

SUMMARY
Many eukaryotes assemble an actin- and myosin-based cytokinetic ring (CR) on the plasma membrane (PM) for cell division, but how it is anchored there remains unclear. In Schizosaccharomyces pombe, the F-BAR protein Cdc15 links the PM via its F-BAR domain to proteins in the CR’s interior via its SH3 domain. However, Cdc15’s F-BAR domain also directly binds formin Cdc12, suggesting that Cdc15 may polymerize a protein network directly adjacent to the membrane. Here, we determine that the F-BAR domain binds Cdc12 using residues on the face opposite its membrane-binding surface. These residues also bind paxillin-like Pxl1, promoting its recruitment with calcineurin to the CR. Mutation of these F-BAR domain residues results in a shallower CR, with components localizing ~35% closer to the PM than in wild type, and aberrant CR constriction. Thus, F-BAR domains serve as oligomeric membrane-bound platforms that can modulate the architecture of an entire actin structure.
Graphical Abstract
In Brief
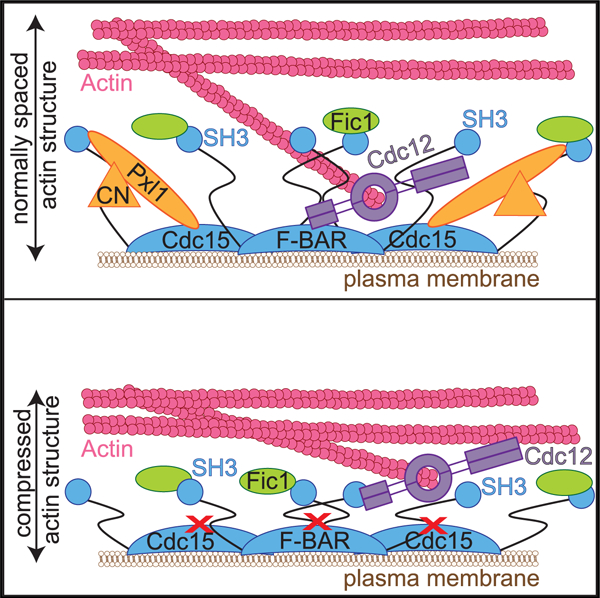
Multiple F-BAR domains link actin structures to membrane. Snider et al. show that the flat Cdc15 F-BAR domain utilizes opposite surfaces to bind the plasma membrane and cytokinetic ring proteins simultaneously. Disrupting Cdc15 F-BAR domain’s interaction with proteins results in an overall compression of the entire cytokinetic ring architecture.
INTRODUCTION
During cytokinesis, many eukaryotes build an actin- and myosin-based cytokinetic ring (CR) that constricts to bring together opposing membranes and physically pinch the cell in two (Cheffings et al., 2016; Gould, 2016). Studies using the fission yeast Schizosaccharomyces pombe have advanced our understanding of the composition, assembly, and function of the CR (Cheffings et al., 2016; Goyal et al., 2011; Mangione and Gould, 2019; Pollard and Wu, 2010; Rincon and Paoletti, 2016). The S. pombe CR is composed of ~40 proteins, many of which are evolution-arily conserved (PomBase; Wood et al., 2012).
More recently, studies using advanced microscopy techniques such as fluorescence photoactivation localization microscopy (fPALM) have revealed finer details of the CR structure (McDonald et al., 2017). The pre-constriction CR is 182 ± 26 nm wide, and its components are stratified into layers relative to the plasma membrane (PM) that extend 293 ± 64 nm into the cytoplasm. A membrane-proximal layer, 0–80 nm from the PM, is composed primarily of membrane-bound scaffolds. An intermediate layer contains a network of signaling and accessory components that localize 80–160 nm from the PM. Finally, a distal layer (160–350 nm from the PM) consists of F-actin, myosin motor domains, and actin-binding proteins. Electron cryotomography analysis demonstrated that there is a ~60-nm gap between the F-actin of constricting CRs and the PM (Swulius et al., 2018), consistent with displacement of the F-actin from the PM in pre-constriction CRs. Though a similar stratification of components has been observed for other actin-based structures built on the PM, like focal adhesions (Kanchanawong et al., 2010), it is unknown if stratification or the overall nanoscale architecture is important for CR function.
Clearly, however, the PM-proximal protein layer is key to connecting the CR to the PM. One component of this layer is the essential Fer/CIP4 homology BAR (F-BAR) protein Cdc15 (Fankhauser et al., 1995; McDonald et al., 2017). Cdc15 is one of the earliest and most abundant CR proteins to arrive at the division site cortex (Wu and Pollard, 2005; Wu et al., 2003), concordant with its role as a core CR scaffold and anchor (Arasada and Pollard, 2014; Carnahan and Gould, 2003; Mangione et al., 2019; McDonald et al., 2015, 2017; Ren et al., 2015; Roberts-Galbraith et al., 2009; Wachtler et al., 2006; Willet et al., 2015). Cdc15 is a conventional F-BAR family member in that it contains an N-terminal F-BAR domain that dimerizes to create a membrane-binding module (McDonald et al., 2015). The F-BAR domain oligomerizes in a tip-to-tip manner, facilitating high-avidity membrane binding; however, it does not tubulate membranes and belongs to a subset of the F-BAR domain family that lacks tubulation activity (McDonald et al., 2015). When oligomerization of the Cdc15 F-BAR domain is disrupted, the CR is unstable and can disassemble, leading to failed cytokinesis (McDonald et al., 2015).
Like other F-BAR family proteins that contain an additional domain or domains for signaling or protein binding, Cdc15 has an SH3 domain (McDonald and Gould, 2016b). Between the N-terminal F-BAR and C-terminal SH3 domains is a predicted intrinsically disordered region (IDR), important for Cdc15 phosphoregulation and recruitment of the protein phosphatase calcineurin (CN) to the CR (Mangione et al., 2019; Roberts-Galbraith et al., 2010). Cdc15 is hyperphosphorylated in interphase predominantly on residues within the IDR (Bhattacharjee et al., 2020; Kettenbach et al., 2015; Lee et al., 2018; Magliozzi et al., 2020; Roberts-Galbraith et al., 2010; Wachtler et al., 2006). This phosphorylation inhibits Cdc15 oligomerization and membrane- and protein-binding activities. Upon mitotic entry, Cdc15 is dephosphorylated, allowing it to oligomerize, bind membrane, and perform its scaffolding functions (Bhattacharjee et al., 2020; Roberts-Galbraith et al., 2010).
In the mature pre-constriction CR, the Cdc15 SH3 domain extends ~150 nm away from the PM (McDonald et al., 2017). There, it interacts with a network of protein partners that promote robust completion of cytokinesis and septation (Arasada and Pollard, 2014; Cortés et al., 2015; Ren et al., 2015; Roberts-Galbraith et al., 2009). Disruption of the SH3 domain network results in CR fragmentation (Roberts-Galbraith et al., 2009).
While most of Cdc15’s protein partners bind to its SH3 domain, the formin Cdc12 binds to the F-BAR domain in the PM-proximal CR layer (Carnahan and Gould, 2003; McDonald et al., 2017; Willet et al., 2015). Other F-BAR family members also interact with partners through their F-BAR domains (Begonja et al., 2015; Garabedian et al., 2018; Kostan et al., 2014; Liu et al., 2019; Oh et al., 2017; Senju et al., 2011; Shoham et al., 2003), but in no case has a complete structural and mechanistic understanding of these interactions at membranes been described.
Here, we show that the Cdc15 F-BAR domain binds membrane and Cdc12 simultaneously, revealing an important mechanism linking the F-actin of the CR to the PM. We also report the crystal structure of the Cdc15 F-BAR domain and identify residues on its cytosolic face, opposite the membrane-binding face, that coordinate binding to the formin Cdc12 and the paxillin-like protein Pxl1. Surprisingly, mutation of the Cdc15 F-BAR domain residues that disrupt the CR recruitment of Pxl1 and its binding partner calcineurin (CN) results in a shallow pre-constriction CR, with intermediate- and distal-layer components localizing closer to each other and ~35% closer to the PM than in wild type. These organizational defects lead to failures in CR constriction. Our results indicate that an F-BAR domain creates a platform on the PM to bring together cytoskeletal and signaling elements necessary to establish the architecture of the CR that, in turn, is required for its function. Our study presents a view of F-BAR domains in which their functions at membranes profoundly influence the overall architecture of an actin-based structure.
RESULTS
The Cdc15 F-BAR Domain Binds Membrane and Cdc12 Simultaneously with Opposite Faces
The Cdc15 F-BAR domain binds membrane and a motif in the formin Cdc12 (McDonald et al., 2015; Willet et al., 2015), but it is unknown if it binds both simultaneously. We generated giant unilamellar vesicles (GUVs), mixed them with purified GFP-Cdc15 F-BAR domain and/or rhodamine (Rh)-conjugated peptide containing the Cdc12 F-BAR binding motif (residues 20–40), and visualized these components by fluorescence microscopy. We observed that the Cdc12 peptide localized to GUVs minimally on its own; however, in combination with Cdc15 F-BAR domain, the two proteins co-localized on vesicles (Figure 1A). These results indicate that the Cdc15 F-BAR domain can position Cdc12 directly at the PM by binding membrane and Cdc12 simultaneously.
Figure 1. The Cdc15 F-BAR Domain Binds Membrane and Cdc12 Simultaneously.
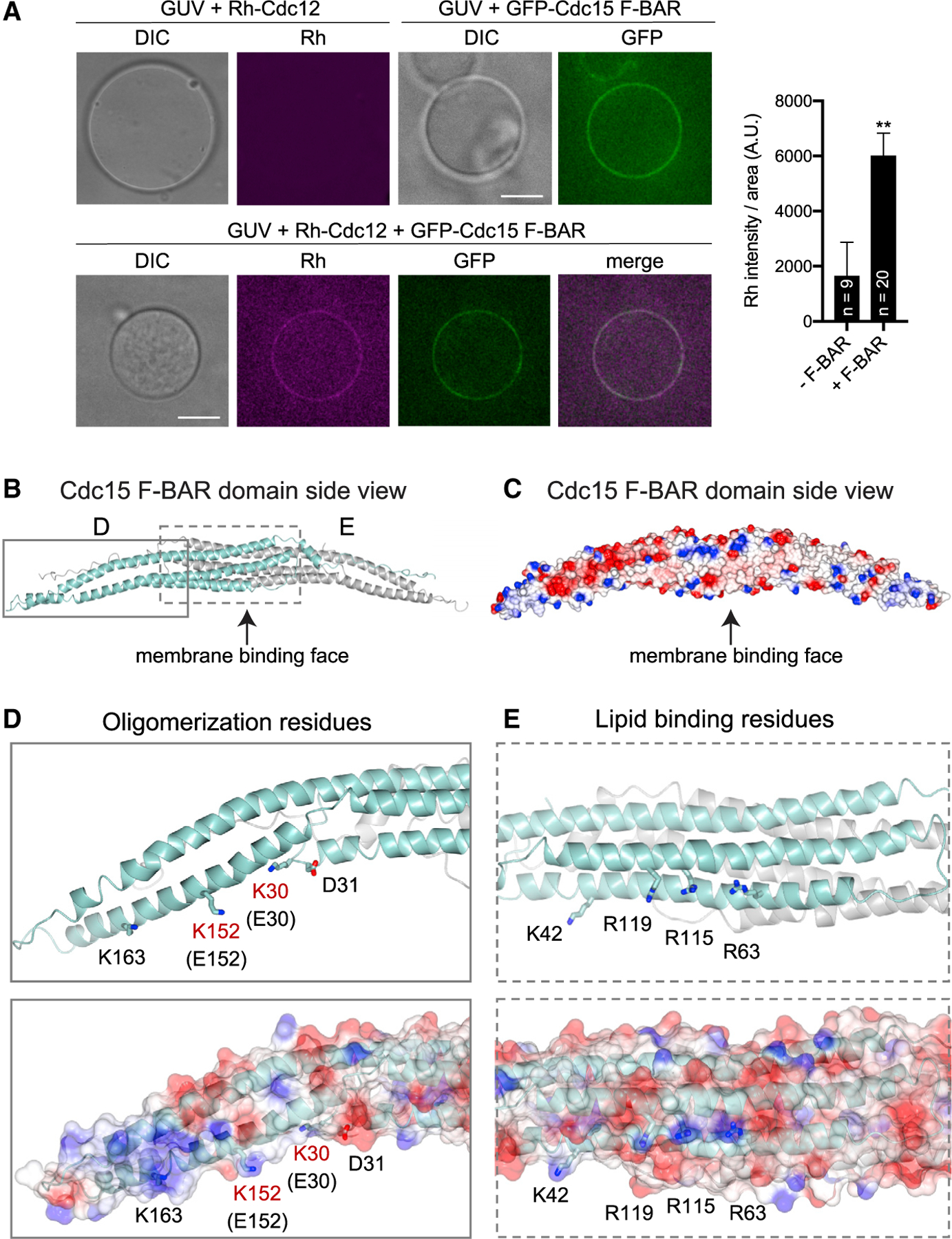
(A) Left: fluorescence micrographs of GUVs to which rhodamine (Rh)-conjugated Cdc12 peptide (aa 20–40) and GFP-Cdc15 F-BAR domain were added as indicated. GUVs were visualized by DIC. Single Z slices are shown, and scale bars = 5 μm. Right: quantification of mean Rh-Cdc12 signal on GUVs in the absence (—F-BAR) and presence (+F-BAR) of GFP-Cdc15 F-BAR domain. Error bars represent SEM, **p < 0.01, Student’s t test. Number of vesicles analyzed (n) is indicated.
