am. J. Epidemiol 154, 193–206 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Wallace DF & Subramaniam VN The global prevalence of HFE and non-HFE hemochromatosis estimated from analysis of next-generation sequencing data. Genet. Med 18, 618–626 (2016). [DOI] [PubMed] [Google Scholar]; This is the first comparative study of the prevalence of HFE-associated and non-HFE-associated haemochromatosis using next-generation sequencing data.
- 7.Walker AR & Arvidsson UB Iron intake and haemochromatosis in the Bantu. Nature 166, 438–439 (1950). [DOI] [PubMed] [Google Scholar]
- 8.Strachan A Haemosiderosis and haemochromatosis in South African natives with a comment on etiology of haemochrmatosis. Thesis, Univ. Glasgow; (1929). [Google Scholar]
- 9.Wallace DF & Subramaniam VM in Iron Physiology and Pathophysiology in Humans (eds Anderson GJ & McLaren GD) 399–416 (Humana Press, New York, 2012). [Google Scholar]
- 10.Gordeuk VR, Boyd RD & Brittenham GM Dietary iron overload persists in rural sub-Saharan Africa. Lancet 1, 1310–1313 (1986). [DOI] [PubMed] [Google Scholar]
- 11.Allen KJ et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N. Engl. J. Med 358, 221–230 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Whitlock EP, Garlitz BA, Harris EL, Beil TL & Smith PR Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Ann. Intern. Med 145, 209–223 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Olynyk JK, Hagan SE, Cullen DJ, Beilby J & Whittall DE Evolution of untreated hereditary hemochromatosis in the Busselton population: a 17-year study. Mayo Clin. Proc 79, 309–313 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Constantine CC et al. A novel association between a SNP in CYBRD1 and serum ferritin levels in a cohort study of HFE hereditary haemochromatosis. Br. J. Haematol 147, 140–149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelucchi S et al. CYBRD1 as a modifier gene that modulates iron phenotype in HFE p. C282Y homozygous patients. Haematologica 97, 1818–1825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLaren CE et al. Exome sequencing in HFE C282Y homozygous men with extreme phenotypes identifies a GNPAT variant associated with severe iron overload. Hepatology 62, 429–439 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barton JC et al. GNPAT p. D519G is independently associated with markedly increased iron stores in HFE p. C282Y homozygotes. Blood Cells Mol. Dis 63, 15–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattman A et al. Transferrin receptor 2 (TfR2) and HFE mutational analysis in nonC282Y iron overload: identification of a novel TfR2 mutation. Blood 100, 1075–1077 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Biasiotto G et al. Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload. Clin. Chem 49, 1981–1988 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Beutler E et al. Polymorphisms and mutations of human TMPRSS6 in iron deficiency anemia. Blood Cells Mol. Dis 44, 16–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T et al. Novel missense mutation in the TMPRSS6 gene in a Japanese female with iron-refractory iron deficiency anemia. Int. J. Hematol 94, 101–103 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Del-Castillo-Rueda A et al. Mutations in the HFE, TFR2, and SLC40A1 genes in patients with hemochromatosis. Gene 508, 15–20 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Barton JC et al. HFE, SLC40A1, HAMP, HJV, TFR2, and FTL mutations detected by denaturing high-performance liquid chromatography after iron phenotyping and HFE C282Y and H63D genotyping in 785 HEIRS Study participants. Am. J. Hematol 84, 710–714 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Wallace DF, Clark RM, Harley HA & Subramaniam VN Autosomal dominant iron overload due to a novel mutation of ferroportin1 associated with parenchymal iron loading and cirrhosis. J. Hepatol 40, 710–713 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Papanikolaou G et al. Mutations in HFE2 cause iron overload in chromosome 1qlinked juvenile hemochromatosis. Nat. Genet 36, 77–82 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Daher R et al. Heterozygous mutations in BMP6 pro-peptide lead to inappropriate hepcidin synthesis and moderate iron overload in humans. Gastroenterology 150, 672–683.e4 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Piubelli C et al. Identification of new BMP6 pro-peptide mutations in patients with iron overload. Am. J. Hematol 92, 562–568 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Stickel F et al. Evaluation of genome-wide loci of iron metabolism in hereditary hemochromatosis identifies PCSK7 as a host risk factor of liver cirrhosis. Hum. Mol. Genet 23, 3883–3890 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Loreal O et al. Liver fibrosis in genetic hemochromatosis. Respective roles of iron and non-iron-related factors in 127 homozygous patients. J. Hepatol 16, 122–127 (1992). [DOI] [PubMed] [Google Scholar]
- 30.Wood MJ, Powell LW, Dixon JL & Ramm GA Clinical cofactors and hepatic fibrosis in hereditary hemochromatosis: the role of diabetes mellitus. Hepatology 56, 904–911 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Wheby MS, Suttle GE & Ford KT III. Intestinal absorption of hemoglobin iron. Gastroenterology 58, 647–654 (1970). [PubMed] [Google Scholar]
- 32.Lombardi-Boccia G, Martinez-Dominguez B & Aguzzi A Total heme and non-heme iron in raw and cooked meats. J. Food Sci 67, 1738–1741 (2002). [Google Scholar]
