

Trisomy 21 induces microglial dysregulation in the Alzheimer’s brain through the perturbation of the ubiquitin-proteasome system.
Abstract
Down syndrome (DS), caused by trisomy of chromosome 21, is the most significant risk factor for early-onset Alzheimer’s disease (AD); however, underlying mechanisms linking DS and AD remain unclear. Here, we show that triplication of homologous chromosome 21 genes aggravates neuroinflammation in combined murine DS-AD models. Overexpression of USP25, a deubiquitinating enzyme encoded by chromosome 21, results in microglial activation and induces synaptic and cognitive deficits, whereas genetic ablation of Usp25 reduces neuroinflammation and rescues synaptic and cognitive function in 5×FAD mice. Mechanistically, USP25 deficiency attenuates microglia-mediated proinflammatory cytokine overproduction and synapse elimination. Inhibition of USP25 reestablishes homeostatic microglial signatures and restores synaptic and cognitive function in 5×FAD mice. In summary, we demonstrate an unprecedented role for trisomy 21 and pathogenic effects associated with microgliosis as a result of the increased USP25 dosage, implicating USP25 as a therapeutic target for neuroinflammation in DS and AD.
INTRODUCTION
Down syndrome (DS), which results from total or partial trisomy of chromosome 21, is the most common cause of intellectual disability, with an incidence of 1 in approximately 800 births worldwide (1, 2). Trisomy 21 is the single most common risk factor for early-onset Alzheimer’s disease (AD) (3). By the age of 40, individuals with DS universally develop neuropathological features associated with AD, including amyloid plaques, neurofibrillary tangles, synaptic dysfunction, and neuroinflammation in vulnerable brain regions (4, 5). However, whether and how chromosome 21 genes influence AD pathogenesis in the context of DS remain largely unknown.
As the major immune cells in the brain, microglia play a key role in maintaining central nervous system (CNS) homeostasis and protecting the brain from infection and injury (6, 7). Microglia transition to a reactive state associated with enhanced proinflammatory cytokine secretion and phagocytic uptake during injury (8–10). Short-term activation of microglia may facilitate debris clearance and tissue repair (11, 12); however, sustained activation of microglia induces chronic release of proinflammatory cytokines, thereby initiating inflammatory cascades and pathogenic neurotoxic effects in neurodegeneration (13–15).
The ubiquitin-proteasome system (UPS) is an essential protein degradation pathway (16), and defects in the UPS induce the accumulation of neurotoxic proteins in various neurodegenerative disorders, such as AD, Parkinson’s disease, and Huntington’s disease (17). The USP25 gene is located on human chromosome 21q11.2 and encodes the ubiquitin-specific protease ubiquitin-specific peptidase 25 (USP25) (18). USP25 was first identified as a negative regulator of interleukin-17 (IL-17)–mediated signaling and the inflammatory response (19). In addition, USP25 deficiency inhibits transcriptional activity of interferon regulatory factor, thereby reducing type I interferon production (20). UPS-mediated protein degradation and neuroinflammation play an important role in neurodegeneration; however, the contribution of USP25 in the pathogenesis of DS and AD remains unclear.
Here, we show that an extra copy of chromosome 21 aggravates neuroinflammation in 5×FAD mice and that the DS-related gene USP25 plays a key role in this process. Bacterial artificial chromosome (BAC)–Tg–USP25 mice displayed neuropathologies resembling those exhibited by DS-AD mice, including microglial activation and impaired synaptic and cognitive function. In addition, genetic deletion of Usp25 in 5×FAD mice markedly attenuated neuroinflammation and cognitive deficits. Pharmacological treatment with USP25 inhibitors restored microglia to a homeostatic transcriptional state and reversed impairments in synaptic and cognitive function in 5×FAD mice. Therefore, our investigation revealed a critical role for USP25 in maintaining microglial homeostasis in neurodegeneration and identified a novel therapeutic avenue for AD and DS.
RESULTS
Trisomy of chromosome 21 genes promotes neuroinflammation
To investigate whether triplication of chromosome 21 genes alters AD pathogenesis, we generated a DS-AD mouse model by crossing the Dp16 DS mouse model (21, 22) with 5×FAD mice (23). We performed whole-genome RNA sequencing (RNA-seq) and subsequent consensus weighted gene coexpression network analyses (WGCNAs) on hippocampal tissue from wild-type (WT), Dp16, 5×FAD, and 5×FAD;Dp16 mice and identified 14 coexpression modules (Fig. 1A). By correlating eigengenes to corresponding genotypes, we found that three modules (M3, M4, and M5) were significantly up-regulated in 5×FAD;Dp16 versus 5×FAD mice (Fig. 1A). Gene Ontology (GO) enrichment analysis revealed that genes within the M3 module (1641 genes) were implicated in pathways related to “cytokine production,” “microglial activation,” and “phagocytosis” (Fig. 1B). Moreover, the expression of microglia-related genes was up-regulated by triplication of murine chromosome 16 (homologous to human chromosome 21) in 5×FAD;Dp16 mice compared with 5×FAD alone (Fig. 1C). Consistent with the transcriptomic profiles, we observed Iba1+ microglial proliferation and activation in hippocampus and cortex from Dp16 and 5×FAD mice (Fig. 1, D to H, and fig. S1, A to E). DS-associated chromosome 16 triplication also enhanced microglial proliferation and activation in 5×FAD;Dp16 mouse brain compared with Dp16 and 5×FAD mouse brain (Fig. 1, D to H, and fig. S1, A to G).
Fig. 1. Trisomy of chromosome 21 genes promotes neuroinflammation.
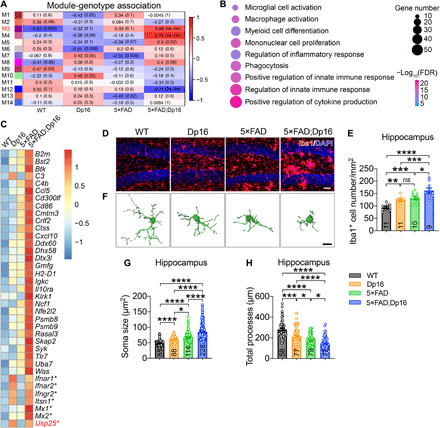
(A to C) Transcriptome analysis of 5-month-old WT, Dp16, 5×FAD, and 5×FAD;Dp16 mice. n = 6 mouse hippocampi. (A) WGCNA analysis of module eigengenes. Rows correspond to modules, and columns correspond to genotypes. Numbers in the heatmap represent Z statistics and corresponding P values of the module eigengene association with the indicated genotypes. (B) GO analysis of M3 genes. (C) Heatmap depicting microglia-related genes that were up-regulated in 5×FAD;Dp16 versus 5×FAD mouse hippocampus. The asterisk indicates homologous genes on human chromosome 21. (D and E) Representative immunostaining (D) and quantification of Iba1+ microglia (E) in 5-month-old WT, Dp16, 5×FAD, and 5×FAD;Dp16 mouse hippocampus. Scale bar, 40 μm. n = 5 to 6 mice per group; n = 9 to 11 slices per group were scored. (F) Representative 3D reconstruction of Iba1+ microglia using Imaris software. Scale bar, 10 μm. (G and H) Quantification of microglial soma size (G) and total processes (H). n = 4 to 6 mice per group; n = 58 to 228 microglia per group were counted. All data represent means ± SEM. P values were determined by one-way ANOVA with Tukey’s post hoc analysis in (E) and by the Kruskal-Wallis test with Dunn’s post hoc analysis in (G) and (H). ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
We then searched for microglia-related differentially expressed genes (DEGs) encoded on chromosome 21 (mouse chromosome 16) that were associated with inflammation and identified USP25 as a potential gene candidate. USP25 was up-regulated in the brains of both human DS patients (fig. S1, H and I) and Dp16 mice (fig. S1, J and K). USP25 is a deubiquitinase that was previously implicated in innate immunity (19). Our previous results indicate that USP25 is ubiquitously expressed in different cell types in the CNS, and show markedly high expression in microglia and astrocytes (fig. S1, L and M).
