

A broad-spectrum heteromultivalent influenza inhibitor is developed by using the host cell membrane on an inverted virus scaffold.
Abstract
Here, we report the topology-matched design of heteromultivalent nanostructures as potent and broad-spectrum virus entry inhibitors based on the host cell membrane. Initially, we investigate the virus binding dynamics to validate the better binding performance of the heteromultivalent moieties as compared to homomultivalent ones. The heteromultivalent binding moieties are transferred to nanostructures with a bowl-like shape matching the viral spherical surface. Unlike the conventional homomultivalent inhibitors, the heteromultivalent ones exhibit a half maximal inhibitory concentration of 32.4 ± 13.7 μg/ml due to the synergistic multivalent effects and the topology-matched shape. At a dose without causing cellular toxicity, >99.99% reduction of virus propagation has been achieved. Since multiple binding sites have also been identified on the S protein of SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), we envision that the use of heteromultivalent nanostructures may also be applied to develop a potent inhibitor to prevent coronavirus infection.
INTRODUCTION
The fast development of broad-spectrum solutions to prevent and inhibit viral infections is of great societal need. COVID-19 (coronavirus disease 2019), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has become a global health problem, with tens of millions of infections and hundreds of thousands of fatalities by now (1, 2). Besides SARS-CoV-2, influenza A virus (IAV) is also a highly infectious pathogen circulating in human beings and many other kinds of animal species (3, 4). Because of a high error rate of the viral RNA polymerase, human IAV evolution is accompanied by unpredictable antigenic drift and may involve the acquisition of mutations conferring resistance to many current antivirals (5, 6). Therefore, methods to develop potent and broad-spectrum viral inhibitors to quickly respond to the unexpected outbreak are urgently required (7, 8).
The viral infection cycle begins with binding to the receptors on the host cell membrane. Therefore, inhibiting viral entry via a binding decoy has been proved to be effective for many types of viruses, e.g., the use of multivalent mannose structures is reported to be effective for inhibiting Ebola virus infection, which is one of few options to fight Ebola infection (9–12). For IAV, two glycoproteins have been identified to be associated with the binding behavior on the cellular membrane, hemagglutinin (HA) and neuraminidase (NA). The multivalent binding between HA and sialic acid receptors on the host cell triggers the viral entry. Therefore, polysialylated structures with synthetic scaffolds have been designed for IAV inhibition (13–15). However, as the HA binding pocket varies in different IAV strains, developing broad-spectrum IAV inhibitors is a challenge. Intertrimer targeting that covers all the strain variations and subtypes of the IAV HA proteins can hardly be achieved with a single synthetic polysialyated structure. However, this task might be accomplished by cellular membrane–based materials. The cellular membrane is a highly stable self-assembled structure with a native display of virus-binding receptors. In principle, the native cell membrane can be a robust IAV binder, as it has the complex, dynamic, and multivalent sialic acid display for the binding of various IAVs. Moreover, the extraction of the cellular membrane is easier than synthesizing multivalent nanostructures matching the sialic acid–binding pockets on HA.
NA acts as a sialidase to cleave sialic acid from the attached host cell surface for the release of progeny virions. As a result of the HA-NA interplay, the interactions between IAV and host cells are transient and need to be dynamically balanced during early and late time points of infection, respectively (16, 17). Therefore, for the potent inhibition of both HA and NA, the heteromultivalent nanostructures are required to block both proteins. This is, however, rarely achieved, as most of the current antiviral strategies rely on homomultivalent engagement of HA (13, 14, 18). Here, we aim for generating a highly potent IAV inhibitor by combining three different bioinspired design incentives: (i) coating the inhibitor surface with a cell-derived membrane that serves as a native source of sialic acids (Fig. 1A); (ii) complementing the cell-derived membrane with zanamivir (Zan), an approved NA inhibitor, that enhances the IAV-membrane interaction by heteromultivalent binding (Fig. 1B); and (iii) using a concave inhibitor topology-matched shape that further enhances the IAV-membrane interaction by maximizing the total interaction area (Fig. 1C).
Fig. 1. Design of heteromultivalent topography-matching nanostructures for virus inhibition.
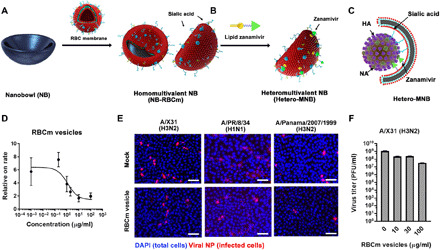
(A and B) Design and synthesis of a heteromultivalent nanobowl (Hetero-MNB) for IAV inhibition, including the coating of red blood cell membranes (RBCm) onto the NB surface and the further modification with lipidic Zan. (C) Proposed binding patterns between IAV and the Hetero-MNB, where sialic acid and Zan bind to HA and NA, respectively, and the bowl shape with facilitating the capping to the surface of the virus particle. (D) Inhibition curves for RBCm vesicles at different concentrations from a TIRF setup. Values are expressed as means ± SD, n = 4. (E) Typical immunofluorescent staining images of viral nucleoprotein (NP) to show the infected cells with the treatment of RBCm vesicles. Scale bars, 50 μm. Multiplicity of infection, 0.1. DAPI, 4′,6-diamidino-2-phenylindole. (F) Propagation of IAV in the presence of RBCm vesicles. Values are expressed as means ± SD, n = 4. PFU, plaque-forming units.
RESULTS
Red blood cell membrane as a potent binder but not inhibitor toward IAV
As most of the human IAV strains bind to red blood cells (RBCs) actively, we first investigated whether membranes extracted from RBCs are efficient IAV binders. Vesicles made of RBC membranes (RBCm) are obtained by disrupting human RBCs and centrifugation, and the obtained RBCm vesicles have the same protein presentation to the RBCs (fig. S1). The binding between RBCm and IAVs is investigated by an assay based on total internal reflection fluorescence (TIRF) microscopy (Fig. 1D) (19). The ganglioside GD1a-containing supported lipid bilayer (SLB) is created at a glass interface, and TIRF microscopy is used to monitor the transient binding of IAVs to this SLB. Subsequent application of single-virus tracking allows for extracting various information about the multivalent IAV–sialic acid interaction, the IAV attachment to the sialic acid–containing SLB, and the IAV mobility and off-rate distributions (19).