- 33.Cook JD Adaptation in iron metabolism. Am. J. Clin. Nutr 51, 301–308 (1990). [DOI] [PubMed] [Google Scholar]
- 34.Cade JE et al. Diet and genetic factors associated with iron status in middle-aged women. Am. J. Clin. Nutr 82, 813–820 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Greenwood DC et al. HFE genotype modifies the influence of heme iron intake on iron status. Epidemiology 16, 802–805 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Liu JM et al. Body iron stores and their determinants in healthy postmenopausal US women. Am. J. Clin. Nutr 78, 1160–1167 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Backstrand JR, Allen LH, Black AK, de Mata M & Pelto GH Diet and iron status of nonpregnant women in rural Central Mexico. Am. J. Clin. Nutr 76, 156–164 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Gordeuk VR et al. Dietary iron intake and serum ferritin concentration in 213 patients homozygous for the HFEC282Y hemochromatosis mutation. Can. J. Gastroenterol 26, 345–349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skoien R, & Powell LW in Iron Physiology and Pathophysiology in Humans (eds Anderson GJ & McLaren GD) 385–398 (Humana Press, New York, 2012). [Google Scholar]
- 40.Fletcher LM, Dixon JL, Purdie DM, Powell LW & Crawford DH Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 122, 281–289 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Barton JC, Preston BL, McDonnell SM & Rothenberg BE Severity of iron overload in hemochromatosis: effect of volunteer blood donation before diagnosis. Transfusion 41, 123–129 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Wood MJ, Powell LW & Ramm GA Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood 111, 4456–4462 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Latour C et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology 59, 683–694 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Aguilar-Martinez P et al. Variable phenotypic presentation of iron overload in H63D homozygotes: are genetic modifiers the cause? Gut 48, 836–842 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aranda N, Viteri FE, Montserrat C & Arija V Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and life-style factors on iron status in a general Mediterranean population from Tarragona, Spain. Ann. Hematol 89, 767–773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finch C Regulators of iron balance in humans. Blood 84, 1697–1702 (1994). [PubMed] [Google Scholar]
- 47.Brissot P & Loreal O Iron metabolism and related genetic diseases: a cleared land, keeping mysteries. J. Hepatol 64, 505–515 (2016). [DOI] [PubMed] [Google Scholar]
- 48.McKie AT et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 5, 299–309 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Donovan A et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Abboud S & Haile DJ A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem 275, 19906–19912 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Park CH, Valore EV, Waring AJ & Ganz T Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem 276, 7806–7810 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Pigeon C et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem 276, 7811–7819 (2001). [DOI] [PubMed] [Google Scholar]; This study demonstrates the link between hepcidin and iron metabolism.
- 53.Nemeth E et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Nicolas G et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl Acad. Sci. USA 98, 8780–8785 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates the link between hepcidin deficiency and the development of iron overload.
- 55.Meynard D et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet 41, 478–481 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Andriopoulos B Jr et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet 41, 482–487 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finberg KE et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet 40, 569–571 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo S et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J. Clin. Invest 123, 1531–1541 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feder JN et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet 13, 399–408 (1996). [DOI] [PubMed] [Google Scholar]; This seminal study establishes the link between haemochromatosis and mutations in HFE.
- 60.Lebron JA et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell 93, 111–123 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Fleming RE et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl Acad. Sci. USA 97, 2214–2219 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roetto A et al. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 97, 2555–2560 (2001). [DOI] [PubMed] [Google Scholar]
- 63.D’Alessio F, Hentze MW & Muckenthaler MU The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol 57, 1052–1060 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Peyssonnaux C et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest 117, 1926–1932 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sonnweber T et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 63, 1951–1959 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Kautz L et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet 46, 678–684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies erythroferrone as an erythroid regulator of iron metabolism.
