

Heat stress–induced survival advantages can be transgenerationally transmitted through the epigenetic marks of N6-mA and H3K9me3.
Abstract
Environmental stress can induce survival advantages that are passed down to multiple generations, representing an evolutionarily advantageous adaptation at the species level. Using the nematode worm Caenorhabditis elegans as a model, we found that heat shock experienced in either parent could increase the longevity of themselves and up to the fifth generation of descendants. Mechanistic analyses revealed that transcription factor DAF-16/FOXO, heat shock factor HSF-1, and nuclear receptor DAF-12/FXR functioned transgenerationally to implement the hormetic stress response. Histone H3K9me3 methyltransferases SET-25 and SET-32 and DNA N6-methyl methyltransferase DAMT-1 participated in transmitting high-temperature memory across generations. H3K9me3 and N6-methyladenine could mark heat stress response genes and promote their transcription in progeny to extend life span. We dissected the mechanisms responsible for implementing and transmitting environmental memories in descendants from heat-shocked parents and demonstrated that hormetic stress caused survival benefits could be transmitted to multiple generations through H3K9me3 and N6-mA modifications.
INTRODUCTION
Physical and behavioral characteristics, including physical appearance (1), energy metabolism (2), behavioral state (3, 4), and longevity (5, 6), have been shown to be influenced by the environment, partially by nongenetic information, known as an epigenetic mark. Epigenetic information can be transmitted from generation to generation through DNA methylation, histone modifications, or small RNAs (7). This phenomenon is known as transgenerational epigenetic inheritance (TEI). TEI has been extensively studied in different model organisms (8), in which a few traits have been characterized, including flower color in plants (9), longevity in worms (5), and heat stress response in Drosophila (10). TEI has been shown to play an important role in physiological responses to stress, such as that caused by pathogenic bacteria (4), osmosis (11), oxidation (11), and starvation (12).
Caenorhabditis elegans has become a powerful model for studying TEI because it has short reproductive cycles, large broods, an easily manipulated germ line, and evolutionarily conserved epigenetic mechanisms. Moreover, using C. elegans, a large number of recent studies have elucidated the molecular mechanisms of TEI in organisms undergoing different environmental experiences. For example, parent mitochondrial stress leads to progeny with the mitochondrial stress adaptation through histone H3K4me3 and DNA N6-methyldeoxyadenine (N6-mA) modifications (6). Starvation-induced TEI in C. elegans is conferred by small RNAs and histone H3K4me3 modification (12, 13). Transgenerationally learned pathogenic avoidance is mediated by Piwi Related Gene (Piwi)/PRG-1 argonaute and H3K9me3 (3, 4). Heritable adaptation to pathogen infection of C. elegans is mediated by cysteine synthases (14). These previous studies revealed that C. elegans has become a powerful model organism with which to elucidate the underlying molecular mechanisms of TEI.
As an important environmental factor, temperature is always experienced by all organisms. Previous studies have demonstrated that mild heat stress can increase life span and stress resistance. This adaptive induction of stress tolerance is called hormesis and has been observed in many model systems, including Drosophila (15), human fibroblasts (16), and C. elegans (17, 18). In C. elegans, that exposure to high environmental temperature–induced hormetic heat stress activates a series of transcription factors, including heat shock factor HSF-1, DAF (abnormal DAuer Formation)–16/FOXO (forkhead box O transcription factor), HLH-30 (Helix Loop Helix)/transcription factor EB (TFEB), and nuclear receptor DAF-12, which, in turn, up-regulate the expression of stress response genes (i.e., hsp-70, hsp-16.2, and hsp-12.6) and autophagy genes (i.e., lgg-1, bec-1, and unc-51), promoting life-span extension and stress tolerance (17, 19). In addition, high temperature–induced TEI effects have been observed in C. elegans (20) and Drosophila (10), in which the high-temperature memory is transmitted from the parental generation to the offspring. However, it is still unclear whether organisms can transmit hormetic heat stress–induced survival advantages to descendants through TEI. If the survival advantages can be inherited nongenetically, then the underlying epigenetic mechanisms have not been characterized yet. Although a recent study showed that in Artemia, parents exposed to nonlethal heat shock could transmit the high-temperature memory to their descendants, leading to increased tolerance toward lethal heat stress and elevated resistance against pathogens (21), many underlying molecular mechanisms remain unknown.
Here, we report that in C. elegans, hormetic heat stress–induced life-span extension can be passed down to progeny for as many as five generations. Moreover, we characterize the critical factors for implementing and transmitting environmental memories in descendants from heat-shocked parents. Our findings also demonstrated that H3K9me3 and N6-mA modifications played important roles in transmitting transgenerational survival advantages induced by hormetic stress.
RESULTS
Heat stress induces a transgenerational inheritable survival advantage
The environment experienced by an animal can influence variations in gene expression for one or a few subsequent generations. A previous study reported that a temperature-mediated environmental change can induce long-lasting epigenetic memory in C. elegans (20). Exposure of C. elegans to hormetic heat stress can increase their life span (17, 18). We hypothesized that C. elegans could transmit the survival advantage induced by hormetic heat stress to their naive, unstressed progeny (Fig. 1A). To test this hypothesis, we randomly selected one wild-type hermaphrodite worm and obtained a population with the same genetic background through its self-fertilization. We then exposed wild-type worms to 35°C for 1 hour on day 1 of adulthood and bleached the parent generation (P0) mothers to obtain the F1 generation. Our results showed that the life span of the heat-shocked parents and their F1 progeny (cultured unimpeded under normal 20°C temperature) is significantly extended (Fig. 1, B and C). The F1 progeny may have been exposed to the temperature stimuli while still inside the mother, in which case, the beneficial longevity effect in the F1 worms would be considered intergenerational inheritance, not transgenerational inheritance. Therefore, we tested for the transgenerational effect and determined the number of generations displaying survival advantage induced by heat stress in the parental worms. We found that preexposure of P0 animals to heat stress extended the F1 generation life span. This life-span extension effect persisted into the F5 generation, although the F1 to F4 generation animals never experienced heat shock. Sixth generation (F6) descendants had a normal life span (Fig. 1, D to F). Here, life-span analyses were conducted with 5-fluoro-2′-deoxyuridine (FUDR). Considering that FUDR might affect TEI because of its function on DNA/RNA synthesis, we tested whether FUDR affected the life-span extension across generations. Life-span analyses revealed no difference between worms treated with or without FUDR, indicating that FUDR did not affect TEI in our settings (fig. S1).
Fig. 1. Longevity induced by heat stress can be inherited.
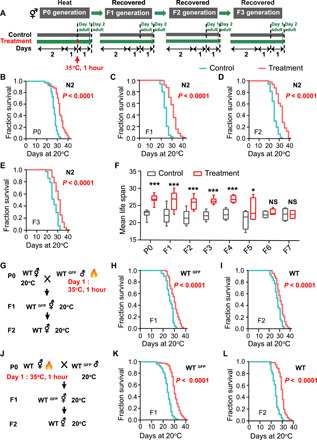
(A to E) Transgenerational inheritance of heat shock induces a survival advantage in C. elegans. Experimental scheme (A) and life-span analyses of with or without treatment P0 (B), F1 (C), F2 (D), and F3 (E). Life span was analyzed using the Kaplan-Meier test; P values were calculated using the log-rank test, and the life-span values of the replicated tests are listed in table S1. (F) Mean life span of generations F1 to F7. Naive progeny from generation F1 to F5 of parents exposed to high temperature had extended life spans; the sixth generation returned to normal life span (mean ± SD, n ≥ 3; *P < 0.05 and ***P < 0.001; NS, not significant; Student’s t test). (G to L) Longevity induced by heat shock is transmitted through both male and female germ lines. Experimental scheme (G and J) and life-span analyses of the progeny of fathers exposed to high temperature (H and I) and the progeny of mothers exposed to high temperature (K and L) (P value by log-rank test). The experiments were repeated at least three times. Detailed life-span values are listed in table S1. WT, wild type.
