

Abstract
Light-harvesting in photosynthesis is accompanied by photoprotective processes. In cyanobacteria, the photoprotective role is played by a specialized complex, the orange carotenoid protein, which is activated by strong blue-green light. This photoactivation involves a unique series of structural changes which terminate with an opening of the complex into two separate domains, one of which acts as a quencher for the light-harvesting complexes. Many experimental studies have tried to reveal the molecular mechanisms through which the energy absorbed by the carotenoid finally leads to the large conformational change of the complex. Here, for the first time, these mechanisms are revealed by simulating at the atomistic level the whole dynamics of the complex through an effective combination of enhanced sampling techniques. On the basis of our findings, we can conclude that the carotenoid does not act as a spring that, releasing its internal strain, induces the dissociation, as was previously proposed, but as a “latch” locking together the two domains. The photochemically triggered displacement of the carotenoid breaks this balance, allowing the complex to dissociate.
1. Introduction
Bacteria are the oldest type of photosynthetic organisms on earth. This makes their investigation even more important as they can be seen as ancestors of modern organisms and they could help us to understand how the photosynthetic process has appeared and evolved over time. Most photosynthetic bacteria have developed anoxygenic forms of photosynthesis, but there is one type, the cyanobacteria, which performs photosynthesis in a manner similar to that of plants using water as an electron donor and releases oxygen. Cyanobacteria have developed a photoprotection system that prevents photodamage when they are illuminated with more light than they can use for photosynthesis, exactly as in plants and algae. Photoprotection necessarily competes with the efficient collection of light energy which is the basis of photosynthesis, and as such, the system has to be switchable and there must be a reversibility between the so-called light-harvesting and photoprotective states. In the latter, the excess energy has to be rapidly dissipated into heat through what is generally known as nonphotochemical quenching (NPQ).1−4 In cyanobacteria, NPQ is rather different with respect to plants and algae, where the same antenna complexes which perform light harvesting can switch to the quenching function.5,6 The antenna of cyanobacteria are in fact phycobilisomes (PBSs), peripheral membrane complexes that incorporate several types of bilins, which are open-chain tetrapyrrole pigments. They are organized into disks which create large rod-like architectures attached radially to a core protein.7 Excitation energy is funneled along the rods to the core which finally can attach to either photosystem I or II. NPQ occurs at the core level and involves a water-soluble protein called orange carotenoid protein (OCP).8−10
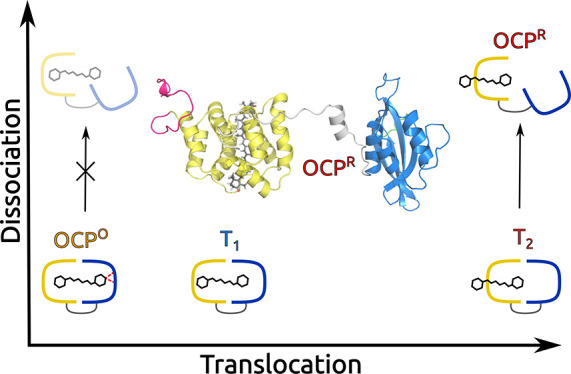
OCP can be found in two interchangeable conformational forms: orange (OCPO) and red (OCPR).11,12 In the former, the embedded keto-carotenoid cofactor is placed in a pocket crossing the two domains (N- and C-terminal domains, or NTD and CTD) which constitute the protein, and it is firmly connected to the CTD domain through two hydrogen bonds with two protein residues (Figure 1). This closed orange form has to switch to the red one to become active in NPQ: this activation requires the absorption of strong blue-green light. The final product (OCPR) is obtained through a translocation of the carotenoid within the NTD domain and a dissociation of the two domains which remain connected by a long, flexible (largely unstructured) loop linker.12−19 The NTD (with its embedded carotenoid) can then interact with the PBS core, allowing the quenching of the bilins.20 The red form slowly back-converts to the orange one in the dark, but the recovery can be accelerated through the interaction with another protein, the fluorescence recovery protein (FRP).21,22
Figure 1.
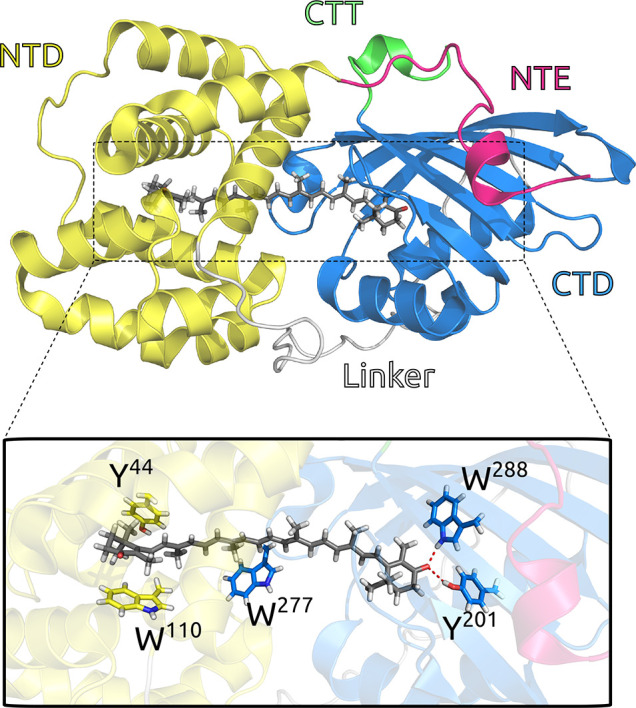
Graphical representation of the OCP:CAN complex from its crystal structure (4XB5).12 Colors are used to show different domains (magenta for NTE, yellow for NTD, gray for the linker, blue for CTD, and green for CTT). CAN and some relevant residues (Y44, W110, Y201, W277, and W288) are shown as licorice.
Experimental techniques have probed the evolution of OCP after the photoactivation with high resolution in either time18,19,23,24 or space.25 Nonetheless, a clear and complete characterization of the molecular mechanisms through which the carotenoid translocates between the two domains and the protein dissociates is not available.
The goal of this article is to reveal such mechanisms through atomistic simulations. This goal poses a major challenge as very different time scales are involved, from the nanosecond to microsecond for the carotenoid to translocate within the protein, to the millisecond for the domain dissociation.18 Moreover, the light-driven nature of the process should require the use of a quantum-mechanical dynamical description that at the present time can deal only with very short time scales.
To achieve such a challenging goal, we exploit a combination of different enhanced sampling techniques26−29 to overcome the initial energy barrier and to efficiently sample the equilibrium configurations visited by the CAN during the translocation, after the initial photochemical event is concluded.
