

FANCA is a multitasking protein with essential functions beyond DNA repair in nucleolar homeostasis and ribosome biogenesis.
Abstract
Fanconi anemia (FA), the most common inherited bone marrow failure and leukemia predisposition syndrome, is generally attributed to alterations in DNA damage responses due to the loss of function of the DNA repair and replication rescue activities of the FANC pathway. Here, we report that FANCA deficiency, whose inactivation has been identified in two-thirds of FA patients, is associated with nucleolar homeostasis loss, mislocalization of key nucleolar proteins, including nucleolin (NCL) and nucleophosmin 1 (NPM1), as well as alterations in ribosome biogenesis and protein synthesis. FANCA coimmunoprecipitates with NCL and NPM1 in a FANCcore complex–independent manner and, unique among the FANCcore complex proteins, associates with ribosomal subunits, influencing the stoichiometry of the translational machineries. In conclusion, we have identified unexpected nucleolar and translational consequences specifically associated with FANCA deficiency that appears to be involved in both DNA damage and nucleolar stress responses, challenging current hypothesis on FA physiopathology.
INTRODUCTION
The nucleolus is a dynamic, membrane-less nuclear substructure visible in interphase that disappears at the onset of mitosis to reappear at the end of telophase. Several hundred proteins are hosted inside the nucleolus together with the genomic sites containing the ribosomal DNA (rDNA) sequences organized in tandemly repeated clusters dispersed on the five human acrocentric chromosomes. Three major events occur in the nucleolus: the RNAPolI-mediated synthesis of the 45S/47S ribosomal RNA (rRNA) precursors and their processing to generate the 28S, 18S, and 5.8S rRNA molecules that will be assembled with the ribosomal proteins (RPs) to form the pre-ribosomes, which are successively processed to form the cytosolic 40S and 60S ribosome subunit. In addition to its canonical role in the biogenesis of the ribosome, the nucleolus acts as a sensor and transducer of cellular stress originating from the nucleolus itself, the nucleus, or the cytosol. It can quickly lose homeostasis by modifying its activity, size, shape, and protein content to adapt cell metabolism to threatening events. The stress-associated transitory loss of nucleolar homeostasis or the nucleolar stress response (NSR), even in the absence of DNA damage and its signaling, is associated with the activation of the p53-p21 axis to restrain cellular proliferation and allow time to recover from stress (1, 2).
The unrestrained activation of the p53-p21 axis has been associated with hematopoietic stem cell (HSC) attrition that characterizes inherited bone marrow failure (iBMF) syndromes, including dyskeratosis congenita (DC), Shwachman-Diamond syndrome (SDS), Diamond-Blackfan anemia (DBA), and Fanconi anemia (FA), the most frequent and genetically heterogeneous iBMF syndrome. In contrast to DC, SDS, and DBA, in which BMF and p53-p21 activation rely on abnormalities in nucleolar homeostasis and/or ribosome biogenesis (3, 4), FA is attributed to abnormalities in the DNA damage response (DDR) (5–7) that lead to p53-p21 activation and BMF (8, 9). The canonical function attributed to the molecular pathway defined by FA proteins (FANC) is to cope with DNA interstrand cross-links (ICLs) and stalled replication forks that can arise from endogenous aldehyde metabolism or following exposure to ICL-inducing agents, such as mitomycin C, cisplatin, or photoactivated psoralens (5–7, 10, 11). The FANC pathway is organized into three distinct functional groups. The FANCcore complex, which comprises FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP20, and FAAP100, is assembled to monoubiquitinate the FANCD2-FANCI (ID2) heterodimer, allowing its accumulation in subnuclear foci required for the optimal work of the third group of FANC proteins, which includes nucleases and homologous recombination proteins (5–7). Among the more than 20 genes involved in FA, FANCA biallelic inactivating mutations account for more than 60% of FA cases worldwide (5–7). However, FANCA is dispensable for optimal FANCD2/FANCI monoubiquitination (12), and several BMF-associated mutations of FANCA do not lead to DNA damage hypersensitivity (13–15), suggesting that FANCA affects BM homeostasis by also participating in pathways other than the DNA repair network. Beyond the DNA repair/DDR, the FANC pathway has been associated with several other cellular and systemic biological activities (16), including proinflammatory cytokine production and responses (17–20), transcription (21–23), mRNA splicing (24), and reactive oxygen species metabolism (25–27), and its failure extends further than BMF to developmental abnormalities, cancer predisposition, and chromosomal instability.
Here, we investigated nucleolar protein localization, rDNA stability, and transcription as well as translational activity in FA-deficient cells. We demonstrated a specific involvement of FANCA in nucleolar homeostasis, ribosome biogenesis, and translation beyond some nucleolar abnormalities likely associated with the DNA repair defect associated with FANC pathway loss of function. Our observations possibly account for the high frequency of FA patients with inactivating mutations in FANCA and for the BMF in FANCA-mutated patients with normal resistance to DNA damage injury (13–15).
RESULTS
FANCA deficiency is associated with nucleolar protein mislocalization
Nucleolar status was monitored looking at the subcellular distribution of key nucleolar proteins such as fibrillarin (FBL), nucleolin (NCL), upstream-binding nucleolar transcription factor 1 (UBF), and nucleophosmin (NPM1 or B23). More than 70% of FANC pathway–proficient HeLa or U2OS cells harbored nucleoli with irregular borders delimited by a thick ring of NCL or NPM1, inside which FBL and UBF were distributed in a fairly homogeneous way (Fig. 1, A to D, and fig. S1, A to C). In contrast, 48 to 96 hours after the small interfering RNA (siRNA)–mediated depletion of FANCA, less than 30% of cells had nucleoli with the previously described distribution of NCL, NPM1, FBL, UBF, and/or irregular borders (Fig. 1, A to D, and fig. S1, A to C). In more than 70% of the FANCA-depleted cells, nucleoli took either a more round and regular shape delimited by an extremely faint ring of NCL or they appeared “empty” with the proteins displaced out of the nucleolus or eventually accumulating in nucleolar peripheral structures called “caps” (Fig. 1, A to D, and fig. S1, A to C). siRNA-mediated depletion of FANCA did not affect the expression of the analyzed nucleolar proteins (fig. S1D) or the number of nucleoli per cell (fig. S1E), and we separately tested the three previously pooled FANCA siRNA sequences with similar results (fig. S1F). Electronic microscopy (EM) analysis supported the occurrence of less organized and/or more condensed and rounded nucleoli in FANCA-depleted cells (Fig. 1E). Compared to the depletion of FANCA, down-regulation of FANCC, belonging as FANCA to the FANCcore complex, FANCG, a direct interactor of FANCA, or FANCD2, a FANCcore complex target, had reduced but still significant consequences on the frequency of cells with nucleolar abnormalities when compared to the FANC pathway–proficient cells (Fig. 1F and fig. S1, C, G, and H). However, whereas the depletion of FANCC, FANCG, or FANCD2 led to a similar frequency of cells with nucleolar abnormalities, this level was significantly lower than that observed when FANCA was depleted (Fig. 1F and fig. S1, G and H). The concomitant down-regulation of FANCA with FANCC, FANCG, or FANCD2 did not modify the frequency of cells with nucleolar abnormalities observed when only FANCA was depleted.
Fig. 1. Nucleolar abnormalities in FANC pathway–deficient cells.
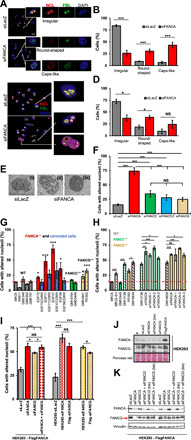
(A and C) Wide fields and single nuclei of HeLa (A) or U2OS (C) cells transfected with the indicated siRNAs and stained with anti-NCL (red) and anti-FBL (green) antibodies. DAPI (4′,6-diamidino-2-phenylindole) staining visualizes nuclei. (B and D) Percentage of HeLa (B) or U2OS (D) cells with canonical nucleoli (irregular shape) or a round or cap-like shape. The mean of six (B) or three (D) independent experiments ± SEM is reported. NS, not significant. (E) Electronic micrographs showing nucleoli as observed in the majority of HeLa cells (i) and the disorganized/condensed nucleoli observed after FANCA depletion (ii and iii). (F) Percentage of HeLa cells ± SEM with altered nucleoli 72 hours following transfection with untargeted (n = 6) (siLacZ) or FANCA-targeted (n = 6), FANCC-targeted n = 4), FANCG-targeted (n = 3), or FANCD2-targeted (n = 4) siRNA. (G and H) Percentage of cells with altered nucleoli in (G) WT, FANCA−/−, and FANCA-corrected, FANCC−/−, and FANCG−/− primary fibroblasts and in (H) WT, MRC5, FANCC−/− (GM00449), and FANCG−/− primary fibroblasts (PD352) and their immortalized counterparts (MRC5-SV, GM13136, and GM16335) under basal conditions or following FANCA depletion. The dotted red line represents the mean of the five primary WT cells. (I) Percentage of cells with altered nucleoli in HEK293- and HEK293-FlagFANCA–expressing cells before or after siRNA-mediated FANCA and/or FANCG depletion. Bars represent the mean of three to six independent experiments ± SEM. (J and K) Western blots showing the expression of the indicated proteins in HEK293 and HEK293-FlagFANCA cells. Extracts from independent experiments or transfections in the same experiment (bis) are shown. Statistics were assessed with two-tailed unpaired Student’s t tests (*P < 0.05, **P < 0.01, and ***P < 0.005).