- 67.Tanno T et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med 13, 1096–1101 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Pietrangelo A et al. STAT3 is required for IL6gp130-dependent activation of hepcidin in vivo. Gastroenterology 132, 294–300 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Verga Falzacappa MV et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109, 353–358 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Wrighting DM & Andrews NC Interleukin6 induces hepcidin expression through STAT3. Blood 108, 3204–3209 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nemeth E et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 101, 2461–2463 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Ikeda Y et al. Estrogen regulates hepcidin expression via GPR30BMP6dependent signaling in hepatocytes. PLoS ONE 7, e40465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levy JE, Jin O, Fujiwara Y, Kuo F & Andrews NC Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet 21, 396–399 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Ohgami RS et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet 37, 1264–1269 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabuchi M, Yoshimori T, Yamaguchi K, Yoshida T & Kishi F Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp2 cells. J. Biol. Chem 275, 22220–22228 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Fleming MD et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet 16, 383–386 (1997). [DOI] [PubMed] [Google Scholar]
- 77.Levi S et al. Mechanism of ferritin iron uptake: activity of the H-chain and deletion mapping of the ferro-oxidase site. A study of iron uptake and ferro-oxidase activity of human liver, recombinant H-chain ferritins, and of two H-chain deletion mutants. J. Biol. Chem 263, 18086–18092 (1988). [PubMed] [Google Scholar]
- 78.Harrison PM & Arosio P The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275, 161–203 (1996). [DOI] [PubMed] [Google Scholar]
- 79.Ford GC et al. Ferritin: design and formation of an iron-storage molecule. Phil. Trans. R. Soc. Lond. B Biol. Sci 304, 551–565 (1984). [DOI] [PubMed] [Google Scholar]
- 80.Hentze MW & Kuhn LC Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl Acad. Sci. USA 93, 8175–8182 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hershko C, Graham G, Bates GW & Rachmilewitz EA Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity. Br. J. Haematol 40, 255–263 (1978). [DOI] [PubMed] [Google Scholar]; This study demonstrates the existence of NTBI.
- 82.Loreal O et al. Determination of non-transferrin-bound iron in genetic hemochromatosis using a new HPLC-based method. J. Hepatol 32, 727–733 (2000). [DOI] [PubMed] [Google Scholar]
- 83.Grootveld M et al. Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy. J. Biol. Chem 264, 4417–4422 (1989). [PubMed] [Google Scholar]
- 84.Le Lan C et al. Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood 105, 4527–4531 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Esposito BP et al. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood 102, 2670–2677 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Koppenol WH The Haber-Weiss cycle — 70 years later. Redox Rep. 6, 229–234 (2001). [DOI] [PubMed] [Google Scholar]
- 87.Bacon BR, Park CH, Brittenham GM, O’Neill R & Tavill AS Hepatic mitochondrial oxidative metabolism in rats with chronic dietary iron overload. Hepatology 5, 789–797 (1985). [DOI] [PubMed] [Google Scholar]
- 88.Volani C et al. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 9, 1634–1644 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Brissot P, Ropert M, Le Lan C & Loreal O Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim. Biophys. Acta 1820, 403–410 (2012). [DOI] [PubMed] [Google Scholar]
- 90.Liuzzi JP, Aydemir F, Nam H, Knutson MD & Cousins RJ Zip14 (Slc39a14) mediates nontransferrin-bound iron uptake into cells. Proc. Natl Acad. Sci. USA 103, 13612–13617 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jenkitkasemwong S et al. SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell. Metab 22, 138–150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oudit GY et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med 9, 1187–1194 (2003). [DOI] [PubMed] [Google Scholar]
- 93.Ludwiczek S et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter1. Nat. Med 13, 448–454 (2007). [DOI] [PubMed] [Google Scholar]
- 94.Brissot P, Bolder U, Schteingart CD, Arnaud J & Hofmann AF Intestinal absorption and enterohepatic cycling of biliary iron originating from plasma non-transferrin-bound iron in rats. Hepatology 25, 1457–1461 (1997). [DOI] [PubMed] [Google Scholar]
