

We describe a nanoparticle platform for brain delivery of nucleic acid–based therapeutics to treat TBI.
Abstract
Small interfering RNA (siRNA)–based therapeutics can mitigate the long-term sequelae of traumatic brain injury (TBI) but suffer from poor permeability across the blood-brain barrier (BBB). One approach to overcoming this challenge involves treatment administration while BBB is transiently breached after injury. However, it offers a limited window for therapeutic intervention and is applicable to only a subset of injuries with substantially breached BBB. We report a nanoparticle platform for BBB pathophysiology–independent delivery of siRNA in TBI. We achieved this by combined modulation of surface chemistry and coating density on nanoparticles, which maximized their active transport across BBB. Engineered nanoparticles injected within or outside the window of breached BBB in TBI mice showed threefold higher brain accumulation compared to nonengineered PEGylated nanoparticles and 50% gene silencing. Together, our data suggest that this nanoparticle platform is a promising next-generation drug delivery approach for the treatment of TBI.
INTRODUCTION
Referred to as a “silent epidemic,” traumatic brain injury (TBI) is a leading cause of death and disability among children and young adults, with millions of people sustaining TBI each year in accidents, sports, and military conflicts (1–3). TBI may lead to potentially long-lasting neurological dysfunctions, memory disturbances, behavioral changes, speech irregularities, and gait abnormalities. It has also been implicated in the development of neurodegenerative disease, particularly chronic traumatic encephalopathy, Alzheimer’s disease, and Parkinson’s disease (4–7). Following primary injury, which is a result of mechanical impact to the brain, secondary injury gradually occurs as a consequence of destructive cellular and molecular events. This can ultimately lead to neuronal and glial cell death, tissue damage, and atrophy (8–10). While prevention strategies offer the best means to mitigate primary injury, strategies targeting secondary injury enable direct intervention by diminishing the cascade of destructive events. However, current treatments for TBI are limited to palliative care, and no effective strategies are available to interrupt the pathological cascade after TBI that leads to neurodegeneration.
Nucleic acid–based therapeutics, especially small interfering RNA (siRNA), can efficiently and specifically silence the expression of target genes, including those that are traditionally considered to be “untreatable” by small-molecule drugs (11). Thus, siRNA-based therapies can target specific pathological pathways to mitigate disease progression and offer a precision medicine approach for TBI treatment. Unfortunately, there are multiple biological barriers toward the delivery of siRNA into brain tissue, the blood-brain barrier (BBB) being the most important one (11–13). One approach to overcoming BBB in TBI involves systemic administration of therapeutics while the BBB is physically breached after injury (14–16). However, physical breaching of BBB in TBI is highly heterogeneous and varies greatly among patients, depending on the extent of primary injury (17–20). Moreover, BBB can self-repair within a few hours to days after injury to restore its integrity (16–20). Hence, physical breaching of BBB offers a limited window for therapeutic interventions, which is not ideal, as the secondary injury can last months and may require repeated dosing beyond this limited time window. There is therefore an unmet need to develop approaches that allow BBB pathophysiology–independent delivery of siRNA in TBI.
Herein, we report a nanoparticle (NP) platform that enables brain delivery of siRNA, independent of BBB pathophysiology in TBI. We achieved this by combined modulation of surface coating chemistry and coating density on NPs, which maximized their active transport through BBB. Incorporating certain surface coatings on NPs has been previously shown to augment their active penetration across BBB in multiple brain diseases (21–24). However, a surface coating that can facilitate active transport of siRNA-loaded NPs across BBB in TBI has not been reported previously. We first aimed to identify a surface coating and coating density that can maximize the active penetration of siRNA-loaded NPs across intact BBB. Specifically, we formulated NPs using poly(lactic-co-glycolic acid) (PLGA), a biodegradable and biocompatible polymer that exists in several Food and Drug Administration–approved products (25, 26) and performed a systematic study to correlate the impact of different surface coatings and their coating densities on the penetration of PLGA NPs across intact, undamaged BBB in healthy mice (Fig. 1A). Brain accumulation of NPs was found to be dependent on the type of surface coating and the coating density, and combined modulation of these two parameters maximized the penetration of NPs across BBB. Surface coating and its coating density also affected the uptake of siRNA-loaded NPs by neural cells and their gene silencing efficiency. Using an established mouse model of weight drop–induced TBI (27, 28), we showed that the optimized NPs delivered siRNA to brain when administered during early or late injury periods, which correspond to physically breached BBB and intact, self-repaired BBB, respectively. Last, intravenous administration of NPs loaded with siRNA against tau (a proof-of-concept target), during early or late injury period in TBI mice, achieved up to 50% reduction in tau expression. Overall, we demonstrate that BBB pathophysiology–independent delivery of siRNA in TBI is feasible and deserves further exploration.
Fig. 1. NPs with different surface coatings were prepared to achieve BBB pathophysiology–independent delivery of siRNA in TBI.
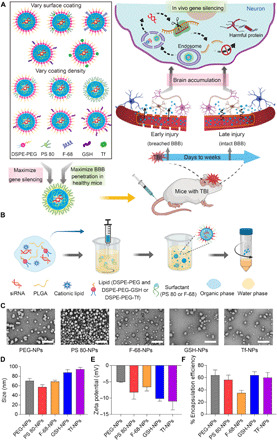
(A) Schematic illustrating the overall study design. siRNA-loaded NPs with different surface coating chemistries and coating densities were compared for their in vitro uptake and gene silencing efficiency in neural cells as well as their ability to cross intact BBB in healthy mice. NPs with maximum gene silencing efficiency and BBB permeability were then evaluated in TBI mice to determine brain accumulation and gene silencing efficiency when administered during early injury or late injury periods, corresponding to physically breached BBB and intact BBB, respectively. Upon neuronal uptake of NPs, siRNA is released and silences the harmful proteins involved in TBI pathophysiology. (B) Schematic for the preparation of siRNA-loaded PLGA NPs by a modified nanoprecipitation method. DSPE-PEG was used to impart stealth character. In addition, polysorbate 80 (PS 80), poloxamer 188 (F-68), DSPE-PEG-glutathione (GSH), or DSPE-PEG-transferrin (Tf) was used to augment BBB penetration. PEG, polyethylene glycol; DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine. (C) Transmission electron microscopy images of siRNA-loaded NPs having different surface coatings. Scale bars, 200 nm. (D and E) Size and zeta potential of siRNA-loaded NPs having different surface coatings, as analyzed by dynamic light scattering. (F) Encapsulation efficiency of siRNA in different NPs. Data in (D) to (F) are means ± SD of technical repeats (n = 3, experiment performed at least twice). The illustrations (A and B) were created with the help of BioRender.com.
RESULTS
Sub–100-nm-sized siRNA NPs were prepared with different surface coatings
PLGA-based siRNA NPs with five different surface coatings were engineered to interact with BBB and neural cells by a variety of mechanisms. Polyethylene glycol (PEG) conjugated to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) was incorporated in NPs for “stealth” effect to extend their systemic circulation time (29). In addition to DSPE-PEG, we also included one of the four different surface coatings that have previously shown to augment active penetration of NPs across BBB in other diseases. Specifically, we chose polysorbate 80 (PS 80), poloxamer 188 (Pluronic F-68), DSPE-PEG-glutathione (GSH), and DSPE-PEG-transferrin (Tf). PS 80 and F-68 have been previously shown to promote drug delivery across BBB by binding to endogenous apolipoproteins and interacting with lipoprotein receptors on BBB (30–32). GSH, a BBB shuttle peptide that has reached phase 2 clinical trials, can transport NPs through BBB via GSH transporters (33, 34). Tf has been shown to facilitate brain delivery of NPs by binding to Tf receptors expressed on brain endothelium (35, 36).
siRNA-loaded NPs with different surface coatings were prepared using a modified nanoprecipitation method (Fig. 1B). In all NPs, irrespective of their surface coating, we used an ethylenediamine core poly(amidoamine) (PAMAM) derivate–based cationic lipid-like molecule for siRNA complexation (37, 38). Because NPs with a small size (<100 nm) were previously demonstrated to exhibit more BBB penetration than larger ones (39, 40), we tuned the size of NPs in this study to <100 nm. Specifically, we tested different organic solvents for preparing NPs. The polarity of organic solvent influences the dispersion of polymer in aqueous solution, thus affecting the size of NPs. As shown by transmission electron microscopy (Fig. 1C) and dynamic light scattering (DLS) analysis (Fig. 1D), spherical NPs with hydrodynamic diameters between 55 and 95 nm were formed with dimethylformamide (DMF), whereas larger NPs were obtained when solvents with lower polarity, such as acetone and tetrahydrofuran, were used (fig. S1). Thus, DMF was chosen to formulate NPs for further studies. All NPs had a narrow size distribution, as suggested by the polydispersity index (fig. S2) and had negative surface charge (Fig. 1E). Encapsulation efficiency of siRNA in different NPs was studied using Cy3-labeled scrambled siRNA. All NPs demonstrated high encapsulation efficiency (55 to 65%) except for NPs coated with F-68, which demonstrated a relatively lower encapsulation efficiency of 35% (Fig. 1F). Because F-68 affected siRNA encapsulation, we did not use F-68–coated NPs for subsequent experiments. Last, NPs showed slow and sustained release of siRNA in phosphate-buffered saline (PBS) (pH 7.4) at 37°C (fig. S3).
Surface coating affects the uptake of NPs by neural cells and gene silencing efficiency
Efficient cellular uptake is the premise for achieving gene silencing. However, cell membrane is a barrier for siRNA delivery as negatively charged siRNA cannot cross the anionic cell membrane by itself. Therefore, we evaluated the uptake of siRNA-loaded NPs by Neuro-2a cells. NPs were loaded with scrambled siRNA labeled with a fluorescence probe (Dy677) to image cellular uptake by a confocal laser scanning microscope (CLSM). Cells were incubated with free siRNA or siRNA-loaded NPs having different surface coatings at 37°C for 2 hours, followed by imaging using a CLSM. Cells incubated with free siRNA showed negligible fluorescence signal, which is expected. PEG-coated NPs (PEG-NPs) and Tf-coated NPs (Tf-NPs) also resulted in a weak fluorescence signal, suggesting that PEG and Tf do not facilitate the interaction between NPs and neural cells (Fig. 2A). Cells treated with GSH-coated NPs (GSH-NPs) or PS 80–coated NPs (PS 80-NPs) showed higher fluorescence signal compared to those treated with free siRNA, PEG-NPs, or Tf-NPs, indicating that GSH and PS 80 facilitated the transport of siRNA-loaded NPs into neural cells. In particular, PS 80-NPs exhibited the highest cellular uptake among all NPs, as suggested by an intense fluorescence signal in cells (Fig. 2A). We also performed flow cytometry to quantify cellular uptake of different NPs (fig. S4). Consistent with CLSM analysis, the intensity of fluorescence signal in cells was found to be dependent on the surface coating chemistry of NPs. Cells treated with PS 80-NPs exhibited the strongest fluorescence signal, which was 20-fold higher than cells treated with PEG-NPs.