Transgenerational inheritance has been shown to be transmitted through both oocytes and sperm (20). To determine whether the survival advantage induced by heat shock was conferred by oocytes, sperm, or both, we conducted reciprocal matings and survival analyses of cross-progeny (Fig. 1, G and J). All matings were set up with wild-type hermaphrodites and green fluorescent protein–positive (GFP+) males to differentiate cross-progeny from self-fertilized progeny. As expected, the F1 progeny of the heat-shocked males mated with non–heat-shocked hermaphrodites and their F2 generation (Fig. 1, H and I), as well as the F1 progeny of non–heat-shocked males mated with heat-shocked hermaphrodites and their F2 descendants (Fig. 1, K and L), all exhibited significant life-span extension, suggesting that both sperm and oocytes can transmit information induced by hormetic heat stress that provides a survival advantage.
Inheritance induced by hormetic heat shock enhances survival of progeny to improve animal fitness
The hormetic heat stress could ameliorate aging, in which loss of protein homeostasis is a pivotal hallmark (22). A recent study reported that hormetic heat stress can significantly reduce the progressive accumulation of protein (17). Because the beneficial hormetic effects of heat stress on longevity can pass transgenerationally to descendants, we speculated that improvement of proteostasis by hormetic heat shock may also transmit to progeny. To test this hypothesis, we examined a transgenic C. elegans strain NL5901, a Parkinson’s disease model, which expresses human α-synuclein–yellow fluorescent protein (YFP) in worms’ body-wall muscle cells of the nematode (23). Exposure of these transgenic worms to hormetic heat stress on day 1 resulted in beneficial hormetic effects in terms of both intergenerational and transgenerational inheritance, which reduced the protein deposition of α-synuclein to maintain protein homeostasis (Fig. 2, A and B).
Fig. 2. Effect of heat stress on C. elegans fitness.
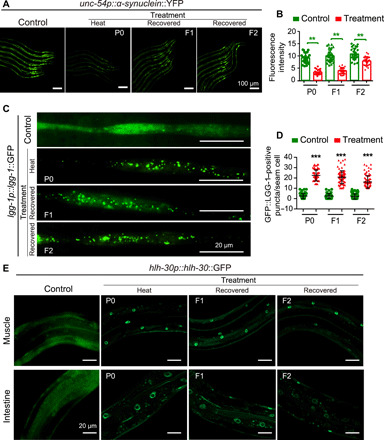
(A and B) Representative images (A) and quantification of muscle α-synuclein aggregates of α-synuclein::YFP worms with or without treatment. (C and D) GFP::LGG-1/Atg8 puncta in the hypodermal seam cells with or without treatment. Means ± SD, n ≥ 30 per condition; P value was calculated by Student’s t test; **P < 0.01 and ***P < 0.001. (E) Representative images of the nuclear localization of HLH-30::GFP in worms with or without treatment. Each experiment was repeated at least three times.
Hormetic heat shock induces the expression of autophagy genes in a HSF-1–dependent manner, which is required for heat shock–mediated survival benefit (17). Therefore, we monitored the autophagy of C. elegans using a GFP-tagged LGG-1 (LC3, GABARAP and GATE-16 family)/Atg8 reporter. Our results showed that heat-shocked P0 worms and recovered F1 and F2 progeny from heat-shocked parents exhibit a marked elevation in the number of GFP::LGG-1 puncta in the hypodermal seam cells (Fig. 2, C and D). These results demonstrated that heat shock–induced autophagy is heritable across generations, which is correlated with the transgenerational inheritance of heat shock–mediated survival benefits.
Heat shock–induced autophagy is regulated by the transcriptional factor HLH-30, which is the ortholog of mammalian TFEB, a master regulator of autophagy and lysosomal genes (17). We found that hormetic heat stress caused translocation of GFP-tagged HLH-30 into the nucleus and elevated protein levels of HLH-30 (fig. S2). This heat stress memory was transmitted to progeny, causing significant elevation protein level and accumulation of HLH-30 in the nuclei of the naive progeny of heat-shocked parents (Fig. 2E and fig. S2). Collectively, these results demonstrated that high-temperature memory leads to up-regulation of autophagy in offspring, which contributes to the TEI effects of hormetic heat stress–induced survival benefits.
HSF-1, DAF-16/FOXO, and DAF-12/FXR are required for the transgenerational inheritance induced by heat shock
To examine the molecular mechanisms underlying hormetic heat stress, we evaluated the contribution of two transcription factors and one nuclear receptor, each of which have well-characterized and important roles in sensing and subsequently responding to heat stress. HSF-1 is a highly conserved transcription factor that functions in the heat stress response (19). Our results showed that loss of hsf-1 caused defects in life-span extension induced by hormetic heat stress in the P0, F1, and F2 generations (Fig. 3A and fig. S3, B, Q, and R). To more definitively characterize the role of hsf-1 in the F1 and F2 generations, we used RNA interference (RNAi) to knock down hsf-1 exclusively in the F1 or F2 generation following parental exposure to heat stress. Here, an RNAi-sensitive rrf-3 mutant was used to elevate the silencing efficiency in all corresponding analyses with RNAi (24). We found that the survival advantage was lost only in the F1 or F2 generations exposed to hsf-1 RNAi (Fig. 3F and fig. S3G), although their heat-shocked mothers retained longevity benefits (Fig. 3E and fig. S3F), suggesting that hsf-1 acts in the progeny to mediate life-span extension. Moreover, consistent with the previous report that HSF-1 was constitutively localized to the nucleus upon heat shock (25), we observed the same localization in parents exposed to heat stress (fig. S4A), while the expression level maintained the same (fig. S4B). Therefore, we deduced that the transgenerational survival advantage was mediated by the heat stress–induced translocation of HSF-1 to the nucleus. We found that naive F1 progeny derived from heat-shocked parents maintained HSF-1 nuclear subcellular localization (fig. S4A). Collectively, these results indicate that HSF-1 is involved not only in the heat stress response of the P0 worms but also in the transgenerational inheritance of hormetic heat effects.
Fig. 3. DAF-16, DAF-12, and HSF-1 are required for transgenerational inheritance.
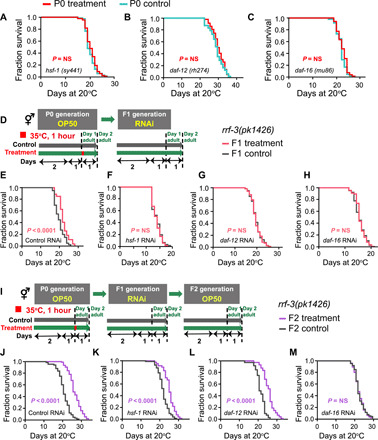
(A to C) Life-span analyses of (A) hsf-1 mutants, (B) daf-12 mutants, and (C) daf-16 mutants exposed or not exposed to high temperature. (D) Experimental scheme. (E to H) P0 worms were heat shocked without RNAi exposure, and then F1 progeny were exposed to (E) control (HT115), (F) hsf-1, (G) daf-12, and (H) daf-16 RNAi to test the requirement for daf-16, daf-12, and hsf-1 in the F1 generation (P value by log-rank test). (I) Experimental scheme. (J to M) P0 animals were heat shocked, and then F1 progeny were subjected to (J) control (HT115), (K) hsf-1, (L) daf-12, and (M) daf-16 RNAi, and life span of F2 progeny were analyzed. The experiments were repeated at least three times. Detailed life-span values are listed in table S1.