To mimic USP25 overexpression in DS in a transgenic mouse model, we used a human USP25 BAC transgenic approach (fig. S2, A and B). To address whether the increased USP25 gene dosage may alter disease-associated microglial function and other AD-related phenotypes, we crossed BAC-Tg-USP25 with 5×FAD mouse lines. We first assessed cognitive function in 5-month-old 5×FAD;BAC-Tg-USP25 mice (early phase of disease progression) using the Y-maze spontaneous alternation task; we observed that 5×FAD;BAC-Tg-USP25 mice displayed impaired spatial memory compared with WT and 5×FAD mice (Fig. 2A). The dendritic spine density in hippocampal neurons was reduced in 5×FAD;BAC-Tg-USP25 compared with 5×FAD mice (Fig. 2B). To confirm whether USP25 overdosage impairs synaptic function, we measured long-term potentiation (LTP) in hippocampal Schaffer collaterals and observed marked LTP impairment in BAC-Tg-USP25, 5×FAD, and 5×FAD;BAC-Tg-USP25 compared with WT mice (Fig. 2, C and D). Furthermore, we observed microglial proliferation and activation in the hippocampus and cortex in BAC-Tg-USP25 and 5×FAD mice (Fig. 2, E to I, and fig. S2, C to I). Moreover, USP25 overexpression further boosted microglial proliferation and activation in 5×FAD;BAC-Tg-USP25 mouse brain (Fig. 2, E to I, and fig. S2, C to I). Together, these results demonstrate that USP25 overexpression phenocopies pathogenic effects associated with DS chromosome 21 triplication.
Fig. 2. Overexpression of USP25 promotes synapse loss and microglial activation in AD mice.
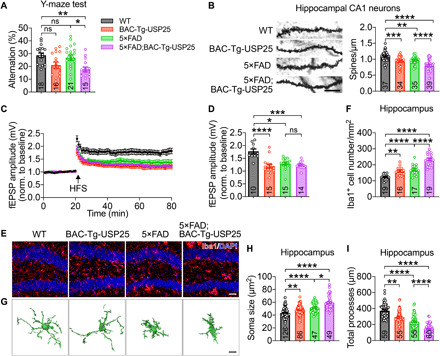
(A) Percentage of spontaneous alternation in the Y-maze. n = 15 to 21 5-month-old mice per group. (B) Golgi staining and quantification of dendritic spines in the hippocampal CA1 regions of 6-month-old WT, BAC-Tg-USP25, 5×FAD, and 5×FAD;BAC-Tg-USP25 mice. Scale bar, 10 μm. n = 4 mice per group; n = 34 to 39 dendrites per group were counted. (C) Hippocampal CA1 LTP recordings from 6-month-old mice. HFS, high-frequency stimulation. (D) fEPSP amplitude quantification during the last 10 min of LTP recording. WT (n = 5 mice, 10 slices), BAC-Tg-USP25 (n = 5 mice, 15 slices), 5×FAD (n = 7 mice, 15 slices), and 5×FAD;BAC-Tg-USP25 (n = 5 mice, 14 slices). (E and F) Representative immunostaining (E) and quantification of Iba1+ microglia (F) in 6-month-old WT, BAC-Tg-USP25, 5×FAD, and 5×FAD;BAC-Tg-USP25 mouse hippocampus. Scale bar, 40 μm. n = 5 to 7 mice per group; n = 16 to 19 slices per group were scored. (G) Representative 3D reconstruction of Iba1+ microglia. Scale bar, 8 μm. (H and I) Quantification of microglial soma size (H) and total processes (I). n = 3 to 5 mice per group; n = 47 to 126 microglia per group were counted. All data represent means ± SEM. P values were determined by one-way ANOVA with Tukey’s post hoc analysis in (A), (B), and (F) and by the Kruskal-Wallis test with Dunn’s post hoc analysis in (D), (H), and (I). ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
Genetic deletion of Usp25 reverses cognitive and synaptic deficits in AD mice
To determine the effects of Usp25 deficiency on AD-related cognitive dysfunction, we assayed learning and memory behavior in 5×FAD;Usp25+/− mice (fig. S3A). Compared with 5×FAD mice, 5×FAD;Usp25+/− mice displayed improved spatial memory in the Y-maze (Fig. 3A) and Morris water maze (MWM) tests (Fig. 3, B and C), as well as improved associative memory in fear conditioning (FC) tests (Fig. 3, D and E). Dendritic spine density was also found to be increased in 5×FAD;Usp25+/− compared with 5×FAD mouse hippocampus (Fig. 3F). To determine whether Usp25 deficiency restores synaptic function in 5×FAD mice, we measured hippocampal LTP and found that it was compromised in 5×FAD compared with WT and Usp25+/− mice; however, 5×FAD-associated LTP impairment was reversed in 5×FAD;Usp25+/− mice (Fig. 3, G and H). Consistent with changes in dendritic spine density and LTP, Usp25 deficiency reduced the miniature inhibitory postsynaptic current (mIPSC) frequency (fig. S3B) and increased the miniature excitatory postsynaptic current (mEPSC) frequency (fig. S3C) in hippocampal granule neurons in 5×FAD mice. Together, these results indicate that disruptions in excitation/inhibition signaling in 5×FAD mouse hippocampus can be restored by Usp25 haploinsufficiency (Fig. 3I), and demonstrate that USP25 down-regulation reverses synaptic and cognitive deficits in 5×FAD mice.
Fig. 3. Genetic deletion of Usp25 enhances synaptic and cognitive function in AD mice.
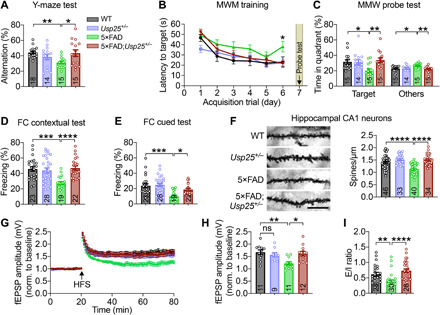
(A) Percentage of spontaneous alternation in the Y-maze. n = 14 to 18 6- to 7-month-old mice per group. (B) MWM test results depicting escape latency, defined as the time taken to find a hidden platform. (C) MWM probe test results. n = 14 to 15 6- to 7-month-old mice per group. (D) Percentage freezing time in contextual FC tests, as a readout of associative memory. (E) Percentage freezing time spent in cued FC tests. n = 19 to 28 6- to 7-month-old mice per group. (F) Golgi staining and quantification of dendritic spines in 9-month-old mouse hippocampus. Scale bar, 10 μm. n = 4 mice per group; n = 33 to 46 dendrites per group were counted. (G) Hippocampal CA1 LTP recordings from 6- to 7-month-old mice. HFS, high-frequency stimulation. (H) Quantification of the last 10 min of LTP recording. WT (n = 6 mice, 11 slices), Usp25+/− (n = 5 mice, 9 slices), 5×FAD (n = 5 mice, 11 slices), and 5×FAD;Usp25+/− (n = 6 mice, 12 slices). (I) Excitation/inhibition (E/I) ratio of hippocampal DG granule cells. n = 4 mice per group; n = 23 to 30 cells. All data represent means ± SEM. P values were determined by the Kruskal-Wallis test with Dunn’s post hoc analysis in (A), (C) to (F), (H), and (I) and by repeated-measures ANOVA with Tukey’s post hoc analysis in (B). ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
USP25 deficiency reduces neuroinflammation in AD mouse brain
To further identify molecular networks that may be selectively targeted by Usp25 deficiency, we performed RNA-seq and subsequent WGCNA analysis on the hippocampi of mice from four groups (WT, Usp25+/−, 5×FAD, and 5×FAD;Usp25+/−) and identified 16 coexpression modules (fig. S4A). Among these modules, M14 was significantly up-regulated in 5×FAD compared with WT and was partially restored in 5×FAD;Usp25+/− mice (fig. S4A). GO enrichment analysis revealed that 4375 genes within the M14 module were implicated in “inflammatory response,” “cytokine production,” and “phagosome” pathways (Fig. 4A). Moreover, expression of microglia-related 5×FAD DEGs was restored by Usp25 haploinsufficiency in 5×FAD;Usp25+/− mice (fig. S4B). In addition, we found that Usp25 haploinsufficiency suppressed mRNA expression of proinflammatory cytokines Il6 and Tnf in 5×FAD mouse hippocampus (fig. S4C).
Fig. 4. Usp25 haploinsufficiency restores microglial homeostasis.