Fig. 2. Surface coating affects the uptake of NPs by neural cells, gene silencing efficiency in vitro, and penetration of NPs across intact BBB.
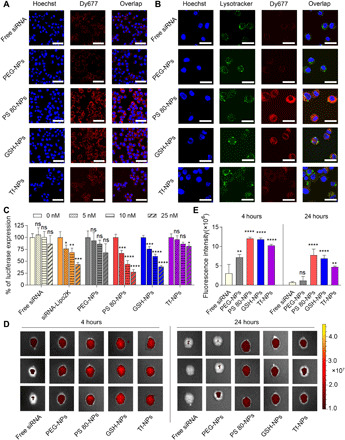
(A) CLSM images of Neuro-2a cells incubated with free siRNA or siRNA-loaded NPs having different surface coatings (PEG-NPs, PS 80-NPs, GSH-NPs, or Tf-NPs) at 37°C for 2 hours. Dy677-labeled scrambled siRNA (red signal) was used. Nuclei were stained with Hoechst 33342 (blue signal). Scale bars, 50 μm. (B) CLSM images showing endosomal escape of siRNA in Neuro-2a cells. Cells were incubated with free siRNA or siRNA-loaded NPs at 37°C for 2 hours. Dy677-labeled siRNA (red signal) was used. Nuclei were stained by Hoechst 33342 (blue signal) and endosomes were stained with LysoTracker Green (green signal). Scale bars, 30 μm. (C) Luciferase expression in Neuro-2a cells. Luciferase-expressing Neuro-2a cells were incubated for 24 hours with medium containing different concentrations of free luciferase siRNA (free siRNA), luciferase siRNA–Lipofectamine 2000 complex (siRNA-Lipo2K), or luciferase siRNA–loaded NPs. Following an additional 48-hour incubation with medium only, luciferase expression was quantified using luminescence assay. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to 0 nM siRNA. ns, not significant. (D) In vivo imaging system (IVIS) images of brains from three representative healthy mice, excised at 4 or 24 hours after intravenous injection of free siRNA or siRNA-loaded NPs (50 nmol siRNA/kg). Dy677-labeled scrambled siRNA was used. (E) Fluorescence intensity measured over excised mice brains using IVIS. **P < 0.01, ****P < 0.0001 compared to free siRNA. Data in (C) are means ± SD of technical repeats (n = 3, experiment performed at least twice). Data in (E) are means ± SD (n = 3 mice/group, experiment performed twice). P values were determined using one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis.
Following cellular uptake, siRNA must escape from endosomes to reach its target RNA in cytoplasm for gene silencing. Endosomal escape is one of the major obstacles for nonviral siRNA delivery, as siRNA trapped in endosomes will eventually undergo enzymatic digestion (41). To evaluate whether siRNA could escape from endosomes, we used LysoTracker Green—a fluorescence probe—to label endolysosomes and observed the colocalization of LysoTracker Green signal with signal from Dy677-labeled siRNA. After 2-hour incubation of cells with NPs, siRNA was found to escape from endolysosomes into cytoplasm, as demonstrated by the separation of red (siRNA) and green (endolysosome) fluorescence signals in the cytoplasm (Fig. 2B). It was noted that siRNA could escape from the endosome in all NP groups, regardless of the surface coating. Endosomal escape of siRNA is attributed to the amino-rich PAMAM derivate–based cationic molecule, which was included in all NP formulations and has previously shown to mediate the release of RNA cage from endosome via proton-sponge effect (37, 38).
We next sought to determine whether surface coating chemistry of NPs affects gene silencing efficiency. Luciferase-expressing Neuro-2a cells were incubated with varying concentrations of luciferase siRNA–loaded NPs having different surface coatings for 24 hours, followed by incubation with medium only for an additional 48 hours. Luciferase expression was quantified using a luminescence assay. Free siRNA and siRNA complexed with a commercially available transfection agent—Lipofectamine 2000 (siRNA-Lipo2K)—were used as controls. While cells treated with free siRNA did not result in any noticeable gene silencing, siRNA-Lipo2K silenced the luciferase gene in a dose-dependent manner (Fig. 2C). Gene silencing efficiency was found to be dependent on the surface coating chemistry of NPs. While PEG-NPs and Tf-NPs showed gene silencing only at the highest concentration of siRNA, PS 80-NPs and GSH-NPs resulted in significant gene silencing at all tested doses. In particular, PS 80-NPs exhibited the highest gene silencing, with 70% knockdown in luciferase expression at a concentration of 25 nM siRNA. Gene silencing efficiency of PS 80-NPs was also demonstrably greater than that mediated by Lipo2K. Notably, none of the NPs resulted in an obvious decrease in the viability of cells compared to cells incubated with culture medium only (fig. S5).
Penetration of NPs across intact BBB depends on surface coating
Penetration of NPs across intact, undamaged BBB in healthy mice could provide a strong and direct evidence of NPs’ ability to cross the fully repaired BBB after TBI. Therefore, we screened siRNA-loaded NPs having different surface coatings in healthy mice. Dy677-labeled scrambled siRNA was loaded into NPs. Free siRNA or siRNA-loaded NPs having different surface coatings were administered intravenously to healthy mice at a siRNA dose of 50 nmol/kg. Mice were euthanized at 4 and 24 hours after injection, and brains were harvested for imaging using in vivo imaging system (IVIS) (Fig. 2, D and E). Free siRNA and siRNA-loaded PEG-NPs showed weak fluorescence signals in the brain, suggesting poor penetration across BBB. PS 80-NPs, GSH-NPs, and Tf-NPs showed significantly higher fluorescence signals in the brain compared to PEG-NPs and free siRNA at both 4- and 24-hour time points. Impressively, brains from mice injected with PS 80-NPs and GSH-NPs exhibited the brightest fluorescence signal compared to mice injected with other formulations. Collectively, our data suggest that surface coating chemistry greatly affects BBB penetration of NPs. For further studies, we chose to evaluate PS 80-NPs and GSH-NPs due to their higher gene silencing efficiency and more effective penetration across intact BBB compared to PEG-NPs and Tf-NPs.
Surface coating density affects cellular uptake of NPs, gene silencing efficiency, and penetration of NPs across intact BBB
We next modulated the surface coating density to understand its impact on the uptake of siRNA-loaded NPs by neural cells and the gene silencing efficiency. NPs coated with low (L), medium (M), or high (H) density of PS 80 or GSH were prepared. Quantitative values for surface coating densities are shown in table S1. DLS analysis showed that the sizes of these NPs were all in the range of 50 to 90 nm (fig. S6A). NPs became more negatively charged with an increase in the coating densities of PS 80 and GSH (fig. S6B) with no impact on the encapsulation efficiency of siRNA (fig. S6C). To study the impact of coating density on cellular uptake, Neuro-2a cells were incubated for 2 hours with Dy677-labeled siRNA-loaded NPs having different coating densities of PS 80 or GSH, followed by imaging using a CLSM. Cellular uptake of PS 80-NPs increased with an increase in the coating density of PS 80 (Fig. 3A). Flow cytometry showed 80-fold higher cellular uptake of PS 80 (H)-NPs compared to PEG-NPs (fig. S7). Cellular uptake of GSH-NPs also increased with an increase in the coating density of GSH, but only modestly (Fig. 3A and fig. S7). Coating density of PS 80 also affected the gene silencing efficiency of NPs (Fig. 3B). Luciferase-expressing Neuro-2a cells were treated for 24 hours with varying concentrations of luciferase siRNA–loaded NPs having different coating densities of PS 80 or GSH. Following an additional 48-hour incubation with medium only, luciferase expression was quantified. We observed a dose-dependent decrease in the expression of luciferase for all the three PS 80-NPs having (L), (M), or (H) coating density. PS 80 (H)-NPs showed highest gene silencing, with 90% reduction of luciferase expression in Neuro-2a cells at a siRNA dose of 10 nM. Although coating density of GSH also affected the gene silencing efficiency, the difference between different coating densities was not statistically significant. None of the NPs resulted in an obvious decrease in the viability of cells compared to cells incubated with medium only (fig. S8).
Fig. 3. Surface coating density affects the uptake of NPs by neural cells, gene silencing efficiency in vitro, and penetration of NPs across intact BBB.
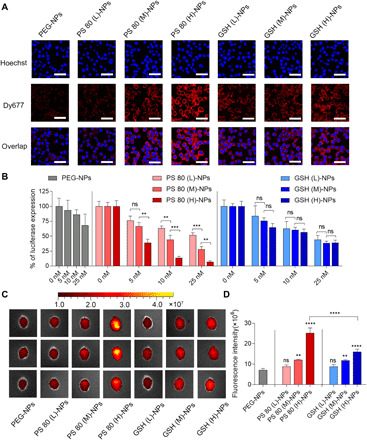
(A) CLSM images of Neuro-2a cells incubated with siRNA-loaded NPs having different coating densities of PS 80 or GSH at 37°C for 2 hours. PEG-NPs were used as control. Dy677-labeled scrambled siRNA (red signal) was loaded into NPs. Nuclei were stained with Hoechst 33342 (blue signal). Scale bars, 50 μm. (B) Luciferase expression in Neuro-2a cells. Luciferase-expressing Neuro-2a cells were incubated for 24 hours with medium containing luciferase siRNA–loaded NPs having different coating densities of PS 80 or GSH at varying concentrations of siRNA. Following an additional 48-hour incubation with medium only, luciferase expression was quantified using a luminescence assay. PEG-NPs were used as control. **P < 0.01 and ***P < 0.001. (C) IVIS images of brains from three representative healthy mice, excised at 4 hours after intravenous injection of siRNA-loaded NPs (50 nmol siRNA/kg) having different coating densities of PS 80 or GSH. PEG-NPs were used as control. Dy677-labeled scrambled siRNA was loaded into NPs. (D) Fluorescence intensity measured over excised mice brains using IVIS. **P < 0.01 and ****P < 0.0001 compared to PEG-NPs. ****P < 0.0001 for PS 80 (H)-NPs versus GSH (H)-NPs. Data in (B) are means ± SD of technical repeats (n = 3, experiment performed at least twice). Data in (D) are means ± SD (n = 3 mice per group, experiment performed twice). P values were determined by one-way ANOVA with Tukey’s post hoc analysis.
To evaluate the impact of coating density on the penetration of NPs across intact BBB, NPs having different coating densities of PS 80 or GSH were intravenously administered to healthy mice at a siRNA dose of 50 nmol/kg. Dy677-labeled scrambled siRNA was loaded into NPs. Brains were collected for IVIS imaging at 4 hours after injection. Increase in the coating density of PS 80 or GSH increased the brain accumulation of NPs (Fig. 3, C and D). Mice injected with PS 80 (H)-NPs exhibited the strongest fluorescence signal, which was about four times higher than PEG-NPs. Increasing the coating density of GSH also enhanced the transport of NPs across BBB, but GSH (H)-NPs showed significantly less BBB penetration than the PS 80 (H)-NPs at the coating densities evaluated in this work.