Using this paradigm, we could reproduce the whole evolution of the complex, from its resting form (OCPO) to its final open form. The simulations demonstrated that the carotenoid acts as a “latch” locking together the two domains in OCPO, by establishing interactions of comparable strength with each of them. This latch, however, is lifted as soon as the carotenoid is allowed to move toward the NTD thanks to the electronic transition, and the dissociation can proceed. Indeed, with our simulations, we showed that only after carotenoid translocation can the dissociation of the two domains take place. Moreover, we could identify two relative free-energy minima in the translocation process that are related to the intermediates revealed by recent spectroscopic studies.18 Finally, we revealed the fundamental role played by water in both the translocation (by stabilizing the intermediates) and the dissociation (through solvation effects), which we showed to occur, leaving the secondary and tertiary structures of the two domains almost unchanged.
2. Results
To achieve a clearer description of the results of the simulations, we divided their presentation into two parts, translocation and dissociation. Finally, we recollected all of the main findings of both parts in a general discussion. In the latter, a direct comparison with available experimental evidence is also reported.
2.1. Translocation
In our simulation of the translocation process, to enhance the sampling of the reaction path we performed an umbrella sampling (US)30 simulation. At each point of the collective variable, to sample perpendicular degrees of freedom, instead of a plain MD, we performed a replica exchange molecular dynamics (REMD) simulation.31 The combination of umbrella sampling and REMD (US-REMD) should allow for an efficient exploration of the transition space along both the main direction and the slow degrees of freedom orthogonal to the collective variable, such as structural rearrangement of the protein or conformational changes of the carotenoid. The collective variable (CV) chosen to represent the translocation process is the distance between the center of mass of the carotenoid and that of the NTD. A detailed discussion of the methods used for the simulation and their analysis is provided in the SI.
To asses the convergence of the simulation, we performed blocking of the data set, and for each block, we computed the free-energy surface (FES) along the CV (details in the Supporting Information). The analysis shows that the second half of the simulation has completely converged; therefore, we analyzed the structures (about 0.8 million) extracted from the second half of the 10 lowest-temperature replicas of our simulations and calculated the FES of the system along the CV. The resulting profile is reported in Figure 2C. To provide a first interpretation of the predicted free-energy minima, for each of them we calculated the average structure and the average RMSD distance of the NTD and CTD using OCP and RCP crystal structures12 as references (Figure 2A,B,D).
Figure 2.

(A) Average structure at CV ≈ 11.5 Å (in blue) superimposed on the crystal structure of OCPO (in orange) and (B) at CV ≈ 2.5 Å superimposed on the RCP crystal structure (in red). (C) Free-energy profile along the CV (in kcal/mol). (D) Average RMSD values (in Å) for the NTD computed using as reference the NTD of the crystal structures of OCPO (orange line) and RCP (red line) and for the CTD computed using as reference the CTD of the crystal structure of OCPO (blue line). Comparison of representative structures of OCPO and (E) T1 and (F) T2 states. A licorice representation is used for CAN and for the side chains of some relevant residues spotted either in the literature (particularly the two H-bonding Y201 and W288 and the π-stacking Y44, W110, and Y111)32 or in this present work (the interface H-bond network E244, R155, N104, and W277). In order to give a better view of the complexes, we used a different orientation in panels A and B (roughly rotated by 180° around the horizontal axis).
In a previous work by our group, 1-μs-long unbiased MD simulations of OCPO and RCP were performed.33 Considering that the selected CV, on this unbiased OCPO trajectory, has an average value of 11.4 Å, we can safely state that the deepest minimum (highlighted in orange in Figure 2C) corresponds to the OCPO state. Indeed, by comparing the RMSD values of the two main domains and the three-dimensional structure of the absolute minimum overlapped with the reference OCPO structure, it is clear that this first state is almost identical to the one known from OCPO crystals (Figure 2A).
Moving to the left of the FES, following the translocation of the carotenoid into the NTD, two other relative minima appear at CV ≈ 7.5 Å (highlighted in blue, T1) and at CV ≈ 2.5 Å (highlighted in red, T2).
According to our simulations, T1 represents an intermediate in the reaction path with the carotenoid only slightly displaced from its original position but still enough to lose its H-bonds with the β1 ionone ring and form other stabilizing interactions with other residues of the pocket, such as W41 and P124–P126 (Figure 2E).
On the contrary, in T2, the position of the carotenoid is strikingly similar to the one found for CAN in RCP (Figure 2B). We stress that the CV here used does not contain any information on the exact binding site in which the carotenoid should reach into the NTD. This finding confirms that the “tunnel” already spotted from the X-ray diffraction structure12 acts as a guide to drive the carotenoid to a new position, which is very similar to the binding mode observed in RCP.
During the process, the NTD undergoes small but significant modifications. These can be quantified by looking at the RMSD of the NTD along the evolution of the CV when calculated with respect to OCPO and RCP (Figure 2D): while the former increases during the process, the latter decreases. It is important to stress here that these results are completely independent of any a priori knowledge of the structure of RCP and that they rely only on the crystal structure of OCPO and on the carotenoid–NTD distance (i.e., very general and unspecific information).
Let us now consider the RMSD of the CTD along the CV from the reference CTD of OCPO. This domain is in general more stable than the NTD during translocation, as it roughly shows a single jump from 1.25 to 1.75 Å at CV ≈ 6.5 Å. Visual inspection reveals that this change in the RMSD can be connected to a small conformational change localized in the highly conserved34 T275–N281 loop, which will be discussed in more detail later. On the basis of these observations, we suggest that T2 is a reasonable representation of the final product of the translocation.
So far, we have explored the evolution of the structure of the system during the translocation process. Now we analyze the same evolution for the interactions among the various components (carotenoid, protein, and solvent).
As a first analysis, we dissected the average energy of the carotenoid in three parts: the interaction with the protein, the interaction with the solvent (i.e., all water molecules), and its internal energy. The values of each part along the CV are reported in Figure 3. We note that the energies reported there do not include the internal energy change of the protein and any entropic contribution to the free energy. As can be seen from Figure 3A, the solvent plays a central role: immediately after the first displacement of the carotenoid from its original binding pocket, strong favorable interactions appear that in T2 largely compensate for the observed decrease in the stabilizing effects of the protein (Figure 3A, green line). The latter behavior can be explained in terms of the loss of the two hydrogen bonds with Y201 and W288 and the appearance of interactions between the water molecules and the polar groups on the β-ionone rings of CAN.
Figure 3.

(A) Interaction energies between CAN and OCP (green line) and CAN and water (blue line), internal energy of CAN (orange line), and total interaction energy (black line): all values are calculated with respect to OCPO. (B) Interaction energies between CTD and CAN (blue lines) and NTD and CAN (yellow lines); interactions among the CTT, linker, and NTE are negligible; dashed lines are the electrostatic interactions, dotted lines the Lennard-Jones interactions, and solid lines are the sum of the two contributions. (C) Electrostatic (ES) and Lennard-Jones (LJ) interaction energies among individual residues of NTD, CTD, and CAN. Values are reported as the difference from the OCPO state. Numerical values for ES and LJ interactions of CAN with the most relevant residues of the protein are reported in Tables S2 and S3, respectively.