In addition to HSF-1, the forkhead transcription factor DAF-16 and a nuclear receptor DAF-12 play essential roles in the heat stress response (26). Similar to HSF-1, both daf-16 and daf-12 loss of function completely suppressed the heat stress–induced life-span extensions in the P0, F1, or F2 generations (Fig. 3, B and C, and fig. S3, C, D, O, and P). To confirm this observation, we also used RNAi specific for F1 or F2 generations to knock down daf-12 and daf-16. Like hsf-1 RNAi, either daf-12 or daf-16 RNAi in the F1 or F2 descendants derived from the heat-stressed P0 parents abrogated the hormetic longevity effect (Fig. 3, G and H, and fig. S3, H and I).
Reportedly, heat stress–activated DAF-16 is translocated to nucleus and regulates the expression of downstream heat stress response genes (27). In agreement with the previous study, upon heat shock, worms had elevated protein levels of DAF-16 and an obvious accumulation in nuclei (fig. S4, C and D). Consistently, naive F1 generation animals from heat-shocked P0 worms had nuclear accumulation of DAF-16 and increased protein levels (fig. S4, C and D). The degree of nuclear aggregation in F1 animals is not as extensive as that of the P0 animals, in which high DAF-16 accumulation was found in various tissues. In the F1 generation, most of the nuclear translocation of DAF-16 was found in the hypodermis, while little accumulation was apparent in the intestine.
To more definitively dissect whether the role of hsf-1, daf-16, and daf-12 is to implement a heat stress response, to transmit heritable memory, or both, we used RNAi to silence hsf-1, daf-12, and daf-16 exclusively in the F1 generation following parental exposure to heat stress and then conducted the life-span analyses of F2 generation (Fig. 3I). If a gene is required for transmitting temperature memory, then the silencing of that gene in the F1 generation will lead to the loss of transgenerational memory transmitters, which will result in the disappearance of the life-extending phenotype of the F2 generation. Alternatively, if a gene only functions in implementing a hormetic response, then the silencing of that gene in the F1 generation should only affect the life-span of F1 progeny so that we would still detect the life-span extension phenotype in F2 generation. One concern is that the RNAi effect might be transmitted to the next generation. To exclude this effect, we fed Dicer (dcr-1) RNAi bacteria to F2 that were born from F1 with RNAi-silenced hsf-1, daf-12, or daf-16 (fig. S3J) (28). The results showed that silencing of daf-12 or hsf-1 in F1 did not abolish the life-span extension of F2 (Fig. 3, J to L, and fig. S3, K to M), suggesting that daf-12 and hsf-1 participated only in implementation but not in transmission; the silencing of daf-16 in F1 caused the loss of the life-span extension of F2 (Fig. 3M and fig. S3N), indicating a role in transmission in addition to implementation. Collectively, in line with reports from a previous study, our results showed that hsf-1, daf-16, and daf-12 contribute to the heat stress response in the P0 generation. In addition, we found that hsf-1 and daf-12 also play roles in implementing heat stress response in the progeny to extend the life-span; however, for DAF-16, it not only implements heat stress response but also transmits heritable memory to progeny.
H3K9me3 histone modification is required for the TEI induced by heat shock
The disappearance of the survival advantage in the F6 generation suggests that heat stress might induce epigenetic changes rather than genetic mutations in descendants. Histone methylation has been implicated in TEI regulation in C. elegans, and a recent study showed that the TEI induced by high-temperature exposure is associated with a decreased H3K9me3 level (20). In results consistent with this research, we found that the loss of set-25 or set-32, genes encoding H3K9me3 methyltransferases, compromised the survival advantage in progeny (Fig. 4, E and G, and fig. S5, I and K). Neither set-25 nor set-32 is required for implementing hormetic heat stress response because their loss-of-function mutants have a similar increased life span under heat-shock condition wild-type animals (Fig. 4, C and D, and fig. S5, H and J). To more definitively exclude the role of set-25 and set-32 in the P0 generation, we also used RNAi to knock down set-25 or set-32 in F1 and F2 progeny derived from heat-stressed P0 worms and found that the life-span advantage of the RNAi-treated progeny was abrogated (fig. S5, A to G). These data suggest that set-25 and set-32 are specifically required for transmitting the memory of heat stress. In addition, other histone modifications, including H3K9me2, H3K4me3, H3K27me3, and H3K36me3, were also reported to mediate the life-span extension and the TEI in C. elegans (29). To characterize the roles of these histone modifications in heat stress–induced TEI of life-span extension, we evaluated their contribution using corresponding mutants. Our results showed that the deletion of the histone modifiers rbr-2 (histone H3K4me3 demethylase), spr-5 (histone H3K4me2 demethylase), set-18 (histone H3K36 dimethyltransferase), and met-2 (histone H3K9me2 methyltransferase) did not abrogate the survival advantage in the F1 or F2 generation (fig. S5, L to P). These results indicated that the TEI induced by heat stress depended on the histone modification H3K9me3.
Fig. 4. Histone H3K9me3 modifiers are required for transgenerational inheritance induced by heat stress.
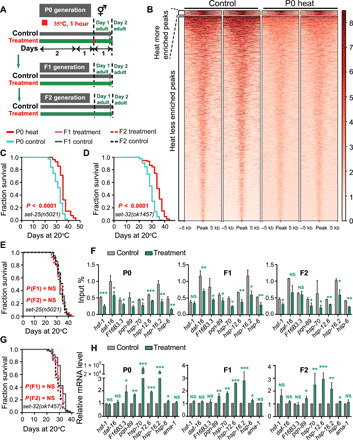
Experimental scheme (A) and transgenerational inheritance tested in (C) set-25 mutants (P0), (D) set-32 mutants (P0), (E) set-25 mutants (recovered F1 and F2), and (G) set-32 mutants (recovered F1 and F2) (P value by log-rank test). The experiment was repeated at least three times. Detailed life-span values were listed in table S1. (B) Heatmap of H3K9me3 ChIP signals approximately 5 kb upstream and downstream of regions that were differentially enriched between the heat shock and control groups. The peaks were ranked in descending order of H3K9me3 intensity within each cluster. (F and H) ChIP-qPCR of H3K9me3 and qPCR of daf-16; hsf-1 and the corresponding target genes of daf-16, hsf-1, and daf-12 in the WT animals with or without treatment (n = 3). The graph data are presented as means ± SD. Statistical analyses were conducted using two-tailed Student’s t test; *P < 0.05, **P < 0.01, and ***P < 0.001.
To determine whether the prolongevity effects induced by heat stress are related to heritable changes in H3K9me3 at specific loci, we conducted RNA sequencing (RNA-seq) and chromatin immunoprecipitation and sequencing (ChIP-seq) to compare changes in the enrichment of H3K9me3 peaks. H3K9me3 peaks were markedly decreased at gene promoters after heat shock (Fig. 4B and fig. S6B). Combining RNA-seq and ChIP-seq data, we found that upon heat shock, the transcription level of many genes was up-regulated; the H3K9me3 level was decreased (fig. S6, A and B). Upon hormetic heat stress, animals activated a series of transcription factors, including HSF-1, DAF-16, and nuclear receptor DAF-12, which subsequently regulate the expression of genes involved in the stress response. Regulation of these genes induces the innate immune response, resets metabolism, and contributes to the maintenance of protein homeostasis, ultimately leading to a survival advantage (19, 26). Using ChIP–quantitative polymerase chain reaction (qPCR), we found that, following heat stress in P0 worms, the number of H3K9me3-occupied sites was obviously decreased in the promoters of heat stress response– and longevity-associated genes, including daf-16, hsf-1, and the respective target genes of daf-16, hsf-1, and daf-12. The decrease observed in P0 was maintained in the F1 and F2 generations (Fig. 4F and fig. S6C). We also found that the heat stress response–related genes had decreased histone H3K9me3 levels but notably elevated transcripts levels (Fig. 4H). Overall, these results indicated that chromatin marks were established in the P0 generation following heat shock, and these marks persisted in the subsequent generations to transmit heritable memories.