We also investigated the mechanism of BBB penetration by PS 80 NPs in an in vitro BBB model developed with mouse brain endothelial cell line, bEnd.3 (fig. S9A). bEnd.3 cells were seeded on Matrigel-coated filter inserts until tight junction formation, as confirmed by tracking the transendothelial electrical resistance (TEER) for the cell monolayer over time until it reached a plateau around 80 ohm·cm2. Dy677-labeled scrambled siRNA–loaded PEG-NPs or PS 80 (H)-NPs suspended in medium were added on the apical side. Following 4-hour incubation, aliquots from the basolateral side of the inserts were collected, and the amount of NPs that crossed the cell layer into the basolateral compartment was quantified by fluorescence. Compared to PEG-NPs, PS 80 (H)-NPs showed significantly higher penetration across the cell layer (fig. S9B), which is consistent with in vivo brain accumulation results. However, when the same experiment was performed in the absence of serum in the culture medium, penetration of PS 80 (H)-NPs reduced significantly (fig. S9C), which suggests that certain serum proteins play a crucial role in the transport of PS 80 (H)-NPs across BBB. It has been reported that PS 80 can adsorb endogenous lipoprotein, such as apolipoprotein E (apoE) or apoA-I to facilitate the transport of NPs across BBB via lipoprotein receptor–mediated transcytosis (30–32). To confirm whether lipoprotein plays a role in mediating the penetration of PS 80 (H)-NPs across BBB, we blocked the lipoprotein receptor [lipoprotein receptor-related protein 1 (LRP1)] by anti-LRP1 antibody and observed a significant decrease in the permeability of PS 80 (H)-NPs (fig. S9C). Our finding is consistent with the previous in vitro and in vivo reports, which have demonstrated that PS 80 coated on NPs can bind to the endogenous apolipoproteins to promote their BBB penetration (30–32).
In vivo circulation profiles of different siRNA formulations were also compared (fig. S10). A longer circulation time allows NPs to have a greater tendency to accumulate in the target organ. Dy677-labeled free siRNA or Dy677-labeled siRNA-loaded PEG-NPs or PS 80 (H)-NPs were injected into mice via tail vein at a siRNA dose of 50 nmol/kg. Blood was withdrawn at the indicated time points for the measurement of fluorescence signal. Free siRNA was eliminated rapidly from the blood and was hardly detected 30 min after injection. PEG-NPs significantly prolonged the circulation time of siRNA in blood, which could be explained by the protection and stealth effect of the PEG layer (42, 43). PS 80 (H)-NPs, which did not have the protective PEG layer on the surface, demonstrated a similar circulation profile to PEG-NPs. This long circulation time is presumably attributable to the hydrophilicity and nonionic structure of PS 80 that reduces the uptake of NPs by the reticuloendothelial system (44). Because PS 80 (H)-NPs showed maximum in vitro gene silencing efficiency, maximum penetration across intact BBB, and long circulation time in blood, we decided to evaluate them further in a mouse model of TBI.
PS 80 (H)-NPs exhibit BBB pathophysiology–independent delivery of siRNA in a mouse model of TBI
Because PS 80 (H)-NPs showed superior in vitro gene silencing and BBB penetration in healthy mice, as compared to GSH-NPs, at coating densities evaluated in this work, we selected PS 80 (H)-NPs for further evaluation in TBI mice. We used the weight drop–induced TBI model (Fig. 4A), which is a clinically relevant murine model of TBI (27, 28). Briefly, mice were anesthetized and subjected to head impact by dropping a 54-g weight from a 60-inch height. We first characterized the time window of physically breached BBB following TBI using Evans blue (EB) penetration assay. EB dye binds to serum albumin in the bloodstream and does not cross the intact BBB under normal physiological conditions but permeates physically breached BBB (45). Healthy mice or mice with TBI were injected with EB via tail vein. In the case of TBI mice, EB was injected at different time points after injury. Two hours after EB administration, animals were perfused extensively with saline, and EB content in brain tissue was quantified. EB dye injected to TBI mice at 6 and 24 hours after injury resulted in significantly higher EB content compared to healthy mice (Fig. 4B). EB dye injected 1 week after injury and later resulted in substantially less brain accumulation compared to 24 hours, indicating a decrease in EB permeability. Collectively, our data suggest that weight drop–induced TBI resulted in physical breaching of BBB that increased over time, until at least 24 hours after injury. BBB was significantly repaired by 1 week, with complete recovery at 2 weeks after injury (Fig. 4B).
Fig. 4. PS 80 (H)-NPs exhibit BBB pathophysiology–independent delivery of siRNA in a mouse model of TBI.
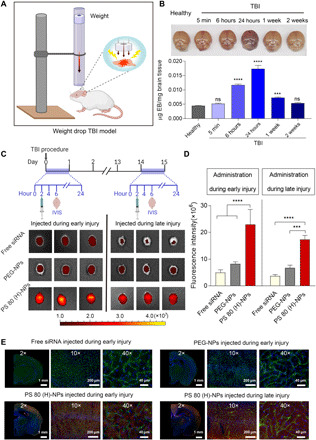
(A) Schematic showing the experimental procedure for the weight drop–induced TBI model. (B) Time window of physically breached BBB following TBI was characterized by Evans blue (EB) penetration assay. EB was intravenously injected into healthy mice or mice with TBI at different time points after injury. Two hours after EB administration, brains were excised and photographed. EB content in brain tissue was also quantified and expressed as micrograms of EB per milligram of brain tissue. ***P < 0.001, ****P < 0.0001 compared to healthy mice. (C) Experimental outline: Free siRNA, siRNA-loaded PEG-NPs, or PS 80 (H)-NPs were intravenously injected into mice after 2 hours or 2 weeks of TBI procedure, corresponding to early and late injury, respectively. Four hours after the administration of siRNA formulations, brains were excised and imaged using IVIS. Dy677-labeled scrambled siRNA was used at a dose of 50 nmol/kg. IVIS images of brains from three representative healthy mice injected during early or late injury are shown. (D) Fluorescence intensity measured over excised mice brains using IVIS. ***P < 0.001, ****P < 0.0001. (E) Representative fluorescence microscopy images of brain sections from mice injected intravenously with free siRNA, siRNA-loaded PEG-NPs, or PS 80 (H)-NPs during early or late injury. Dy677-labeled scrambled siRNA (red signal) was used. Nuclei were stained with Hoechst 33342 (blue signal), and blood vessels were labeled with fluorescein isothiocyanate lectin (green signal). Data in (B) and (D) are means ± SD (n = 3 mice per group, experiment performed twice). P values were determined by one-way ANOVA with Tukey’s post hoc analysis.
Next, we investigated the penetration of Dy677-labeled siRNA-loaded PS 80 (H)-NPs in the weight drop–induced TBI model (Fig. 4C). Free siRNA and siRNA-loaded PEG-NPs were used as control. To characterize penetration across physically breached BBB during early injury, siRNA-loaded NPs or free siRNA at a scrambled siRNA dose of 50 nmol/kg was injected at 2 hours after injury. Brains were collected for IVIS imaging at 4 hours after injection (Fig. 4C). Weak fluorescence signal was detected in the brains of mice injected with free siRNA or siRNA-loaded PEG-NPs, indicating low brain accumulation (Fig. 4, C and D). By contrast, we observed a strong fluorescence signal in the brains of mice injected with siRNA-loaded PS 80 (H)-NPs, with a fivefold and threefold higher fluorescence intensity when compared with free siRNA and siRNA-loaded PEG-NP groups, respectively (Fig. 4, C and D). To evaluate siRNA delivery across intact BBB during late injury, free siRNA or siRNA-loaded NPs were administered intravenously 2 weeks after injury, when BBB was self-repaired (Fig. 4C). Again, mice administered with siRNA-loaded PS 80 (H)-NPs displayed the highest fluorescence signal in the brain (Fig. 4, C and D). These results indicate that PS 80 (H)-NPs can efficiently deliver siRNA into the brain, when administered during early or late injury period after TBI. This holds great potential for down-regulating pathological targets involved in the secondary brain injury, a process that usually lasts weeks to months. To specifically localize the delivered siRNA within brain tissue, blood vessels were labeled with fluorescein isothiocyanate (FITC)–lectin by injecting it into mice via tail vein 10 min before euthanasia. Brains from mice were sectioned and observed with a fluorescence microscope. There was nearly no detectable siRNA fluorescence signal in the brain of mice injected with free siRNA or siRNA-loaded PEG-NPs (Fig. 4E). However, in the brain sections from mice treated with siRNA-loaded PS 80 (H)-NPs, we observed substantial red fluorescence signal from Dy677-labeled siRNA in the cortex (Fig. 4E). Notably, we observed minimum colocalization of red fluorescence signal from Dy677-labeled siRNA with green fluorescence signal from FITC-lectin–stained brain vessels. This indicates that most NPs accumulated in the extravascular brain tissue, which confirms the ability of PS 80 (H)-NPs to cross brain vasculature. Similarly, NPs administered at 2 weeks after injury also showed penetration into the extravascular brain tissue.
Tau siRNA–loaded PS 80 (H)-NPs suppress tau expression in murine primary neuronal cells
Among various harmful pathways, tau pathology has been found to be highly associated with chronic neuroinflammation, neurodegeneration, and cognitive impairment caused by TBI (46–48). Tau is a microtubule-associated protein found mostly in neurons. Following TBI, tau becomes hyperphosphorylated and dissociates from microtubules, forming abnormal aggregates that are strongly linked to TBI-associated chronic traumatic encephalopathy and Alzheimer’s disease (27, 46–48). Prior studies showed that the reduction of tau protein attenuated its aggregation by decreasing the amount of tau available for phosphorylation (48–50), representing potential interventions for TBI treatment. We therefore sought to investigate whether PS 80 (H)-NPs could efficiently deliver tau siRNA into the brains of TBI mice to suppress tau expression.
