

Specific patterns of DNA mutations in normal skin show history of UV exposure and association with cancer risk.
Abstract
In ultraviolet (UV) radiation–exposed skin, mutations fuel clonal cell growth. The relationship between UV exposure and the accumulation of clonal mutations (CMs) and the correlation between CMs and skin cancer risk are largely unexplored. We characterized 450 individual-matched sun-exposed (SE) and non-SE (NE) normal human skin samples. The number and relative contribution of CMs were significantly different between SE and NE areas. Furthermore, we identified hotspots in TP53, NOTCH1, and GRM3 where mutations were significantly associated with UV exposure. In the normal skin from patients with cutaneous squamous cell carcinoma, we found that the cancer burden was associated with the UV-induced mutations, with the difference mostly conferred by the low-frequency CMs. These findings provide previously unknown information on UV’s carcinogenic effect and pave the road for future development of quantitative assessment of subclinical UV damage and skin cancer risk.
INTRODUCTION
Ultraviolet (UV) light is responsible for more than 5 million cases of skin cancer annually in the United States, which is more human malignancies than all other environmental carcinogens combined (1, 2). In mammals, nucleotide excision repair eliminates UV-mediated DNA lesions, but this mechanism of repair is error prone, resulting in frequent mutations (3). The preferential location of UVB-induced DNA lesions results in a specific pattern of so-called UV signature mutations (USMs) at dipyridine sites (C>T and CC>TT) (4). In most skin cancers, including cutaneous squamous cell carcinoma (cSCC), the burden of UV signature driver mutations is high (4, 5). While some cSCC arise from visible precancerous lesions known as actinic keratoses (AKs), many cSCC arise in apparently “normal” skin areas from precursors that are clinically invisible (6), and only a small fraction of AKs ever progress to invasive carcinoma (7). Moreover, AKs and other clinical signs of photoaging arise decades after the initiation of the photocarcinogenic process and almost exclusively in elder population (7) and, therefore, are unsuitable for early assessment of photocarcinogenesis. Additional precision in assessment of skin cancer risk is therefore required to appropriately direct screening and prevention efforts.
TP53 mutations are among the most common driver mutations in cSCC and are also detected by immunohistochemistry in aged normal skin (8, 9). These UV-induced TP53 mutations facilitate clonal expansion of cells harboring them and therefore behave as early clonal mutations (CMs) (10). For two decades, TP53 mutant keratinocyte cell clones were considered the earliest manifestations of skin carcinogenesis (8, 9, 11). Because p53 clonal immunopositivity could not be efficiently quantified in human skin, detection of mutant TP53 for assessment of photocarcinogenesis in clinical dermatology practice has been unattainable. The low relative abundance of clonal DNA previously limited efficient detection of early mutated cell groups. However, with improved high-throughput sequencing technology, we have finally reached the lower end of this threshold and efficient detection of rare mutations in normal tissue is becoming feasible in recent studies by others and us using deep bulk sequencing or single-cell DNA sequencing (12–17). In exploratory analyses, CMs were found to be abundant in clinically normal skin from sun-exposed (SE) sites in NOTCH1, NOTCH2, FAT1 and several other genes besides TP53 (13). Prior attempts to establish a quantitative method for assessing photodamage and skin cancer risk had limited success (18, 19). A method that enables quantitative evaluation of early photodamage is expected to help optimize personalized sun-protective measures and may also serve as a tool for assessing the need and efficacy of early preventative treatment interventions.
In the current work, we developed an ultradeep sequencing–based method to identify CMs in clinically normal epidermis and show differences in CMs between SE and non-SE (NE) skin areas. We then correlated CMs with skin cancer burden in another independent cohort of cSCC patients and found that mutational features in normal skin are significantly associated with cancer risk burden.
RESULTS
Ultradeep sequencing of epidermal samples using customized focused panels
To generate a focused sequencing panel targeting the most commonly mutated sequences in normal human skin, we selected an area of focus based on a previous dataset (13). All previous mutations were assigned to 100–base pair (bp) genomic segments. After sorting the segments by number of mutations, we designed a panel to capture the top 55 most frequently mutated segments from 12 genes (5.5 kb in total; table S1). The majority (65%) of the targeted segments came from the following three genes: NOTCH1, NOTCH2, and TP53. When summarized by coding regions, 79% of the targeted segments lie in protein-coding regions, and the remaining segments were mostly in introns. In the previous dataset (13), 87% of the samples harbored at least one mutation within this panel. Thus, as designed, this panel captured the most frequently mutated genomic regions in SE skin and was highly focused for efficient deep sequencing to identify low-frequency mutations.
The primary cohort was sequenced using the focused panel in two batches. We first sequenced a discovery cohort of 374 human skin samples from 13 postmortem donors: 360 epidermal samples, equally acquired from both SE and NE regions using 1-, 2-, 3-, 4-, or 6-mm punch sizes. From the same 13 donors, DNA from bulk NE dermis (n = 14, 1 donor contributed two samples) was isolated for germline controls. After initial analysis to determine the optimal punch size, we then tested a separate validation cohort of 90 epidermal samples from 9 of the 13 donors using the most effective punch size (2 mm, as detailed in the “The effect of punch size on USM detection” section). In total, the dataset contains 464 samples: 225 SE, 225 NE, and 14 dermal samples as controls (table S2A) from 13 individuals. After sequencing, 85% of samples reached a minimum of 10,000× coverage in at least 80% of the targeted region. The median of average coverage across all samples was 64,730× (table S3A), with only one sample exclusion (NE sample) due to sequencing failure. This unique design of ultradeep sequencing from individual-matched SE/NE samples enabled us to discriminate between the mutational profiles of SE and NE skin samples.
To better define the clinical relevance of CMs, we sequenced an extended cohort of SE skin samples from human patients with cSCC. Twenty 2-mm punch biopsy specimens were obtained from surgically excised skin from eight individuals, including 16 normal skin samples and 4 samples of cSCC. For this extended cohort, a custom sequencing panel was designed to encompass the complete protein-coding region of 12 genes with frequently reported mutations in UV-exposed skin (NOTCH1, NOTCH2, NOTCH3, TP53, CDKN2A, BRAF, HRAS, KRAS, NRAS, KNSTRN, FAT1, and FGFR3), and 1 control gene without expected functional significance in skin (VHL). This sequencing panel encompassed 59.5 kb. After sequencing, all samples have at least 80% of the targeted region covered by a minimum of 10,000× coverage. The median value of average coverages across all samples was 47,158× (table S3B). This extended cohort from cSCC patients would allow us to correlate the features of CMs to patient clinical outcomes.
Delineate the mutational patterns associated with UV exposure
To identify the mutations solely caused by UV exposure, we characterized the mutational profiles of individual-matched SE/NE epidermal samples. In addition, we compared the epidermal samples to patient-matched dermal samples, followed by an in silico error suppression to remove germline polymorphisms and low-frequency technical artifacts. Dinucleotide and other complex mutations were identified by revisiting the raw reads using a program that we previously developed (20). Together, a total of 638 mutations were identified, predominantly single-nucleotide variants (SNVs; n = 614 or 96.2%) or dinucleotide variants (DNVs; n = 20 or 3.1%) (table S4). The median variant allele fraction (VAF) of all mutations was 2.1% (range, 0.1 to 36.6%), and only 3% mutations reached a VAF greater than 10%.