Next, we extended our observations to human primary and SV-40 immortalized fibroblasts. In FANC pathway–proficient primary fibroblasts, the frequency of cells with a noncanonical nucleolar distribution of NCL and/or FBL ranged between approximately 5 and 18% (Fig. 1G). With respect to the mean value of the FANC pathway–proficient cells (12%; red dotted line in Fig. 1G), all analyzed FANCA-mutated primary fibroblasts presented a significant, albeit variable, increase in the frequency of cells with altered nucleoli, a frequency that was invariably reduced by the ectopic expression of the wild-type (WT) FANCA gene (Fig. 1, G and H). Frameshift and/or nonsynonymous mutations compromising the C-terminal part of FANCA were identified in FANCA primary fibroblasts (table S1), and FANCA protein in primary fibroblasts was hardly detectable by Western blot (WB) (fig. S1I). Thus, even if we cannot exclude that the observed variability in the frequency of cells with nucleolar abnormalities may be linked to the different mutations affecting FANCA or the residual expression level of mutated proteins, it could be due to other characteristics specific to each cell line or to their different adaptation to the culture conditions. The WT cell lines also exhibit substantial variations in the frequency of cells with nucleolar abnormalities that are proportionally similar to those of FANCA-deficient cells (i.e., a difference of approximately three times between the lowest and the highest, 4.8% versus 18.4% and 21.7% versus 68.3% in WT and FANCA−/− cells, respectively). Compared to the WT, two analyzed FANCC−/− primary cell lines were indistinguishable, whereas in both FANCG-deficient fibroblasts, the frequency of cells with nucleolar abnormalities was more elevated (Fig. 1G). SV40-mediated immortalization increases the frequency of cells with nucleolar abnormalities regardless of their FANC pathway status (Fig. 1H). However, immortalized FANCC−/− and FANCG−/− cell lines demonstrated a significant increase in the frequency of cells with mislocalized nucleolar proteins compared with FANC pathway–proficient MRC5-SV cells. Moreover, FANCA depletion in FANCC−/− or FANCG−/− immortalized fibroblasts led to an additional increase in cells with nucleolar abnormalities (Fig. 1H). An increased level of nucleolar abnormalities was also observed in primary fibroblasts from Fanca−/− mice (fig. S1, J to L).
We observed a high level of nuclear abnormalities in FANCG−/− primary cells (Fig. 1G). FANCG directly interacts with FANCA, forming FAAP20, one of the three subgroups of the FANCcore complex (5–7). Because the depletion of one protein in a complex is expected to affect the level of its partner(s), we decided to analyze the nucleolar consequence of FANCG depletion in human embryonic kidney (HEK) 293 cells stably expressing a Flag-tagged FANCA, which should maintain a satisfactory level of FANCA even in the absence of FANCG (Fig. 1, I to K). As in HeLa and U2OS cells (Fig. 1F and fig. S1, G and H), the frequency of cells with altered nucleoli in the parental HEK293 cell line increased following FANCA or FANCG depletion, with the loss of FANCA being more effective than FANCG (Fig. 1I). As expected for directly interacting proteins, FANCA depletion impinges on FANCG stability, reducing its expression, and vice versa, FANCG depletion affects FANCA expression (Fig. 1J). Notably, when FANCG was depleted in the Flag-FANCA–overexpressing cells, the level of FANCA appeared nearly unaltered (Fig. 1K), and accordingly, the frequency of cells with altered nucleoli was lower compared to the parental HEK293 cell line (Fig. 1I). Thus, it seems that the increased consequences of FANCG loss of function clearly observed in some settings (Fig. 1, G and I) but not in others (Fig. 1F) could be due to both FANC pathway inactivation (as observed in FANCC- and FANCD2-deficient cells) and the impact of FANCG depletion on the expression of its direct partner, FANCA.
Overall, our data pinpoint the presence of mislocalized nucleolar proteins in FANC pathway–deficient cells, mainly in association with FANCA (or FANCA-FANCG heterodimer) loss of function.
Increased nucleolar stress in FANCA-deficient cells is independent of DNA damage, ATM/ATR signaling, or accumulation of RNA-DNA hybrids
The FANC pathway has key roles in DNA repair, replication rescue, and coordinating replication and transcription cellular functions (28). This raises the question of whether the nucleolar abnormalities observed in FANCA-deficient cells were an additional consequence of alterations in the previous process, i.e., the outcome of increased rDNA damage or instability, the result of altered DNA damage signaling, and/or the consequence of defective coordination between transcription and replication machineries.
Not surprisingly, in light of their role in genomic stability maintenance, we identified by cell fractionation the presence of FANC proteins inside the nucleolus (Fig. 2A). The presence of FANCA inside the nucleolus was validated by immunofluorescence analysis of a yellow fluorescent protein (YFP)–FANCA construct (Fig. 2B) and that of FANCD2 by chromatin immunoprecipitation (ChIP) sequencing analysis. Depletion of FANCA or FANCD2 led to a slightly significant (P < 0.05) increased frequency of cells with nucleoli presenting γ-H2AX foci, a widely accepted readout of the presence of DNA breaks, in association with the induction of rDNA rearrangements as determined by polymerase chain reaction (PCR) analysis (Fig. 2, C and D). DNA breakage–induced ataxia telangiectasia mutated (ATM) signaling participates in the redistribution of the nucleolar proteins in the nucleoplasm, where some of them, including NCL and NPM1, participate in the DDR (29–33). However, even if ATM signaling is known to be constitutively active in FANC pathway–deficient cells (34, 35), the inhibition or depletion of ATM and/or of its related kinase, ataxia telangiectasia and Rad3 related (ATR), led only to a slight and not statistically significant reduction in the frequency of FANCA-deficient cells with nucleolar abnormalities (Fig. 2E).
Fig. 2. Nucleolar fragility and DNA damage signaling in FANCA-deficient cells.
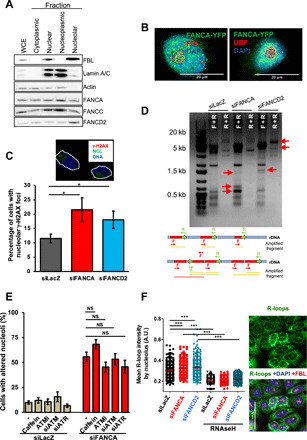
(A) Subcellular localization of FANCA, FANCC, and FANCD2. WCE, whole-cell extract. FBL and lamin A/C identify nucleolar and nuclear/nucleoplasmic fractions, respectively. (B) FANCA cellular localization as evaluated by confocal microscopy on cells transiently expressing a YFP-tagged FANCA construct. Cells were counterstained with FBL or UBF to identify nucleoli and DAPI to stain DNA. (C) Percentage of HCT116 cells with γ-H2AX foci–positive nucleoli. Cells were transfected with the indicated siRNAs and analyzed at 72 hours. Bars represent the mean of three independent experiments ± SEM. (D) Genomic PCR analysis of rDNA units 72 hours following FANCA or FANCD2 depletion in HeLa cells by using forward (F)–reverse (R) or reverse-reverse primers (see inset). Red arrows indicate new bands, suggestive of rDNA rearrangements. (E) Percentage of HeLa cells with altered nucleoli following transfection with untargeted (siLacZ) or FANCA-targeted siRNAs and exposure to caffeine, an ATM-specific inhibitor (ATMi), or cotransfected with ATM or ATR siRNA. Bars represent the mean of three independent experiments ± SEM. (F) Images of HeLa cells costained with anti–R-loops (green), anti-FBL (red), and DAPI (blue). Dots in the diagram represent the intensity of R-loop staining measured using CellProfiler software inside the FBL-positive region for each cell transfected with the indicated siRNAs. Cells incubated with RNaseH, which eliminates DNA-RNA hybrids, validate antibody specificity. A representative experiment of three is shown. At least 100 cells were scored for each condition. Statistical significance was assessed with the Z (normal distribution) test (*P < 0.05 and ***P < 0.005). A.U., Arbitrary Unit.
Next, as previously published genome wide (36), we observed that the level of the nucleolar RNA-DNA hybrid structures or R-loops, readout of badly managed transcription-replication conflicts, is increased as a consequence of FANCA depletion but to a similar extent as observed in FANCD2-depleted cells (Fig. 2F).
Overall, the above data validated that siRNA-mediated depletion of FANCA recapitulates key cellular features of FA to a similar extent as FANCD2 depletion, which, in contrast to FANCA down-regulation, presents fewer signs of nucleolar abnormalities (Fig. 1F). Thus, although the increased level of DNA damage and/or of its signaling associated with FANC pathway deficiency can undoubtedly participate in the observed nucleolar abnormalities, the stronger consequences of FANCA loss of function on nucleolar protein localization suggest that this protein (or its FANCA-FANCG-FAAP20 subcomplex) could have additional roles in nucleolar homeostasis maintenance.