Tau silencing ability of tau siRNA–loaded PS 80 (H)-NPs was first evaluated in vitro in primary neurons of mice. Unlike cell lines, primary neurons maintain certain phenotypic features and functions of nervous systems, thereby representing a more relevant in vitro model for neuroscience research. The primary cortical neurons were prepared from freshly isolated mouse embryonic cortical tissues. After 10 days of culture, primary neurons displayed a high degree of neurite outgrowth and branching (Fig. 5A). At that point, neurons were treated for 24 hours with medium only or medium containing free tau siRNA, tau siRNA–loaded PEG-NPs, tau siRNA–loaded PS 80 (H)-NPs, or scrambled (control) siRNA–loaded PS 80 (H)-NPs at 15 nM concentration of siRNA. Following an additional 48-hour incubation with medium only, Western blot analysis was performed to measure the expression of tau in primary neurons. Tau siRNA–loaded PS 80 (H)-NPs markedly down-regulated tau expression in neurons with an approximately 70% decrease of tau expression (Fig. 5B). In contrast, treatment with free tau siRNA, tau siRNA–loaded PEG-NPs, or control siRNA-loaded PS 80 (H)-NPs resulted in negligible reduction of tau expression (Fig. 5B). Tau knockdown by tau siRNA–loaded PS 80 (H)-NPs was also shown to be dose dependent (Fig. 5C). Inhibition of tau was further confirmed by immunofluorescence imaging (Fig. 5D). Compared with all the other groups, cells treated with tau siRNA–loaded PS 80 (H)-NPs showed much weaker green fluorescence, suggesting lower expression levels of tau protein.
Fig. 5. Tau siRNA–loaded PS 80 (H)-NPs suppress tau expression in murine primary neuronal cells.
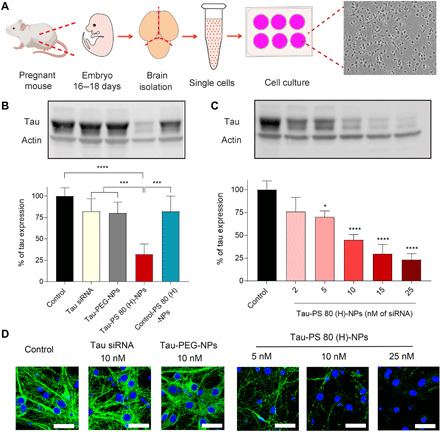
(A) Schematic illustration depicting the isolation of primary neuronal cells from mouse embryos and bright-field image of primary cells after 10 days in culture. Pregnant female C57BL/6 mice at E18 were euthanized, and the cortex was dissected from embryo brains and enzymatically digested to obtain single cell suspension. Last, cells were cultured on plates precoated with poly-l-lysine. (B) Western blots and quantification of tau expression in primary neuronal cells. Cells were incubated for 24 hours with only medium (control) or medium containing free tau siRNA (Tau siRNA), tau siRNA–loaded PEG NPs (Tau-PEG-NPs), tau siRNA–loaded PS 80 (H)-NPs [Tau-PS 80 (H)-NPs], or scrambled siRNA–loaded PS 80 (H)-NPs [Control-PS 80 (H)-NPs]. The dose of siRNA was 15 nM. Following an additional 48-hour incubation with medium only, Western blot analysis was performed. ***P < 0.001, ****P < 0.0001. (C) Western blot and quantification of tau expression in primary neuronal cells treated with tau siRNA–loaded PS 80 (H)-NPs at different concentrations of siRNA. *P < 0.05, ****P < 0.0001 compared to control. (D) Immunofluorescence images of tau expression in primary neuronal cells. Cells were treated for 24 hours with medium only (control) or medium containing free tau siRNA (10 nM siRNA), tau-PEG-NPs (10 nM siRNA), or tau-PS 80 (H)-NPs (5, 10, or 25 nM siRNA). Following an additional 48-hour incubation with medium only, immunofluorescence staining was performed. Nuclei were stained with Hoechst 33342 (blue signal), and tau was stained with the anti-tau primary antibody followed by Alexa Fluor 488–labeled secondary antibody (green signal). Scale bars, 30 μm. Data in (B) and (C) are means ± SD of technical repeats (n = 3, experiment performed twice). P values were determined by one-way ANOVA with Tukey’s post hoc analysis.
Tau siRNA–loaded PS 80 (H)-NPs silence tau expression during early and late injury phases with no systemic toxicity
We next investigated the tau silencing ability of tau siRNA–loaded PS 80 (H)-NPs in the weight drop–induced model of TBI (Fig. 6, A and B). To investigate tau silencing during the early injury phase, mice were intravenously administered with PBS, free tau siRNA, or PS 80 (H)-NPs loaded with either scrambled (control) siRNA or tau siRNA at a siRNA dose of 75 nmol/kg per day. Treatments were administered at 2 hours and 1 day after injury. On day 4, brains were harvested, and the cortex was isolated and processed for analysis of tau expression using Western blotting. Free siRNA treatment resulted in negligible tau knockdown, whereas tau siRNA–loaded PS 80 (H)-NPs markedly reduced the expression of tau by about 50% (Fig. 6A). Scrambled siRNA–loaded PS 80 (H)-NPs did not reduce tau expression. Even when administered during the late injury period, i.e., 2 weeks after the injury, tau siRNA–loaded PS 80 (H)-NPs could block around 40% tau expression in the cortex (Fig. 6B). Immunohistochemical staining analysis further confirmed the robust tau silencing ability of tau siRNA–loaded PS 80 (H)-NPs, when administered during the early or late injury period (Fig. 6C).
Fig. 6. Tau siRNA–loaded PS 80 (H)-NPs silence tau expression in vivo during early and late injury.
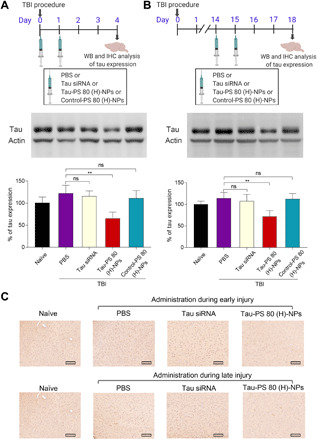
(A) Experimental outline: To evaluate tau silencing efficiency during early injury, mice received tail vein injection with PBS, free tau siRNA (Tau siRNA), tau siRNA–loaded PS 80 (H)-NPs [Tau-PS 80 (H)-NPs], or scrambled siRNA–loaded PS 80 (H)-NPs [Control-PS 80 (H)-NPs] at 2 hours and 1 day after injury. siRNA dose was 75 nmol/kg per day. Brains were dissected to isolate cortex on day 4 for quantification of tau expression using Western blot. Representative Western blots and quantification of tau expression are shown. Naïve animals were healthy mice with no treatment. **P < 0.01. (B) Experimental outline: To evaluate tau silencing efficiency during late injury, mice received tail vein injection of PBS, tau siRNA, tau-PS 80 (H)-NPs, or control-PS 80 (H)-NPs at days 14 and 15 after injury. siRNA dose was 75 nmol/kg per day. Brains were harvested on day 18 for quantification of tau expression using Western blot. Representative Western blot analysis and quantification of tau expression were shown. **P < 0.01. (C) Immunohistochemical staining of tau expression in brain tissue sections of naïve mice (healthy mice with no treatment) or TBI mice treated with PBS, tau siRNA, or tau-PS 80 (H)-NPs. Treatments were performed during the early or late injury phase at a siRNA dose of 75 nmol/kg per day. Scale bars, 150 μm. Data in (A) and (B) are means ± SD (n = 3 mice per group, experiment performed twice). P values were determined by one-way ANOVA with Tukey’s post hoc analysis.
We also evaluated the in vivo safety profile and systemic toxicity of PS 80 (H)-NPs. Healthy mice were injected with PBS or PS 80 (H)-NPs loaded with scrambled (control) siRNA or tau siRNA at a siRNA dose of 75 nmol/kg per day for two consecutive days. None of the treatment groups showed significant body weight reduction over 2 weeks after injection (fig. S11). In addition, hematoxylin and eosin (H&E) staining did not show any notable pathological changes in major organs, including lung, heart, liver, spleen, and kidney, after 3 days of injection (fig. S12). Hematology markers for mice that received PS 80 (H)-NPs loaded with tau siRNA or scrambled siRNA were within normal range and not significantly different from the PBS-injected group at 3 days and 2 weeks after the last injection (fig. S13). Last, multiple blood biochemical parameters, including alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine, and blood urea nitrogen (BUN), were evaluated for assessing effects on liver and kidney (fig. S13). All biochemical parameters for mice injected with siRNA-loaded NPs were not significantly different from PBS-injected mice. These results collectively indicate the safety of PS 80 (H)-NPs as siRNA nanocarriers.
DISCUSSION
To date, more than 30 clinical trials have investigated treatment options for TBI; however, none have successfully proceeded to phase 3 trials (51). siRNA represents a powerful therapeutic tool against TBI, yet the utility of siRNA for TBI treatment is hindered by multiple barriers, especially because of its inability to cross BBB. Direct injection of siRNA into brain tissue has been explored to bypass BBB (52), and few other efforts focused on the use of chemical/biological agents or physical stimuli to artificially induce the opening of BBB for siRNA transport. Although effective, these methods are either invasive or nonselective, which may add further complications to the injury. Recently, several NP platforms have been explored to deliver therapeutics across BBB for TBI treatment (14–16). However, these NPs usually rely on injury-induced physical breaching of BBB for brain accumulation, which may lead to highly variable therapeutic response due to the heterogeneous nature of physical breaching of BBB in TBI. Moreover, because physical breaching of BBB is transient, these approaches necessitate administration of the treatment within an extremely narrow time window after TBI, which is difficult to achieve in practice and has been an important contributing factor in the failure of several previous clinical trials (52, 53). In addition, because secondary injury lasts for months, repeated treatments beyond the transient window of physically breached BBB might be required. Hence, NPs that do not rely on BBB pathophysiology for brain accumulation are highly desired for reliable and long-term treatment of TBI.
This is the first reported example of BBB pathophysiology–independent delivery of siRNA-loaded NPs in TBI. We achieved this by combined modulation of surface coating chemistry and coating density, which maximized the active transport of NPs across BBB. While modulation of surface coating chemistry alone has been previously shown to affect BBB penetration of NPs, herein, we demonstrate that combined modulation of surface coating chemistry along with coating density can further augment the active penetration of NPs through BBB. Our initial screening studies in healthy mice showed that at coating densities evaluated in this work, PS 80 (H)-NPs showed the best penetration across intact BBB and the best neural cell uptake compared to other surface coatings and coating densities. Penetration of PS 80 (H)-NPs across intact, undamaged BBB in healthy mice was also a strong and direct evidence of their ability to penetrate across the fully repaired BBB after TBI. This motivated us to further evaluate PS 80 (H)-NPs in TBI mice, which showed significant brain accumulation of PS 80 (H)-NPs when administered during early or late injury periods. Intravenous injection of PS 80 (H)-NPs, within or outside the window of physically breached BBB in TBI mice, resulted in threefold higher brain accumulation compared to conventional PEGylated NPs. Fluorescence microscopy images of brain sections in our study revealed that PS 80 (H)-NPs accumulate primarily in the cortex, which has been previously shown to play a crucial role in TBI pathogenesis (51). Although the mechanism of this preferential distribution is unknown at this time, the topic could potentially shed light on improving therapeutic targeting for TBI and merits more exploration in future studies.