Among the 55 targeted genomic segments, mutations were detected in 50 segments with an average of 7.1 and 4.7 mutations per segment in SE and NE samples, respectively (Fig. 1A). Two segments were significantly [false discovery rate (FDR) P < 0.001] associated with UV exposure status, approximately corresponding to p53 p.227-261 (“TP53-3,” mutations in SE/NE = 38/0) and Notch1 p.449-481 (“NOTCH1-9,” mutations in SE/NE = 30/4). Mutations in an adjacent region in Notch1 p.419-449 (“NOTCH1-10”) were not associated with UV exposure (mutations in SE/NE = 48/40), although “NOTCH1-10” was the most frequently mutated segment in the current study. In addition, mutations were marginally enriched in SE samples (FDR P < 0.1) in three other segments: two in NOTCH1 (“NOTCH1-14” and “NOTCH1-19”) and one in GRM3 (“GRM3-2”). On the gene level, mutations in SE samples were only significantly enriched in TP53 (FDR P < 0.001) and marginally significant in GRM3 (FDR P < 0.1). Overall, the numbers of mutations in SE samples were 6.3 times higher than NE samples in TP53 and 4.3 times in GRM3 (Fig. 1B). Mutations identified in nine other genes did not exhibit significant association with sun-exposure status either on the gene or segment level: NOTCH2, ARID1A, SALL1, SCN1A, ERBB4, FAT4, FGFR3, ADGRB3, and PPP1R3A. These findings indicate a highly genomic region–specific pattern of the accumulation of UV-induced somatic mutations.
Fig. 1. Region-specific enrichment of somatic mutations in SE skin.
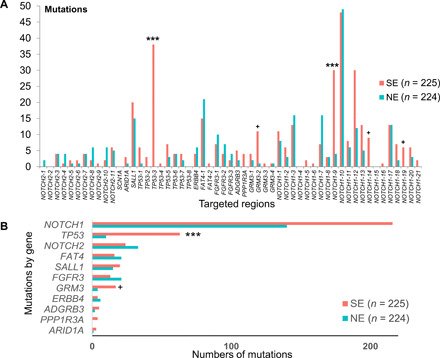
(A) The graph shows the number of mutations identified within each 100-bp genomic target window grouped by SE and NE skin types. (B) The overall gene-level number of mutations from SE and NE samples. Asterisks indicate the segments or genes where mutations are significantly enriched in the SE samples (FDR P values: ***P < 0.001 and +P < 0.1).
We next investigated potential hotspots and mutations associated with UV exposure. After sorting all mutations by their genomic locations, one specific region in TP53 (corresponding to p53 p.217-280), appeared to be “mutation exempt” in comparison to surrounding regions in NE samples. In contrast, this region was highly mutated in SE samples (Fig. 2A). We reanalyzed a recent study involving RNA sequencing (RNA-seq) of both SE and NE normal skin samples (12) and found four mutations in this region, all from SE samples (table S5). To identify mutations associated with UV exposure, we focused on highly recurrent mutations (present in five or more samples; n = 18). By comparing the frequency in SE and NE skin samples, we identified six mutations significantly enriched in SE samples—TP53 R248W, NOTCH1 P460L, NOTCH1 S385F, NOTCH1 E424K, TP53 G245D, and NOTCH1 P460S—and nearly all of them were exclusively found in SE samples (FDR P < 0.05; Fig. 2B). No mutation was significantly enriched in NE samples. Five of the six SE-enriched mutations were found in both discovery and validation cohorts, indicating that they were unlikely to be caused by batch effect. Unexpectedly, one specific mutation (NOTCH1 E424K) was associated with significantly elevated VAFs (median, 10%; P < 0.001, Wilcoxon test), about fivefold higher than other mutations (median VAF, 2.1%; Fig. 2, A and B). Through protein structure modeling (Fig. 2C), we found that the NOTCH1 E424K mutation is predicted to disrupt the binding of NOTCH1 to delta-like canonical ligand 4 (DLL4), a negative regulator of the Notch signaling pathway (12). By prohibiting formation of a salt bridge between NOTCH1 E424 and DLL4 K189/R191, the mutation E424K creates a repulsive force that inhibits DLL4 binding (21). On the basis of the biological role of DLL4 and NOTCH1, the NOTCH1 E424 mutation is expected to promote epithelial proliferation (22, 23). The overall prevalence of the NOTCH1 E424K mutation in our dataset is 2.7%. For comparison, in GENIE cBioPortal (24), NOTCH1 E424K is mutated in 1.3% of cSCCs and 0.04% in melanomas and is rarer in other cutaneous or noncutaneous malignancies (table S6).
Fig. 2. Hotspots and mutations associated with UV exposure.
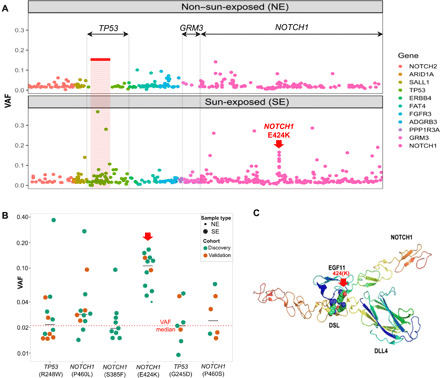
(A) All mutations are ordered by their genomic locations. x axis: The order of the mutation’s genomic location. y axis: Variant allele fraction (VAF) of individual mutations. The color depicts the gene harboring the mutations. The three genes demonstrating significant difference between SE and NE, either on the gene level or segment level, were labeled on top (TP53, GRM3, and NOTCH1). One specific mutation with elevated VAFs (NOTCH1 E424K) is indicated with a red arrow. (B) The VAF of the six individual mutations that are significantly enriched in SE versus NE epidermis in the primary discovery (green) and validation (orange) datasets. The dotted red line represents median VAF of all mutations, and black lines indicate the median of each group. (C) The predicted protein complex structure of NOTCH1 and DLL4 to show the position of the mutant E424K and the interacting partners, DLL4 K189/R191, in wild type.
USMs exclusively account for the elevated mutation burdens in SE skin
We next intercorrelated the identified mutations with previously known USMs, i.e., C>T transition at dipyrimidines (4). Among all 638 mutations in SE and NE samples, 298 were USMs. Of these 298 USMs, 76% were present in SE samples. USMs were significantly enriched in SE compared to NE samples (n = 226 and 72, respectively, P < 0.001, Fisher’s exact test). Especially among the high-VAF mutations, 18 of 19 mutations with VAFs above 0.1 were from SE samples, and most (13 of 18) were USMs. Conversely, non-USMs (NUSMs) were approximately equally present (n = 159 and 181, not significant, Fisher’s exact test) in SE and NE skin types (Fig. 3A), indicating that these mutations may not be directly associated with UV exposure.
Fig. 3. UV-induced DNA damage assessed by USMs.
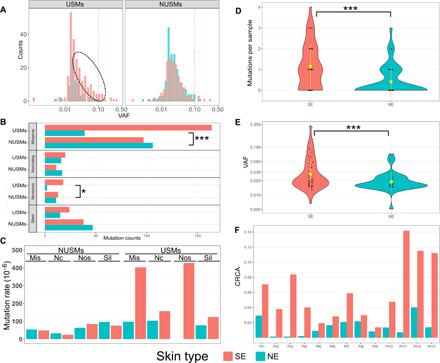
(A) Only USMs are associated with sun-exposure status. Left: Higher numbers of USMs are in SE than NE skin. Right: NUSMs are almost equally presented in SE and NE samples. The red dotted line indicates high VAF (>0.1). The black dotted ellipse indicates additional USMs in SE compared with NE skin. (B) The numbers of mutations by each amino acid–change type in SE/NE, grouped by USMs and NUSMs. (C) Normalized mutation rate of each effect group, measured as the numbers of observed mutations per sample per million possible mutations within the current panel. Effect groups by amino acid–change type: Mis, missense; Nc, noncoding; Nos, nonsense; Sil, silent. Overall distribution of the numbers of USMs per sample (D) and the VAFs of the mutations (E) using the 2-mm punch size. Inside the violin plots: black dots, individual samples; yellow dot with bar, averaged value with SD. SE samples are associated with higher numbers of USMs, as well as higher VAFs indicating potential larger clones. (F). Cumulative relative clonal areas (CRCAs) were higher in SE than NE skin of all 13 patients, with the ratios of SE/NE ranging from 1.4 to 55.0 (mean, 11.2). Statistical tests used: (B) Fisher’s exact test with multiple test correction implemented using the FDR method and (C and D) Wilcoxon test; *P < 0.05 and ***P < 0.001.