- 95.Craven CM et al. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: a rodent model for hemochromatosis. Proc. Natl Acad. Sci. USA 84, 3457–3461 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brissot P, Wright TL, Ma WL & Weisiger RA Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J. Clin. Invest 76, 1463–1470 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feder JN et al. The haemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression J. Biol. Chem 272, 14025–14028 (1997). [DOI] [PubMed] [Google Scholar]
- 98.Bridle KR et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 361, 669–673 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Gehrke SG et al. Expression of hepcidin in hereditary hemochromatosis: evidence for a regulation in response to serum transferrin saturation and non-transferrin-bound iron. Blood 102, 371–376 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Roetto A et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet 33, 21–22 (2003). [DOI] [PubMed] [Google Scholar]
- 101.Porto G et al. A Portuguese patient homozygous for the −25G>A mutation of the HAMP promoter shows evidence of steady-state transcription but fails to upregulate hepcidin levels by iron. Blood 106, 2922–2923 (2005). [DOI] [PubMed] [Google Scholar]
- 102.Island ML et al. A new mutation in the hepcidin promoter impairs its BMP response and contributes to a severe phenotype in HFE related hemochromatosis. Haematologica 94, 720–724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kawabata H et al. Expression of hepcidin is downregulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 105, 376–381 (2005). [DOI] [PubMed] [Google Scholar]
- 104.Camaschella C et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet 25, 14–15 (2000). [DOI] [PubMed] [Google Scholar]
- 105.Biasiotto G et al. Identification of new mutations of hepcidin and hemojuvelin in patients with HFE C282Y allele. Blood Cells Mol. Dis 33, 338–343 (2004). [DOI] [PubMed] [Google Scholar]
- 106.Merryweather-Clarke AT et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet 12, 2241–2247 (2003). [DOI] [PubMed] [Google Scholar]
- 107.Jacolot S et al. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood 103, 2835–2840 (2004). [DOI] [PubMed] [Google Scholar]
- 108.Le Gac G et al. The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Hum. Mol. Genet 13, 1913–1918 (2004). [DOI] [PubMed] [Google Scholar]
- 109.Hamdi-Roze H et al. Rare HFE variants are the most frequent cause of hemochromatosis in nonc282y homozygous patients with hemochromatosis. Am. J. Hematol 91, 1202–1205 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Njajou OT et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet 28, 213–214 (2001). [DOI] [PubMed] [Google Scholar]; This study identifies ferroportin-associated hemochromatosis.
- 111.Drakesmith H et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 106, 1092–1097 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Fernandes A et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood 114, 437–443 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Detivaud L et al. Ferroportin diseases: functional studies, a link between genetic and clinical phenotype. Hum. Mutat 34, 1529–1536 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Le Gac G et al. Structure-function analysis of the human ferroportin iron exporter (SLC40A1): effect of hemochromatosis type 4 disease mutations and identification of critical residues. Hum. Mutat 34, 1371–1380 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Preza GC et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Invest 121, 4880–4888 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pietrangelo A et al. Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene. N. Engl. J. Med 341, 725–732 (1999). [DOI] [PubMed] [Google Scholar]
- 117.Montosi G et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J. Clin. Invest 108, 619–623 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates ferroportin disease.
- 118.Mayr R et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J. Hepatol 53, 941–949 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sabelli M et al. Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology 65, 1512–1525 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brissot P Optimizing the diagnosis and the treatment of iron overload diseases. Expert Rev. Gastroenterol. Hepatol 10, 359–370 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Brissot P, Bardou-Jacquet E, Jouanolle AM & Loreal O Iron disorders of genetic origin: a changing world. Trends Mol. Med 17, 707–713 (2011). [DOI] [PubMed] [Google Scholar]