A decrease in the IAV attachment rate for increasing RBCm vesicle concentration is observed, indicating that RBCm vesicles can bind with IAVs and block their interaction with GD1a. The IC50TIRF (half maximal inhibitory concentration from TIRF) value from this measurement is 2.9 ± 1.2 μg/ml. From the hemagglutination inhibition assays of A/X31 (H3N2), A/Panama/2007/1999 (H3N2), A/PR/8/34 (H1N1), and A/Bayern/63/2009 (H1N1pdm), the broad-spectrum IAV binding ability of the RBCm vesicles is confirmed with an inhibition constant (KiHAI) in the nanomolar range (Table 1). The cellular infection assay, where the Madin-Darby canine kidney II (MDCK-II) cells, an epithelial host cell for IAV infection derived from canine kidney, are infected by influenza A/X31 (H3N2) in the presence of RBCm vesicles (100 μg/ml), revealed the broad-spectrum inhibition potential of the RBCm vesicles. However, the inhibitory activity of RBCm vesicle is not ideal; only a small decrease in infected cells is noticed, as shown in Fig. 1E. The reduction of virus titer for RBCm vesicles is two orders of magnitude (Fig. 1F).
Table 1. Inhibition constants of RBCm vesicles of different IAV strains from hemagglutination inhibition assay.
Values are expressed as means ± SD, n = 4.
| KiHAI (μg/ml) | KiHAI (nM sialic acid) | |
| A/X31 (H3N2) | 41.6 ± 20.6 | 8.4 ± 4.2 |
| A/PR/8/34 (H1N1) | 23.4 ± 9.0 | 4.8 ± 1.8 |
| A/Panama/2007/1999 (H3N2) |
27.3 ± 7.8 | 5.5 ± 1.7 |
It should be noted that the sialic acids for RBCm can be different from those for MDCK-II cells, especially the glycan structures (20, 21). The membrane of MDCK-II cells is obtained, and its binding to influenza A/X31 (H3N2) is studied via hemagglutination inhibition assays. The MDCK-II cell membrane exhibits a KiHAI of 1.6 ± 0.5 nM, which is lower than that of RBCm in the same assay (8.4 ± 4.2 nM). As a binding decoy, the binding to the inhibitor should outperform the binding to the actual receptor to achieve potent inhibition (22). When applying RBCm to MDCK-II cells, the virions may still bind to the actual sialic acid receptor on MDCK-II cells because of the higher binding affinity and then start the infection. Therefore, to develop a potent IAV inhibitor with RBCm, the virus binding ability must be further improved.
Heteromultivalent surface for enhanced virus binding
Two concepts are introduced for improving the virus binding ability of RBCm: (i) supplementing additional NA inhibitors to further promote the viral binding to the RBCm and (ii) transferring the RBCm to a nanoparticle surface with matched topology to the IAV virion to maximize the interaction area (Fig. 1, B and C). To prove the first concept that heteromultivalent targeting of both HA and NA on IAV can further promote virus binding, hybrid vesicles of RBCm with an approved NA inhibitor, Zan, are obtained. Zan is initially covalently coupled to lipid and then formed into a lipid vesicle (figs. S2 and S3). The hybrid RBCm-Zan vesicle is obtained via cosonication. The binding of IAV virions to the vesicles is checked by hemagglutination inhibition assays (Table 2). The addition of free Zan decreases the KiHAI value for RBCm vesicles, indicating that inhibiting NA can improve virion binding due to the inhibition of HA–sialic acid cleavage. The NA activity for RBCm-Zan–treated virions is similar to those treated by “RBCm + Free Zan” (fig. S4), but a lower KiHAI value is noticed. This means that besides the NA inhibitory activity, Zan may also provide an additional binding possibility for the virion, which is analyzed in detail via TIRF microscopy using single-virus tracking and the equilibrium fluctuation analysis (23, 24).
Table 2. Inhibition constants of samples against A/X31 (H3N2) from hemagglutination inhibition assay.
Values are expressed as means ± SD, n = 4.
| KiHAI (μg/ml) | KiHAI (nM sialic acid) | |
| RBCm | 41.6 ± 20.6 | 8.4 ± 4.2 |
| MDCK-II membrane | 6.8 ± 1.9 | 1.6 ± 0.5 |
| RBCm vesicles and free Zan |
11.7 ± 4.5 | 2.4 ± 0.9 |
| RBCm-Zan hybrid vesicles |
5.9 ± 2.3 | 1.2 ± 0.5 |
| NB-RBCm | 51.9 ± 23.8 | 3.1 ± 1.4 |
| NB-RBCm and Zan | 19.5 ± 7.8 | 1.2 ± 0.5 |
| Hetero-MNB | 13.7 ± 3.9 | 0.8 ± 0.2 |
The TIRF-based assay is now operated using SLBs that have been fused with RBCm vesicles, Zan-conjugated lipids, or a mixture of both, allowing to determine changes in the IAV binding dynamics that are caused by the presence of different glycostructures (RBCm-originating sialic acid, Zan-conjugated lipids, or a combination of both; Fig. 2). In this analysis, RBCm vesicles are fused with POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)–based vesicles, allowing to form “hybrid” SLBs containing RBCm material and serving as a homomultivalent structure (25, 26). A heteromultivalent SLB membrane is generated by incorporating lipidic Zan into the RBCm hybrid SLB (RBCm-Zan).
Fig. 2. Heteromultivalent surfaces for the interaction with IAV virions.
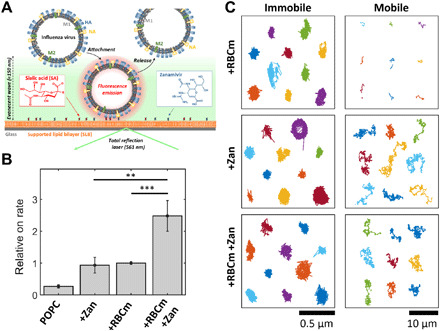
(A) TIRF-based assay for assessing the interaction of IAVs with homo- or heteromultivalent membranes. The corresponding glycostructures (either sialic acids or lipid-conjugated Zan) are incorporated in an SLB, and the IAV-SLB interaction is recorded using TIRF microscopy. Application of single-virus tracking allows for extracting changes of (B) the rate of IAV attachment to the membranes and (C) the motion of SLB-bound IAVs, when using the different homo- and heteromultivalent structures. Each color represents the track of a single virion. Values in (B) are expressed as means ± SEM, n = 10. **P < 0.01 and ***P < 0.001 by Student’s t test. Note the different scale bars used for the immobile and mobile IAVs in (C).