To explore specific community enrichment patterns in different mutational function groups, we classified all 638 mutations into four effect groups: nonsense, missense, silent, and noncoding. Inside each effect group, we correlated the mutational properties (USM versus NUSM) with the matched samples’ sun-exposure statuses (SE versus NE) (Fig. 3B). Significant enrichment of USMs was observed in two of four effect groups by Fisher’s exact test: nonsense (FDR P < 0.05) and missense (FDR P < 0.001). Specifically, nonsense mutations were 9 times more frequently occurring in SE skins than in NE skins and similarly enriched by 4.2 times for missense mutations. To control for differences in the resulting effect group between the mutations caused by USMs and NUSMs, we simulated all possible mutations, including SNVs and CC>TT DNVs, within the current panel. There were a lower fraction of missense mutations and a higher fraction of silent mutations among all possible mutations caused by USMs compared with the ones caused by NUSMs (fig. S1). After adjusting by the total possible mutations for each effect group, the normalized mutation rate per sample were below 100 mutations per million possible mutations for all effect groups of NUSMs in either SE or NE skin. For USMs in SE skin, the normalized mutation rate was markedly increased to 403 and 427 mutations per million possible mutations for missense and nonsense mutations, respectively (Fig. 3C). In contrast, the silent or noncoding USMs in SE skin, as well as all effect groups of USMs in NE skin, were no more than 160 mutations per million possible mutations. These findings indicate that the mutations initiated by UV radiation are further selected by the host system or interclonal competition (25), in which the mutations with functional impacts give the clone greater competitive fitness.
Quantification of UV-induced DNA damage level by USMs
We next investigated the feasibility of using CMs to quantify UV-induced DNA damage. This was based on the hypothesis that SE samples harbor more CMs and are associated with higher VAFs compared to NE samples. Since our analyses indicated that NUSMs were not correlated with UV exposure, only USMs were used for quantifying UV-induced DNA damage. To avoid the potential bias introduced by different punch sizes, initially, only the most abundant size of 2 mm (n = 90 and 89, SE and NE, respectively) (Fig. 3D) was analyzed. A threefold difference was observed in the average USMs per sample between SE (mean, 1.2) and NE (mean, 0.4), which was significantly higher (P < 0.001, Wilcoxon test). Multiple USMs were found in 33% of SE samples but only 9% of NE samples (table S7). In addition, the identified USMs had significantly higher VAFs in SE (mean, 3.7%) than NE (mean, 2.1%) samples (P < 0.001, Wilcoxon test), indicating the presence of larger clones in SE samples (Fig. 3E). We further extended the analysis to include all punch sizes and found that the pattern was consistent with 34% of SE and only 6% of NE samples having multiple USMs and threefold higher average USMs per sample in SE (1.0) than NE (0.3) samples (P < 0.001, Wilcoxon test). These findings of increased USMs and elevated VAFs in SE than NE skin would then serve as the cornerstones for the quantification of UV-induced DNA damage.
To overcome the heterogeneity between samples, we developed cumulative relative clonal area (CRCA) as a single metric to assess the overall patient-level burden of CMs. The CRCA was defined as the overall percentage of biopsied skin area covered by USMs in a patient skin punch, which accounts for both the number of USMs and their VAFs (Fig. 3F). It is worth mentioning that our data did not allow us to distinguish whether mutations occurred independently or were present in the same clone. Hence, CRCA does not provide an exact measure of the mutated cell population but rather serves as an index of the mutation burden in the sampled area. To minimize the potential chance for repeated counting of co-occurring mutations in the same cells, co-occurring mutations were identified, primarily dinucleotide CC>TT mutations, and consolidated. When counted separately by sun-exposure status, the median CRCA across the 13 patients was 6.1% (range, 1.4 to 14.2%) in SE and 1.4% (range, 0.1 to 4.0%) in NE sites. On individual patient level, the CRCAs were higher in SE than the matched NE skin in all patients, with the average ratio of 11.2-fold higher (range, 1.4- to 55.0-fold). These CRCAs were calculated using only USMs. If all CMs were included, the CRCA would be only 2.2-fold higher (range, 0.8 to 5.6-fold) in SE than NE skin. On the basis of these results, CRCA may have the potential to be used as an objective measurement of the level of UV-induced DNA damage. To measure the variability of different samples obtained from the same skin area of one patient, we recalculated the CRCA on the sample level for the most abundant size of 2-mm punches. The SDs of the sample-level CRCAs within a patient were greater in the SE than NE skin samples (P = 2.062 × 10−9; median, 0.077 and 0.024 in SE and NE skin samples, respectively), suggesting a relatively larger variability of mutation patterns in SE than NE skin samples.
The effect of punch size on USM detection
In the discovery cohort, we sought to evaluate different punch sizes to determine the most efficient one for detecting USMs. Theoretically, although larger punches likely contain more clones, they tend to become less effective for detecting smaller clones because of a dilutional effect by other clones harboring no or different mutations (Fig. 4A). Overall, across all five punch sizes, USMs were detected in 54% of the SE, which was significantly higher than the 21% of the NE (P < 0.001, Fisher’s exact test). Between different punch sizes, 2-mm punches were found to have the highest positive rate of 64% and with the most significant difference between SE and NE samples (P < 0.0001; Fig. 4B). Thus, only 2-mm punches were collected in the 90-sample validation cohort and the extended cohort from cSCC patients. In the validation cohort, similarly, we found that the SE samples had higher numbers of USMs and the positive rate of USMs (69%) was similar to the discovery cohort (64%).
Fig. 4. Optimization of punch size for detecting USMs.
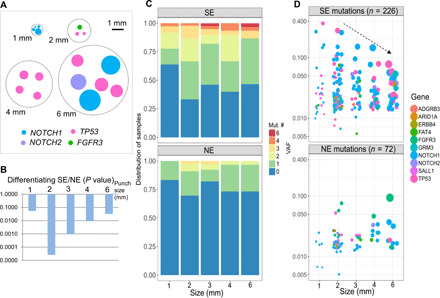
(A) A representative figure showing one representative punch of each collection size. We selected the sample with the highest number of mutations under each size for easy illustration. Every mutation is plotted as a dot with its size calculated to match the clonal area harboring the mutation. One punch size, 3 mm, was not shown, as it was obtained by cutting a 6-mm punch into quarters. (B) In the discovery cohort, 2 mm was found to be the most efficient size in differentiating CRCA from SE and NE skin samples by P value. (C) Distribution of numbers of USMs per sample at each punch size, after combining both the discovery and validation cohorts. (D) VAF of USM detected in different punch sizes. The size of the dot indicates the approximate relative area of cells containing the mutation. In SE samples, VAFs of USMs detected from larger punches are associated with smaller variations.
When combining the discovery and validation cohorts, the SE samples had the highest positive rate of 67% for USMs in 2-mm samples and were significantly higher than NE samples (P < 0.001), followed by 60% in 4 mm (P < 0.05) and 54% in 3 mm (P < 0.05). The USM positive rates were relatively lower in the largest punch size of 6 mm (53%) and the smallest punch size of 1 mm (36%). In all NE samples, positive USM rates ranged from 17 to 30% (Fig. 4C). Moreover, the punch size also affected the detected VAFs of the mutations. Specifically, in SE samples, larger punches were associated with smaller VAFs. The VAFs’ SD was the highest in 1-mm punches (8.9%) and decreased with punch size: 2 mm (4.3%), 3 mm (2.8%), 4 mm (2.6%), and 6 mm (1.7%). This trend, between VAF range and punch size, was not present in NE samples (Fig. 4D). These results suggested that the most effective punch size in detecting USMs under the current sequencing condition was 2 mm.
