

Abstract
Cancer vaccines targeting patient-specific neoantigens have emerged as a promising strategy for improving responses to immune checkpoint blockade. However, neoantigenic peptides are poorly immunogenic and inept at stimulating CD8+ T cell responses, motivating a need for new vaccine technologies that enhance their immunogenicity. The stimulator of interferon genes (STING) pathway is an endogenous mechanism by which the innate immune system generates an immunological context for priming and mobilizing neoantigen-specific T cells. Owing to this critical role in tumor immune surveillance, a synthetic cancer nanovaccine platform (nanoSTING-vax) was developed that mimics immunogenic cancer cells in its capacity to efficiently promote co-delivery of peptide antigens and the STING agonist, cGAMP. The co-loading of cGAMP and peptides into pH-responsive, endosomolytic polymersomes promoted the coordinated delivery of both cGAMP and peptide antigens to the cytosol, thereby eliciting inflammatory cytokine production, costimulatory marker expression, and antigen cross-presentation. Consequently, nanoSTING-vax significantly enhanced CD8+ T cell responses to a range of peptide antigens. Therapeutic immunization with nanoSTING-vax, in combination with immune checkpoint blockade, inhibited tumor growth in multiple murine tumor models, even leading to complete tumor rejection and generation of durable antitumor immune memory. Collectively, this work establishes nanoSTING-vax as a versatile platform for enhancing immune responses to neoantigen-targeted cancer vaccines.
Keywords: neoantigen, cancer vaccine, immune checkpoint blockade, immunotherapy, polymer nanoparticle
Graphical Abstract

Immune checkpoint inhibitors are transforming the treatment of an expanding number of tumor types, yet immune checkpoint blockade still benefits only a minority of cancer patients.1, 2 While resistance to immune checkpoint blockade is complex and multifaceted,3 poor clinical responses can, in part, be ascribed to an insufficient number and/or poor function of endogenously-generated, pre-existing T cells that recognize tumor antigens.4–6 This challenge has created an urgent need for new strategies to bolster the magnitude, breadth, and quality of the antitumor T cell response, including a revitalized interest in therapeutic cancer vaccines.7–10 While the clinical impact of cancer vaccines over the past several decades has been largely disappointing,11 the discovery that neoantigens—peptides derived from cancer-specific mutations—are the primary antigenic targets for antitumor T cells has fueled a revolution in the development of personalized cancer vaccines targeting patient-specific mutanomes.12 Mutations unique to an individual’s cancer are identified via whole exosome sequencing, advanced immunopeptidomic methods are employed to determine which mutations are most likely to generate neoepitopes, and neoantigenic peptides are then synthesized and administered to the patient as a personalized vaccine product. Peptide antigens, however, are typically weakly immunogenic and, consequently, many cancer vaccine formulations being explored clinically do not elicit robust tumor antigen-specific CD8+ T cell responses, which are critical for effective antitumor immunity.13, 14 This can be mostly attributed to several interrelated barriers, including inefficient accumulation in vaccine site draining lymph nodes (LNs), poor intracellular uptake by antigen presenting cells (APCs), low levels of antigen cross-presentation, and suboptimal choice and/or delivery of immunostimulatory adjuvants.7, 15 This long-standing challenge in synthetic vaccine design has motivated the development of a wide-range of materials-based strategies (e.g., nanoparticles, microparticles, scaffolds, hydrogels) to augment cellular immunity to protein and peptide antigens.16–18 However, despite these advances there has been relatively little investigation into the design of particle-based platforms for enhancing the performance of neoantigen-targeted cancer vaccines for personalized immunotherapy.19–24
The stimulator of interferon genes (STING) pathway plays a critical role in initiating and propagating endogenous mechanisms of antitumor T cell immunity.25–27 Upon activation by 2′,5′–3′5′ cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), STING triggers a type-I interferon (IFN-I)-driven inflammatory response that stimulates dendritic cell (DC) cross-presentation of tumor antigens, leading to mobilization of tumor-specific CD8+ T cells. This indispensable role for STING in cancer immune surveillance has recently motivated the study of cGAMP and related cyclic dinucleotides (CDN) as cancer vaccine adjuvants to more closely replicate the natural inflammatory cues that underlie and drive generation of antitumor immunity.28–32 Notably, Fu et al. demonstrated the capacity of STING agonists to enhance immune responses to an autologous cancer cell vaccine,28 and Kinkead et al. have recently leveraged CDNs as an adjuvant in a pancreatic cancer vaccine.29 While such pre-clinical studies highlight the potential of STING agonists as cancer vaccine adjuvants, they have co-delivered antigens and CDN adjuvants as a soluble mixture. This formulation strategy has been widely demonstrated to yield less potent and effective immune responses relative to the use of particle-based carriers that mimic pathogens through their capacity to promote the co-delivery of antigen and adjuvant.33, 34 This challenge is further exacerbated by the high water solubility, low molecular weight, and poor drug-like properties of CDNs, which results in their rapid clearance from the injection site with minimal accumulation in the draining LN and inefficient intracellular uptake by APCs,35–39 critical processes that underlie effective antigen-specific T cell activation. To overcome these barriers, several groups have developed nanocarrier platforms to enhance the intracellular delivery of CDNs, including several liposomal carriers,40–44 polymeric systems,45, 46 and inorganic nanostructures.47 Our group has recently described the development of polymer vesicles (polymersomes) with pH-responsive, membrane-destabilizing activity that enhances intracellular uptake and cytosolic delivery of CDNs, resulting in a dramatic enhancement in their immunostimulatory activity.38 At physiologic pH, the membrane-destabilizing block is sequestered in the polymersome bilayer, shielded by a poly(ethylene glycol) corona. Upon endocytosis and endosomal acidification, the nanoparticles rapidly disassemble to reveal the membrane-interactive segments, resulting in the release of CDNs into the cytosol. Here, we leverage this technology for the development of nanoSTING-vax – a platform for neoantigen-targeted cancer vaccines based on endosomolytic nanoparticles designed to enhance and coordinate the intracellular delivery of neoantigenic peptides and CDN STING agonists (Figure 1). Using synthetic long peptide antigens containing neoepitopes, we demonstrate that nanoSTING-vax can promote the dual-delivery of peptides and CDNs to the cytosol, resulting in enhanced antigen cross-presentation and dendritic cell maturation, while also promoting accumulation in the vaccine site draining lymph node. Consequently, nanoSTING-vax enhanced the CD8+ T cell responses to a diversity of peptide antigens, resulting in a dramatic improvement in the response to immune checkpoint blockade in two murine tumor models.
Figure 1. NanoSTING-vax – a platform for dual-delivery of peptide antigens and cyclic dinucleotide STING agonists to enhance responses to neoantigen-targeted cancer vaccines.
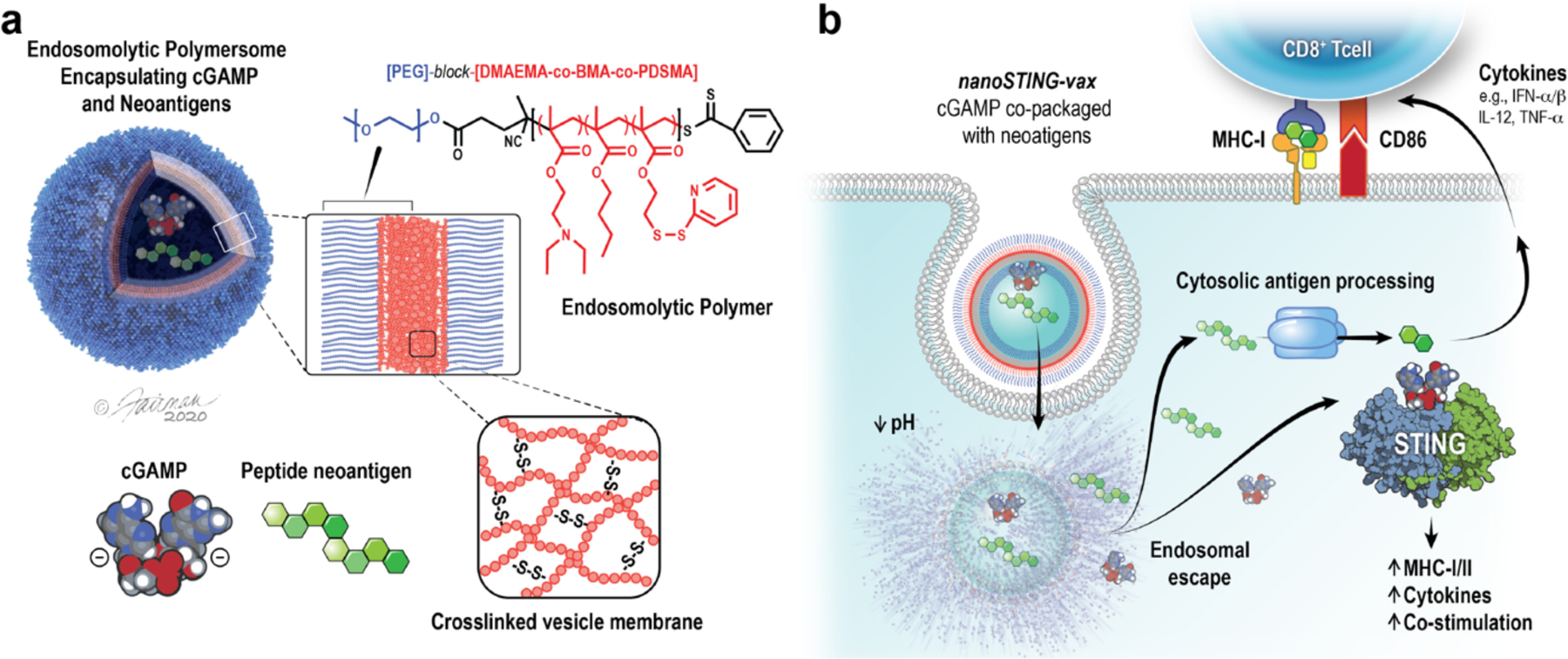
(a) Schematic of nanoSTING-vax structure. Peptide antigens and cyclic dinucleotide STING agonists (e.g., cGAMP) are co-loaded into pH-responsive polymersomes comprised of endosomolytic diblock polymers. (b) NanoSTING-vax enables uptake of peptides and cGAMP by antigen presenting cells and facilitates cytosolic co-delivery of neoantigenic peptides and cGAMP via endosomal escape. Cytosolic delivery of antigen promotes MHC class I presentation while cytosolic cGAMP delivery enhances its immunostimulatory adjuvant capacity, collectively resulting in enhanced CD8+ T cell priming and activation. Credit: ©Fairman Studios, LLC, 2020.