Nucleolar stress in FANCA-depleted cells delays rDNA transcription
As the production of rRNAs is one of the main nucleolar activities, we asked whether the observed abnormalities in nucleolar protein localization could be a cause or consequence of abnormalities in rDNA transcription. Pulse-chase experiments with [32P]orthophosphate followed by electrophoretic separation of the pre-rRNA species (Fig. 3B and fig. S2A, left) or Northern blot analysis (Fig. 3C) revealed some anomalies in rDNA transcription/processing in FANCA down-regulated cells. The 45S/47S rRNA precursors are produced at a reduced rate in FANCA-deficient relative to FANC pathway–proficient cells, and their processing, qualitatively unaltered, is slowed down in the absence of FANCA. Thus, at steady state, a consequent increased level of both 45S/47S and 30S rRNA precursors is observed (Fig. 3B and fig. S2A), suggesting a delay in pre-rRNA transcription/processing that appears unchanged in FANCD2-depleted cells (fig. S2A, right). Last, nascent RNA synthesis was also evaluated by the quantification of the incorporation of the modified RNA precursor 5-ethynyl uridine (EU) (37). However, fluorescence-activated cell sorting (FACS) analysis of EU incorporation showed a marginal, if any, increase in FANCA-depleted cells, probably due to the confounding incorporation of EU in all cell transcripts (fig. S2B).
Fig. 3. rDNA transcription and rRNA processing in FANCA-deficient cells.
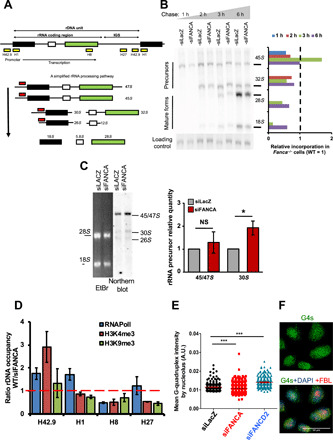
(A) Top, rDNA repeat organization. Yellow boxes indicate regions amplified by ChIP-qPCR analysis: promoter, H42.9; starting codon, H1; and final part, H8, of the RNAPolI-transcribed region, and the inter-rDNA gene sequence H27 (IGS). Bottom, pre-rRNA processing paths. Red boxes indicate probes used in the Northern blot analysis. (B) Experiment showing precursor and mature rRNAs. At 72 hours after transfection, HeLa cells were labeled (20 min) with [32P]orthophosphate and chased with cold orthophosphate for the indicated time. The EtBr-stained gel is shown as a loading control. Right, relative level of different rRNA forms in siFANCA-transfected cells normalized versus the FANC-proficient cells. (C) Northern blot analysis performed with the probe indicated in (A). RNAs were isolated 72 hours after siRNA transfection of HeLa cells. The EtBr-stained gel is shown as a loading control. The quantity measured in FANCA-depleted cells was adjusted to that in FANCA-proficient cells, which was set to 1. Bars represent the mean of four independent experiments ± SEM. Statistical significance was assessed with one-tailed Student’s t tests (*P < 0.05). (D) Ratio WT/siFANCA of the distribution of RNAPolI, H3K9me3, and H3K4me3 on the rDNAs of HeLa cells, as determined by ChIP-qPCR analysis. ChIP was performed 48 hours after siRNA transfection. Bars represent the mean of three experiments ± SEM. (E and F) Cells costained with anti-G4 (green), anti-FBL (red), and DAPI (blue). Dots in the diagram represent the intensity of G4 staining measured using CellProfiler software inside the nucleoli for each cell 48 hours after transfection with the indicated siRNAs. At least 100 cells were scored for each condition. Statistical significance was assessed with a Z (normal distribution) test (***P < 0.005).
ChIP–quantitative PCR (qPCR) analysis demonstrated that FANCA depletion leads to a lower association of RNAPolI in the rDNA promoter region (H42.9 and H1) in parallel with its increased accumulation on the terminal part of the rRNA coding region (H8) (Fig. 3, A and D). Accordingly, we also observed a reduced level of the transcription-activating H3K4me3 histone mark on the promoter region. Last, immunofluorescence analysis revealed that the level of nucleolar G4s, which are secondary DNA structures that facilitate transcription progression (38–42) and that are regulated by NCL and NPM1, was lower in FANCA-deficient than in FANC pathway–proficient or FANCD2-deficient cells (Fig. 3, E and F).
Together, our data are consistent with the presence of some perturbations in rDNA transcription in FANCA-depleted cells, associated with an increased level of R-loops (Fig. 2F) and a lower level of G4s (Fig. 3E), which cooperatively may slow RNAPolI progression. However, their limited magnitude suggests that they are more likely a consequence of FANCA depletion–associated loss of the nucleolar proteins and not a cause of it.
FANCA coimmunoprecipitates with NPM1 and NCL
The consequence of FANCA loss of function on nucleolar protein localization seems to be due to a still-undetermined role of the protein in cell physiology and not an additional consequence of its loss of function in DNA processes such as DNA repair, replication, recombination, and/or transcription.
To shed light on this role, we performed FANCA immunoprecipitation by using three different antibodies directed against different epitopes of FANCA in protein extracts isolated from HSC93, U2OS, or HEK293 cells (Fig. 4A). The three antibodies successfully coimmunoprecipitated FANCA with its well-known partner FANCG. Conversely, FANCA was identified among the FANCG-associated proteins (Fig. 4B). Successively, we examined the presence of NPM1 and NCL in FANCA- and FANCG-immunoprecipitated cell extracts. Both nucleolar proteins were undoubtedly and repeatedly retrieved in FANCA but not in FANCG immunoprecipitations (Fig. 4, A and B). A yeast two-hybrid screen with FANCA as bait retrieved FANCG and FAAP20, its two key direct interactors, but failed to identify NPM1 or NCL among its potential partners. Thus, still unidentified proteins or RNAs could mediate the observed FANCA-NCL-NPM1 coimmunoprecipitation. Treatment of the immunoprecipitated protein complexes with benzonase, a DNA/RNA dual-specific nuclease, affected the immunoprecipitation of NCL but had minor consequences on FANCA-NPM1 and FANCA-FANCG coimmunoprecipitation (Fig. 4C). Thus, at least part of the FANCA-NCL coimmunoprecipitation might depend on RNA, for which FANCA has high affinity (43). Moreover, whereas the coimmunoprecipitation between FANCA and FANCG was maintained at a higher salt concentration (300 mM), NPM1 and NCL were no longer found in association with FANCA in such a setting (Fig. 4D). Last, we immunoprecipitated FANCA with NCL/NPM1 in both FANCG- and FANCC-mutated lymphoblasts, suggesting that FANC pathway loss had no major impact on the coimmunoprecipitation between FANCA and NCL and NPM1 (Fig. 4E and fig. S3A). Next, we tried the reverse, i.e., coimmunoprecipitated FANCA with antibodies directed against NPM1 or NCL. In both cases, even loading on the gel two times more immunoprecipitated proteins than for the previous analysis, it appeared very hard to detect the presence of FANCA, if any, in NCL or NPM1 immunoprecipitates (Fig. 4F; see the long exposure image). As expected, the immunoprecipitation of one of the two nucleolar proteins coimmunoprecipitated with the other, even if the quantity of NPM1 coimmunoprecipitated with NCL and that of the latter coimmunoprecipitated with NPM1 were small (Fig. 4F). Inversely, because the relative quantity of FANCA is largely inferior to that of NCL or NPM1 (it is barely detectable when 5 μg of total protein extract was used as input; Fig. 4, B to F), our results suggest that only a very low proportion of NCL or NPM1 eventually coimmunoprecipitates with FANCA, whereas a more substantial proportion of FANCA coimmunoprecipitates with NCL or NPM1.
Fig. 4. FANCA immunoprecipitates with NPM1 and NCL.
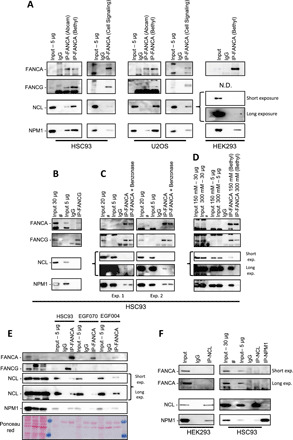
(A) Anti-FANCA rabbit antibodies from Abcam, Bethyl, and Cell Signaling laboratories were used to immunoprecipitate FANCA in cell extracts from exponentially growing human lymphoblasts (HSC93), human bone osteosarcoma cells (U20S), or human embryonic kidney cells (HEK293). Immunoblotting was performed with anti-FANCA Bethyl or Abcam antibodies and antibodies against FANCG, NCL, and NPM1. N.D., Not Done. (B) Immunoprecipitation (IP) with a FANCG antibody followed by immunoblot with antibodies against the indicated proteins. Different quantities of input fractions were analyzed to clearly visualize FANCA and FANCG. (C and D) Immunocomplexes isolated by the FANCA antibody were treated with benzonase [(C), two independent experiments], 150 nM NaCl, or 300 nM NaCl (D) before immunoblot analysis. Different quantities of input fractions were analyzed to clearly visualize FANCA and FANCG. (E) Immunoprecipitation with a FANCA antibody in cell extracts from HSC93 (WT), EGF070 (FANCG−/−), and EGF004 (FANCG−/−) cells followed by immunoblot with antibodies against the indicated proteins. Different quantities of input fractions were loaded. (F) NCL or NPM1 were immunoprecipitated from HEK293 or HSC93 cells. Immunoblot showing the coimmunoprecipitation of the indicated proteins when cell extracts were immunoprecipitated with anti-NCL or anti-NPM1 antibodies.