Mutation nucleotide contexts enriched with UV exposure
We next assessed the enrichment of different mutation nucleotide contexts in SE skin. The mutation nucleotide contexts were defined by each SNV’s trinucleotide and DNV’s dinucleotide contexts. A total of 83 contexts were identified from current mutations, including 13 contexts matching to previously described USMs (4). None of the remaining 70 NUSM contexts were enriched in SE or NE samples (Fig. 5A). The 13 previously defined USM contexts were not equally enriched in SE samples. After multiple test correction, only 5 of the 13 contexts were significantly enriched in SE samples (FDR P < 0.05), including the dinucleotide CC>TT context, which was exclusively found in SE samples (Fig. 5B). The most significant mutation context enriched in SE samples was T[C>T]C (FDR P = 0.00013), which was in consonance with the previously defined “mutational signature #7” in skin cancers (26). The remaining eight UV signature contexts were not significantly enriched in SE samples. G[C>T]C, which was the most abundant context by total number of mutations, appeared to be equally presented in SE and NE skin samples and, therefore, not associated with sun exposure.
Fig. 5. Mutational contexts associated with UV exposure.
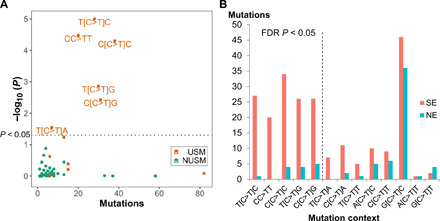
(A) Each dot represents a specific mutation context of SNVs and DNVs. x axis: The total numbers of mutations of each context. y axis: P value of the context for differentiating SE and NE skin, shown as log10(P). The dotted line indicate P < 0.05 (the above area). None of the NUSM contexts was significant. (B) Further refinement of USM contexts by depicting the numbers of mutations in SE and NE skin for all current USM contexts. Mutation contexts are ordered by the P value of SE versus NE in an increasing order from left to right. Multiple test correction was implemented using the FDR method. The dotted line indicates FDR P < 0.05 (the left side).
CMs are correlated with cSCC burden
To define the clinical significance of CMs and investigate the potential association with skin cancer risk, we sequenced an extended cohort of 20 samples (16 SE normal skin samples and 4 cSCC samples; table S2B) from eight patients with cSCC using a 59.5-kb customized panel as described above. Four individuals (including eight normal skin samples and two cSCC samples from face, scalp, and arm) had a low burden of skin cancer with only a single diagnosis of cSCC and few AKs (low-cSCC). Four individuals (including eight normal skin samples and two cSCC samples from face, hand, and lower leg) had a high burden of skin cancer with severe UV damage, multiple prior cSCC (range, 3 to 10), and many AKs (high-cSCC). Low-cSCC and high-cSCC patients were matched for age (mean age, 76.5 and 79.3, respectively). Normal skin samples were all SE and obtained a linear distance of either 1 or 6 mm from the clear surgical margin of the excised cSCC, allowing for analysis of CMs arising in skin subjected to carcinogenic UV radiation. Samples from three of four low-cSCC patients were on the head (one of four on SE extremity), and samples from two of four high-cSCC patients were on the head (two of four on SE extremity). None of the patients had received prior treatment with radiation or chemotherapy. Visible AKs were not present in normal skin samples. A total of 535 somatic mutations were identified (table S8), with a median VAF of 1.2%. Only 15 mutations had VAF greater than 10%, most of which (10 of 15) were from the cSCC tumor samples (Fig. 6A). The median numbers of mutations per sample in each group were 22 and 17.5 for the high- and low-cSCC normal skin samples (marginally significant, P = 0.078, Wilcoxon) and 41.5 for the cSCC samples. The overall mutation rates in normal skin were 0.45 and 0.29 mutations per million bases, in high- and low-cSCC patients, respectively. The latter was comparable to the rate of SE normal skin of noncancer patients in the primary cohort (0.31 mutations per MB) despite the technical differences between the two cohorts such as sequencing depth, targeted regions, and punch sizes.
Fig. 6. CMs are correlated with cSCC burden.
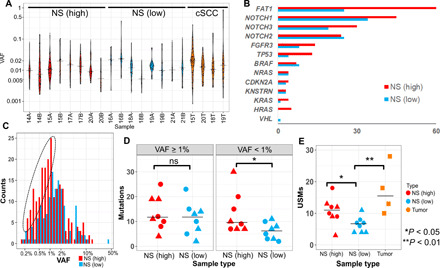
(A) Violin plots depicting the overall distribution of somatic mutations in each sample, ordered by sample type. (B) Mutation numbers by genes in the normal skin. NS (high), normal skin from high-cSCC patients; NS (low), normal skin from low-cSCC patients. (C) High-cSCC patients are associated with increased low-VAF (<1%) mutations. Histogram depicting the distribution of VAFs of the detected mutations in normal skin separated by cSCC burden. The dotted oval highlights the increased low-VAF mutations in the normal skin of high-cSCC patients compared with low-cSCC patients. (D) Number of mutations per sample in normal skin, separated by high (≥1%) and low (<1%) VAFs. ns, not significant. (E) Number of USMs per sample in high- and low-cSCC normal skin (NS) and cSCC tumors. The shape indicates the two normal skin samples from each patient, taken either 1 mm (circle) or 6 mm (triangle) from the surgical margin.
The frequently mutated genes in normal skin (more than two mutations per gene on average) included FAT1, NOTCH1, NOTCH2, NOTCH3, FGFR3, and TP53 (Fig. 6B). Two of the genes were mutated at least twice as frequently in the normal skin of high-cSCC patients as that of low-cSCC patients: TP53 (ratio, 3.25) and FAT1 (ratio, 2.4). In addition, two less frequently mutated genes, KRAS and HRAS, were almost exclusively mutated in high-cSCC patients (9 of 10). None of these differences reached statistical significance after multiple test correction, indicating that larger cohorts will be needed to further explore these potential associations.
Although the normal skin of high-cSCC patients contains more mutations per sample, unexpectedly, these mutations were associated with significantly lower VAFs (median, 1.0%) than the normal skin of low-cSCC patients (median, 1.3%; P = 0.011, Wilcoxon). We found that this overall reduction in VAF resulted from a higher number of low-frequency mutations in high-cSCC patients (Fig. 6C). For mutations with VAF greater than 1%, the mutations were equally present in high- and low-cSCC patients. However, for low-VAF mutations (defined as <1%), the numbers of mutations per sample were significantly higher in high-cSCC (median, 9.5) than low-cSCC patients (median, 6; P = 0.032, Wilcoxon; Fig. 6D).
We next further refined the analysis by focusing on USMs. There were a total of 206 USMs, including 8 CC>TT DNVs. We observed a significantly greater number of USMs in the high-cSCC normal skin samples (median, 11) than the low-cSCC ones (median, 6.5; FDR P = 0.015) (Fig. 6E). The tumor samples were found to harbor even higher numbers of USMs (median, 15.5). The CRCA values, as defined in the primary cohort, were significantly higher in the tumor than the normal skin samples (FDR P = 0.03) in the extended cohort. The normal skin samples from high-cSCC patients had slightly higher CRCAs (median, 0.37) than low-cSCC patients (median, 0.31), but the difference was not statistically significant (FDR P = 0.16). The CRCA is essentially the sum of VAF values for all detected mutations, normalized for biopsy size. The lack of a significant difference between CRCA values for high-cSCC and low-cSCC skin samples is likely due to the observation that the increased mutations present in high-cSCC samples were enriched for low-frequency mutations. For example, if we calculate CRCA by only including low-frequency mutations (VAF < 1%), then the CRCA values would be significantly higher in high-cSCC than low-cSCC samples (FDR P = 0.04; table S9). In addition, we found no significant difference in overall mutation burden, VAF, USMs, or CRCA between normal skin samples collected at 1 mm versus 6 mm from the surgical margin. Last, almost all mutations (>99%) were present only in one of two skin samples from the same patient. The absence of shared recurrent mutations across different samples from the same individual indicates that the identified mutations arose independently.
