

Highly accurate SR structures and damage-free XFEL structures provide detailed insight into catalytic mechanism of CuNiR.
Abstract
Copper-containing nitrite reductases (CuNiRs), encoded by nirK gene, are found in all kingdoms of life with only 5% of CuNiR denitrifiers having two or more copies of nirK. Recently, we have identified two copies of nirK genes in several α-proteobacteria of the order Rhizobiales including Bradyrhizobium sp. ORS 375, encoding a four-domain heme-CuNiR and the usual two-domain CuNiR (Br2DNiR). Compared with two of the best-studied two-domain CuNiRs represented by the blue (AxNiR) and green (AcNiR) subclasses, Br2DNiR, a blue CuNiR, shows a substantially lower catalytic efficiency despite a sequence identity of ~70%. Advanced synchrotron radiation and x-ray free-electron laser are used to obtain the most accurate (atomic resolution with unrestrained SHELX refinement) and damage-free (free from radiation-induced chemistry) structures, in as-isolated, substrate-bound, and product-bound states. This combination has shed light on the protonation states of essential catalytic residues, additional reaction intermediates, and how catalytic efficiency is modulated.
INTRODUCTION
Understanding catalysis by enzymes underpinned by high-resolution three-dimensional structures has attracted major attention from structural enzymologists who have catalyzed the advances in experimental capabilities by continuing to push the capabilities of synchrotron radiation (SR) crystallography improving brilliance of x-ray beams and detectors and have recently opened the possibility of obtaining damage-free, free from radiation-induced chemistry (FRIC), structures. We have applied these advanced methods to obtain the most accurate [SR structures at resolutions of ~1 Å enabling unrestrained, SHELX refinement for any copper-containing nitrite reductase (CuNiR)] and damage-free x-ray free-electron laser (XFEL)–FRIC structures, in the as-isolated, substrate-bound, and product-bound states, again to high resolutions of 1.3 Å. For all of these structures, optical spectra were collected on single crystals. This combination has led to a real scientific advance in the understanding of the catalytic mechanism of NiRs, which are part of the nitrogen cycle.
The global nitrogen cycle has seen a marked imbalance over the years as a result of anthropogenic factors, primarily from an increased use of artificial nitrogenous fertilizers and the burning of fossil fuels (1, 2). Major contributors to the nitrogen cycle are several α-proteobacteria of the order Rhizobiales, collectively Rhizobia, which uniquely form specific symbiotic relationships with leguminous plants such as soybean (3). In addition to nitrogen fixation, Rhizobia are also able to use fixed nitrogen, in the form of nitrate, as a replacement for dioxygen during respiratory adenosine 5′-triphosphate synthesis through a dissimilatory denitrification process (4, 5). In doing so, they also contribute considerably to the removal of bioavailable fixed nitrogen from terrestrial sources and are responsible for a substantial balance in the world’s fixed nitrogen supply (3, 6, 7).
Denitrification involves the sequential reduction of nitrate (NO3−) and/or nitrite (NO2−) to dinitrogen (N2) through gaseous intermediates nitric oxide (NO) and nitrous oxide (N2O) in a four-step process involving specific reductases: NO3− → NO2− → NO → N2O → N2 (4, 5). A major by-product of denitrification is the greenhouse gas N2O, the largest and more potent ozone-depleting substance than CO2 (8, 9). In addition to their contribution to nitrogen fixation in worldwide agriculture, Rhizobia also make a substantial contribution to total denitrification and, consequently, atmospheric N2O production and hence are of major importance to both environment and agriculture (7, 10).
CuNiRs, encoded by the nirK gene, are key enzymes in the denitrification pathway, as it is at this point that the bioavailable fixed nitrogen gets recycled back to the atmosphere. These redox-active metalloenzymes are responsible for catalyzing the one-electron and two-proton reduction of NO2− to gaseous product NO in the first committed step of this pathway (NO2− + 2H+ + e−→NO + H2O) (4). CuNiRs are a highly conserved enzyme family that use cupredoxin-like domains to assemble a catalytic core made up of type 1 Cu (T1Cu) and type 2 Cu (T2Cu) (11). Structures of the well-characterized homotrimeric two-domain CuNiRs show the catalytic T2Cu center located at the interface of two monomers and is linked to the electron-donating T1Cu center by a 12 Å Cys-His bridge. Two invariant active-site pocket residues AspCAT and HisCAT are involved in substrate binding and provision of protons during catalysis, and a conserved IleCAT residue provides steric control of ligand binding to the T2Cu (12–14). Depending on the organism, the oxidized enzymes are blue or green, arising from differences in the geometry of the copper ligation of the T1Cu, often accompanied by a different physiological donor. These enzymes are located in the periplasm, and each class receives an electron from different partner proteins either cytochrome or a cupredoxin (15). To date, structural studies at atomic resolution of as-isolated, substrate-bound, and product-bound species are restricted to the green CuNiRs from Achromobacter cycloclastes (AcNiR) and Alcaligenes faecalis (AfNiR) (16–18). In contrast, ligand-bound species of blue NiRs such as Achromobacter xylosoxidans (AxNiR) have proved intractable to even high-resolution studies and are known to very limited resolutions of 2.34 and 2.8 Å (19, 20).
We have recently identified a blue CuNiR from Bradyrhizobium ORS 375 of the order Rhizobiales (Br2DNiR) (21). The catalytic efficiency of Br2DNiR was found to be substantially lower than two of the well-studied two-domain CuNiRs representing the blue (AxNiR) and green (AcNiR) subclasses, despite a sequence identity of ~70% (21). The blue crystals of Br2DNiR proved amenable to soaking of substrate and yielded highly diffracting crystals, providing a unique opportunity to obtain atomic-resolution structures of up to 1 Å with synchrotron x-rays and damage-free FRIC structures using XFEL, to ~1.3 Å resolution of as-isolated, nitrite- and nitric oxide–bound states. The significantly higher resolution of synchrotron structures allowed unrestrained SHELX refinement, while the XFEL structures of as-isolated and ligand-bound states allowed us to define these catalytically critical states without any x-ray–induced damage and unintended X-ray induced redox chemistry. We have generated the product in situ by using substrate-soaked crystals with full occupancy of substrate at the catalytic T2Cu site and treating them with a chemical reductant to initiate the turnover. This has allowed characterization of the stable nitrosyl intermediate with “side-on” binding unequivocally refuting the hypothesis that “the proton-coupled electron transfer to the nitrite-bound T2Cu leads to reduction of nitrite to form NO, which is released without forming a Cu-nitrosyl species” (22–24). Together, these results highlight structural aspects that are important for defining and regulating catalysis in CuNiR and thus should be considered before designing synthetic chemical systems that are capable of performing this widespread biological function in a variety of environmental and biomedical applications (25–27).
RESULTS
Structure of Br2DNiR at 1.1 Å atomic resolution using synchrotron-radiation crystallography
To characterize this new two-domain CuNiR enzyme, we obtained a 1.1 Å atomic-resolution structure in its as-isolated state using conventional synchrotron-radiation crystallography (SRX) at 100 K. To provide rigor and reduce the ambiguities in the assignments of multiple conformations of active-site ligands and define SDs of individual bond distances and angles of residues in the catalytic core, we used SHELX-97 (tables S2 to S4) [estimated standard deviation (e.s.d.) values available from SHELX are given in brackets] (28). A notable observation was the presence of two waters: (W1) and (W2) concurrently bound to the catalytic T2Cu site in one conformation (Fig. 1, A and B) and an alternative water (W3) in second conformation (Fig. 1, A and C). Two waters, W1 and W2, with equal occupancies (0.67) are 2.49 Å apart and bound to the T2Cu atom at distances of 1.940(3) Å and 2.162(3) Å, respectively (table S3). The occupancies of ~0.7 for these two waters strongly argue for their coexistence rather than alternate conformations of the same water. An alternative third water, W3, bound at 2.018(3) Å with ideal tetrahedral geometry to T2Cu with an occupancy of 0.33 represents the site in molecules where both W1 and W2 are absent (Fig. 1, A and C). The catalytically important AspCAT residue (Asp92) is present in both the main (occupancy, 0.67) and distorted (occupancy, 0.33) conformations of its proximal position, as first seen in XFEL structures of AcNiR (29), with these facing toward the T2Cu site (Fig. 1). Analysis of the carboxyl group bond lengths (Oδ1─Cγ and Oδ2─Cγ) in these conformations with the ideal bond length (fig. S1, D and E, and table S4) suggests the possibility that proximal position is negatively charged [1.25 Å (3)] and [1.26 Å (3)], while the carboxyl group in distorted proximal position is protonated [1.25 Å (7)] and [1.29 Å (8)] and neutral. Although seen together in our atomic-resolution structure, these two AspCAT conformations do not occur simultaneously but are only present either alongside W1 and W2 (main conformation) (Fig. 1B) or alongside W3 (distorted conformation) (Fig. 1C). In its main conformation, the carboxyl oxygen (Oδ2) atom of AspCAT is hydrogen bonded with water W1 and a substrate channel water (W4) with 0.5 occupancy at distances 2.52 and 2.45 Å, respectively, while Oδ2 atom of the distorted conformation is bound to W3 (3.13 Å). W4 can also hydrogen bond with W2, while a second position at 0.5 occupancy (W5) can hydrogen bond with W3 only at same distances (2.98 Å). All hydrogen bond contact distances are listed in table S5.