RESULTS AND DISCUSSION
Inspired by endogenous mechanisms of antitumor T cell immunity,26, 27, 48–50 we sought to develop a vaccine platform that mimicked an immunogenic cancer cell based on the reductionistic design concept of a vesicular particle encapsulating both peptide antigens and cGAMP (Figure 1a). To accomplish this, we leveraged pH-responsive, endosomolytic polymer vesicles (i.e., polymersomes) previously described by our group that enable the efficient cytosolic delivery of cGAMP.38 We hypothesized that this class of neoantigen-targeted vaccine, referred to herein as nanoSTING-vax, could enhance tumor antigen-specific T cell responses and, therefore, mitigate resistance to immune checkpoint inhibitors, via several mechanisms. First, nanoparticle co-delivery of cGAMP and peptide neoantigen increases the probability that both antigen and adjuvant are delivered to the same APC, allowing antigen processing and presentation to occur in an appropriate pro-inflammatory context, while minimizing the potential for T cell anergy or tolerance due to antigen presentation by immature APCs.51, 52 Second, polymersomes are designed to promote efficient endosomal escape of cargo to the cytosol, allowing cGAMP to access STING while also promoting cytosolic antigen delivery and processing via the classical MHC-I antigen presentation pathway (Figure 1b), which has been shown to enhance priming of antigen-specific CD8+ T cells.53, 54 Finally, polymersomes have the potential to enhance LN accumulation and uptake by APCs due to their nanoscale properties.55
Co-loading of a bisphosphorothioate analog of cGAMP and six unique peptide sequences, ranging from 9 to 27 amino acids, into polymersomes had no or minimal impact on the size (Figure 2a; Figure S1a) or neutral zeta potential (Figure S1b) of self-assembled particles, which transmission electron microscopy revealed were predominantly of vesicular morphology, though micelles and filamentous structures were also observed (Figure 2b; Figure S2), species that we have previously demonstrated are inefficient at enhancing cGAMP delivery.38 Importantly, the potent STING activation that we have previously described can be achieved via loading of cGAMP into endosomolytic nanoparticles was maintained when both peptides and cGAMP were co-loaded into polymersomes (Figure 2c; Figure S1c). Notably, peptides of variable length, charge, and hydrophobicity could be loaded into polymersomes, albeit with variable encapsulation efficiency (Table S1). Interestingly, we observed a statistically significant correlation between peptide hydrophobicity and loading efficiency (Figure S3), potentially reflecting a preferential association of peptides with the vesicle membrane or aqueous core during the self-assembly process based on their relative water solubility. Hence, the biphasic structure inherent to polymersomes may offer an important advantage for the delivery of peptide neoantigens, which are inherently personalized and, therefore, span a wide range of properties.
Figure 2. Endosomolytic nanoparticles enhance dual-delivery of cGAMP and peptide antigens to the cytosol.
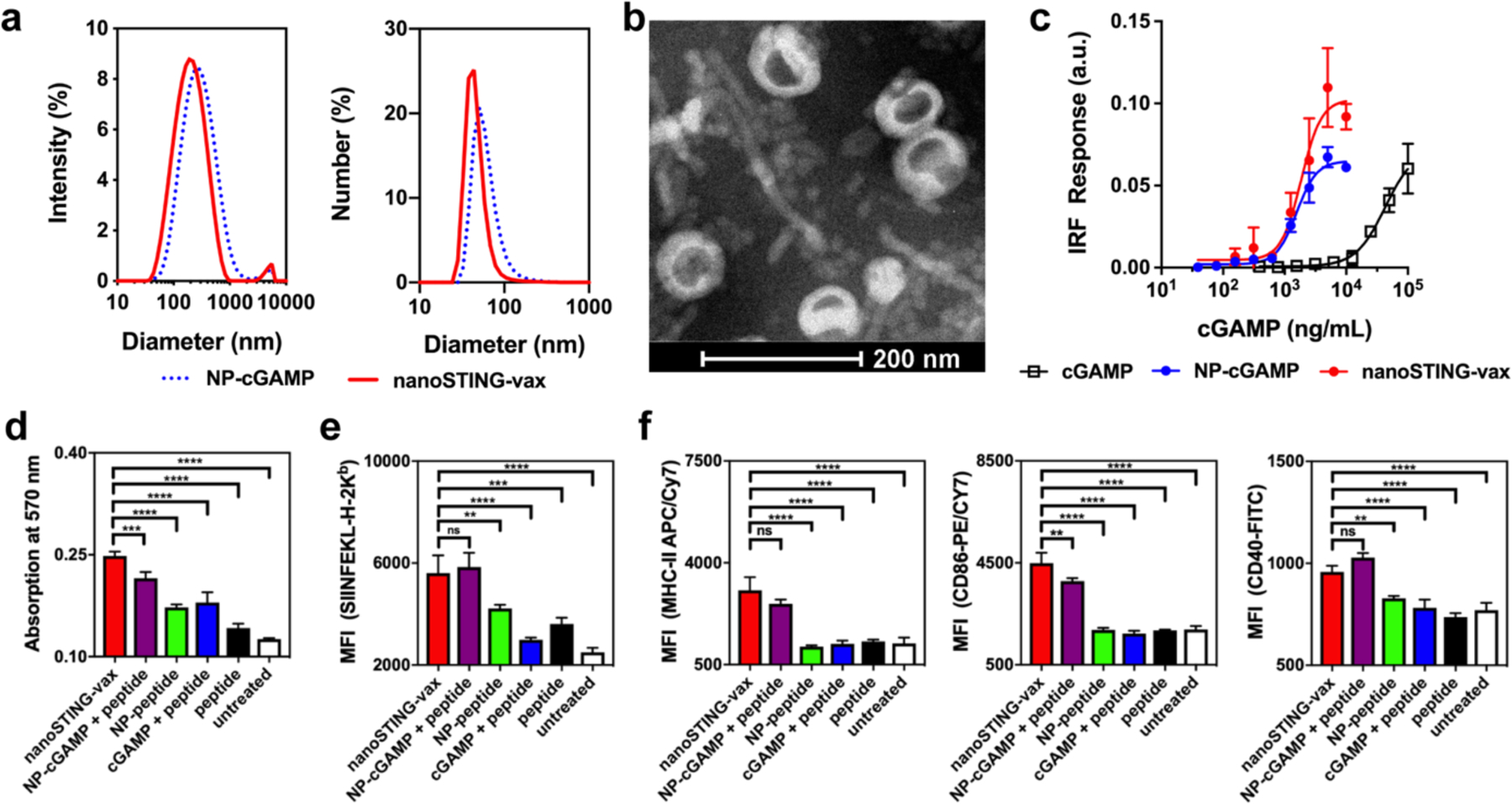
(a) Dynamic light scattering analysis (intensity and number average size distributions) of cGAMP-loaded polymersomes (NP-cGAMP) and polymersomes loaded with both cGAMP and the Ova-derived peptide SGLEQLESIINFEKL (nanoSTING-vax). (b) Representative transmission electron micrograph of nanoSTING-vax, here loaded with cGAMP and the peptide SGLEQLESIINFEKL. (c) Dose–response curves of the IFN-I response elicited by indicated cGAMP-containing formulations in RAW 264.7 cells with an IFN regulatory factor (IRF)-inducible reporter construct (n = 3 biologically independent samples). (d) B3Z T cell response to DC2.4 dendritic cells treated with the indicated formulation (n = 4 biologically independent samples). (e) Flow cytometric analysis of median fluorescent intensity (MFI) of BMDCs treated with the indicated formulation and stained with an antibody (25-D1.16) specific to the SIINFEKL/H-2Kb complex (n = 3 biologically independent samples). (f) Flow cytometric quantification (MFI) of MHC-II, CD86, and CD40 expression by BMDCs treated with indicated formulation (n=3 biologically independent samples). Statistical data are presented as mean ± s.d. Statistical significance between nanoSTING-vax and all other formulations are shown; **P<0.01, ***P<0.001, ****P<0.0001 by one-way ANOVA with Tukey post-hoc test.