DISCUSSION
Most cancers are initiated by accumulation of somatic mutations (27, 28). However, early mutations in normal tissues are difficult to detect because of the low abundance and random patterns. Several recent studies demonstrated the feasibility of detecting CMs using high-throughput sequencing in various tissue types (12, 13, 29). However, the contribution of these CMs to cancer remains unclear in several ways: how they are generated, what types of mutations are generated by which exogenous and endogenous carcinogens, how the CMs are accumulated and selected by the host microenvironment and interclonal competition (25), and which mutations contribute or lead to the development of cancer. All types of tissues are under the influence of multiple intrinsic and extrinsic factors that vary greatly by individual’s lifestyle and environment. Therefore, studying the CMs generated by one specific carcinogen requires comparative studies of matched sample types.
In the SE normal skin, although previous studies (12, 13, 16, 30–33) provided a rich body of evidence about the abundance of CMs, one fundamental question that has remained unanswered was which of these mutations are caused by UV, rather than aging or other environmental and endogenous factors. This question is also clinically important since the majority of skin cancers are associated with UV exposure. The current study was designed to tackle this question by sequencing a large number of individual-matched SE and NE skin samples; furthermore, all our samples were obtained from the same body sites (SE skin from the left dorsal forearm and NE skin from the left medial buttock area), which helped minimize the potential variation between different body areas. This unique design allowed us to precisely characterize CMs introduced by UV while minimizing the effects of other environmental or endogenous factors. The current study yielded the following previously unknown findings: (i) the existence of mutation-exempt genomic regions that are never mutated in normal NE skin; (ii) the highly region-specific pattern of UV mutation enrichment in TP53 and NOTCH1; (iii) the known USMs might be subclassified by different nucleotide contexts, which showed differential association with UV exposure; (iv) the punch size used in clinic can have a marked effect on the detection of CMs. In our study, we found that 2-mm punch was the optimal size for capturing CMs; and (v) in our extended cohort, the low-frequency CMs from smaller clones, but not the ones from the expanded clones, were associated with cSCC burden.
Although mutations frequently occur across most of the sequenced regions in NE skin, presumably because of metabolism and aging related factors, no detectable mutations were found in the current mutation-exempt regions. It is unclear whether the absence of mutations in these genomic regions is caused by an active protection or a passive selection mechanism involving altered clone fitness. The mutation-exempt property of these regions appears to be altered upon exposure to UV radiation, and these regions become highly mutable. Theoretically, the mutation-exempt property of these genomic regions suggests that either mutations are less likely to occur in these regions because of the topological properties of genome organization (34) or early mutations (without other driver mutations or the cellular metabolic changes triggered by UV exposer) in these regions lead to cellular demise or mutations in these regions are under strong negative selection pressure by the immune system (35) in lieu of local chronic tissue effects of chronic sun exposure. There are very few published studies on mutations in NE normal skin. In recent studies using RNA-seq (12) or deep-WES (whole-exome sequencing) (30), small numbers of mutations were reported in the mutation-exempt region. In these studies, almost all identified mutations were from SE skin, with the only exception of one single mutation reported in a “sun-protected area” (the right lateral chest, rather than the current NE buttock area) from an iatrogenically immunosuppressed patient with a history of multiple skin cancers (30). Unfortunately, none of the abovementioned studies were powered and designed to establish differences in the patterns of mutations between SE and NE areas (12, 30). However, Muradova et al. (30) in two immunosuppressed individuals on five large (0.8 cm by 1.8 cm) epidermal samples at 3000× sequencing depth identified mutation pattern differences in line with our findings. Future studies are warranted to explore the mechanism of mutation-exempt regions and how this mechanism is abrogated by UV radiation.
We identified six mutations that were almost exclusively mutated in SE skin. All six mutations had been previously reported in human cSCCs in the cBioPortal (24). Among these mutations, TP53 R248W and G245D were highly recurrent with hundreds of occurrences reported in COSMIC (36), indicating that the presence of these mutations may be representative of an early phase of carcinogenesis. One of these six SE-enriched mutations, NOTCH1 E424K, was present in 11 of 225 SE samples and was also associated with an average of fivefold increase of VAFs. The elevated VAFs could be the result from two different mechanisms: single-clone expansion or enrichment of multiple clones. For the latter, after being normalized by the median VAFs of all mutations and rounded up, these 11 SE samples would contain a total of 56 normal-size clones. If all clones developed randomly, then the chance of these 56 clones only occurring in 11 of 225 SE samples would be extremely small (less than 5.8 × 10−56). Here, we assume that all clones occur randomly and therefore cannot exclude the possibility that these clones might be driven by any systematic factor, such as predisposition by certain genetic variants in some patients. Furthermore, the lower presence of NOTCH1 E424K in SCC than in the SE normal skin suggests that early clonal growth is not the only determinant of malignant potential. Therefore, this finding argues that some clones may carry greater and some lower weight in risk of later SCC formation. Future studies using human and mouse models are warranted to determine the contribution of individual mutations to skin cancer risk.
Consistent with the current finding that UV exposure results in higher USM burden and the known knowledge that UV exposure directly correlates with the risk of cSCC (37), the results of our extended cohort of cSCC patients provided direct evidence that elevated USM burdens are associated with increased burden of cSCC. Presumably, this burden correlates with risk of future cSCC as well. Unexpectedly, we further discovered that most mutational differences between normal skin of high- and low-cSCC patients derived from low-frequency clones (VAF < 1%) but not the “expanded” clones (VAF ≥ 1%). It remains unclear why such difference was not seen in the expanded clones. It has been previously reported that the immune system may suppress expansion of CMs, with immunosurveillance selectively targeting larger (expanded) clones (35). The low-frequency clones, in contrast, may be less actively monitored by the immune system and more truthfully represent the level of ongoing mutational or genomic instability in patients with multiple cSCCs. Additional investigation is needed to verify this hypothesis. In any case, the total USM burden in SE skin of patients with cSCC may be a more accurate measure of skin cancer risk than VAF or clonal area.
Our approach was directed by future clinical utilities, focusing on quantitative measurement of UV-induced DNA damage for sun protection, and cSCC patient risk stratification. These results demonstrate the feasibility of using a small panel of genomic regions (5.5 kb) to quantitatively measure UV-induced CMs. We established CRCA as a combined measure of mutation burden and relative abundance, which was strongly correlated with sun-exposure status, but not with cSCC burden in SE skin. In the current study, we found the most effective punch size for capturing CMs was 2 mm, which is also clinically favorable, as it leaves relatively smaller scars due to the small diameter punch. In future, a noninvasive skin sampling method may provide even wider accessibility to epidermal sampling. Furthermore, the optimal punch size is likely dependent on clone size distribution, which may vary between different body areas. The current SE skin samples were all collected from left dorsal forearm. In the future, precisely determining the consistency of mutational profiles across multiple areas of the body warrants further investigation. These future human studies will also include noninvasive sampling (30) to help clinical translation of CM detection. In addition, the efficiency of this panel is related to the performance of sequencing method and mutation calling algorithm, which will likely be improved with adoption of more sensitive future methods focusing on the genomic hotspots that are sensitive to UV exposure.