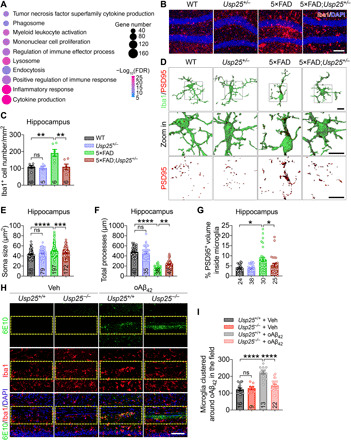
(A) GO analysis of M14 genes in fig. S4A. (B and C) Representative immunostaining (B) and quantification of Iba1+ microglia (C) in 6- to 7-month-old WT, Usp25+/−, 5×FAD, and 5×FAD;Usp25+/− mouse hippocampus. Scale bar, 100 μm. n = 5 to 6 mice per group. (D) Representative 3D reconstruction and rendering of PSD95 puncta internalized in Iba1+ microglia (PSD95/Iba1). Scale bar, 10 μm. (E to G) Quantification of microglial soma size (E), total processes (F), and percentage of PSD95+ volume within microglia (G) in (D). n = 3 to 6 mice per group; n = 24 to 197 microglia per group were counted. (H) Representative immunostaining of oAβ42-expressing regions in 7-month-old Usp25+/+ and Usp25−/− mouse brain 16 hours after oAβ42 injection. Scale bar, 100 μm. (I) Quantification of Iba1+ microglia clustered in oAβ42-expressing regions. n = 3 mice per group; n = 9 to 22 fields [selected area with yellow lines in (H)] were quantified. All data represent means ± SEM. P values were determined by one-way ANOVA with Tukey’s post hoc analysis in (C), (F), and (I) and by the Kruskal-Wallis test with Dunn’s post hoc analysis in (E) and (G). ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Consistent with our transcriptomic analyses, genetic deletion of Usp25 in a 5×FAD background suppressed Iba1+ microglial proliferation (Fig. 4, B and C, and fig. S4, D and E). Quantitative morphometric analysis revealed that 5×FAD microglia featured an increased soma size and reduced branch complexity and that these morphogenic changes were reversed with Usp25 haploinsufficiency (Fig. 4, D to F, and fig. S4, F to H). In addition, quantification of the PSD95+ puncta volume internalized in Iba1+ microglia suggested that Usp25 haploinsufficiency attenuated microglia-mediated engulfment of synapses in 5×FAD mouse brain (Fig. 4, D and G, and fig. S4, F and I). To confirm the effects of USP25 in AD-associated microglial dysfunction, we performed in vitro phagocytosis assays using cultured primary microglia and pH-sensitive dye-labeled synaptosomes. We found that oligomeric Aβ42 (oAβ42) treatment markedly increased phagocytosis of synaptosomes in Usp25+/+, but not in Usp25−/−, microglia (fig. S4, J and K). To further determine whether Usp25 deficiency affects clustering of microglia in vivo, we stereotactically injected oAβ42 into hippocampus of adult Usp25+/+ and Usp25−/− mice and quantified the number of microglia proximal to oAβ42. oAβ42 increased the accumulation of activated microglia around the injection site in Usp25+/+ mouse brain; however, microglia largely failed to home to oAβ42 in Usp25−/− mice (Fig. 4, H and I).
USP25 deficiency increases proteasomal degradation of WDFY1 and ATP6V0C
To determine the underlying molecular mechanism by which USP25 regulates microglial function, we characterized protein expression profiles from WT, 5×FAD, and 5×FAD;Usp25−/− mouse cerebrum using tandem mass tag (TMT)–based quantitative proteomics. We identified 192 up-regulated and 193 down-regulated differentially expressed proteins (DEPs; fold change > 1.1; P < 0.05) in 5×FAD;Usp25−/− relative to those in 5×FAD mouse brain (Fig. 5A). WDFY1 and ATP6V0C were the top down-regulated proteins identified by proteomic analysis (Fig. 5A). GO enrichment analysis revealed an enrichment of proteins related to the immune system and neuroinflammation among the down-regulated DEPs (Fig. 5B), as well as neuroinflammation and learning/memory-related pathways among the up-regulated DEPs (fig. S5A). Given that USP25 is a deubiquitinase, down-regulated DEPs in response to Usp25 deletion such as WDFY1 and ATP6V0C could potentially represent USP25 targets. WDFY1 and ATP6V0C have been previously implicated in inflammation and pathways associated with lysosomal function. WDFY1 promotes Toll-like receptor 3 (TLR3)– and TLR4-mediated activation of nuclear factor κB (NF-κB) and enhances inflammatory cytokine production (24). Our results indicated that depletion of ATP6V0C markedly attenuated oAβ42-induced microglial phagocytosis of synaptosomes (Fig. 5C and fig. S5B). We further verified the colocalization between USP25 and ATP6V0C or WDFY1 by immunocytochemistry (fig. S5, C and D) and their interaction with USP25 by coimmunoprecipitation (Fig. 5, D and E). We demonstrated that USP25 knockdown reduced HA-ATP6V0C and HA-WDFY1 expression (fig. S5, E and F) and increased polyubiquitinated HA-ATP6V0C and HA-WDFY1 levels (Fig. 5, F and G). We next found that enhanced ATP6V0C and WDFY1 degradation associated with USP25 depletion was reversed by inhibition of proteasomal protein degradation and, to a smaller extent, by inhibition of lysosomal protein degradation (fig. S5, G and H).
Fig. 5. USP25 deubiquitinates and stabilizes WDFY1 and ATP6V0C.
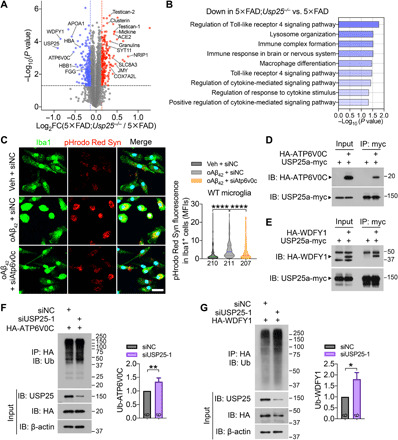
(A and B) Quantitative proteomic analysis of the mouse cerebrum. (A) Proteomic analysis of the mouse cerebrum and a volcano plot illustrating down-regulated (blue) and up-regulated (red) proteins in the cerebra of 6-month-old 5×FAD;Usp25−/− versus 5×FAD mice (fold change > 1.1, P < 0.05). n = 3 samples per group; each sample is a pool of three cerebra from the same genotype. (B) GO analysis of down-regulated proteins in 5×FAD;Usp25−/− mice relative to 5×FAD mice. (C) Primary microglia from WT mice were transfected with siAtp6v0c or control siRNA (siNC) for 48 hours and subsequently treated with 10 μM oAβ42 or vehicle control along with pHrodo Red–labeled synaptosomes (Syn) for another 24 hours, and the phagocytosis abilities of microglia were quantified on the basis of the pHrodo Red intensity in Iba1+ cells. Scale bar, 25 μm. n = 3 independent experiments; n = 207 to 211 cells per group were counted. (D and E) Coimmunoprecipitation (IP) between exogenously expressed USP25a-myc and HA-ATP6V0C (D) or HA-WDFY1 (E) proteins. (F and G) Immunoblot (IB) analysis of polyubiquitinated HA-ATP6V0C (F) and HA-WDFY1 (G) in HEK293T cells upon USP25 knockdown. n = 6. All data represent means ± SEM. P values were determined by the Kruskal-Wallis test with Dunn’s post hoc analysis in (C) and by the Mann-Whitney test in (F) and (G). *P < 0.05; **P < 0.01; ****P < 0.0001.
USP25 inhibition ameliorates neurological dysfunction in AD mice
To assess the functional role of USP25 inhibition in vivo, we evaluated the pharmacokinetic properties of the USP25 inhibitor AZ1 in mice (Fig. 6A) (25). We found that at 0.5 hour after a single administration of AZ1 by intragastric gavage, AZ1 reached a maximum concentration in the brain that was maintained for up to 8 hours (Fig. 6B). Subchronic administration of AZ1 (20 mg kg−1) did not show any apparent toxic effects, as indicated by body weight gain (Fig. 6C and fig. S6A) and metabolic profiling (fig. S6, B to L). Subchronic injection of AZ1 in 5×FAD mice markedly ameliorated memory deficits in cued FC (Fig. 6D) and MWM tests (Fig. 6, E and F). In addition, we measured hippocampal LTP and found that LTP impairment was reversed through long-term AZ1 administration (Fig. 6G).
Fig. 6. USP25 inhibition ameliorates AD-related impairments in 5×FAD mice.