Application of the TIRF assay indicates that the rate of IAV attachment to the heteromultivalent surface of RBCm-Zan shows threefold higher values with respect to Zan- or RCBm-SLB (Fig. 2B). Furthermore, single-virus tracking also provides information about the motion of IAV while being SLB bound. On RBCm-SLBs, most of the mobile tracks are very short, indicating that the viruses stay at the surface for less than 1 s before being released (Fig. 2C, fig. S5A, and movie S1). This behavior is qualitatively similar to IAVs interacting with GD1a-containing SLBs (19). The addition of Zan-conjugated lipids, however, strongly increases the duration of the mobile population, which now exhibits very long tracks, and thus the IAV residence time being much larger than 1 s (Fig. 2C, fig. S5B, and movie S2). This behavior is observed for both membranes, the homomultivalent Zan-SLBs and the heteromultivalent RBCm-Zan-SLB. Hence, while IAVs show only short residence times to sialic acid–containing SLBs, the NA-Zan interaction generates very large IAV residence times to Zan-containing SLBs. These findings are in line with the observation that the binding between Zan and NA is predominant to the binding between sialic acid and IAV (27) and with the concept that the inhibitory effect of Zan on NA plays a beneficial role for permanent binding, as NA is able to cleave the binding between HA and sialic acid as sialidase. Hence, while a notable fraction of IAVs exhibit only short residence times to sialic acid–containing SLBs and can, therefore, bind to a small number of sialic acids, the addition of lipid-Zan strongly extends the IAV residence time by the formation of NA-Zan interactions. In the heteromultivalent SLB, this behavior keeps the IAVs sufficiently long at the membrane to allow for binding a large number of sialic acids, thereby promoting the IAV attachment rate and IAV entrapment by the membrane. Thus, the heteromultivalent membrane provides a much better binding compared to homomultivalent ones.
Nanobowl with a matched surface toward IAV virion for virus binding
To prove the second concept that matching topology can benefit the virus binding, bioinert and biostable nanoparticles with sphere [nanosphere (NS)] and bowl-like [nanobowl (NB)] morphologies are synthesized, as shown in Fig. 3A and figs. S6 and S7 (28). The nanoparticle is homomultivalently coated with RBCm (figs. S8 and S9), named as NS-RBCm and NB-RBCm, respectively. The coating is carried out via cosonicating an RBCm vesicle with NS and NB (29, 30). A cellular membrane is a highly dynamic lipid system with the ability to deform and adapt to the surrounding environment. In the coating process, the high-power sonication drives the membrane vesicle to follow the curvature of the NB. After the sonication, the membrane is stabilized on the surface of the nanoparticle via hydrophobic interactions. Last, free membrane vesicles are removed by centrifugation.
Fig. 3. Topography-matching principle for maximized virus interaction.

(A) High-resolution transmission electron microscopy (HR-TEM) images for NS and NB with RBCm coating. Scale bars, 50 nm. (B) IC50infection inhibition values for the homomultivalent NS-RBCm and NB-RBCm. Values are expressed as means ± SD, n = 4. (C) Geometry analysis for a virion binding to a convex inhibitor and concave inhibitor. The actual contact between them is marked with θ1; the steric shielding by the inhibitor is marked with purple lines with θ2. Steric shielding is estimated by placing inhibitors with the same curvature. (D) Negatively stained HR-TEM image for the IAV particle binding with the Hetero-MNB. Scale bar, 50 nm. Images for other samples are shown in fig. S11.
The prototypic IAV virion is a nanosized sphere around 100 nm in diameter, and in our synthesis, an NB with a concave structure of around 120 nm is selected, as it matches the size of the IAV to benefit multivalent interactions (Fig. 3A). The binding for the NS-RBCm and NB-RBCm is studied by a prophylactic infective assay (fig. S10). Less infected cells and higher cell viability are noticed for NB-RBCm than for NS-RBCm. The IC50infection inhibition value for NB-RBCm is about four times lower than that of NS-RBCm, as shown in Fig. 3B. The better inhibition by the NB-RBCm should be attributed to its matched shape to the IAV virion, which can promote the binding of the virus, as proved by our binding test in fig. S11.
Geometry analysis of the virus binding to NS-RBCm and NB-RBCm is performed with the model of a convex virion binding to a convex or concave inhibitor, respectively, as shown in Fig. 3C. The IAV virion is illustrated as a particle of around 100 nm. The steric shielding by the inhibitor is estimated by placing inhibitors with the same curvature, which are marked with dashed lines (31). The actual contact between the virion and the inhibitor is marked white with an angle of θ1, while the steric shielding by the inhibitor is marked purple with an angle of θ2. In these two cases, the actual contact area is quite similar, but the steric shielding is different. A concave inhibitor creates a much larger steric shielding than a convex inhibitor; therefore, it shows better virus inhibition than the convex one. Another difference between them is the edge distance. The concave inhibitor is much closer to the virion than the convex one. Monovalent sialic acid–HA interaction is weak with a dissociation constant of 1 to 20 mM (17, 32). When binding to a surface, e.g., cellular membrane, the virion remains mobile in the beginning. This is what we have observed from TIRF that IAV virions remain mobile when binding to RBCm. The virion is transiently interacting with RBCm and then stabilizes on the surface via multivalent interactions. Many other studies also reported that influenza virions are mobile when binding at the biological interfaces (17, 33–35). Therefore, we presume that in the case of a convex inhibitor (NS), a virion could be released by crawling and gliding, especially when NA is constantly cleaving the sialic acid–HA interaction. For a concave inhibitor (NB), the small edge distance enables a virion to more easily recruit binding ligands for multivalent interaction and subsequent stabilization.
Heteromultivalent NB as a potent and broad-spectrum IAV inhibitor
Last, concepts (i) and (ii) are combined into a heteromultivalent NB (Hetero-MNB) for the in vitro viral inhibition studies, which is obtained by inserting the lipid-Zan to NB-RBCm. The negatively stained high-resolution transmission electron microscopy (HR-TEM) images in Fig. 3D reveal the binding between IAV virion and Hetero-MNB. The hemagglutination inhibition assay (Table 2) reveals that the Hetero-MNB is more predominant than NB-RBCm for viral binding. As a result of heteromultivalent inhibition of HA and NA, the IAV virion binding to MDCK-II cells is notably inhibited, as shown in Fig. 4 (A to C). Little-to-no virus particles are detected on the surface of MDCK-II cells by flow cytometry and fluorescence microscopy, indicating that the inhibitors can effectively block the entry of the virus.