The current study focused on the most frequently mutated regions in SE skin samples defined by the mutations in a previous study (13). However, we note that many of these regions are mutated in both SE and NE skin samples, indicating that many mutations in these regions were unrelated to UV exposure. Only 6 of 55 original regions were found to harbor significantly enriched mutations in SE samples. Future studies, including much larger targeted regions, are needed to systematically identify UV-sensitive genomic regions. The skin samples were collected at the same time; therefore, they do not provide longitudinal information about clone initiation and progression. While our analyses of the extended cohort indicate that the burdens of CMs in normal skin are correlated with cancer risk in cSCC patients, this initial study was not powered to fully elucidate all the factors that contribute to CM burden and skin cancer development. Additional studies, with larger cohorts of patients, will allow for clarification of the contribution of cumulative sun exposure, patient age, anatomic location, genetic predisposition, and prior treatment or prevention strategies on the development and progression of skin cancer over time.
MATERIALS AND METHODS
Experimental design
The current study mainly consists of two cohorts of normal human skin samples: (i) A primary cohort of individual-matched SE and NE skin samples from mostly non–skin cancer patients to compare the profiles of CMs in SE and NE normal skin areas. A total of 450 epidermal samples were collected from 13 postmortem donors in two batches: a discovery cohort of 360 samples collected using five different punch sizes for comparing the efficiency of different punch sizes for detecting CMs and a validation cohort of 90 samples collected using only the best punch size as determined in the discovery cohort. An additional 14 dermal samples were collected for control purposes. (ii) An extended cohort of normal skin samples obtained from patients with high or low burden of cSCCs, to identify potential patterns of CMs in normal skin associated with the cSCC burden. A small number of cSCC samples were also collected for comparing the tumor mutational profiles with the CMs in the normal skin.
Sample collection
A total of 464 normal human skin samples were collected from 13 Caucasian postmortem donors over the age of 55 years using Roswell Park’s Rapid Tissue Acquisition Program under a Roswell Park–approved IRB (Institutional Review Board) protocol within 24 hours of death. The SE skin pieces (at least 5 cm by 7 cm) were first taken from left dorsal forearm and similar sized SE (NE) skin pieces were obtained from the left medial buttock area. Small epidermal punch samples were randomly from these large (>35 cm2) skin pieces. Exclusion criteria included any visible skin abnormalities in the tissue areas. Eligible donors were identified, and clinically normal appearing skin was harvested. Skin samples were kept in tissue preservation medium, Belzer UW cold storage solution (Bridge to Life, USA) at 4°C until processed. All samples that could be processed within 36 hours or less after death were included in the study. The mean age of the donors was 72.3 years (SD, ±8.2 years; range, 60 to 80 years). The male-to-female gender ratio was 7:6, and 12 of 13 donors had no history of skin cancer.
The adipose tissue was removed from each human skin sample using sterile scissors. The samples were cut into strips wide enough to harvest 6-mm punches. The epidermis was separated from the dermis by placing the strips in tubes containing 10 ml of Dispase II (5 U/ml; Stem Cell Technologies, USA) and incubated at 4°C overnight and at 37°C for 2 to 3 hours. After Dispase digestion, the specimens were placed in a petri dish containing a small amount of 1× DPBS (Dulbecco’s Phosphate Buffered Saline) (Corning, USA), and using sterile tweezers, the epidermis was carefully removed from the dermis. Using disposable biopsy punches, 1-, 2-, 3-, 4-, and 6-mm-diameter epidermal pieces were taken from the epidermal sheets and punched epidermal pieces were placed into sterile 1.5-ml vials. In addition to the epidermal punches, large bulk pieces of dermis were also removed from the skin samples using a disposable #15 blade and placed into a sterile 1.5-ml vial for use as a germline control.
For the extended cohort of the study, 20 human skin samples were obtained in a deidentified manner from eight undergoing surgeries for cSCC. The mean age of the donors was 77.9 years (SD, ±12.3 years; range, 54 to 92 years). The male-to-female gender ratio was 1:1. The study was granted exemption by the Yale University Human Investigation Committee (protocol 1509016421). All individuals had biopsy-confirmed cSCC that was completely excised by Mohs micrographic surgery with intraoperative histologic verification of clear surgical margins. Immediately following excision of cSCC, adjacent normal skin was excised to facilitate surgical repair, and samples for sequencing were immediately harvested. From each individual, two skin samples at a fixed linear distance from the cSCC were obtained from the adjacent, SE normal skin. One sample was obtained at a distance of 1 mm from the cSCC surgical margin, and one sample was obtained at a distance of 6 mm from the surgical margin. From four patients, a tumor sample from grossly visible cSCC was also obtained at the time of surgery. All samples were obtained with a 2-mm punch biopsy to a depth of approximately 1 mm, including epidermis and superficial dermis.
DNA isolation
DNA samples from the primary cohort were extracted using PureLink Genomic DNA Mini Kit (Invitrogen, USA). Epidermal samples were digested using proteinase K at 55°C heating block overnight following the manufacturer’s recommendations. For the extended cohort of samples, skin biopsies were similarly digested using proteinase K and DNA was purified with phenol-chloroform extraction and ethanol precipitation. DNA was eluted with 28 μl of Molecular Biology Grade Water (Corning, USA) for 1- and 2-mm punches or 36 μl of Molecular Biology Grade Water for 3-, 4-, and 6-mm punches. The isolated genomic DNA was stored at −20°C, and the DNA concentration of each extraction was measured using a Qubit fluorometer or a Quant-iT PicoGreen kit (Invitrogen, USA).
Ultradeep targeted sequencing
The sequencing libraries were generated using the TruSeq Custom Amplicon Kit (Illumina, USA) using 10 to 50 ng of genomic DNA (gDNA). Amplicons of ~150 bp (primary cohort) or ~250 bp (extended cohort) in length were designed using Illumina DesignStudio software. Custom oligo capture probes that flank the regions of interest were hybridized to the gDNA. A combined extension/ligation reaction completed the region of interest between these flanking custom oligo probes. Polymerase chain reaction (PCR) was then performed to add indices and sequencing adapters. The amplified final libraries were cleaned up using AMPure XP beads (Beckman Coulter). Purified libraries were run on a TapeStation DNA 1000 ScreenTape chip to verify desired size distribution, quantified by KAPA quantitative PCR (KAPA Biosystems), and pooled equal molar in a final concentration of 2 nM. Pooled libraries were loaded on an Illumina HiSeq Rapid Mode V2 flow cell following standard protocols for 2× 100 cycle sequencing (primary cohort) or Illumina NextSeq for 2× 150 cycle sequencing (extended cohort).
Bioinformatics analysis
High-quality paired-end reads passing Illumina RTA filter were initially processed against the National Center for Biotechnology Information (NCBI) human reference genome (GRCh37) using public available bioinformatics tools (38, 39) and Picard (http://picard.sourceforge.net/). The coverage quality control required at least 80% of the targeted region covered by a minimum of 1000× coverage. Putative mutations, including SNVs and small insertions/deletions (indels), were initially identified by running variation detection module of Strelka (40) on each SE or NE epidermis sample paired with the matched dermal sample. From the detected SNVs, DNVs or composite SNVs (CSNV) were recognized by running Multi-Nucleotide Variant Annotation Corrector (MAC) (20) on the original sequences. The putative mutations detected from all samples were consolidated into a list of unique mutations. Every unique mutation was revisited in all samples to calculate the numbers of mutant/wild-type reads, as well as VAF in each sample as previously described (14).