(B) Ribbon diagram of the crystal structure of the Cdc15 F-BAR domain (E30K E152K). Chain A = cyan, Chain B = gray.
(C) Electrostatic surface potential of the Cdc15 F-BAR domain. Red = negative, blue = positive surface charge.
(D) Inset shows a close-up of oligomerization residues (McDonald et al., 2015) on ribbon (top) and surface (bottom) diagrams with wild-type residues in black; two residues shown in red (K30, K152) were mutated to facilitate crystallization.
(E) Inset highlights lipid-binding residues (McDonald et al., 2015) on ribbon (top) and surface (bottom) diagrams.
We next took a hybrid structural and biophysical approach to understand how the Cdc15 F-BAR domain coordinates Cdc12 at the PM. Attempts to determine the X-ray crystal structure of wild-type Cdc15 F-BAR domain were unsuccessful. Therefore, we focused on a well-characterized mutant, E30K E152K, which prevents F-BAR domain oligomerization (McDonald et al., 2015). We determined the X-ray crystal structure of the Cdc15 F-BAR domain (residues 19–312; E30K E152K) at 3.5-A resolution using molecular replacement in Phaser with final Rwork and Rfree values of 26.7% and 31.0%, respectively (Figures 1B and 1C; Table S1). The S. pombe Imp2 F-BAR domain (PDB: 5C1F) (McDonald et al., 2016) served as the initial search model (33% sequence identity). Crystals were of space group P1211 with two molecules in the asymmetric unit. Both copies show well-ordered electron density for residues 35–297. We could not clearly trace electron density at the N and C termini of either copy. Specifically, we lack density for chain A residues 19–34 and 299–312 and chain B residues 19–22 and 297–312. The final model exhibits good overall geometry, with 92.5% residues favored and 7.3% residues allowed in the Ramachandran plot.
The Cdc15 F-BAR domain has four a helices (Figures S1A and S1B). We resolved four helices in chain A and three in chain B because the C terminus exhibits more disorder. As expected (Henne et al., 2007; Peter et al., 2004), the Cdc15 F-BAR domain formed a dimer. The two copies interacted over an extensive solvent-accessible surface area (4,279.5 Å2 calculated in AREAIMOL), primarily mediated by van der Waals contacts (data not shown). The structure reveals a central core containing a six-α-helical dimer bundle, in which each F-BAR domain contributes three extended and partly kinked α helices (α1/α2/α3). The Cdc15 F-BAR domain has an α helix at its C terminus. We observe density for residues in helix α4 (270–279) in both chains, but it is better ordered in chain A. The ends of the F-BAR domain have extended wings that adopt a slightly bent conformation compared to the core. The C termini of both chains (chain A residues 278–299; chain B residues 272–297) lack secondary structure but maintain extensive contact with helices α2 and α3 from the symmetry copy. Previously defined residues required for oligomerization were at the tips of the F-BAR domain dimer (Figure 1D), and membrane-binding residues were on the concave face of the dimer (Figure 1E), as predicted (McDonald et al., 2015).
The two closest structural homologs in the Protein Data Bank are the S. pombe Imp2 and S. cerevisiae Hof1 F-BAR domains (McDonald et al., 2016; Moravcevic et al., 2015); these exhibit 1.8 Å and 2.9 Å root-mean-square deviation (RMSD), respectively. Structural comparison of the Cdc15 and Imp2 F-BAR domains, however, reveals the Cdc15 F-BAR domain is flatter and wider overall (Figures S1C–S1G). We also compared our structure with the human GAS7 F-BAR domain (PDB: 6IKN) (Hanawa-Suetsugu et al., 2019). These two F-BAR domains are approximately the same length: the Cdc15 F-BAR domain measures 211 Å from tip to tip, while the GAS7 F-BAR domain measures 212 Å. GAS7 F-BAR appears slightly flatter than the Cdc15 F-BAR domain (Figures S1C–S1G), but they are generally similar in form. Considering that GAS7, like Cdc15, is a non-tubulating F-BAR domain (Hanawa-Suetsugu et al., 2019; McDonald et al., 2015), it is logical that the Cdc15 F-BAR domain is more similar in shape to Gas7 than to the Imp2 F-BAR domain (Figures S1C–S1G), which can tubulate membranes (McDonald et al., 2016).
We next determined where on the Cdc15 F-BAR domain Cdc12 binds. The Cdc12 F-BAR-binding motif is rich in basic amino acids (Willet et al., 2015), and mutation of an acidic patch on the non-membrane-binding face of the F-BAR domain (Cdc15-3A: D255, E256, E259; Figure 2A) to alanines eliminated the F-BAR interaction with Cdc12 peptide (residues 20–40) (Figure 2B, left). Although the Imp2 F-BAR domain is similar to the Cdc15 F-BAR domain and has acidic patches on its non-membrane-binding face (Figure S1H), it did not interact with the Cdc12 peptide (Figure 2B, right). The Cdc15 (E30K E152K) F-BAR domain exists as a dimer in solution (McDonald et al., 2015), and using isothermal titration calorimetry (ITC), we found that the Cdc12 peptide binds Cdc15 (E30K E152K) F-BAR with low micromolar affinity and calculated stoichiometry (n) of 0.5 (Kd =5 μM; Figure 2C; Table S2). We interpret these results to mean that one peptide binds two molecules of Cdc15 F-BAR, or one dimer. Together, these results establish that one side of the Cdc15 F-BAR domain binds membrane, while the opposite face binds a single molecule of Cdc12, thus linking the key actin nucleator of the CR to the PM. It will be interesting to determine if utilization of non-membrane-binding surfaces for protein interaction is a general feature of other F-BAR domains.
Figure 2. Cdc12 Binds the Cytosolic Face of the Cdc15 F-BAR Domain.
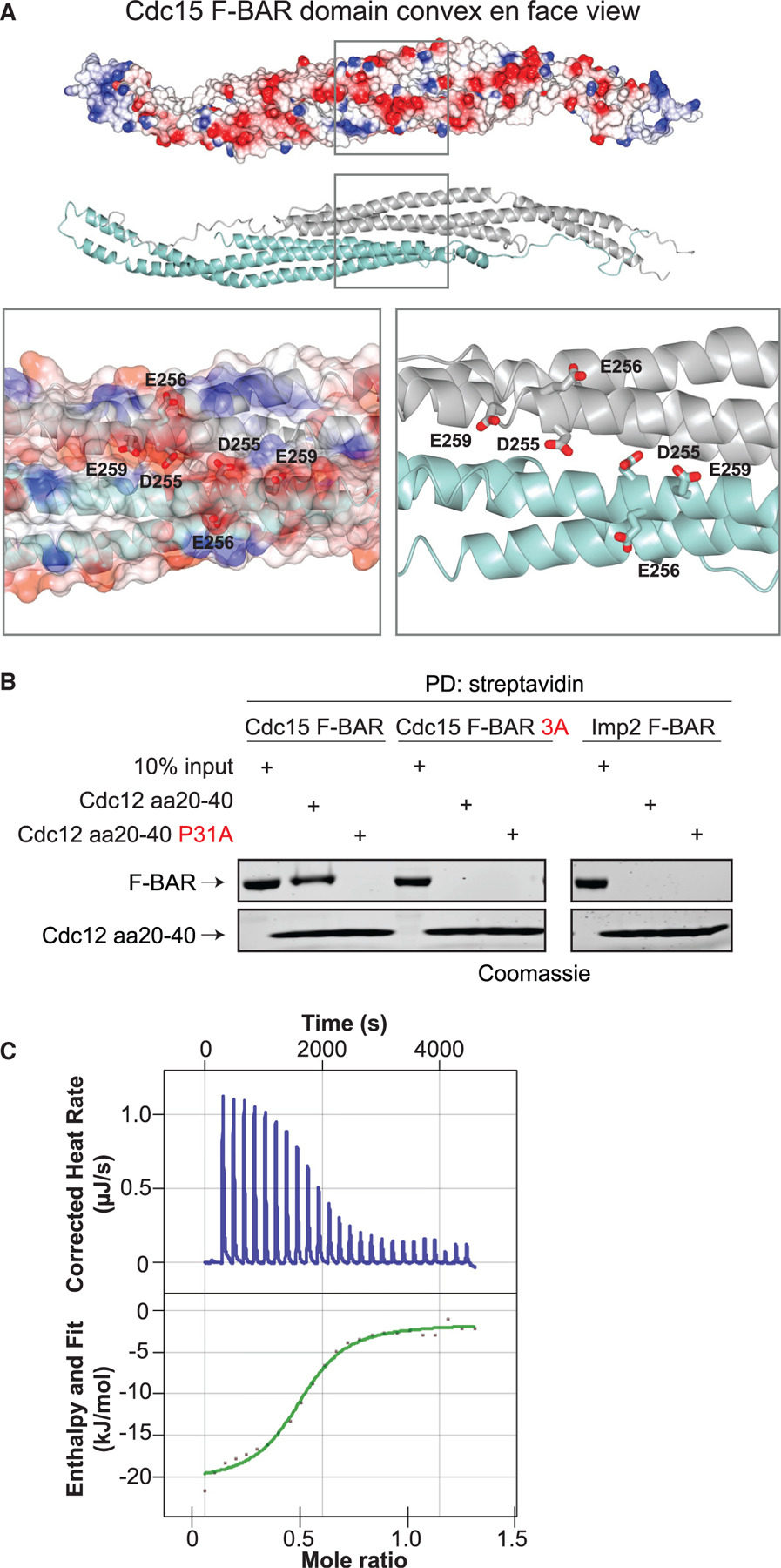
(A) Top: electrostatic surface potential of the Cdc15 F-BAR domain cytosolic face. Red = negative, blue = positive surface charge. Middle: ribbon diagram of the same face. Bottom: close-up of residues mutated in Cdc15-3A.