- 122.Pietrangelo A Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 139, 393–408.e2 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Powell LW, Seckington RC & Deugnier Y Haemochromatosis. Lancet 388, 706–716 (2016). [DOI] [PubMed] [Google Scholar]
- 124.Beaumont-Epinette MP et al. Hereditary hypotransferrinemia can lead to elevated transferrin saturation and, when associated to HFE or HAMP mutations, to iron overload. Blood Cells Mol. Dis 54, 151–154 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Miyajima H Aceruloplasminemia. Neuropathol. 35, 83–90 (2015). [DOI] [PubMed] [Google Scholar]
- 126.Wood JC Estimating tissue iron burden: current status and future prospects. Br. J. Haematol 170, 15–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.St Pierre TG et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood 105, 855–861 (2005). [DOI] [PubMed] [Google Scholar]
- 128.Gandon Y et al. Non-invasive assessment of hepatic iron stores by MRI. Lancet 363, 357–362 (2004). [DOI] [PubMed] [Google Scholar]
- 129.d’Assignies G et al. Non-invasive measurement of liver iron concentration using 3Tesla magnetic resonance imaging: validation against biopsy. Eur. Radiol 10.1007/s00330-017-5106-3 (2017). [DOI] [PubMed]
- 130.Franca M et al. Optimizing the management of hereditary haemochromatosis: the value of MRI R2* quantification to predict and monitor body iron stores. Br. J. Haematol 10.1111/bjh.14982 (2017). [DOI] [PubMed]
- 131.Deugnier YM et al. Liver pathology in genetic hemochromatosis: a review of 135 homozygous cases and their bioclinical correlations. Gastroenterology 102, 2050–2059 (1992). [DOI] [PubMed] [Google Scholar]
- 132.Porter JB & Garbowski M The pathophysiology of transfusional iron overload. Hematol. Oncol. Clin. North Am 28, 683–701 (2014). [DOI] [PubMed] [Google Scholar]
- 133.Musallam KM, Cappellini MD, Wood JC & Taher AT Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Rev. 26 (Suppl. 1), S16–S19 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Kautz L & Nemeth E Molecular liaisons between erythropoiesis and iron metabolism. Blood 124, 479–482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Badens C & Guizouarn H Advances in understanding the pathogenesis of the red cell volume disorders. Br. J. Haematol 174, 674–685 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Ribeiro S, Belo L, Reis F & Santos-Silva A Iron therapy in chronic kidney disease: recent changes, benefits and risks. Blood Rev. 30, 65–72 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Deugnier Y, Bardou-Jacquet E & Laine F Dysmetabolic iron overload syndrome (DIOS). Presse Med. 46, e306–e311 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Harrison-Findik DD et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J. Biol. Chem 281, 22974–22982 (2006). [DOI] [PubMed] [Google Scholar]
- 139.Porto G et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur. J. Hum. Genet 24, 479–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper contains practical guidelines for the molecular genetic diagnosis of haemochromatosis.
- 140.Cezard C et al. Phenotypic expression of a novel C282Y/R226G compound heterozygous state in HFE hemochromatosis: molecular dynamics and biochemical studies. Blood Cells Mol. Dis 52, 27–34 (2014). [DOI] [PubMed] [Google Scholar]
- 141.Brissot P et al. Current approach to hemochromatosis. Blood Rev. 22, 195–210 (2008). [DOI] [PubMed] [Google Scholar]
- 142.Barton JC et al. Increased risk of death from iron overload among 422 treated probands with HFE hemochromatosis and serum levels of ferritin greater than 1000 mug/L at diagnosis. Clin. Gastroenterol. Hepatol 10, 412–416 (2012). [DOI] [PubMed] [Google Scholar]
- 143.Le Lan C et al. Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology 140, 1199–1207.e2 (2011). [DOI] [PubMed] [Google Scholar]
- 144.de Graaff B et al. Cost-effectiveness of different population screening strategies for hereditary haemochromatosis in Australia. Appl. Health Econ. Health Policy 15, 521–534 (2017). [DOI] [PubMed] [Google Scholar]
- 145.Grosse SD, Gurrin LC, Bertalli NA & Allen KJ Clinical penetrance in hereditary hemochromatosis: estimates of the cumulative incidence of severe liver disease among HFE C282Y homozygotes. Genet. Med 10.1038/gim.2017.121 (2017). [DOI] [PMC free article] [PubMed]
- 146.McLaren GD & Gordeuk VR Hereditary hemochromatosis: insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematology Am. Soc. Hematol. Educ. Program 2009, 195–206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Adams PC & Barton JC How I treat hemochromatosis. Blood 116, 317–325 (2010). [DOI] [PubMed] [Google Scholar]