We next investigated the effect of co-delivery of peptide antigen and cGAMP on MHC-I antigen presentation using a model synthetic long peptide containing the immunodominant CD8+ T cell epitope from ovalbumin (Ova), SIINFEKL. We first evaluated the SIINFEKL-specific CD8+ T cell response in a co-culture assay comprising DC2.4 dendritic cells and a B3Z CD8+ T cell hybridoma that produces β-galactosidase upon recognition of SIINFEKL in complex with the H-2Kb molecule (Figure 2d). Co-delivery of antigen and cGAMP in polymersomes (i.e., nanoSTING-vax) enhanced B3Z T cell activation to a greater extent than nanoparticles loaded only with peptide (NP-peptide), free peptide, or a mixture of soluble cGAMP and peptide. This is consistent with the ability of endosomolytic materials to promote cytosolic delivery of antigen, which enhances presentation on MHC-I,53, 54 a process that was further augmented via induction of STING signaling. Similar results were observed in bone marrow derived DCs (BMDCs) using an antibody against the H-2Kb/SIINFEKL complex (Figure 2e). Interestingly, in these studies, a mixture of free peptide and nanoparticles loaded only with cGAMP (NP-cGAMP + peptide) enhanced SIINFEKL presentation to a similar extent as nanoSTING-vax. This may reflect the capacity of STING signaling and IFN-I to promote antigen cross-presentation and increase surface expression of MHC-I48, 56, 57 and/or a spontaneous physical association between the peptide and polymersomes post-assembly that resulted in enhanced intracellular uptake and antigen presentation. Consistent with their capacity to enhance cGAMP activity, we also found that all polymersomes loaded with cGAMP (i.e., nanoSTING-vax and NP-cGAMP) increased expression of the DC maturation marker MHC-II and the costimulatory molecules CD40 and CD86 to a greater extent than free cGAMP or nanoparticle formulations lacking cGAMP (Figure 2f), reflecting the relatively weak intrinsic adjuvant activity of the nanoparticle and the need for co-delivery of cGAMP. Collectively, these data demonstrate the capacity of endosomolytic polymersomes to mediate cytosolic dual-delivery of CDN STING agonists and peptide antigens, resulting in coordinated DC activation and antigen presentation, which, in turn, enhances CD8+ T cell activation.
An attractive feature of nanoparticle vaccines is their ability to promote the biodistribution of vaccine components to LNs, with attendant enhancement in antigen presentation, APC maturation, and T cell priming.55, 58 To evaluate the distribution of vaccine components to the LN in vivo, vaccine formulations containing fluorescently-labeled polymersomes (Cy5-labeled) and peptide (Alexa Fluor 700-labeled SGLEQLESIINFEKL) were administered subcutaneously to allow for monitoring of carrier and cargo distribution to a vaccine site draining LN (inguinal) and uptake by leukocytes in the LN. Fluorescent imaging of inguinal LNs isolated 18 h following injection demonstrated that loading of peptide antigen into polymersomes significantly increased antigen accumulation in the LN (Figure 3a). Additionally, a significant increase in the expression of Ifnb1 in the inguinal LN was observed 4 h after administration (Figure 3b), demonstrating the ability of nanoSTING-vax to enhance cGAMP delivery to vaccine site draining LNs. Flow cytometric analysis of LNs 24 h post-immunization revealed that both peptide and polymer accumulated primarily in CD11b+F4/80+ macrophages and CD11c+ DCs (Figure 3c,d; Figure S4b), which play direct roles in antigen presentation to T cells; minimal uptake of peptide or polymer was observed in NK cells, B cells, or T cells. Consistent with the data from in vitro experiments, nanoSTING-vax also resulted in increased expression of CD80 and CD86 costimulatory molecules on CD11c+ DCs in the LN (Figure 3e; Figure S4c). Collectively, these data demonstrate the ability of the nanoSTING-vax platform to enhance peptide antigen and CDN delivery to APCs residing in draining LNs, a critical process in stimulation of cellular adaptive immunity.
Figure 3. NanoSTING-vax improves delivery of cGAMP and peptide antigens to vaccine site draining lymph nodes.
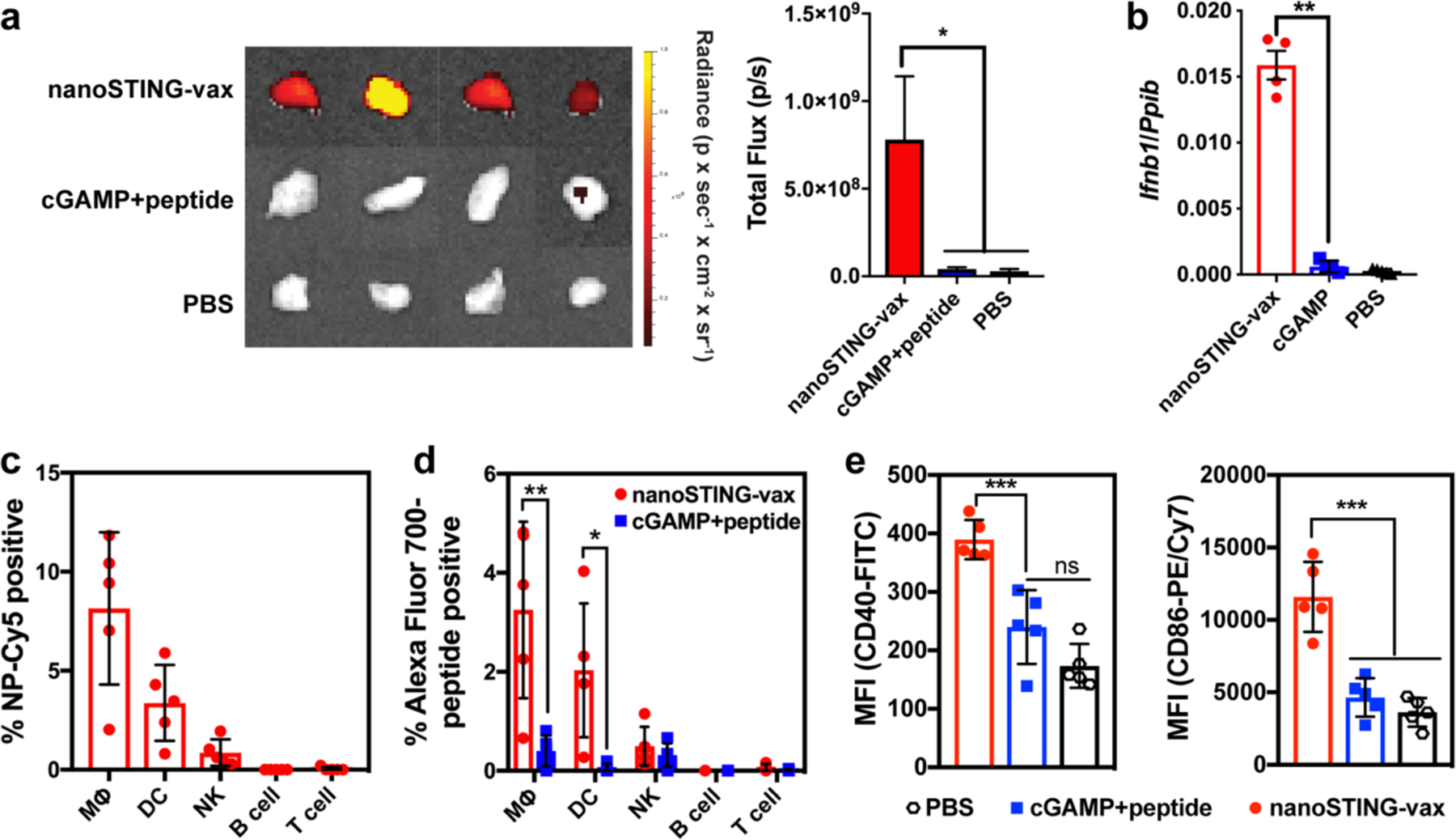
(a) Representative images (left) and IVIS quantification of fluorescence (right) of the vaccine site draining inguinal LN 18 h following subcutaneous administration of nanoSTING-vax containing an Alexa Fluor 700-labeled peptide or a soluble mixture of Alexa Fluor 700-peptide and cGAMP (mean +/− s.e.m; n = 8–10 mice/group; *P<0.05; one-way ANOVA with Tukey post-hoc test). (b) Ifnb1 expression in the inguinal LN 4 h following administration of indicated vaccine formulation (mean +/− s.e.m; n = 4–5 mice/group; ****P<0.001; one-way ANOVA with Tukey post-hoc test). (c) Percentage of NP-Cy5+ cells among cell populations in the inguinal LN following administration of the labeled nanoparticle (n = 5 mice/group). MΦ, macrophage; DC, dendritic cell; NK, natural killer cell. (d) Percentage of Alexa Fluor 700-peptide+ cells among cell populations in the inguinal LN following administration of the indicated formulation (n = 5 mice/group; two-tailed Student’s t-test; *P<0.05, **P<0.01). MΦ, macrophage; DC, dendritic cell; NK, natural killer cell. (e) Flow cytometric quantification (MFI) of CD86 and CD40 expression by CD11c+ dendritic cells in the inguinal LN in response to immunization with the indicated formulation (n = 5 mice/group; ***P<0.001, ****P<0.0001; one-way ANOVA with Tukey post-hoc test).
We next evaluated the capacity of nanoSTING-vax to enhance CD8+ T cell responses in vivo using a synthetic long peptide (SGLEQLESIINFEKL) containing the H-2Kb-restricted Ova epitope, SIINFEKL. Mice were administered nanoSTING-vax, a soluble mixture of peptide and cGAMP, peptide only, or PBS (vehicle), and boosted on day 14 and 24 (Figure 4a). Additionally, based on the data from in vitro experiments demonstrating that mixing soluble peptide with cGAMP-loaded NPs (NP-cGAMP + peptide) could enhance antigen presentation, we also included this formulation as an additional control in these studies. On day 31, peptide/MHC-I tetramer staining was used to evaluate the magnitude of the SIINFEKL-specific CD8+ T cell response in peripheral blood. NanoSTING-vax generated the highest antigen-specific CD8+ T cell response of all of formulations tested, resulting in ~8% SIINFEKL-specific CD8+ T cells in the blood, whereas free peptide and a soluble mixture of cGAMP and peptide elicited responses undetectable from baseline (Figure 4b). Similar to in vitro findings, a slight, but statistically insignificant, increase in the percentage of tetramer-positive CD8+ T cells was observed for free peptide mixed with cGAMP-loaded NPs. As a functional validation of the CD8+ T cell response, we challenged immunized mice on day 32 with a subcutaneous inoculation of Ova-expressing B16.F10 murine melanoma cells (B16-Ova) and evaluated tumor growth. Consistent with tetramer staining, only immunization with nanoSTING-vax resulted in significant inhibition of tumor growth (Figure 4c). Collectively, these experiments demonstrate that co-delivery of cGAMP and synthetic long peptides with endosomolytic polymersomes can significantly enhance the immunogenicity of peptide vaccines.