Fig. 1. T2Cu site in as-isolated Br2DNiR: SRX v SF-ROX structures.
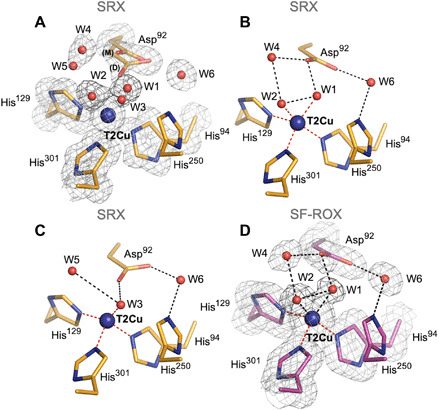
(A) In the 1.1 Å resolution SRX structure (orange), five waters (W1 to W5) and two conformations of Asp92: proximal (main and distorted) are clearly observed. (B) In one active-site conformation, positions of W1 and W2 at the T2Cu site are correlated with the main proximal conformation of Asp92. (C) In alternative conformation, water W3 is correlated with the distorted conformation of Asp92. (D) T2Cu site in the 1.3 Å XFEL FRIC structure (magenta) clearly shows two full-occupancy waters (W1 and W2) and Asp92 in main proximal conformation only. 2Fo − Fc electron density map, contoured at 1σ level, colored in gray. Water molecules are shown as red spheres, and T2Cu is shown as a blue sphere. Distances for possible bonding are shown as black dashed lines.
The catalytically important HisCAT residue (His250) has its imidazole ring rotated toward Glu274 forming a strong hydrogen bond by its Nδ1 atom with the main-chain carbonyl oxygen of Glu274 at 2.67 Å. While its Nδ1 atom is connected to AspCAT via water W6 (Figs. 1 and 2A). C─N─C angles associated with Nδ1 and Nε2 atoms of HisCAT are 112.5°(1.6) and 106.4°(1.5), respectively, which corresponds to protonated Nδ1 and nonprotonated Nε2 (fig. S1, A and B, and table S4) (30, 31) when compared with imidazole groups in the Cambridge Structural Database (32).
Fig. 2. Rotation of His250 (HisCAT) imidazole in each structure.
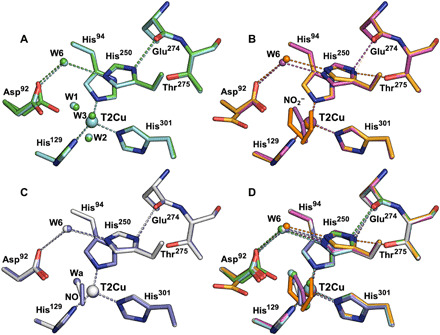
(A) Nδ1 atom of HisCAT in as-isolated Br2DNiR SRX (green) v SF-ROX (cyan) structures, both forming a strong hydrogen bond with main-chain O atom of Glu274. (B) HisCAT in nitrite-bound Br2DNiR SRX (orange) v SF-ROX (magenta) structures; Nδ1 atom of HisCAT forms a strong hydrogen bond with main-chain O atom of Glu274 in nitrite-bound SF-ROX structure but is rotated in nitrite-bound SRX structure to form a new hydrogen bond with Oγ1 of Thr275. (C) Nδ1 atom of HisCAT in NO-bound/T1Cu-reduced Br2DNiR SRX (white) v SF-ROX (purple) structures forms a strong hydrogen bond with main-chain O atom of Glu274. (D) Superimposition of all Br2DNiR structures, HisCAT rotation and change in hydrogen bond position, is only observed in atomic-resolution SRX nitrite-bound structure.
Damage-free XFEL FRIC structure of Br2DNiR at 1.3 Å resolution
To understand the nature of two waters seen bound to T2Cu in the atomic-resolution SRX structure, we obtained a 1.3 Å XFEL FRIC structure of Br2DNiR in its fully oxidized as-isolated state by using serial femtosecond rotational crystallography (SF-ROX) (33) technique at 100 K using XFEL. The oxidation state of the crystal was confirmed by ultraviolet-visible (UV/vis) microspectrophotometry before irradiation of one of the crystals at SACLA (SPring-8 Angstrom Compact free-electron LAser) (fig. S2). This damage-free FRIC structure obtained with pulses of SACLA of less than 10-fs duration (a time frame in which all of the atoms are frozen in space and time, as it is shorter than the rotational and vibrational frequencies) revealed the presence of two clear full-occupancy waters (W1) and (W2) bound to T2Cu, at distances of 2.05 and 1.94 Å, respectively, that are 2.51 Å apart. AspCAT is in main proximal conformation, and its Oδ2 atom forms similar hydrogen bonds as in the SRX structure with full-occupancy waters W4 and W1 at distances of 2.81 and 2.39 Å, respectively (Fig. 1D). In a recent damage-free XFEL structure of AfNiR (18), a rotation of the HisCAT imidazole ring was reported when compared to its SRX structure. This rotation was suggested to be catalytically relevant, with HisCAT having the role of a redox-coupled proton switch, essential for CuNiR catalysis. However, our results differ with no change seen between conformation of HisCAT in the SRX and XFEL (FRIC) structures of as-isolated Br2DNiR (Fig. 3A). The imidazole ring of HisCAT residue in the XFEL’s FRIC structure is rotated toward Glu274 forming a hydrogen bond between HisCAT Nδ1 atom and Glu274 O at 2.64 Å (Fig. 2A). We note that in the XFEL structure of AfNiR (18), a chloride present in the purification and crystallization stages had replaced the usual water ligand of the T2Cu. In the case of AxNiR, SF-ROX structure had revealed dioxygen was ligated to T2Cu (34).
Fig. 3. T2Cu site in nitrite-bound Br2DNiR: SRX v SF-ROX structures.
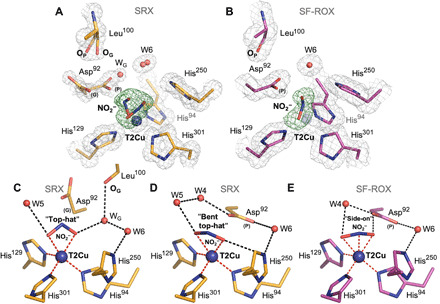
(A) The 1 Å SRX structure (orange) showing two top-hat conformations of nitrite and Asp92 (proximal and gatekeeper). A partial water (WG) and movement of Leu100 oxygen atom (OP > OG) are also seen. (B) The 1.3 Å XFEL FRIC structure (magenta) showing single nitrite in side-on position and Asp92 in proximal conformation. (C) SRX T2Cu site with bidentate-bonded top-hat nitrite present. (D) Alternative T2Cu of SRX structure with “bent top-hat” conformation of nitrite. (E) SF-ROX structure with horizontal side-on coordination of nitrite. Bonding and coordination of waters and atoms are shown for all. OMIT Fo − Fc electron density maps around nitrite molecules are contoured at 5σ level and colored green. 2Fo − Fc electron density maps are contoured at 1σ level in gray. Water molecules are shown as red spheres, and T2Cu is shown as a blue sphere. Hydrogen bonds are shown as black dashed lines, and coordination distances to T2Cu are shown as red dashed lines.
The electron-donating T1Cu site in both structures of Br2DNiR is similar to that of other two-domain CuNiRs, except the conformation of Met136, which is the terminal residue in an adjacent loop termed the “proton trigger loop” (Pro132-Met136). This loop connects to the proton-gated Cys-His electron transfer bridge. In Br2DNiR, Met136 is seen in only one conformation (flipped 180° away from His140) when compared to other two-domain CuNiRs, allowing a 2.79 Å hydrogen bond to form between T1Cu residue His140 and a new water (W10) as part of an ordered water network (fig. S3).