- 148.Adams PC The natural history of untreated HFE-related hemochromatosis. Acta Haematol. 122, 134–139 (2009). [DOI] [PubMed] [Google Scholar]
- 149.Andersen RV, Tybjaerg-Hansen A, Appleyard M, Birgens H & Nordestgaard BG Hemochromatosis mutations in the general population: iron overload progression rate. Blood 103, 2914–2919 (2004). [DOI] [PubMed] [Google Scholar]
- 150.Beutler E Natural history of hemochromatosis. Mayo Clin. Proc 79, 305–306 (2004). [DOI] [PubMed] [Google Scholar]
- 151.Husar-Memmer E, Stadlmayr A, Datz C & Zwerina J HFE-related hemochromatosis: an update for the rheumatologist. Curr. Rheumatol. Rep 16, 393 (2014). [DOI] [PubMed] [Google Scholar]
- 152.Brissot P, Ball S, Rofail D, Cannon H & Jin VW Hereditary hemochromatosis: patient experiences of the disease and phlebotomy treatment. Transfusion 51, 1331–1338 (2011). [DOI] [PubMed] [Google Scholar]
- 153.Lynch SR, Skikne BS & Cook JD Food iron absorption in idiopathic hemochromatosis. Blood 74, 2187–2193 (1989). [PubMed] [Google Scholar]
- 154.Rombout-Sestrienkova E et al. Course of iron parameters in HFE-hemochromatosis patients during initial treatment with erythrocytapheresis compared to phlebotomy. J. Clin. Apher 31, 564–570 (2016). [DOI] [PubMed] [Google Scholar]
- 155.Adams PC, Kertesz AE & Valberg LS Rate of iron reaccumulation following iron depletion in hereditary hemochromatosis. Implications for venesection therapy. J. Clin. Gastroenterol 16, 207–210 (1993). [DOI] [PubMed] [Google Scholar]
- 156.Adams PC Factors affecting the rate of iron mobilization during venesection therapy for genetic hemochromatosis. Am. J. Hematol 58, 16–19 (1998). [DOI] [PubMed] [Google Scholar]
- 157.Levstik M & Adams PC Eligibility and exclusion of hemochromatosis patients as voluntary blood donors. Can. J. Gastroenterol 12, 61–63 (1998). [DOI] [PubMed] [Google Scholar]
- 158.West KA & Eder AF Accepting hereditary hemochromatosis blood donors: ask not why, ask why not. Transfusion 56, 2907–2909 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Bardou-Jacquet E et al. Worse outcomes of patients with HFE hemochromatosis with persistent increases in transferrin saturation during maintenance therapy. Clin. Gastroenterol. Hepatol 15, 1620–1627 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Rombout-Sestrienkova E, van Kraaij MG & Koek GH How we manage patients with hereditary haemochromatosis. Br. J. Haematol 175, 759–770 (2016). [DOI] [PubMed] [Google Scholar]
- 161.Ong SY et al. Reduction of body iron in HFE-related haemochromatosis and moderate iron overload (MiIron): a multicentre, participant-blinded, randomised controlled trial. Lancet Haematol. 4, e607–e614 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Barton JC Should we treat individuals homozygous for HFE p. Cys282Tyr with ferritin 300–1000 mug/L? Lancet Haematol. 4, e569–e570 (2017). [DOI] [PubMed] [Google Scholar]
- 163.Phatak P et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 52, 1671–1779 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Olsson KS, Vaisanen M, Konar J & Bruce A The effect of withdrawal of food iron fortification in Sweden as studied with phlebotomy in subjects with genetic hemochromatosis. Eur. J. Clin. Nutr 51, 782–786 (1997). [DOI] [PubMed] [Google Scholar]
- 165.Kaltwasser JP et al. Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis. Gut 43, 699–704 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vanclooster A, van Deursen C, Jaspers R, Cassiman D & Koek G Proton pump inhibitors decrease phlebotomy need in HFE hemochromatosis: double-blind randomized placebo-controlled trial. Gastroenterology 153, 678–680.e2 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Barton JC & Acton RT Hemochromatosis and Vibrio vulnificus wound infections. J. Clin. Gastroenterol 43, 890–893 (2009). [DOI] [PubMed] [Google Scholar]
- 168.Crawford DH et al. Patient and graft survival after liver transplantation for hereditary hemochromatosis: implications for pathogenesis. Hepatology 39, 1655–1662 (2004). [DOI] [PubMed] [Google Scholar]
- 169.Ludwig J, Hashimoto E, Porayko MK, Moyer TP & Baldus WP Hemosiderosis in cirrhosis: a study of 447 native livers. Gastroenterology 112, 882–888 (1997). [DOI] [PubMed] [Google Scholar]
- 170.Bardou-Jacquet E et al. Liver transplantation normalizes serum hepcidin level and cures iron metabolism alterations in HFE hemochromatosis. Hepatology 10.1002/hep.26570 (2013). [DOI] [PubMed]; This study is a clinical demonstration that the liver is the key organ in haemochromatosis, not only as a target of iron overload but also as the cause of iron excess through hepcidin deficiency.