(B) In vitro binding assays with biotin-labeled Cdc12 peptide (aa 20–40) and purified Cdc15 F-BAR domain (left; wild type and 3A) or Imp2 F-BAR domain (right). F-BAR domain associated with streptavidin resin (PD, pull down) was analyzed by SDS-PAGE followed by Coomassie staining. Cdc12(aa 20–40 P31A) was a negative control (Willet et al., 2015).
(C) Representative ITC experiment with Cdc12 (aa 20–40) peptide and Cdc15 F-BAR domain (E30K E152K). Top panel shows raw data; bottom panel shows normalized integrated data.
See also Table S2.
The Cdc15 F-BAR Domain Has Additional Functions
When the Cdc12-Cdc15 F-BAR domain interaction is disrupted by mutating P31 in the Cdc12 F-BAR-binding motif (cdc12-P31A), Cdc12-P31A recruitment to the CR is reduced (Willet et al., 2015). We expected that if we disrupted the Cdc12-binding site on the F-BAR domain, Cdc12 localization to the CR would be similarly reduced. Thus, we introduced the 3A mutations into the endogenous S. pombe Cdc15 locus to construct cdc15-3A and examined Cdc12-mNeonGreen (mNG) localization (Figure 3A). As predicted, Cdc12-mNG fluorescence intensity at the CR was reduced by ~40% in cdc15-3A compared to wild type, similar to the reduction measured for Cdc12-P31A-mNG (Figure 3A). As expected for two mutations that disrupt the same interaction, the effect of cdc15-3A on Cdc12-P31A-mNG CR localization was non-additive (Figure 3A). However, unlike cdc12-P31A, ~30% of cdc15-3A cells had a strikingly abnormal morphology, with defects including additional cell poles, bulges, or swells and heterogeneous lengths (Figure 3B). These results indicated that cdc15-3A disrupted another F-BAR domain function in addition to interfering with Cdc12 recruitment to the CR.
Figure 3. The Cdc15 F-BAR Domain Cytosolic Face Has Functions in Addition to Scaffolding Cdc12.
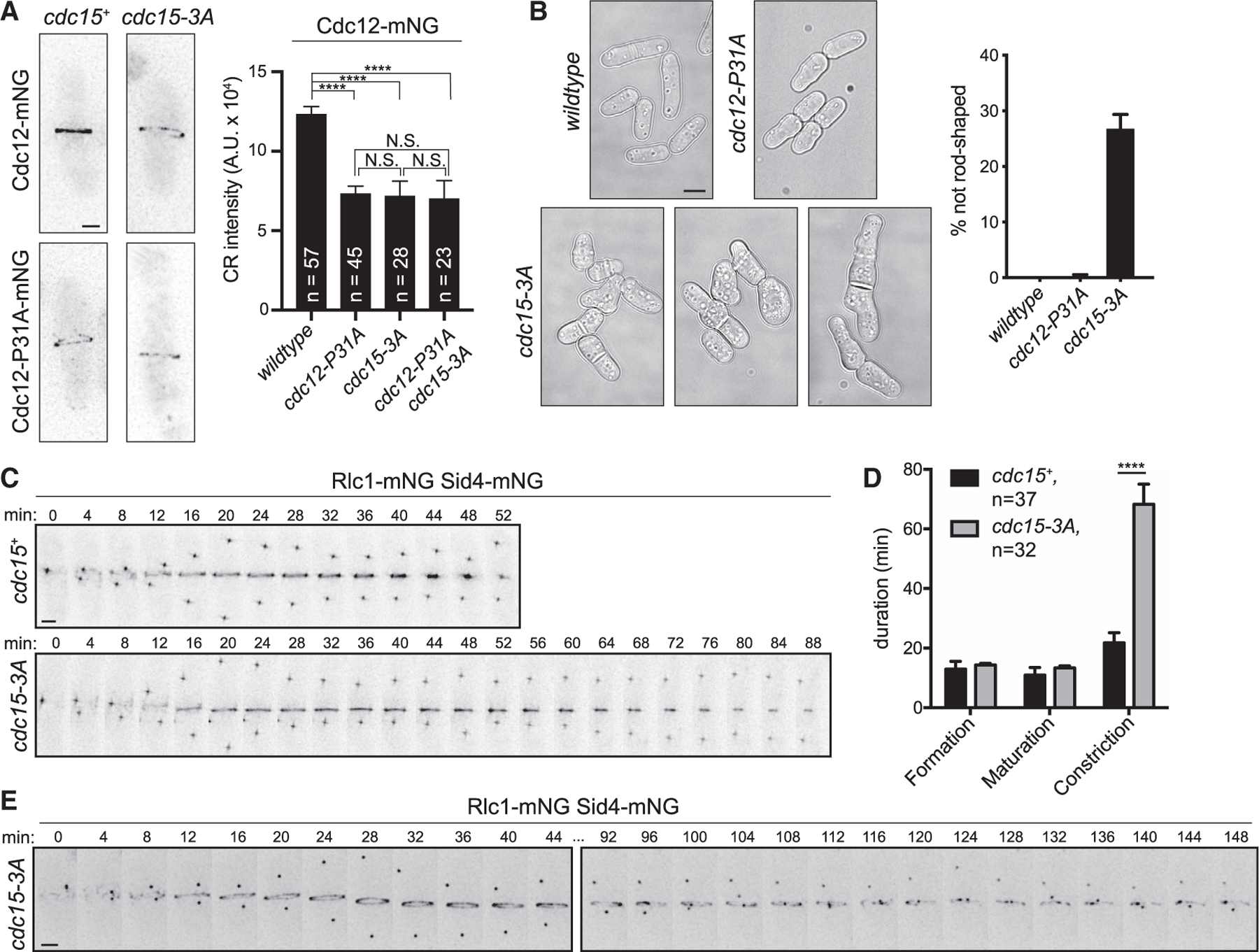
(A) Left: representative live-cell images; deconvolved max projections are shown. Right: quantification of mean Cdc12-mNG CR intensity. Error bars represent SEM. ****p < 0.0001, ***p < 0.001, NS, not significant; p > 0.99; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. Total number of cells (n) from three experiments is indicated. Scale bar = 2 µm.
(B) Left: DIC images of the indicated strains grown at 25°C. Single Z slices are shown, and scale bar = 5 µm. Right: quantification of morphology defects in indicated strains. Wild type, n = 305 cells; cdc12-P31A, n = 341 cells; cdc15-3A, n = 436 cells; three experiments, error bars = SEM.
(C) Representative montages from time-lapse imaging. Minutes elapsed since spindle pole body separation are indicated. Max projections are shown. Scale bar = 2 µm. (D) Quantification of the mean duration of CR events, related to (C). ****p < 0.0001, Student’s t test. Error bars represent SEM, and number of cells analyzed (n) is indicated.
(E) Example montage of cdc15-3A cell with aberrant CR constriction. Ellipses denote frames removed for space, and max projections are shown. Scale bar = 2 µm.
We tested if mutating the F-BAR domain surface resulted in an overall loss of protein folding or defects in the known F-BAR domain functions of oligomerization and membrane binding. Recombinant Cdc15-3A F-BAR domain was not defective in overall tertiary structure, as determined by circular dichroism (Figure S2A), and was oligomerization competent, as shown by negative stain electron microscopy (Figure S2B). To assay Cdc15-3A membrane binding, we produced Cdc15 or Cdc15-3A in bacteria with kinase Pom1, as previously described (Bhattacharjee et al., 2020). Mutations that abolish F-BAR domain oligomerization (E30K E152K) were included to allow robust purification (Bhattacharjee et al., 2020; McDonald et al., 2015). As expected, we found that phosphorylated Cdc15-3A (E30K E152K) did not bind membrane in a liposome co-pelleting assay (Figure S2C), as previously reported for phosphorylated Cdc15 (E30K E152K) (Bhattacharjee et al., 2020). But dephosphorylated Cdc15-3A (E30K E152K) co-pelleted with liposomes to a similar extent as Cdc15 (E30K E152K), indicating that the 3A mutations do not prevent Cdc15 from binding membrane (Figure S2C). Next, we confirmed that Cdc15-3A was produced (Figure S2D) and localized to the CR at wild-type levels in cells (Figure S2E). Thus, there is no significant defect in any known function ascribed to the Cdc15 F-BAR domain except the binding of Cdc12.
Because Cdc15 is most well known as a cytokinesis protein (Fankhauser et al., 1995), we examined cytokinesis dynamics in cdc15-3A. Cytokinesis occurs in three stages marked by (1) formation of the CR, the time from spindle pole body separation until cytokinetic nodes coalesce into a coherent ring; (2) CR maturation, the time from which the CR is fully formed until constriction initiates; and (3) CR constriction, the time during which the CR decreases in diameter to a single point. Live-cell time-lapse imaging of wild-type and cdc15-3A cells expressing markers of the CR (Rlc1-mNG) and the spindle pole body (Sid4-mNG) to monitor progression through cytokinesis and mitosis, respectively, showed that while the duration of CR formation and maturation were comparable to wild type in cdc15-3A, CR constriction was significantly prolonged (Figures 3C and 3D). In fact, we were unable to evaluate all cells because they did not complete constriction before photo-bleaching of the fluorescent probes or cell death (12.5% of cdc15-3A failed constriction, n = 32; 0% of wild type failed constriction, n = 37). We also saw evidence of CR instability manifested as CR sliding events (CR sliding frequency = 51% ± 8% for cdc15-3A, n = 38 cells; 0% for wild type, n = 30 cells; two experiments) and aberrant constriction (Figure 3E). We concluded that cdc15-3A cells are defective in the constriction phase of cytokinesis; based on previous results, this cannot be explained by reduced recruitment of Cdc12 alone (Willet et al., 2015).
The Cdc15 F-BAR Domain Drives the Nanoscale Architecture of the CR
Because Cdc15 scaffolds a cohort of proteins through its F-BAR domain, SH3 domain, and IDR and spans multiple layers of the CR, it is a candidate driver of CR architecture. To examine whether the cytokinesis defects in cdc15-3A cells reflected a global change in CR architecture, we used fPALM (McDonald et al., 2017). In wild-type pre-constriction CRs, the Cdc15 F-BAR domain is positioned at the PM, while its C-terminal SH3 domain extends into the intermediate layer (McDonald et al., 2017). To determine whether this overall organization is retained in Cdc15-3A, we compared the distances to the PM (dcenter) of the fluorophore mMaple3 fused to the Cdc15 N or C terminus. We used the membrane dye mCLING-ATTO647N to label the PM (McDonald et al., 2017; Revelo et al., 2014). Consistent with our in vitro results that there is no apparent deficit in Cdc15-3A membrane binding or oligomerization, we found that mMaple3-Cdc15-3A (F-BAR) was membrane proximal, like wild type (Figure 4A; wild type: dcenter = 54 ± 33 nm; 3A: dcenter = 51 ± 41 nm). While in wild-type cells Cdc15-mMaple3 (SH3 domain) is extended into the intermediate layer of the CR, Cdc15-3A-mMaple3 was on average ~40 nm closer to the PM than wild type (wild type: dcenter = 140 ± 51 nm; 3A: dcenter = 98 ± 60 nm) (Figure 4A). Considering that the cdc15-3A mutations are on the F-BAR domain, a position that is ordinarily spatially separated from the Cdc15 SH3 domain, this result was surprising.
Figure 4. The Cdc15 F-BAR Domain Coordinates Other Binding Partners Required for Proper CR Architecture.