To distinguish mutations from background errors, we modeled each mutation’s background error rate distributions using VAFs from all control (dermal) samples. For each mutation, we started by fitting a Weibull distribution to VAFs from all control samples following a previously published method (41); then, every SE or NE epidermal sample’s VAF was compared to the fitted distribution. A positive sample was defined if a mutation’s VAF in that sample was significantly above background (P < 0.05, after Bonferroni correction). In the extended cohort where the control samples were not available, we adapted a dynamic control strategy, based on the assumption that any somatic mutation cannot be recurrent in more than 10% of all samples at the same site. In the previous primary cohort, all recurrent mutations were within 5% of all samples. For each potential mutation, we first cluster the VAFs of the mutation in all samples. Subsequently starting from the cluster with lowest VAF, we transferred all samples of each cluster to the control cohort until at least 90% of all samples are in the control cohort. After mutation calling, all identified mutations including SNVs, DNVs, CSNVs, and indels were annotated using a customized program with NCBI RefSeq database.
CRCA, defined as the overall percentage of biopsied skin area covered by USMs in a patient skin punch, was calculated as follows
where n is the total number of punches collected in the patient, ri is the size (radius) of each punch, mi is the number of mutations in punch i, and VAFj is the variant allele fraction of a specific mutation j. Here, the calculation of CRCA was based on the assumption that all mutations occur in one chromosome of regular diploid genomic regions. In addition, although we did not consider the situation when multiple mutations occur in the same cell, we did identify mutations that occur on the same reads and combined them into one mutation using MAC (20).
Statistical analysis
The overall mutation numbers and VAFs between two groups, including SE and NE in the primary cohort and the high- and low-cSCC burden in the extended cohort, were evaluated using a Wilcoxon test. Group-specific markers, including mutations, genes, regions, and signatures, were identified using a Fisher’s exact test, where the two variables in the contingency table were the samples’ sun-exposure status (SE versus NE, in cohort #1) or cSCC burden (high versus low, in cohort #2) and mutational status. Multiple testing correction was implemented using the FDR approach as indicated.
Supplementary Material
Acknowledgments
We thank the excellent technical help provided by P. Pera, assistance with design of the 13-gene custom sequencing panel for the extended cohort provided by Y. Zhang (Yale University School of Medicine), and assistance with library generation and sequencing of the extended cohort provided by M. Zhong (Yale Stem Cell Center). We dedicate our work to Dr. Oscar Colegio, who passed away suddenly on 14 June 2020. Oscar was not just a colleague and co-author; he was a passionate and exceptionally empathetic physician, a brilliant researcher, and a thoughtful friend. Oscar was among the leading transplant dermatologists in the world. He had a captivating personality and a unique ability to connect ideas and people. This manuscript is a testament to Oscar’s ability to bring people together. As we continue our collaboration, his insight, mentorship, wit, and hard work will be greatly missed. Funding: This work was mainly supported by the Roswell Park Alliance Foundation. L.W. and S.L. were supported, in part, by NIH grant U24CA232979. The used Genomics and Bioinformatics Shared Resources and Rapid Tissue Acquisition Program at Roswell Park Comprehensive Cancer Center was supported by NCI grant P30CA016056. L.W. and J.X. were supported, in part, by a travel grant from NIH 5U24ES026465. S.R.C. was supported by a Career Development Award from the Dermatology Foundation. Ethics statement: All specimens in the primary cohort were collected from postmortem donors collected in collaboration with Buffalo’s local organ procurement organization (ConnectLife, formerly Unyts), the Roswell Park’s Rapid Tissue Acquisition Program under a Roswell Park–approved IRB protocol. Specimens in the expanded cohort were collected from discarded surgical tissue under a Yale University Human Investigation Committee–approved protocol. Author contributions: Primary cohort: L.W., W.J.H., B.A.F., and G.P. conceptualized and designed the study. M.E.F., W.J.H., B.A.F., and G.P. collected and processed the samples. L.W., M.E.F., J.G., N.D.H., C.Z., Z.H., Q.H., F.Z., J.X., J.Z., S.L., E.R., E.G., O.R.C., M.B., J.X., W.J.H., B.A.F., and G.P. analyzed the data. Extended cohort: S.R.C. and H.L. conceptualized and designed the study and collected the samples. L.W., S.R.C., Q.H., and G.P. analyzed the data. L.W., S.R.C., W.J.H., B.A.F., and G.P. wrote and revised the manuscripts with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The raw sequences of all ultradeep sequencing data for the primary cohort can be provided by Roswell Park Comprehensive Cancer Center pending scientific review and a completed material transfer agreement. Requests for the raw sequences should be submitted to Lei.Wei@RoswellPark.org and Gyorgy.Paragh@RoswellPark.org. The raw sequences of the ultradeep sequencing data for the extended cohort can be requested from Sean.Christensen@Yale.edu. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd7703/DC1
REFERENCES AND NOTES
- 1.Rogers H. W., Weinstock M. A., Feldman S. R., Coldiron B. M., Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 151, 1081–1086 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Koh H. K., Geller A. C., Miller D. R., Grossbart T. A., Lew R. A., Prevention and early detection strategies for melanoma and skin cancer. Current status. Arch. Dermatol. 132, 436–443 (1996). [PubMed] [Google Scholar]
- 3.Marteijn J. A., Lans H., Vermeulen W., Hoeijmakers J. H., Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465–481 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Brash D. E., UV signature mutations. Photochem. Photobiol. 91, 15–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wikonkal N. M., Brash D. E., Ultraviolet radiation induced signature mutations in photocarcinogenesis. J. Investig. Dermatol. Symp. Proc. 4, 6–10 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Marks R., Rennie G., Selwood T. S., Malignant transformation of solar keratoses to squamous cell carcinoma. Lancet 1, 795–797 (1988). [DOI] [PubMed] [Google Scholar]
- 7.Siegel J. A., Korgavkar K., Weinstock M. A., Current perspective on actinic keratosis: A review. Br. J. Dermatol. 177, 350–358 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Ling G., Persson A., Berne B., Uhlén M., Lundeberg J., Ponten F., Persistent p53 mutations in single cells from normal human skin. Am. J. Pathol. 159, 1247–1253 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brash D. E., Cancer. Preprocancer. Science 348, 867–868 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Urano Y., Asano T., Yoshimoto K., Iwahana H., Kubo Y., Kato S., Sasaki S., Takeuchi N., Uchida N., Nakanishi H., Arase S., Itakura M., Frequent p53 accumulation in the chronically sun-exposed epidermis and clonal expansion of p53 mutant cells in the epidermis adjacent to basal cell carcinoma. J. Invest. Dermatol. 104, 928–932 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Williams C., Pontén F., Ahmadian A., Ren Z. P., Ling G., Rollman O., Ljung A., Jaspers N. G., Uhlen M., Lundeberg J., Pontén J., Clones of normal keratinocytes and a variety of simultaneously present epidermal neoplastic lesions contain a multitude of p53 gene mutations in a xeroderma pigmentosum patient. Cancer Res. 58, 2449–2455 (1998). [PubMed] [Google Scholar]