If we analyze more in detail the profile of the interactions of CAN with the protein, then we see complex behavior. After the initial destabilizing effect due to the loss of electrostatic effects from the broken H-bonds, we see a first minimum roughly corresponding to T1 and a second shallow one corresponding to T2. To understand more clearly, we can further dissect the interaction energy into nonelectrostatic, namely, repulsion and dispersion as described by the Lennard-Jones (LJ) potential, and electrostatic (ES) contributions from the two main domains (Figure 3B). Contrary to the chemical intuition, in OCPO the carotenoid is bound somewhat more tightly to the NTD than to the CTD, despite the two H-bonds. Therefore, it is not surprising that, when the carotenoid is placed outside the H-bonding range of W288 and Y201, it is much more strongly bound to the NTD than to the CTD (about 20 kcal mol–1). This large effect has a strong contribution from new LJ interactions with the NTD and partially also from breaking those with the CTD.
The evolution of the LJ and ES interaction energies of CAN with single residues along the CV is reported in Figure 3C. Many residues in the NTD enhance their LJ interactions with CAN, passing from OCPO to T1. The most relevant ones are W41 (−2.1 kcal mol–1), R155 (−2.0 kcal mol–1), I125 (−1.5 kcal mol–1), and P124 (−1.2 kcal mol–1) (Figure 2E). This seems to indicate that the translocation process is not driven by a few specific residues but instead the whole region of the protein around the carotenoid contributes. It is also interesting that after the loss of the two H-bonds, no other strong electrostatic interactions arise in state T1.
As the translocation proceeds further and the carotenoid has moved deeper into the NTD, its LJ interactions with this domain keep increasing. In particular, we observe that a group of residues has a large stabilization effect (E34, L37, W41, T80, N104, W110, Y111, I125, P126, and Y129). The set of these residues defines a mainly apolar/aromatic binding pocket for the carotenoid. As expected, two of the tightest interactions between CAN and NTD that were present in OCPO state (the ones with W110 and R155) are considerably looser in T2 because of the new geometry of the complex in which the carotenoid is rearranged in a new binding site (Figure 2F). Moreover, two noticeable electrostatic interactions appear in T2. They can be classified as (relatively loose) H-bonds with the highly conserved W277 and T275 residues belonging to the CTD. The formation of these two H-bonds is connected to the largest local conformational change observed in the protein, corresponding to a rearrangement of the T275–N281 loop (Figure S9), which is one of the constituents of the NTD/CTD interface. To understand the importance of this conformational change, we should note that in the RCP crystal structure, as in those obtained from previous MD simulations,33 a stable H-bond between R155 (involved in a salt bridge between NTD and CTD) and N104 is observed. In OCPO and in T1, N104 is stably bound to W277; after the rearrangement of loop T275–N281, this last H-bond breaks up and N104 can establish a new stabilizing long-range interaction for R155.
We finally note that the β1 ionone ring can form H-bond interactions with various residues belonging to both NTD (G57 and S60 (OH in the side chain)) and CTD domains (W277 (NH in the side chain and NH in the backbone) and T275 (OH in the side chain)). As T2 is expected to evolve into a dissociated state, it is reasonable to expect that the carotenoid could interact with different sites and obtain a global stabilization effect (clearly visible in Figure 3A), without forming very tight connections that could inhibit the further evolution of the system.
As already pointed out, water plays a central role in stabilizing intermediates T1 and T2. It is well known that the β1 carbonyl of CAN in OCPO is stably bound with Y201 and W288 in a solvent-excluded region of the protein, and it does not form any relevant interaction with water. However, as the system evolves toward T1, the number of waters that can form H-bonds with CAN starts increasing (Figure S4A). This is likely due to the combination of different effects: the rupture of the two stable H-bonds of CAN with the protein residues, the motion of CAN toward a more solvent-exposed region, and also the entrance of water in the pocket of the CTD that initially was occupied by the CAN. We investigated the latter process by calculating the change in the solvent-accessible surface (SAS) for each residue along the CV (Figure S4B in the SI).
Many CTD residues increase their solvent accessibility during the process. With few exceptions, this change takes place starting from T1. This is an indication that the first H-bonds between β1 carbonyl and solvent are formed with water molecules that are already present within the protein pocket. In the second part of the translocation process (from T1 to T2) instead, a great increase in the SAS of CTD residues is observed together with a significant global decrease in the SAS of NTD residues. This fact suggests that the motion of the carotenoid in the protein tunnel displaces the water molecules present in the NTD and simultaneously allows them to enter in the CTD cavity, probably through the NTD/CTD interface.
As a final important analysis of the translocation energetics, we note that the gain in the internal energy of the carotenoid from OCPO to T2 is small, and at T1, the CAN has a larger internal energy than in OCPO (Figure 3A). On the contrary, the interactions with solvent and protein provide a substantial stabilization of the carotenoid in T2 with respect to OCPO. All of these findings contradict a previously proposed hypothesis35 that the distortion of the carotenoid acts as a spring that pushes the system from OCPO to OCPR.
To reach a more complete analysis of the role of internal geometrical changes, we have investigated the conformational freedom of the terminal rings of the carotenoid during the translocation. To do so, we have plotted the FES on the CV vs β1 and β2 dihedral angles (Figure 4A,B) and extracted some relevant conformations of the carotenoid from each minimum. It should be noted that since the torsion constants of the force field have been fine-tuned against DFT data, the terminal rings are normally able to rotate on the nanosecond time scale (Figure S8 and SI of ref (33)). The conformational rigidity observed in some states of the system is therefore determined by intermolecular interactions between the carotenoid and protein/solvent and does not depend on the force field parametrization itself.
Figure 4.

Plot of the free-energy surface projected on the (A) CV-β1 and (B) CV-β2 subspaces obtained from reweighting of the US-REMD simulations. (C) Overlap of 10 configurations of CAN in T2, T1, and OCPO together with a single representation of the protein environment as a spatial reference. The upper panels show the β1 side of the carotenoid, and the lower ones, the β2 side.
Looking at β1, it is clear that this angle remains locked into an s-trans-like conformation until the system reaches T2. This conformation is the same as the one seen in the OCPO crystal structure (Figure 4C). No other relevant conformation of this dihedral angle is seen until the completion of the translocation, consistent with the picture of Konold et al.18 where the photoproducts before translocation have the β1 ring in an s-trans conformation. β2 behaves similarly, but since in T1 this ring of the carotenoid is already outside the pocket defined by Y44 and W110, it has a larger conformational freedom that allows it to switch between the two s-cis conformations. Once the system is in T2, both terminal rings are able to rotate with remarkable freedom, as the disorder found in the configurations extracted from the MD clearly demonstrates. Nonetheless, we can notice a slight preference of β1 for s-cis conformations and of β2 for s-trans conformations. This fact is coherent with the picture of a very different conformational population of the carotenoid in RCP and OCPO, as already shown by our previous simulations and confirmed by subsequent experimental findings.33,36
2.2. Dissociation
There is general agreement on the fact that the dissociation is by far the slowest process in the photoactivation of OCP. This step is expected to have large entropic contributions (due to the larger conformational freedom of the two dissociated domains in OCPR) that compensate for the loss of binding interactions between minor and major interfaces. In light of these observations, we used well-tempered metadynamics (wt-MetaD) to enable a more efficient exploration of the region relevant to the dissociation process. The choice of the CV for wt-MetaD is based on the experimental evidence that upon dissociation both the minor and major interfaces of OCPO break up. We therefore defined a contact map, restricted to the specific contacts relevant to the interfaces: by definition the CV is zero when both major and minor interfaces are formed and reaches one when the dissociation is complete. (A rigorous definition of this variable is provided in the SI.) The wt-MetaD simulations were run by biasing both the radius of gyration (Rg), which is expected to increase, and the interface contact map.