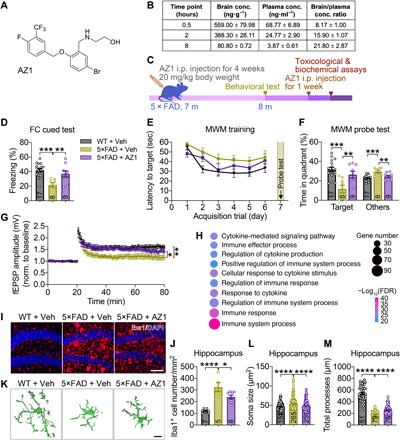
(A) Chemical structure of AZ1. (B) Brain concentrations of AZ1 detected by LC-MS/MS after intragastric gavage at body weight (10 mg kg−1). (C) Timeline of the AZ1 injection experiments. Three treatment groups in this study included the following: WT + vehicle, 5×FAD + vehicle, and 5×FAD + AZ1 groups. Seven-month-old mice were intraperitoneally (i.p.) injected with vehicle or AZ1 (20 mg kg−1 day−1) for 4 weeks and subjected to behavioral and pathological analyses. (D) Characterizing therapeutic effects associated with AZ1 on cued fear memory. WT + vehicle (n = 17 mice), 5×FAD + vehicle (n = 8 mice), and 5×FAD + AZ1 (n = 9 mice). (E) Escape latency in MWM tests. (F) MWM probe test results. (G) Hippocampal CA1 LTP recordings and quantification of the last 10 min of the LTP recording. WT + vehicle (n = 5 mice, 10 slices), 5×FAD + vehicle (n = 4 mice, 7 slices), and 5×FAD + AZ1 (n = 7 mice, 12 slices). (H) GO analysis of overlapping genes in fig. S7A. (I and J) Representative immunostaining (I) and quantification of Iba1+ microglia (J) in WT + vehicle, 5×FAD + vehicle, and 5×FAD + AZ1 mouse hippocampus. Scale bar, 100 μm. n = 5 to 8 mice per group. (K) Representative 3D reconstruction of Iba1+ microglia. Scale bar, 10 μm. (L and M) Quantification of microglial soma size (L) and total processes (M). n = 5 mice per group; n = 51 to 265 microglia per group were counted. All data represent means ± SEM. P values were determined by one-way ANOVA with Dunnett’s post hoc analysis in (D), (F), (J), (L), and (M); by repeated-measures ANOVA with Tukey’s post hoc analysis in (E); and by one-way ANOVA with Holm-Sidak’s post hoc analysis in (G). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
We next investigated the mechanisms underlying AZ1-dependent neuroprotection through transcriptomic profiling in 5×FAD animals treated with AZ1. A total of 2349 genes were up-regulated in the 5×FAD hippocampus compared with the WT hippocampus (fold change > 1.2; P < 0.05); among up-regulated DEGs identified, expression of 446 genes was partially restored with AZ1 (fig. S7A). GO analysis revealed that genes implicated in immune- and inflammation-related pathways were enriched in 5×FAD DEGs restored with AZ1 treatment (Fig. 6H), with a large number of DEGs featuring microglia-specific expression (fig. S7B). In addition, AZ1 suppressed the expression of proinflammatory cytokines Il1b and Il6 in 5×FAD mouse hippocampus, as quantified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (fig. S7C). Consistent with our transcriptomic analysis, we found that AZ1 administration attenuated microglial proliferation and activation in the hippocampus (Fig. 6, I to M) and cortex (fig. S7, D to H) of 5×FAD mice. To validate whether AZ1 regulates microglial function in a USP25-dependent manner, we treated Usp25+/+ and Usp25−/− microglia with AZ1 and found that AZ1 markedly attenuated oAβ42-induced phagocytosis of synaptosomes in Usp25+/+ microglia, with little or no effect in Usp25−/− microglia (fig. S7, I and J). Together, these results indicated that AZ1 ameliorated AD neuropathology by attenuating microglial activation.
DISCUSSION
Emerging evidence supports a pivotal role for innate immunity and neuroinflammation in DS and AD (26–30). Here, we show that a key component of UPS, USP25, is a critical regulator of CNS immune homeostasis in DS and AD. Overexpression of human USP25 recapitulated major neurological phenotypes in 5×FAD;Dp16 mice, including neuroinflammation and microglial activation. In addition, either genetic ablation or pharmacological inhibition of USP25 reversed neuroinflammatory, synaptic, and cognitive deficits in 5×FAD mice through restoration of microglial homeostasis. Prolonged USP25 inhibition restored proinflammatory transcriptional signatures associated with AD microglia and enhanced cognitive function in 5×FAD mice.
Although our focus here characterized a role for microglial USP25 in neurodegeneration, USP25 is known to be widely expressed in the CNS and peripheral nervous system (31). Our results using cultured microglia from Usp25 knockout mice showed that USP25 deficiency attenuated Aβ-induced phagocytosis of synapses, implicating a crucial role for USP25 in microglia-dependent synaptic uptake. However, there is still a possibility that USP25 in other cell types in the brain may also affect the CNS. Future studies characterizing the effects of conditional Usp25 deletion in various cell types will be required to decipher cell type–specific effects of USP25 perturbation in the CNS. Triplication of human chromosome 21 genes has been found to promote Aβ burden in a mouse model of DS-AD independently of an extra copy of APP (32). In addition to APP and USP25, the contribution of several chromosome 21–encoded genes and noncoding RNAs to AD pathogenesis has been reported, e.g., transcription factor ETS2 (33), small ubiquitin-related modifier 3 (SUMO3) (34), dual-specificity tyrosine-phosphorylation regulated kinase 1 A (DYRK1A) (35), β-site APP-cleaving enzyme 2 (BACE2) (36), cysteine protease cathepsin B (CSTB) (37), and microRNA miR-155 (38). Therefore, whether other chromosome 21 genes, individually or together with USP25, contribute to neuroinflammation in AD/DS pathogenesis remains to be further determined.
Multiple mechanisms may contribute to the protective effects associated with USP25 deficiency in 5×FAD mice. The induced proinflammatory cytokines TNFα (tumor necrosis factor α), IL-1β, and IL-6 are potentially neurotoxic and likely induce downstream neurodegenerative pathways (39, 40). Thus, suppression of cytokine production in the AD brain may inhibit neuroinflammation and associated pathogenic effects (41, 42). In addition, enhanced synaptic function may be directly attributed to decreased synaptic uptake in pathogenically activated microglia in AD brain. Our proteomic analysis identified WDFY1 and ATP6V0C as substrates of USP25. It has been reported that WDFY1 promotes TLR3- and TLR4-mediated activation of NF-κB and inflammatory cytokine production (24). We also found that depletion of ATP6V0C markedly attenuated microglia-mediated phagocytosis of synapses. Together, these results indicate that USP25 deficiency suppresses microglia-mediated proinflammatory cytokine production and synapse elimination by targeting WDFY1 and ATP6V0C, respectively (fig. S8).
In conclusion, our results establish a critical role for USP25 in reprogramming microglial homeostasis in AD and DS. Given that ubiquitination and neuroinflammation have been found to play a crucial role in neurodegenerative disorders, further investigation of the function of USP25 may provide insights into therapeutic strategies to prevent or treat various neurodegenerative diseases through the restoration of microglial homeostasis.
MATERIALS AND METHODS
Study design
This study was performed to explore the underlying mechanism of chromosome 21–encoded deubiquitinase USP25 in the pathogenesis of AD, especially to verify the therapeutic potential of USP25 inhibitors for treating cognitive deficits and neuroinflammation in a mouse model of AD. In addition, human fetal brain tissues from the patients with DS and normal control were used to determine USP25 expression. To investigate whether trisomy 21 alters AD pathogenesis, we generated a DS-AD mouse model by crossing Dp16 mice with 5×FAD mice. In addition, BAC-Tg-USP25 mice that overexpress human USP25 gene were used to determine the pathological role of USP25 overdosage in AD pathogenesis. To discern the therapeutic potential of USP25, genetic deletion and pharmacological inhibition of USP25 were conducted in 5×FAD mice. All animal studies were performed in mice according to the protocols approved by the Institutional Animal Care and Use Committee of Xiamen University. Mice were randomly grouped by genotype, and age-matched littermates were used as controls. Experiments were conducted and analyzed in a double-blind manner, with replicates described in the figure legends. All collected data are included in the figures or supplementary figures.