Figure 4. NanoSTING-vax enhances CD8+ T cell responses to peptide antigens.

(a) Administration, analysis, and tumor challenge scheme for mice immunized with nanoSTING-vax or indicated control formulations containing Ova peptide. (b) Quantification of the frequency of SIINFEKL-specific CD8+ T cells in peripheral blood via peptide/MHC tetramer staining (n = 15 mice/group. Statistical significance between nanoSTING-vax and all other formulations are shown; ***P<0.001, ****P<0.0001; one-way ANOVA with Tukey post-hoc test). (c) Average tumor volume following challenge of mice immunized with indicated vaccine formulations with B16-Ova cells (n = 8–15; mice/group; **P<0.01, ****P<0.0001; one-way ANOVA with Tukey post-hoc test on day 20). (d) Administration scheme for mice immunized with nanoSTING-vax containing Reps1 and Adpgk neoantigenic peptides or indicated control formulations. (e) Percentage of IFN-γ+TNF-α+ CD8α+ T cells in peripheral blood after ex vivo restimulation with Reps1 and Adpgk epitopes and intracellular cytokine staining following by flow cytometric analysis (n = 7–8 mice/group; *P<0.05, ***P<0.001, ****P<0.0001; one-way ANOVA with Tukey post-hoc test). Statistical data are presented as mean ± s.d unless otherwise indicated.
We next evaluated the capacity of nanoSTING-vax to enhance CD8+ T cell responses to two established tumor neoantigenic peptides arising from mutations in the proteins Reps1 (AQLANDVVL) and Adpgk (ASMTNMELM) in the MC38 murine colon adenocarcinoma cell line.59 In these studies, polymersomes were co-loaded with cGAMP and a synthetic long peptides containing either the Reps1 or Adpgk neoepitope, and the two formulations were then mixed to generate a vaccine containing both neoantigenic targets at equal peptide doses, as was done in recent clinical trials of multi-epitope cancer vaccines.60 First, non-tumor bearing mice were immunized with nanoSTING-vax or indicated control formulations and boosted on days 8 and 16 (Figure 4d). The magnitude and functionality of the neoantigen-specific T cell response was evaluated via peptide restimulation of peripheral blood mononuclear cells (PBMCs) followed by intracellular cytokine staining for IFNγ and TNFα. Consistent with findings using SIINFEKL as a model antigen, nanoSTING-vax enhanced the frequency of polyfunctional (IFNγ+TNFα+) antigen-specific CD8+ T cells relative to a mixture of synthetic long peptide and free cGAMP (Figure 4e). Interestingly, a mixture of free peptide and cGAMP-loaded NPs (NP-cGAMP + peptide) resulted in a statistically significant increase in the percentage of IFNγ+TNFα+ CD8+ T cells for the Adpgk antigen, but not for Reps1, potentially reflecting a differential capacity of different peptides to spontaneously associate with polymersomes post-assembly. To further investigate this, we used size exclusion chromatography to evaluate the extent to which the peptides employed in this study spontaneously associated with pre-formed polymersomes (Figure S5). While variable levels of interaction were observed, SGLEQLESIINFEKL and Reps1 displayed the highest level of association, whereas the Adpgk peptide interacted only minimally. While stability or nature of such interactions remains to be elucidated, such data demonstrating that a mixture of free peptide and nanoparticles loaded with cGAMP (NP-cGAMP + peptide) can enhance immune responses to some synthetic long peptides highlights a potential opportunity to design peptide antigens that can spontaneously and efficiently integrate with pre-assembled cGAMP-loaded NPs. Designing peptides that enable such a “mix-and-go” nanoSTING-vax formulation merits future investigation as a strategy to further streamline the just-in-time manufacturing of personalized cancer vaccine products.9, 20, 54 Nonetheless, these studies validate the ability of the nanoSTING-vax platform to enhance CD8+ T cell responses to multiple neoantigenic peptides.
Based on the capacity of nanoSTING-vax to enhance neoantigen-specific CD8+ T cell responses, we next evaluated whether this could be leveraged to improve responses to immune checkpoint blockade in a therapeutic vaccine setting. Many cancers can evade immune recognition through the expression of PD-L1 in response to secretion of IFN-γ by infiltrating T cells, resulting in inhibition of cytotoxic T cell function via binding to PD-1 on T cells.2 Therefore, maximizing the efficacy of cancer vaccines often requires blockade of PD-1, PD-L1, or other immune checkpoint molecules (e.g., CTLA-4) to circumvent this resistance mechanism.10 We first used the MC38 murine colorectal adenocarcinoma model, which has known neoantigens and expresses PD-L1, but is largely resistant to PD-L1/PD-1 blockade owing to a highly immunosuppressive microenvironment.61 Mice were inoculated subcutaneously with MC38 cells and vaccinated starting on day 8 with nanoSTING-vax or control formulations (Figure 5a). Additionally, nanoSTING-vax and selected controls were combined with systemic (i.p) administration of anti-PD-1 antibody (αPD-1) which has minimal therapeutic effect when delivered as monotherapy. While nanoSTING-vax conferred minimal therapeutic benefit alone in this model, we observed a dramatic improvement in the response to αPD-1, resulting in a ~70% (5/7 mice) complete response rate as indicated by the absence of any outward evidence of tumor growth up to 200 days post-vaccination (Figure 5b–d). Importantly, eliminating peptide antigens from the vaccine formulation (cGAMP-NP + αPD-1) nearly entirely abrogated therapeutic benefit, indicating that a vaccine-induced, antigen-specific T cell response was critical to the efficacy observed. To evaluate the capacity of nanoSTING-vax in combination with αPD-1 to generate immunological memory with potential to prevent disease recurrence, we re-challenged complete responders ~180 days following the last vaccine treatment. Several months after cessation of treatment and in mice of a more advanced age (~36 weeks), the combination of nanoSTING-vax and αPD-1 resulted in significant protection from tumor re-challenge, with 4/5 mice remaining tumor free for at least 30 days (Figure 5e).
Figure 5. NanoSTING-vax enhances response to immune checkpoint blockade.
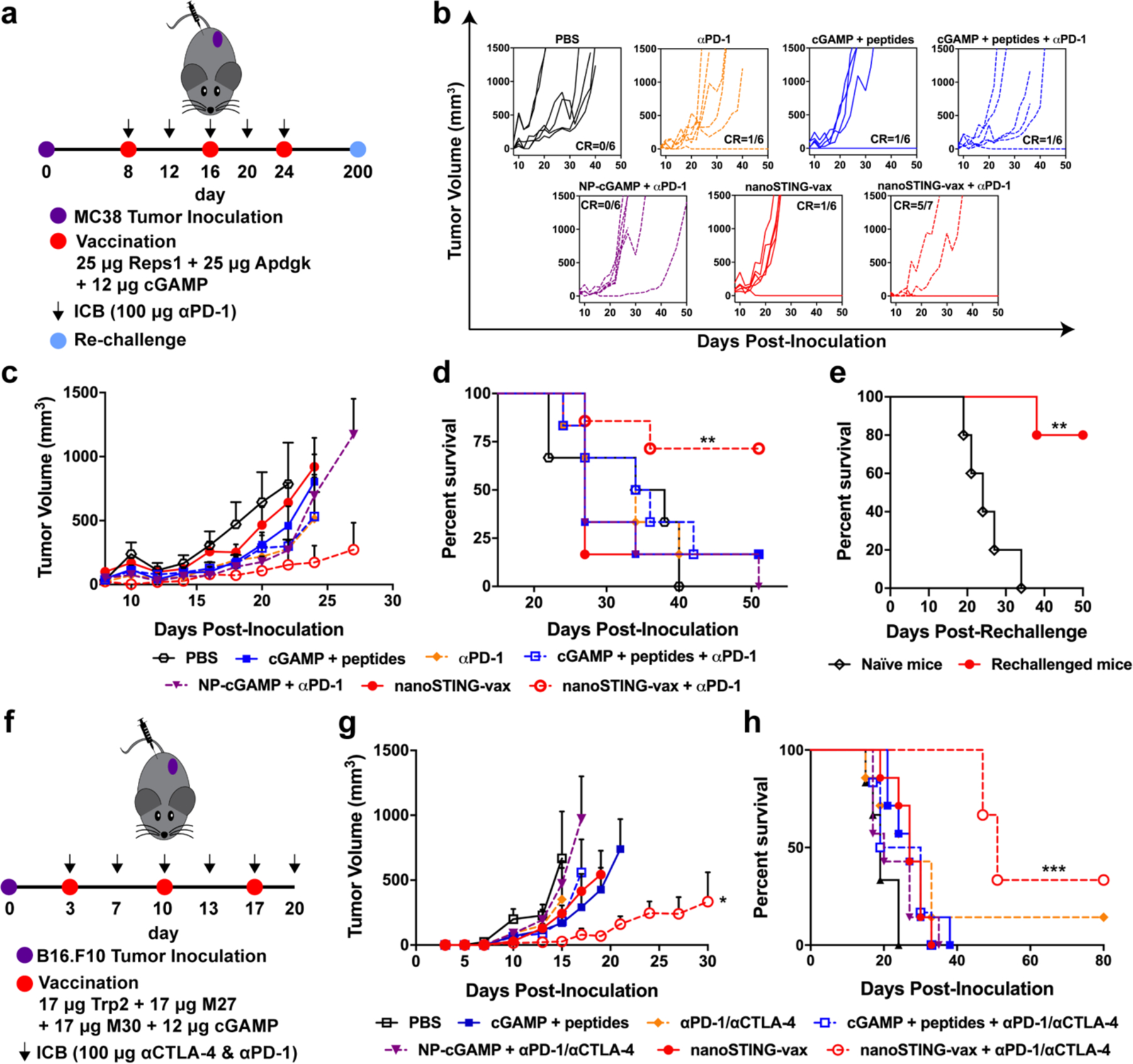
(a) Tumor inoculation, therapeutic vaccination, immune checkpoint blockade regimen, and re-challenge scheme for mice with subcutaneous MC38 tumors. (b) Spider plots of individual tumor growth curves, with the numbers of complete responders denoted. (c) Average MC38 tumor volume in response to indicated treatment (n = 6–7 mice/group; *P<0.05; unpaired t-test of nanoSTING-vax + αPD-1 vs. NP-cGAMP + αPD-1 on day 27). (d) Kaplan–Meier survival curves of mice growing MC38 tumors treated with the indicated formulation using a 1,500 mm3 tumor volume as the endpoint criteria (n = 6–7 mice/group; **P<0.01; two-tailed Mantel–Cox test). (e) Kaplan–Meier survival curves for treatment-naïve and mice demonstrating complete responses to nanoSTING-vax + αPD-1 after challenge with MC38 cells on the contralateral flank after 200 d without any further treatment (n = 5 mice; **P<0.01; two-tailed Mantel–Cox test). (f) Tumor inoculation, therapeutic vaccination, immune checkpoint blockade regimen for mice with subcutaneous B16.F10 tumors. (g) Average B16.F10 tumor volume in response to indicated treatment (n = 6–7 mice/group; *P<0.05; unpaired t-test of nanoSTING-vax + αPD-1 + αCTLA-4. cGAMP + peptides on day 21). (h) Kaplan–Meier survival curves of mice growing B16.F10 tumors treated with the indicated formulation using a 1,500 mm3 tumor volume as the endpoint criteria (n = 6–7 mice/group; ***P<0.001; two-tailed Mantel–Cox test). All statistical data are presented as mean ± s.e.m.