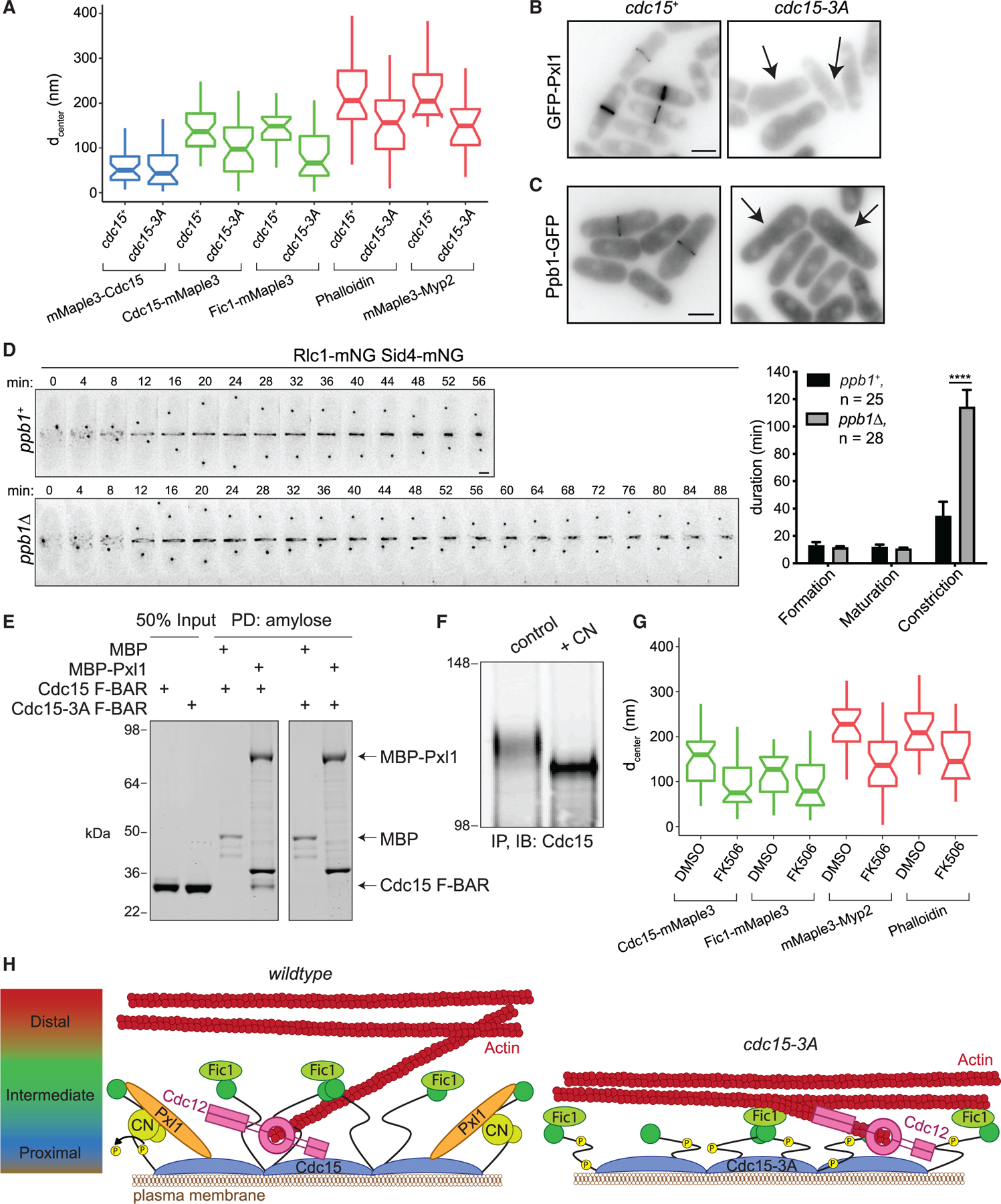
(A) Distance from the PM (dcenter) of the indicated CR components calculated from fPALM. Components of the CR membrane-proximal layer are in blue, intermediate in green, and distal in red. Boxplots depict first and third quartiles and median; whiskers, minimum and maximum; notches, 95% confidence intervals. See also Table S4.
(B and C) Representative images of GFP-Pxl1 (B) and Ppb1-GFP (C). Sum projections are shown, and scale bars = 5 µm. Arrows indicate cells without CR signal.
(D) Left: representative time-lapse montages. Minutes elapsed since spindle pole body separation are indicated; deconvolved max projections are shown. Scale bar = 2 µm. Right: quantification of the mean duration of CR events, ****p < 0.0001, Student’s t test. Error bars represent SEM, and number of cells analyzed (n) is indicated.
(E) Coomassie-stained SDS-PAGE of proteins PD with amylose resin after in vitro binding assay. Both F-BAR constructs also contain E30K E152K mutations. Experiment was repeated with similar results.
(F) Immunoblot (IB) of Cdc15 after immunoprecipitation (IP) from asynchronous cells and treatment with either a control or recombinant calcineurin (CN) phosphatase reaction. Numbers indicate position of molecular weight markers in kDa. Experiment was repeated with similar results.
(G) Distance from the PM (dcenter) of the indicated CR components in cells treated with DMSO or FK506, as in (A). See also Table S4.
(H) Model for the role of the Cdc15 F-BAR domain in driving nanoscale architecture of the CR. Left: the Cdc15 F-BAR domain recruits the formin Cdc12, but also Pxl1, which is required for CN localization. CN dephosphorylates Cdc15 to promote its open conformation. Right: in cdc15-3A, the protein network established by the Cdc15 F-BAR domain is disrupted, resulting in a shallower CR. Model not drawn to scale.
Consistent with the altered localization of Cdc15-3A-mMa-ple3, a second normally intermediate-layer protein and Cdc15 SH3-binding partner, Fic1, also localized closer to the PM in cdc15-3A (wild type: dcenter = 144 ± 43 nm; cdc15-3A: dcenter = 79 ± 59 nm). Unexpectedly, two components of the distal layer, the myosin 2 Myp2 motor head and F-actin, also localized closer to the PM (Figure 4A) (mMaple3-Myp2 wild type: dcenter = 230 ± 75 nm; cdc15-3A: dcenter = 150 ± 70 nm; Phalloidin-Atto488 wild type: dcenter = 216 ± 83 nm; cdc15-3A: dcenter = 150 ± 85 nm). On average, these intermediate and distal components localize 35% closer to the PM in cdc15-3A compared to wild type. Altogether, these results indicate that there is an overall disruption in the nanoscale architecture of the CR in cdc15-3A, resulting in a shallower pre-constriction CR. Considering the defects in CR constriction in cdc15-3A, this implies that proper distance of the CR from the PM and distribution of its components relative to one another is required for CR constriction.
The Cdc15 F-BAR Domain Coordinates Partners Required for CR Architecture
The disruption of CR architecture, subsequent failures in CR constriction, and morphological defects in cdc15-3A support the idea that the cytosolic face of the Cdc15 F-BAR domain does more than recruit Cdc12 and likely binds additional factors required for assembling a CR of the proper depth. To define these factors, we examined the localization of 32 proteins in cdc15-3A (Table S3). Of these, we found that neither paxillin-like protein GFP-Pxl1 (Figure 4B) nor the catalytic subunit of the protein phosphatase CN Ppb1-GFP (Figure 4C) localized to the CR in cdc15-3A. Notably, the morphological defects of cdc15-3A resemble those of pxl1Δ and ppb1Δ (Fujita et al., 2002; Ge and Balasubramanian, 2008; Lu et al., 2002; Martıń-Garcıá et al., 2018; Pinar et al., 2008; Yoshida et al., 1994), and CR constriction takes longer in ppb1Δ (Figure 4D; Martín-García et al., 2018), similar to cdc15-3A (Figures 3C and 3D). Therefore, the absence of Pxl1 and CN at the CR likely accounts for the morphological and CR defects of cdc15-3A.
As the other 30 proteins examined localized to the division site in cdc15-3A (Table S3), we focused on the possibility that the Cdc15 F-BAR domain binds Pxl1 and/or CN. Although we did not detect binding between the purified Cdc15 F-BAR domain and recombinant CN, MBP-Pxl1 bound the Cdc15 F-BAR domain (Figure 4E). This binding interaction was abrogated by the Cdc15-3A mutation (Figure 4E), indicating that Pxl1 utilizes the same F-BAR-binding site as Cdc12. Because Pxl1 associates with CN and is required for its CR localization (Martıń-Garcıá et al., 2018), we reasoned that loss of CN from the CR in cdc15-3A is likely an indirect effect of Pxl1 loss.
We next tested if Pxl1 and/or CN influences Cdc15 conformation in the pre-constriction CR. We wanted to assay Cdc15-mMaple3 position in pxl1Δ by fPALM, but a negative genetic interaction with the tagged cdc15 allele and pxl1Δ prevented cell growth in liquid culture. Thus, we focused on CN. Cdc15 phosphostate changes when cells are treated with the CN inhibitor FK506 (Martıń-Garcıá et al., 2018), and we confirmed that Cdc15 is a direct substrate of recombinant CN in vitro (Figure 4F). Because phosphorylated and dephosphorylated forms of Cdc15 have different conformations when visualized by negative stain electron microscopy, one model for Cdc15 regulation posits that dephosphorylation allows Cdc15 to adopt a more open conformation (Roberts-Galbraith et al., 2010; Wachtler et al., 2006). Therefore, we considered if loss of CN in cdc15-3A CRs underlies the defects in placement of the Cdc15 C terminus away from the PM. As predicted, after treatment with the CN inhibitor FK506, the Cdc15 C terminus was not extended away from the PM to the same extent as in DMSO-treated cells, similar to the defect in cdc15-3A (Figure 4G; DMSO: dcenter = 152 ± 55 nm; FK506: dcenter = 94 ± 53 nm). Furthermore, we found that Fic1 and the two distal layer components F-actin and the Myp2 motor head localized closer to the PM in FK506-treated cells (Figure 4G; Fic1-mMaple3 DMSO: dcenter = 114 ± 49 nm, FK506: dcenter = 95 ± 61 nm; mMaple3-Myp2 DMSO: dcenter = 222 ± 62 nm, FK506: dcenter = 136 ± 67 nm; Phalloidin-Atto488 DMSO: dcenter = 216 ± 63 nm, FK506: dcenter = 153 ± 67 nm). This indicates that CN activity is required for proper Cdc15 conformation and nanoscale organization of the pre-constriction CR.
DISCUSSION
We have demonstrated here that the dimeric Cdc15 F-BAR domain forms a membrane-binding module of shallow curvature. Although multiple F-BAR domain structures have been determined, only one prior study described a crystal structure of an F-BAR domain from the subset that does not tubulate membranes, GAS7 (Hanawa-Suetsugu et al., 2019). Given that both Cdc15 and GAS7 F-BAR crystal structures reveal shallow curvature, we suggest that this structural feature may underlie their inability to tubulate and distinguish them from other family members. Together, these structural and functional studies provide further evidence that the primary function of F-BAR domains is to serve as oligomeric membrane-binding platforms for the assembly of actin-rich structures (McDonald and Gould, 2016b; Traub, 2015).
Our results demonstrate that an F-BAR domain can bind membrane and a protein partner simultaneously and establish that the Cdc15 F-BAR does so using opposite faces. Because the PM-bound Cdc15 F-BAR domain is abundant within the CR (Wu and Pollard, 2005), it is intuitive that the large amount of F-BAR domain surface area facing the cytoplasm could be utilized for placing other proteins at the PM. Considering that Cdc15 is in vast excess to its known partner Cdc12 within the mature CR (16,100 ± 2,300 Cdc15 molecules versus 300 ± 50 Cdc12 molecules; Wu and Pollard, 2005), it seems likely that other proteins (e.g., Pxl1) can be accommodated on this surface without competing for a binding site. Although several F-BAR domains from other proteins can substitute for the F-BAR of Cdc15 to support cell viability, cells producing these F-BAR-Cdc15 fusions display morphological defects similar to cdc15-3A (Mangione et al., 2019). It is likely that this phenotype arises from the inability of these other F-BAR domains to bind Cdc12 and Pxl1, as we have determined here is the case for the Imp2 F-BAR domain. It will be fascinating to discern how generally F-BAR domains serve to create binding platforms on membranes for specialized components of complex actin structures.