In conclusion, our data revealed that FANCA participates in complex assembly of NPM1 and NCL, even if the relative weakness of the interaction, i.e., its absence in yeast two-hybrid analysis and its partial or total loss following exposure to an RNA/DNA nuclease or to high salt, advocates for an indirect association. Thus, our data suggest that FANCA could exist in two separate complexes or aggregates: one with FANCG and another with NPM1 and NCL, the first involved in DDR and the second associated with nucleolar homeostasis.
FANCA deficiency associates altered ribosome profiles with reduced protein synthesis
Ribosome biogenesis is the other main process that takes place in the nucleolus. We reasoned that FANCA loss of function altering the nucleolar localization of proteins such as NCL and NPM1 could affect ribosome biogenesis and, consequently, the translation process, which, in turn, would amplify the nucleolar abnormalities observed in FANCA-depleted cells. Thus, to determine whether this was the case, we quantified nascent protein synthesis by measuring OP-Puro incorporation in FANCA−/− (HSC72 and HSC99), FANCC−/− (HSC536), and FANCG−/− (EGF004 and EGF070) lymphoblasts as well as in FANC pathway–proficient cells [the FANCA- and FANCC-corrected counterparts (HSC72CORR and HSC536CORR) and the WT HSC93 and GM3657 cell lines]. Flow cytometry analysis (fig. S3B) revealed a diminished OP-Puro signal indicative of a reduced rate of protein synthesis in FANCA- and FANCG-deficient cells (Fig. 5, A to C). Similar results were observed in FANCA-depleted versus FANCC-depleted, FANCD2-depleted, and FANC pathway–proficient HeLa cells, indicating that the observed anomalies are essentially associated with the loss of function of the FANCA-FANCG subgroup of the FANCcore complex.
Fig. 5. FANCA deficiency leads to an altered ribosome profile and reduced protein synthesis.
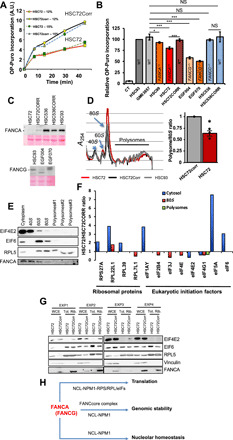
(A) Time-course incorporation of OP-Puro assessed by FACS in FANCA lymphoblasts (HSC72) and their corrected counterpart (HSC72Corr) cultured in 12 or 15% FCS. (B) Relative levels of OP-Puro incorporation in FANC-proficient (HSC93 and GM3657), FANCA−/− (HSC72 and HSC99), FANCC−/− (HSC536), or FANC-corrected lymphoblasts. Bars represent the mean of three to five independent experiments ± SEM. The value observed in HSC93 cells was set to 1 in each individual experiment. Statistical significance was assessed with a two-tailed Student’s t test against the GM03657 cell line (*P < 0.05 and ***P < 0.005). (C) Western blot showing the expression of FANCA and FANCG in the indicated cell lines. (D) Polysome profiling in FANCA-deficient HSC72 and FANCA-proficient HSC72corr and HSC93 lymphoblasts and quantification of the polysome/monosome ratio in HSC72 relative to HSC72corr cells. Bars represent the mean of four independent experiments ± SEM. The value of the HSC72corr cell was set to 1 in each individual experiment. (E) Western blots showing FANCA presence in the ribosomal-enriched cytoplasmic, 40S, 60S, and monosome (80S) fractions. (F) Histograms representing the mean expression level of the indicated proteins in the cytosol, 80S, and polysomes in FANCA-deficient cells compared with their corrected counterparts. Data are from MS analysis. (G) Western blots showing the expression of the indicated proteins in whole-cell (WCE) and ribosomal-enriched (Tot. Rib.) extracts in HSC72 and HSC7CCorr cells in four experiments. (H) Simplified diagram illustrating the several roles of FANCA in cell physiology.
To further analyze the involvement of FANCA in protein synthesis, we performed polysome profiling, which allows a rough evaluation of translation process proficiency (44). Such analysis revealed a higher 80S monosome/polysome ratio in FANCA-deficient cells than in FANCA-proficient cells (Fig. 5D), indicative of a delay in translation initiation. Notably, the expression of WT complementary DNA (cDNA) in the patient-derived FANCA-deficient cell line completely recovered the observed abnormality. The level of FANCA expression in HSC72CORR cells was not beyond the level observed in FANCA-proficient cells (Fig. 5C). Thus, the observed polysome profile of the HSC72CORR cells perfectly overlays that of the FANC pathway–proficient HSC93 cells, validating the rescuing of the FANCA activity associated with the efficiency of the translational machinery.
Next, we performed a liquid chromatography–tandem mass spectrometry (LC-MS/MS)–based analysis of the cytosolic, ribosomal, and polysomal fractions enriched in RPs and translational factors. MS and successive WB analysis identified FANCA as the sole FANCcore complex protein present in the cytosolic and ribosomal fractions (40S, 60S, and 80S) and revealed its absence in polysomes (Fig. 5E). Notably, among all the FANC proteins, our MS analysis retrieved only FANCI and FANCD2, which were present in the 80S and polysomal fractions.
Next, we focused our analysis on the key components of the translational initiation process identified by MS, i.e., the RPs of the small and large subunits (RPS and RPL, respectively) and the eukaryotic translation initiation factors (eIFs). Globally, MS analysis in FANCA-deficient and FANCA-proficient cells identified all the known RPSs, RPLs, and eIFs. The analysis of the cytosols enriched in RPs and translational factors by MS demonstrated imbalances in the stoichiometry of several RPs and translation factors in each individual experiment, suggesting a requirement for FANCA for the optimal assembly of the translation machinery. Successively, for robustness, we looked for proteins retrieved in three of the four experiments on the cytosolic fractions or in both analyzed experiments performed on the 80S and polysomal fractions with an HSC72/HSC72corr ratio of >1.5 (higher level in FANCA−/− cells) or <0.66 (higher level in WT-FANCA–corrected cells) (Fig. 5F). In the cytosolic fraction, the levels of RPS27A, RPL22L1, and RPL39 were substantially increased in FANCA-mutated cells. The level of RPL22L1 is also increased in the 80S monosomal fraction, which additionally presents a deficiency in RPL7L1. Last, in the cytosolic fraction, FANCA deficiency associates with an increased level of eIF4Y, eIF5A, and eIF6 and a deficit in both eIF4E2 and eIF4G1. In the 80S fraction from FANCA−/− cells, several eukaryotic initiation factors, including eIF2B4, eIF3J, eIF4E, and eIF4G1, were underrepresented. In the polysomal fraction, only the level of eIF4G1 was altered between FANCA-deficient and FANCA-proficient cells, supporting the hypothesis that FANCA affects translation initiation efficiency but not progression. Therefore, despite undeniable variability, two factors appeared to be robustly affected by the absence of FANCA: RPL22L1, overrepresented in the cytosolic and 80S ribosomal enriched fractions, and eIF4G1, the only underrepresented in all the analyzed fractions. To validate the MS data, we analyzed RPL5, eIF4E2, and eIF6 expression in the same extracts by WB (Fig. 5G).
Together, the previous data provide two unexpected evidence. First, FANCA, unique among the FANCcore complex proteins, cosegregates with isolated ribosome subunits and with the 80S monosome associated with mRNA. Second, its loss of function perturbs the physiology of the ribosome and of the translational machinery, affecting the translation rate of the cells.
DISCUSSION
Our present work unveiled new phenotypic traits associated with the loss of function of FANCA, known to be responsible for 60 to 70% of FA cases worldwide. FANCA-deficient cells harbor several hallmarks of nucleolar stress, including delocalization of proteins from the nucleolus to the nucleus and the cytosol, impairment of ribosome biogenesis, and reduced translation efficiency (Fig. 5H). Supporting their dependence on FANCA inactivation, nucleolar and translational abnormalities were observed following siRNA-mediated depletion of FANCA in HeLa, U2OS, and HEK293 cells as well as in primary and immortalized cells issued from FA-A patients, where they are rescued by the ectopic expression of WT FANCA. We provided several robust pieces of evidence demonstrating that the observed nucleolar and translational abnormalities in FANCA-deficient cells represent the readout of new FANCA-associated function(s) on the cellular physiology and not an additional consequence of the canonical defects in DDR associated with the loss of function of the FANC pathway.
We showed here that the depletion of FANCA, which, like FANCC or FANCD2, is localized inside the nucleolus, has the same consequences on nucleolar DNA damage, rDNA instability, and R-loop accumulation observed following FANCD2 depletion. However, FANCC-, FANCG-, or FANCD2-depleted or mutant cells show lower levels of nucleolar stress than in the absence of FANCA. Moreover, FANCA depletion in FANCC−/− or FANCG−/− cell lines significantly increased the frequency of cells with nucleolar abnormalities. Thus, even if part of the observed nucleolar anomalies can effectively depend on the role of the FANC pathway in the DDR, our observations represent a first step in supporting the existence of a FANCA “private” role in the maintenance of nucleolar homeostasis.
Next, we demonstrated that FANCA, but not its partner FANCG, coimmunoprecipitates two of the major nucleolar proteins, NCL and NPM1, and that their coimmunoprecipitation is independent of FANCG or FANCC. Thus, together, the results of our immunoprecipitations revealed that FANCA associates with (at least) two separate and independent complexes: one with FANCG, known to be involved in DDR and participating in FANCD2/FANCI monoubiquitination and subnuclear foci assembly, and another with NCL and NPM1. Our data strengthen several observations demonstrating specific roles of the individual FANC proteins and of FANCA in particular, regardless of their participation in the FANC pathway (13–15). Although two-thirds of FA patients present biallelic FANCA mutations, the activity of the protein is not mandatory for FANCD2 monoubiquitination and resistance to DNA damage (12, 13, 15), meaning that the BMF and the preleukemic status in such patients might be partially unrelated to altered DDR and the associated genetic instability.