PS 80 is a nonionic surfactant that has been widely used in pharmaceutical and food industries. PS 80–coated NPs have been previously used to promote drug delivery into brain for multiple applications, including glioblastoma (32, 44). However, it is difficult to directly compare brain accumulation observed in our study with literature owing to the differences in particle type, drug type, and disease/animal model (21). Nevertheless, to our knowledge, the use of PS 80–coated NPs for brain delivery of siRNA in TBI treatment has never been explored previously. In addition, we demonstrated that PS 80 coating density is a critical parameter to maximize penetration of PS 80-coated NPs across BBB.
Compared to protein or peptide-based approaches to augment BBB penetration, PS 80 has the following advantages: low cost, high stability, and low immunogenicity. PS 80-NPs developed here also demonstrated small size (about 60 nm), high siRNA encapsulation efficiency, and long circulation profile, comparable to PEG-NPs. Moreover, they were prepared by a robust one-step method, which greatly enhances the platform’s potential for large-scale manufacturing and clinical translation. These features collectively suggest that PS 80 could serve as a promising tool for brain delivery of siRNA for TBI treatment.
As a proof of concept, we investigated the efficacy of PS 80 (H)-NPs to silence tau expression after TBI. Tau pathology is strongly linked to the TBI-caused neurodegeneration and brain dysfunction (27, 46–48). Therefore, therapies that reduce tau level or inhibit the phosphorylation or aggregation of tau represent potential interventions for TBI treatment. Although a few small-molecule drugs have been reported to reduce tau level (54, 55), they can potentially interact with other unintended and unknown targets. siRNA is a promising strategy to modulate tau levels because of its high efficacy and specificity. siRNA-mediated reduction of tau protein has been shown to attenuate tau aggregation and neuronal death by decreasing the amount of tau available for phosphorylation (49, 50). However, to our knowledge, systemic delivery of tau siRNA has not been investigated for the treatment of TBI. We showed that tau siRNA–loaded PS 80 (H)-NPs not only could markedly knock down tau expression in cultured primary neural cells but also achieved 40 to 50% tau silencing in TBI mice, when administered within or outside the window of physically breached BBB, which correspond to early and late injury, respectively. This opens up exciting possibilities in TBI research by enabling long-term, noninvasive mitigation of harmful pathways.
There are some limitations in our study. First, although we clearly demonstrated the significance of surface coating density to improve BBB penetration of NPs, we have limited our experiments to three different coating densities of PS 80 and GSH. Future studies could explore a wider range of coating densities to further establish the extent to which BBB penetration can be enhanced. Second, because the concentrations of PS 80, as chosen based on the literature to prepare PS 80-NPs, were higher than the concentrations of GSH, the resulting coating densities for PS 80 ended up being higher than GSH. Future studies could evaluate higher coating densities for GSH or compare GSH and PS 80 at equal coating densities. Third, although we only proceeded with PS 80 (H)-NP formulation for evaluation in TBI mice, we acknowledge that GSH-NPs also showed substantial accumulation across intact BBB and merit further exploration. Last, due to the complexity of TBI pathobiology, there is as of yet a limited understanding of the pathways that lead to neurobehavioral sequelae of TBI. One of these pathways almost certainly involves tau protein, which we have chosen as the target for our proof-of-concept study. However, suppressing tau pathology alone may not be sufficient to result in a significant change in functional outcomes. For this reason, we have not included functional or behavior evaluations in this study. Future studies could explore siRNA against additional pathobiological pathways, such as cell apoptosis, neuroinflammation, edema, calcium overload, or coagulation, as well as larger animal models of TBI (such as rabbit or pig) and evaluate more clinically relevant therapeutic benefits, including functional outcomes and performance in behavioral tasks. Moreover, because siRNAs against other targets could be readily incorporated into this NP platform, it also presents a potential research tool for the validation and modulation of previously unidentified therapeutic targets for neurological diseases, particularly those that are considered as “undruggable” targets.
In conclusion, our work elucidates a clinically relevant approach for developing siRNA therapeutics to prevent the long-term effects of TBI. In addition, we demonstrate that combined modulation of surface chemistry and coating density can be an efficient tool to tune BBB penetration of NPs. Our NP approach also increases the potential of siRNA as a therapeutic tool against other neurological diseases, such as Alzheimer’s and Parkinson’s diseases, and therefore holds great promise for the design and translation of precision therapeutics for brain disorders.
MATERIALS AND METHODS
Experimental design
The objective of this study was to develop an NP platform for BBB pathophysiology–independent delivery of siRNA in TBI. To achieve this, we first modulated surface coating chemistry and coating density to maximize the active penetration of siRNA-loaded NPs across intact BBB. Specifically, we formulated siRNA-loaded NPs using PLGA and studied the impact of different surface coatings and their coating densities on the cellular uptake of siRNA-loaded NPs and gene silencing efficiency in Neuro-2a cells, as well as the penetration of NPs across intact BBB in healthy mice. Next, we evaluated the in vivo brain accumulation of the optimal NPs, when administered during early or late injury periods in a murine model of weight drop–induced TBI. Last, we assessed the in vitro gene silencing efficiency of the optimal NPs against tau protein in murine primary neuronal cells, followed by in vivo evaluation of gene silencing efficiency in the weight drop–induced model of TBI.
Synthesis of DSPE-PEG-GSH and DSPE-PEG-Tf
Tf (Sigma-Aldrich) was first thiolated using 2-iminothiolane (Traut’s reagent) as previously described (56). Thiolated Tf was then immediately used for conjugation with DSPE-PEG-maleimide with PEG molecular weight 3400 (DSPE-PEG-MAL) (NANOCS) via thiol-Michael addition reaction (56, 57). DSPE-PEG-Mal was first dissolved in dry DMF, and then 10-fold molar excess of DSPE-PEG-Mal solution was added to the Tf solution. The reaction proceeded in PBS buffer (pH 7.0) for 2 hours at room temperature (RT) under mild stirring. Similarly, GSH (Sigma-Aldrich) and DSPE-PEG-MAL were incubated at a 2:1 molar ratio for 2 hours at RT (34). DSPE-PEG-Tf or DSPE-PEG-GSH was purified by dialysis.
Preparation and characterization of NPs coated with different surface chemistries
siRNA-loaded PLGA NPs with different surface coatings were prepared by a modified nanoprecipitation method. Briefly, the organic phase was prepared by mixing 20 μl of siRNA (4 nmol in water) with 1 ml of PLGA (Durect Corporation) (5 mg/ml in acetone, DMF, or tetrahydrofuran) and 200 μl of the cationic lipid-like molecule (5 mg/ml), which was synthesized by reacting the ethylenediamine core PAMAM generation 0 dendrimer (Sigma-Aldrich) and 1,2 epoxytetradecane (Sigma-Aldrich) using a ring opening reaction, as described previously (58). The siRNA sequences used in this study include the following: luciferase siRNA: 5′-CUU ACG CUG AGU ACU UCG AdTdT-3′ (sense strand) and 5′-UCG AAG UAC UCA GCG UAA GdTdT-3′ (antisense strand); tau siRNA: 5′-CCU AGA AAU UCC AUG ACG AUU-3′ (sense strand) and 5′-UCG UCA UGG AAU UUC UAG GUU-3′ (antisense strand); scrambled siRNA: 5′-UUC UCC GAA CGU GUC ACG UUU-3′ (sense strand) and 5′-ACG UGA CAC GUU CGG AGA AUU-3′ (antisense strand); Cy3 or Dy677-labeled siRNAs were synthesized by labeling the 5′-end of both the sense and antisense strands of scrambled siRNA with Cy3 and Dy677. Under vigorous stirring, the mixture was added slowly into a 15-ml aqueous solution. Various coating materials were added to either the organic phase or water phase to endow the surface of NPs with different coatings. For PEG-NPs, the organic phase contained DSPE-PEG (2 mg/ml) (PEG molecular weight 3000) (Avanti Polar Lipids). For GSH-NPs, 5, 10, or 25 mole percent (mol %) of DSPE-PEG was replaced with DSPE-PEG-GSH to yield GSH (L)-NPs, GSH (M)-NPs, and GSH (H)-NPs, respectively. For Tf-NPs, 10 mol % of DSPE-PEG was replaced with DSPE-PEG-Tf. For F-68-NPs, the organic phase contained DSPE-PEG (1 mg/ml), and the water phase contained Pluronic F-68 (1 mg/ml) (Sigma-Aldrich). For PS 80 (L)-NPs, the organic phase contained DSPE-PEG (1.5 mg/ml), and water phase contained PS 80 (0.5 mg/ml) (Sigma-Aldrich); for PS 80 (M)-NPs, the organic phase contained DSPE-PEG (1 mg/ml), and water phase contained PS 80 (1 mg/ml); for PS 80 (H)-NPs, the water phase contained PS 80 (2 mg/ml). Concentrations of surface coating materials were based on previously published reports (31, 34, 35). PS 80-NPs and GSH-NPs used for evaluating the impact of surface coating chemistry on cellular uptake, gene silencing, and BBB penetration in healthy mice were formulated with medium coating densities of PS 80 and GSH. NPs with medium coating densities of PS 80 or GSH were prepared by using their respective solutions at concentrations that have been previously used to formulate BBB-penetrating NPs. Lower and higher concentrations compared to the one chosen from literature were then used to prepare NPs with low and high coating densities of PS 80 and GSH. The resulting NP dispersions were transferred to centrifuge filters with a 300-kDa molecular weight cutoff (MWCO) (Sartorius Vivaspin) and collected via centrifugation and washed with cold water three times. Last, they were dispersed in PBS. Particle size distribution and surface charge of NPs were measured by DLS (Brookhaven Instruments Corporation). NPs were stained with 1% uranyl acetate, and their morphologies and size were observed under the Tecnai G2 Spirit BioTWIN transmission electron microscope (FEI Company).
To determine siRNA encapsulation efficiency, Cy3-labeled scrambled siRNA–loaded NPs were prepared as described above. Five microliters of NP solution was removed and mixed with 95 μl of dimethyl sulfoxide (DMSO). Fluorescence intensity was analyzed using a Synergy HT Multi-Mode Microplate Reader (BioTek Instruments). Release kinetics of siRNA from NPs was also evaluated using Cy3-labeled scrambled siRNA–loaded NPs. Suspension of NPs in PBS was placed in dialysis tubing (float-a-lyzer G2 dialysis device, MWCO 100 kDa, Spectrum Labs) and dialyzed against PBS (pH 7.4) at 37°C with a shaking speed of 150 rpm. At different time points, an aliquot of the NP suspension was withdrawn and dissolved in DMSO, and the fluorescence intensity of siRNA was analyzed. To determine PS 80 coating density on PS 80–coated NPs, PS 80-NP suspension was added to DMSO in a 1:10 ratio to dissolve the NPs and the concentration of PS 80 in the solution was analyzed using a high-performance liquid chromatography evaporative light scattering detector (Agilent 1260 Infinity II; Column: Zorbax 300SB-C18, 3 × 150 mm, 3.5 μm; flow rate: 1 ml/min; PS 80 peak elution time: 7.8 min in a water-acetonitrile gradient). To determine GSH coating density on NPs, GSH-NP suspension was concentrated by centrifugation and added to methanol in a 1:9 ratio. Concentration of GSH in the solution was analyzed using the Micro BCA (bicinchoninic acid) Protein Assay Kit (Thermo Fisher Scientific). Briefly, GSH was quantified from the dissolved GSH-NPs solution following 30-min incubation with BCA reagents at 37°C, in accordance with the manufacturer’s protocol. All analyses were performed in triplicate.