Fig. 4. Inhibition of IAV infection by the heteromultivalent topography-matching nanostructure.
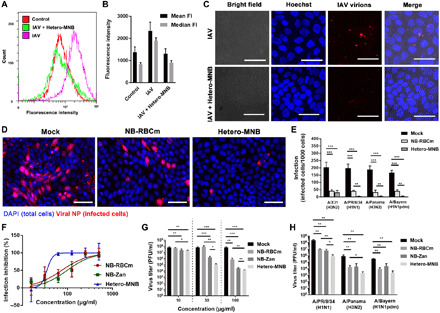
(A) Flow cytometry for the virus attachment to MDCK-II cells and (B) corresponding mean and median fluorescence intensities (FI) for the samples. The error bars are generated by the CytExpert Acquisition and Analysis Software Version 2.3. Fluorescence intensity is measured in arbitrary units. (C) Projection confocal laser scanning microscopy images for the viral attachment to MDCK-II cells in the presence of Hetero-MNB. Scale bars, 20 μm. (D) Typical immunofluorescence images to show the IAV [A/X31 (H3N2)] infection in the presence of the inhibitors. Other images are shown in figs. S12 and S13. Scale bars, 100 μm. (E) Detection of viral NP in cells infected with four typical human IAV strains, including A/X31 (H3N2), A/PR/8/34 (H1N1), A/Panama/2007/1999 (H3N2), and A/Bayern/63/2009 (H1N1pdm) in the presence or absence of inhibitors. (F) Inhibition of A/X31 (H3N2) of the inhibitors at different dosages revealed by cell viability. (G) Inhibition toward IAV propagation [A/X31 (H3N2)] for the inhibitors. (H) Inhibition toward IAV propagation for the inhibitors against three other IAV strains, including A/PR/8/34 (H1N1), A/Panama/2007/1999 (H3N2), and A/Bayern/63/2009 (H1N1pdm). The inhibitors are used after the first cycle of infection. Inhibitor dosage, 100 μg/ml. Values are expressed as means ± SD, n = 4. *P < 0.05, **P < 0.01, and ***P < 0.001 by Student’s t test, respectively.
Next, we investigated whether the Hetero-MNB is able to inhibit the virus infection at the entry step by counting cells expressing IAV nucleoprotein (NP) (Fig. 4, D and E, and figs. S12 and S13). The treatment of NB-RBCm and Hetero-MNB significantly reduces the infectivity of A/X31 (H3N2), and Hetero-MNB shows better inhibitory effects than NB-RBCm. We have tested the inhibition toward four typical human IAV strains [A/X31 (H3N2), A/PR/8/34 (H1N1), A/Panama/2007/1999 (H3N2), and A/Bayern/63/2009 (H1N1pdm)]. The Hetero-MNB shows broad-spectrum inhibition, as little-to-no infected cells are observed in these four tests. The cell viability test further verifies the inhibitory effects of Hetero-MNB (Fig. 4E), whereas the cells are more viable after the treatment by Hetero-MNB. Here, Hetero-MNB reveals an IC50infection inhibition value of 32.4 ± 13.7 μg/ml. It should be noted that Hetero-MNB shows no clear toxicity in MDCK-II cells (fig. S14). The CC50 (50% cytotoxic concentration) value is 2.4 ± 0.5 mg/ml, corresponding to a selective index of 74.1.
In addition to the cellular infections, the ability to reduce viral propagation is also studied by plaque assay for the samples, as shown in Fig. 4G. In this test, to investigate whether they can be used as a therapeutic agent, the inhibitors are used after the cells have been infected by the virus. Both the NB-RBCm and Hetero-MNB reduced viral titers. Hetero-MNB outperforms the homomultivalent ones with four orders of magnitude of reduction, which corresponds to inhibitory effects higher than 99.99%. Figure 4H further illustrates that the Hetero-MNB is active against the typical IAV strains circulating in humans, revealing its potential to be a broad-spectrum IAV inhibitor.
IAV mutation with Zan resistance has been noticed recently (36, 37). We also tested Hetero-MNB for the inhibition of a Zan-resisting strain (influenza A/BLN/28/2012) via cellular infection assays (fig. S15) (38). Hetero-MNB can inhibit viral replication with two orders of magnitude reduction of virus titer. However, in this case, because NA has mutated, NB-Zan does not show any inhibitory effects; Hetero-MNB and NB-RBCm show similar activities. Considering the inhibitory effects of RBCm, NB-RBCm and Hetero-MNB can be one possible solution to inhibit Zan-resisting IAV strains. The use of Zan is a proof of concept for heteromultivalency; other NA inhibitors (e.g., oseltamivir and NA antibodies) may also be used for heteromultivalency via similar methods (39–41).
The inhibitory activity of RBCm-based structures might be lower than the one of recently reported, rationally designed polysialylated structures (14, 15, 42), but compared with their complicated synthetic process and high costs, the approach in this study is much simpler, sparing the efforts of preparing and screening a big library of candidate compounds. If a mutation that IAV requires a different sialic acid density occurs, then the synthetic structures with the fixed sialic acid display may lose their activity. Membrane-based compounds may be less prone to this pitfall since membrane fluidity and the possibility of lateral diffusion of membrane proteins enable attachment factors to adjust and possibly even be recruited by virions as needed. The easy preparation of RBCm-based inhibitors makes it an excellent candidate to prepare in case they do lose activity after all. Even if IAV acquires mutations to not bind RBC anymore, we still have the option of taking the membrane from its host cells, e.g., A549 cells and MDCK-II cells for inhibitor development. It should be noted that the extraction of A549 and MDCK-II cell membranes takes more effort and needs further optimization for large-scale production.
DISCUSSION
The development of heteromultivalent RBCm inhibitors with a topology-matched design is believed to expand the toolbox for fighting pathogens as alternatives for the rapidly emerging resistance to the current drugs and therapies. In our study, the combination of the specific binding capabilities of sialic acid and Zan provide a unique heteromultivalent dynamic viral binding decoy to achieve potent interactions with IAV, which is much better than the conventional homomultivalent ones. When coated onto a fitting bowl-shaped nanostructure, the heteromultivalent membrane exhibits 99.99% viral inhibition due to the synergistic multivalent effects and topology-matched shape at a dose, causing no cellular toxicity. The IC50infection inhibition value is 32.4 ± 13.7 μg/ml (1.8 ± 0.8 nM sialic acid) with a selective index of 74.1. We also proved that Hetero-MNB could serve as a broad-spectrum binding decoy and entry inhibitors for human IAV strains.