We propose a model wherein the Cdc15 F-BAR domain itself establishes a protein interaction network critical for the integrity of the CR (Figure 4H). In addition to Cdc12, we discovered that Pxl1 and CN are part of this network. Although a second Pxl1-binding site exists within Cdc15, consisting of the SH3 domain and a portion of the IDR (Bhattacharjee et al., 2020), we found that an intact F-BAR domain-binding site is required for Pxl1 recruitment to the CR. Because both Pxl1 and the Cdc15 IDR are required for CN localization to the CR (Mangione et al., 2019; Martín-García et al., 2018), we propose that Pxl1 recruitment by the Cdc15 F-BAR domain allows Pxl1 and the Cdc15 IDR to cooperatively recruit CN.
Disruption of the network established by the Cdc15 F-BAR domain on the PM has surprisingly long-range effects on the placement of the Cdc15 C terminus and the rest of the CR’s architecture. Our results suggest that there is either a defect in extension of CR components away from the PM during ring assembly, or there is a collapse or compression of CR components toward the membrane over time, and it will be interesting to distinguish between these possibilities in future studies.
Although the complete cohort of CN substrates involved in proper CR architecture is unknown, Cdc15 is likely a major target. Because CN does not localize to the CR until it is fully formed, Cdc15 is likely dephosphorylated by other phosphatases first to allow CR formation. Indeed, one model of Cdc15 regulation suggests that dephosphorylation and the subsequent change in conformation occur in two steps, with the first dependent upon the Cdc14 family phosphatase, Clp1 (Roberts-Galbraith et al., 2010; Wachtler et al., 2006). We propose that in the absence of CN, Cdc15 is only partially dephosphorylated, resulting in an intermediate conformation that allows F-BAR domain membrane binding but not full extension of the remainder of the protein away from the PM (Figure 4H). In accord, when cells are treated with FK506, Cdc15 is dephosphorylated as cells enter mitosis, but not to the same extent as in the absence of inhibitor (Martín-García et al., 2018). In support of a multi-step activation model for F-BAR proteins, more than one stimulus is required for relief of Nwk autoinhibition, including binding to PI(4,5)P2 and multiple proteins (Del Signore et al., 2020).
In summary, our results establish that F-BAR domain oligomers can serve as an extensive platform to link numerous proteins in actin-based structures to the PM. Moreover, disruption of F-BAR domain function as a protein landing pad can have long-range ramifications to the architecture and functionality of cytoskeletal structures.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kathleen Gould (Kathy.Gould@vanderbilt.edu)
Materials availability
Strains and plasmids generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The genotypes and sources of S. pombe strains used in this study are detailed in Table S5. S. pombe strains were grown in yeast extract (YE) media and all strains for live-cell imaging were grown at 25°C. Standard methods were used for genetic crosses and tetrad analyses.
METHOD DETAILS
Yeast methods
cdc15 mutants were integrated at the endogenous locus by rescuing cdc15+/cdc15::ura4+ diploids with pIRT2-cdc15 mutant constructs containing 5ʹ and 3ʹ noncoding flanks. Haploid integrants resistant to 5-fluorourotic acid (5-FOA) were isolated and the cdc15 mutations were verified by PCR and sequencing. Transformations of S. pombe cells were performed with a lithium acetate method (Keeney and Boeke, 1994).
Plasmids
Cdc15-3A mutant constructs (D255A E256A E259A) were generated by site-directed mutagnesis and sequenced to confirm. To coexpress the calcineurin subunits, ppb1, cnb1, and cam1 were PCR amplified from S. pombe cDNA. In pET-Duet-1, ppb1 was assembed in the BamHI/NotI sites in frame with the 6X-His tag and Cnb1 was assembled in the NdeI/KpnI sites by Gibson assembly. Cam1 was assembled in the NcoI/NotI sites of pRSF-Duet-1.
Recombinant protein expression and purification
The pET15b plasmid encoding 6X-His fusion Cdc15 F-BAR domain (residues 19–312 E30K E152K) was transformed into BL21(DE3)/pLysS E. coli cells and expressed in 2XTY broth at 37°C until the O.D. (A600) reached 0.8. The cultures were induced with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) at 20°C overnight, and cells were harvested by centrifugation (6000 × g, 10 min, 4°C). The cell pellet was resuspended in lysis buffer (20 mM Tris pH 8.0, 300 mM NaCl, 50 mg/ml of benzamidine, 100 units DNase I, and 2 mM β-mercaptoethanol). The cells were lysed by mechanical disruption at 30 kpsi using a cell disrupter (Constant Systems). The lysate was clarified by centrifugation at 50,000 × g for 30 min at 4°C. Proteins were purified using affinity chromatography from the clarified lysate. The purification was performed on a Ni-NTA (GE healthcare) gravity column. The column was thoroughly washed with lysis buffer containing 100–500 mM salt. Finally, the protein of interest was eluted using a linear gradient of imidazole (100–300 mM) in 20mM Tris pH 8.0, 300 mM NaCl. The eluted affinity purified 6X-His tagged Cdc15 F-BAR domain was finally subjected to size exclusion chromatography using a superdex-200 16/600 HiLoad column, pre-equilibrated with 20 mM Tris (pH 8.0), 100 mM NaCl and 2 mM DTT, attached to an AKTA pure (GE Healthcare). The purified protein was concentrated to 10 mg/ml using a Centricon Ultra-10 kDa centrifugal filter (Millipore, USA) for crystallization.
Cdc15 F-BAR domains (residues 19–312) for all experiments other than crystallography were expressed as a 6xHis fusion in E. coli Rosetta2(DE3)pLysS cells grown to log phase in terrific broth, induced overnight at 17°C with 0.1 mM IPTG. Cells were lysed by sonication and F-BAR domains were purified on cOmplete His-Tag resin (Roche) according to manufacturer’s protocol. His tags were then cleaved by thrombin digestion and F-BARs were further purified on a HiTrap Q SP anion exchange column (Cytiva Life Sciences) and concentrated with Amicon Ultra Centrifugal Filters (EMD-Millipore).
Full-length Cdc15 constructs were produced as previously described (Bhattacharjee et al., 2020) by co-expressing FLAG-Cdc15(E30K E152K, with or without the 3A mutations) with the kinase Pom1 in Rosetta2(DE3)pLysS cells grown to log phase in terrific broth, induced overnight at 17°C with 0.1 mM IPTG. Cells were lysed in 50 mM Tris pH 7.4, 150 mM NaCl, 0.1% NP-40 by sonication. Cleared lysate was incubated with Anti-FLAG-M2 magnetic resin for 2 h at 4°C. Beads were then washed extensively in 50 mM Tris pH 7.4, 150 mM NaCl before elution in 200 µg/mL 3x-FLAG peptide for 20 min at room temperature. Eluates were then concentrated and cleared by spinning at high speed. For de-phosphorylated samples, 1 µL lambda phosphatase (New England Biolabs) and 1 mM MnCl2 was added and incubated for 15 min at room temperature.
Pxl1 was expressed as an MBP fusion as previously described (Bhattacharjee et al., 2020) in the pMAL-c2 vector in Rosetta2(DE3) pLysS cells, which were grown to log phase in terrific broth + 150 µM ZnCl2 (to increase production), induced overnight at 17°C with 0.1 mM IPTG. Cells were harvested, flash frozen, and lysed by sonication in 20 mM Tris pH 7.4, 150 mM NaCl, 0.1% NP-40, and cOmplete EDTA-free protease inhibitor cocktail (Roche). Lysate was cleared at high speed then incubated with amylose resin (New England BioLabs) for 2 hr at 4°C. Resin was washed thoroughly in lysis buffer. After washing, buffer was added to the resin to create a 1:1 slurry of buffer and resin-bound protein. The concentration of MBP-Pxl1 fusion on resin was calculated by running a sample on SDS-PAGE alongside BSA standards and staining with Coomassie blue.
Calcineurin was produced by co-expressing Ppb1 as a 6xHis fusion (catalytic subunit), Cnb1 (regulatory subunit), and Cam1 (calmodulin) in Rosetta2(DE3)pLysS cells. Cells were lysed by sonication in 50 mM Tris pH 7.4, 150 mM NaCl, 0.1% NP-40 and cleared lysate was incubated with cOmplete His Tag resin (Roche). Resin was washed thoroughly in lysis buffer and then eluted in lysis buffer + 200 mM imidazole. Eluted protein was then dialyzed into 50 mM Tris pH 7.4, 150 mM NaCl, 0.1% NP-40, 2 mM CaCl2, 10 mM MgCl2.
Crystallization and crystallography
Cdc15 F-BAR domain was supplemented with 10 mM DTT before setting up 96-well sitting-drop crystallization screens using a Mosquito liquid handling robot (TTP LabTech). Crystallization plates were incubated at 20°C in a Rock-imager storage hotel (Formulatrix). Cdc15 F-BAR domain at 10 mg/ml was crystallized in 100 mM HEPES (pH 7.0), 1.1 M Sodium Malonate Dibasic Monohydrate (pH 7.0) and 0.5% Jeffamine ED 2003 (pH 7.0) by mixing equal volumes of protein with reservoir solution. Optimized diffraction-quality crystals of Cdc15 F-BAR domain were obtained using streak seeding in hanging drop vapor diffusion 24 well plates at 20°C by mixing equal volumes of protein with reservoir solution. The optimized crystallization condition was 100 mM HEPES (pH 7.0), 1.1 M Sodium Malonate Dibasic Monohydrate (pH 7.0) and 1% Jeffamine ED 2003 (pH 7.0). Diffraction data were collected at the Life Sciences Collaborative Access Team (LS-CAT) facility on beamline 21-ID-D at a wavelength of 0.979Å. To prevent radiation damage, crystals were equilibrated in a cryo-protectant containing 20% Glycerol (v/v) and then flash frozen in a 100 K Nitrogen stream. Cdc15 F-BAR crystal diffracted to 3.5 Å resolution. The dataset for Cdc15 F-BAR domain was merged and scaled in the Primitive Monoclinic space group, P1211, using the HKL2000 software suite (Otwinowski and Minor, 1997). The structure of Cdc15 F-BAR domain was solved using molecular replacement in Phaser (McCoy et al., 2007) with the crystal structure of Imp2 F-BAR (PDB ID: 5C1F) as the search model. After several rounds of refinement in PHENIX and model building in COOT (Emsley et al., 2010), the final model consists of two molecules in the asymmetric unit with a Rfactor and Rfree of 26% and 31% respectively. Structure images were prepared using PYMOL (pymol.org). Stereochemistry and quality of the structure was monitored after each round of refinement using PROCHECK (Laskowski et al., 1993) and Molprobity (Chen et al., 2010). Data collection and refinement statistics are summarized in Table S1. The structural coordinates and structure factors of the Cdc15 F-BAR domain have been deposited in the RCSB Protein Data Bank (PDB: 6XJ1).
Structural analyses and comparison of F-BAR structures
Visualizations and structural superpositions of structures, including Cα overlay traces presented in Figure S1, were carried out in CCPMG (McNicholas et al., 2011). F-BAR domain length distances were measured in Coot. Solvent-accessible surface area was calculated using CCP4 suite (Winn et al., 2011) program AREAIMOL (Lee and Richards, 1971) with a 1.4 Å radius sphere as a probe. Buried surface area was calculated with the PISA server (Krissinel and Henrick, 2007) (https://www.ebi.ac.uk/pisa).
Peptides
The Cdc12 synthetic peptide (residues 20–40, TPTPSARRTIGPRAPKSKTTY) used for ITC was synthesized by Genscript. Cdc12 peptide was dissolved in 20 mM Tris, pH 8, 100 mM NaCl and 2 mM DTT for use in ITC binding experiments and crystal screening with Cdc15 F-BAR domain protein. The Cdc12 (residues 20–40) and Cdc12-P31A (residues 20–40) peptides used for binding assays were synthesized with an N-terminal biotin modification and Cdc12 (residues 20–40) peptide for GUV binding assays was synthesized with an N-terminal rhodamine, also by GenScript.