- 171.Liu J, Sun B, Yin H & Liu S Hepcidin: a promising therapeutic target for iron disorders: a systematic review. Medicine 95, e3150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vyoral D & Jiri P Therapeutic potential of hepcidin — the master regulator of iron metabolism. Pharmacol. Res 115, 242–254 (2017). [DOI] [PubMed] [Google Scholar]
- 173.Ware JE Jr SF36 health survey update. Spine 25, 3130–3139 (2000). [DOI] [PubMed] [Google Scholar]
- 174.Ware J et al. User’s Manual for the SF36v2 Health Survey 2nd edn (QualityMetric, Lincoln RI, 2007). [Google Scholar]
- 175.Mainous AG III et al. Elevated transferrin saturation, health-related quality of life and telomere length. Biometals 27, 135–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Allen KJ et al. HFE Cys282Tyr homozygotes with serum ferritin concentrations below 1000 microg/L are at low risk of hemochromatosis. Hepatology 52, 925–933 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.de Graaff B, Neil A, Sanderson K, Yee KC & Palmer AJ Quality of life utility values for hereditary haemochromatosis in Australia. Health Qual. Life Outcomes 14, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Adams PC & Speechley M The effect of arthritis on the quality of life in hereditary hemochromatosis. J. Rheumatol 23, 707–710 (1996). [PubMed] [Google Scholar]
- 179.Barg A, Elsner A, Hefti D & Hintermann B Total ankle arthroplasty in patients with hereditary hemochromatosis. Clin. Orthop. Relat. Res 469, 1427–1435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Meiser B, Dunn S, Dixon J & Powell LW Psychological adjustment and knowledge about hereditary hemochromatosis in a clinic-based sample: a prospective study. J. Genet. Counsel 14, 453–463 (2005). [DOI] [PubMed] [Google Scholar]
- 181.Drummond M, Sculpher M, Torrance G, O’Brine B & Stoddart G Methods for the Economic Evaluation of Health Care Programmes 3rd edn (Oxford Univ. Press, 2005). [Google Scholar]
- 182.Rombout-Sestrienkova E et al. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE hemochromatosis patients: results from a randomized crossover trial. Transfusion 56, 261–270 (2016). [DOI] [PubMed] [Google Scholar]
- 183.Weinberg ED Iron availability and infection. Biochim. Biophys. Acta 1790, 600–605 (2009). [DOI] [PubMed] [Google Scholar]
- 184.Pietrangelo A Pathogens, metabolic adaptation, and human diseasesan iron-thrifty genetic model. Gastroenterology 149, 834–838 (2015). [DOI] [PubMed] [Google Scholar]
- 185.Hollerer I, Bachmann A & Muckenthaler MU Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica 102, 809–817 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ellervik C, Mandrup-Poulsen T, Tybjaerg-Hansen A & Nordestgaard BG Total and cause-specific mortality by elevated transferrin saturation and hemochromatosis genotype in individuals with diabetes: two general population studies. Diabetes Care 37, 444–452 (2014). [DOI] [PubMed] [Google Scholar]
- 187.Steinbicker AU et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 118, 4224–4230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wu XG et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 124, 1335–1343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schmidt PJ & Fleming MD Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor2. Am. J. Hematol 87, 588–595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Goswami T & Andrews NC Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem 281, 28494–28498 (2006). [DOI] [PubMed] [Google Scholar]
- 191.Rishi G, Crampton EM, Wallace DF & Subramaniam VN In situ proximity ligation assays indicate that hemochromatosis proteins Hfe and transferrin receptor 2 (Tfr2) do not interact. PLoS ONE 8, e77267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Waheed A et al. Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum. Proc. Natl Acad. Sci. USA 96, 1579–1584 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Montosi G et al. Wild-type HFE protein normalizes transferrin iron accumulation in macrophages from subjects with hereditary hemochromatosis. Blood 96, 1125–1129 (2000). [PubMed] [Google Scholar]
- 194.Drakesmith H et al. The hemochromatosis protein HFE inhibits iron export from macrophages. Proc. Natl Acad. Sci. USA 99, 15602–15607 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Makui H, Soares RJ, Jiang W, Constante M & Santos MM Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood 106, 2189–2195 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Garuti C et al. Hepcidin expression does not rescue the iron-poor phenotype of Kupffer cells in Hfe-null mice after liver transplantation. Gastroenterology 139, 315–322.e1 (2010). [DOI] [PubMed] [Google Scholar]