We compared two equivalent wt-MetaD simulations starting from a representative structure of OCPO and T2 states. This comparison will allow us to understand if the translocation of the carotenoid facilitates the dissociation and the changes in the interactions that are responsible for this opening.
The results of the two simulations are collected in Figure 5: panels A and B show the configurations of the system sampled in each wt-MetaD simulation projected in the subspace of the biased collective variables, and panels C and D show the estimated free-energy surfaces for the dissociation process.
Figure 5.

(A, B) Configurations of the system sampled in the wt-MetaD simulation projected in the subspace of the biased collective variables: the radius of gyration (Rg) and the interface contact map (CM). (C, D) Free-energy surfaces for the dissociation process projected in the subspace of the biased collective variables. Plots A and C refer to the simulation started from OCPO, and plots B and D refer to the one started from T2.
At a qualitative level, we note a clearly different behavior in the two simulations. Starting from OCPO we do not see any dissociation event even if the bias accumulated during the wt-MetaD simulation is very large (about 80 kcal mol–1); we recall that this bias can be seen as an artificial lowering of the free-energy barriers encountered by the system along the CVs. Since under these simulation conditions the system remains trapped in a set of configurations very close to the original minimum, the well-known stability of the closed form of the system is confirmed. On the contrary, the T2 state dissociates rather quickly: it explores a transition region where the two interfaces are only partially formed, and then it reaches the dissociated region (CM ≈ 1) where the two interfaces are broken and the system is able to move almost freely in the Rg dimension.
This result is not unexpected from what was reported in the previous section. According to our results, in fact, the interaction of the carotenoid with each of the domains of the protein (roughly 40 kcal mol–1) was almost perfectly balanced in OCPO and was fundamental to the stabilization of the closed form of OCP. Once the carotenoid is translocated into the NTD, the interaction energy with the CTD is largely reduced (less than 10 kcal mol–1) and the dissociation process becomes much easier, as the only barrier for the process is the cleavage of the inter-residue interactions at major and minor interfaces. From a thermodynamic point of view, we expect that the energy loss due to the disruption of the two interfaces can be compensated for by the solvation of the interfaces themselves and from the entropy gain of the dissociated state.
According to this model, the interactions of the carotenoid with NTD and CTD are crucial to the stabilization of the “closed”, globular form of the protein. To verify this hypothesis, we performed an additional wt-MetaD simulation under the same conditions detailed above, but starting from OCPO without the CAN molecule inside the cavity (details in the SI). In this case, the two domains of OCP are able to dissociate as they do in the simulation starting from T2 (Figure S5). This fact further proves that the dissociation is mainly enabled by a change in carotenoid–protein interactions and not by a modification of the protein structure induced by the photochemistry of the carotenoid itself.
Unfortunately, simulating transitions with such a large entropy production remains an extremely challenging task even when using advanced enhanced sampling techniques. In fact, even when extending the simulation starting from T2 up to 1 μs, no back-conversion is observed. Since the ergodic regime was not attained, the simulation cannot be used to estimate the relative free energy of the closed and open states. In spite of these limitations, our wt-MetaD simulations clearly show that the dissociation of the two domains follows the translocation of the carotenoid, confirming the hypothesis of the most recent working model for the photoactivation process.18,24,25 Moreover, it becomes clear that this order of events is a consequence of the fact that the modification of the carotenoid–protein interactions caused by the translocation plays a great role in enabling the subsequent dissociation.
To gain information on the dissociated state, we analyzed the data collected after the dissociation event which involve several transitions from small and large Rg.
In particular, we focused on the structural stability of the different domains of the protein. To do so, we calculated the RMSD of structured domains over the wt-MetaD trajectory. Furthermore, for each structure extracted from the trajectory, we computed a structure assignment using the DSSP algorithm.37,38 Considering the secondary structure data (Figure 6A), we note that the two NTD and CTD domains are quite stable; this is also confirmed by the fact that the RMSD of these domains remains around 2 Å for the whole simulation (Figure 6B) and by the direct observation of the structures extracted from the simulation (Figure 6C,D). A minor instability in CTD is observed in the region corresponding to the T275–N281 loop (Figure 6A,D). This loop, in the globular form of the protein, is localized at the interface between NTD and CTD, and its structure is already influenced by the translocation process. Therefore, it is not surprising that, upon dissociation, it can experience increased conformational flexibility. On the other hand, the structure of the two minor domains, namely, CTT and NTE, seems to be deeply influenced by the dissociation: the RMSD of these two domains already indicates a notable structural change with respect to the globular form of the protein (Figure 6B). Moreover, looking directly at the structures extracted from the simulations, it seems that the relative orientation of NTD and NTE (represented respectively in yellow and pink in Figure 6C) is not fixed after the dissociation and the NTE behaves as a disordered coil. This finding suggests that the formation of the minor interface is essential to the stability of the structure of the NTE.
Figure 6.

(A) Secondary structure of OCP calculated on the frame extracted from wt-MetaD simulation. (B) RMSD of NTE (purple), NTD (yellow), CTD (blue), and CTT (green) for the structures extracted from wt-MetaD. Each RMSD was calculated using OCPO as a reference and aligning the domain to the reference structure.Superposition of 80 frames of the wt-MetaD simulation, showing NTE and NTD with (C) CAN inside and (D) CTD. (E) Sample structure of the full dissociated OCPO with the α-helix already formed in the linker region.
As a final remark, we note that the relative position of the carotenoid inside the NTD remains almost unchanged with respect to the T2 state, which closely resembles the binding site of RCP, as shown in Figure 2. This fact strongly supports the validity of RCP as a model for the photoactivated form of OCP.
A last interesting fact that arises from the analysis of the dissociated state is that a new α-helix motif appears in the region of the unstructured linker of the two domains (Figure 2E, with the linker region represented in gray). This motif seems to be quite stable during our simulation, suggesting that, even if it corresponds to a low conservation zone of the protein, it could have biological relevance.