Two putative proton channels, one involving Asn90 and the other His254 (AxNiR numbering), have been proposed with the Asn90 channel as the probable main proton channel, based on mutational and kinetic studies (20). We have examined these two proton channels in both our 1.1 Å SRX and 1.3 Å XFEL FRIC structures in relation to AxNiR [Protein Data Bank (PDB): 10E1] and AcNiR (PDB: 2BW4) atomic-resolution SRX structures and the 1.5 Å XFEL damage-free structure of AcNiR (PDB: 6GSQ). The Asn90 channel in both SRX and XFEL structures of Br2DNiR shows no marked differences but is more similar to AcNiR structures (fig. S4A) than AxNiR structures (fig. S4B), with similar waters and hydrogen bonds constituting the network. Unusually, a half-occupancy water from the substrate channel (W7a) is favored in Br2DNiR Asn90 channel compared to AcNiR and AxNiR channels (W3/W2) to maintain strong bonding distances and efficient proton transfer (fig. S4, A and B). In Br2DNiR, Leu107 forms the entrance to the Asn90 channel in contrast to Asn107 (AxNiR) and Gln113 (AcNiR), which, similarly to green NiRs, results in a key hydrogen bond to Asn90 (as in blue AxNiR) being abolished. Residue Gly103 substitutes this by allowing a hydrogen bond to be formed between its carbonyl oxygen atom and Asn90 as well as a water molecule within the channel (fig. S4C). The surface residue Ala307 forms the entrance of the substrate channel to the Asn90 channel, substituting a bulky phenylalanine residue present in other two-domain CuNiRs; this allows more water molecules into the channel compared to other CuNiRs. The His254 channel is conserved in all structures of Br2DNiR, AxNiR, and AcNiR. The overall preservation of both proton channels suggests that these channels provide alternative routes, allowing the enzyme to use either of them depending on the denitrifying conditions including pH and/or nitrite concentration.
SRX structure of nitrite-bound enzyme at 1 Å atomic resolution
A 1 Å resolution structure of Br2DNiR with nitrite bound was obtained using one of the sophisticated SR crystallographic beamlines at the Diamond Light Source I03 equipped with DECTRIS PILATUS3 6M detector and crystal maintained at 100 K. The very high resolution was used to obtain the most accurate structural information by deploying SHELX. The OMIT (Fo − Fc) electron density map clearly shows that a nitrite molecule binds to T2Cu in two different “top-hat” conformations (Fig. 3A). The oxygen atoms of the first top-hat orientated nitrite are coordinated to T2Cu at distances of 2.13(6) and 1.98(2) Å (Fig. 3C), while a “bent top-hat” nitrite is bound at slightly longer distances of 2.22(5) Å and 2.13(2) Å for O1 and O2 atoms, respectively (Fig. 3D). Unrestrained refinement with estimated standard deviations (e.s.d.’s) analysis also showed that geometries of the two nitrite molecules are slightly different, the bond lengths of the top-hat nitrite are longer [1.30(4) Å] with a bond angle of 119.6°(3.5), while in the bent top-hat nitrite, they are shorter and slightly asymmetric [O1─N 1.25(6)] and [O2─N 1.27(3)] with a bond angle of 121.10°(4.0) (table S4). AspCAT is present in two different conformations, a proximal position, as seen in the as-isolated structures of Br2DNiR, and a gatekeeper position that was first reported in an atomic resolution study for AcNiR (16). This is the first observation of a gatekeeper position in a blue NiR, allowed by the 1 Å resolution of the structure with increased confidence provided by unrestrained SHELX refinement. The proximal and gatekeeper positions correlate well with the two conformations of bound nitrite (bent top-hat and top-hat), with refined AspCAT occupancies of (0.60) and (0.40) for proximal and gatekeeper positions, respectively. In addition, two conformations of the main-chain oxygen atom on residue Leu100 (named here for clarity as OP and OG), located in the Asn90 channel allow a suitable hydrogen bond to be formed with a partial-occupancy water (WG) that is now observed instead of the second carboxyl oxygen (Oδ1) atom of AspCAT proximal, with each coinciding with the presence and occupancy of the gatekeeper conformation (Fig. 3, A and C).
The position of WG allows a suitable hydrogen bond to form with top-hat nitrite O2 atom (2.73 Å), but Oδ2 atom of proximal AspCAT is situated too close (1.92 Å), emphasizing that proximal AspCAT and top-hat nitrite ligand are unlikely to be present at the same time. The proximal AspCAT Oδ2 atom can form weak hydrogen bonding with the bent top-hat nitrite O2 atom (3.42 Å). Analysis of the carboxyl groups bond lengths (Oδ1─Cγ and Oδ2─Cγ) in these conformations suggests that they are both negatively charged [the values are 1.25(3) and 1.26(3) for gatekeeper and 1.25(2) and 1.26(2) for the proximal position] (table S4). A partial-occupancy water (W5) present in the vicinity of T2Cu in line with O1 atoms in both conformations of nitrite is likely to be an important factor in stabilizing the substrate. Another notable feature of this structure is the visible rotation of the HisCAT imidazole ring toward the hydroxyl oxygen atom (Oϒ1) of Thr275 with equal hydrogen bonds for HisCAT Nδ1 atom (~2.7 Å) with both Thr275 and Glu274 (Fig. 2B). The C─N─C angles associated with Nδ1 and Nε2 atoms of HisCAT are 109.1°(5) and 109.2°(9), respectively, which corresponds to full residue protonation (fig. S1C and table S4) (31). The Nε2 atom of HisCAT is positioned within hydrogen bonding distance (3.06 Å) to bent top-hat nitrite O2 atom only (Fig. 3D), and W6, bridging HisCAT with AspCAT, is seen in two positions correlating with proximal and gatekeeper positions of AspCAT. The waters around the gatekeeper conformation of AspCAT are disordered, with multiple conformations associated with the two conformations of AspCAT.
XFEL FRIC structure of nitrite-bound enzyme at 1.3 Å resolution
As many or all diffraction images are collected from the same crystal volume in an SRX structure determination, nitrite-soaked CuNiR crystals undergo radiation-induced chemistry and turnover using electrons generated from the x-rays. To obtain a structure of nitrite-bound Br2DNiR FRIC structures, we obtained a 1.3 Å XFEL structure of Br2DNiR in its nitrite-bound state [confirmed by UV/vis microspectrophotometry on one of the crystals before irradiation at SACLA (fig. S2)] by using the SF-ROX technique (33) at 100 K. In SF-ROX, a single diffraction image is obtained from a single shot of <10-fs XFEL pulse per spot of a crystal, ensuring that data are captured before any x-ray–induced photochemistry can occur.
The most notable observation in this substrate-bound XFEL FRIC structure was the nitrite molecule at the T2Cu2+ site in a single side-on conformation with all three atoms O1, N, and O2 binding by almost identical distances of 2.12, 2.02, and 1.92 Å at an occupancy of 0.7 (Fig. 2B). This is the first time that a single side-on nitrite has been observed at the T2Cu2+ site in an XFEL FRIC structure from any CuNiR. The positions of O1 and O2 atoms are very close to top-hat conformation of nitrite in the atomic-resolution SRX structure. The side-on nitrite in this FRIC structure is now in an ideal position to the full-occupancy proximal AspCAT, with its O2 atom 2.25 Å away from AspCAT Oδ2 atom (Fig. 3, B and E). Half-occupancy water W4 is now bonded with O1 atom of nitrite. HisCAT has its imidazole ring and its Nδ1 atom rotated toward residue Glu274 carbonyl oxygen again with a shorter hydrogen bond of 2.52 Å in comparison with the atomic-resolution nitrite-bound SRX structure (Fig. 2B) and both structures of Br2DNiR in its as-isolated state. Nε2 atom of HisCAT is positioned 3.17 Å away from side-on nitrite O2 atom and 2.94 Å from W6.
Low-dose (0.5 MGy) nitrite-bound SRX structure at 1.48 Å resolution
To confirm that the structural differences of HisCAT in our nitrite-bound structures were due to x-ray induced electron transfer after substrate binding in the 1 Å SRX structure, we obtained a low-dose [0.5 MGy] SRX dataset to 1.48 Å resolution. It showed a single “L-shaped” nitrite with coordination distances to T2Cu of 2.33, 1.78, and 1.92 Å for O1, N, and O2 atoms, respectively (fig. S5A). AspCAT is again present in both its main (occupancy, 0.6) and distorted conformation (occupancy, 0.2) of its proximal position, as seen in the as-isolated SRX structure. Distorted conformation Oδ2 atom is 1.95 Å away from the O2 atom of nitrite, and main conformation Oδ2 atom is 2.36 Å away. W6 is seen in two positions correlating with main proximal and distorted proximal positions of AspCAT. No change in HisCAT position is observed in comparison with the XFEL FRIC structure (fig. S5B), providing clear evidence that rotation of the HisCAT imidazole ring toward the hydroxyl oxygen (Oϒ1) atom of Thr275 seen in the 1 Å SRX structure is due to the initiation of the catalytic reaction induced by x-ray–generated electrons reaching the nitrite-bound T2Cu.