Furthermore, this unique strategy using bioinspired heteromultivalent topology-matching nanostructures is a general approach to fabricate broad virus inhibitors and might compete with previously unknown or mutated viruses better than specifically designed drugs. Recent studies have pointed out the in vivo safety of RBCm-coated nanoparticles, of which the intravenous injection does not induce any adverse effects in mice (29, 43). They have been used for the development of an in vivo drug delivery vehicle, toxin absorber, and vaccine platform (43–45). The potential antigenicity of a foreign cell membrane can be addressed by taking the RBCs from the host. The protocol for membrane extraction is simple and can be finished within 1 day. Therefore, it is believed that cell membranes can serve as a promising platform to develop virus inhibitors.
There are many similarities between SARS-CoV-2 and IAV, not only in disease symptoms but also in the infection cycles. SARS-CoV-2 uses its spike protein (S protein) to interact with the angiotensin-converting enzyme 2 (ACE2) on host cells’ surface to mediate cellular entry. Structural analysis of S protein has revealed several binding sites for potential inhibitor design (46, 47). On the head of S protein (S1 domain) locates the receptor binding domain (RBD) that interacts with ACE2. The stalk domain (S2 domain) contains a fusion peptide that triggers the virus fusion with host cells to release the viral genome for replication. A furin cleavage site is identified at the S1/S2 region, the cleavage of which, by furin or TMPRSS2, might contribute to activate RBD interaction with ACE2. It has also been revealed by a recent study that these domains can be targeted separately by noncompeting antibodies. Antibody cocktails showed a substantially improved virus neutralization ability compared to monovalent antibodies (48, 49). Taking together the information from this study, it is believed that the heteromultivalent display of ligands that target different domains on S proteins can result in a potent SARS-CoV-2 inhibitor (50, 51). The membrane of the host cells, e.g., Vero E6 cell, may be used as one target to the RBD (52). For the design of such an inhibitor, the platform supporting the ligands should be rationally designed to facilitate multiple interactions with heteromultivalency.
MATERIALS AND METHODS
Materials and characterizations
All chemicals and solvents are of high-performance liquid chromatography or reagent grade and purchased from Sigma-Aldrich (Steinheim, Germany) unless stated otherwise. The deionized water (minimum resistivity of 18.0 megohms∙cm) is obtained from a Millipore water purification system. The human RBCs are obtained from Haema Blutspendezentrum, Berlin, Germany. Characterization methods are listed in the Supplementary Materials.
Viruses and cells
MDCK-II epithelial cells are maintained in monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM) [with 10% fetal calf serum, 2 mM l-glutamine, streptomycin (100 mg/ml), and penicillin (100 U/ml)] at 37°C and 5% CO2. IAV strains A/Panama/2007/1999 (H3N2), A/X31 (H3N2), A/PR/8/34 (H1N1), A/Bayern/63/2009 (H1N1), and A/BLN/28/2012 are taken from the strain collection of Unit 17 at the Robert Koch-Institut (Berlin, Germany) and propagated in 10-day-old embryonated chicken eggs. The stock virus is quantitated by plaque titration on MDCK-II cells.
Extraction of RBCm
The RBCm is obtained by a hypotonic treatment and ultracentrifugation process. The human RBCs are first washed with phosphate-buffered saline (PBS) (pH 7.4) at 4°C to remove the residual platelets and blood proteins and then treated overnight with deionized water–diluted PBS buffer (0.25×) at 4°C. The RBCm is collected by 5-min centrifugation at 10, 000 rpm, then washed with PBS buffer extensively, and then frozen at −20°C for storage.
Extraction of MDCK-II cell membrane
The membrane of MDCK-II cells is extracted according to earlier protocols (53, 54). The MDCK-II cells are harvested by trypsin and collected by 5-min centrifugation at 1000 rpm. The cells are washed three times with PBS and then placed in cold tris buffer (pH 7.4) containing 10 mM tris, 10 mM MgCl2, and 1× EDTA-free protease inhibitor. The cells are then disrupted by homogenizing with a dounce homogenizer on ice. The solution is centrifuged at 5000 rpm for 5 min; the supernatant containing membrane vesicles is then centrifuged at 100,000g for 2 hours (Optima L-100XP, Beckman Coulter). The pellet is washed with PBS and centrifuged again at 100,000g for 2 hours. The resuspended cell membrane vesicles are extruded through 400-nm polycarbonate membranes (Millipore) and stored at −80°C.
Hemagglutination inhibition assay
The samples are diluted in 25 μl of PBS twofold in a U-shaped 96-well plate. Twenty-five microliters of IAV solution (in PBS, containing 4 HA units) is mixed with the samples by pipetting and incubated for 30 min at room temperature. Afterward, 50 μl of 1% chicken RBCs is added to each well and incubated for another 30 min at room temperature. The hemagglutination is observed by tilting the plates. The sample concentration of the last well showing inhibition of hemagglutination is recorded as the binding constant KiHAI.
TIRF microscopy
TIRF microscopy is used to quantify the interaction of single, R18-labeled IAV with different glycostructures embedded in SLBs with different compositions, as described (19). The glycostructures originated from native RBCm vesicles, Zan-conjugated lipids, or both. This method provides information about various properties of the IAV-membrane interaction, such as the attachment rate to the membrane, IAV mobility, and off-rate distributions regarding the different membrane compositions. The conduction of measurements is shown in the Supplementary Materials.
Synthesis of spherical and bowl-like nanoparticles
First, the resorcinol-formaldehyde–coated SiO2 nanoparticle is synthesized via a one-step reaction according to an earlier report with slight modifications (28). Briefly, 2.5 ml of ammonia aqueous solutions [30 weight % (wt %)] is added into 100 ml of ethanol/water (v/v, 2/1) and stirred for 1 hour at room temperature. With vigorous stirring, 3 ml of TEOS (tetraethoxysilane) (19.2 mmol), 0.2 g of resorcinol (4.5 mmol), and 0.7 ml of formaldehyde solution (37 wt % in H2O; 9 mmol) are added sequentially. The reaction is ceased by 10-min centrifugation at 10,000 rpm after 3, 6, and 24 hours, respectively. The final products are washed five times with water and collected via 10-min centrifugation at 10,000 rpm and then lyophilized. The nanocomposites are carbonized at 600°, 700°, and 800°C (5°C/min) for 1 hour with argon, respectively. The SiO2 core is then removed by etching in 2 M NaOH for 2 days at 60°C, and different types of hollow nanocarbons, including the spherical and bowl-like nanoparticles, can be obtained via 10-min centrifugation at 10,000 rpm and washed extensively by water. Afterward, the nanoparticles are carbonized again at 1100°C (5°C/min) for 1 hour with argon to fix the morphology.