While including peptide neoantigens into the formulation was critical to achieving complete responses and long-term survival, it was notable that a slight, but statistically significant (P<0.01), decrease in tumor volume was observed in the nanoSTING-vax + αPD-1 group compared to and PBS on day 10, only two days after initial vaccination and too early for such effects to be mediated by vaccine-induced T cells. We have previously demonstrated that intravenous administration of cGAMP-NP could inhibit tumor growth and improve response to immune checkpoint blockade,38 and it has also been shown that peripherally administered nanoparticles can access the circulation via lymphatic drainage.62 We therefore investigated the possibility that subcutaneous vaccination with nanoSTING-vax may induce systemic STING activation. Mice were administered cGAMP-NP subcutaneously and serum levels of several established STING-driven cytokines (IFN-α, TNFα, IL-6) were measured (Figure S6). We found that peripheral administration of cGAMP-NP resulted in a rapid and transient elevation of serum cytokines, consistent with systemic nanoparticle distribution. Therefore, it is possible that the observed tumor suppression at early time points, while modest, may result from a subset of nanoparticles that distribute systemically and exert direct effects on the tumor. This may be important for controlling tumor burden during priming and expansion of neoantigen-specific T cells as well as for inhibiting immunosuppression in the tumor microenvironment, both of which would be anticipated to enhance the efficacy of T cells elicited via vaccination.7 A similar concept was demonstrated by Zhang et al. who intravenously administered immunostimulatory nanoparticles to reduce tumor burden and inhibit immunosuppression to improve the ability of CAR T cells to infiltrate solid tumors.63 The possibility that nanoSTING-vax may act both via generation of neoantigen-specific T cells and by mediating direct effects on the tumor microenvironment that enhance vaccine efficacy merits future exploration. Additionally, while we have previously demonstrated that intravenous administration of cGAMP-NP is well-tolerated,38 the stimulation of a systemic cytokine response nonetheless raises important questions regarding toxicity and safety that will need to be addressed. It is notable that an mRNA-based cancer vaccine that was administered intravenously in humans also induced a similar type of systemic cytokine response, with patients experiencing only transient flu-like symptoms.64 Nonetheless, additional research is necessary to optimize nanoSTING-vax dose, to modulate the extent of systemic distribution, and to further understand and manage potential toxicities.
Finally, we evaluated the nanoSTING-vax platform in an aggressive and poorly immunogenic B16.F10 melanoma model, which is highly resistant to immune checkpoint inhibitors and is difficult to treat using conventional cancer vaccines.19 Again, we utilized a mixture of nanoparticles, each loaded with a synthetic long peptide containing an established B16.F10 T cell epitope; here, we used two neoantigens, the MHC class I epitope M27 and the MHC-II antigen M30, as well as the MHC-I restricted epitope Trp2180–188 from the melanoma-associated antigen tyrosinase-related protein 2 (TRP2).19, 65 This strategy of combining shared tumor-associated antigens (e.g. TRP2) with individualized neoantigens has recently been explored in a clinical trial for glioblastoma.66 Mice were vaccinated with nanoSTING-vax comprising a pool of all three peptides at an equal dose (Figure 5f). Similar to our results in the MC38 model, vaccination with nanoSTING-vax alone did not confer significant therapeutic benefit, likely a consequence of the highly immunosuppressive microenvironment that is rapidly established in these tumor models.61 Nonetheless, nanoSTING-vax in combination with αPD-1 + αCTLA-4, the most aggressive immune checkpoint inhibitor regimen used clinically, significantly inhibited tumor growth and extended mean survival time, leading to complete rejection in ~30% (2/6) of treated mice (Figure 5g,h). As in the MC38 model, minimal tumor suppression was observed in all other groups, including an analogous vaccine formulation lacking peptide antigens, further indicating that the induced antigen-specific T cell response is critical to enhancing responses to immune checkpoint blockade.
CONCLUSION
Immune checkpoint blockade continues to expand the treatment of diverse cancer types. Nonetheless, a growing body of clinical evidence has demonstrated that complete and durable responses to immune checkpoint inhibitors are still the exception rather than the rule. This disappointing outcome is largely attributed to an insufficient antitumor T cell response that can be reinvigorated by immune checkpoint inhibitors, fueling the clinical exploration of neoantigen vaccines targeting patients’ tumor-specific mutations. Inspired by endogenous mechanisms of tumor immune surveillance, here we describe a platform for personalized cancer vaccines, nanoSTING-vax. NanoSTING-vax elicits robust antigen-specific CD8+ T cell responses via dual-delivery of peptide antigens and CDN STING agonists to the cytosol. Owing to its nanoscale properties, ability to enhance antigen cross-presentation, and potent immunostimulatory capacity, nanoSTING-vax is an enabling technology for increasing the immunogenicity of peptide antigens, resulting in the generation of antigen-specific T cell responses capable of rejecting pre-established, poorly immunogenic tumors when administered in combination with immune checkpoint blockade antibodies. Furthermore, nanoSTING-vax enables co-loading of CDNs and a wide range of peptide antigens of variable length and composition, offering a versatile vaccine delivery system that is well-suited for integration into current neoantigen vaccine production pipelines. Additionally, due to their vesicular structure, polymersomes are amenable to loading molecules of diverse physiochemical properties and, hence, may also provide a versatile template for coordinating the delivery of adjuvant combinations that can act in synergy with CDNs67–69 to augment or shape cellular immunity to personalized cancer vaccines. In summary, nanoSTING-vax – endosomolytic nanoparticles designed for dual-delivery of cyclic dinucleotide STING agonists and peptide antigens – is a promising platform for improving responses to personalized cancer vaccines, particularly in combination with immune checkpoint inhibitors.
MATERIALS AND METHODS
NanoSTING-vax fabrication:
Poly[(ethylene glycol)-block-[(2-diethylaminoethyl methacrylate)-co-(butyl methacrylate)-co-(pyridyl disulfide ethyl methacrylate)]] (PEG-DBP) was synthesized and characterized as previously described and detailed in the Supporting Information.38 A phosphorothioated cGAMP analog (RpRp dithio 2’3’cGAMP) was synthesized using a method adapted from Gaffney et. al. and described in detail in the Supporting Information.70 Synthetic long peptides containing established epitopes were purchased from Elim Biopharmaceuticals. Synthetic long peptides used in this study include: Ova (SGLEQLESIINFEKL), Reps1 (RVLELFRAAQLANDDVVLQIMELC), Adpgk (GIPVHLELASMTNMELMSSIVHQQVF), TRP2 (SVYDFFVWL), M27 (REGVELCPGNKYEMRRHGTTHSLVIHD), and M30 (PSKPSFQEFVDWENVSPELNSTDQPFL). NanoSTING-vax were formulated using polymersome self-assembly methods as previously described38 with minor modification to allow for cGAMP and peptide co-encapsulation. PEG-DBP was dissolved in a small quantity of ethanol to a concentration of 1,250 mg/ml followed by addition of a solution of 25 mg/mL cGAMP in DI H2O and 50 mg/mL of water-soluble peptides (Ova) to drive polymer phase separation and cargo encapsulation. Less water-soluble peptides (Reps1, Adpgk, M27, M30, TRP2) were dissolved in DMSO (50 mg/ml) and directly incorporated into the organic ethanolic phase prior to addition of the aqueous cGAMP solution. The volume of cGAMP added to the polymer solution was varied based on average encapsulation efficiency of each peptide to ensure all formulations had the same peptide to cGAMP mass ratio of 4:1. Gradual dilution into DI H2O with sonication dispersed the polymer gel into colloidally stable nanoparticles, whereupon the formulation was crosslinked via addition of 0.5 equivalents of dithiothreitol (DTT) relative to PDSMA groups. Unencapsulated cargo was removed via ultracentrifugal filtration through a 50,000 MWCO membrane to yield nanoSTING-vax nanoparticles. Control particles were formulated via the exact same method by simply omitting the addition of either the cGAMP or peptide solution. To quantify cGAMP and peptide loading, an aliquot was removed and diluted in pH 5.8 PBS to disassemble the vesicles, and then analyzed by high-performance liquid chromatography with a gradient mobile phase from 0.1% trifluoroacetic acid (TFA) in water to 0.1% TFA in acetonitrile over 10 minutes. cGAMP concentration was measured at 260 nm and peptide concentration was measured at 214 nm. Nanoparticle size distribution and zeta potential were measured by diluting particles in PBS (pH 7.4) and characterized using a Malvern Nano ZS. For transmission electron microscopy, particles were drop cast onto carbon Type-B support grid (Ted Pella), stained with NANO-W negative stain (Nanoprobes) and imaged on a 200 kV Osiris Transmission Electron Microscope in high contrast mode with a 20 μm objective aperture.