Enzymatically generated nitrosyl species and its SRX structure at 1.19 Å resolution
We have used a new approach to obtain the catalytically formed NO-bound species by chemically inducing NiR turnover in the crystals. The nitrite soaking of crystals that ensured full nitrite occupancy as demonstrated above was followed by a time-dependent soak with the strong reductant dithionite to initiate the catalytic reaction by chemical reduction of T1Cu with the expectation of capturing a NO-bound state through the catalytic conversion of bound nitrite to nitric oxide, with NO remaining bound to T2Cu as an essential reaction intermediate. The total reduction of T1Cu was monitored by obtaining UV/vis microspectrophotometry data on the colorless crystals (fig. S2).
A 1.19 Å structure from a reductant-treated nitrite-soaked crystal allowed us to perform unrestrained refinement with SHELX. A NO molecule could be placed at the T2Cu site with occupancy (0.73) refined by SHELX (Fig. 4, A and B). It is clearly coordinated in a side-on manner, with slightly asymmetric distances of 2.17(4) and 2.08(3) Å for Cu─N and Cu─O, respectively. The O atom of NO is positioned 2.48 Å away from Oδ2 of AspCAT in proximal position. Analysis of the carboxyl group bond lengths of AspCAT 1.24(3) Å for Oδ1─Cγ and 1.28(3) Å for Oδ2─Cγ suggests that AspCAT could be protonated at Oδ2 (table S4). W6 bridging water is connecting Oδ1 atom of AspCAT with HisCAT Nε2 at ~2.9 Å. HisCAT imidazole is in its usual position, forming a hydrogen bond between HisCAT Nδ1 and Glu274 carbonyl oxygen at 2.62 Å, similar to the as-isolated SRX structure. HisCAT is only protonated at Nδ1 atom, as C─N─C angle associated with Nδ1 atom is 113.5°(2.4), while similar angle associated with Nε2 is much smaller 105.0°(2.2), which corresponds to nonprotonated state (fig. S1A and table S4) (31).
Fig. 4. T2Cu site in NO-bound/T1Cu-reduced Br2DNiR: SRX v SF-ROX structures.
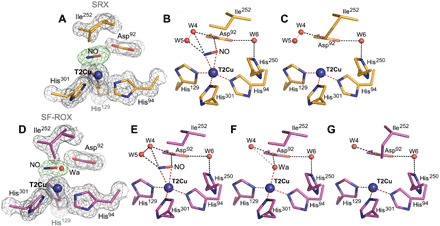
(A) SRX NO-bound structure (1.19 Å; orange) has two conformations of the active site: (B) one with NO present and (C) with three coordinated T2Cu site. (D) In the 1.3 Å XFEL FRIC structure (magenta), there are three alternative conformations of the T2Cu site: (E) one with NO, (F) second with water, and (G) third with three coordinated T2Cu site where NO has vacated the T2Cu site with alternative Ile252 conformation of CD atom flipping down to fill the space. Water molecules are shown as red spheres, and T2Cu is shown as a blue sphere. Distances for possible hydrogen bonding are shown as black dashed lines; gray dashed lines represent unlikely bonding due to steric restraints, and coordination distances to T2Cu are shown as red dashed lines. OMIT Fo − Fc electron density maps around T2Cu ligands are contoured at 5σ level and colored green. Gray 2Fo − Fc electron density map is contoured at 1σ level.
Enzymatically generated NO-bound XFEL FRIC structure determined at 1.3 Å resolution
We were successful in obtaining a damage-free XFEL structure of Br2DNiR in its NO-bound state at 100 K using the SF-ROX technique (33) using the dithionite-reduced nitrite-soaked crystals. We were able to capture three distinct species with structures representing stages of bound NO (resulting from in situ nitrite reduction) and its dissociation under these turnover conditions. The data are consistent with a mixture of enzymatically produced NO-bound, a ligand-free, and water-ligated T2Cu states (Fig. 4D). In the first conformation of the active site, NO was modeled bound to the T2Cu site with an occupancy of 0.4 (Fig 4, D and E), and it was coordinated to T2Cu with even greater asymmetry in the distances of 2.47 and 2.12 Å for Cu─N and Cu─O, respectively, with the O atom forming a hydrogen bond with Oδ2 atom of proximal AspCAT (2.56 Å) (Fig. 4E). In the second species, a single water molecule with occupancy of 0.3 was assigned at the T2Cu site (Fig. 4, D and F). This water coordinated to T2Cu at a distance of 2.07 Å is also able to hydrogen bond to Oδ2 of AspCAT by 2.16 Å (Fig. 4F). In the third species, the active site had a previously unseen conformation of the conserved IleCAT residue (Ile252) where the side chain is flipped to occupy the space in the T2Cu active-site cavity with an occupancy of 0.25 (Fig. 4, D and G). Again, because of steric restraints, this conformation can only exist when neither the NO nor the water ligand is present at the active site (Fig. 4G) and represents a new intermediate representing the status of catalytic Cu where NO is released but the T2Cu is tricoordinate and has not yet acquired the water ligand required for returning to the resting state. This Ile position is similar to Cu+(His)3 coordination formed on chemical reduction of the resting state of CuNiR, where the T2Cu is devoid of the water ligand, resulting in loss of activity (29, 35). As in the SRX NO-bound structure, the HisCAT imidazole ring is rotated toward Glu274 carbonyl oxygen atom at 2.61 Å (Fig. 2C). The reduction of T1Cu site, as confirmed by the absence of bands in optical spectra (fig. S2), results in slight elongation of electron density of the T1Cu atoms and surrounding residues, consistent with subtle changes expected to accompany Cu2+/Cu+ redox change for cupredoxin.
DISCUSSION
Accuracy needed for defining chemistry of catalytic pocket residues in resting and nitrite-bound enzyme
It has been shown previously that at atomic resolution, the bond lengths’ e.s.d.s. can be defined with an accuracy better than 0.008 Å (36). Such accuracy allowed us to determine the protonation states of some of the catalytically important residues from an analysis of the bond lengths in the catalytic coordination sphere. To remove influence of the crystallographic dictionary restraints on the structure, we used unrestrained refinement at the final step in SHELXL and determined e.s.d.s. for each of the bond lengths, including T1Cu and T2Cu coordinating spheres (tables S2 and S3). We also evaluated the protonation states of the catalytically important AspCAT (Asp92) and HisCAT (His250) residues. When aspartate is nonprotonated, the bond lengths between Cγ─Oδ1 and Cγ─Oδ2 are expected to be equal to 1.256(15) Å; upon protonation, one bond will increase to 1.310(17) Å and the other will become a double bond with a distance of 1.210(16) Å [data are from the Cambridge Structural Database (www.ccdc.cam.ac.uk)] (fig. S1, D and E). Analysis of the bond lengths for the carboxyl group of AspCAT in the SRX as-isolated structure at 1.1 Å resolution is consistent with the protonation, only when this residue is in the distorted position. In contrast, in the SRX nitrite-bound structure at 1 Å atomic resolution, AspCAT is negatively charged in both gatekeeper and proximal conformations. The SRX NO-bound structure at 1.19 Å resolution shows deviation in the bond lengths [1.24(3) Å for Oδ1─Cγ and 1.28(3) Å for Oδ2─Cγ)] and possible protonation of Oδ2 atom. The double conformations of AspCAT in both as-isolated and nitrite-bound SRX structures result in slightly reduced accuracy but are somewhat compensated by low-temperature factors and ~70% occupancy of the main conformation of AspCAT associated with two waters present in the active site (Fig. 1, A and B, and table S4). The analysis of bond angles for the HisCAT residue imidazole ring in SRX structures (table S4) suggests its full protonation only in the 1 Å resolution SRX nitrite-bound structure, where rotation of the imidazole ring is seen.