The observed interaction of FANCA with NPM1 and the nuclear/cytoplasmic delocalization of NPM1 in FANCA-depleted cells are of particular interest. The nucleolar exclusion of NPM1 is a known consequence of somatic heterozygous mutations that abolish a nucleolar localization signal (45, 46) and/or create an additional nuclear exclusion signal (47) that sequesters the mutated protein, NPM1c or NPM1mut, in the cytoplasm, where it has been immunoprecipitated with FANCA (48), an interaction that fosters NPM1c stability. The NPM1c mutation is a key event observed in more than one-third of acute myeloid leukemia (AML) cases (49, 50). DNA damage induction in NPM1c-mutated AML allows ATM-dependent nuclear/nucleolar import of the mutated protein (51). On the basis of previous work, we speculate that FANCA, whose activity in DDR is ATM/ATR signaling dependent (52, 53), contributes to the correct spatiotemporal localization and activity of NPM1 (and even NPM1c) in different settings, and consequently, the observed mislocalization of NPM1 outside the nucleolus in FANCA-deficient cells could participate in both the BMF and the preleukemic status of FA patients.
Both NPM1 and NCL are abundant multifunctional proteins with roles in several unrelated cellular processes, including the DDR and DNA repair (54). Thus, FANCA behaves like a hub protein, mediating the trafficking/localization of several other proteins and complexes. In accordance with such a role as a hub protein, we unveiled the presence of FANCA, unique among the FANCcore complex proteins, in the cytosolic fraction enriched in ribosomal subunits, as well as in the 40S, 60S, and 80S fractions. A survey on the BioGRID database (https://thebiogrid.org/) revealed eight proteins as potential common interactors of FANCA, NCL, and NPM1, including the RP RPL18 that we retrieved in our immunoprecipitations with anti-FANCA antibody (preliminary observation; fig. S3C). Although FANCA depletion did not alter the stoichiometry of RPL18 in the analyzed ribosomal fractions in our MS analysis, FANCA deficiency was clearly associated with alterations in the composition of the ribosomal subunits and in translation. In particular, FANCA deficiency affects the levels of RPL22L1, eIF6, eIF4G1, and eIF4E, alterations that can participate or amplify the FA phenotypes. First, the RPL22L1/RPL22 balance is critical for HSC emergence and maintenance (55). Second, the observed increased association of eIF6 with the total ribosomal fraction and the reduced translation rate observed in FANCA-deficient cells are reminiscent of the phenotypic hallmarks of another rare BMF disease, SDS, resulting from downstream consequences of mutations in the SBDS or EFL1 genes (56, 57). Third, the observed alterations in eIF4G1 and eIF4E levels could contribute to both the reduced translation rate and altered DDR in FANCA-deficient cells. eIF4G1 and eIF4E are key factors for mRNA scanning and AUG selection (58), rate-limiting steps in translational initiation that appeared altered in FANCA-depleted cells. As we observed in FANCA-deficient cells, eIF4G1 down-regulation leads to a decrease in overall protein synthesis of approximately 20% (58, 59) and to an increased cellular sensitivity to DNA damage.
To underline its specificity in ribosome biogenesis and translation, FANCA was the sole FANCcore complex protein present in the fraction enriched in ribosomal subunits. A possibility is that its presence can be mediated by interactions with rRNAs and/or mRNAs. As also reported for FANCD2 (60), FANCA has been shown to have a stronger affinity for RNA and single-stranded DNA (ssDNA) than for double-stranded DNA (dsDNA), with a Kd (dissociation constant) significantly better for single-stranded RNA (ssRNA) (2.8 nM) than for ssDNA (11.1 nM) (43). The RNA binding activity of FANCA could be involved in nucleolar and/or ribosome biogenesis homeostasis, which we discovered as perturbed in its absence. More generally, in such fractions, among the more than 20 proteins associated with the FANC pathway, we retrieved only FANCI and FANCD2.
The presence of FANCI in the ribosomal fractions validated previous data demonstrating that the functions of FANCI, such as FANCA, encompass ribosome biogenesis and DNA repair. As reported here for FANCA, Sondalle et al. (61) demonstrated that FANCI depletion perturbs rDNA transcription and processing slightly but consistently (in both cases, delayed formation of the mature forms was observed) and affects global translation. As in the current study, Sondalle et al. (61) reported that FANCD2 depletion did not perturb rDNA transcription, rRNA processing, or translation.
Last, we cannot ignore that the loss of function of FANCG, contrary to that observed in cells deficient in FANCC or FANCD2, leads to impaired translation (Fig. 5B) and, in some settings, affects the localization of nucleolar proteins more than a deficiency in FANCC or FANCD2 (Fig. 1, F and I). FANCG with FAAP20 forms a trimeric complex with FANCA, and depletion of FANCG strongly affects the expression of FANCG (Fig. 1, J and K). However, FANCG did not coimmunoprecipitate with NCL or NPM1, it was not identified as FANCA in the ribosomal fractions analyzed by MS, and, in its absence, FANCA still coimmunoprecipitated with NCL and NPM1 (Fig. 4F). Thus, we support the hypothesis that the observed consequences of FANCG deficiency on nucleolar and ribosomal homeostasis are due to its consequences on FANCA expression more likely than on its direct involvement in such processes.
In conclusion, our observations extend the phenotypic landscape associated with FANCA loss of function and drive FA closer to the group of iBMF syndromes called ribosomopathies that include DC, DBA, and SDS, in which BMF is associated with mutations in protein-coding genes for which the loss of function directly leads to alteration in nucleolar homeostasis and ribosome biogenesis, supporting the centrality of nucleolar metabolism in the maintenance of bone marrow functionality. Last, our observations unveil a key role for FANCA in two stress response pathways, the DDR and the NSR networks, whose activity is required for the steady-state maintenance of the BM and for raising proficient cancer barriers.
MATERIALS AND METHODS
Cell lines, culture, and reagents
Cell lines are listed in table S1. Adherent cells (HeLa, U2OS, HCT116, HEK293, and human and mouse fibroblasts) were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies) supplemented with 10% fetal calf serum (FCS), penicillin (0.5 mg/ml), streptomycin (100 μg/ml), and 1 mM pyruvate at 37°C and 5% CO2. Human lymphoblasts were grown in RPMI medium (Life Technologies) supplemented with 12% FCS, penicillin (0.5 mg/ml), and streptomycin (100 μg/ml) at 37°C and 5% CO2. Cell cultures were regularly tested for the absence of mycoplasma.
ATM and/or ATR inhibitors, 1 mM caffeine (Sigma-Aldrich), 10 μM KU-55933 (Abcam), and 100 nM VE-822 (Sellchem) were added to the culture medium 6 hours after transfection. The addition was repeated every 24 hours until the end of the experiment.
siRNA and plasmid transfection
siRNA sequences are listed in table S2. siRNAs were transfected at 20 to 40 nM using calcium phosphate diluted at 6.25 mM in Hanks’ Balanced Salt Solution (HBSP) buffer [750 μM Na2HPO4, 5 mM KCl, 140 mM NaCl, 25 mM Hepes (pH 7)] or with INTERFERin (Polyplus). The FANCA-YFP plasmid (a gift from K. J. Patel, Cambridge, UK) was transfected using jetPRIME (Polyplus) in HeLa cells or Fugene HD (Promega) in U2OS cells. A FANCA-Flag vector (a gift of A. Constantinou, CNRS, Montpellier, France) was transfected using jetPRIME (Polyplus) in HEK293 cells, and a stable expressing cell population was isolated under antibiotic selection.
Immunofluorescence and image quantification
Antibodies are listed in table S3. Samples were fixed 48 to 72 hours after siRNA transfection. For FBL, NCL, NPM1, and UBF, cells were washed with phosphate-buffered saline (PBS), fixed in 4% formaldehyde (Sigma-Aldrich) for 5 min at room temperature, and permeabilized in PBS with 0.5% Triton (Sigma-Aldrich) for 5 min at room temperature. For R-loops and G-quadruplexes, cells were washed with PBS, fixed in methanol at −20°C for 5 min, and then blocked in PBS, 0.05% Tween 20, and 3% bovine serum albumin for 1 hour at room temperature and stained with primary antibodies followed by secondary antibodies in the same buffer. Epifluorescence images were acquired with a Zeiss microscope at ×63 magnification 1.4 NA (numerical aperture). Quantification was performed on at least 100 cells. Nucleolar and nucleoplasmic intensity quantification of R-loops and G-quadruplexes was performed on epifluorescence images using CellProfiler software and FBL staining as a nucleolar marker. For colocalization assessment, image acquisition was performed using a confocal Leica TCS Sp8 microscope at ×63 magnification 1.4 NA (with magnification to a resolution of approximately 40 nm per pixel). Deconvolution was performed using Huygens software. A negative control was performed by acquiring each signal separately.