Isothermal titration calorimetry (ITC)
ITC measurements were conducted on a Nano-ITC instrument (TA Instruments) in buffer consisting of 20 mM Tris (pH 8.0), 100 mM NaCl and 2 mM DTT. The sample cell was filled with 300 µL of Cdc15 F-BAR domain protein, while the syringe contained 50 µL of the Cdc12 synthetic peptide (TPTPSARRTIGPRAPKSKTTY). All solutions were degassed prior to being loaded into the cell. Aliquots (2 µL) of 750 µM Cdc12 synthetic peptide solutions were injected into a 100 µM Cdc15 F-BAR domain protein solution at 25°C with an interval gap of 3 min and the syringe rotating at 150 rpm to ensure proper mixing. The heat of dilution was measured by injecting the Cdc12 synthetic peptide into the buffer solution and subtracted from the heat of interaction. Data were analyzed using Nanoanalyser software to extract the thermodynamic parameters, ΔH°, Kd (1/Ka), and stoichiometry, n. The dissociation constant (Kd), enthalpy of binding (ΔH°), and stoichiometry (n) were obtained after fitting the integrated and normalized data to a single-site binding model. The stoichiometry was refined initially, and the value obtained was close to 1; then, n was set to 1.0 for calculation. The apparent binding free energy (ΔG°) and entropy (ΔS°) were calculated from the relationships ΔG° = RTln(Kd) and ΔG° = ΔH°–TΔS°. Experiments were performed in triplicate to check for reproducibility of the data.
Binding assays
Binding assays with Cdc15 F-BAR domains and synthetic biotinylated Cdc12 peptides were performed by mixing F-BAR domain at 0.05 μM and peptide at 0.10 μM with Pierce streptavidin UltraLink resin and incubating for 1 hr at 4°C in binding buffer (50 mM Tris pH 7.4, 250 mM NaCl, 0.1% NP-40). After thorough washing, samples were separated by SDS-PAGE, then stained with Coomassie blue and documented using an Odyssey CLx (Li-Cor Biosciences).
Binding assays with Cdc15 F-BAR domains and MBP/MBP-Pxl1 were performed by mixing 1 μM MBP or MBP-Pxl1 on amylose resin with 5 mg Cdc15 F-BAR domain in 50 mM Tris pH 7.4, 250 mM NaCl, 0.1% NP-40. All F-BAR domain constructs contained the mutations E30K E152K alone or in addition to 3A mutations. Binding assays were incubated for 1 hr at 4°C. Resin was then washed thoroughly and separated by SDS-PAGE. Gels were then stained with Coomassie blue and documented using an Odyssey CLx.
Western blot of cell lysates
To assay Cdc15 expression levels, pellets were collected from asynchronous populations and snap frozen. Cells were lysed by glass bead disruption in NP-40 buffer (Gould et al., 1991). Lysates were denatured by boiling at 95°C for 1 min then lysate samples were separated by SDS-PAGE, transferred to PVDF membrane (Immobilon-FL) and probed with anti-Cdc15 serum (VU326 1:10,000) (Roberts-Galbraith et al., 2009) and anti-PSTAIRE (1:10,000, Sigma-Aldrich) for loading control. Secondary antibodies conjugated to IRdye680/800 were utilized for detection with the Odyssey CLx.
Calcineurin phosphatase assay
Pellets collected from asynchronous cells were flash frozen then lysed by glass bead disruption in a modified NP-40 buffer lacking EDTA. Lysates were denatured by boiling at 95°C for 1 min. Cdc15 was immunoprecipitated from denatured lysate using anti-Cdc15 serum (VU326) and protein A Sepharose (GE Healthcare 17–5280–04) incubated for two h at 4°C. Sepharose was washed three times with modified NP-40 buffer then washed two times with calcineurin buffer (50 mM Tris pH 7.4, 150 mM NaCl, 0.1% NP-40, 2 mM CaCl2, 10 mM MgCl2). 20 mg recombinant calcineurin or calcineurin buffer for control was then added to the Sepharose and incubated for 30 min at 30°C with shaking. Samples were then resolved by SDS-PAGE, transferred to PVDF, and probed with anti-Cdc15 serum (VU326). Secondary antibodies conjugated to IRdye680/800 were utilized for detection with the Odyssey CLx.
Circular dichroism
For Near-UV circular dichroism (CD) spectrometry, purified Cdc15 F-BAR domains were analyzed using a Jasco J-810 spectropolarimeter (Jasco Analytical Instruments, Easton, MD). Near-UV data were collected at a protein concentration of 1.05 mg/ml for Cdc15 F-BAR domain and 1.10mg/ml for Cdc15-3A F-BAR domain. Near-UV CD spectra were collected in a 1-cm quartz cuvette with an average time of 4 s for each point and a step size of 10 nm/min from a wavelength of 250 to 330 nm. For both Cdc15 and Cdc15-3A F-BAR domains, four spectra were collected for Near-UV data and background corrected against a buffer blank. Data were converted to mean residue ellipticity [θ]m (degrees cm2dmol—1) using the formula [θ]m = (100*signal in mdeg)/(Cnl), where [θ]m is the mean residue ellipticity, C is the molar concentration of protein in millimolar, n is the number of residues/chain, and l is the cell path length in cm.
Liposome assays
Folch fraction lipids were obtained from Sigma-Aldrich, all others were obtained from Avanti Polar Lipids. Liposomes were formed as previously described (Itoh et al., 2005; McDonald et al., 2015). Briefly, CHCl3 lipid stocks were evaporated in a glass tube under N2 stream, vacuum dried for 1 h, rehydrated in 20 mM Tris pH 7.4, 150 mM NaCl buffer before vortexing, 10 cycles of freeze-thawing, and extrusion to 400 nm with an Avanti mini-extruder. Liposome co-pelleting assays were performed as previously described (McDonald and Gould, 2016a), where 100 uL of 1 mg/mL liposomes were mixed with 100 uL of FLAG-Cdc15 (10 µg total) for 15 min at room temperature before centrifugation at 150,000 xg in an Optima TL ultracentrifuge for 15 min at 25°C. Supernatant and pellet fractions were separated by SDS-PAGE and visualized by staining with Coomassie blue.
Giant Unilamellar Vesicles (GUVs) were formed as previously described (McDonald and Gould, 2016a) by drying 10 µL of 10 mg/mL CHCl3 lipids (69% DOPC, 15% DOPE, 10% DOPS, 5% PI(4)P) on Indium-Tin-Oxide-coated (ITO) glass coverslips (Sigma-Aldrich) under N2 stream followed by vacuum for 1 h. A 2 mm chamber was assembled between the coverslips and filled with a 20 mM HEPES pH 7.4 and 500 mM sucrose buffer through which a 10 Hz, 2.5 V sinusoidal current was passed for 2 ours. After formation, glucose was added to the GUV solution to 500 mM, forming an isomolar solution. Recombinant GFP-F-BAR domain and Rhodamine-Cdc12(residues 20–40) solutions were mixed with GUVs at a final concentration of 10 µM before imaging in a 0.5 mm chamber on the DeltaVision system described below.
Microscopy methods
Yeast for live-cell imaging were grown at 25°C. Live-cell images of S. pombe cells were acquired using a Personal DeltaVision (Cytiva Life Sciences) that includes a microscope (IX71; Olympus), 60 × NA 1.42 Plan Apochromat and 100 3 NA 1.40 U Plan S Apochromat objectives, fixed and live-cell filter wheels, a camera (CoolSNAP HQ2; Photometrics), and softWoRx imaging software (Cytiva Life Sciences). Z sections were spaced at 0.5 mm. Images for quantification of S. pombe CR intensity were not deconvolved and were sum projected. Intensity measurements were made with FIJI (Schindelin et al., 2012). For CR measurements, a region of interest (ROI) was created to measure the intensity around the CR and background was subtracted by creating a ROI in the same image in an area containing no cells (Waters, 2009). For measurement of Rh-Cdc12 intensity on GUVs, a single Z-slice through the middle of the GUV was analyzed, determined as the slice where the diameter of the GUV was the greatest. The intensity was then measured for two concentric circles per GUV, one encompassing the entire GUV and one just below the surface of the membrane. To calculate the intensity of Rh-Cdc12 on the membrane only, the intensity of the inner circle was subtracted from the intensity of the outer circle, a background subtraction was performed, and the subtracted intensity was divided by the number of pixels (outer circle area subtracted by the inner circle area) to normalize for GUV size.
Time-lapse imaging was performed on log-phase cells at 29°C using an ONIX microfluidics perfusion system that allows for flow of YE media through the imaging chamber at 5 psi throughout the time-lapse. Time-lapse images were deconvolved with 10 iterations and visualized as maximum projections.
S. pombe samples for fPALM imaging were prepared as previously described (McDonald et al., 2017). Briefly, samples were prepared by enriching for cells with CRs by isolating newly separated short cells from a 7%–30% lactose gradient, grown for 80 min in YE, labeled with 0.5 mM mCLING-ATTO647N (Revelo et al., 2014) (Synaptic Systems, Goettingen, Germany) for 10 min, and fixed in 3.7% formaldehyde for 15 min. Cells were washed 6x with PBS. For actin staining, fixed cells were incubated with 3.3 mM Phalloidin-Atto488, 0.01% NP-40 for 30 min and washed 3 times with PBS. Cells were resuspended with a small volume of PBS containing 1:100 diluted 80 nm gold particles (Microspheres-Nanospheres, Cold Spring, NY) and mounted on a 3% agar pad to prevent drift. For samples treated with inhibitor, DMSO or 20 µg/mL FK506 (LC Laboratories) was added to media immediately after extraction from the lactose gradient, resulting in a treatment time of 90 min prior to fixation.
As described previously (McDonald et al., 2017), fPALM imaging was performed on a Nikon dSTORM 4.0 system including a Nikon Eclipse Ti microscope, 405, 488, 561, and 647 nm solid state lasers, a Hamamatsu ORCA-Flash4.0 camera, and a 100x CFI HP Apochromat TIRF 1.49NA objective with a 1.5x tubelens (110 nm pixel size). A 0°C laser angle focused at the center of cells in Z was used. The Nikon Perfect Focus system was engaged to minimize drift. The mMaple3 channel was imaged with simultaneous 0.1%–0.5% 405 nm activation and 7.5% 561 nm excitation lasers, filtered through a polychroic mirror (ZT405/488/561/647rpc, Chroma, Bellows Falls, VT) and emission filter (ET585/65 m, Chroma), and captured with 30 ms exposures of 10–20k frames. The ATTO647N channel was imaged with simultaneous 0.2% 405 nm activation and 2% 647 nm excitation lasers, filtered through a polychroic mirror (ZT405/488/561/647rpc, Chroma) and emission filter (ET705/75 m, Chroma), and captured with 10 ms exposures over 15k frames. For Phalloidin-Atto488 imaging, simultaneous 0.2% 405nm activation and 2% 488 nm excitation lasers were used, filtered through a polychroic mirror (ZT405/488/561/647rpc, Chroma) and emission filter (ET525/50 m, Chroma), and captured with 10 ms exposures over 15k frames. Laser powers and exposure times were optimized for single photoactivated localizations per ring per frame.
fPALM images were analyzed as previously described (McDonald et al., 2017) using the ImageJ ThunderSTORM plugin (Ovesnýet al., 2014). Images were pre-filtered with a wavelet B-spline filter and molecules were approximately localized with the 8-neighborhood local maximum method. Molecules were localized using the elliptical Gaussian weighted least-squares method to identify precise lateral and Z positions. 0.1 mm Tetraspek beads were used for a Z-calibration (ThermoFisher). Fully formed pre-constriction CRs were identified in processed and aligned images as previously described (McDonald et al., 2017). The PM edge was identified using an automated threshold method to determine where mCLING-ATTO647N signal drops to 5% of its maximum PM intensity (McDonald et al., 2017). Distances of each individual mMaple3 or Phalloidin-Atto488 localization to this edge were determined and these data were fit with a Gaussian curve with the MASS package in R (Kanchanawong et al., 2010; McDonald et al., 2017; Venables and Ripley, 2002). Distances from the PM (dcenter), vertical width parameter (svert or FWHM), and horizontal width parameter (swidth or FWHM) were determined. dcenter from multiple CRs analyzed were plotted using ggplot2 in R (McDonald et al., 2017).