Coating of RBCm onto the nanoparticle surface
The RBCm is coated onto the nanoparticle surface with horn sonication (Sonopuls Ultrasonic Homogenizers HD 2200, BANDELIN electronic GmbH & Co. KG, Berlin, Germany) at 4°C for 30 min and then the RBCm-coated nanocarbons are isolated by centrifugation at 5000 rpm for 5 min and washed with PBS buffer. The morphology of the RBCm-coated nanoparticles is studied by scanning electron microscopy and TEM. For a better view of RBCm, the sample is stained with 1% sodium phosphotungstate before the measurement. The protein content is studied by the Pierce BCA assay kit (23225; Thermo Fisher Scientific, USA) according to the manual instructions. The RBCm coating weight ratio is then estimated by the protein contents following the equation
For SDS–polyacrylamide gel electrophoresis, 10 μg of samples is loaded to 10% polyacrylamide (PAM) gel, and the gel is run at 200 V for 1 hour and then stained with 10% (v/v) Coomassie blue solution and then imaged by the ChemiDoc MP imaging system (Bio-Rad). The sialic acid content for the RBCm-coated nanocarbons is studied by Sialic Acid (NANA) Assay Kit (Abcam) according to the instructions by the provider.
Virus binding tests
For the Western blot test, 5 μl of concentrated A/X31 (H3N2) virus solution (protein content, 0.72 mg/ml) is incubated with 15 μl of solution with different nanoinhibitors (1 mg/ml) for 45 min at 37°C. Then, the mixture is centrifuged at 10,000 rpm for 10 min to collect the virus-bound nanoinhibitors. The precipitant is washed with PBS and loaded to a 10% PAM gel. After running at 200 V for 1 hour, the proteins are transferred onto the polyvinylidene difluoride membrane, which is then blocked by 3 wt % of milk powder. The NP is marked by influenza A NP monoclonal antibody (Invitrogen) and horseradish peroxidase–conjugated secondary antibody (Invitrogen). Then, chemiluminescent detection is performed using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). The intensities of the bands are analyzed by ImageJ.
Virus attachment and inhibition
To study virus attachment, 100 μl of A/X31 (H3N2) solution (protein content, 0.73 mg/ml) is incubated with 10 μl of 20 μM octadecyl rhodamine B chloride (R18; Thermo Fisher Scientific) in PBS for 45 min first. The free R18 is removed with a spinning column (3300 rpm, 2 min; Protein A HP SpinTrap, GE Healthcare). Then, 10 μl of labeled virus is incubated with a 90-μl nanoinhibitor solution (100 μg/ml) for 45 min at 37°C. The mixture is then incubated with MDCK-II cells on ice for 1 hour. The unbound virions are removed by washing with PBS. Afterward, the cells are stained with 4′,6-diamidino-2-phenylindole (DAPI) and visualized by fluorescence microscopy. For a quantitative analysis of virus attachment, the mixture of labeled virus and nanoinhibitor is incubated with 100,000 MDCK-II cells for 1 hour, and then the unbounded virus is removed by centrifugation at 1500 rpm for 2 min. The cells are washed with PBS and analyzed by flow cytometry (CytoFLEX S, Beckman Coulter).
For the infection inhibition test, 0.2 × 106 plaque-forming units (PFU) of A/X31 (H3N2) virus is treated with the nanoinhibitors for 45 min at 37°C and then incubated with MDCK-II cells. After 45 min, the cells are washed twice by PBS and cultured in DMEM [0.1% bovine serum albumin (BSA), streptomycin (1100 mg/ml), and penicillin (100 U/ml)]. After 24 hours, the cells are fixed with 4% formaldehyde and permeabilized with 0.5% Triton X-100. The infected cells are marked by influenza A NP monoclonal antibody (Invitrogen) and Alexa Fluor 594–coupled secondary antibody (Invitrogen). The cell nucleus is stained using DAPI. The infection is estimated by counting infected cells from at least 10,000 cells in total and expressed as the number of infected cells per 1000 cells.
Multicyclic viral replication inhibition test
MDCK-II cells are first infected with IAV at the multiplicity of infection of 0.01 and cultured in an infection medium [DMEM, 0.1% BSA, 1% l-glutamine, 1% penicillin-streptomycin, and TPCK(N-tosyl-L-phenylalanine chloromethyl ketone)-trypsin (1 μg/ml)] containing the nanoinhibitors for 24 hours. The viruses in the supernatants are then titered by plaque assay on MDCK-II cells and are expressed as PFU/ml.