N6-mA DNA modification mediates the TEI induced by heat shock
In addition to its role in histone modification, methylation is also known to modify DNA to render it a carrier of inheritable information (30). Recent studies have revealed that N6-mA–modified DNA mediates TEI in C. elegans (6, 31). Notably, using dot blot analysis, we found a clear elevation of N6-mA in animals subjected to heat stress (Fig. 5C). The elevation of N6-mA–modified DNA was maintained in naive progeny from heat-shocked mothers through four generations, a finding that was correlated with the survival advantage returning to normal levels in the F6 generation (Fig. 5C). The N6-mA methyltransferase mutants damt-1 (tm6887) and damt-1(gk961032), as well as the N6-mA demethylase nmad-1 (ok3133) mutants, failed to elevate the levels of N6-mA following heat stress (fig. S8, A to C). In addition, the results from survival analyses of the damt-1 and nmad-1 deletion mutants showed that loss of function of both damt-1 and nmad-1 abolished the survival advantage of heat-stressed P0 and their recovered F1 and F2 progeny (Fig. 5, B and E, and figs. S7, A to F, and S8, I to K). To more definitively characterize the role of damt-1 in heat-induced TEI, we conducted two sets of RNAi experiments similar to that performed in the above-mentioned treatments (hsf-1, daf-12, and daf-16 RNAi). The first experiment was to feed damt-1 RNAi to F1 or F2 worms from heat-shocked P0 and measure their life span, respectively. We found that silencing damt-1 in F1 or F2 progeny abolished the hormetic heat stress benefits (fig. S7, G to K), suggesting that damt-1 acts in the progeny to extend life span. The other silencing experiment was to feed damt-1 RNAi to F1 exclusively and measure the life span of F2. Our results showed that F2 progeny without RNAi [or with dcr-1 RNAi to exclude the possible RNAi inheritance (28)] from F1 with RNAi-silenced damt-1 also abolished the hormetic heat stress benefits (fig. S7, L and M), suggesting that damt-1 contributed to transmitting heat stress memories.
Fig. 5. DNA N6-mA mediates the transgenerational inheritance induced by heat stress.
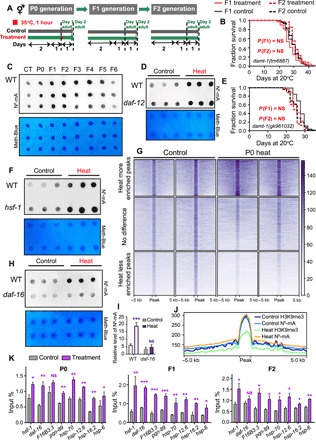
(A) Experimental scheme. (C) Representative dot blot analysis from three biological replicates used to analyze the global N6-mA level in genomic DNA (gDNA) in control and P0-F6 worms. Methylene blue detected DNA loading. (B and E) Transgenerational inheritance tested in (B) the damt-1 (tm6887) mutants (recovered F1 and F2) and (E) the damt-1 (gk961032) mutants (recovered F1 and F2) (P value by log-rank test). Detailed life-span values are listed in table S1. (D, F, H, and I) Dot blots analyses from heat-shocked and non–heat-shocked WT animals and (D) daf-12 mutants, (F) hsf-1 mutants, and (H and I) daf-16 mutants (means ± SD; two-tailed Student’s t test, ***P < 0.001). Experiment was repeated at least three times. Methylene blue was used as a DNA loading control. (G) Heatmap of N6-mA MeDIP signals in the heat-shocked and non–heat-shocked groups. The peaks were ranked in descending order of N6-mA intensity within each cluster. (J) Profile plot demonstrating the overlapping N6-mA peaks and H3K9me3-associated domains in the exposed and nonexposed WT animals. (K) MeDIP-qPCR analyses of N6-mA occupancy of daf-16, hsf-1, and corresponding target genes of daf-16, hsf-1, and daf-12 in the WT animals (n = 3). Statistical analyses were conducted using two-tailed Student’s t test; *P < 0.05, **P < 0.01, and ***P < 0.001.
Notably, the basal level of N6-mA in the damt-1 mutant is comparable to that of the wild-type N2 strain (fig. S8, A and B), while the level of N6-mA in the nmad-1 mutant was significantly elevated (fig. S8C). These results were consistent with those reported previously (6, 31). For nmad-1 loss-of-function worms (heat-shocked P0, recovered F1, or F2), no change was detected for both N6-mA levels and life span compared with corresponding controls (fig. S8, C, J, and K), likely because the nmad-1 mutant, which has no demethylase activity, maintains a high level of N6-mA (fig. S8C); the mild heat shock could not induce a significant increase of the N6-mA to transmit heritable memory.
To determine which N6-mA–modified genes are affected when animals are subjected to heat stress, we immunoprecipitated N6-mA–modified DNA and conducted subsequent sequencing (MeDIP-seq). MeDIP-seq showed that the N6-mA DNA modification of some genes was significantly increased following heat stress, and the modification of some other genes was decreased (Fig. 5G). A recent study reported that over 80% of N6-mA DNA–marked regions intersected with regions occupied by heterochromatic histone modifications (H3K9me3 and/or H3K27me3) (32). We speculated that relationship existed between the DNA methylation–modified regions and the histone methylation–modified regions under heat stress. Notably, N6-mA and H3K9me3 marks were found at the same loci in the genome (Fig. 5J), and the ratio of the marks appeared to be associated with gene transcription activity. MeDIP-qPCR analyses revealed that, for heat stress response–related genes, reduced H3K9me3 (Fig. 4F) and elevated N6-mA (Fig. 5K) increased transcription levels (Fig. 4H). Under heat stress, the N6-mA elevation and H3K9me3 reduction collaboratively promote transcription of stress response genes to extend life span.
To determine whether the factors that respond to heat stress–induced life-span extension, including DAF-12, DAF-16, and HSF-1, may establish or maintain changes within the chromatin landscape following acute heat stress, we performed dot blot analyses on the corresponding mutants. We found that daf-16 regulated N6-mA level following heat stress (Fig. 5, H and I, and fig. S8, E, F, and H). By contrast, daf-12 and hsf-1 did not affect the increase in N6-mA level under heat stress (Fig. 5, D and F). However, inactivating the H3K9me3 methyltransferase genes set-25 and set-32 moderately increased N6-mA level in unstressed animals, but heat stress did not cause an extra increase in the N6-mA level in set-25 and set-32 animals (fig. S8, D and G). These results agreed with our observation that set-25 and set-32 mutations abolished the TEI effect induced by hormetic heat stress, and they further indicated that N6-mA was colocalized with H3K9me3 mark. In addition, in daf-16 loss-of-function worms, no difference of H3K9me3 modification levels was observed between animals with or without heat shock (fig. S9).
Together, our observation demonstrated that TEI induced by hormetic heat stress required daf-16, H3K9me3, and N6-mA modification. Upon hormetic heat stress, several genes are activated to respond to heat stress and prolong life span. At the same time, high-temperature memory will be passed down to the offspring, in which stress response genes and autophagy-related genes will be up-regulated in offspring to contribute to the TEI effects of hormetic heat stress–induced survival benefits. We also detected the effect of daf-16, H3K9me3, and N6-mA modification on hormetic heat shock–induced TEI of animal fitness improvement. Our observations showed that loss of daf-16, set-25, and damt-1 abolished transgenerational inheritance of the α-synuclein deposition of reduction (fig. S10, A to C), the GFP::LGG-1 puncta elevation in the hypodermal seam cells (fig. S10, D to F), and the nuclear translocation of HLH-30 (fig. S10, G to I). Here, we applied damt-1 RNAi in LGG-1::GFP analyses, as we failed to obtain damt-1(tm6887); lgg-1::GFP double mutant by numerous matings. In addition, we also found the up-regulation of heat stress response genes (i.e., hsp-6, hsp-70, hsp-12.6, and hsp-16.2) to be eliminated because of the loss of set-32, set-25, and damt-1 (fig. S10, J to L). These results indicated that daf-16, H3K9me3, and N6-mA modification played important roles in transmitting heritable memories.