EM analysis
Ultrastructural study: Seventy-two hours after siRNA transfection, cell monolayers were fixed in 2% glutaraldehyde in 0.1 M Sörensen phosphate buffer for 1 hour at 4°C. The cells were scraped off during fixation and centrifuged. The fixed pellets were rinsed for 1 hour in ice-cold phosphate buffer, postfixed with 2% aqueous osmium tetroxide, and dehydrated in increasing concentrations of ethanol before Epon embedding. Polymerization was carried out for 48 hours at 60°C. Ultrathin sections were stained with standard uranyl acetate and lead citrate before observation with an FEI Tecnai Spirit transmission electron microscope at 80 kV. Digital images were taken with a SIS MegaView III charge-coupled device (CCD) camera.
Protein extraction, immunoprecipitation, and Western blotting
Antibodies are listed in table S3. Cellular extracts for immunoprecipitation were prepared from exponentially growing lymphoblasts or adherent cells. Cells were lysed on ice for 30 min in NaCl, EDTA, Tris, NP-40 (NETN) buffer [150 mM/300 mM NaCl, 50 mM tris (pH 8.0), 1% NP-40, 1 mM EDTA] supplemented with phosphatase inhibitor PhosSTOP (Roche) and cOmplete ULTRA EDTA-Free Protease inhibitors (Roche). Cells were sonicated for 2 × 10 s at 30% (Vibracell 75042, Bioblock) and spun down for 5 min at 13,000 rpm. Supernatants were quantified using the Bradford assay (BioPhotometer, Eppendorf). One milligram of protein extract was used per immunoprecipitation. For each immunoprecipitation, antibodies (3 μg) were coupled to 20 μl of magnetic beads (Dynabeads Protein G Magnetic Beads, Thermo Fisher Scientific). Beads washed in 1× PBS (two times) were resuspended in 300 μl of 1× PBS and incubated with antibodies for at least 2 hours in a roll shaker in a cold room. To preclear the protein extracts, 10 μl of beads washed in PBS and in NETN buffer was added to each whole protein extract in the presence of 0.5 μg of control immunoglobulin G (IgG) and incubated for at least 2 hours in a roll shaker in a cold room. Beads coupled with IgG were collected on a magnetic support and discarded. Supernatants were resuspended in beads coupled to specific antibodies. A small amount of the precleared protein extract was kept as input. Precleared supernatants were incubated with the magnetic beads coupled to the antibodies overnight in a roll shaker in a cold room. When necessary, cells were incubated for 10 min at room temperature after the addition of benzonase (Merck, 1 mM) and 1 mM MgCl2 to the lysate. Beads coupled with antibodies were captured on a magnetic support and washed four times with NETN buffer before being resuspended in 40 μl of 2× Laemmli and heated for 5 min at 70°C to dissociate proteins and antibodies from the beads. The supernatants were transferred into a new tube, 1 μl of μ-mercaptoethanol was added, and half of this immunoprecipitation was loaded on an acrylamide gel after 5 min of heating at 98°C. For analysis of protein expression, cells were lysed in 50 mM tris-HCl (pH 7.5), 125 mM NaCl, 1 mM MgCl, and 0.1% SDS buffer supplemented with benzonase (Merck, 1:1000), phosphatase inhibitor PhosSTOP (Roche), and cOmplete ULTRA EDTA-Free Protease inhibitors (Roche). Proteins were resuspended in Laemmli’s blue [16 mM tris-HCl (pH 6.8), 10% glycerol, 2.5% SDS] supplemented with β-mercaptoethanol (Sigma-Aldrich, 1:10). Proteins from immunoprecipitation and cellular extracts were separated by electrophoretic migration performed in 2.5 mM tris, 19.2 mM glycine, and 0.01% SDS buffer. Semidry transfer was performed with a TransBlot cell apparatus (Bio-Rad) in transfer buffer composed of 2.5 mM tris-HCl (pH 8.3), 19.2 mM glycine, 0.01% SDS, and 20% isopropanol for 1 hour and 30 min at 20 V. Liquid transfer was performed in an XCell II module (Invitrogen) in 2.5 mM tris-HCl (pH 8.3), 19.2 mM glycine, 0.04% SDS, and 20% methanol buffer at 15 V overnight at 4°C. Nitrocellulose membranes (Protran 0.2 mm, Amersham) were blocked for at least 1 hour in 0.1% PBS–Tween 20 and 5% milk and incubated with primary antibodies in PBS/0.1% Tween 20/5% milk. Visualization was performed using ECL (Life), Western Bright ECL (Advansta), West Dura ECL, or West Femto ECL (Thermo Fisher Scientific) developer. Images were acquired using a CCD camera (General Electric) or Amersham Imager 600 (GE Healthcare).
PCR-based analysis of rDNA rearrangement
Seventy-two hours after transfection, cells were lysed, and DNA was extracted using the Maxwell RSC Blood DNA Kit (Promega). PCR was performed on 100 ng of DNA using Phusion polymerase (0.02 U/ml), 0.2 mM deoxynucleotide triphosphate (dNTP), and 0.5 mM of the following probes: forward: 5′-AGTCGGGTTGCTTGGGAATGC-3′; reverse: 5′-GGACAAACCCTTGTGTCGAGG-3′ (predicted position in gene: nucleotides 8204 to 12970). The following amplification program was used: 98°C, 30 s; 35× (98°C, 10 s; 62°C, 30 s; 72°C, 4 min); 72°C, 10 min.
Metabolic labeling
Seventy-two hours after siRNA transfection, nonconfluent HeLa cells were incubated for 1 hour at 37°C and 5% CO2 in phosphate-free DMEM containing 10% dialyzed FCS. They were then pulsed for 20 min with 15 mCi of [32P]orthophosphate (PerkinElmer). After two washes in culture medium, they were incubated for a chase period of 1 to 6 hours. RNA was extracted using TRIzol reagent, and 1 mg of RNA was loaded on a denaturing gel with Ethidium Bromide (EtBr). The EtBr signal was acquired after migration. The gel was dried, and the radioactive signal was acquired using a Typhoon FLA9500 scanner (GE Healthcare). Signals were quantified using Fiji software.
Northern blot
Seventy-two hours after siRNA transfection, 1 mg of TRIzol-extracted RNA was loaded on a denaturing gel with BET. RNA was passively transferred on a nitrocellulose membrane (Amersham) in SSC 10× buffer [1.5 M NaCl, 0.15 M sodium citrate (pH 7)]. The BET signal was acquired after transfer. Membranes were blocked in 3× SSC, 5× Denhardt’s, 0.5% SDS, and transfer RNA (tRNA) (0.5 mg/ml) for 1 hour at 45°C. Northern blot probes (described in table S4) were labeled with 25 mCi of [32P]ATP (adenosine triphosphate) (PerkinElmer) using T4 polynucleotide kinase (NEB). Incubation was performed overnight at 45°C. Radioactive signals were acquired using a Typhoon FLA9500 scanner (GE Healthcare).
EU and OP-Puro labeling
EU (BaseClick) and O-propargyl-puromycin (Jena Bioscience) were from the same kit using 5-FAM (Lumiprobe) as a fluorophore. Seventy-two hours after transfection, labeling was performed by adding EU (1 mM) for 15 min and OP-Puro (50 mM) for 15 to 45 min. Labeling was analyzed by flow cytometry (C6, BD Accuri apparatus).
Chromatin immunoprecipitation
HeLa cells (10 × 106 to 15 × 106) were grown in petri dishes (confluency of approximately 70 to 80%). Forty-eight hours after transfection, the medium was replaced with fresh medium, and fixation was performed as recommended by the ChIP-IT High Sensitivity Kit (Active Motif) (15 min in 1.1% paraformaldehyde fixation). Cells were then processed using the ChIP-IT High Sensitivity Kit, and immunoprecipitation was performed using 4 μg of the antibodies listed in table S3.
Chromatin immunoprecipitation–quantitative PCR
Probes are listed in table S5. For rDNA ChIP-qPCR, the following program was used: 50°C, 2 min; 95°C, 10 min; 45× (95°C, 15 s; 58°C, 1 min/30 s); dissociation stage (95°C, 15 s; 60°C, 1 min; 95°C, 15 s; 60°C, 15 s).
Enriched ribosome fraction isolation
Cells were lysed in 20 mM tris (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT), cycloheximide (100 μg/ml), and 1% Triton and triturated 10 times through a 26-gauge needle. The lysate was clarified by centrifugation at 10 min for 20,000g at 4°C, and the supernatant was loaded on a 1 M sucrose cushion prepared in 20 mM tris (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, and cycloheximide (100 μg/ml). Ribosomes were recovered after centrifugation at 4 hours for 70,000 rpm at 4°C in a TLA100.3 rotor (Beckman) and washed in PBS followed by 1 hour at 70,000 rpm at 4°C (62).
Polysomal fractionation
Cycloheximide (100 μg/ml) was added for 5 min to cell cultures and maintained in PBS washes. Cells were fractionated in 5 mM tris (pH 7.5), 2.5 mM MgCl2, and 1.5 mM KCl buffer supplemented with cOmplete ULTRA EDTA-free protease inhibitors (Roche), cycloheximide (100 μg/ml), RNase inhibitor (Promega RNasin 0.2 U/μl), 2 mM DTT, 0.5% Triton, and 0.5% sodium deoxycholate, and nuclei were discarded after a 7-min 16,000g centrifugation. The cytoplasmic optical density (OD) at 260 nm was assessed, and the lysate was diluted up to 10 to 20 OD (40 OD for MS). Then, 500 μl was loaded on a 5 to 50% sucrose gradient [20 mM Hepes (pH 7.6), 100 mM KCl, 5 mM MgCl2, 10 μM cycloheximide, 1/10 protease inhibitors, and RNase inhibitor (10 U/ml)] and centrifuged for 2 hours at 36,000 rpm at 4°C in a Beckman SW41Ti rotor. The A254 of the fractions was determined using a UA-6 UV/VIS detector (63).