Mouse strains
Sanger sequencing of PCR products was used to characterize the USP25 BAC clone [National Center for Biotechnology Information (NCBI) Clone DB, clone no. RP11-840D8] containing the human USP25 gene. Purified BAC DNA was microinjected into pronuclei of C57BL/6 fertilized oocytes and transplanted into pseudopregnant foster mothers. The transgenic offspring were screened by PCR, and BAC-Tg-USP25 mice were maintained in a C57BL/6 background. Usp25−/− mice were generated as previously described (19). Both Dp(16)1Yey/+ mice (stock no. 013530, referred to as Dp16 mice in this study) and 5×FAD mice (stock no. 34840-JAX) were obtained from the Jackson Laboratory (Ellsworth, ME, USA). Age-matched littermate male mice were used in the experiments except the quantitative proteomics; age-matched littermate female mice were used in the quantitative proteomics. All experiments involving animals were performed under the guidelines of the Institutional Animal Care and Use Committee of Xiamen University.
Human brain specimens
Human DS fetal brain specimens were collected and curated by the Women and Children’s Hospital, School of Medicine, Xiamen University. Human studies were approved with informed consent by the ethical review board at the School of Medicine, Xiamen University (project no. XDYX2020002). Donors consisted of pregnant women who discontinued due to congenital heart defects or trisomy 21, which was confirmed by karyotype analysis. For specimen information, see table S1.
RNA interference
Small interfering RNAs (siRNAs) were ordered from RiboBio (Guangzhou, China). The human USP25 siRNA target sequences were as follows: siUSP25-1: 5′-GTGAGCGATTTGCCCGAAT-3′; siUSP25-2: 5′-GCATCAGGATTATAGGAAA-3′. The mouse Atp6v0c siRNA target sequence was as follows: siAtp6v0c: 5′-GTCCCGTTGTCCTAGCTCG-3′. The control siRNA (siN0000001-1-5) was provided by RiboBio. siRNA was transfected into human embryonic kidney (HEK) 293T cells or primary microglia using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Carlsbad, CA, USA; 13778100).
Cell culture and transfection
Primary microglial cultures were prepared as previously described (43). Briefly, mixed glial cultures were acquired from WT or Usp25−/− mice on postnatal days 1 to 2, plated in flasks coated with poly-l-lysine, and grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Granulocyte-macrophage colony-stimulating factor (25 ng ml−1; R&D Systems, Minneapolis, MN, USA; 415-ML-050) was added to the cultures after 3 days. Primary microglia were harvested by shaking (200 rpm, 30 min) 10 to 12 days after plating and once every 3 days thereafter (up to four harvests). HEK293T or HeLa cells were cultured in DMEM containing 10% FBS. Once they reached an appropriate confluence, HEK293T or HeLa cells were transfected with the indicated constructs using TurboFect Transfection Reagent (Thermo Fisher Scientific, R0534).
RNA isolation and quantitative reverse transcription PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, 15596026), and 1 μg of total RNA was reverse-transcribed into complementary DNA (cDNA) using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan; FSQ-201). Real-time PCR was performed on the LightCycler 480 System (Roche, Mannheim, Germany) using the FastStart Universal SYBR Green Master (Roche, 04913850001). The primer sequences of target genes are included in table S2. The 2–ΔΔCt method was used to calculate relative gene expression after normalization to the Actb internal control.
Immunoblot analysis
Immunoblot analysis was performed as previously described (44). Antibodies were used as follows: anti-USP25 (Abcam, Cambridge, MA, USA; ab187156, 1:1000), anti-APP (Millipore, Billerica, MA, USA; MAB348, 1:1000), anti-Iba1 (Wako Pure Chemical, Osaka, Japan; 016-20001, 1:500), anti-GFAP (glial fibrillary acidic protein) (Cell Signaling Technology, Danvers, MA, USA; 3670, 1:1000), anti–β-III-tubulin (Abcam, ab18207, 1:1000), anti-HA (hemagglutinin) (Sigma-Aldrich, St. Louis, MO, USA; H6908, 1:1000), anti-ubiquitin (Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc-8017, 1:500), anti–β-actin (Xmbcss, Xiamen, China; bc001, 1:2000), and horseradish peroxidase (HRP)–conjugated secondary antibodies (Thermo Fisher Scientific, 31430 or 31460; 1:3000).
Coimmunoprecipitation
Transfected HEK293T cells were lysed in lysis buffer [20 mM tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40] supplemented with the Complete Protease Inhibitor Cocktail (Roche, 04693132001). Cell lysates were subjected to immunoprecipitation with antibody against Myc (Thermo Fisher Scientific, 132500, 1:200) or HA (Sigma-Aldrich, H6908, 1:200) and incubation with Dynabeads Protein G (Thermo Fisher Scientific, 10004D) followed by immunoblot analysis.
Pharmacological treatment with proteasomal and lysosomal inhibitors
HEK293T cells transfected with siUSP25 or control siRNA were incubated with lysosomal inhibitor leupeptin (100 μg ml−1) (MCE, Monmouth Junction, NJ, USA; HY-18234A) or 10 μM proteasomal inhibitor MG132 (MCE, HY-13259). After 8 hours of treatment, the cells were harvested and subjected to immunoblot analysis.
Stereotactic injection of Aβ42 oligomer
Stereotactic injection of oAβ42 was performed as previously described (45, 46). Seven-month-old Usp25−/− mice and littermate Usp25+/+ mice were anesthetized, and oAβ42 (a total of 1.5 μg in 2 μl; AnaSpec, Fremont, CA, USA; AS-20276) or control vehicle was injected bilaterally into the hippocampus using an automated stereotaxic injection apparatus (RWD Life Science, Shenzhen, China) at the following coordinates: anteroposterior, −2.2 mm from the bregma; mediolateral, ±2.0 mm; dorsoventral, −2.0 mm. Sixteen hours after injection, the mice were sacrificed and subjected to immunohistochemistry to quantify microglial migration.
Administration of AZ1
AZ1 (TubePharm, Shanghai, China) was dissolved in 5% dimethyl sulfoxide (DMSO) and +95% corn oil. Five-month-old (for LTP recordings) or 7-month-old (for behavioral tests and biochemical analyses) male 5×FAD and littermate WT mice were intraperitoneally injected with AZ1 (20 mg kg−1) every day for 28 consecutive days. An equivalent amount of vehicle solvent was administered to the control group.
Pharmacokinetics of AZ1
WT ICR mice were given a single intragastric gavage of AZ1 (10 mg kg−1) dissolved in 0.5% sodium carboxymethyl cellulose (CMC-Na) [containing 1% Tween 80 (pH 4.0)]. Thereafter, whole blood and brain tissues were collected at 0.5, 2, and 8 hours (n = 3 per group). One gram of brain tissue was homogenized in 5 ml of methanol:water (1:4, v:v), and whole blood was collected in EDTA-K2 anticoagulant tubes and centrifuged at 1500g for 10 min at 4°C. Afterward, the supernatant was collected as plasma. Twenty-microliter aliquots of plasma and brain samples were diluted in 400 μl of acetonitrile containing internal standard (IS) [verapamil (5 ng ml−1) and glibenclamide (50 ng ml−1)]. The mixtures were vortexed thoroughly and centrifuged at 13,000 rpm for 8 min, and then 70 μl of each supernatant was diluted in 70 μl of water. Five-microliter aliquots of the mixtures were injected into an API 4000 liquid chromatography–tandem mass spectrometry (LC-MS/MS) system (AB SCIEX, Concord, Ontario, Canada) for analysis. Positive multiple reaction monitoring (MRM) mode was used to scan the ion transitions [mass/charge ratio (m/z) 422.1→361.1 for AZ1 and m/z 455.2→165.1 for IS). Chromatographic separation was performed using a ZORBAX XDB-C18 column (2.1 × 50-mm internal diameter and 5-μm particle size; Agilent Technologies, Santa Clara, CA, USA; column no. 50-282) at a flow rate of 0.40 ml min−1 in the atmospheric pressure chemical ionization (APCI) source. Mobile phase A was an aqueous solution with 0.1% formic acid; mobile phase B was acetonitrile with 0.1% formic acid. The LC gradient was as follows: 0 to 0.50 min, 35% B; 0.50 to 1.20 min, 35 to 98% B; 1.20 to 2.20 min, 98% B; 2.20 to 2.21 min, 98 to 35% B; 3.50 min, stop.