Analysis of spontaneous association of peptides with nanoparticle:
In order to measure the ability of each peptide to spontaneously associate with the pre-formulated polymersomes, each peptide was mixed with the polymersomes and then run down a Sephadex G-50 column equilibrated with PBS. The polymersomes were collected and any peptide that co-eluted with the polymersomes was measured using fluoraldehyde o-phthaldiadlehyde reagent solution. Separately, the same volume and quantity of peptide alone was run down the column, the same fraction was collected, and the amount of peptide was again quantified. The amount of peptide in this control was subtracted from the amount of peptide associated with the polymersome, and the percent associated peptide was calculated.
In vitro evaluation of cGAMP activity:
cGAMP activity was measured using RAW-Blue ISG cells (InvivoGen) that were cultured in DMEM supplemented with 10% FBS, 4.5 g/L glucose, 2mM L-Glutamine, and 100 μg/mL Normacin. Zeocin (200 μg/mL) was added every other passage to maintain selection pressure. RAW-Blue ISG cells were seeded at a density of 50,000 cells/well in a 96 well plate and then treated with the indicated formulations and concentrations for 24 hours. Relative expression of IFN-I was measured using QUANTI-Blue reagent (InvivoGen).
Evaluation of dendritic cell antigen presentation, activation, and cross-priming:
Bone marrow cells were harvested from the tibias of female C57BL/6J mice by flushing them with PBS and passing the cell suspension through a 70 μm cell strainer. Cells were then cultured on non-cell culture treated plates in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% heat inactivated FBS, 0.4 mM sodium pyruvate, 50 μM β-mercaptoethanol, and 20 ng/mL GM-CSF to induce differentiation into BMDCs. Fresh media was added on days 4 and 7, and on day 8 cells were re-plated in 12 well plates at 105 cells per well. Cells were then treated with vaccine formulations containing 500 nM SGLEQLESIINFEKL peptide and/or 100 ng/mL cGAMP for 24 hours. Following incubation, cells were scraped, washed with cold PBS, and stained with antibodies against SIINFEKL/H-2Kb and markers of dendritic cell activation (MHC-II, CD40, CD86) followed by flow cytometric analysis (Amnis CellStream, Luminex). The following antibodies were used for these studies: αCD40: (30-F11, FITC, BioLegend), αCD86 (GL-1, PE/Cy7, BioLegend), αOva257–264-H-2Kb (PE, eBioscience), αI-A/I-E (M5/114.15.2, APC/Cy7, BioLegend).
To evaluate cross-priming of T cells, a co-culture model comprising DC2.4 dendritic cells and a B3Z T cell hybridoma that produces β-galactosidase upon recognition of SIINFEKL/H-2Kb was used.71 The mouse dendritic cell line DC2.4 (H-2Kb-positive) was kindly provided by K. Rock (University of Massachusetts Medical School) and B3Z T cells were a generous gift from Nilabh Shastri (UC Berkeley). DC2.4 cells were cultured in RPMI 1640 media supplemented with 10% FBS, 1% penicillin and streptomycin, 2 mM L-glutamine, 10mM HEPES, 1x non-essential amino acids, and 55 μM β-mercaptoethanol in 96 well plates at a density of 10,000 cells/well. Cells were treated with vaccine formulations in a working concentration of 500 nM SGLEQLESIINFEKL peptide and/or 100 ng/mL cGAMP for 24 hours. Following incubation, media was aspirated, and 105 B3Z cells in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin/100 μg/mL streptomycin, 50 μM 2-mercaptoethanol, and 1 mM sodium pyruvate were added to cell culture wells and cultured for 24h. Cells were then pelleted and media were aspirated and replaced with lysis buffer consisting of 0.1% Triton, 0.15 mM chlorophenol red-β-d-galactopyranoside (Sigma), 9 mM MgCl, and 100 μM β-mercaptoethanol. After incubation for 90 minutes at 37 °C, the magnitude of antigen recognition was evaluated through absorbance measurements (λ = 570 nm).
In vivo analysis of lymph node accumulation, cellular uptake, and dendritic cell activation:
Female C57BL/6J mice (6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained at the animal facilities of Vanderbilt University Medical Center under conventional conditions. All animal experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC). To evaluate LN accumulation and cellular uptake, C57BL/6J mice were injected subcutaneously at the base of tail with formulations containing Alexa Fluor 700-labeled SGLEQLESIINFEKL (Elim Biopharmaceuticals) and Cy5-labeled polymersomes, which were generated by incorporating Cy5-labeled PEG-DBP that was synthesized via partial reduction of PDSMA groups followed by reaction with Cy5-maleimide (Abcam). After 18 h, inguinal LNs were harvested, and the Alexa Fluor 700 fluorescence signal was measured with IVIS optical imaging system (Caliper Life Sciences). Following imaging of the LN, single cell suspensions were prepared and stained with a panel of the antibodies BV650-αCD45 (30-F11), PE/Cy5-αCD11b (M1/70), PE/Cy5-αCD11c (N418), PE-αNK1.1 (PK136), PE/Cy7-αF4/80 (BM8), PE/Cy7-αCD86 (GL-1), BV510-αCD3 (17A2) (BioLegend). Cells were then washed and analyzed on a BD LSR Fortessa flow cytometer. Representative flow cytometry data and gating strategies for defining cell populations and determining cellular uptake are shown in Figure S4.
Ifnb1 expression in the lymph node:
C57BL/6J mice were injected subcutaneously at the base of tail with PBS, a mixture of peptide and cGAMP, or nanoSTING-vax. After 4 h, the inguinal LNs were harvested, and placed in RLT lysis buffer (Qiagen) supplemented with 2% β-mercaptoethanol (Sigma) in a gentleMACS M tube with mechanical disruption using an OctoMACS tissue dissociator (Miltenyi). Tumor RNA was isolated with a RNeasy RNA isolation kit (Qiagen) with the RNase-free DNase Set (Qiagen), used according to manufacturer’s specifications. Complementary DNA (cDNA) was synthesized with the Bio-Rad iScript cDNA kit and analysed via qPCR using the appropriate TaqMan kits (Thermo Fisher Scientific). The TaqMan gene expression kits were: Mm00439552_s1 for mouse Ifnb1 and Mm00478295-m1 for mouse Ppib.
In vivo immunization and analysis of T cell response:
To evaluate the CD8+ T cell response elicited by formulations containing SGLEQLESIINFEKL, mice (6–8 week) were immunized via subcutaneous injection at the base of the tail on days 0, 14, and 24 with formulations containing 50 μg peptide and 12 μg cGAMP in PBS. At day 31, whole blood was treated with ACK lysis buffer (Gibco), washed and resuspended in cold PBS supplemented with 2% FBS and 50 μM dasatinib, and stained with PE-labeled peptide-MHC tetramer (H-2Kb-restricted SIINFEKL, NIH Tetramer Core Facility, Atlanta, GA) and following antibodies: αCD45(30-F11, PE/Cy5, BioLegend), αCD3 (17A2, APC, BioLegend), αCD8 (KT15, FITC, ThermoFisher). After 1 h, cells were washed with PBS supplemented with 2% FBS and 50 μM dasatinib and stained with propidium iodide (BD Biosciences) to discriminate live from dead cells. Flow cytometry (BD LSRII) was used to determine the frequency of SIINFEKL-specific CD8+ T cells. Representative flow cytometry data and gating strategies for determining the frequency of tetramer+CD8+ T cells are shown in Figure S6.
To evaluate responses to vaccines containing Reps1and Adpgk, mice were immunized on days day 0, 8, and 16 via subcutaneous injection at the base of the tail. In all studies where multiple peptides were used, independent nanoSTING-vax preparations were prepared for each peptide and then pooled prior to administration. On day 23, blood was harvested, treated with ACK lysis buffer, and plated at a density of 2×106 cells per well in 12 well plates in RPMI 1640 supplemented with 10% FBS. Cells were treated for 6 hours with exact peptides for mutant Reps1 (AQLANDVVL) or Adpgk (ASMTNMELM) at a concentration of 10 μg/mL for 6 hrs. At the second hour, media was supplemented with brefeldin A (Biolegend) according to manufacturer specifications. Following incubation, cells were harvested and stained with αCD3 (17A2, APC, BioLegend), αCD8a (53.6.7, PE/Cy5, BioLegend), and αCD4 (RM4–5, APC/Cy7, BioLegend). Cells were then washed and fixed using fixation buffer (BioLegend), permeabilized with intracellular staining permeabilization wash buffer (BioLegend), and stained with αIFNγ (XMG1.2, Alexafluor488, BioLegend) and αTNFα (MP6-XT22, PE, BioLegend) before flow cytometric quantification of the percentage of TNFα+ and IFNγ+ CD8+ T cells (BD LSR II). Representative flow cytometry data and gating strategies for determining the frequency of TNFα+ and IFNγ+ CD8+ T cells are shown in Figure S7.