3. Discussion
In our simulations, we cannot see the ultrafast photochemical steps after carotenoid excitation. However, irrespective of the mechanism involved, these steps lead the complex to a first intermediate where the hydrogen bonds are broken. This is an out-of-equilibrium state that is thermally not accessible, which should resemble the geometry of T1. The nonequilibrium population of this state should be able, at least in part, to overcome the barrier and evolve toward T2. On the contrary, if the system was equilibrated in state T1, then, considering the relative heights of the barriers found in our simulations, we would observe a fall back to OCPO. This is what happens during OCPR → OCPO thermal reversion, which follows a different path with respect to the photoactivation.17,25
The most recent experiments also support the fact that the translocation process precedes the dissociation of the NTD and CTD domains by orders of magnitude.18,24,25 These findings are fully confirmed by our results. In fact, our wt-MetaD simulations show that the T2 state has a remarkable preference to undergo dissociation with respect to OCPO. In addition, our results indicate that the carotenoid interactions with the NTD and CTD are finely balanced in the dark-adapted state (Figure 3B), stabilizing the closed OCPO form. Similarly, carotenoids were shown to be essential to the stabilization of cyanobacterial PSII.39 Instead, as soon as the carotenoid is displaced along its conjugation axis, toward the NTD, an imbalance in these interactions appears, slackening the binding of the two domains (Figure 3B). This change in the interaction energy with both domains allows them to break apart in the ensuing entropically driven thermal process.
This picture is in contrast with the model of a dissociation process pushed forward by a released strain of the carotenoid.12,35 Indeed, according to our results, the internal energy of the carotenoid only slightly changes during the translocation, showing that the geometrical relaxation of the chromophore cannot be the driver of the process. Instead, here we propose that after excitation the carotenoid relaxes back to its ground state inside the cavity of an out-of-equilibrium system resembling the T1 state. In such a state, the structure of the protein is almost unchanged, the two H-bonds are already broken, but the carotenoid is still less than 5 Å away from its original binding site. Part of this population will then evolve into an equilibrium state (T2), where the carotenoid is in its final location and the NTD has rearranged, together with the T275–N281 loop. This hypothesis allows us to link the calculated FES found for the translocation process with recent experimental studies.11,18,24 In particular, by focusing on the model proposed by Konold et al.,18 we note a remarkable resemblance between our T1 state and the P2 proposed by the authors. In addition, the global rearrangement of the NTD and the loop that we observe along the translocation, between T1 and T2, could be responsible for the changes observed in the time-resolved IR spectra of the complex 0.5–10 μs after the photoexcitation.18 The structure of the NTD shows several changes (Figure 2D) which reflect more than one rearrangement taking place along the translocation. Finally, it is easy to link our T2 state to the P3 intermediate spectroscopically characterized by the same authors.
We note that the largest component of the carotenoid–protein binding energy is nonelectrostatic in character (Figure 3C). This is not unexpected, as it was noted that many highly conserved residues with hydrophobic/aromatic side chains are found in the carotenoid binding pockets of both OCPO and RCP.34 The importance of these interactions in the photoactivity of the protein has been demonstrated by several mutagenesis experiments. In fact, mutating aromatic residues, even absolutely conserved ones, around the binding tunnel preserve the photoactivity of the system as long as these are substituted by other aromatic residues.12,34 On the other hand, all NTD single mutations affect the relative stability of OCPO/OCPR to some extent, enhancing the thermal recovery rate and reducing the photoactivation yield. A combination of these mutations, which taken alone are not able to switch off the OCPO photoactivity, makes the protein nonphotoresponsive.12 This evidence, together with our results, leads us to propose that the photoactivity of OCP and its ability to thermally reform the orange state are the results of the fine-tuning of electrostatic and nonelectrostatic interactions of the carotenoid with the two main domains.
Another important open question that cannot be easily answered, even by exploiting the most advanced experimental techniques, is which structural changes of the protein are induced by the translocation. Whereas it is possible to gain general knowledge about the identity of structural rearrangements from IR studies, the insight obtained is far away from atomic-level resolution. On the other hand, XF-MS experiments, which have an intrinsically high spatial resolution, cannot provide time-resolved data at the required accuracy level.12,14,24,25 Our model, on the contrary, provides a detailed atomistic description of the structural changes of the protein along the whole process. Surprisingly, localized changes in the NTD, such as helix unfolding, were not observed in our simulation. Instead, we found a global rearrangement of this domain, mostly affecting the relative orientation of helices αC, αD, αE, and αG and of the N-terminal side of αI (using the nomenclature proposed in ref (34)). In the CTD, we observed a localized rearrangement of the T275–N281 loop that connects the β3 and β4 strands. These changes in the tertiary structure of the protein allow for a rearrangement of the H-bond network at the interfaces of the two domains (Figure 3). Moreover, the change in side-chain orientation of some residues, such as W277, provides stabilizing interactions for the carotenoid along the translocation. Remarkably, a very recent paper indeed found that the mutation of W277 with a phenylalanine results in a nonphotoactive complex, while the W277H mutation does not disrupt the photoactivity.40 This result indirectly supports the importance of residue W277 not only for π-stacking interactions with the carotenoid moiety but also for its H-bond donor properties that allow its interaction with the carbonyl group of the carotenoid itself.
In our simulations, without any a priori bias in this direction, we observe a change in the NTD that makes it more similar to the crystal structure of RCP (Figure 2B,D). However, our simulations do not see the “bending” of helices αC and αD which is evidenced by the comparison of crystal structures of OCPO:CAN and RCP:CAN. This discrepancy can be explained as either an artifact of the crystal structure or a change that takes place after the dissociation of the CTD. To support the first hypothesis, we also note that the two crystal structures of the RCP:CAN complex (PDB IDs 4XB4 and 5FCX) show slightly different structures in αC and αD helices (Figure S7).
Moving to the dissociation step, our data provide a clear indication of the relative stability of the different domains of the protein after their separation. As expected, the structures of NTD and CTD do not change significantly upon dissociation, as their average RMSD values remain at around 2 Å. The main instability is located in CTD, and corresponds to a relative flexibility of the T275–N281 loop (Figure 6D). This loop was already noted to play an important role during the dissociation and is an active constituent of the NTD/CTD interface. It is therefore not surprising that its structure is less stable after the unbinding of the NTD.
Upon dissociation of the NTE, this polypeptide chain is not able to maintain its helical structure and appears disordered both in its secondary structure and in its relative position with the NTD. This result is indeed expected from IR light-minus-dark data provided by Konold et al. for WT-OCP and ΔNTE-OCP.18 Our results support the fact that the presence of the minor interface is essential to stabilizing the helical structure of NTE.
On the other hand, we noted that the linker has a certain tendency to form an α-helical structure after dissociation. We think that the presence of this structural element in a region that has always been thought to be unstructured in both OCPO and OCPR should be considered when interpreting IR spectra. Our data do not allow a quantitative assessment of the stability of this helix. This fact, combined with the low conservation of the primary structure of this region, suggests that a more in-depth study is needed to asses the importance of the aforementioned folding in the actual OCPR solution structure.
4. Conclusions
By simulating for the first time, with atomistic techniques, the evolution of OCP after the photoactivation event has taken place, this study has shed light on the many still unclear features of this unique light-driven process, clarifying its most important structural and energetic aspects.