Comprehensive metabolic panel
Mice were anaesthetized with isoflurane. Then, whole blood was collected without anticoagulant and centrifuged at 1500g for 10 min at 4°C after incubation at room temperature for 30 min. The supernatant serum was collected and then processed for the following biochemical assays: aspartate aminotransferase [International Federation of Clinical Chemistry (IFCC) method; Mindary, Shenzhen, China; 105-000443-00], alanine aminotransferase (IFCC method, Mindary, 105-000442-00), alkaline phosphatase [2-amino-2-methyl-1-propanol (AMP) buffer method, Mindary, 105-000444-00], albumin [bromocresol green (BCG) method, Mindary, 105-000450-00], total protein (biuret method, Mindary, 105-000451-00), urea [ultraviolet (UV) method, Mindary, 105-000452-00], creatinine (sarcosine oxidase method, Mindary, 105-000457-00), total cholesterol [cholesterol oxidase-peroxidase (CHOD-POD) method, Mindary, 105-000448-00], triglycerides [glycerol 3-phosphate oxidase-peroxidase (GPO-POD) method, Mindary, 105-000449-00], creatine kinase (IFCC method, Mindary, 105-000458-00), and glucose (GPO-POD method, Mindary, 105-000949-00). Spectrophotometric readings were obtained with the BS-240 Clinical Chemistry Analyzer (Mindary).
Immunocytochemistry
Cells were plated on coverslips coated with poly-l-lysine, fixed with 4% paraformaldehyde at room temperature for 20 min, and then permeabilized with 0.2% Triton X-100 in phosphate-buffered saline for 5 min. After blocking, the cells were subjected to immunostaining with the indicated primary antibodies at 4°C overnight, stained with Alexa Fluor 488– and Alexa Fluor 594–conjugated secondary antibodies (Thermo Fisher Scientific, A11001, A11005, A11008, and A11012; 1:500), and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, D95542; 1 μg ml−1). The following primary antibodies were used: anti-Iba1 (Wako Pure Chemical, 019-19741; 1:200), anti-Myc (Thermo Fisher Scientific, 132500; 1:500), and anti-HA (Sigma-Aldrich, H6908; 1:500). Confocal images were acquired with a Leica SP8 confocal microscope and subjected to quantification with ImageJ software [National Institutes of Health (NIH)].
Immunohistochemistry
Perfusion and brain slice preparation were performed as previously described (44). Brain slices were stained using the following primary antibodies: anti-Iba1 (Wako Pure Chemical, 019-19741; 1:500), anti-CD68 (Bio-Rad, Hercules, CA, USA; MCA1957, 1:100), anti-PSD95 (Millipore, MAB1596; 1:100), and 6E10 (BioLegend, San Diego, CA, USA; 803001, 1:400). Alexa Fluor 488– and Alexa Fluor 594–conjugated secondary antibodies (Thermo Fisher Scientific, A11001, A11005, A11008, or A11012; 1:500) were used, and then the slices were counterstained with DAPI (Sigma-Aldrich, D95542; 1 μg ml−1). Z-stack confocal images were acquired with a Leica SP8 confocal microscope. The number and somatic size of Iba1+ cells were manually counted using ImageJ software (NIH), and lengths of Iba1+ cell processes were analyzed using the Filament function in the Imaris software (Bitplane, Belfast, UK; version 9.2.0). To analyze synapse elimination by microglia, z-stack images were acquired from slices coimmunostained with anti-PSD95 and anti-Iba1 antibodies, and the volumes of microglia and PSD95+ synapses were reconstructed using the Surfaces function in the Imaris software (Bitplane, version 9.2.0).
Golgi staining
Mice were anesthetized, and brains were collected for Golgi staining using the FD Rapid Golgi Stain Kit (FD Neuro Technologies, Columbia, MD, USA; PK401), according to the manufacturer’s protocol. Z-stack images were acquired with a laser scanning confocal microscope (Olympus FV1000), and spine density was quantified using ImageJ software (NIH).
Synaptosome purification and phagocytosis assay
Synaptosomes were purified as previously described (47) and conjugated with pHrodo Red dye (Thermo Fisher Scientific, P36600) in Na2CO3 at room temperature for 2 hours (48). Unbound pHrodo Red dye was washed out with Dulbecco’s phosphate-buffered saline (DPBS). Usp25+/+ and Usp25−/− microglia were plated at a density of 2 × 105 cell per well and then incubated with 5 μl of pHrodo Red dye–conjugated synaptosomes in the presence or absence of 10 μM oAβ42 (AnaSpec, AS-20276). Twenty-four hours after incubation, microglia were subjected to immunocytochemistry.
RNA-seq analysis
Total RNA was extracted from hippocampus using TRIzol reagent (Thermo Fisher Scientific, 15596026). After qualification using an Agilent 2100 Bioanalyzer system (Agilent Technologies), the total RNA was subjected to sequencing library preparation using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA; E7530) according to the manufacturer’s recommendations, followed by clustering, which was performed on the cBot Cluster Generation System (Illumina, San Diego, CA, USA) using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, PE-401-3001). Sequencing was performed on the Illumina NovaSeq 6000 or HiSeq 4000 System, and 150–base pair (bp) paired-end reads were generated.
Raw RNA-seq reads were aligned to the Ensembl mouse reference genome GRCm38 (mm10) using HISAT2 (version 2.0.5) (49). Gene expression was subsequently quantified using featureCounts (version 1.5.0-p3) (50). Then, the raw gene counts were processed with the DESeq2 package (version 1.26.0) (51) in R (version 3.6.1) to analyze DEGs. GO enrichment analysis of DEGs was performed in R using the package clusterProfiler (version 3.14.3, qvalueCutoff = 0.05) (52).
The WGCNA (53) systematic biological method for describing the correlation patterns of genes among different samples was performed using the WGCNA package (version 1.69) (54) to identify unsigned gene coexpression networks and modules. First, gene modules were identified with similar gene expression patterns using hierarchical clustering. For each gene module, the expression matrix of the genes contained in the module was extracted and then the first principal component (PCA1) was calculated as “module eigengene.” In addition, the Pearson correlation between the module eigengene of each sample and different genotypes of each sample were analyzed. Numbers in the heatmap represent the Pearson correlation and corresponding P values. Sample traits, including among the sample groups, were correlated to genes and modules, and significantly correlated modules were determined by P < 0.05. Eigengenes in each module that were significantly correlated with sample traits were selected by P < 0.05.
TMT-based quantitative proteomics
The cerebral cortices and hippocampi of 6-month-old female 5×FAD, 5×FAD;Usp25−/−, and littermate WT mice were lysed in 6 M guanidine hydrochloride, homogenized with a homogenizer (MP Biomedicals, Irvine, CA, USA), sonicated, and then boiled for 10 min. After centrifugation, the supernatants were processed for filter-aided sample preparation (FASP) digestion using trypsin, and then 100 μg of the peptide mixture from each sample was labeled using TMT 10plex Isobaric Label Reagent (Thermo Fisher Scientific, 90110) according to the manufacturer’s instructions. The TMT-labeled digested samples were separated into 15 fractions using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific, 84868). Each fraction was separated with EASY-nLC 1000 Liquid Chromatograph (Thermo Fisher Scientific) and then subjected to LC-MS/MS analysis on Q Exactive HF-X Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) for 90 min. The spectra were analyzed by Proteome Discoverer (Thermo Fisher Scientific, version 1.4) and then subjected to a database search using the MASCOT search engine (Matrix Science, Boston, MA, USA; version 2.2) for peptide identification. All identified proteins were determined using a false discovery rate (FDR) threshold of <0.01. After retrieval from the UniProtKB database and searching against the SwissProt database (mouse), the top BLAST hits were processed for GO enrichment analysis using Blast2GO (BioBam, Valencia, Spain; version 3.3.5).
Electrophysiology
Electrophysiology was performed as previously described (44). Mice were anesthetized with isoflurane, and then the brains were rapidly removed, placed in ice-cold artificial cerebrospinal fluid [ACSF; 120 mM sucrose, 2.5 mM KCl, 10 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM d-glucose, 64 mM NaCl, and 0.5 mM CaCl2 (pH 7.4), ~310 mOsm], and bubbled with carbogen (95% O2 + 5% CO2). Brain slices were cut (400 μm thick) using a Leica VT1200S vibratome, incubated in ACSF [3.5 mM KCl, 120 mM NaCl, 1.3 mM MgSO4, 10 mM d-glucose, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 2.5 mM CaCl2 (pH 7.4), ~300 mOsm], and bubbled with carbogen (95% O2 + 5% CO2) at 32°C for 1 hour. The slices recovered at room temperature for at least 1 hour before recording. For LTP recording, the Schaffer collateral inputs to the CA1 region were stimulated with a bipolar tungsten electrical stimulating electrode, while field excitatory postsynaptic potentials (fEPSPs) were recorded from the dendritic layer in the Schaffer collateral pathway. Baseline responses were acquired every 20 s with a stimulation intensity that yielded 30% of the maximum response. After a 20-min stable baseline recording, LTP was induced by high-frequency stimulation (two trains of 100-Hz stimuli with an interval of 30 s), followed by continued recording for 60 min.