Tumor studies:
For prophylactic tumor challenge studies, mice vaccinated with formulations containing 50 µg SGLEQLESIINFEKL, as described above, were challenged 7 days following the final vaccination by subcutaneous flank injection of 5×105 B16-Ova cells, generously provided by Amanda Lund (Oregon Health Sciences University). Tumor volume was measured every other day via caliper measurements using the formula V=LxWxH/2. Mice were euthanized at a tumor burden endpoint of 1,500 mm.3 For the MC38 therapeutic model, mice were inoculated subcutaneously on the flank with 106 MC38 cells (generously provided by Daniel Beauchamp, Vanderbilt University) and vaccinated on day 8, 16, and 24 via subcutaneous injection at the base of the tail. Vaccines consisted of 12 μg cGAMP and 25 μg each of Reps1 and Adpgk synthetic long peptides. Mice were administered αPD-1 (Clone, BioXCell) on days 8, 12, 16, 20, and 24 intraperitoneally, and tumor growth was monitored as described above. For the B16.F10 therapeutic vaccination model, mice were inoculated via subcutaneous flank injection with 5×105 B16.F10 cells (generously provided by Ann Richmond, Vanderbilt University). Mice were then vaccinated as described above on days 3, 10, and 17, with formulations containing 17 μg of the TRP2, M27, and M30 neoantigen peptides and 10 μg of cGAMP. αCTLA-4 and αPD-1 antibody (100 μg each) were administered intraperitoneally on days 7, 10, 13, 17, and 20. Tumor growth was monitored as indicated above.
Analysis of serum cytokines:
C57BL/6J mice were injected subcutaneously at the base of the tail with either PBS or nanoSTING-vax. At various time points (6, 24, 48, 72 h), blood was collected and allowed to clot. Serum was analyzed for amounts of TNF-α, IFN-α, and IL-6 via the LEGENDplex Multi-Analyte Flow Assay Kit (BioLegend) following manufacturer’s instructions for the assay using a V-bottom plate. A 1:4 dilution was used for 6 and 24 h timepoints while a 1:2 dilution was used for 48 and 72 h timepoints. Data were collected on a CellStream Flow Cytometer (Luminex) equipped with 405, 488, 561, and 642 nm lasers and analyzed with LEGENDplex Data Analysis software v8.0 (VigeneTech).
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Kenneth Rock for providing DC2.4 cells, N. Shastri for providing B3Z T cells, A. Lund for providing B16-Ova cells, D. Beauchamp for providing MC38 cells, Ann Richmond for providing B16.F10 cells, L. Glickman for consultation on peptide antigens, and C. Duvall for the use of gel permeation chromatography equipment and IVIS imaging system. The following reagent was obtained through the NIH Tetramer Core Facility: PE-labeled SIINFEKL/H-2Kb tetramer. We thank the core facilities of the Vanderbilt Institute of Nanoscale Sciences and Engineering (VINSE) for use of dynamic light scattering and electron microscopy, and the VUMC Flow Cytometry Shared Resource, supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). 2’3’-cGAMP was provided by the Vanderbilt Institute of Chemical Biology Chemical Synthesis Core. This research was supported by grants from the National Science Foundation CBET-1554623 (JTW), a Vanderbilt Ingram Cancer Center (VICC) Ambassador Discovery Grant (JTW), a VICC-Vanderbilt Center for Immunobiology Pilot Grant (JTW), the National Institutes of Health (NIH) 5R21AI121626 (JTW) and 5R01DE027749 (SJ), the Vanderbilt University Discovery Grant Program (JTW, SJ), a Veteran’s Affairs Merit Award BX001444 (SJ), and also supported by a Stand Up To Cancer Innovative Research Grant, Grant Number SU2C-AACR-IRG 20–17 (JTW). Stand Up To Cancer (SU2C) is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. MW acknowledges a postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR). CSC acknowledges a National Science Foundation Graduate Research Fellowship under grant numbers DGE-1445197 and DGE-1937963.
REFERENCES
- 1.Ribas A; Wolchok JD Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P; Allison JP The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell JS; Teng MWL; Smyth MJ Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat Rev Clin Oncol 2019, 16, 151–167. [DOI] [PubMed] [Google Scholar]
- 4.Binnewies M; Roberts EW; Kersten K; Chan V; Fearon DF; Merad M; Coussens LM; Gabrilovich DI; Ostrand-Rosenberg S; Hedrick CC; Vonderheide RH; Pittet MJ; Jain RK; Zou W; Howcroft TK; Woodhouse EC; Weinberg RA; Krummel MF Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat Med 2018, 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen DS; Mellman I Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [DOI] [PubMed] [Google Scholar]
- 6.Fridman WH; Zitvogel L; Sautes-Fridman C; Kroemer G The Immune Contexture in Cancer Prognosis and Treatment. Nat Rev Clin Oncol 2017, 14, 717–734. [DOI] [PubMed] [Google Scholar]
- 7.Shae D; Baljon JJ; Wehbe M; Becker KW; Sheehy TL; Wilson JT At the Bench: Engineering the Next Generation of Cancer Vaccines. J Leukoc Biol 2019. [DOI] [PubMed]
- 8.Scheetz L; Park KS; Li Q; Lowenstein PR; Castro MG; Schwendeman A; Moon JJ Engineering Patient-Specific Cancer Immunotherapies. Nat Biomed Eng 2019, 3, 768–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tureci O; Lower M; Schrors B; Lang M; Tadmor A; Sahin U Challenges towards the Realization of Individualized Cancer Vaccines. Nat Biomed Eng 2018, 2, 566–569. [DOI] [PubMed] [Google Scholar]
- 10.Ott PA; Wu CJ Cancer Vaccines: Steering T Cells Down the Right Path to Eradicate Tumors. Cancer Discov 2019, 9, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romero P; Banchereau J; Bhardwaj N; Cockett M; Disis ML; Dranoff G; Gilboa E; Hammond SA; Hershberg R; Korman AJ; Kvistborg P; Melief C; Mellman I; Palucka AK; Redchenko I; Robins H; Sallusto F; Schenkelberg T; Schoenberger S; Sosman J; et al. The Human Vaccines Project: A Roadmap for Cancer Vaccine Development. Sci Transl Med 2016, 8, 334ps339. [DOI] [PubMed] [Google Scholar]
- 12.Schumacher TN; Schreiber RD Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [DOI] [PubMed] [Google Scholar]
- 13.Melief CJM; van der Burg SH Immunotherapy of Established (Pre)Malignant Disease by Synthetic Long Peptide Vaccines. Nat Rev Cancer 2008, 8, 351–360. [DOI] [PubMed] [Google Scholar]
- 14.Yewdell JW Designing CD8+ T Cell Vaccines: It’s Not Rocket Science (Yet). Curr Opin Immunol 2010, 22, 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehta NK; Moynihan KD; Irvine DJ Engineering New Approaches to Cancer Vaccines. Cancer Immunol Res 2015, 3, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eppler HB; Jewell CM Biomaterials as Tools to Decode Immunity. Adv Mater 2019, e1903367. [DOI] [PMC free article] [PubMed]
- 17.Feng X; Xu W; Li Z; Song W; Ding J; Chen X Immunomodulatory Nanosystems. Adv Sci (Weinh) 2019, 6, 1900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J; Kroll AV; Holay M; Fang RH; Zhang L Biomimetic Nanotechnology toward Personalized Vaccines. Adv Mater 2019, e1901255. [DOI] [PMC free article] [PubMed]
- 19.Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat Mater 2017, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li AW; Sobral MC; Badrinath S; Choi Y; Graveline A; Stafford AG; Weaver JC; Dellacherie MO; Shih TY; Ali OA; Kim J; Wucherpfennig KW; Mooney DJ A Facile Approach to Enhance Antigen Response for Personalized Cancer Vaccination. Nat Mater 2018. [DOI] [PMC free article] [PubMed]
- 21.Luo M; Wang H; Wang Z; Cai H; Lu Z; Li Y; Du M; Huang G; Wang C; Chen X; Porembka MR; Lea J; Frankel AE; Fu YX; Chen ZJ; Gao J A STING-Activating Nanovaccine for Cancer Immunotherapy. Nat Nanotechnol 2017, 12, 648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu C; Nam J; Hong H; Xu Y; Moon JJ Positron Emission Tomography-Guided Photodynamic Therapy with Biodegradable Mesoporous Silica Nanoparticles for Personalized Cancer Immunotherapy. ACS Nano 2019, 13, 12148–12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu G; Mei L; Vishwasrao HD; Jacobson O; Wang Z; Liu Y; Yung BC; Fu X; Jin A; Niu G; Wang Q; Zhang F; Shroff H; Chen X Intertwining DNA-RNA Nanocapsules Loaded with Tumor Neoantigens as Synergistic Nanovaccines for Cancer Immunotherapy. Nat Commun 2017, 8, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynn GM; Sedlik C; Baharom F; Zhu Y; Ramirez-Valdez RA; Coble VL; Tobin K; Nichols SR; Itzkowitz Y; Zaidi N; Gammon JM; Blobel NJ; Denizeau J; de la Rochere P; Francica BJ; Decker B; Maciejewski M; Cheung J; Yamane H; Smelkinson MG; et al. Peptide-TLR-7/8a Conjugate Vaccines Chemically Programmed for Nanoparticle Self-Assembly Enhance CD8 T-Cell Immunity to Tumor Antigens. Nat Biotechnol 2020. [DOI] [PMC free article] [PubMed]