- 197.Vanoaica L, Darshan D, Richman L, Schumann K & Kuhn LC Intestinal ferritin h is required for an accurate control of iron absorption. Cell. Metab 12, 273–282 (2010). [DOI] [PubMed] [Google Scholar]
- 198.Kato J et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet 69, 191–197 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pietrangelo A Hemochromatosis: an endocrine liver disease. Hepatology 46, 1291–1301 (2007). [DOI] [PubMed] [Google Scholar]
- 200.Benyamin B et al. Novel loci affecting iron homeostasis and their effects in individuals at risk for hemochromatosis. Nat. Commun 5, 4926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.de Tayrac M et al. Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis. J. Hepatol 62, 664–672 (2015). [DOI] [PubMed] [Google Scholar]
- 202.Bardou-Jacquet E, de Tayrac M, Mosser J & Deugnier Y GNPAT variant associated with severe iron overload in HFE hemochromatosis. Hepatology 62, 1917–1918 (2015). [DOI] [PubMed] [Google Scholar]
- 203.Osaki S, Johnson DA & Frieden E The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem 241, 2746–2751 (1966). [PubMed] [Google Scholar]
- 204.Zhao L et al. Cp/Heph mutant mice have iron-induced neurodegeneration diminished by deferiprone. J. Neurochem 135, 958–974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jiang B et al. Hephaestin and ceruloplasmin facilitate iron metabolism in the mouse kidney. Sci. Rep 6, 39470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bartnikas TB Known and potential roles of transferrin in iron biology. Biometals 25, 677–686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Iolascon A et al. Natural history of recessive inheritance of DMT1 mutations. J. Pediatr 152, 136–139 (2008). [DOI] [PubMed] [Google Scholar]
- 208.Bardou-Jacquet E et al. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis 47, 243–248 (2011). [DOI] [PubMed] [Google Scholar]
- 209.Doyard M et al. Decreased bone formation explains osteoporosis in a genetic mouse model of hemochromatosiss. PLoS ONE 11, e0148292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Aslan D, Crain K & Beutler E A new case of human atransferrinemia with a previously undescribed mutation in the transferrin gene. Acta Haematol. 118, 244–247 (2007). [DOI] [PubMed] [Google Scholar]
- 211.Guggenbuhl P, Brissot P & Loreal O Miscellaneous non-inflammatory musculoskeletal conditions. Haemochromatosis: the bone and the joint. Best Pract. Res. Clin. Rheumatol 25, 649–664 (2011). [DOI] [PubMed] [Google Scholar]
- 212.Gunshin H et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488 (1997). [DOI] [PubMed] [Google Scholar]
- 213.McKie AT et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291, 1755–1759 (2001). [DOI] [PubMed] [Google Scholar]
- 214.Vulpe CD et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet 21, 195–199 (1999). [DOI] [PubMed] [Google Scholar]
- 215.Harris ZL, Durley AP, Man TK & Gitlin JD Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl Acad. Sci. USA 96, 10812–10817 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Crielaard BJ et al. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov 16, 400–423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rausa M et al. Bmp6 expression in murine liver non parenchymal cells: a mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS ONE 10, e0122696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Canali S et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 129, 405–414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Silvestri L et al. Defective targeting of hemojuvelin to plasma membrane is a common pathogenetic mechanism in juvenile hemochromatosis. Blood 109, 4503–4510 (2007). [DOI] [PubMed] [Google Scholar]
- 220.Xia Y, Babitt JL, Sidis Y, Chung RT & Lin HY Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 111, 5195–5204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang RH et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell. Metab 2, 399–409 (2005). [DOI] [PubMed] [Google Scholar]
- 222.Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW & Muckenthaler MU Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med 87, 471–480 (2009). [DOI] [PubMed] [Google Scholar]
- 223.Truksa J, Lee P & Beutler E Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 113, 688–695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Verga Falzacappa MV, Casanovas G, Hentze MW & Muckenthaler MU A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2mediated hepatic hepcidin expression and its response to IL6 in cultured cells. J. Mol. Med 86, 531–540 (2008). [DOI] [PubMed] [Google Scholar]
- 225.Ramey G, Deschemin JC & Vaulont S Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 94, 765–772 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]