Cdc15 F-BAR domain oligomerization was observed by negative stain electron microscopy as described previously (McDonald et al., 2015), 5 uL of 1 ug/mL Cdc15 F-BAR solution diluted into 50 mM NaCl, 20 mM Tris-HCl pH 7.4 was adsorbed to a glow discharged 200-mesh copper grid covered with carbon-coated collodion film (Electron Microscopy Services, Hatfield, PA). Grids were washed in two drops of water to remove unbound sample and stained with two drops of 0.75% uranyl formate (Ohi et al., 2004). Samples were imaged on a FEI Morgagni electron microscope operated at 100 kV and images were captured at 18,000–36,000X on a 1K x 1K CCD camera (AMT, Woburn, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to pre-determine sample size. Details of statistical tests performed are described in the relevant figures, legends, and tables.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Cdc15 | (Roberts-Galbraith et al., 2009) | RRID: AB_2877106 |
| Mouse monoclonal PSTAIRE (anti-Cdc2) | Sigma-Aldrich | Cat# P7962; RRID: AB_261183 |
| IRDye 680RD Goat anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–68071; RRID: AB_10956166 |
| IRDye 800CW Goat anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32211; RRID: AB_621843 |
| IRDye 680RD Goat anti-Mouse IgG antibody | LI-COR Biosciences | Cat# 926–68070; RRID: AB_10956588 |
| IRDye 800CW Goat anti-Mouse IgG antibody | LI-COR Biosciences | Cat# 926–32210; RRID: AB_621842 |
| Bacterial and Virus Strains | ||
| E. coli Rosetta2(DE3)pLysS competent cells | Novagen | Cat#70954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DOPC | Avanti Polar Lipids | Cat#850375C |
| DOPE | Avanti Polar Lipids | Cat#850725C |
| DOPS | Avanti Polar Lipids | Cat#840035C |
| PI4P | Avanti Polar Lipids | Cat#840045X |
| Rhodamine-Cdc12(aa20–40) | Genscript | N/A |
| Biotin-Cdc12(aa20–40) | Genscript | N/A |
| Biotin-Cdc12(aa20–40) P31A | Genscript | N/A |
| Cdc12(aa20–40) | Genscript | N/A |
| mCLING-ATTO 647N-labeled | Synaptic Systems GmbH | Cat#710006AT1(SY) |
| Amylose Resin | New England Biolabs | Cat#E8021 |
| cOmplete His-Tag Purification Resin | Roche | Cat#5893682001 |
| Brain Extract from bovine brain, Type I, Folch Fraction I | Sigma-Aldrich | Cat#B1502 |
| Lambda Protein Phosphatase | New England BioLabs | Cat#P0753 |
| Isopropyl-β-D-thiogalactopyranoside (IPTG) | Fisher Scientific | Cat#BP1755–1, CAS367–93-1 |
| Anti-FLAG M2 Magnetic Beads | Millipore Sigma | Cat#M8823 |
| 3x FLAG peptide | Millipore Sigma | Cat#F4799 |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Roche | Cat#05056489001 |
| 1.0 M HEPES pH 7.0 | Hampton Research | Cat#HR2–931-03 |
| Sodium malonate pH 6.0 3.4 M solution | Hampton Research | Cat#HR2–751 |
| Jeffamine ED 2003 pH 7.0 titrated with conc. HCl solution | Molecular Dimensions | Cat#MD2–100-134 |
| Pierce Streptavidin UltraLink Resin | Thermo Scientific | Cat#53114 |
| Protein A Sepharose | GE Healthcare | Cat#17–5280–04 |
| FK506 | LC Laboratories | Cat#F4900 |
| Phalloidin-Atto488 | Sigma-Aldrich | Cat#49409 |
| Deposited Data | ||
| Crystal structure of the Cdc15 F-BAR domain | This study | PDB: 6XJ1 |
| Structure of the Imp2 F-BAR domain | (McDonald et al., 2016) | PDB: 5C1F |
| Crystal structure of the GAS7 F-BAR domain | (Hanawa-Suetsugu et al., 2019) | PDB: 6IKN |
| Crystal structure of the Hof1p F-BAR domain | (Moravcevic et al., 2015) | PDB: 4WPE |
| Experimental Models: Organisms/Strains | ||
| Fission yeast strains used in this study are described in Table S5 | N/A | N/A |
| Oligonucleotides | ||
| Cdc15 D255A E256A E259A forward (for site-directed mutagenesis: CCTATGCAAATATAATATCTACTGCTTGTGTAAAAGATGCTGCATCTTGTGCAAAGATTCGACTCACACTCGAAAACAC) | This study | N/A |
| Cdc15 D255A E256A E259A reverse (for site-directed mutagenesis: GTGTTTTCGAGTGTGAGTCGAATCTTTGCACAAGATGCAGCATCTTTTACACAAGCAGTAGATATTATATTTGCATAGG) | This study | N/A |
| pET-Duet-1 Ppb1 For (for Gibson assembly into BamHI/NotI sites: catcaccatcatcaccacagccaggatccgATGACTTCGGGTCCTCATAATTTAGAA) | This study | N/A |
| pET-Duet-1 Ppb1 Rev (for Gibson assembly into BamHI/NotI sites: ttctgttcgacttaagcattatgcggccgcCTACAAAGAGCTTTTCTTATCT GCAAAGC) | This study | N/A |
| pET-Duet-1 Cnb1 For (for Gibson assembly into NdeI/KpnI sites: agttaagtataagaaggagatatacatatgATGGGTCAGTCGCAGTCGC) | This study | N/A |
| pET-Duet-1 Cnb1 Rev (for Gibson assembly into NdeI/KpnI sites: agcggtttctttaccagactcgagggtaccTCAAAAAGAATCGAGTGTCATGCTACT) | This study | N/A |
| pRSF-Duet-1 Cam1 For (for Gibson assembly into NcoI/NotI sites: tgtttaactttaataaggagatataccatgACTACCCGTAACCTTACAGATGAGC) | This study | N/A |
| pRSF-Duet-1 Cam1 Rev (for Gibson assembly into NcoI/NotI sites: ttctgttcgacttaagcattatgcggccgcCTACTTGGAAGAAATGACACGAGAGA) | This study | N/A |
| Recombinant DNA | ||
| pET15b GFP-Cdc15(aa19–312) | (McDonald et al., 2015) | N/A |
| pET15b Cdc15(aa19–312, E30K E152K) | (McDonald et al., 2015) | N/A |
| pET15b Cdc15(aa19–312) | (McDonald et al., 2015) | N/A |
| pET15b Cdc15(aa19–312, D255A E256A E259A) | This study | N/A |
| pET15b Cdc15 (aa19–312, D255A E256A E259A E30K E152K) | This study | N/A |
| pMAL-c2-Pxl1 | (Bhattacharjee et al., 2020) | N/A |
| pET-Duet-1 Ppb1, Cnb1 | This study | N/A |
| pRSF-Duet-1 Cam1 | This study | N/A |
| pET-Duet-1 Flag-Cdc15(E30K, E152K), Pom1 | (Bhattacharjee et al., 2020) | N/A |
| pET-Duet-1 Flag-Cdc15(E30K, E152K, D255A, E256A, E259A), Pom1 | This study | N/A |
| pIRT2 Cdc15(D255A E256A E259A) + 500 bp 5ʹ and 3ʹ flanks | This study | N/A |
| Software and Algorithms | ||
| FIJI | (Schindelin et al., 2012) | https://imagej.net/Fiji |
| Prism 8 | GraphPad Software | https://www.graphpad.com |
| SoftWoRx | Cytiva Life Sciences | https://cytivalifesciences.com |
| NIS-Elements | Nikon Instruments | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| HKL2000 | (Otwinowski and Minor, 1997) | https://www.hkl-xray.com/ |
| Phaser | (McCoy et al., 2007) | https://www.phenix-online.org/documentation/reference/phaser_mr.html |
| PHENIX | (Afonine et al., 2012) | https://www.phenix-online.org/ |
| COOT | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PYMOL | Schrodinger LLC | https://pymol.org/2/ |
| PROCHECK | (Laskowski et al., 1993) | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| Molprobity | (Chen et al., 2010) | http://molprobity.biochem.duke.edu/ |
| CCPMG | (McNicholas et al., 2011) | https://www.ccp4.ac.uk/ |
| CCP4 | (Winn et al., 2011) | https://www.ccp4.ac.uk/ |
| AREAIMOL | (Lee and Richards, 1971) | https://www.ccp4.ac.uk/ |
| PISA | (Krissinel and Henrick, 2007) | https://www.ebi.ac.uk/pdbe/pisa/ |
| RStudio | RStudio, PBC | https://rstudio.com/ |
| Other | ||
| CellASIC ONIX plate for haploid yeast cells (4 chamber, 3.5–5 micron) | Millipore | Cat#Y04C-02–5PK |
| Immobilon FL PVDF | Fisher Scientific | Cat#IPFL00010 |
| Indium tin oxide coated glass slide, square | Sigma-Aldrich | Cat#703184 |
Highlights.
Cdc15 F-BAR domain uses opposite faces to bind membrane and proteins simultaneously
F-BAR non-membrane-binding faces create extensive surfaces for binding partners
Cdc15 F-BAR organizes both structural and signaling components for cytokinesis
F-BAR domains can dictate nanoscale spacing and function of actin-based structures
ACKNOWLEDGMENTS
We thank Gould lab members for critical reading of this manuscript and Dr. Michael Tackenberg for assistance with R. fPALM was performed via the Vanderbilt Cell Imaging Shared Resource (NIH grants CA68485, DK20593, DK58404, DK59637, EY08126, and S10 OD012324). M.C. and L.P.J. are supported by NIH R35GM119525 to L.P.J. We acknowledge support from T32GM008554-21 (C.E.S.); American Heart Association grants 14PRE19740000 (A.H.W.), 15PRE21780003 (N.A.M.), and 17PRE33410245 (C.E.S.); and NIH grants R01GM101035 and R35GM131799 (to K.L.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108526.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Data and code availability
The structural coordinates and structure factors of the Cdc15 F-BAR domain have been deposited in the RCSB Protein Data Bank with accession code PDB: 6XJ1. All data and code are available from the Lead Contact by request.