For mEPSC and mIPSC recordings, brain slices were incubated in ACSF [2.5 mM KCl, 126 mM NaCl, 2.4 mM MgCl2, 1.2 mM CaCl2, 1.2 mM NaH2PO4, 11 mM d-glucose, and 18 mM NaHCO3 (pH 7.4), ~300 mOsm], bubbled with carbogen (95% O2 + 5% CO2), and kept at 32°C. Tetrodotoxin (1 μM) was added to the perfusion ACSF to block sodium channels. A glass pipette was filled with a solution containing 2 mM MgCl2·6H2O, 140 mM CsCH3SO3, 5 mM TEA-Cl, 1 mM EGTA, 2.5 mM Mg-ATP (adenosine triphosphate), and 0.3 mM Na-GTP (guanosine triphosphate) (pH 7.3, ~290 mOsm), and the resistance of the pipette was 5 to 8 megohms. The pipette was placed in dentate gyrus (DG) granule neurons, and recordings were obtained at a holding potential of −70 mV for mEPSCs and 0 mV for mIPSCs. The resistance of the pipette was 5 to 8 megohms. Data were acquired with a Multiclamp 700B patch-clamp amplifier (Molecular Devices, San Jose, CA, USA), filtered at 2 kHz, and sampled at 10 kHz, followed by processing for analysis using pClamp software (Molecular Devices, version 10.6).
Y-maze test
Experimental mice were placed in the center of a Y-shaped maze with three arms at 120° from each other and allowed to freely explore the three arms for 5 min. The percentage of spontaneous alternation was calculated automatically using Smart Video Tracking Software 3.0 (Panlab, Harvard Apparatus, Holliston, MA, USA).
MWM test
MWM tests were conducted in a circular tank (diameter of 120 cm) filled with opaque water kept at 22°C, using a modified protocol (55). Four bright and contrasting shapes, which served as reference cues, were affixed to the walls surrounding the tank. A fixed platform (diameter of 10 cm) was submerged 1 cm below the surface of the water in the target quadrant. On the training days, the mice were placed into the maze at one of four random points, and two trials were performed every day for six consecutive days. Mice were allowed to search for the hidden platform for 60 s. If a mouse was unable to find the platform within 60 s, it was guided to the platform and kept there for another 10 s. The latency to reach the hidden platform was scored by Smart Video Tracking Software 3.0 (Panlab, Harvard Apparatus). On day 7 after training, the platform was removed, and a probe test was performed. The time spent in each quadrant was recorded.
FC test
FC was performed using a modified protocol (56, 57). Experimental mice were placed in a conditioning chamber (Panlab, Harvard Apparatus) and allowed to freely explore the chamber for 2 min. Thereafter, a 60-dB white noise stimulus was presented for 30 s as a conditioned stimulus (CS), and a 0.5-mA foot shock was given to the mice as an unconditioned stimulus (US) during the last 2 s of the noise. The CS-US pair was presented three times at 1-min intervals. Mice were removed 1 min after the last CS-US pair. For the contextual test, the mice were placed in the same conditioning chamber 24 hours after training, and freezing behaviors were scored for 5 min. For the cued test, the mice were placed in another testing chamber with a novel contextual environment for 3 min, and thereafter, a CS was presented for 3 min.
Statistical analysis
All the data were collected and analyzed in a double-blind manner. Statistical analyses were performed using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA; version 8). The data distribution was assessed by the Shapiro-Wilk normality test. The statistical methods used to analyze the quantitative data are described in the figure legends. All data represent means ± SEM. P values of <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank T. Huang and L. Jiang for helpful discussion and F. Zeng, T. Guo, Y. Zeng, Q. Liu, J. Huang, X. You, and C. Zhang for technical assistance. We also thank Beijing Genomics Institute and Novogene Co. Ltd. for technical assistance in transcriptomic analysis, Shanghai Applied Protein Technology Co. Ltd. for technical assistance in proteomic analysis, and 3D BioOptima for technical assistance in pharmacokinetic analysis. Funding: This work was supported by the National Natural Science Foundation of China (31871077, 81822014, and 81571176 to X.W.; 81701130 to Q.Z.), the National Key R&D Program of China (2016YFC1305900 to X.W.), the Natural Science Foundation of Fujian Province of China (2017J06021 to X.W.), the Fundamental Research Funds for the Chinese Central Universities (20720150061 to X.W.), and the BrightFocus Foundation (A2018214F to Yingjun Zhao). Author contributions: X.W. and Q.Z. designed the study and wrote the manuscript. Q.Z., G.L., K.L., Ying Zhou, L. Zhang, and X.W. performed data analyses. Q.Z. performed biochemical experiments. G.L. performed morphological analyses. Q.Z., G.L., M.W., A.D., and H.Z. performed behavioral analyses. S.W. and Q.D. performed LTP recording. Y.G., L. Zhu, and H.S. performed whole-cell recording. L. Zheng and Yulin Zhou collected human fetal brain tissues. C.D. generated Usp25 knockout mice. H.X. and Yingjun Zhao discussed and edited the manuscript. X.W. supervised the project. Competing interests: X.W., Q.Z., Q.D., G.L., and S.W. are inventors on a Chinese patent application related to this work filed by Xiamen University (no. 2020100711437, filed on 21 January 2020). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The RNA-seq data have been deposited into the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with the accession number CNP0001317. The TMT-based quantitative proteomics data have been deposited into the iProX, an official member of ProteomeXchange Consortium, with the dataset identifier PXD021637.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabe1340/DC1
REFERENCES AND NOTES
- 1.Antonarakis S. E., Skotko B. G., Rafii M. S., Strydom A., Pape S. E., Bianchi D. W., Sherman S. L., Reeves R. H., Down syndrome. Nat. Rev. Dis. Primers 6, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonarakis S. E., Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 18, 147–163 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Wiseman F. K., Al-Janabi T., Hardy J., Karmiloff-Smith A., Nizetic D., Tybulewicz V. L. J., Fisher E. M. C., Strydom A., A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisniewski K. E., Dalton A. J., McLachlan C., Wen G. Y., Wisniewski H. M., Alzheimer’s disease in Down’s syndrome: Clinicopathologic studies. Neurology 35, 957–961 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Leverenz J. B., Raskind M. A., Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: A regional quantitative analysis. Exp. Neurol. 150, 296–304 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Li Q., Barres B. A., Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Colonna M., Butovsky O., Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong S., Beja-Glasser V. F., Nfonoyim B. M., Frouin A., Li S., Ramakrishnan S., Merry K. M., Shi Q., Rosenthal A., Barres B. A., Lemere C. A., Selkoe D. J., Stevens B., Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heckmann B. L., Teubner B. J. W., Tummers B., Boada-Romero E., Harris L., Yang M., Guy C. S., Zakharenko S. S., Green D. R., LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell 178, 536–551.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeTure M. A., Dickson D. W., The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou X., Wahane S., Friedl M.-S., Kluge M., Friedel C. C., Avrampou K., Zachariou V., Guo L., Zhang B., He X., Friedel R. H., Zou H., Microglia and macrophages promote corralling, wound compaction and recovery after spinal cord injury via Plexin-B2. Nat. Neurosci. 23, 337–350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otxoa-de-Amezaga A., Miró-Mur F., Pedragosa J., Gallizioli M., Justicia C., Gaja-Capdevila N., Ruíz-Jaen F., Salas-Perdomo A., Bosch A., Calvo M., Marquez-Kisinousky L., Denes A., Gunzer M., Planas A. M., Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 137, 321–341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S. V., Vieira-Saecker A., Schwartz S., Albasset S., McManus R. M., Tejera D., Griep A., Santarelli F., Brosseron F., Opitz S., Stunden J., Merten M., Kayed R., Golenbock D. T., Blum D., Latz E., Buée L., Heneka M. T., NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett F. C., Liddelow S. A., Microglia metabolic breakdown drives Alzheimer’s pathology. Cell Metab. 30, 405–406 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Gratuze M., Leyns C. E. G., Holtzman D. M., New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13, 66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pohl C., Dikic I., Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Zheng Q., Huang T., Zhang L., Zhou Y., Luo H., Xu H., Wang X., Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 8, 303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valero R., Marfany G., González-Angulo O., González-González G., Puelles L., González-Duarte R., USP25, a novel gene encoding a deubiquitinating enzyme, is located in the gene-poor region 21q11.2. Genomics 62, 395–405 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Zhong B., Liu X., Wang X., Chang S. H., Liu X., Wang A., Reynolds J. M., Dong C., Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 13, 1110–1117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong B., Liu X., Wang X., Liu X., Li H., Darnay B. G., Lin X., Sun S.