Supplementary Material
Acknowledgments
P. Winchester is acknowledged for language-polishing the manuscript. We would like to acknowledge the assistance of the Core Facility BioSupraMol in Freie Universität Berlin supported by the Deutsche Forschungsgemeinschaft (DFG) and Zhongkebaice Technology Service Co. Ltd., Beijing, China for materials characterizations. Funding: The authors gratefully acknowledge financial support from DFG through grants from the Collaborative Research Center (SFB) 765. S. Bl. acknowledges the support of DFG through grant BL1514/1. C.N. acknowledges the support from the China Scholarship Council (CSC). C.C. acknowledges the support of the National Key R&D Program of China (2019YFA0110600 and 2019YFA0110601), the Science and Technology Project of Sichuan Province (2020YFH0087 and 2020YJ0055), Special Funds for Prevention and Control of COVID-19 of Sichuan University (2020scunCoV-YJ-20005), and SKLFPM, Donghua University (YJ202005), State Key Laboratory of Polymer Materials Engineering (grant no. sklpme2019-2-03), Fundamental Research Funds for the Central Universities, Ten Thousand Youth Talents Plan, and Alexander von Humboldt Fellowship. Author contributions: C.N. and C.C. synthesized the materials. T.W. provided the IAV strains and supervised the virus infection tests. C.N. and M.S. performed the virus infection inhibition tests. M.W., Y.K., and S.Bl. designed and performed TIRFM analysis. B.P. and S.Bh. synthesized functionalized Zan molecules. V.A. performed MALDI-TOF analysis. C.C., T.W., and R.H. designed and supervised the project. C.N., C.C., T.W., and R.H. wrote and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd3803/DC1
REFERENCES AND NOTES
- 1.Wang C., Horby P. W., Hayden F. G., Gao G. F., A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W.; China Novel Coronavirus Investigating and Research Team , A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taubenberger J. K., Morens D. M., The pathology of influenza virus infections. Annu. Rev. Pathol. 3, 499–522 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson W. W., Shay D. K., Weintraub E., Brammer L., Bridges C. B., Cox N. J., Fukuda K., Influenza-associated hospitalizations in the United States. JAMA 292, 1333–1340 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Bloom J. D., Gong L. I., Baltimore D., Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328, 1272–1275 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrat F., Flahault A., Influenza vaccine: The challenge of antigenic drift. Vaccine 25, 6852–6862 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Laursen N. S., Friesen R. H. E., Zhu X., Jongeneelen M., Blokland S., Vermond J., van Eijgen A., Tang C., van Diepen H., Obmolova G., van der Neut Kolfschoten M., Zuijdgeest D., Straetemans R., Hoffman R. M. B., Nieusma T., Pallesen J., Turner H. L., Bernard S. M., Ward A. B., Luo J., Poon L. L. M., Tretiakova A. P., Wilson J. M., Limberis M. P., Vogels R., Brandenburg B., Kolkman J. A., Wilson I. A., Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 362, 598–602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Dongen M. J. P., Kadam R. U., Juraszek J., Lawson E., Brandenburg B., Schmitz F., Schepens W. B. G., Stoops B., van Diepen H. A., Jongeneelen M., Tang C., Vermond J., van Eijgen-Obregoso Real A., Blokland S., Garg D., Yu W., Goutier W., Lanckacker E., Klap J. M., Peeters D. C. G., Wu J., Buyck C., Jonckers T. H. M., Roymans D., Roevens P., Vogels R., Koudstaal W., Friesen R. H. E., Raboisson P., Dhanak D., Goudsmit J., Wilson I. A., A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 363, eaar6221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muñoz A., Sigwalt D., Illescas B. M., Luczkowiak J., Rodríguez-Pérez L., Nierengarten I., Holler M., Remy J.-S., Buffet K., Vincent S. P., Rojo J., Delgado R., Nierengarten J.-F., Martín N., Synthesis of giant globular multivalent glycofullerenes as potent inhibitors in a model of Ebola virus infection. Nat. Chem. 8, 50–57 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ramos-Soriano J., Reina J. J., Illescas B. M., de la Cruz N., Rodríguez-Pérez L., Lasala F., Rojo J., Delgado R., Martín N., Synthesis of highly efficient multivalent disaccharide/[60]fullerene nanoballs for emergent viruses. J. Am. Chem. Soc. 141, 15403–15412 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Rodríguez-Pérez L., Ramos-Soriano J., Pérez-Sánchez A., Illescas B. M., Muñoz A., Luczkowiak J., Lasala F., Rojo J., Delgado R., Martín N., Nanocarbon-based glycoconjugates as multivalent inhibitors of ebola virus infection. J. Am. Chem. Soc. 140, 9891–9898 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Illescas B. M., Rojo J., Delgado R., Martín N., Multivalent glycosylated nanostructures to inhibit Ebola virus infection. J. Am. Chem. Soc. 139, 6018–6025 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Kwon S.-J., Na D. H., Kwak J. H., Douaisi M., Zhang F., Park E. J., Park J.-H., Youn H., Song C.-S., Kane R. S., Dordick J. S., Lee K. B., Linhardt R. J., Nanostructured glycan architecture is important in the inhibition of influenza A virus infection. Nat. Nanotechnol. 12, 48–54 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Bhatia S., Lauster D., Bardua M., Ludwig K., Angioletti-Uberti S., Popp N., Hoffmann U., Paulus F., Budt M., Stadtmüller M., Wolff T., Hamann A., Böttcher C., Herrmann A., Haag R., Linear polysialoside outperforms dendritic analogs for inhibition of influenza virus infection in vitro and in vivo. Biomaterials 138, 22–34 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Lauster D., Klenk S., Ludwig K., Nojoumi S., Behren S., Adam L., Stadtmüller M., Saenger S., Zimmler S., Hönzke K., Yao L., Hoffmann U., Bardua M., Hamann A., Witzenrath M., Sander L. E., Wolff T., Hocke A. C., Hippenstiel S., De Carlo S., Neudecker J., Osterrieder K., Budisa N., Netz R. R., Böttcher C., Liese S., Herrmann A., Hackenberger C. P. R., Phage capsid nanoparticles with defined ligand arrangement block influenza virus entry. Nat. Nanotechnol. 15, 373–379 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Wagner R., Matrosovich M., Klenk H.-D., Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 12, 159–166 (2002). [DOI] [PubMed] [Google Scholar]
- 17.de Vries E., Du W., Guo H., de Haan C. A. M., Influenza A virus hemagglutinin–neuraminidase–receptor balance: Preserving virus motility. Trends Microbiol. 28, 57–67 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lauster D., Glanz M., Bardua M., Ludwig K., Hellmund M., Hoffmann U., Hamann A., Böttcher C., Haag R., Hackenberger C. P. R., Herrmann A., Multivalent peptide–nanoparticle conjugates for influenza-virus inhibition. Angew. Chem. Int. Ed. 56, 5931–5936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller M., Lauster D., Wildenauer H. H. K., Herrmann A., Block S., Mobility-based quantification of multivalent virus-receptor interactions: New insights into influenza A virus binding mode. Nano Lett. 19, 1875–1882 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Hiono T., Matsuda A., Wagatsuma T., Okamatsu M., Sakoda Y., Kuno A., Lectin microarray analyses reveal host cell-specific glycan profiles of the hemagglutinins of influenza A viruses. Virology 527, 132–140 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Meischel T., Villalon-Letelier F., Saunders P. M., Reading P. C., Londrigan S. L., Influenza A virus interactions with macrophages: Lessons from epithelial cells. Cell. Microbiol. 