REFERENCES
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arasada R, and Pollard TD (2014). Contractile ring stability in S. pombe depends on F-BAR protein Cdc15p and Bgs1p transport from the Golgi complex. Cell Rep 8, 1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begonja AJ, Pluthero FG, Suphamungmee W, Giannini S, Christensen H, Leung R, Lo RW, Nakamura F, Lehman W, Plomann M, et al. (2015). FlnA binding to PACSIN2 F-BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood 126, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee R, Mangione MC, Wos M, Chen J-S, Snider CE, Roberts-Galbraith RH, McDonald NA, Presti LL, Martin SG, and Gould KL (2020). DYRK kinase Pom1 drives F-BAR protein Cdc15 from the membrane to promote medial division. Mol. Biol. Cell 31, 917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnahan RH, and Gould KL (2003). The PCH family protein, Cdc15p, recruits two F-actin nucleation pathways to coordinate cytokinetic actin ring formation in Schizosaccharomyces pombe. J. Cell Biol 162, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheffings TH, Burroughs NJ, and Balasubramanian MK (2016). Actomyosin Ring Formation and Tension Generation in Eukaryotic Cytokinesis. Curr. Biol 26, R719–R737. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés JCG, Pujol N, Sato M, Pinar M, Ramos M, Moreno B, Osumi M, Ribas JC, and Pérez P (2015). Cooperation between Paxillin-like Protein Pxl1 and Glucan Synthase Bgs1 Is Essential for Actomyosin Ring Stability and Septum Formation in Fission Yeast. PLoS Genet 11, e1005358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Signore SJ, Kelley CF, Messelaar EM, Lemos T, Marchan MF, Ermanoska B, Mund M, Kaksonen M, and Rodal AA (2020). Autoregulation clamps the synaptic membrane-remodeling machinery and promotes productive actin-dependent endocytosis. bioRxiv 10.1101/2020.03.06.981076. [DOI] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fankhauser C, Reymond A, Cerutti L, Utzig S, Hofmann K, and Simanis V (1995). The S. pombe cdc15 gene is a key element in the reorganization of F-actin at mitosis. Cell 82, 435–444. [DOI] [PubMed] [Google Scholar]
- Fujita M, Sugiura R, Lu Y, Xu L, Xia Y, Shuntoh H, and Kuno T (2002). Genetic interaction between calcineurin and type 2 myosin and their involvement in the regulation of cytokinesis and chloride ion homeostasis in fission yeast. Genetics 161, 971–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garabedian MV, Stanishneva-Konovalova T, Lou C, Rands TJ, Pollard LW, Sokolova OS, and Goode BL (2018). Integrated control of formin-mediated actin assembly by a stationary inhibitor and a mobile activator. J. Cell Biol 217, 3512–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W, and Balasubramanian MK (2008). Pxl1p, a paxillin-related protein, stabilizes the actomyosin ring during cytokinesis in fission yeast. Mol. Biol. Cell 19, 1680–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould GW (2016). Animal cell cytokinesis: the role of dynamic changes in the plasma membrane proteome and lipidome. In Seminars in Cell & Developmental Biology, Volume 53 (Elsevier; ), pp. 64–73. [DOI] [PubMed] [Google Scholar]
- Gould KL, Moreno S, Owen DJ, Sazer S, and Nurse P (1991). Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J 10, 3297–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A, Takaine M, Simanis V, and Nakano K (2011). Dividing the spoils of growth and the cell cycle: The fission yeast as a model for the study of cytokinesis. Cytoskeleton (Hoboken) 68, 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa-Suetsugu K, Itoh Y, Ab Fatah M, Nishimura T, Takemura K, Takeshita K, Kubota S, Miyazaki N, Wan Mohamad Noor WNI, Inaba T, et al. (2019). Phagocytosis is mediated by two-dimensional assemblies of the F-BAR protein GAS7. Nat. Commun 10, 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Kent HM, Ford MGJ, Hegde BG, Daumke O, Butler PJG, Mittal R, Langen R, Evans PR, and McMahon HT (2007). Structure and analysis of FCHo2 F-BAR domain: a dimerizing and membrane recruitment module that effects membrane curvature. Structure 15, 839–852. [DOI] [PubMed] [Google Scholar]
- Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, and De Camilli P (2005). Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev. Cell 9, 791–804. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, and Waterman CM (2010). Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney JB, and Boeke JD (1994). Efficient targeted integration at leu1–32 and ura4–294 in Schizosaccharomyces pombe. Genetics 136, 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Deng L, Wu Y, Baldissard S, Adamo ME, Gerber SA, and Moseley JB (2015). Quantitative phosphoproteomics reveals pathways for coordination of cell growth and division by the conserved fission yeast kinase pom1. Mol. Cell. Proteomics 14, 1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostan J, Salzer U, Orlova A, Törö I, Hodnik V, Senju Y, Zou J, Schreiner C, Steiner J, Meriläinen J, et al. (2014). Direct interaction of actin filaments with F-BAR protein pacsin2. EMBO Rep 15, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, and Thornton JM (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291. [Google Scholar]
- Lee B, and Richards FM (1971). The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Lee ME, Rusin SF, Jenkins N, Kettenbach AN, and Moseley JB (2018). Mechanisms Connecting the Conserved Protein Kinases Ssp1, Kin1, and Pom1 in Fission Yeast Cell Polarity and Division. Curr. Biol 28, 84–92.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McDonald NA, Naegele SM, Gould KL, and Wu J-Q (2019). The F-BAR Domain of Rga7 Relies on a Cooperative Mechanism of Membrane Binding with a Partner Protein during Fission Yeast Cytokinesis. Cell Rep 26, 2540–2548.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Sugiura R, Yada T, Cheng H, Sio SO, Shuntoh H, and Kuno T (2002). Calcineurin is implicated in the regulation of the septation initiation network in fission yeast. Genes Cells 7, 1009–1019. [DOI] [PubMed] [Google Scholar]
- Magliozzi JO, Sears J, Cressey L, Brady M, Opalko HE, Kettenbach AN, and Moseley JB (2020). Fission yeast Pak1 phosphorylates anillin-like Mid1 for spatial control of cytokinesis. J. Cell Biol 219, e201908017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangione MC, and Gould KL (2019). Molecular form and function of the cytokinetic ring. J. Cell Sci 132, jcs226928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangione MC, Snider CE, and Gould KL (2019). The intrinsically disordered region of the cytokinetic F-BAR protein Cdc15 performs a unique essential function in maintenance of cytokinetic ring integrity. Mol. Biol. Cell 30, 2790–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-García R, Arribas V, Coll PM, Pinar M, Viana RA, Rincón SA, Correa-Bordes J, Ribas JC, and Pérez P (2018). Paxillin-Mediated Recruitment of Calcineurin to the Contractile Ring Is Required for the Correct Progression of Cytokinesis in Fission Yeast. Cell Rep 25, 772–783.e4. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NA, and Gould KL (2016a). Characterization of Cytokinetic F-BARs and Other Membrane-Binding Proteins. In Yeast Cytokinesis: Methods and Protocols, Sanchez-Diaz A and Perez P, eds. (Springer; ), pp. 181–189. [DOI] [PubMed] [Google Scholar]
- McDonald NA, and Gould KL (2016b). Linking up at the BAR: Oligomerization and F-BAR protein function. Cell Cycle 15, 1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NA, Vander Kooi CW, Ohi MD, and Gould KL (2015). Oligomerization but Not Membrane Bending Underlies the Function of Certain F-BAR Proteins in Cell Motility and Cytokinesis. Dev. Cell 35, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NA, Takizawa Y, Feoktistova A, Xu P, Ohi MD, Vander Kooi CW, and Gould KL (2016). The Tubulation Activity of a Fission Yeast F-BAR Protein Is Dispensable for Its Function in Cytokinesis. Cell Rep 14, 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NA, Lind AL, Smith SE, Li R, and Gould KL (2017). Nanoscale architecture of the Schizosaccharomyces pombe contractile ring. eLife 6, e28865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS, and Noble MEM (2011). Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr 67, 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moravcevic K, Alvarado D, Schmitz KR, Kenniston JA, Mendrola JM, Ferguson KM, and Lemmon MA (2015). Comparison of Saccharomyces cerevisiae F-BAR domain structures reveals a conserved inositol phosphate binding site. Structure 23, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Schreiter JH, Okada H, Wloka C, Okada S, Yan D, Duan X, and Bi E (2017). Hof1 and Chs4 Interact via F-BAR Domain and Sel1-like Repeats to Control Extracellular Matrix Deposition during Cytokinesis. Curr. Biol 27, 2878–2886.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, and Walz T (2004). Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol. Proced Online 6, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology, Volume 276 (Academic Press; ), pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Ovesný M, Kŕíźek P, Borkovec J, Švindrych Z, and Hagen GM (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30, 2389–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, and McMahon HT (2004). BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303, 495–499. [DOI] [PubMed] [Google Scholar]
- Pinar M, Coll PM, Rincón SA, and Pérez P (2008). Schizosaccharomyces pombe Pxl1 is a paxillin homologue that modulates Rho1 activity and participates in cytokinesis. Mol. Biol. Cell 19, 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, and Wu J-Q (2010). Understanding cytokinesis: lessons from fission yeast. Nat. Rev. Mol. Cell Biol 11, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L, Willet AH, Roberts-Galbraith RH, McDonald NA, Feoktistova A, Chen JS, Huang H, Guillen R, Boone C, Sidhu SS, et al. (2015). The Cdc15 and Imp2 SH3 domains cooperatively scaffold a network of proteins that redundantly ensure efficient cell division in fission yeast. Mol. Biol. Cell 26, 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelo NH, Kamin D, Truckenbrodt S, Wong AB, Reuter-Jessen K, Reisinger E, Moser T, and Rizzoli SO (2014). A new probe for super-resolution imaging of membranes elucidates trafficking pathways. J. Cell Biol 205, 591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon SA, and Paoletti A (2016). Molecular control of fission yeast cytokinesis. Semin. Cell Dev. Biol 53, 28–38. [DOI] [PubMed] [Google Scholar]
- Roberts-Galbraith RH, Chen J-S, Wang J, and Gould KL (2009). The SH3 domains of two PCH family members cooperate in assembly of the Schizosaccharomyces pombe contractile ring. J. Cell Biol 184, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Galbraith RH, Ohi MD, Ballif BA, Chen J-S, McLeod I, McDonald WH, Gygi SP, Yates JR 3rd, and Gould KL (2010). Dephosphorylation of F-BAR protein Cdc15 modulates its conformation and stimulates its scaffolding activity at the cell division site. Mol. Cell 39, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju Y, Itoh Y, Takano K, Hamada S, and Suetsugu S (2011). Essential role of PACSIN2/syndapin-II in caveolae membrane sculpting. J. Cell Sci 124, 2032–2040. [DOI] [PubMed] [Google Scholar]
- Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA, and Kastner DL (2003). Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA 100, 13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swulius MT, Nguyen LT, Ladinsky MS, Ortega DR, Aich S, Mishra M, and Jensen GJ (2018). Structure of the fission yeast actomyosin ring during constriction. Proc. Natl. Acad. Sci. USA 115, E1455–E1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM (2015). F-BAR/EFC Domain Proteins: Some Assembly Required. Dev. Cell 35, 664–666. [DOI] [PubMed] [Google Scholar]
- Venables WN, and Ripley BD (2002). Modern Applied Statistics with S (Springer-Verlag; ), p. 498. [Google Scholar]
- Wachtler V, Huang Y, Karagiannis J, and Balasubramanian MK (2006). Cell cycle-dependent roles for the FCH-domain protein Cdc15p in formation of the actomyosin ring in Schizosaccharomyces pombe. Mol. Biol. Cell 17, 3254–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JC (2009). Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol 185, 1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willet AH, McDonald NA, Bohnert KA, Baird MA, Allen JR, Davidson MW, and Gould KL (2015). The F-BAR Cdc15 promotes contractile ring formation through the direct recruitment of the formin Cdc12. J. Cell Biol 208, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, Harris MA, McDowall MD, Rutherford K, Vaughan BW, Staines DM, Aslett M, Lock A, Bähler J, Kersey PJ, and Oliver SG (2012). PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res 40, D695–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J-Q, and Pollard TD (2005). Counting cytokinesis proteins globally and locally in fission yeast. Science 310, 310–314. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Kuhn JR, Kovar DR, and Pollard TD (2003). Spatial and temporal pathway for assembly and constriction of the contractile ring in fission yeast cytokinesis. Dev. Cell 5, 723–734. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Toda T, and Yanagida M (1994). A calcineurin-like gene ppb1+ in fission yeast: mutant defects in cytokinesis, cell polarity, mating and spindle pole body positioning. J. Cell Sci 107, 1725–1735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.