-C., Dong C., Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci. Signal. 6, ra35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z., Yu T., Morishima M., Pao A., LaDuca J., Conroy J., Nowak N., Matsui S.-I., Shiraishi I., Yu Y. E., Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 16, 1359–1366 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Yu T., Liu C., Belichenko P., Clapcote S. J., Li S., Pao A., Kleschevnikov A., Bechard A. R., Asrar S., Chen R., Fan N., Zhou Z., Jia Z., Chen C., Roder J. C., Liu B., Baldini A., Mobley W. C., Yu Y. E., Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 1366, 162–171 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R., Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y.-H., Zhang Y., Jiang L.-Q., Wang S., Lei C.-Q., Sun M.-S., Shu H.-B., Liu Y., WDFY1 mediates TLR3/4 signaling by recruiting TRIF. EMBO Rep. 16, 447–455 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wrigley J. D., Gavory G., Simpson I., Preston M., Plant H., Bradley J., Goeppert A. U., Rozycka E., Davies G., Walsh J., Valentine A., McClelland K., Odrzywol K. E., Renshaw J., Boros J., Tart J., Leach L., Nowak T., Ward R. A., Harrison T., Andrews D. M., Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem. Biol. 12, 3113–3125 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., Herrup K., Frautschy S. A., Finsen B., Brown G. C., Verkhratsky A., Yamanaka K., Koistinaho J., Latz E., Halle A., Petzold G. C., Town T., Morgan D., Shinohara M. L., Perry V. H., Holmes C., Bazan N. G., Brooks D. J., Hunot S., Joseph B., Deigendesch N., Garaschuk O., Boddeke E., Dinarello C. A., Breitner J. C., Cole G. M., Golenbock D. T., Kummer M. P., Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilcock D. M., Griffin W. S. T., Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation 10, 84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo T., Zhang D., Zeng Y., Huang T. Y., Xu H., Zhao Y., Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 15, 40 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandey R. S., Graham L., Uyar A., Preuss C., Howell G. R., Carter G. W., Genetic perturbations of disease risk genes in mice capture transcriptomic signatures of late-onset Alzheimer’s disease. Mol. Neurodegener. 14, 50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D. K., Park J., Han D., Yang J., Kim A., Woo J., Kim Y., Mook-Jung I., Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 13, 2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosch-Comas A., Lindsten K., Gonzàlez-Duarte R., Masucci M. G., Marfany G., The ubiquitin-specific protease USP25 interacts with three sarcomeric proteins. Cell. Mol. Life Sci. 63, 723–734 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiseman F. K., Pulford L. J., Barkus C., Liao F., Portelius E., Webb R., Chávez-Gutiérrez L., Cleverley K., Noy S., Sheppard O., Collins T., Powell C., Sarell C. J., Rickman M., Choong X., Tosh J. L., Siganporia C., Whittaker H. T., Stewart F., Szaruga M., Murphy M. P., Blennow K., de Strooper B., Zetterberg H., Bannerman D., Holtzman D. M., Tybulewicz V. L. J., Fisher E. M. C., Trisomy of human chromosome 21 enhances amyloid-β deposition independently of an extra copy of APP. Brain 141, 2457–2474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolvetang E. W., Bradfield O. M., Tymms M., Zavarsek S., Hatzistavrou T., Kola I., Hertzog P. J., The chromosome 21 transcription factor ETS2 transactivates the β-APP promoter: Implications for Down syndrome. Biochim. Biophys. Acta 1628, 105–110 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Li Y., Wang H., Wang S., Quon D., Liu Y.-W., Cordell B., Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc. Natl. Acad. Sci. U.S.A. 100, 259–264 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryoo S. R., Cho H.-J., Lee H.-W., Jeong H. K., Radnaabazar C., Kim Y.-S., Kim M.-J., Son M.-Y., Seo H., Chung S.-H., Song W.-J., Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: Evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 104, 1333–1344 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Mok K. Y., Jones E. L., Hanney M., Harold D., Sims R., Williams J., Ballard C., Hardy J., Polymorphisms in BACE2 may affect the age of onset Alzheimer’s dementia in Down syndrome. Neurobiol. Aging 35, 1513.e1-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueller-Steiner S., Zhou Y., Arai H., Roberson E. D., Sun B., Chen J., Wang X., Yu G., Esposito L., Mucke L., Gan L., Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 51, 703–714 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Readhead B., Haure-Mirande J.-V., Mastroeni D., Audrain M., Fanutza T., Kim S. H., Blitzer R. D., Gandy S., Dudley J. T., Ehrlich M. E., miR155 regulation of behavior, neuropathology, and cortical transcriptomics in Alzheimer’s disease. Acta Neuropathol. 140, 295–315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glass C. K., Saijo K., Winner B., Marchetto M. C., Gage F. H., Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thome A. D., Faridar A., Beers D. R., Thonhoff J. R., Zhao W., Wen S., Pascual B., Masdeu J. C., Appel S. H., Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol. Neurodegener. 13, 61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Litvinchuk A., Wan Y.-W., Swartzlander D. B., Chen F., Cole A., Propson N. E., Wang Q., Zhang B., Liu Z., Zheng H., Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 100, 1337–1353.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heppner F. L., Ransohoff R. M., Becher B., Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Zhong L., Xu Y., Zhuo R., Wang T., Wang K., Huang R., Wang D., Gao Y., Zhu Y., Sheng X., Chen K., Wang N., Zhu L., Can D., Marten Y., Shinohara M., Liu C.-C., Du D., Sun H., Wen L., Xu H., Bu G., Chen X.-F., Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 10, 1365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng F., Ma X., Zhu L., Xu Q., Zeng Y., Gao Y., Li G., Guo T., Zhang H., Tang X., Wang Z., Ye Z., Zheng L., Zhang H., Zheng Q., Li K., Lu J., Qi X., Luo H., Zhang X., Wang Z., Zhou Y., Yao Y., Ke R., Zhou Y., Liu Y., Sun H., Huang T., Shao Z., Xu H., Wang X., The deubiquitinase USP6 affects memory and synaptic plasticity through modulating NMDA receptor stability. PLOS Biol. 17, e3000525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y., Wu X., Li X., Jiang L.-L., Gui X., Liu Y., Sun Y., Zhu B., Piña-Crespo J. C., Zhang M., Zhang N., Chen X., Bu G., An Z., Huang T. Y., Xu H., TREM2 is a receptor for β-Amyloid that mediates microglial function. Neuron 97, 1023–1031.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong L., Wang Z., Wang D., Wang Z., Martens Y. A., Wu L., Xu Y., Wang K., Li J., Huang R., Can D., Xu H., Bu G., Chen X.-F., Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 13, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunkley P. R., Jarvie P. E., Robinson P. J., A rapid Percoll gradient procedure for preparation of synaptosomes. Nat. Protoc. 3, 1718–1728 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Byun Y. G., Chung W.-S., A novel in vitro live-imaging assay of astrocyte-mediated phagocytosis using pH indicator-conjugated synaptosomes. J. Vis. Exp. 132, 56647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L., Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu G., Wang L.-G., Han Y., He Q.-Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang B., Horvath S., A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Langfelder P., Horvath S., WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seo J., Giusti-Rodriguez P., Zhou Y., Rudenko A., Cho S., Ota K. T., Park C., Patzke H., Madabhushi R., Pan L., Mungenast A. E., Guan J.-S., Delalle I., Tsai L.-H., Activity-dependent p25 generation regulates synaptic plasticity and Aβ-induced cognitive impairment. Cell 157, 486–498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C.-C., Tsai C.-W., Deak F., Rogers J., Penuliar M., Sung Y. M., Maher J. N., Fu Y., Li X., Xu H., Estus S., Hoe H.-S., Fryer J. D., Kanekiyo T., Bu G., Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron 84, 63–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shoji H., Takao K., Hattori S., Miyakawa T., Contextual and cued fear conditioning test using a video analyzing system in mice. J. Vis. Exp. 85, 50871 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabe1340/DC1