Cell culture
Neuro-2a cells (ATCC CCL-131) were purchased from the American Type Culture Collection (ATCC). Cells were maintained in Eagle’s minimum essential medium (EMEM) (ATCC) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin-streptomycin antibiotic (Thermo Fisher Scientific). Luciferase-expressing Neuro-2a cells were generated by the transduction of cells with luciferase expression vector. The lentiviral vector pLenti CMV Puro LUC encoding firefly luciferase was transfected with ViraPower Lentiviral Packaging Mix to 293T cells (ATCC CRL-3216) using Lipofectamine 2000 (Invitrogen). After 48 hours, lentiviral supernatant was collected and added into 20 to 40% confluent Neuro-2a cells. Polybrene (8 μg/ml) was added during the transduction. Two days after the transduction, Neuro-2a cells were selected by puromycin (2 μg/ml) and were maintained in EMEM containing 10% FBS and puromycin (1 μg/ml). All cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 in a cell culture incubator.
Cellular uptake and endosomal escape of NPs
For cellular uptake study, Neuro-2a cells were seeded into the eight-well chambered coverglass (Nunc Lab-Tek Chambered Coverglass, Thermo Fisher Scientific) at a density of 15,000 cells per well. Cells were allowed to attach for 24 hours. After that, cells were incubated with Dy677-labeled scrambled siRNA (free) or Dy677-labeled siRNA-loaded NPs having different surface coatings or coating densities. siRNA concentration was 15 nM. After incubation at 37°C for 2 hours, cells were washed twice with cold PBS buffer. Nuclei of cells were stained with Hoechst 33342 (Sigma-Aldrich) (1 μg/ml), and cells were imaged using a CLSM (Olympus, FV1200). For endosomal escape, cells were incubated according to the procedures described above, except that before nuclear staining, cells were treated with green LysoTracker Green (Life Technologies) at a final concentration of 60 nM for 30 min at 37°C. Cells were then stained with Hoechst 33342 and observed under a CLSM.
Uptake of NPs by Neuro-2a cells was quantified by flow cytometry. Neuro-2a cells were seeded in six-well plates at 50,000 cells per well. After incubation at 37°C for 2 days, cells were treated with Cy3-labeled scrambled siRNA (free) or Cy3-labeled siRNA-loaded NPs having different surface coatings or coating densities at final siRNA concentration of 15 nM. After 2 hours, cells were washed twice with PBS buffer and then harvested by trypsin treatment for flow cytometry quantitative analysis (BD Biosciences FACS flow cytometer).
In vitro gene silencing efficiency of NPs
Luciferase-expressing Neuro-2a cells were seeded in 96-well plates at 5000 cells per well. Cells were incubated at 37°C in a CO2 incubator overnight followed by treatment with luciferase siRNA–loaded NPs having different surface coatings and coating densities at a final siRNA concentration of 0, 5, 10, and 25 nM. As controls, different concentrations of free luciferase siRNA or luciferase siRNA complexed with Lipofectamine 2000 (Invitrogen) were used. siRNA-Lipo2K was prepared according to the manufacturer’s suggested protocols. Medium was replaced after 24 hours of incubation, and cells were continuously cultured in medium only for another 48 hours. Cell viability was determined by quantifying cellular metabolic activity using alamarBlue assay (Thermo Fisher Scientific). Metabolic activity for each group was normalized to the metabolic activity of cells treated with medium only. Steady-Glo Luciferase Assay Kit (Promega) was used to measure the expression of luciferase in cells. Luciferase expression for each group was normalized to the luciferase expression of cells treated with medium only. Fluorescence intensity (for alamarBlue assay) and luminescence (for luciferase assay) of cells were recorded using a microplate reader.
In vitro BBB penetration of NPs
bEnd.3 cells (ATCC) were grown on 1% gelatin-coated flasks at 37°C, 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS and 1% penicillin-streptomycin. Then, the cells were seeded on 2% growth factor–reduced Matrigel-coated 12-mm inserts with 0.4-μm pore polycarbonate membrane (Corning Transwell, New York, NY) at a density of 80,000 cells per well. Medium was replaced every 2 to 3 days. TEER was measured using an epithelial volt/ohm meter resistance meter (World Precision Instruments, Sarasota, FL). We tracked TEER for the cell monolayer over time until the TEER reached a plateau around 80 ohm·cm2 after 1 week, at which point permeability experiments were performed. Medium containing Dy677-labeled scrambled siRNA–loaded NPs with PEG or PS 80 (H) coating was added to the apical compartment. After 4 hours, filter inserts were withdrawn from the receiver compartment. Aliquots from the basolateral compartments were collected and fluorescence was quantified using an Infinite 200 PRO plate reader (Tecan). Empty filters without cells were used as control to determine the 100% penetration of NPs. Penetration of Dy677-labeled siRNA-loaded PS 80 (H)-NPs was also studied in the absence of serum in the cell culture medium. Anti–low-density LRP1 antibody (CST, Danvers, MA) was further used to block the lipoprotein receptor. Both compartments of the insert were incubated with 200 μM anti-LRP1 antibody in DMEM before NP introduction. After 1 hour, NPs were introduced to the apical compartment. Penetration assay was then performed as described above.
Animals
All in vivo experiments, except the isolation of primary neuronal cells, were performed in 6- to 10-week-old male C57BL/6J mice (The Jackson Laboratory). For isolation of primary neural cells, female, pregnant C57BL/6J mice at E18 were used as donors. Group sizes for each experimental design were determined based on the minimal number of animals needed to achieve a significant difference of P < 0.05 between experimental groups for the primary outcome of the individual experiment using statistical analysis indicated in each figure legend. We randomly selected mice from the cage to assign them to different experimental groups. Experiments were performed in specific pathogen–free animal facilities at Boston Children’s Hospital (BCH). Mice were housed under standard 12-hour light/12-hour dark conditions with ad libitum access to water and chow. Animal care and handling procedures complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal protocols were approved by the Institutional Animal Care and Use Committee at BCH.
Penetration of NPs across intact BBB in healthy mice
Dy677-labeled scrambled siRNA (free) or Dy677-labeled siRNA-loaded NPs having different surface coatings or coating densities were intravenously administered into healthy male C57BL/6 mice via tail vein at a siRNA dose of 50 nmol/kg. At 4 or 24 hours after the injection, mice were perfused with saline and euthanized. Brains were collected for ex vivo imaging using the IVIS spectrum imaging system (Xenogen Imaging Technologies) at excitation and emission wavelengths of 675 and 740 nm, respectively.
Pharmacokinetics of NPs
Healthy male C57BL/6 mice were intravenously injected with Dy677-labeled scrambled siRNA (free) or Dy677 siRNA-loaded PEG-NPs or PS 80 (H)-NPs via tail vein at a siRNA dose of 50 nmol/kg. At predetermined time points, blood was withdrawn from the tail artery and Dy677 siRNA fluorescence signal in blood was measured using a microplate reader (BioTek) at excitation and emission wavelengths of 670 and 710 nm, respectively.
Isolation of primary neuronal cells and tau silencing in vitro
Primary neuronal cells were isolated from the cerebral cortex of mouse embryos. Briefly, pregnant female C57BL/6 mice at E18 were euthanized, the cortex was dissected from embryo brains, and meninges were removed thoroughly. Tissues were collected in a 15-ml conical tube and enzymatically digested at 37°C for 20 min. The obtained cell solution was then passed through a 40-μm cell strainer to remove tissue debris and centrifuged at 300g for 4 min to collect the cells. Last, cells were resuspended in B-27 Plus Neurobasal Medium (Thermo Fisher Scientific) and plated on culture plates precoated with poly-l-lysine (Sigma-Aldrich). Half of the medium was changed every 3 days. After 10 days in culture, primary neuronal cells were incubated for 24 hours with medium only (control) or medium containing free tau siRNA, tau siRNA–loaded PEG-NPs, scrambled (control) siRNA–loaded PS 80 (H)-NPs, or tau siRNA–loaded PS 80 (H)-NPs. Following an additional 48-hour incubation with fresh medium, cells were lysed with lysis buffer (Cell Signaling Technology) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific) for 1 hour on ice. Cell lysate was centrifuged at 14,000 rpm for 10 min, and supernatant was collected for measuring tau protein expression using Western blot analysis. For Western blotting, cell lysates from different treatment groups were resolved on SDS–polyacrylamide gel electrophoresis gels (Thermo Fisher Scientific) and transferred to polyvinylidene difluoride membrane. Blots were blocked with 3% bovine serum albumin (BSA) in TBST (tris-buffered saline with Tween 20) for 1 hour and then incubated with mouse anti-tau (Abcam) and rabbit anti-beta actin (Abcam) primary antibody at 4°C overnight. After further incubation with horseradish peroxidase–conjugated goat anti-mouse immunoglobulin G (IgG) H&L and goat anti-rabbit IgG H&L (Thermo Fisher Scientific) for 1 hour, the protein bands were then visualized using Enhanced Chemiluminescence (ECL) detection reagent (Cell Signaling Technology) and imaged using a Multi-Application Gel Imaging System (Syngene). For quantification of tau expression, bands were analyzed using the Gels tool in ImageJ. The relative expression level of tau (tau/beta actin) was normalized to the control group.
For immunofluorescence staining, primary neuronal cells were incubated with medium only (control) or medium containing free tau siRNA (10 nM siRNA), tau siRNA–loaded PEG-NPs (10 mM siRNA), or tau siRNA–loaded PS 80 (H)-NPs (5, 10, or 25 nM siRNA) for 24 hours. Cells were further cultured in fresh medium for another 48 hours, followed by fixation with 4% paraformaldehyde (PFA) for 10 min at RT. Cells were then permeabilized using 0.1% Triton X-100 and blocked by 1% BSA and 10% goat serum for 1 hour. Cells were then incubated with mouse anti-tau primary antibody (10 μg/ml in 1% BSA) overnight at 4°C, followed by incubation with Alexa Fluor 488 goat anti-mouse IgG H&L (Abcam) (2 μg/ml) for 1 hour at RT in the dark. Nuclei were counterstained with Hoechst 33342. Last, cells were imaged using a CLSM (Olympus, FV1200).