LC-MS/MS acquisition
Protein extracts (30 μg) were precipitated with acetone at −20°C for 3 hours and incubated with 20 μl of 25 mM NH4HCO3 containing sequencing-grade trypsin (12.5 μg/ml; Promega) overnight at 37°C. The resulting peptides were desalted using ZipTip μ-C18 Pipette Tips (Millipore) and analyzed in technical triplicates by a Q-Exactive Plus coupled to a Nano-LC Proxeon 1000 equipped with an easy spray ion source (all from Thermo Fisher Scientific). Peptides were separated by chromatography with the following parameters: Acclaim PepMap100 C18 precolumn (2 cm, 75 μm inside diameter, 3 μm, 100 Å), Pepmap-RSLC Proxeon C18 column (50 cm, 75 μm inside diameter, 2 μm, 100 Å), flow rate (300 nl/min), gradient from 95% solvent A (water, 0.1% formic acid) to 35% solvent B (100% acetonitrile, 0.1% formic acid) over a period of 98 min, followed by column regeneration for 23 min, giving a total run time of 2 hours. Peptides were analyzed in the Orbitrap cell, in full ion scan mode, at a resolution of 70,000 [at mass/charge ratio (m/z) 200], with a mass range of m/z 375 to 1500 and an automatic gain control (AGC) target of 3 × 106. Fragments were obtained by high collision-induced dissociation activation with a collisional energy of 30% and a quadrupole isolation window of 1.4 Da. MS/MS data were acquired in the Orbitrap cell in Top20 mode at a resolution of 17,500, with an AGC target of 2 × 105, and a dynamic exclusion of 30 s. MS/MS of most intense precursors was first acquired. Peptides with unassigned charge states or monocharged peptides were excluded from the MS/MS acquisition. The maximum ion accumulation times were set to 50 ms for MS acquisition and 45 ms for MS/MS acquisition.
Quantitative analysis of protein abundance variations
Label-free relative quantification was performed in between subject analysis for each biological independent experiment, condition by condition, with raw data acquired in technical replicates, using Progenesis-Qi software 3.0 (Nonlinear Dynamics Ltd., Newcastle, UK). The procedure was as follows: (i) chromatogram alignment, (ii) peptide abundance normalization, which was performed on a set of 76 RPs, (iii) statistical analyses of features, and (iv) peptide identification. For this last step, all MS and MS/MS data were processed with Proteome Discoverer software (Thermo Fisher Scientific, version 2.1) and with the Mascot search engine (Matrix Science, version 5.1). The mass tolerance was set to 7 ppm for precursor ions and 0.02 Da for fragments. The maximum number of missed cleavages was limited to two for the trypsin protease. The following variable modifications were allowed: oxidation (Met) and phosphorylation (Ser and Thr). The SwissProt database (02/17) with the Homo sapiens taxonomy was used for the MS/MS identification step. Peptide identifications were validated using a 1% false discovery rate threshold calculated with the Percolator algorithm. Abundance variation measured for each protein by label-free quantification was validated if the calculated analysis of variance (ANOVA) P values were <0.05.
Supplementary Material
Acknowledgments
We thank all the members of the UMR9019-CNRS research unit for helpful discussions; the Proteomics platform of the Jacques Monod Institute (UMR7592 CNRS/Paris Diderot University) for the quantitative proteomics experiments; the Imaging and Cytometry Platform, UMS 23/3655, Gustave Roussy Cancer Campus, University Paris Saclay, Villejuif, France; P. Pasero for the gift of the S9.6 hybridoma–purified antibody and for sharing protocols; K. J. Patel and A. Constantinou for the gift of the FANCA-YFP and Flag-FANCA vectors, respectively; the Fanconi Anemia Research Fund (FARF) for the gift of antibodies; and A. Londono for the gift of the G4 antibody. Funding: A.G. was a fellow of the INSERM School Liliane Bettencourt and was supported by a “Course of Excellence in Oncology—Fondation Philanthropia” award. This work was funded by a grant from La Ligue Contre Le Cancer to F.R. Author contributions: A.G., F.M.-C., G.R., and S.S.-B. performed experiments. A.G. and F.R. conceived and designed experiments. S.A. and J.-J.D. provided expertise on nucleolar metabolism and ribosome biogenesis. A.G., S.A., J.-J.D., and F.R. analyzed the data. A.G. and F.R. wrote the paper. F.R. financed the study. Figure contributions: A.G.: Figs. 1, A to D and F; 2, A and C to F; 3, B to G; and 5, A, B, and D to G; F.M.-C.: Figs. 1, I to K; 4, A to F; and 5C; G.R.: Figs. 2, B and F, and 3G; S.S.-B.: Fig. 1E. Competing interests: The authors declare that they have no competing interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabb5414/DC1
REFERENCES AND NOTES
- 1.Tsekrekou M., Stratigi K., Chatzinikolaou G., The nucleolus: In genome maintenance and repair. Int. J. Mol. Sci. 18, 1411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulon S., Westman B. J., Hutten S., Boisvert F.-M., Lamond A. I., The nucleolus under stress. Mol. Cell 40, 216–227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J. M., Ellis S. R., Ribosomes and marrow failure: Coincidental association or molecular paradigm? Blood 107, 4583–4588 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Ruggero D., Shimamura A., Marrow failure: A window into ribosome biology. Blood 124, 2784–2792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bogliolo M., Surrallés J., Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 33, 32–40 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Ceccaldi R., Sarangi P., D’Andrea A. D., The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 17, 337–349 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Gueiderikh A., Rosselli F., Neto J. B. C., A never-ending story: The steadily growing family of the FA and FA-like genes. Genet. Mol. Biol. 40, 398–407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceccaldi R., Parmar K., Mouly E., Delord M., Kim J. M., Regairaz M., Pla M., Vasquez N., Zhang Q. S., Pondarre C., Peffault de Latour R., Gluckman E., Cavazzana-Calvo M., Leblanc T., Larghero J., Grompe M., Socie G., D’Andrea A. D., Soulier J., Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 11, 36–49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter D., Lier A., Geiselhart A., Thalheimer F. B., Huntscha S., Sobotta M. C., Moehrle B., Brocks D., Bayindir I., Kaschutnig P., Muedder K., Klein C., Jauch A., Schroeder T., Geiger H., Dick T. P., Holland-Letz T., Schmezer P., Lane S. W., Rieger M. A., Essers M. A. G., Williams D. A., Trumpp A., Milsom M. D., Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Rosado I. V., Langevin F., Crossan G. P., Takata M., Patel K. J., Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 18, 1432–1434 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Langevin F., Crossan G. P., Rosado I. V., Arends M. J., Patel K. J., Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58 (2011). [DOI] [PubMed] [Google Scholar]
- 12.van Twest S., Murphy V. J., Hodson C., Tan W., Swuec P., O’Rourke J. J., Heierhorst J., Crismani W., Deans A. J., Mechanism of ubiquitination and deubiquitination in the Fanconi anemia pathway. Mol. Cell 65, 247–259 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Adachi D., Oda T., Yagasaki H., Nakasato K., Taniguchi T., D’Andrea A. D., Asano S., Yamashita T., Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum. Mol. Genet. 11, 3125–3134 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Nepal M., Ma C., Xie G., Jia W., Fei P., Fanconi anemia complementation group C protein in metabolic disorders. Aging 10, 1506–1522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benitez A., Liu W., Palovcak A., Wang G., Moon J., An K., Kim A., Zheng K., Zhang Y., Bai F., Mazin A. V., Pei X. H., Yuan F., Zhang Y., FANCA promotes DNA double-strand break repair by catalyzing single-strand annealing and strand exchange. Mol. Cell 71, 621–628.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaddar T., Carreau M., Fanconi anemia proteins and their interacting partners: A molecular puzzle. Anemia 2012, 425814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Briot D., Mace-Aime G., Subra F., Rosselli F., Aberrant activation of stress-response pathways leads to TNF-alpha oversecretion in Fanconi anemia. Blood 111, 1913–1923 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Rosselli F., Sanceau J., Gluckman E., Wietzerbin J., Moustacchi E., Abnormal lymphokine production: A novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood 83, 1216–1225 (1994). [PubMed] [Google Scholar]