Role of distorted proximal conformation of AspCAT
The lower distorted proximal rotamer of AspCAT was first reported in the SF-ROX XFEL structure of the as-isolated green AcNiR (29), where it was suggested to shorten the hydrogen bond between the Oδ2 atom of AspCAT and the T2Cu coordinated W1, compared to main proximal position. However, as the same conformation in our structure appears to have an association only with the latent W3 within the T2Cu active site of SRX structure and also appears to be protonated, where its main conformation is not, suggesting a more important role. We may have either observed a flexible position of proximal AspCAT, which has a role in chaperoning T2Cu waters from the substrate channel/Asn90 proton channel to their coordinated positions within the T2Cu site or have captured a unique conformation where a proton is soon to be abstracted by AspCAT to a T2Cu ligand in as-isolated state. As distorted conformation is also observed in the low-dose nitrite-bound structure (0.5 MGy), its role as a conformation prepped for proton abstraction is more probable.
Mode of nitrite binding (side-on versus top-hat configuration) in NiR
The 1 Å SRX and 1.3 Å XFEL structures of nitrite-bound Br2DNiR allow a detailed comparison of nitrite binding in a blue CuNiR with those of green CuNiRs. It also helps to consolidate these high-resolution structural data to reach a consensus scheme. The two different conformations of the bidentate top-hat binding modes for nitrite in the SRX structure correlating with gatekeeper and proximal positions of AspCAT indicate that the mobility of this essential residue is critical in controlling the passage of the substrate and its anchoring in all CuNiRs (16, 29, 37, 38). The top-hat vertical bidentate position can only be present with AspCAT in its gatekeeper position in Br2DNiR because of steric constraints. The single horizontal side-on position of nitrite with coordination of all atoms to a fully oxidized Cu2+ site of Br2DNiR has been seen for the first time in an XFEL FRIC structure of a CuNiR. The clear observation of full-occupancy side-on horizontal coordination of nitrite provides unambiguous evidence that the bidentate oxygen binding mode of nitrite to a fully oxidized T2Cu2+ in contrast to the monodentate binding being proposed previously (39). It also indicates that the conformational change from top-hat to side-on position is not a result of reduction (29). As indicated earlier, Br2DNiR has a more open substrate channel compared to other CuNiRs, resulting in more waters being present in the vicinity of the T2Cu in the nitrite-bound structure compared to the XFEL structures of green AfNiR and AcNiR.
Enzymatically produced copper nitrosyl species defined
Our new approach to obtain NO-bound intermediate generated in turnover of a nitrite-soaked crystals of a CuNiR provides insight into binding of NO that is enzymatically generated. The observation of an in situ produced copper nitrosyl species in both the SRX and XFEL FRIC structures at ~1.2 Å unambiguously demonstrates this as a catalytically relevant reaction intermediate, refuting the hypothesis that “the proton-coupled electron transfer (PCET) to the nitrite-bound T2Cu leads to reduction of nitrite to form NO, which is released without forming a Cu-nitrosyl species” (22–24). The side-on binding copper-nitrosyl coordination observed in all of the crystallographic structural studies irrespective of the organisms (16, 37, 38, 40), including the current in situ enzymatically generated species, is at variance with “bent” or “end-on” binding modes suggested on the basis of synthesis of chemical compounds and computational studies (15, 25, 27, 41–44) and needs to be taken into account if the synthetic biomimetic systems are to prove effective NiRs.
Lower activity of Br2DNiR compared to AcNiR and AxNiR
Our as-isolated SRX and XFEL FRIC structure provides insight into the much lower catalytic efficiency of Br2DNiR compared to two of the well-studied two-domain CuNiRs represented by the blue (AxNiR) and green (AcNiR) subclasses (21). A notable feature of the as-isolated Br2DNiR compared with other CuNiRs is the presence of two water ligands simultaneously bound to T2Cu confirmed by both atomic-resolution SRX and XFEL FRIC structures. The XFEL FRIC structure clearly shows two full-occupancy waters coordinated to T2Cu. This contrasts from other CuNiRs where a single water is observed and is the likely origin of lower specific activity of this enzyme. We note that two ligands were also seen coordinated to T2Cu (partial waters or chloride and water) in CuNiR from Geobacillus thermodenitrificans (GtNiR), but contaminants close to the active site, including a sodium ion and/or the coordinated chloride ion and an extra copper ion at HisCAT, made the functional significance of these observations less clear (39, 45). A high-resolution neutron crystallographic structure of GtNiR suggested that one of these waters is actually a hydroxide ion (OH−) in its resting state (23), but again, the study revealed excess Cu (at least four) in the structure. Another case, where an extra water is seen close to the T2Cu site of a two-domain blue CuNiR, is on an F306C mutation of AxNiR, a surface residue at the mouth of the substrate access channel (46) some 12 Å from the T2Cu site. This mutation also showed considerably decreased catalytic efficiency, assigned to the second water, making nitrite binding energetically less favorable because of extra hydrogen bonding to the water bound to the T2Cu. Similarly, to this mutant, Br2DNiR has an alanine residue at the mouth of the substrate channel instead of a bulky phenylalanine seen in other well-studied two-domain CuNiRs (AxNiR, AcNiR, and AfNiR). This results in the active site being more accessible to solvent and is likely to be the reason for an extra water bound at T2Cu active site of as-isolated structure. The increased accessibility to solvent and two strongly coordinated waters may contribute to a much lower NiR catalytic activity in Br2DNiR when compared to AxNiR and AcNiR. Activity data for GtNiR have not been published.
Proposed catalytic reaction
In addition to being a substrate in the reaction catalyzed by CuNiRs, protons also have a role in gating electron transfer from the T1Cu site to the catalytic T2Cu site, both in the presence and absence of nitrite (20, 47). Our SHELX analysis of substrate- and product-bound intermediates allows the protonation states of two essential active site residues, AspCAT and HisCAT, to be defined throughout the catalytic cycle, allowing us to refine the proposed mechanism for CuNiRs. We suggest the following catalytic mechanism based on previous structural and biophysical studies of CuNiR and incorporating the new data presented here: The resting state of the oxidized enzyme has water (two waters in the case of Br2DNiR) bound to T2Cu site (Fig. 5A). AspCAT is in the proximal position, and neither AspCAT nor HisCAT Nε1 is protonated as seen in both SRX and XFEL FRIC structures and neutron crystallography of AcNiR, the only neutron study where only T1/T2Cu were present and no exogenous ligand was bound to the T2Cu (29). Initially, nitrite binds side-on with all three atoms ligated to the T2Cu site by displacement of the water ligand(s), as seen in the nitrite-bound XFEL FRIC structure (Fig. 5B), followed by protonation of HisCAT and the proposed Cu-NOOH intermediate (Fig. 5C), triggering proton-coupled electron transfer from T1Cu site and reduction of the T2Cu site. This then leads to reduction of nitrite via OH─N─O bond breakage to nitric oxide and release of water (Fig. 5, D1/D2). The release of water leaves NO coordinated in side-on manner (Fig. 5, D1/D2), consistent with a rigorous Density functional study (48). Release of nitric oxide leaves the T2Cu site tricoordinate (Fig. 5E), ready to capture a water molecule and return to the resting state (Fig. 5F) and enter the next cycle of nitrite binding and reduction.
Fig. 5. Proposed structure-based catalytic mechanism of Br2DNiR.
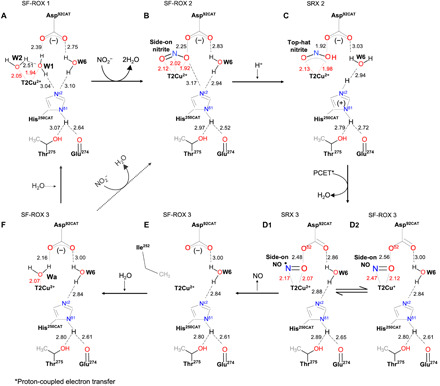
Solid arrows show the reaction sequence based on the structures of as-isolated and ligand-bound species of Br2DNiR described here (A to F). The further reaction of species (F) in CuNiRs that have a single water ligand of T2Cu in the resting state is shown as a dashed line. (A) Two waters bound in as-isolated SF-ROX structure 1 and in conformation 1 of as-isolated SRX structure 1. (B) Side-on nitrite bound in nitrite-bound SF-ROX structure 2. (C) Intermediate state with top-hat nitrite bound in nitrite-bound SRX structure 2. (D1) and (D2) represent two slightly different NO bound conformations in the SF-ROX 3 (D2) and SRX 3 (D1) structures; in D1, N and O of NO are almost equidistant. (E) Tricoordinate T2Cu site in NO-bound SF-ROX structure 3 where NO is no longer at the T2Cu site, resulting in the flipping of Ile252 into the space vacated from loss of fourth ligand. (F) Single water bound in NO-bound SF-ROX structure 3 where the water has been captured by the T2Cu site following the release of NO. The step from (F) to (B) represents the well-studied CuNiRs, such as AcNiR, AfNiR, and AxNiR, while (F) to (A) represents the return of Br2DNiR to its resting state.