We found that only after carotenoid translocation could the dissociation of the two domains take place. Moreover, the latter process leaves the secondary and tertiary structures almost unchanged. We identified two relative free-energy minima in the translocation process, namely, T1 and T2, that are related to the intermediate products revealed by recent spectroscopic studies.18
The atomistic detail of our simulations allowed us to understand the reason for the reduced or absent photoactivity of OCP mutants recently reported in the literature.40 In fact, we highlighted the crucial role played by W277, which stabilizes the T2 state via H-bonding to the carotenoid, and by many aromatic residues in the NTD cavity. Mutations of these residues are expected to alter the stability of the intermediate, making it short-lived and more likely to return back to OCPO. Therefore, nanosecond transient absorption of NTD mutants, such as W277F, would reveal the spectral response of the early translocation intermediates only. Comparing these transient absorption spectra among the mutants and with the wild type will then help to identify the intermediates along the photoactivation path.
On the basis of our findings, we can conclude that the carotenoid locks the two domains together in the resting form of the complex (OCPO) by establishing comparably strong interactions with both of them. The photochemically triggered displacement of the carotenoid breaks this balance, allowing the complex to dissociate. In this model, the carotenoid does not act as a “spring” that, releasing its internal strain, induces the dissociation, as was previously proposed.35 Instead, the carotenoid seems to act as a “latch” that, when moving into the NTD, releases the binding interactions between the two domains, allowing them to split up.
5. Methods
We model OCP starting from the final structure of the 1 μs plain dynamics described in our previous work33 and based on the high-resolution crystal structure of CAN-binding OCP (PDB: 4XB5).12 All simulations were run with GROMACS 2019.4 suite using the classical MM parameter from the ff14SB AMBER force field.41 CAN was described using modified GAFF parameters reported in a previous article.33 Water was described using the TIP3P model. Unless differently specified, all of the simulations were run using a 2 fs integration time step, together with the LINCS algorithm to constrain every bond between a heavy atom and a hydrogen. A cutoff was applied after 12 Å on the LJ interactions, while long-range electrostatics was treated with a particle mesh Ewald algorithm. The simulation were performed in the NVT ensemble using the modified velocity rescaling thermostat,42 applied to the whole system, with a coupling constant of 0.1 ps. Periodic boundary conditions were applied in the three dimensions.
5.1. US-REMD Simulations
We performed 11 REMD simulations, each restrained with a different umbrella potential as described in Table S1. Each REMD simulation was performed with 50 replicas in the temperature range of 300–400 K (details in the Supporting Information). This allows for a significantly improved exploration of the orthogonal degrees of freedom. The harmonic potential on the reaction coordinate was applied using PLUMED 2.5.43 Each replica of the REMD simulation was first minimized and then equilibrated in the NVT ensemble (100 ps) and NPT ensemble (100 ps). The production simulation was then started. An exchange attempt was performed every 500 steps (1 ps). Average exchange probabilities of around 0.1 between neighboring replicas were attained (details on replica exchange results in the SI). Every 5 ps we saved the structure of the system for further analysis. Simulation lengths are reported in the SI. To generate the initial structures for the US-REMD simulations, a short ratchet-and-pawl MD (rMD) run was performed.44,45 In an rMD simulation, the system evolves freely whenever it spontaneously approaches the target state (defined according to a suitable CV). Conversely, a harmonic biasing force switches on any time the system attemps to backtrack to the initial state. This simulation was started from a structure of the OCPO complex (taken from a previous unbiased simulation33) equilibrated in an appropriate dodecahedron solvent box (14 763 water molecules, 37 Na+, and 30 Cl– needed to obtain a neutral system with a salt concentration of 0.1 M). The rMD simulation was run for 10 ns. The harmonic ratchet force was applied to the same CV as already described for the US-REMD simulation (the distance between NTD’s and CAN’s centers of mass). The target value was set to 1.0 Å, and the force constant was set to 10 000 J mol–1 nm–2. The closest frame to the center of the umbrella potential was used as the initial configuration for the subsequent simulation.
5.2. wt-MetaD Simulations
wt-MetaD simulations were performed using PLUMED to apply the bias.43,46 We used σ = 0.05 on the CM dimension and σ = 0.2 on the Rg dimension. The hill height was set to 0.5 kcal/mol, and the γ factor was set to 15. A hill was deposited every 2000 steps (4 ps). To speed up the bias calculation, the potential was calculated on a grid spanning from −0.1 to 1.1 in the CM dimension and from 10 to 60 Å in the Rg dimension; 50 points were used in each dimension. Two simulations were started from representative structures of OCPO and T2. Both of these structures were extracted from the previous US-REMD simulations. In order to avoid self-interaction of the system once it underwent dissociation, a larger cubic box was used with respect to the US-REMD simulations. To do so, we re-equilibrated the system in a larger cubic box (20 Å from the box walls instead of about 10 Å used before) composed of 40 850 water molecules and 86 Na+ and 79 Cl– ions. Before wt-MetaD was started, the system was equilibrated with harmonic restraints on the initial position of the heavy atoms of the complex in the NVT ensemble (100 ps) and in the NPT ensemble (100 ps) and finally for 10 ns without any restraint in the NVT ensemble. To further guarantee that self-interactions were avoided, harmonic upper and lower wall potentials were used to limit the system in a reasonable region of the phase space on the Rg dimension. The upper wall was placed at 45 Å, and the lower one was placed at 15 Å. Both harmonic potentials have a force constant of 5 kcal/mol Å–2.
5.3. Analysis of the Trajectories
An analysis of the MD trajectories was performed using in-house python scripts. Data manipulation was performed by exploiting numpy47,48 and scipy49 libraries, and graphics were generated with matplotlib50 libraries. Direct manipulation of the MD trajectory (e.g., extraction of relevant configurations, calculation of geometrical properties, and interaction energies) was performed directly with either GROMACS, PLUMED,43 or the MDAnalysis library.51 To efficiently solve the MBAR/UWHAM equations in the analysis of the US-REMD trajectory, we used the fastMBAR52 library, which allows us to use a modern GPU-accelerated computer to solve these equations with limited computational cost. Utilities from the PyEMMA53 library were also used for trajectory binning and CV discretization. All graphical representations of the system’s structures were rendered with the open source version of PyMol.
Acknowledgments
The authors thank Dr. Gianluca Lattanzi from the University of Trento for fruitful discussions. M.B., L.C., and B.M. acknowledge funding by the European Research Council under grant ERC-AdG-786714 (LIFETimeS). P.F. is a cofounder and shareholder of Sibylla Biotech SRL, a company involved in early-stage rational drug discovery.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10461.