Traumatic brain injury
Mouse model of TBI was established using the weight drop method as reported previously (27). Male C57BL/6 mice were anesthetized using 4% isoflurane in oxygen and placed on a delicate task wiper (Kimwipes). Mice were then held by their tail and their heads were placed directly under a hollow guide tube. A 54-g metal bolt was dropped from a 60-inch height, which delivered an impact to the dorsal aspect of the skull and led to a rotational acceleration of head through Kimwipes. All mice were allowed to recover in room air after injury and no mortality was found.
Assessment of BBB permeability after TBI
BBB permeability of mice following weight drop–induced TBI was assessed by EB penetration assay. EB dye (2% w/v) was intravenously injected into healthy mice or mice with TBI at a dose of 4 ml/kg. Mice with TBI received EB at different time points (5 min, 6 hours, 24 hours, 1 week, and 2 weeks) after injury. Two hours after EB administration, animals were perfused extensively with saline and then euthanized to collect the whole brain. Photos of brains were taken with a digital camera. To quantify the amount of EB in the brain, brain tissue was cut into small pieces and homogenized. EB was extracted from brain homogenate by treatment with 60% trichloroacetic acid. Fluorescence intensity of EB was recorded using a plate reader at an excitation wavelength of 620 nm and an emission wavelength of 680 nm.
Penetration of NPs across BBB in TBI mice
Dy677-labeled scrambled siRNA (free) or Dy677-labeled siRNA-loaded PEG-NPs or PS 80 (H)-NPs were administered intravenously into TBI mice at a siRNA dose of 50 nmol/kg, either at 2 hours or at 2 weeks after injury. At 4 hours after injection, mice were anesthetized and perfused with saline. Brains were harvested and imaged with IVIS Spectrum Imaging System (Xenogen). To specifically localize delivered siRNA within brain tissue, blood vessels were labeled with FITC-lectin (Vector Laboratories) by injecting it into mice via tail vein 10 min before euthanasia (59). Mice were then anesthetized and perfused with saline and 4% PFA. Next, brains were harvested and fixed in 4% PFA. Brains were further equilibrated in 30% sucrose solution and transferred to tissue base mold. After covering the tissue block with optimal cutting temperature compound (Fisher Scientific), base molds containing tissue blocks were immersed into liquid nitrogen until the tissue was frozen completely. Frozen brain tissues were cut into 40-μm sections and counterstained with Hoechst 33342, followed by imaging with a fluorescence microscope.
Tau silencing in vivo
The TBI-inducing weight drop procedure was performed on day 0. To evaluate tau silencing efficiency during early injury, mice received tail vein injection of PBS, free tau siRNA, tau siRNA–loaded PS 80 (H)-NPs, or scrambled (control) siRNA-loaded PS 80 (H)-NPs at a siRNA dose of 75 nmol/kg per day at 2 hours and 1 day after injury. To evaluate tau silencing efficiency during late injury, a separate group of mice received tail vein injections on days 14 and 15 after injury. Three days after the last injection in each injury period, mice were euthanized, and brains were harvested to determine tau expression in the cortex region using Western blot. For 10 mg of cortex tissue, 500 μl of ice-cold lysis buffer was added. Tissues were homogenized, and the homogenate was centrifuged for 20 min at 14,000 rpm at 4°C. The supernatant was carefully collected and analyzed by Western blotting as described above. The relative expression level of tau (tau/beta actin) was normalized to the naïve animal group (healthy mice with no treatment).
For immunohistochemistry staining, harvested brain tissues were subjected to PFA-fixed paraffin-embedded sections. The paraffin-embedded sections (5 μm thick) were deparaffinized with xylene and rehydrated by immersing the slides in a series of different concentrations of alcohol. Following antigen retrieval using Dako target retrieval solution, samples were incubated with Dako peroxidase blocking buffer at RT for 10 min to quench endogenous peroxidase activity. Slides were then incubated with the anti-tau primary antibody solution, followed by incubation with peroxidase-labeled polymer conjugated to secondary antibodies for 30 min. Slides were lastly stained with DAB+ substrate-chromogen solution and hematoxylin and imaged using Aperio digital slide scanner (20×).
In vivo safety
Healthy C57BL/6 mice were intravenously injected with PBS (control) or PS 80 (H)-NPs loaded with scrambled (control) siRNA or tau siRNA at a siRNA dose of 75 nmol/kg per day for two consecutive days. Main organs including lung, heart, liver, spleen, and kidney were collected 3 days after the last dose and fixed in 4% PFA, followed by embedding in paraffin and sectioning for H&E staining. H&E sections (5 μm) were imaged using the Aperio digital slide scanner (20×). In addition to H&E staining, the hematology markers and multiple biochemical parameters in blood were also evaluated. Specifically, 3 days or 2 weeks after the last dose, mice were euthanized, and blood was drawn by a retro-orbital puncture. One milliliter of blood was collected in the Microvette blood collection tube and analyzed within 1 hour using an automated hematology analyzer. Hematological parameters examined in this study included white blood cells, red blood cells, hemoglobin (HGB), hematocrit, mean corpuscular HGB, platelets, mean platelet volume, red cell distribution width, mean corpuscular HGB concentration, and mean corpuscular volume. For biochemical analysis, serum was collected by the Microvette serum gel tube and analyzed for ALP, ALT, AST, creatinine, and BUN.
Statistics
Statistical analysis and graphing were done with GraphPad Prism. The two-tailed Student’s t test was used to compare two experimental groups, and one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis was used for comparing more than two groups. A value of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Institutes of Health grant HL095722 (to J.M.K.) and Fundação para a Ciência e a Tecnologia through MIT-Portugal-TB/ECE/0013/2013 (to J.M.K.). This work was also conducted with the support of the Football Players Health Study at Harvard (to R.M.). The Football Players Health Study is funded by a grant from the National Football League Players Association. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Medical School, Harvard University or its affiliated academic health care centers, the National Football League Players Association, Boston Children’s Hospital, or the Brigham and Women’s Hospital. Author contributions: W.L., J.Q., R.M., J.M.K., and N.J. conceived the study and designed experiments. W.L., J.Q., X.-L.L., S.A., J.Z., G.C., J.X., J.J., and H.L. performed the experiments and analyzed the data with N.J. and J.M.K. W.L. crafted all the figures and wrote the manuscript. X.-L.L. performed all the statistical analysis. R.L., R.M., J.M.K., and N.J. edited and revised the manuscript and supervised the research. Competing interests: J.M.K. has been a paid consultant and or equity holder for multiple biotechnology companies (listed here: https://www.karplab.net/team/jeff-karp). The interests of J.M.K. were reviewed and are subject to a management plan overseen by his institutions in accordance with its conflict of interest policies. N.J., J.M.K., W.L., R.M., J.Q., and R.L. have one unpublished patent based on the nanoparticle work presented in this manuscript. For a list of entities with which R.L. is involved, compensated or uncompensated, see www.dropbox.com/s/yc3xqb5s8s94v7x/Rev%20Langer%20COI.pdf?dl=0. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd6889/DC1
REFERENCES AND NOTES
- 1.Roozenbeek B., Maas A. I. R., Menon D. K., Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 9, 231–236 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Simon D. W., McGeachy M. J., Bayır H., Clark R. S. B., Loane D. J., Kochanek P. M., The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13, 171–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meabon J. S., Huber B. R., Cross D. J., Richards T. L., Minoshima S., Pagulayan K. F., Li G., Meeker K. D., Kraemer B. C., Petrie E. C., Raskind M. A., Peskind E. R., Cook D. G., Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci. Transl. Med. 8, 321ra6 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Goldstein L. E., Fisher A. M., Tagge C. A., Zhang X.-L., Velisek L., Sullivan J. A., Upreti C., Kracht J. M., Ericsson M., Wojnarowicz M. W., Goletiani C. J., Maglakelidze G. M., Casey N., Moncaster J. A., Minaeva O., Moir R. D., Nowinski C. J., Stern R. A., Cantu R. C., Geiling J., Blusztajn J. K., Wolozin B. L., Ikezu T., Stein T. D., Budson A. E., Kowall N. W., Chargin D., Sharon A., Saman S., Hall G. F., Moss W. C., Cleveland R. O., Tanzi R. E., Stanton P. K., McKee A. C., Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, 134ra160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKee A. C., Stein D., Nowinski C. J., Stern R. A., Daneshvar D. H., Alvarez V. E., Lee H.-S., Hall G., Wojtowicz S. M., Baugh C. M., Riley D. O., Kubilus C. A., Cormier K. A., Jacobs M. A., Martin B. R., Abraham C. R., Ikezu T., Reichard R. R., Wolozin B. L., Budson A. E., Goldstein L. E., Kowall N. W., Cantu R. C., The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith D. H., Johnson V. E., Stewart W., Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat. Rev. Neurol. 9, 211–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeKosky S. T., Blennow K., Ikonomovic M. D., Gandy S., Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat. Rev. Neurol. 9, 192–200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blennow K., Hardy J., Zetterberg H., The neuropathology and neurobiology of traumatic brain injury. Neuron 76, 886–899 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Sharp D. J., Scott G., Leech R., Network dysfunction after traumatic brain injury. Nat. Rev. Neurol. 10, 156–166 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Yoo D., Magsam A. W., Kelly A. M., Stayton P. S., Kievit F. M., Convertine A. J., Core-cross-linked nanoparticles reduce neuroinflammation and improve outcome in a mouse model of traumatic brain injury. ACS Nano 11, 8600–8611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitehead K. A., Langer R., Anderson D. G., Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowdy S. F., Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 35, 222–229 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Zheng M., Tao W., Zou Y., Farokhzad O. C., Shi B., Nanotechnology-based strategies for siRNA brain delivery for disease therapy. Trends Biotechnol. 36, 562–575 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Kwon E. J., Skalak M., Bu R. L., Bhatia S. N., Neuron-targeted nanoparticle for siRNA delivery to traumatic brain injuries. ACS Nano 10, 7926–7933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang J., Joo J., Kwon E. J., Skalak M., Hussain S., She Z.-G., Ruoslahti E., Bhatia S. N., Sailor M. J., Self-sealing porous silicon-calcium silicate core–shell nanoparticles for targeted siRNA delivery to the injured brain. Adv. Mater. 28, 7962–7969 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bharadwaj V. N., Lifshitz J., Adelson P. D., Kodibagkar V. D., Stabenfeldt S. E., Temporal assessment of nanoparticle accumulation after experimental brain injury: Effect of particle size. Sci. Rep. 6, 29988 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong Y., Mahmood A., Chopp M., Animal models of traumatic brain injury. Nat. Rev. Neurosci. 14, 128–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chodobski A., Zink B. J., Szmydynger-Chodobska J., Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2, 492–516 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuriakose M., Rao K. V. R., Younger D., Chandra N., Temporal and spatial effects of blast overpressure on blood-brain barrier permeability in traumatic brain injury. Sci. Rep. 8, 8681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahyouni R., Gutierrez P., Gold E., Robertson R. T., Cummings B. J., Effects of concussion on the blood–brain barrier in humans and rodents. J. Concussion. 1, 10.1177/2059700216684518, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nowak M., Helgeson M. E., Mitragotri S., Delivery of nanoparticles and macromolecules across the blood brain barrier. Adv. Therap. 3, 1900073 (2019). [Google Scholar]