- 12.Yizhak K., Aguet F., Kim J., Hess J. M., Kubler K., Grimsby J., Frazer R., Zhang H., Haradhvala N. J., Rosebrock D., Livitz D., Li X., Arich-Landkof E., Shoresh N., Stewart C., Segrè A. V., Branton P. A., Polak P., Ardlie K. G., Getz G., RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364, eaaw0726 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D. C., Fullam A., Alexandrov L. B., Tubio J. M., Stebbings L., Menzies A., Widaa S., Stratton M. R., Jones P. H., Campbell P. J., Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei L., Hussein A. A., Ma Y., Azabdaftari G., Ahmed Y., Wong L. P., Hu Q., Luo W., Cranwell V. N., Bunch B. L., Kozlowski J. D., Singh P. K., Glenn S. T., Smith G., Johnson C. S., Liu S., Guru K. A., Accurate quantification of residual cancer cells in pelvic washing reveals association with cancer recurrence following robot-assisted radical cystectomy. J. Urol. 201, 1105–1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei L., Papanicolau-Sengos A., Liu S., Wang J., Conroy J. M., Glenn S. T., Brese E., Hu Q., Miles K. M., Burgher B., Qin M., Head K., Omilian A. R., Bshara W., Krolewski J., Trump D. L., Johnson C. S., Morrison C. D., Pitfalls of improperly procured adjacent non-neoplastic tissue for somatic mutation analysis using next-generation sequencing. BMC Med. Genomics 9, 64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang J., Fewings E., Chang D., Zeng H., Liu S., Jorapur A., Belote R. L., McNeal A. S., Yeh I., Arron S. T., Judson-Torres R. L., Bastian B. C., Shain A. H., The genomic landscapes of individual melanocytes from human skin. Nature 586, 600–605 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huss W. J., Hu Q., Glenn S. T., Gangavarapu K. J., Wang J., Luce J. D., Quinn P. K., Brese E. A., Zhan F., Conroy J. M., Paragh G., Foster B. A., Morrison C. D., Liu S., Wei L., Comparison of sureselect and nextera exome capture performance in single-cell sequencing. Hum. Hered. 83, 153–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamble R. G., Asdigian N. L., Aalborg J., Gonzalez V., Box N. F., Huff L. S., Barón A. E., Morelli J. G., Mokrohisky S. T., Crane L. A., Dellavalle R. P., Sun damage in ultraviolet photographs correlates with phenotypic melanoma risk factors in 12-year-old children. J. Am. Acad. Dermatol. 67, 587–597 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creidi P., Vienne M. P., Ochonisky S., Lauze C., Turlier V., Lagarde J.-M., Dupuy P., Profilometric evaluation of photodamage after topical retinaldehyde and retinoic acid treatment. J. Am. Acad. Dermatol. 39, 960–965 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Wei L., Liu L. T., Conroy J. R., Hu Q., Conroy J. M., Morrison C. D., Johnson C. S., Wang J., Liu S., MAC: Identifying and correcting annotation for multi-nucleotide variations. BMC Genomics 16, 569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luca V. C., Jude K. M., Pierce N. W., Nachury M. V., Fischer S., Garcia K. C., Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science 347, 847–853 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanpain C., Lowry W. E., Pasolli H. A., Fuchs E., Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 20, 3022–3035 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefort K., Dotto G. P., Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin. Cancer Biol. 14, 374–386 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N., Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colom B., Alcolea M. P., Piedrafita G., Hall M. W. J., Wabik A., Dentro S. C., Fowler J. C., Herms A., King C., Ong S. H., Sood R. K., Gerstung M., Martincorena I., Hall B. A., Jones P. H., Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat. Genet. 52, 604–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Aparicio S. A., Behjati S., Biankin A. V., Bignell G. R., Bolli N., Borg A., Børresen-Dale A.-L., Boyault S., Burkhardt B., Butler A. P., Caldas C., Davies H. R., Desmedt C., Eils R., Eyfjörd J. E., Foekens J. A., Greaves M., Hosoda F., Hutter B., Ilicic T., Imbeaud S., Imielinski M., Jäger N., Jones D. T., Jones D., Knappskog S., Kool M., Lakhani S. R., López-Otín C., Martin S., Munshi N. C., Nakamura H., Northcott P. A., Pajic M., Papaemmanuil E., Paradiso A., Pearson J. V., Puente X. S., Raine K., Ramakrishna M., Richardson A. L., Richter J., Rosenstiel P., Schlesner M., Schumacher T. N., Span P. N., Teague J. W., Totoki Y., Tutt A. N., Valdés-Mas R., van Buuren M. M., van ‘t Veer L., Vincent-Salomon A., Waddell N., Yates L. R.; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain, Zucman-Rossi J., Futreal P. A., McDermott U., Lichter P., Meyerson M., Grimmond S. M., Siebert R., Campo E., Shibata T., Pfister S. M., Campbell P. J., Stratton M. R., Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu S., Powers S., Zhu W., Hannun Y. A., Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomasetti C., Vogelstein B., Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martincorena I., Fowler J. C., Wabik A., Lawson A. R. J., Abascal F., Hall M. W. J., Cagan A., Murai K., Mahbubani K., Stratton M. R., Fitzgerald R. C., Handford P. A., Campbell P. J., Saeb-Parsy K., Jones P. H., Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muradova E., Patel N., Sell B., Bittencourt B. B., Ojeda S. S., Adelmann C. H., Cen L., Cheng C.-H., Shen J., Davis C. M., Ehli E. A., Newberg J. Y., Cherpelis B., Black M. A., Mann M. B., Mitragotri S., Tsai K. Y., Noninvasive assessment of epidermal genomic markers of UV exposure in skin. J. Invest. Dermatol. 10.1016/j.jid.2020.05.093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lynch M. D., Lynch C. N. S., Craythorne E., Liakath-Ali K., Mallipeddi R., Barker J. N., Watt F. M., Spatial constraints govern competition of mutant clones in human epidermis. Nat. Commun. 8, 1119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abyzov A., Tomasini L., Zhou B., Vasmatzis N., Coppola G., Amenduni M., Pattni R., Wilson M., Gerstein M., Weissman S., Urban A. E., Vaccarino F. M., One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin. Genome Res. 27, 512–523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saini N., Roberts S. A., Klimczak L. J., Chan K., Grimm S. A., Dai S., Fargo D. C., Boyer J. C., Kaufmann W. K., Taylor J. A., Lee E., Cortes-Ciriano I., Park P. J., Schurman S. H., Malc E. P., Mieczkowski P. A., Gordenin D. A., The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLOS Genet. 12, e1006385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akdemir K. C., Le V. T., Kim J. M., Killcoyne S., King D. A., Lin Y.-P., Tian Y., Inoue A., Amin S. B., Robinson F. S., Nimmakayalu M., Herrera R. E., Lynn E. J., Chan K., Seth S., Klimczak L. J., Gerstung M., Gordenin D. A., O’Brien J., Li L., Deribe Y. L., Verhaak R. G., Campbell P. J., Fitzgerald R., Morrison A. J., Dixon J. R., Andrew Futreal P., Somatic mutation distributions in cancer genomes vary with three-dimensional chromatin structure. Nat. Genet. 52, 1178–1188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gejman R. S., Chang A. Y., Jones H. F., DiKun K., Hakimi A. A., Schietinger A., Scheinberg D. A., Rejection of immunogenic tumor clones is limited by clonal fraction. eLife 7, e41090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forbes S. A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J. W., Futreal P. A., Stratton M. R., The catalogue of somatic mutations in cancer (COSMIC). Curr. Protoc. Hum. Genet. Chapter 10, Unit 10.11.1–Unit10.11.26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson T. M., Rowe D. E., Nelson B. R., Swanson N. A., Squamous cell carcinoma of the skin (excluding lip and oral mucosa). J. Am. Acad. Dermatol. 26, 467–484 (1992). [DOI] [PubMed] [Google Scholar]
- 38.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Q., Hu Q., Yao S., Kwan M. L., Roh J. M., Zhao H., Ambrosone C. B., Kushi L. H., Liu S., Zhu Q., SeqSQC: A Bioconductor package for evaluating the sample quality of next-generation sequencing data. Genomics Proteomics Bioinformatics 17, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saunders C. T., Wong W. S., Swamy S., Becq J., Murray L. J., Cheetham R. K., Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Newman A. M., Lovejoy A. F., Klass D. M., Kurtz D. M., Chabon J. J., Scherer F., Stehr H., Liu C. L., Bratman S. V., Say C., Zhou L., Carter J. N., West R. B., Sledge G. W. Jr., Shrager J. B., Loo B. W. Jr., Neal J. W., Wakelee H. A., Diehn M., Alizadeh A. A., Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd7703/DC1