Details of the protocol used to perform US-REMD simulation; information on the mixing and exchange probability of REMD simulations; histogram of the conformation sampled by US; details on the application of the MBAR/UWHAM method used for the analysis of US-REMD; details on the statistical analysis of the US-REMD simulation; mathematical definition of the contact map collective variable; details of the wt-MetaD simulation of OCPO without CAN; a graphical analysis of H-bond interactions of the β1 carbonyl group of CAN with some protein residue; change in the solvent-accessible surface from state OCPO divided into residues contributions and computed along the CV; superposition of crystal structures 4XB4 and 5FCX with helixes αC and αD highlighted; distribution of β1 and β2 dihedral angles of CAN computed from a simulation in water; graphical representation of the structural change spotted in loop T275–N281; detailed figures of the representative frames of T1 and T2; and data used to construct Figure 3C in tabular form (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Croce R.; van Amerongen H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 2014, 10, 492–501. 10.1038/nchembio.1555. [DOI] [PubMed] [Google Scholar]
- Niyogi K. K.; Truong T. B. Evolution of flexible non-photochemical quenching mechanisms that regulate light harvesting in oxygenic photosynthesis. Curr. Opin. Plant Biol. 2013, 16, 307–314. 10.1016/j.pbi.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Derks A.; Schaven K.; Bruce D. Diverse mechanisms for photoprotection in photosynthesis. Dynamic regulation of photosystem II excitation in response to rapid environmental change. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 468–485. 10.1016/j.bbabio.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Magdaong N. C. M.; Blankenship R. E. Photoprotective, excited-state quenching mechanisms in diverse photosynthetic organisms. J. Biol. Chem. 2018, 293, 5018–5025. 10.1074/jbc.TM117.000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruban A. V. Light harvesting control in plants. FEBS Lett. 2018, 592, 3030–3039. 10.1002/1873-3468.13111. [DOI] [PubMed] [Google Scholar]
- Pinnola A.; Bassi R. Molecular mechanisms involved in plant photoprotection. Biochem. Soc. Trans. 2018, 46, 467–482. 10.1042/BST20170307. [DOI] [PubMed] [Google Scholar]
- Grossman A. R.; Schaefer M. R.; Chiang G. G.; Collier J. L. The phycobilisome, a light-harvesting complex responsive to environmental conditions. Microbiol. Rev. 1993, 57, 725–749. 10.1128/MR.57.3.725-749.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirilovsky D.; Kerfeld C. A. Cyanobacterial photoprotection by the orange carotenoid protein. Nat. Plants 2016, 2, 16180. 10.1038/nplants.2016.180. [DOI] [PubMed] [Google Scholar]
- Sonani R. R.; Gardiner A.; Rastogi R. P.; Cogdell R.; Robert B.; Madamwar D. Site, trigger, quenching mechanism and recovery of non-photochemical quenching in cyanobacteria: recent updates. Photosynth. Res. 2018, 137, 171–180. 10.1007/s11120-018-0498-8. [DOI] [PubMed] [Google Scholar]
- Muzzopappa F.; Kirilovsky D. Changing Color for Photoprotection: The Orange Carotenoid Protein. Trends Plant Sci. 2020, 25, 92–104. 10.1016/j.tplants.2019.09.013. [DOI] [PubMed] [Google Scholar]
- Wilson A.; Punginelli C.; Gall A.; Bonetti C.; Alexandre M.; Routaboul J.-M.; Kerfeld C. A.; van Grondelle R.; Robert B.; Kennis J. T. M.; Kirilovsky D. A photoactive carotenoid protein acting as light intensity sensor. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 12075–12080. 10.1073/pnas.0804636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz R. L.; Sutter M.; Wilson A.; Gupta S.; Thurotte A.; de Carbon C. B.; Petzold C. J.; Ralston C.; Perreau F.; Kirilovsky D.; Kerfeld C. A. A 12 A carotenoid translocation in a photoswitch associated with cyanobacterial photoprotection. Science 2015, 348, 1463–1466. 10.1126/science.aaa7234. [DOI] [PubMed] [Google Scholar]
- Leverenz R. L.; Jallet D.; Li M.-D.; Mathies R. A.; Kirilovsky D.; Kerfeld C. A. Structural and Functional Modularity of the Orange Carotenoid Protein: Distinct Roles for the N- and C-Terminal Domains in Cyanobacterial Photoprotection. Plant Cell 2014, 26, 426–437. 10.1105/tpc.113.118588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Guttman M.; Leverenz R. L.; Zhumadilova K.; Pawlowski E. G.; Petzold C. J.; Lee K. K.; Ralston C. Y.; Kerfeld C. A. Local and global structural drivers for the photoactivation of the orange carotenoid protein. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E5567–E5574. 10.1073/pnas.1512240112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Zhang H.; Orf G. S.; Lu Y.; Jiang J.; King J. D.; Wolf N. R.; Gross M. L.; Blankenship R. E. Dramatic Domain Rearrangements of the Cyanobacterial Orange Carotenoid Protein upon Photoactivation. Biochemistry 2016, 55, 1003–1009. 10.1021/acs.biochem.6b00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T.; Leverenz R. L.; Nagamine M.; Kerfeld C. A.; Unno M. Raman Optical Activity Reveals Carotenoid Photoactivation Events in the Orange Carotenoid Protein in Solution. J. Am. Chem. Soc. 2017, 139, 10456–10460. 10.1021/jacs.7b05193. [DOI] [PubMed] [Google Scholar]
- Maksimov E. G.; Sluchanko N. N.; Slonimskiy Y. B.; Slutskaya E. A.; Stepanov A. V.; Argentova-Stevens A. M.; Shirshin E. A.; Tsoraev G. V.; Klementiev K. E.; Slatinskaya O. V.; Lukashev E. P.; Friedrich T.; Paschenko V. Z.; Rubin A. B. The photocycle of orange carotenoid protein conceals distinct intermediates and asynchronous changes in the carotenoid and protein components. Sci. Rep. 2017, 7, 15548. 10.1038/s41598-017-15520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konold P. E.; van Stokkum I. H. M.; Muzzopappa F.; Wilson A.; Groot M.-L.; Kirilovsky D.; Kennis J. T. M. Photoactivation Mechanism, Timing of Protein Secondary Structure Dynamics and Carotenoid Translocation in the Orange Carotenoid Protein. J. Am. Chem. Soc. 2019, 141, 520–530. 10.1021/jacs.8b11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimov E. G.; Protasova E. A.; Tsoraev G. V.; Yaroshevich I. A.; Maydykovskiy A. I.; Shirshin E. A.; Gostev T. S.; Jelzow A.; Moldenhauer M.; Slonimskiy Y. B.; Sluchanko N. N.; Friedrich T. Probing of carotenoid-tryptophan hydrogen bonding dynamics in the single-tryptophan photoactive Orange Carotenoid Protein. Sci. Rep. 2020, 10, 11729. 10.1038/s41598-020-68463-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L.; van Stokkum I. H. M.; Koehorst R. B. M.; Jongerius A.; Kirilovsky D.; van Amerongen H. Site, Rate, and Mechanism of Photoprotective Quenching in Cyanobacteria. J. Am. Chem. Soc. 2011, 133, 18304–18311. 10.1021/ja206414m. [DOI] [PubMed] [Google Scholar]