DISCUSSION
Animals exposed to hormetic heat stress have the ability to activate an innate immune response, to extend life span, to increase stress resistance, and to maintain proteostasis (19). In this study, we found that parental generation could transmit information of survival benefit induced by hormetic heat shock to descendants to extend their life span. In agreement with a previous study (20), we found that high-temperature memory from parents can be transmitted to progeny to induce a series of genes expression, in which H3K9me3 marks were involved in the underlying mechanisms. New evidence in the present study suggested that the exposure to hormetic heat stress established H3K9me3-dependent and N6-mA–dependent transgenerational inheritance networks to confer gene expression and behaviors changes, which ultimately lead to a survival advantage in the progeny. Hormetic heat stress in the parent generation activates daf-16, hsf-1, and daf-12 to extend life span, and daf-16 regulates the N6-mA and H3K9me3 levels and establishes these epigenetic marks in descendants. In turn, H3K9me3 and N6-mA mark the progeny genome to promote the recruitment of stress response–related genes (i.e., daf-16, hsf-1, and target genes for daf-12, daf-16, and hsf-1) and facilitate the activation of the heat stress response, ultimately leading to the TEI of the survival benefit (Fig. 6).
Fig. 6. Working model of the transgenerational inheritance of survival advantages induced by hormetic heat stress.
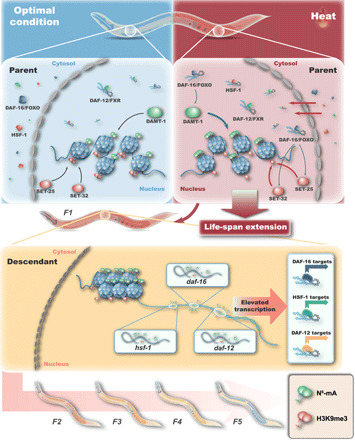
Findings in this study suggested that the functions of hsf-1, daf-16, and daf-12 were required for TEI induced by hormetic heat stress. Among them, hsf-1 and daf-12 were responsible for implementing heat stress response, while daf-16 acted in both implementation and transmission. Notably, daf-16 itself is critical for elevating the N6-mA levels and reducing the H3K9me3 levels to transmit the transgenerational memories. In addition, we also observed the autophagy induction, the nuclear translocation of HLH-30, and reduced protein aggregation in the naive offspring from heat-shocked parents. These findings demonstrate that daf-16, hsf-1, daf-12, and hlh-30 function in implementation, transmission, or both under thermal stress conditions to realize hormetic stress–induced survival benefit in both parent and descendant generations.
In present study, we found that the H3K9 methyltransferases set-25 and set-32 were both required for efficiently propagating high-temperature memory in all subsequent generations. The role of set-25 as the methyltransferase of H3K9me3 was identified by Zeller et al. (33), as no H3K9me3 was detectable in set-25 knockout worms. However, the role of set-32 is more complicated. Woodhouse et al. (34) found no difference in the levels of H3K9me3 between set-32 mutant with wild type, but others reports a correlation between loss of set-32 and a decrease in H3K9me3 modification (35, 36). The discrepancy reported by different researchers indicates that the role of set-32 in H3K9me3 modification might be different at specific loci or development stages or in response to particular stimuli.
Moreover, we found that set-25 and set-32 are both required for maintenance of TEI induced by hormetic heat stress in all generations. It seems to be different from what Woodhouse et al. (34) reported, in which they demonstrated that SET-25 and SET-32 are required for the establishment of a transgenerational silencing signal but not for long-term maintenance of this signal across subsequent generations. These interesting findings were not in contradiction of our results, as Klosin et al. (20) reported that the inactivation of RNAi components [such as Heritable RNAi Deficient (HRDE-1) or Nuclear RNAi DEfective (NRDE-2)] does not affect the H3K9me3-mediated epigenetic transmission of high-temperature memory.
Notably, met-2 deletion did not abrogate the survival advantage in the F1 or F2 generation (fig. S5P), suggesting that the TEI of heat stress–induced survival benefits does not require histone METhyltransferase-like (MET-2)/SETDB1. This makes our findings distinct from other TEI studies in C. elegans that were focused on the inheritance of exogenously induced silencing in the germ line (5, 29). In our settings, it was implied that the life-span extension in progeny had to do with their soma, although the signals must travel through the germ line to the next generation. This might bring up the possibility that SET-32, whose role remains variable in different studies (34–36), might play context-specific roles when MET-2 was not involved.
Recent studies have revealed interplay between N6-mA and H3K4me3 in C. elegans (6, 31), but interaction between H3K9me3 and N6-mA in C. elegans has not been reported. In our study, we found that a large fraction of genes marked by N6-mA overlapped with H3K9me3 occupancy genes. In C. elegans, it was reported that N6-mA is associated with active transcription (6), while H3K9me3 is well known as a transcription-repressive modification. Therefore, we hypothesized that, in our hormetic heat shock model, cross-talk between N6-mA–modified DNA and the H3K9me3 mark leads to a reduction in H3K9me3 occupancy and an increase in N6-mA level, thereby collaboratively promoting the expression of stress response genes to enhance resistance to stress and prolong life span. Note that the TEI induced by hormetic heat stress is exhibited in only a limited number of generations. A possible explanation is that, upon heat shock, the H3K9me3 level is reduced, while the N6-mA level and the heat stress response genes expression levels are elevated; in the later generations grown under normal conditions, these changes are gradually restored because of heterochromatin remodeling.
However, the mechanism by which DAF-16 induces epigenetic alterations in response to heat stress, as shown in this study, remains unclear. In the present study, we observed that N6-mA responds to heat stress and the occupancy of the promoters in heat stress response genes to promote life-span extension. However, how N6-mA methyltransferase DAMT-1 (DNA N6 Adenine MethylTransferase) recognizes and modifies the heat stress response genes needs to be investigated. It will be interesting to study the molecular mechanism that integrates and collaborates with this regulatory network consisting of DAF-16, DAF-12, HSF-1, HLH-30, H3K9me3, and N6-mA. It is also crucial to discover how epigenetic marks are gradually erased and restored.
In summary, we characterized the inheritance of hormetic heat effects across generations and identified the factors involved in the memory transmission. Our study provided a framework for further understanding the underlying molecular mechanism of the TEI of the effects induced by hormetic heat stress.
MATERIALS AND METHODS
C. elegans strain and cultivation
Strains were obtained from the Caenorhabditis Genetics Center and the National BioResource Project. Worms were maintained on nematode growth media (NGM) in plates at 20°C with Escherichia coli OP50 bacteria, as described previously unless otherwise stated (37). The following strains were used in this study: Bristol N2 wild type, CF1038 daf-16(mu86) I, PS3551 hsf-1(sy441) I, AA89 daf-12(rh274) X, tm5092 spr-5 (tm5092) I, RB1941 rbr-2(ok2544) IV, VC767 set-18(gk334) I, MT17463 set-25(n5021) III, VC967 set-32(ok1457) I, tm6887 damt-1 (tm6887) II, VC40319 damt-1 (gk961032) II, VC2552 nmad-1(ok3133) III, MT13293 met-2(n4256) III, NL2099 rrf-3(pk1426) I, OG532 drSi13 [hsf-1p::hsf-1::GFP::unc-54 3′UTR II + Cbr-unc-119(+)], TJ356 zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su1006)], NL5901 pkIs2386 [unc-54p::α-synuclein::YFP + unc-119(+)], DA2123 adIs2122 [lgg-1p::GFP::lgg-1 + rol-6(su1006)], MAH240 sqIs17 [hlh-30p::hlh-30::GFP + rol-6(su1006)], and MT18143 nIs286 [mir-71(+) + sur-5::GFP] X.