- 22.Furtado D., Björnmalm M., Ayton S., Bush A. I., Kempe K., Caruso F., Overcoming the blood–brain barrier: The role of nanomaterials in treating neurological diseases. Adv. Mater. 30, 1801362 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Anraku Y., Kuwahara H., Fukusato Y., Mizoguchi A., Ishii T., Nitta K., Matsumoto Y., Toh K., Miyata K., Uchida S., Nishina K., Osada K., Itaka K., Nishiyama N., Mizusawa H., Yamasoba T., Yokota T., Kataoka K., Glycaemic control boosts glucosylated nanocarrier crossing the BBB into the brain. Nat. Commun. 8, 1001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J.-L., Jiang G., Song Q.-X., Gu X., Hu M., Wang X.-L., Song H.-H., Chen L.-P., Lin Y.-Y., Jiang D., Chen J., Feng J.-F., Qiu Y.-M., Jiang J.-Y., Jiang X.-G., Chen H.-Z., Gao X.-L., Lipoprotein-biomimetic nanostructure enables efficient targeting delivery of siRNA to Ras-activated glioblastoma cells via macropinocytosis. Nat. Commun. 8, 15144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hrkach J., Von Hoff D., Ali M. M., Andrianova E., Auer J., Campbell T., De Witt D., Figa M., Figueiredo M., Horhota A., Low S., McDonnell K., Peeke E., Retnarajan B., Sabnis A., Schnipper E., Song J. J., Song Y. H., Summa J., Tompsett D., Troiano G., Van Geen Hoven T., Wright J., LoRusso P., Kantoff P. W., Bander N. H., Sweeney C., Farokhzad O. C., Langer R., Zale S., Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differenciated pharmacological profile. Sci. Transl. Med. 4, 128ra39 (2012). [DOI] [PubMed] [Google Scholar]
- 26.McHugh K. J., Nguyen T. D., Linehan A. R., Yang D., Behrens A. M., Rose S., Tochka Z. L., Tzeng S. Y., Norman J. J., Anselmo A. C., Xu X., Tomasic S., Taylor M. A., Lu J., Guarecuco R., Langer R., Jaklenec A., Fabrication of fillable microparticles and other complex 3D microstructures. Science 357, 1138–1142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondo A., Shahpasand K., Mannix R., Qiu J., Moncaster J., Chen C.-H., Yao Y., Lin Y.-M., Driver J. A., Sun Y., Wei S., Luo M.-L., Albayram O., Huang P., Rotenberg A., Ryo A., Goldstein L. E., Pascual-Leone A., McKee A. C., Meehan W., Zhou X. Z., Lu K. P., Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mannix R., Meehan W. P., Mandeville J., Grant P. E., Gray T., Berglass J., Zhang J., Bryant J., Rezaie S., Chung J. Y., Peters N. V., Lee C., Tien L. W., Kaplan D. L., Feany M., Whalen M., Clinical correlates in an experimental model of repetitive mild brain injury. Ann. Neurol. 74, 65–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suk J. S., Xu Q., Kim N., Hanes J., Ensign L. M., PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 99, 28–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J., Cai P., Shalviri A., Henderson J. T., He C., Foltz W. D., Prasad P., Brodersen P. M., Chen Y., Costa R. D., Rauth A. M., Wu X. Y., A multifunctional polymeric nanotheranostic system delivers doxorubicin and imaging agents across the blood–brain barrier targeting brain metastases of breast cancer. ACS Nano 8, 9925–9940 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Koffie R. M., Farrar C. T., Saidi L.-J., William C. M., Hyman B. T., Spires-Jones T. L., Nanoparticles enhance brain delivery of blood–brain barrier-impermeable probes for in vivo optical and magnetic resonance imaging. Proc. Natl. Acad. Sci. U.S.A. 108, 18837–18842 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.M. Naito, T. Yokoyama, K. Hosokawa, K. Nogi, Nanoparticle Technology Handbook (Elsevier, ed. 3, 2018), 471 pp. [Google Scholar]
- 33.Maussang D., Rip J., van Kregten J., van den Heuvel A., van der Pol S., van der Boom B., Reijerkerk A., Chen L., de Boer M., Gaillard P., de Vries H., Glutathione conjugation dose-dependently increases brain-specific liposomal drug delivery in vitro and in vivo. Drug Discov. Today Technol. 20, 59–69 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Rip J., Chen L., Hartman R., van den Heuvel A., Reijerkerk A., van Kregten J., van der Boom B., Appeldoorn C., de Boer M., Maussang D., de Lange E. C. M., Gaillard P. J., Glutathione PEGylated liposomes: Pharmacokinetics and delivery of cargo across the blood–brain barrier in rats. J. Drug Target. 22, 460–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam F. C., Morton S. W., Wyckoff J., Han T.-L. V., Hwang M. K., Maffa A., Balkanska-Sinclair E., Yaffe M. B., Floyd S. R., Hammond P. T., Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 9, 1991 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oller-Salvia B., Sánchez-Navarro M., Giralt E., Teixidó M., Blood–brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 45, 4690–4707 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Islam M. A., Xu Y., Tao W., Ubellacker J. M., Lim M., Aum D., Lee G. Y., Zhou K., Zope H., Yu M., Cao W., Oswald J. T., Dinarvand M., Mahmoudi M., Langer R., Kantoff P. W., Farokhzad O. C., Zetter B. R., Shi J., Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2, 850–864 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Sun A.-n., Liu Y.-j., Zhang W.-j., Pang N., Cheng S.-x., Qi X.-r., Amphiphilic dendrimer engineered nanocarrier systems for co-delivery of siRNA and paclitaxel to matrix metalloproteinase-rich tumors for synergistic therapy. NPG Asia Mater. 10, 238–254 (2018). [Google Scholar]
- 39.Saraiva C., Praça C., Ferreira R., Santos T., Ferreira L., Bernardino L., Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Control. Release 235, 34–47 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Hoshyar N., Gray S., Han H., Bao G., The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 11, 673–692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahay G., Querbes W., Alabi C., Eltoukhy A., Sarkar S., Zurenko C., Karagiannis E., Love K., Chen D., Zoncu R., Buganim Y., Schroeder A., Langer R., Anderson D. G., Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 31, 653–658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu J. P. J., Cheng B., Roffler S. R., Lundy D. J., Yen C. Y. T., Chen P., Lai J. J., Pun S. H., Stayton P. S., Hsieh P. C. H., Reloadable multidrug capturing delivery system for targeted ischemic disease treatment. Sci. Transl. Med. 8, 365ra160 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Nance E. A., Woodworth G. F., Sailor K. A., Shih T.-Y., Xu Q., Swaminathan G., Xiang D., Eberhart C., Hanes J., A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci. Transl. Med. 4, 149ra119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambruosi A., Khalansky A. S., Yamamoto H., Gelperina S. E., Begley D. J., Kreuter J., Biodistribution of polysorbate 80-coated doxorubicin-loaded [14C]-poly(butyl cyanoacrylate) nanoparticles after intravenous administration to glioblastoma-bearing rats. J. Drug Target. 14, 97–105 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Jaffer H., Adjei I. M., Labhasetwar V., Optical imaging to map blood-brain barrier leakage. Sci. Rep. 3, 3117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shahim P., Tegner Y., Wilson D. H., Randall J., Skillbäck T., Pazooki D., Kallberg B., Blennow K., Zetterberg H., Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 71, 684–692 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Cheng J. S., Craft R., Yu G.-Q., Ho K., Wang X., Mohan G., Mangnitsky S., Ponnusamy R., Mucke L., Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLOS ONE 9, e115765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeVos S. L., Miller R. L., Schoch K. M., Holmes B. B., Kebodeaux C. S., Wegener A. J., Chen G., Shen T., Tran H., Nichols B., Zanardi T. A., Kordasiewicz H. B., Swayze E. E., Bennett C. F., Diamond M. I., Miller T. M., Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9, eaag0481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu H., Rösler T. W., Carlsson T., de Andrade A., Fiala O., Höllerhage M., Oertel W. H., Goedert M., Aigner A., Höglinger G. U., Tau silencing by siRNA in the P301S mouse model of tauopathy. Curr. Gene Ther. 14, 343–351 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Chiasseu M., Vargas J. L. C., Destroismaisons L., Velde C. V., Leclerc N., Polo A. D., Tau accumulation, altered phosphorylation, and missorting promote neurodegeneration in glaucoma. J. Neurosci. 36, 5785–5798 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carron S. F., Alwis D. S., Rajan R., Traumatic brain injury and neuronal functionality changes in sensory cortex. Front. Syst. Neurosci. 10, 47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda A. M., Badaut J., siRNA treatment: “A sword-in-the-Stone” for acute brain injuries. Genes 4, 435–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marklund N., Hillered L., Animal modelling of traumatic brain injury in preclinical drug development: Where do we go from here? Br. J. Pharmacol. 164, 1207–1229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calcul L., Zhang B., Jinwal U. K., Dickey C. A., Baker B. J., Natural products as a rich source of tau-targeting drugs for Alzheimer’s disease. Future Med. Chem. 4, 1751–1761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siahaan A. M. P., Japardi I., Rambe A. S., Indharty R. S., Ichwan M., Turmeric extract supplementation reduces Tau protein level in repetitive traumatic brain injury model. J. Med. Sci. 6, 1953–1958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng C., Ma C., Bai E., Yang K., Xu R., Transferrin and cell-penetrating peptide dual-functioned liposome for targeted drug delivery to glioma. Int. J. Clin. Exp. Med. 8, 1658–1668 (2015). [PMC free article] [PubMed] [Google Scholar]
- 57.Liu H., Moynihan K. D., Zheng Y., Szeto G. L., Li A. V., Huang B., Egeren D. S. V., Park C., Irvine D. J., Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu X., Xie K., Zhang X.-Q., Pridgen E. M., Park G. Y., Cui D. S., Shi J., Wu J., Kantoff P. W., Lippard S. J., Langer R., Walker G. C., Farokhzad O. C., Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc. Natl. Acad. Sci. U.S.A. 110, 18638–18643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robertson R. T., Levine S. T., Haynes S. M., Gutierrez P., Baratta J. L., Tan Z., Longmuir K. J., Use of labeled tomato lectin for imaging vasculature structures. Histochem. Cell Biol. 143, 225–234 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd6889/DC1