CONCLUSIONS
We have undertaken a comprehensive structural study of a new two-domain blue CuNiR from Bradyrhizobium sp. ORS 375 using SR and XFEL to obtain the most accurate (SR structures at resolutions of ~1 Å, enabling unrestrained SHELX refinement, not undertaken for any CuNiR). We determined damage-free (XFEL) FRIC structures, in the as-isolated, substrate-bound, and product-bound states, to high resolutions of 1.3 Å. For all of the structures, optical spectra were collected on single crystals. This combination of structural approaches has allowed characterization of the stable nitrosyl intermediate with side-on binding, unequivocally refuting the hypothesis that the proton-coupled electron transfer to the nitrite-bound T2Cu leads to reduction of nitrite to form NO, which is released without forming a Cu-nitrosyl species. We demonstrate that during enzyme turnover, the mobility of AspCAT to adopt the gatekeeper and proximal positions is critical in controlling the passage of the substrate and its anchoring at the T2Cu site in both blue and green CuNiRs. The single horizontal side-on position of nitrite with coordination of all atoms to T2Cu2+ site of a fully oxidized of Br2DNiR enzyme crystals has been seen for the first time in any XFEL FRIC structure of a CuNiR. The ability to enzymatically generate the product in situ allowed us to capture “the act of product release” that leaves the T2Cu site tricoordinate. Our results illustrate that the combined approach of very-high-resolution SRX structures with unrestrained SHELX refinement together with FRIC structures on crystals using XFEL in well-defined states supported by single crystal spectroscopy has wider applicability to structural enzymology. It is likely to be particularly helpful in elucidating catalytic mechanisms of metalloenzymes, which would gain further boost from the availability of higher-energy x-ray pulses from XFEL that should allow even higher resolution structures approaching ~1 Å.
MATERIALS AND METHODS
Br2DNiR expression and purification for both as-isolated structures and atomic-resolution SRX nitrite-bound structure
Recombinant plasmid for wild-type (WT) two-domain BrNiR [pET-26b(+)-WTBr2DNiR-TEV-6xHis] was purchased from GenScript. Escherichia coli BL21 (DE3) cells harboring the plasmid were added to 500 ml of LB broth supplemented with kanamycin (50 μg/ml) and incubated at 37°C until an optical density at 600 nm of ~0.6 was reached. Overexpression of Br2DNiR was induced with 500 μM isopropyl-β-d-thiogalactopyranoside with 1 mM CuSO4 added, followed by incubation of cell culture for 18 hours at 180 rpm before cells were harvested by centrifugation. Cell pellet was collected and resuspended in 100 mM tris-HCl buffer (pH 8.0), containing 500 mM NaCl (buffer A) supplemented with lysozyme (50 μg/ml) and protease inhibitor and disrupted by sonication. Cleared cell lysate, collected by centrifugation, was dialyzed overnight against 1 mM CuSO4 in buffer A at 4°C. Blue lysate was loaded onto a HisTrap HP Ni affinity column (GE Healthcare), equilibrated with 10 mM imidazole in buffer A. Protein was eluted with 250 mM imidazole in the same buffer and consequently dialyzed overnight against 100 mM tris-HCl, 500 mM NaCl, and 10% (v/v) glycerol (pH 8) (buffer B) before loading onto a HiLoad Superdex 200 16/600 pg size exclusion chromatography (SEC) column (GE Healthcare) equilibrated with the same buffer B. A final overnight dialysis against 1 mM CuSO4 in buffer B at 4°C was required to achieve full copper loading of the T2Cu site. Before crystallization, protein was buffer-exchanged into 10 mM Hepes (pH 6.5).
Br2DNiR expression and purification for all other structures
As above but with the following changes, cleared cell lysate, collected by centrifugation, was loaded onto the same HisTrap HP column, equilibrated with 10 mM imidazole in buffer A. Protein was eluted with 250 mM imidazole in the same buffer, dialyzed against buffer B overnight, and then loaded onto to the same SEC column equilibrated with the same buffer. Elution was again dialyzed overnight against 1 mM CuSO4 in buffer B at 4°C and applied to a SEC column, as before. A final overnight dialysis was required to achieve full copper loading of the T2Cu site, with overnight dialysis against 1 mM CuSO4 in buffer B at 4°C. The solution was applied to a SEC column and then buffer-exchanged into 10 mM Hepes (pH 6.5) before crystallization.
Crystallization and crystal treatment
All Br2DNiR crystals were grown using the vapor diffusion hanging drop method at room temperature, with a protein concentration of 30 mg/ml mixed 1:1 with reservoir solution composed of either 1.6 or 1.8 M ammonium sulfate with 50 mM Hepes buffer (pH 5 or pH 5.5, respectively). Crystals in space group P213 grew within a few days.
Synchrotron-radiation crystallography
For SRX as-isolated experiment, the crystal was soaked into a cryoprotectant solution composed of 3.6 M ammonium sulfate and 50 mM Hepes buffer (pH 5.5), while SRX nitrite-bound crystals were soaked in a solution composed of 100 mM NaNO2, 2.4 M ammonium sulfate, and 50 mM Hepes buffer (pH 5.5) for 30 s and then transferred into a cryoprotectant solution composed of 100 mM NaNO2, 3.6 M ammonium sulfate, and 50 mM Hepes buffer (pH 5.5) before both were cryocooled by plunging into liquid nitrogen and stored in pucks for data collection. Crystals for SRX NO-bound structure was soaked in a nitrite solution composed of 200 mM NaNO2, 2.5 M ammonium sulfate, and 50 mM Hepes buffer (pH 5.5) for 30 s and then transferred into a cryoprotectant solution composed of 200 mM NaNO2, 3.3 M ammonium sulfate, 20.3% sucrose, and 50 mM Hepes buffer (pH 5.5) before lastly being soaked into a dithionite containing solution composed of 100 mM sodium dithionite, 3.3 M ammonium sulfate, 20.3% sucrose, and 50 mM Hepes buffer (pH 5.5) until the crystals became completely colorless before being cryocooled by plunging into liquid nitrogen and stored in pucks for data collection.
Serial femtosecond rotational crystallography
For SF-ROX as-isolated structure, crystals were soaked into a cryoprotectant solution composed of 3.3 M ammonium sulfate, 20.3% sucrose, and 50 mM Hepes buffer (pH 5.5). SF-ROX nitrite-bound crystals were soaked into a nitrite-containing solution composed of 200 mM NaNO2, 2.5 M ammonium sulfate, and 50 mM Hepes buffer (pH 5.5) for 30 s and then transferred for 30 s into a cryoprotectant solution composed of 200 mM NaNO2, 3.3 M ammonium sulfate, 20.3% sucrose, and 50 mM Hepes buffer (pH 5.5) for the same time before being cryocooled by plunging into liquid nitrogen. SF-ROX NO-bound crystals were prepared in the same way as for the SRX NO-bound structure.
Data collection, processing, and refinement
Synchrotron-radiation crystallography
Crystallographic data for atomic-resolution as-isolated and nitrite-bound structures were collected at 100 K using a DECTRIS PILATUS3 6M detector on I03 beamline at Diamond Light Source, UK at a wavelength of 0.97634 and 0.86999 Å, respectively. For NO-bound, data collection was carried out on BL41XU at SPring-8 using a DECTRIS EIGER X 16M detector at a wavelength of 0.80000 Å. All Diamond datasets were processed using DIALS (49) with xia2 (50) through Diamond’s automated processing pipeline. Data at SPring-8 were processed using XDS (X-ray Detector Software) (51) and CCP4 (The Collaborative Computational Project Number 4) packages using the automated data processing pipeline KAMO (52). Using structure 6THF as a starting model, SRX as-isolated, nitrite-bound, and NO-bound structures were refined with REFMAC5 (53) in the CCP4 program suite (54) with anisotropic B-factors and riding hydrogen atoms. For atomic-resolution structures, as-isolated, nitrite-bound, and NO-bound SRX, REFMAC5 refinement was followed up by refinement with SHELX-97 (28), where refinement of anisotropic B-factors, hydrogen positions, occupancies of the double conformation of the side chains, and T2Cu ligands was used. At the final stage of the refinement, one cycle of unrestrained block-matrix least-squares refinement was implemented to estimate the SDs (e.s.d.s.) of coordinates and derived parameters (bond lengths and angles). Each block consisted of 54 residues with 5 residue overlap. The information on the Cu ligands bonding with e.s.d.s. is shown in tables S2 and S3. Manual model rebuilding was performed using Coot (55), with ligands added to weighted election density maps and waters added after each stage of refinement. Data collection and refinement statistics are shown in table S1.