RNA interference
RNAi experiments were performed using the standard feeding protocol as previously described (38). Bacterial clones expressing the control (empty vector, pL4440) construct and double-stranded RNA targeting C. elegans genes were obtained from the Ahringer library (Source Bioscience, Nottingham, UK). All RNAi clones were verified before use. RNAi bacteria were grown at 37°C in LB with ampicillin (100 μg/ml) and then freshly spotted on NGM plates containing 1 mM isopropyl-β-d-thiogalactoside (IPTG) and ampicillin (100 μg/ml). RNAi silencing was conducted by transferring synchronized L1 larvae to fresh plates with gene-specific RNAi bacteria.
Environmental stressor conditions
For heat stress induction, ~500 animals were transferred to a single non-prewarmed OP50-seeded 90-mm NGM plate and put into a 35°C incubator on day 1 for 1 hour. To obtain F1 progeny, ~500 stressed P0 worms were shifted back from 35° to 20°C to for 8 hours before they were bleached. As a control, ~500 unstressed gravid adults at the same age were also bleached to obtain F1 animals. Afterward, the eggs were hatched overnight in 10 ml of M9 buffer, and the larvae were subsequently cultured at 20°C on NGM plates for 3 days. F2 to Fn generations were obtained using the same protocol, except that they were not stressed.
Life-span assay
All life-span assays were performed at 20°C on the basis of standard protocols, as previously described (38). Briefly, ~100 late L4 larvae or young adults were transferred to NGM plates with or without 10 μM FUDR (Sigma-Aldrich) seeded with heat-inactivated OP50. RNAi life-span assays were performed similarly, except that worms were fed with live RNAi bacteria on IPTG-containing NGM plates. The day when the worms were transferred to the plates was defined as day 1. Death events were scored daily. The experiments were repeated at least twice. The mean, SEM, P, and life-span values are summarized in table S1.
Mating protocol
To distinguish the cross-progeny from the self-fertilized ones, we used a GFP+ strain MT18143. Spontaneous males were picked out and propagated. Before mating, day 1 worms were picked onto non-prewarmed OP50-seeded NGM plates to heat shock at 35°C for 1 hour. Worms (a ratio of males and females of 3:1) were put onto OP50-seeded NGM plates to mate for 24 hours at 20°C. Two days after mating, at least 60 GFP+ F1 hermaphrodites were picked out for life-span assays, and another ~500 GFP+ hermaphrodites were bleached to obtain F2 descendants.
Quantitative reverse transcription PCR assay
Total RNA was extracted using RNAiso Plus (TaKaRa) on the basis of the phenol-chloroform extraction method from frozen day 1 animals. Reverse transcription was performed using a cDNA reverse transcription kit (TaKaRa or ABclonal) following the manufacturer’s protocol. qPCR was conducted with a CFX96 real-time system (Bio-Rad) using SYBR Premix Ex Taq II (TaKaRa) or SYBR Green select master mix (ABclonal). For each gene, triplicate biological samples and three technical replicates were conducted. The relative mRNA levels were calculated by the 2–ΔΔCt method after normalization to the reference gene cdc-42. P values were calculated using two-tailed Student’s t tests. The primers used in this study are summarized in table S2.
Western blot analysis
Animals were grown synchronously to day 1 adults and collected in M9 buffer. After three washes, pellets were snap-frozen in liquid nitrogen and stored at −80°C. To lyse the worms, pellets were put in radioimmunoprecipitation assay buffer, ground twice using a TissueLyser at 75 Hz for 6 min at low temperature, and then centrifuged at 10,000g at 4°C to collect the supernatant. Protein concentrations were measured using the BCA Protein Assay Kit. Proteins were separated using SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The membranes were blocked in 5% milk and then incubated with primary antibodies to H3K9me3 (1:1000; Abcam, ab176916), H3 antibody (1:10,000; Cell Signaling Technology, H9715), GFP (1:5000; Roche, 11814460001), or actin (1:5000; Sigma-Aldrich, A1978). Then, protein was visualized using horseradish peroxidase–conjugated anti-rabbit (1:5000) or anti-mouse (1:5000) secondary antibody and ECL Western Blotting Substrate.
H3K9me3 ChIP-qPCR
ChIP experiments were conducted as previously described (39). Embryo pellets (50 to 100 μl) were washed with M9 buffer three times and incubated in ChIP cross-link buffer [M9 containing 2% formaldehyde (Sigma-Aldrich)] at room temperature for 30 min, and then the reaction was terminated with 2.5 M glycine rotated at room temperature for another 5 min. After cross-link termination, the samples were washed and resuspended in 2 ml of FA buffer [50 mM Hepes/KOH (pH 7.5), 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, and 150 mM NaCl] with a proteinase inhibitor cocktail (Roche) and sonicated for 13 cycles (each cycle, 30-s on and 30-s off) with a Bioruptor 200 sonication system (Diagenode). The lysates were precleared and then immunoprecipitated with 5 μl of H3K9me3 antibody (Abcam, ab8898) at 4°C for 12 to 16 hours. Then, 40 μl of preblocked SureBeads starter kit protein G (Bio-Rad) was added and rotated at 4°C for 2 hours. Then, the beads were washed sequentially with 1 ml of each of the following buffers as indicated: twice with FA buffer (5 min each time), once with 1 M NaCl FA buffer (10 min), twice with 500 mM NaCl FA buffer (10 min each time), twice with TEL buffer [0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, and 10 mM tris-HCl (pH 8.0); 10 min each time], and lastly, twice with 1× TE buffer (5 min each time). Then, the pellets were eluted with 200 μl of elution buffer (1% SDS in TE with 250 mM NaCl). The supernatant and the input samples were treated with proteinase K (0.1 μg/ml) for 2 hours at 50°C and then overnight at 65°C. The DNA was purified using the phenol/chloroform/isoamyl extraction method for qPCR. ChIP-qPCR data on the targeted genes were normalized to those of the coimmunoprecipitated ama-1. The sequences of qPCR primers are listed in table S2. For the data on each gene, triplicate biological samples and three technical replicates were used.
H3K9me3 ChIP-seq
ChIP assays were performed according to the previously described, with few modifications (6). ChIP-seq libraries were prepared and sequenced by Wuhan IGENEBOOK Biotechnology Co. Ltd. Briefly, 3 g of C. elegans were harvested and washed with cold phosphate-buffered saline buffer and then cross-linked with formaldehyde at a final concentration of 1%. Afterward, samples were lysed on ice to obtain chromatins. Chromatins were sonicated to get soluble sheared chromatin [average DNA length of 200 to 500 base pairs (bp)]. One part of the soluble chromatin was saved at −20°C for input DNA, and the remainder was used for immunoprecipitation by H3K9me3 antibody (Abcam, ab8898).
Immunoprecipitated DNA was used to construct next-generation sequencing libraries following the protocol of the Illumina TruSeq ChIP Sample Prep Set A and then sequenced using Illumina X Ten with PE 150. Trimmomatic (version 0.38) was used to filter out low-quality reads. Clean reads were mapped to the C. elegans genome by BWA (Burrows-Wheeler alignment tool) (version 0.7.15) (6), allowing up to two mismatches. After removing potential PCR duplicates by using SAM Tools (version 1.3.1), the peaks were identified by using MACS2 peak caller software (version 2.1.1.20160309) with default parameters, and then wig files produced by MACS software were used for data visualization using Integrative Genomics Viewer (version 2.3.91). The diffpeak was identified using DiffBind (version 1.16.3). Gene ontology (GO) enrichment analysis was performed using the EasyGO gene ontology enrichment analysis tool (http://bioinformatics.cau.edu.cn/easygo/). The GO term enrichment was calculated using hypergeometric distribution with a P value cutoff of 0.01. KEGG (Kyoto Encyclopedia of Genes and Genomes; http://genome.jp/kegg/) enrichment analysis was performed using clusterProfiler (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) in R package. The heatmaps, reads distribution of diffpeak, and all peaks were obtained using deepTools (version 3.2.1) (40).