22, e13170 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Parveen N., Rydell G. E., Larson G., Hytönen V. P., Zhdanov V. P., Höök F., Block S., Competition for membrane receptors: Norovirus detachment via lectin attachment. J. Am. Chem. Soc. 141, 16303–16311 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Bally M., Gunnarsson A., Svensson L., Larson G., Zhdanov V. P., Höök F., Interaction of single viruslike particles with vesicles containing glycosphingolipids. Phys. Rev. Lett. 107, 188103 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Lee D. W., Hsu H.-L., Bacon K. B., Daniel S., Image restoration and analysis of influenza virions binding to membrane receptors reveal adhesion-strengthening kinetics. PLOS ONE 11, e0163437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pace H., Simonsson Nyström L., Gunnarsson A., Eck E., Monson C., Geschwindner S., Snijder A., Höök F., Preserved transmembrane protein mobility in polymer-supported lipid bilayers derived from cell membranes. Anal. Chem. 87, 9194–9203 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Castellana E. T., Cremer P. S., Solid supported lipid bilayers: From biophysical studies to sensor design. Surf. Sci. Rep. 61, 429–444 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen F., Wan X.-F., Influenza neuraminidase: Underrated role in receptor binding. Trends Microbiol. 27, 477–479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H., Yu M., Song H., Noonan O., Zhang J., Yang Y., Zhou L., Yu C., Self-organized mesostructured hollow carbon nanoparticles via a surfactant-free sequential heterogeneous nucleation pathway. Chem. Mater. 27, 6297–6304 (2015). [Google Scholar]
- 29.Ben-Akiva E., Meyer R. A., Yu H., Smith J. T., Pardoll D. M., Green J. J., Biomimetic anisotropic polymeric nanoparticles coated with red blood cell membranes for enhanced circulation and toxin removal. Sci. Adv. 6, eaay9035 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang R. H., Kroll A. V., Gao W., Zhang L., Cell membrane coating nanotechnology. Adv. Mater. 30, e1706759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vonnemann J., Liese S., Kuehne C., Ludwig K., Dernedde J., Böttcher C., Netz R. R., Haag R., Size dependence of steric shielding and multivalency effects for globular binding inhibitors. J. Am. Chem. Soc. 137, 2572–2579 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Cuellar-Camacho J. L., Bhatia S., Reiter-Scherer V., Lauster D., Liese S., Rabe J. P., Herrmann A., Haag R., Quantification of multivalent interactions between sialic acid and influenza A virus spike proteins by single-molecule force spectroscopy. J. Am. Chem. Soc. 142, 12181–12192 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Hamming P. H., Overeem N. J., Huskens J., Influenza as a molecular walker. Chem. Sci. 11, 27–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vahey M. D., Fletcher D. A., Influenza A virus surface proteins are organized to help penetrate host mucus. eLife 8, e43764 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakadamyali M., Rust M. J., Babcock H. P., Zhuang X., Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. U.S.A. 100, 9280–9285 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurt A. C., Holien J. K., Parker M., Kelso A., Barr I. G., Zanamivir-resistant influenza viruses with a novel neuraminidase mutation. J. Virol. 83, 10366–10373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trebbien R., Pedersen S. S., Vorborg K., Franck K. T., Fischer T. K., Development of oseltamivir and zanamivir resistance in influenza A(H1N1)pdm09 virus, Denmark, 2014. Eurosurveillance 22, 30445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rath B., Chen X., Spies V., Muehlhans S., Obermeier P., Tief F., Seeber L., Karsch K., Milde J., Skopnik H., Prospective surveillance of antiviral resistance in hospitalized infants less than 12 months of age with A (H3N2) influenza infection and treated with oseltamivir. Antivir. Ther. 22, 515–522 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Hayden F. G., Atmar R. L., Schilling M., Johnson C., Poretz D., Paar D., Huson L., Ward P., Mills R. G., Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. N. Engl. J. Med. 341, 1336–1343 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Chen Y.-Q., Wohlbold T. J., Zheng N.-Y., Huang M., Huang Y., Neu K. E., Lee J., Wan H., Rojas K. T., Kirkpatrick E., Henry C., Palm A.-K. E., Stamper C. T., Lan L. Y.-L., Topham D. J., Treanor J., Wrammert J., Ahmed R., Eichelberger M. C., Georgiou G., Krammer F., Wilson P. C., Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429.e410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stadlbauer D., Zhu X., McMahon M., Turner J. S., Wohlbold T. J., Schmitz A. J., Strohmeier S., Yu W., Nachbagauer R., Mudd P. A., Wilson I. A., Ellebedy A. H., Krammer F., Broadly protective human antibodies that target the active site of influenza virus neuraminidase. Science 366, 499–504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waldmann M., Jirmann R., Hoelscher K., Wienke M., Niemeyer F. C., Rehders D., Meyer B., A nanomolar multivalent ligand as entry inhibitor of the hemagglutinin of avian influenza. J. Am. Chem. Soc. 136, 783–788 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Luk B. T., Fang R. H., Hu C.-M. J., Copp J. A., Thamphiwatana S., Dehaini D., Gao W., Zhang K., Li S., Zhang L., Safe and immunocompatible nanocarriers cloaked in RBC membranes for drug delivery to treat solid tumors. Theranostics 6, 1004–1011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu C.-M. J., Fang R. H., Copp J., Luk B. T., Zhang L., A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 8, 336–340 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu C.-M. J., Fang R. H., Luk B. T., Zhang L., Nanoparticle-detained toxins for safe and effective vaccination. Nat. Nanotechnol. 8, 933–938 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walls A. C., Park Y.-J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baum A., Fulton B. O., Wloga E., Copin R., Pascal K. E., Russo V., Giordano S., Lanza K., Negron N., Ni M., Wei Y., Atwal G. S., Murphy A. J., Stahl N., Yancopoulos G. D., Kyratsous C. A., Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 369, 1014–1018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., Gong Y., Xiao H., Fan Z., Tan S., Wu G., Tan W., Lu X., Fan C., Wang Q., Liu Y., Zhang C., Qi J., Gao G. F., Gao F., Liu L., A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L., Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia S., Yan L., Xu W., Agrawal A. S., Algaissi A., Tseng C.-T. K., Wang Q., Du L., Tan W., Wilson I. A., Jiang S., Yang B., Lu L., A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 5, eaav4580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Q., Honko A., Zhou J., Gong H., Downs S. N., Vasquez J. H., Fang R. H., Gao W., Griffiths A., Zhang L., Cellular nanosponges inhibit SARS-CoV-2 infectivity. Nano Lett. 20, 5570–5574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han X., Shen S., Fan Q., Chen G., Archibong E., Dotti G., Liu Z., Gu Z., Wang C., Red blood cell–derived nanoerythrosome for antigen delivery with enhanced cancer immunotherapy. Sci. Adv. 5, eaaw6870 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun H., Su J., Meng Q., Yin Q., Chen L., Gu W., Zhang P., Zhang Z., Yu H., Wang S., Li Y., Cancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumors. Adv. Mater. 28, 9581–9588 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd3803/DC1