- Boulay C.; Wilson A.; D’Haene S.; Kirilovsky D. Identification of a protein required for recovery of full antenna capacity in OCP-related photoprotective mechanism in cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 11620–11625. 10.1073/pnas.1002912107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter M.; Wilson A.; Leverenz R. L.; Lopez-Igual R.; Thurotte A.; Salmeen A. E.; Kirilovsky D.; Kerfeld C. A. Crystal structure of the FRP and identification of the active site for modulation of OCP-mediated photoprotection in cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10022–10027. 10.1073/pnas.1303673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re E. D.; Schlau-Cohen G. S.; Leverenz R. L.; Huxter V. M.; Oliver T. A. A.; Mathies R. A.; Fleming G. R. Insights into the Structural Changes Occurring upon Photoconversion in the Orange Carotenoid Protein from Broadband Two-Dimensional Electronic Spectroscopy. J. Phys. Chem. B 2014, 118, 5382–5389. 10.1021/jp502120h. [DOI] [PubMed] [Google Scholar]
- Mezzetti A.; Alexandre M.; Thurotte A.; Wilson A.; Gwizdala M.; Kirilovsky D. Two-Step Structural Changes in Orange Carotenoid Protein Photoactivation Revealed by Time-Resolved Fourier Transform Infrared Spectroscopy. J. Phys. Chem. B 2019, 123, 3259–3266. 10.1021/acs.jpcb.9b01242. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Sutter M.; Remesh S. G.; Dominguez-Martin M. A.; Bao H.; Feng X. A.; Chan L.-J. G.; Petzold C. J.; Kerfeld C. A.; Ralston C. Y. X-ray radiolytic labeling reveals the molecular basis of orange carotenoid protein photoprotection and its interactions with fluorescence recovery protein. J. Biol. Chem. 2019, 294, 8848–8860. 10.1074/jbc.RA119.007592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barducci A.; Bonomi M.; Parrinello M. Metadynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 826–843. 10.1002/wcms.31. [DOI] [Google Scholar]
- Abrams C.; Bussi G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2014, 16, 163–199. 10.3390/e16010163. [DOI] [Google Scholar]
- Bernardi R. C.; Melo M. C. R.; Schulten K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 872–877. 10.1016/j.bbagen.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Kamiya M.; Oshima H.; Re S. In Methods in Molecular Biology; Bonomi C., Massimilianoand C., Eds.; Springer: New York, 2019; pp 155–177. [DOI] [PubMed] [Google Scholar]
- Torrie G.; Valleau J. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Earl D. J.; Deem M. W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 2005, 7, 3910. 10.1039/b509983h. [DOI] [PubMed] [Google Scholar]
- Wilson A.; Punginelli C.; Couturier M.; Perreau F.; Kirilovsky D. Essential role of two tyrosines and two tryptophans on the photoprotection activity of the Orange Carotenoid Protein. Biochim. Biophys. Acta, Bioenerg. 2011, 1807, 293–301. 10.1016/j.bbabio.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Bondanza M.; Cupellini L.; Lipparini F.; Mennucci B. The Multiple Roles of the Protein in the Photoactivation of Orange Carotenoid Protein. Chem. 2020, 6, 187–203. 10.1016/j.chempr.2019.10.014. [DOI] [Google Scholar]
- Wilson A.; Kinney J. N.; Zwart P. H.; Punginelli C.; D’Haene S.; Perreau F.; Klein M. G.; Kirilovsky D.; Kerfeld C. A. Structural Determinants Underlying Photoprotection in the Photoactive Orange Carotenoid Protein of Cyanobacteria. J. Biol. Chem. 2010, 285, 18364–18375. 10.1074/jbc.M110.115709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara S.; Ren Z.; Lu L.; Zeng X.; Shin H.; Zhao K.-H.; Yang X. Photoactivation mechanism of a carotenoid-based photoreceptor. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 6286–6291. 10.1073/pnas.1700956114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T.; Dominguez-Martin M. A.; Šímová I.; Fuciman M.; Kerfeld C. A.; Polívka T. Excited-State Properties of Canthaxanthin in Cyanobacterial Carotenoid-Binding Proteins HCP2 and HCP3. J. Phys. Chem. B 2020, 124, 4896–4905. 10.1021/acs.jpcb.0c03137. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Touw W. G.; Baakman C.; Black J.; te Beek T. A. H.; Krieger E.; Joosten R. P.; Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakar T.; Laczko-Dobos H.; Toth T. N.; Gombos Z.. Carotenoids Assist in Cyanobacterial Photosystem II Assembly and Function. Front. Plant Sci. 2016, 7, 10.3389/fpls.2016.00295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimov E. G.; Protasova E. A.; Tsoraev G. V.; Yaroshevich I. A.; Maydykovskiy A. I.; Shirshin E. A.; Gostev T. S.; Jelzow A.; Moldenhauer M.; Slonimskiy Y. B.; Sluchanko N. N.; Friedrich T. Probing of carotenoid-tryptophan hydrogen bonding dynamics in the single-tryptophan photoactive Orange Carotenoid Protein. Sci. Rep. 2020, 10, 11729. 10.1038/s41598-020-68463-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Paci E.; Karplus M. Forced unfolding of fibronectin type 3 modules: an analysis by biased molecular dynamics simulations. J. Mol. Biol. 1999, 288, 441–459. 10.1006/jmbi.1999.2670. [DOI] [PubMed] [Google Scholar]
- Tiana G.; Camilloni C. Ratcheted molecular dynamics simulations identify efficiently the transition state of protein folding. J. Chem. Phys. 2012, 137, 235101. 10.1063/1.4769085. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Oliphant T. E.A Guide to NumPy; Trelgol Publishing: 2006; Vol. 1. [Google Scholar]
- Van Der Walt S.; Colbert S. C.; Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22. 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- Virtanen P.; Gommers R.; Oliphant T. E.; Haberland M.; Reddy T.; Cournapeau D.; Burovski E.; Peterson P.; Weckesser W.; Bright J.; van der Walt S. J.; Brett M.; Wilson J.; Millman K. J.; Mayorov N.; Nelson A. R. J.; Jones E.; Kern R.; Larson E.; Carey C. J.; Polat İ.; Feng Y.; Moore E. W.; VanderPlas J.; Laxalde D.; Perktold J.; Cimrman R.; Henriksen I.; Quintero E. A.; Harris C. R.; Archibald A. M.; Ribeiro A. H.; Pedregosa F.; van Mulbregt P. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J. D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X.; Vilseck J. Z.; Brooks C. L. Fast Solver for Large Scale Multistate Bennett Acceptance Ratio Equations. J. Chem. Theory Comput. 2019, 15, 799–802. 10.1021/acs.jctc.8b01010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M. K.; Trendelkamp-Schroer B.; Paul F.; Pérez-Hernández G.; Hoffmann M.; Plattner N.; Wehmeyer C.; Prinz J.-H.; Noé F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. 10.1021/acs.jctc.5b00743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.