Serial femtosecond rotational crystallography
For each dataset, a total of 40 (as-isolated), 59 (nitrite-bound), and 55 (NO-bound) crystals of sizes ranging from 0.4 to 0.7 mm were frozen and used for collection. Data collection was performed on BL2/EH3 at SACLA at 100 K, as described previously (33), with the following parameters of XFEL pules: pulse duration (<10 fs), pulse energy (10 keV), and beam size at sample position [6.2 μm (H) by 2.5 μm (V)] for all. The crystals were rotated 0.1° and translated 50 μm between each shot. The diffraction images were collected on a MX300-HS detector (Rayonix) with a camera length of 90 mm and a pixel size of 78.2 μm. The same data collection and processing procedure were applied for all three datasets; twenty-eight crystals were selected for processing for nitrite-bound, and all crystals were selected for processing for both as-isolated (40) and NO-bound (55). Indexing and integration were performed using CrystFEL version 0.6.3 (56) with the inner, middle, and outer integration radii set to of 6, 7, and 8 pixels respectively. The spot-finding algorithm peakfinder8 was used, and indexing was carried out using DirAx (57) and Mosflm (58). After the resolution of the indexing ambiguity using ambigator, Bragg intensities were merged using the Monte Carlo integration with linear frame scaling. When merging, the high-resolution limit (d−1) in each pattern was extended by 1.7 nm−1 (as-isolated and nitrite-bound) or 1.9 nm−1 (NO-bound) using --push-res option.
In both cases, hydrogen bond contact distances were measured using CONTACT in the CCP4 program suite (54) and manually in Coot. All distances are listed in table S5.
UV/vis microspectrophotometry of samples in crystalline state
Optical absorption spectra for single uncollected crystals prepared for each SF-ROX dataset were obtained at 100 K using a fiber-optic microspectrophotometer with linear charge-coupled device array detector (Ocean Optics, SD2000).
Supplementary Material
Acknowledgments
The SR experiments at SPring-8 were performed at BL41XU (proposal nos. 2019A2575 and 2019B2575), and the XFEL experiments were performed at BL2 of SACLA (proposal no. 2019B8009), both with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). We would also like to thank Diamond Light Source, UK (MX15991 BAG) for access to I03 beamline and its staff for support. Funding: This work was supported by the Biotechnology and Biological Sciences Research Council, UK (grant no. BB/N013972/1 to S.S.H./S.V.A.). S.L.R. was supported by the RIKEN-Liverpool Partnership awarded to M.Y. and S.S.H. Author contributions: S.V.A., R.R.E., M.Y., and S.S.H. conceived and designed the project. S.L.R. and D.S. expressed and purified Br2DNiR. S.L.R. and S.V.A. crystallized Br2DNiR. S.L.R., S.V.A., K.H., G.U., H.A., T.T., and S.S.H. collected the SF-ROX data. S.L.R. and K.Y. performed the SF-ROX data processing. S.L.R. performed the SF-ROX structure determination and refinement. S.V.A. performed SHELX structure refinement. S.L.R. and S.V.A. collected the synchrotron crystallographic data. S.L.R., S.V.A., R.R.E., and S.S.H. wrote the manuscript with contributions from all the authors. All authors have given approval to the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. SRX atomic-resolution coordinates of the as-isolated, substrate-bound (nitrite-bound), and product-bound (NO-bound) Br2DNiR structures have been deposited to the PDB under accession codes 6ZAR, 6ZAT, and 6ZAV, respectively. XFEL FRIC coordinates of the as-isolated, substrate-bound (nitrite-bound), and product-bound (NO-bound) Br2DNiR structures have been deposited to the PDB under accession codes 6ZAS, 6ZAU, and 6ZAW, respectively. SRX low-dose (0.5 MGy) coordinates of the substrate-bound (nitrite-bound) Br2DNiR structure have been deposited to the PDB under accession code 6ZAX. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd8523/DC1
REFERENCES AND NOTES
- 1.Gruber N., Galloway J. N., An Earth-system perspective of the global nitrogen cycle. Nature 451, 293–296 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Canfield D. E., Glazer A. N., Falkowski P. G., The evolution and future of earth’s nitrogen cycle. Science 330, 192–196 (2010). [DOI] [PubMed] [Google Scholar]
- 3.O’Hara G. W., Daniel R. M., Rhizobial denitrification: A review. Soil Biol. Biochem. 17, 1–9 (1985). [Google Scholar]
- 4.Zumft W. G., Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 61, 533–616 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eady R. R., Hasnain S. S., Denitrification. Compr. Coord. Chem. II 8, 759–786 (2004). [Google Scholar]
- 6.O’Hara G. W., Daniel R. M., Steele K. W., Bonish P. M., Nitrogen losses from soils caused by Rhizobium-dependent denitrification. Soil Biol. Biochem. 16, 429–431 (1984). [Google Scholar]
- 7.M. A. J. Delgado, S. Casella, E. J. Bedmar, Denitrification in rhizobia-legume symbiosis, in Biology of the Nitrogen Cycle, H. Bothe, S. J. Ferguson, W. E. Newton, Eds. (Elsevier, 2007), pp. 83–91. [Google Scholar]
- 8.Firestone M. K., Firestone R. B., Tiedje J. M., Nitrous oxide from soil denitrification: Factors controlling its biological production. Science 208, 749–751 (1980). [DOI] [PubMed] [Google Scholar]
- 9.Ravishankara A. R., Daniel J. S., Portmann R. W., Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 326, 123–125 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Hidalgo-García A., Torres M. J., Salas A., Bedmar E. J., Girard L., Delgado M. J., Rhizobium etli produces nitrous oxide by coupling the assimilatory and denitrification pathways. Front. Microbiol. 10, 980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.M. Nojiri, Structure and function of copper nitrite reductase, in Metalloenzymes in Denitrification: Applications and Environmental Impacts, I. Moura, J. Moura, S. Pauleta, L. Maia, Eds. (The Royal Society of Chemistry, 2016). pp 91–113. [Google Scholar]
- 12.Kataoka K., Furusawa H., Takagi K., Yamaguchi K., Suzuki S., Functional analysis of conserved aspartate and histidine residues located around the type 2 copper site of copper-containing nitrite reductase. J. Biochem. 127, 345–350 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Boulanger M. J., Murphy M. E. P., Directing the mode of nitrite binding to a copper-containing nitrite reductase from Alcaligenes faecalis S-6: Characterization of an active site isoleucine. Protein Sci. 12, 248–256 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulanger M. J., Kukimoto M., Nishiyama M., Horinouchi S., Murphy M. E. P., Catalytic roles for two water bridged residues (Asp-98 and His-255) in the active site of copper-containing nitrite reductase. J. Biol. Chem. 275, 23957–23964 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Merkle A. C., Lehnert N., Binding and activation of nitrite and nitric oxide by copper nitrite reductase and corresponding model complexes. Dalton Trans. 41, 3355–3368 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Antonyuk S. V., Strange R. W., Sawers G., Eady R. R., Hasnain S. S., Atomic resolution structures of resting-state, substrate- and product-complexed Cu-nitrite reductase provide insight into catalytic mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 12041–12046 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blakeley M. P., Hasnain S. S., Antonyuk S. V., Sub-atomic resolution x-ray crystallography and neutron crystallography: Promise, challenges and potential. IUCrJ 2, 464–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuda Y., Tse K. M., Nakane T., Nakatsu T., Suzuki M., Sugahara M., Inoue S., Masuda T., Yumoto F., Matsugaki N., Nango E., Tono K., Joti Y., Kameshima T., Song C., Hatsui T., Yabashi M., Nureki O., Murphy M. E. P., Inoue T., Iwatae S., Mizohata E., Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proc. Natl. Acad. Sci. U.S.A. 113, 2928–2933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dodd F. E., Hasnain S. S., Abraham Z. H. L., Eady R. R., Smith B. E., Structures of a blue-copper nitrite reductase and its substrate-bound complex. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 406–418 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Leferink N. G. H., Han C., Antonyuk S. V., Heyes D. J., Rigby S. E. J., Hough M. A., Eady R. R., Scrutton N. S., Hasnain S. S., Proton-coupled electron transfer in the catalytic cycle of Alcaligenes xylosoxidans copper-dependent nitrite reductase. Biochemistry 50, 4121–4131 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Sasaki D., Watanabe T. F., Eady R. R., Garratt R. C., Antonyuk S. V., Hasnain S. S., Reverse protein engineering of a novel 4-domain copper nitrite reductase reveals functional regulation by protein-protein interaction. FEBS J. doi:10.1111/febs.15324 , (2020). [DOI] [PubMed] [Google Scholar]