- 19.Oppezzo A., Bourseguin J., Renaud E., Pawlikowska P., Rosselli F., Microphthalmia transcription factor expression contributes to bone marrow failure in Fanconi anemia. J. Clin. Invest. 130, 1377–1391 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Kozono D. E., O’Connor K. W., Vidal-Cardenas S., Rousseau A., Hamilton A., Moreau L., Gaudiano E. F., Greenberger J., Bagby G., Soulier J., Grompe M., Parmar K., D’Andrea A. D., TGF-beta inhibition rescues hematopoietic stem cell defects and bone marrow failure in Fanconi anemia. Cell Stem Cell 18, 668–681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epanchintsev A., Shyamsunder P., Verma R. S., Lyakhovich A., IL-6, IL-8, MMP-2, MMP-9 are overexpressed in Fanconi anemia cells through a NF-kappaB/TNF-alpha dependent mechanism. Mol. Carcinog. 54, 1686–1699 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Matsushita N., Endo Y., Sato K., Kurumizaka H., Yamashita T., Takata M., Yanagi S., Direct inhibition of TNF-alpha promoter activity by Fanconi anemia protein FANCD2. PLOS ONE 6, e23324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zanier R., Briot D., Dugas du Villard J. A., Sarasin A., Rosselli F., Fanconi anemia C gene product regulates expression of genes involved in differentiation and inflammation. Oncogene 23, 5004–5013 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Moriel-Carretero M., Ovejero S., Gerus-Durand M., Vryzas D., Constantinou A., Fanconi anemia FANCD2 and FANCI proteins regulate the nuclear dynamics of splicing factors. J. Cell Biol. 216, 4007–4026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du W., Adam Z., Rani R., Zhang X., Pang Q., Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid. Redox Signal. 10, 1909–1921 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pagano G., Talamanca A. A., Castello G., d’Ischia M., Pallardo F. V., Petrovic S., Porto B., Tiano L., Zatterale A., From clinical description, to in vitro and animal studies, and backward to patients: Oxidative stress and mitochondrial dysfunction in Fanconi anemia. Free Radic. Biol. Med. 58, 118–125 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Pagano G., Degan P., d’Ischia M., Kelly F. J., Nobili B., Pallardo F. V., Youssoufian H., Zatterale A., Oxidative stress as a multiple effector in Fanconi anaemia clinical phenotype. Eur. J. Haematol. 75, 93–100 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Schwab R. A., Nieminuszczy J., Shah F., Langton J., Lopez Martinez D., Liang C. C., Cohn M. A., Gibbons R. J., Deans A. J., Niedzwiedz W., The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 60, 351–361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kruhlak M., Crouch E. E., Orlov M., Montano C., Gorski S. A., Nussenzweig A., Misteli T., Phair R. D., Casellas R., The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447, 730–734 (2007). [DOI] [PubMed] [Google Scholar]
- 30.van Sluis M., McStay B., A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 29, 1151–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kidiyoor G. R., Kumar A., Foiani M., ATR-mediated regulation of nuclear and cellular plasticity. DNA Repair 44, 143–150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warmerdam D. O., van den Berg J., Medema R. H., Breaks in the 45S rDNA lead to recombination-mediated loss of repeats. Cell Rep. 14, 2519–2527 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Ma H., Pederson T., The nucleolus stress response is coupled to an ATR-Chk1-mediated G2 arrest. Mol. Biol. Cell 24, 1334–1342 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kennedy R. D., Chen C. C., Stuckert P., Archila E. M., De la Vega M. A., Moreau L. A., Shimamura A., D’Andrea A. D., Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J. Clin. Invest. 117, 1440–1449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guervilly J. H., Mace-Aime G., Rosselli F., Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum. Mol. Genet. 17, 679–689 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Rubio M. L., Perez-Calero C., Barroso S. I., Tumini E., Herrera-Moyano E., Rosado I. V., Aguilera A., The Fanconi anemia pathway protects genome integrity from R-loops. PLOS Genet. 11, e1005674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jao C. Y., Salic A., Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U.S.A. 105, 15779–15784 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cammas A., Millevoi S., RNA G-quadruplexes: Emerging mechanisms in disease. Nucleic Acids Res. 45, 1584–1595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rhodes D., Lipps H. J., G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 43, 8627–8637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richard P., Manley J. L., R Loops and links to human disease. J. Mol. Biol. 429, 3168–3180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos-Pereira J. M., Aguilera A., R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 16, 583–597 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Hall A. C., Ostrowski L. A., Pietrobon V., Mekhail K., Repetitive DNA loci and their modulation by the non-canonical nucleic acid structures R-loops and G-quadruplexes. Nucleus 8, 162–181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan F., Qian L., Zhao X., Liu J. Y., Song L., D’Urso G., Jain C., Zhang Y., Fanconi anemia complementation group A (FANCA) protein has intrinsic affinity for nucleic acids with preference for single-stranded forms. J. Biol. Chem. 287, 4800–4807 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chasse H., Boulben S., Costache V., Cormier P., Morales J., Analysis of translation using polysome profiling. Nucleic Acids Res. 45, e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falini B., Bolli N., Liso A., Martelli M. P., Mannucci R., Pileri S., Nicoletti I., Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: Molecular basis and clinical implications. Leukemia 23, 1731–1743 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Falini B., Bolli N., Shan J., Martelli M. P., Liso A., Pucciarini A., Bigerna B., Pasqualucci L., Mannucci R., Rosati R., Gorello P., Diverio D., Roti G., Tiacci E., Cazzaniga G., Biondi A., Schnittger S., Haferlach T., Hiddemann W., Martelli M. F., Gu W., Mecucci C., Nicoletti I., Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 107, 4514–4523 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Bolli N., Nicoletti I., De Marco M. F., Bigerna B., Pucciarini A., Mannucci R., Martelli M. P., Liso A., Mecucci C., Fabbiano F., Martelli M. F., Henderson B. R., Falini B., Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res. 67, 6230–6237 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Du W., Li J., Sipple J., Chen J., Pang Q., Cytoplasmic FANCA-FANCC complex interacts and stabilizes the cytoplasm-dislocalized leukemic nucleophosmin protein (NPMc). J. Biol. Chem. 285, 37436–37444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falini B., Mecucci C., Tiacci E., Alcalay M., Rosati R., Pasqualucci L., La Starza R., Diverio D., Colombo E., Santucci A., Bigerna B., Pacini R., Pucciarini A., Liso A., Vignetti M., Fazi P., Meani N., Pettirossi V., Saglio G., Mandelli F., Lo-Coco F., Pelicci P. G., Martelli M. F.; GIMEMA Acute Leukemia Working Party , Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352, 254–266 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Meani N., Alcalay M., Role of nucleophosmin in acute myeloid leukemia. Expert Rev. Anticancer Ther. 9, 1283–1294 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Bailey G. D., Qutob H. M. H., Akhtar A., Russell N. H., Seedhouse C. H., DNA damage corrects the aberrant cytoplasmic localisation of nucleophosmin in NPM1 mutated acute myeloid leukaemia. Br. J. Haematol. 186, 343–347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collins N. B., Wilson J. B., Bush T., Thomashevski A., Roberts K. J., Jones N. J., Kupfer G. M., ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 113, 2181–2190 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taniguchi T., Garcia-Higuera I., Xu B., Andreassen P. R., Gregory R. C., Kim S. T., Lane W. S., Kastan M. B., D’Andrea A. D., Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109, 459–472 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Scott D. D., Oeffinger M., Nucleolin and nucleophosmin: Nucleolar proteins with multiple functions in DNA repair. Biochem. Cell Biol. 94, 419–432 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y., Duc A. C., Rao S., Sun X. L., Bilbee A. N., Rhodes M., Li Q., Kappes D. J., Rhodes J., Wiest D. L., Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev. Cell 24, 411–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finch A. J., Hilcenko C., Basse N., Drynan L. F., Goyenechea B., Menne T. F., Gonzalez Fernandez A., Simpson P., D’Santos C. S., Arends M. J., Donadieu J., Bellanne-Chantelot C., Costanzo M., Boone C., McKenzie A. N., Freund S. M., Warren A. J., Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 25, 917–929 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan S., Kermasson L., Hoslin A., Jaako P., Faille A., Acevedo-Arozena A., Lengline E., Ranta D., Poiree M., Fenneteau O., Ducou le Pointe H., Fumagalli S., Beaupain B., Nitschke P., Bole-Feysot C., de Villartay J. P., Bellanne-Chantelot C., Donadieu J., Kannengiesser C., Warren A. J., Revy P., EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood 134, 277–290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haimov O., Sehrawat U., Tamarkin-Ben Harush A., Bahat A., Uzonyi A., Will A., Hiraishi H., Asano K., Dikstein R., Dynamic interaction of eukaryotic initiation factor 4G1 (eIF4G1) with eIF4E and eIF1 underlies scanning-dependent and -independent translation. Mol. Cell. Biol. 38, e00139-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Badura M., Braunstein S., Zavadil J., Schneider R. J., DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc. Natl. Acad. Sci. U.S.A. 109, 18767–18772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang Z., Liang F., Teng Y., Chen X., Liu J., Longerich S., Rao T., Green A. M., Collins N. B., Xiong Y., Lan L., Sung P., Kupfer G. M., Binding of FANCI-FANCD2 complex to RNA and R-loops stimulates Robust FANCD2 monoubiquitination. Cell Rep. 26, 564–572.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sondalle S. B., Longerich S., Ogawa L. M., Sung P., Baserga S. J., Fanconi anemia protein FANCI functions in ribosome biogenesis. Proc. Natl. Acad. Sci. U.S.A. 116, 2561–2570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ingolia N. T., Brar G. A., Rouskin S., McGeachy A. M., Weissman J. S., The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7, 1534–1550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandin V., Sikstrom K., Alain T., Morita M., McLaughlan S., Larsson O., Topisirovic I., Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J. Vis. Exp. 17, 51455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grandori C., Gomez-Roman N., Felton-Edkins Z. A., Ngouenet C., Galloway D. A., Eisenman R. N., White R. J., c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 7, 311–318 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabb5414/DC1