Purification of genomic DNA from the worms
The purification of genomic DNA (gDNA) from the worms was performed as previously described (31). Young adult animals were harvested with M9 and lysed with worm gDNA lysis buffer [200 mM NaCl, 100 mM tris-HCl (pH 8.5), 50 mM EDTA (pH 8.0), 0.5% SDS, and proteinase K (0.1 mg/ml)]. The lysate was then incubated at 65°C for 1 hour with periodic vortexing. Then, the samples were incubated at 95°C for 20 min to inactivate the proteinase K. After that, ribonuclease A (RNase A) was added and incubated at 37°C for 1 hour. Then, an equal volume of phenol:chloroform:isopentanol (25:24:1) was added to the sample, mixed, and centrifuged (13,000 rpm, room temperature) for 5 min. The aqueous phase was transferred into a new tube, in which 0.1 volume of sodium acetate and 2 volumes of 100% EtOH were added. The mixtures were incubated at −20°C overnight. Then, the samples were spun at 16,000g at 4°C for 10 min to obtain the DNA pellet, which was washed with 70% EtOH, air-dried, and resuspended in TE buffer. Then, the purified gDNA was treated with RNase A/T1 (Thermo Fisher Scientific) and RNase H (New England Biolabs) for 1 hour at 37°C, followed by repurification of the gDNA, starting at the phenol/chloroform/isopentanol extraction step.
Dot blot analysis
Dot blots were performed as previously described (6). gDNA samples were denatured at 95°C for 10 min and cooled on ice for 5 min. For each sample, 300 ng of DNA was spotted on a membrane (Amersham, Hybond-N+, GE) and air-dried for 5 min, followed by baking at 80°C for 1 hour and ultraviolet cross-linking. The membrane was blocked with 5% nonfat milk in tris-buffered saline containing 0.1% Tween-20 for 2 hours at room temperature and then incubated overnight with N6-mA antibodies (1:1000; Synaptic Systems, 202-003) at 4°C. After three washes, the membrane was incubated with secondary antibody for 1 hour at room temperature. Signals were detected with a Bio-Rad ChemiDoc MP imaging system.
MeDIP-qPCR
MeDIP-qPCR was performed as previously described (6). Briefly, worm gDNA was fragmented to approximately 400 bp using a Bioruptor 200 sonication system (Diagenode). The fragmented DNA (15 μg) was immunoprecipitated with 5 μl of N6-mA antibody (Synaptic Systems, 202-003) at 4°C in a final volume of 500 μl containing MeDIP buffer [10 mM sodium phosphate (pH 7.0), 140 mM NaCl, and 0.05% Triton X-100]. Then, 40 μl of SureBeads starter kit protein G (Bio-Rad) was added and incubated at 4°C for 4 hours. Afterward, the beads were washed with MeDIP buffer (five times) and then digested with proteinase K digestion buffer [50 mM tris-HCl (pH 8.0), 10 mM EDTA, 0.5% SDS, and proteinase K (2.5 mg/ml)] for 3 hours at 50°C. The DNA was purified and used for qPCR. The sequences of the qPCR primers are listed in table S2. For each gene, triplicate biological samples and three technical replicates were conducted.
MeDIP-seq
Worm gDNA was sonicated to 200 to 300 bp with a Bioruptor 200 sonication system (Diagenode). Then, adaptors were ligated to gDNA using a NEBNext Ultra II DNA library prep kit (Illumina). The ligated DNA was denatured at 95°C for 10 min. A portion of 10 μl of DNA was saved as input. Then, the gDNA was pulled down according to the MeDIP-qPCR protocol. After that, the N6-mA–enriched DNA fragments were purified. Then, immunoprecipitated DNA and input DNA were amplified by PCR with NEBNext Multiplex Oligos for Illumina following the manufacturer’s protocol and were then subjected to sequencing with Illumina HiSeq 4000 sequencing. MeDIP-seq was sequenced by Wuhan IGENEBOOK Biotechnology Co. Ltd. The adapter and low-quality reads were filtered out through Trimmomatic (version 0.38). Clean reads were mapped to the C. elegans genome by BWA (version 0.7.15), allowing up to two mismatches. Then, the data analysis was performed similarly to ChIP-seq using the same pipeline and software.
RNA-seq data analysis
Total RNA was extracted using the RNAprep Pure Kit DP432 (TIANGEN Biotech Co. Ltd., Beijing, China), following the manufacturer’s instructions. All RNA samples were assessed for their integrity using Qsep1 instrument. After polyadenylate-selected RNA extraction, RNA fragmentation, random hexamer-primed reverse transcription, 100-nucleotide paired-end sequencing was performed by BGI 500. Differentially expressed genes were identified using DESeq2 with a filter threshold of adjusted q value < 0.05 and |log2 fold change| > 1. GO and KEGG analysis were performed using the same method as ChIP-seq.
DAF-16, HLH-30, and HSF-1 localization
DAF-16, HLH-30, and HSF-1 localization was determined using the worm strains TJ356, MAH240, and OG532, respectively. For each assay, at least 30 worms were anesthetized in M9 containing sodium azide (50 mM), mounted on 2% agarose pads, and directly observed under a Zeiss Axio Imager Z2 with an Apotome.2 with a 63× oil immersion/1.4–numerical aperture objects. Muscle or intestine cells were identified on the basis of the morphology and size of nuclei. Z position was selected so that nuclei were clearly in focus. Each experiment was repeated at least three times.
α-Synuclein aggregation assay
The aggregation of α-synuclein protein was analyzed using the NL5901 strain. Animals were raised on NGM plates at 20°C and treated as described in the “Environmental stressor conditions” section to obtain P0, F1, and F2 generation animals. Day 1 animals were transferred to 2% agarose pads after paralyzing using 10 μM levamisole and imaged using a Nikon Ti2-U epifluorescence microscope with 20× air objectives. To demonstrate the fluorescence intensity, six to seven randomly selected worms were put together to take images. For quantification purpose, each worm was imaged individually. To eliminate bias caused by different focal planes, we took images right through the middle of worms. The whole-animal fluorescence images were containing an entire worm taken and outlined to quantify the total fluorescence intensity of each worm. Aggregate α-synuclein protein levels were measured by mean fluorescence intensity (total intensity/area) using ImageJ. Flat-field and auto-fluorescence correction was performed on the basis of the methods as previously reported by Klosin et al. (20). Nematodes that overlap or are at the edge of the image were not analyzed. Two-tailed Student’s t test was performed using GraphPad Prism. At least 30 animals were used for each experiment, and each experiment was repeated at least three times.
Autophagy measurements
Autophagy was determined by counting the GFP-positive LGG-1 puncta in the seam cells of strain DA2123. For imaging and puncta quantification, the animals were mounted on a 2% agarose pad, and GFP::LGG-1 puncta were counted in the hypodermal seam cells with a Zeiss Axio Imager Z2 with an Apotome.2 microscope at 630-fold magnification. At least 30 animals were imaged for each condition, and two to three seam cells were counted for each worm. The results from three independent assays were combined, and the statistical analyses were performed using Student’s t tests.