- 22.Ghosh S., Dey A., Sun Y., Scholes C. P., Solomon E. I., Spectroscopic and computational studies of nitrite reductase: Proton induced electron transfer and backbonding contributions to reactivity. J. Am. Chem. Soc. 131, 277–288 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda Y., Hirano Y., Kusaka K., Inoue T., Tamada T., High-resolution neutron crystallography visualizes an OH-bound resting state of a copper-containing nitrite reductase. Proc. Natl. Acad. Sci. U.S.A. , 201918125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedison T. M., Shanmugam M., Heyes D. J., Edge R., Scrutton N. S., Active intermediates in copper nitrite reductase reactions probed by a cryotrapping-electron paramagnetic resonance approach. Angew. Chem. Int. Ed. 59, 13936–13940 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cioncoloni G., Roger I., Wheatley P. S., Wilson C., Morris R. E., Sproules S., Symes M. D., Proton-coupled electron transfer enhances the electrocatalytic reduction of nitrite to NO in a bioinspired copper complex. ACS Catal. 8, 5070–5084 (2018). [Google Scholar]
- 26.Moore C. M., Szymczak N. K., Nitrite reduction by copper through ligand-mediated proton and electron transfer. Chem. Sci. 6, 3373–3377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bower J. K., Sokolov A. Y., Zhang S., Four-coordinate copper halonitrosyl {CuNO}10 complexes. Angew. Chem. Int. Ed. 58, 10225–10229 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Sheldrick G. M., A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 64, 112–122 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Halsted T. P., Yamashita K., Gopalasingam C. C., Shenoy R. T., Hirata K., Ago H., Ueno G., Blakeley M. P., Eady R. R., Antonyuk S. V., Yamamoto M., Hasnain S. S., Catalytically important damage-free structures of a copper nitrite reductase obtained by femtosecond X-ray laser and room-temperature neutron crystallography. IUCrJ 6, 761–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malinska M., Dauter M., Kowiel M., Jaskolski M., Dauter Z., Protonation and geometry of histidine rings. Acta Crystallogr. Sect. D Biol. Crystallogr. 71, 1444–1454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liebschner D., Dauter M., Brzuszkiewicz A., Dauter Z., On the reproducibility of protein crystal structures: Five atomic resolution structures of trypsin. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1447–1462 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen F. H., The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 58, 380–388 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Hirata K., Shinzawa-Itoh K., Yano N., Takemura S., Kato K., Hatanaka M., Muramoto K., Kawahara T., Tsukihara T., Yamashita E., Tono K., Ueno G., Hikima T., Murakami H., Inubushi Y., Yabashi M., Ishikawa T., Yamamoto M., Ogura T., Sugimoto H., Shen J. R., Yoshikawa S., Ago H., Determination of damage-free crystal structure of an x-ray-sensitive protein using an XFEL. Nat. Methods 11, 734–736 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Halsted T. P., Yamashita K., Hirata K., Ago H., Ueno G., Tosha T., Eady R. R., Antonyuk S. V., Yamamoto M., Hasnain S. S., An unprecedented dioxygen species revealed by serial femtosecond rotation crystallography in copper nitrite reductase. IUCrJ 5, 22–31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strange R. W., Murphy L. M., Dodd F. E., Abraham Z. H. L., Eady R. R., Smith B. E., Hasnain S. S., Structural and kinetic evidence for an ordered mechanism of copper nitrite reductase. J. Mol. Biol. 287, 1001–1009 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Fisher S. J., Blakeley M. P., Cianci M., McSweeney S., Helliwell J. R., Protonation-state determination in proteins using high-resolution x-ray crystallography: Effects of resolution and completeness. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 800–809 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Horrell S., Antonyuk S. V., Eady R. R., Hasnain S. S., Hough M. A., Strange R. W., Serial crystallography captures enzyme catalysis in copper nitrite reductase at atomic resolution from one crystal. IUCrJ 3, 271–281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horrell S., Kekilli D., Sen K., Owen R. L., Dworkowski F. S. N., Antonyuk S. V., Keal T. W., Yong C. W., Eady R. R., Hasnain S. S., Strange R. W., Hough M. A., Enzyme catalysis captured using multiple structures from one crystal at varying temperatures. IUCrJ 5, 283–292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukuda Y., Tse K. M., Lintuluoto M., Fukunishi Y., Mizohata E., Matsumura H., Takami H., Nojiri M., Inoue T., Structural insights into the function of a thermostable copper-containing nitrite reductase. J. Biochem. 155, 123–135 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Tocheva E. I., Rosell F. I., Mauk A. G., Murphy M. E. P., Side-on copper-nitrosyl coordination by nitrite reductase. Science 304, 867–870 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Periyasamy G., Sundararajan M., Hillier I. H., Burton N. A., McDouall J. J. W., The binding of nitric oxide at the Cu(I) site of copper nitrite reductase and of inorganic models: DFT calculations of the energetics and EPR parameters of side-on and end-on structures. Phys. Chem. Chem. Phys. 9, 2498–2506 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Sundararajan M., Hillier I. H., Burton N. A., Mechanism of nitrite reduction at T2Cu centers: Electronic structure calculations of catalysis by copper nitrite reductase and by synthetic model compounds. J. Phys. Chem. B 111, 5511–5517 (2007). [DOI] [PubMed] [Google Scholar]
- 43.De Marothy S. A., Blomberg M. R. A., Siegbahn P. E. M., Elucidating the mechanism for the reduction of nitrite by copper nitrite reductase—A contribution from quantum chemical studies. J. Comput. Chem. 28, 528–539 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Li Y., Hodak M., Bernholc J., Enzymatic mechanism of copper-containing nitrite reductase. Biochemistry 54, 1233–1242 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Fukuda Y., Tse K. M., Suzuki M., Diederichs K., Hirata K., Nakane T., Sugahara M., Nango E., Tono K., Joti Y., Kameshima T., Song C., Hatsui T., Yabashi M., Nureki O., Matsumura H., Inoue T., Iwata S., Mizohata E., Redox-coupled structural changes in nitrite reductase revealed by serial femtosecond and microfocus crystallography. J. Biochem. 159, 527–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leferink N. G. H., Antonyuk S. V., Houwman J. A., Scrutton N. S., Eady R. R., Hasnain S. S., Impact of residues remote from the catalytic centre on enzyme catalysis of copper nitrite reductase. Nat. Commun. 5, 4395 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brenner S., Heyes D. J., Hay S., Hough M. A., Eady R. R., Hasnain S. S., Scrutton N. S., Demonstration of proton-coupled electron transfer in the copper-containing nitrite reductases. J. Biol. Chem. 284, 25973–25983 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hough M. A., Conradie J., Strange R. W., Antonyuk S. V., Eady R. R., Ghosh A., Hasnain S. S., Nature of the copper-nitrosyl intermediates of copper nitrite reductases during catalysis. Chem. Sci. 10.1039/d0sc04797j (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winter G., Waterman D. G., Parkhurst J. M., Brewster A. S., Gildea R. J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I. D., Sauter N. K., Evans G., DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 74, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter G., Xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 (2010). [Google Scholar]
- 51.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamashita K., Hirata K., Yamamoto M., KAMO: Towards automated data processing for microcrystals. Acta Crystallogr. Sect. D Struct. Biol. 74, 441–449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 56.White T. A., Mariani V., Brehm W., Yefanov O., Barty A., Beyerlein K. R., Chervinskii F., Galli L., Gati C., Nakane T., Tolstikova A., Yamashita K., Yoon C. H., Diederichs K., Chapman H. N., Recent developments in CrystFEL. J. Appl. Crystallogr. 49, 680–689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duisenberg A. J. M., Indexing in single-crystal diffractometry with an obstinate list of reflections. J. Appl. Crystallogr. 25, 92–96 (1992). [Google Scholar]
- 58.Powell H. R., Johnson O., Leslie A. G. W., Autoindexing diffraction images with iMosflm. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1195–1203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeldin O. B., Gerstel M., Garman E. F., RADDOSE-3D: Time- and space-resolved modelling of dose in macromolecular crystallography. J. Appl. Crystallogr. 46, 1225–1230 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd8523/DC1