Supplementary Material
Acknowledgments
We thank L. He and S. Guang from the University of Science and Technology of China and B. Harry from the University of Colorado for advising and proofreading the manuscript. We thank X. Lai for technical help on the qPCR procedure. We thank the Caenorhabditis Genetic Center (CGC) and the National BioResource Project (NBRP) for providing the worm strains, which is funded by the NIH Office of Research Infrastructure Programs (P40OD010440). Funding: This work was supported by the National Key R&D Program of China (2018YFC2002000), the Program of Introducing Talents of Discipline to Universities (111 Project, no. B16021), the Natural Science Foundation of Guangdong Province, China (2018A0303131003), the China Postdoctoral Science Special Foundation (2018M633288), and the Science and Technology Plan Project of Guangzhou, China (202002030021). Author contributions: Q.Z. and Q.-L.W. designed the study. Q.-L.W., X.M., W.D., Z.L., C.W., X.F., J.Y., and Q.Y. conducted the experiments. Q.Z., Q.-L.W., X.M., and W.D. analyzed the data. Q.-L.W. wrote the manuscript. Q.Z. reviewed and edited the manuscript. All the authors commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabc3026/DC1
REFERENCES AND NOTES
- 1.Cavalli G., Paro R., The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell 93, 505–518 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Han S., Schroeder E. A., Silva-Garcia C. G., Hebestreit K., Mair W. B., Brunet A., Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 544, 185–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charlesworth A. G., Seroussi U., Claycomb J. M., Next-gen learning: The C. elegans approach. Cell 177, 1674–1676 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Moore R. S., Kaletsky R., Murphy C. T., Piwi/PRG-1 argonaute and TGF-β mediate transgenerational learned pathogenic avoidance. Cell 177, 1827–1841.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greer E. L., Maures T. J., Ucar D., Hauswirth A. G., Mancini E., Lim J. P., Benayoun B. A., Shi Y., Brunet A., Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479, 365–371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma C., Niu R., Huang T., Shao L. W., Peng Y., Ding W., Wang Y., Jia G., He C., Li C. Y., He A., Liu Y., N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat. Cell Biol. 21, 319–327 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Skvortsova K., Iovino N., Bogdanovic O., Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 19, 774–790 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Perez M. F., Lehner B., Intergenerational and transgenerational epigenetic inheritance in animals. Nat. Cell Biol. 21, 143–151 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Cubas P., Vincent C., Coen E., An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Seong K. H., Li D., Shimizu H., Nakamura R., Ishii S., Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145, 1049–1061 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Kishimoto S., Uno M., Okabe E., Nono M., Nishida E., Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat. Commun. 8, 14031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demoinet E., Li S., Roy R., AMPK blocks starvation-inducible transgenerational defects in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 114, E2689–E2698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rechavi O., Houri-Ze’evi L., Anava S., Goh W. S. S., Kerk S. Y., Hannon G. J., Hobert O., Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 158, 277–287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton N. O., Riccio C., Dallaire A., Price J., Jenkins B., Koulman A., Miska E. A., Cysteine synthases CYSL-1 and CYSL-2 mediate C. elegans heritable adaptation to P. vranovensis infection. Nat. Commun. 11, 1741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hercus M. J., Loeschcke V., Rattan S. I., Lifespan extension of Drosophila melanogaster through hormesis by repeated mild heat stress. Biogerontology 4, 149–156 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Rattan S. I., Ali R. E., Hormetic prevention of molecular damage during cellular aging of human skin fibroblasts and keratinocytes. Ann. N. Y. Acad. Sci. 1100, 424–430 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Kumsta C., Chang J. T., Schmalz J., Hansen M., Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat. Commun. 8, 14337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Rosa M. J., Veuthey T., Florman J., Grant J., Blanco M. G., Andersen N., Donnelly J., Rayes D., Alkema M. J., The flight response impairs cytoprotective mechanisms by activating the insulin pathway. Nature 573, 135–138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas P. M., Baird N. A., Simic M. S., Uhlein S., McCormick M. A., Wolff S. C., Kennedy B. K., Dillin A., Heterotypic signals from neural HSF-1 separate thermotolerance from longevity. Cell Rep. 12, 1196–1204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klosin A., Casas E., Hidalgo-Carcedo C., Vavouri T., Lehner B., Transgenerational transmission of environmental information in C. elegans. Science 356, 320–323 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Norouzitallab P., Baruah K., Vandegehuchte M., Van Stappen G., Catania F., Vanden Bussche J., Vanhaecke L., Sorgeloos P., Bossier P., Environmental heat stress induces epigenetic transgenerational inheritance of robustness in parthenogenetic Artemia model. FASEB J. 28, 3552–3563 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Otin C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Ham T. J., Thijssen K. L., Breitling R., Hofstra R. M., Plasterk R. H., Nollen E. A., C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLOS Genet. 4, e1000027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simmer F., Tijsterman M., Parrish S., Koushika S. P., Nonet M. L., Fire A., Ahringer J., Plasterk R. H., Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr. Biol. 12, 1317–1319 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Morton E. A., Lamitina T., Caenorhabditis elegans HSF-1 is an essential nuclear protein that forms stress granule-like structures following heat shock. Aging Cell 12, 112–120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horikawa M., Sural S., Hsu A. L., Antebi A., Co-chaperone p23 regulates C. elegans lifespan in response to temperature. PLOS Genet. 11, e1005023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henderson S. T., Johnson T. E., daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 11, 1975–1980 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S. D., Quiros P. M., Steffen K. K., Williams E. G., Mouchiroud L., Tronnes S. U., Murillo V., Wolff S. C., Shaw R. J., Auwerx J., Dillin A., Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greer E. L., Maures T. J., Hauswirth A. G., Green E. M., Leeman D. S., Maro G. S., Han S., Banko M. R., Gozani O., Brunet A., Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooney C. A., Dave A. A., Wolff G. L., Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 132, 2393S–2400S (2002). [DOI] [PubMed] [Google Scholar]
- 31.Greer E. L., Blanco M. A., Gu L., Sendinc E., Liu J., Aristizabal-Corrales D., Hsu C. H., Aravind L., He C., Shi Y., DNA methylation on N6-adenine in C. elegans. Cell 161, 868–878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie Q., Wu T. P., Gimple R. C., Li Z., Prager B. C., Wu Q., Yu Y., Wang P., Wang Y., Gorkin D. U., Zhang C., Dowiak A. V., Lin K., Zeng C., Sui Y., Kim L. J. Y., Miller T. E., Jiang L., Lee C. H., Huang Z., Fang X., Zhai K., Mack S. C., Sander M., Bao S., Kerstetter-Fogle A. E., Sloan A. E., Xiao A. Z., Rich J. N., N6-methyladenine DNA modification in glioblastoma. Cell 175, 1228–1243.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeller P., Padeken J., van Schendel R., Kalck V., Tijsterman M., Gasser S. M., Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 48, 1385–1395 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Woodhouse R. M., Buchmann G., Hoe M., Harney D. J., Low J. K., Larance M., Boag P. R., Ashe A., Chromatin modifiers SET-25 and SET-32 are required for establishment but not long-term maintenance of transgenerational epigenetic inheritance. Cell Rep. 25, 2259–2272.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Kalinava N., Ni J. Z., Peterman K., Chen E., Gu S. G., Decoupling the downstream effects of germline nuclear RNAi reveals that H3K9me3 is dispensable for heritable RNAi and the maintenance of endogenous siRNA-mediated transcriptional silencing in Caenorhabditis elegans. Epigenetics Chromatin 10, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lev I., Gingold H., Rechavi O., H3K9me3 is required for inheritance of small RNAs that target a unique subset of newly evolved genes. eLife 8, e40448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenner S., The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan Q.-L., Meng X., Fu X., Chen B., Yang J., Yang H., Zhou Q., Intermediate metabolites of the pyrimidine metabolism pathway extend the lifespan of C. elegans through regulating reproductive signals. Aging 11, 3993–4010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu S. G., Pak J., Guang S., Maniar J. M., Kennedy S., Fire A., Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat. Genet. 44, 157–164 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iberg-Badeaux A., Collombet S., Laurent B., van Oevelen C., Chin K.-K., Thieffry D., Graf T., Shi Y., A transcription factor pulse can prime chromatin for heritable transcriptional memory. Mol. Cell. Biol. 37, e00372-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabc3026/DC1