- 25.Woo SR; Corrales L; Gajewski TF The STING Pathway and the T Cell-Inflamed Tumor Microenvironment. Trends Immunol 2015, 36, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woo SR; Fuertes MB; Corrales L; Spranger S; Furdyna MJ; Leung MY; Duggan R; Wang Y; Barber GN; Fitzgerald KA; Alegre ML; Gajewski TF STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng L; Liang H; Xu M; Yang X; Burnette B; Arina A; Li XD; Mauceri H; Beckett M; Darga T; Huang X; Gajewski TF; Chen ZJ; Fu YX; Weichselbaum RR STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu J; Kanne DB; Leong M; Glickman LH; McWhirter SM; Lemmens E; Mechette K; Leong JJ; Lauer P; Liu W; Sivick KE; Zeng Q; Soares KC; Zheng L; Portnoy DA; Woodward JJ; Pardoll DM; Dubensky TW Jr.; Kim Y STING Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to PD-1 Blockade. Sci Transl Med 2015, 7, 283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinkead HL; Hopkins A; Lutz E; Wu AA; Yarchoan M; Cruz K; Woolman S; Vithayathil T; Glickman LH; Ndubaku CO; McWhirter SM; Dubensky TW Jr.; Armstrong TD; Jaffee EM; Zaidi N Combining STING-Based Neoantigen-Targeted Vaccine with Checkpoint Modulators Enhances Antitumor Immunity in Murine Pancreatic Cancer. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutjahr A; Papagno L; Nicoli F; Kanuma T; Kuse N; Cabral-Piccin MP; Rochereau N; Gostick E; Lioux T; Perouzel E; Price DA; Takiguchi M; Verrier B; Yamamoto T; Paul S; Appay V The STING Ligand cGAMP Potentiates the Efficacy of Vaccine-Induced CD8+ T Cells. JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z; Celis E STING Activator C-Di-Gmp Enhances the Anti-Tumor Effects of Peptide Vaccines in Melanoma-Bearing Mice. Cancer Immunol Immunother 2015, 64, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandra D; Quispe-Tintaya W; Jahangir A; Asafu-Adjei D; Ramos I; Sintim HO; Zhou J; Hayakawa Y; Karaolis DK; Gravekamp C STING Ligand C-Di-Gmp Improves Cancer Vaccination against Metastatic Breast Cancer. Cancer Immunol Res 2014, 2, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irvine DJ; Swartz MA; Szeto GL Engineering Synthetic Vaccines Using Cues from Natural Immunity. Nat Mater 2013, 12, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swartz MA; Hirosue S; Hubbell JA Engineering Approaches to Immunotherapy. Sci Transl Med 2012, 4, 148rv149. [DOI] [PubMed] [Google Scholar]
- 35.Liu H; Moynihan KD; Zheng Y; Szeto GL; Li AV; Huang B; Van Egeren DS; Park C; Irvine DJ Structure-Based Programming of Lymph-Node Targeting in Molecular Vaccines. Nature 2014, 507, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dubensky TW Jr.; Kanne DB; Leong ML Rationale, Progress and Development of Vaccines Utilizing STING-Activating Cyclic Dinucleotide Adjuvants. Ther Adv Vaccines 2013, 1, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanson MC; Crespo MP; Abraham W; Moynihan KD; Szeto GL; Chen SH; Melo MB; Mueller S; Irvine DJ Nanoparticulate STING Agonists Are Potent Lymph Node-Targeted Vaccine Adjuvants. J Clin Invest 2015, 125, 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shae D; Becker KW; Christov P; Yun DS; Lytton-Jean AKR; Sevimli S; Ascano M; Kelley M; Johnson DB; Balko JM; Wilson JT Endosomolytic Polymersomes Increase the Activity of Cyclic Dinucleotide STING Agonists to Enhance Cancer Immunotherapy. Nat Nanotechnol 2019, 14, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbell JA; Swartz MA Trojan Horses for Immunotherapy. Nat Nanotechnol 2019, 14, 196–197. [DOI] [PubMed] [Google Scholar]
- 40.Koshy ST; Cheung AS; Gu L; Graveline AR; Mooney DJ Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Advanced Biosystems 2017, 1, 1600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng N; Watkins-Schulz R; Junkins RD; David CN; Johnson BM; Montgomery SA; Peine KJ; Darr DB; Yuan H; McKinnon KP; Liu Q; Miao L; Huang L; Bachelder EM; Ainslie KM; Ting JP A Nanoparticle-Incorporated STING Activator Enhances Antitumor Immunity in PD-L1-Insensitive Models of Triple-Negative Breast Cancer. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura T; Miyabe H; Hyodo M; Sato Y; Hayakawa Y; Harashima H Liposomes Loaded with a STING Pathway Ligand, Cyclic Di-Gmp, Enhance Cancer Immunotherapy against Metastatic Melanoma. J Control Release 2015, 216, 149–157. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y; Crowe WN; Wang L; Lu Y; Petty WJ; Habib AA; Zhao D An Inhalable Nanoparticulate STING Agonist Synergizes with Radiotherapy to Confer Long-Term Control of Lung Metastases. Nat Commun 2019, 10, 5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J; Li P; Yu Y; Fu Y; Jiang H; Lu M; Sun Z; Jiang S; Lu L; Wu MX Pulmonary Surfactant-Biomimetic Nanoparticles Potentiate Heterosubtypic Influenza Immunity. Science 2020, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin LC; Huang CY; Yao BY; Lin JC; Agrawal A; Algaissi A; Peng BH; Liu YH; Huang PH; Juang RH; Chang YC; Tseng CT; Chen HW; Hu CJ Viromimetic STING Agonist-Loaded Hollow Polymeric Nanoparticles for Safe and Effective Vaccination against Middle East Respiratory Syndrome Coronavirus. Adv Funct Mater 2019, 29, 1807616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson DR; Sen R; Sunshine JC; Pardoll DM; Green JJ; Kim YJ Biodegradable STING Agonist Nanoparticles for Enhanced Cancer Immunotherapy. Nanomedicine 2018, 14, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.An M; Yu C; Xi J; Reyes J; Mao G; Wei WZ; Liu H Induction of Necrotic Cell Death and Activation of STING in the Tumor Microenvironment via Cationic Silica Nanoparticles Leading to Enhanced Antitumor Immunity. Nanoscale 2018, 10, 9311–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahn J; Xia T; Rabasa Capote A; Betancourt D; Barber GN Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 2018, 33, 862–873 e865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitai Y; Kawasaki T; Sueyoshi T; Kobiyama K; Ishii KJ; Zou J; Akira S; Matsuda T; Kawai T DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J Immunol 2017, 198, 1649–1659. [DOI] [PubMed] [Google Scholar]
- 50.Kwon J; Bakhoum SF The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 2019. [DOI] [PMC free article] [PubMed]
- 51.Manicassamy S; Pulendran B Dendritic Cell Control of Tolerogenic Responses. Immunol Rev 2011, 241, 206–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Audiger C; Rahman MJ; Yun TJ; Tarbell KV; Lesage S The Importance of Dendritic Cells in Maintaining Immune Tolerance. J Immunol 2017, 198, 2223–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knight FC; Gilchuk P; Kumar A; Becker KW; Sevimli S; Jacobson ME; Suryadevara N; Wang-Bishop L; Boyd KL; Crowe JE; Joyce S; Wilson JT Mucosal Immunization with a pH-Responsive Nanoparticle Vaccine Induces Protective CD8+ Lung-Resident Memory T Cells. ACS Nano 2019, 13, 10939–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu F; Becker KW; Knight FC; Baljon JJ; Sevimli S; Shae D; Gilchuk P; Joyce S; Wilson JT Poly(propylacrylic Acid)-Peptide Nanoplexes as a Platform for Enhancing the Immunogenicity of Neoantigen Cancer Vaccines. Biomaterials 2018, 182, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas SN; Schudel A Overcoming Transport Barriers for Interstitial-, Lymphatic-, and Lymph Node-Targeted Drug Delivery. Curr Opin Chem Eng 2015, 7, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lirussi D; Ebensen T; Schulze K; Trittel S; Duran V; Liebich I; Kalinke U; Guzman CA Type I IFN and Not TFN, Is Essential for Cyclic Di-Nucleotide-Elicited CTL by a Cytosolic Cross-Presentation Pathway. EBioMedicine 2017, 22, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H; Hu S; Chen X; Shi H; Chen C; Sun L; Chen ZJ cGAS Is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc Natl Acad Sci U S A 2017, 114, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reddy ST; van der Vlies AJ; Simeoni E; Angeli V; Randolph GJ; O’Neil CP; Lee LK; Swartz MA; Hubbell JA Exploiting Lymphatic Transport and Complement Activation in Nanoparticle Vaccines. Nat Biotech 2007, 25, 1159–1164. [DOI] [PubMed] [Google Scholar]
- 59.Yadav M; Jhunjhunwala S; Phung QT; Lupardus P; Tanguay J; Bumbaca S; Franci C; Cheung TK; Fritsche J; Weinschenk T; Modrusan Z; Mellman I; Lill JR; Delamarre L Predicting Immunogenic Tumour Mutations by Combining Mass Spectrometry and Exome Sequencing. Nature 2014, 515, 572–576. [DOI] [PubMed] [Google Scholar]
- 60.Ott PA; Hu Z; Keskin DB; Shukla SA; Sun J; Bozym DJ; Zhang W; Luoma A; Giobbie-Hurder A; Peter L; Chen C; Olive O; Carter TA; Li S; Lieb DJ; Eisenhaure T; Gjini E; Stevens J; Lane WJ; Javeri I, et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taylor MA; Hughes AM; Walton J; Coenen-Stass AML; Magiera L; Mooney L; Bell S; Staniszewska AD; Sandin LC; Barry ST; Watkins A; Carnevalli LS; Hardaker EL Longitudinal Immune Characterization of Syngeneic Tumor Models to Enable Model Selection for Immune Oncology Drug Discovery. J Immunother Cancer 2019, 7, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kourtis IC; Hirosue S; de Titta A; Kontos S; Stegmann T; Hubbell JA; Swartz MA Peripherally Administered Nanoparticles Target Monocytic Myeloid Cells, Secondary Lymphoid Organs and Tumors in Mice. PLoS One 2013, 8, e61646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang F; Stephan SB; Ene CI; Smith TT; Holland EC; Stephan MT Nanoparticles That Reshape the Tumor Milieu Create a Therapeutic Window for Effective T-Cell Therapy in Solid Malignancies. Cancer Res 2018, 78, 3718–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kranz LM; Diken M; Haas H; Kreiter S; Loquai C; Reuter KC; Meng M; Fritz D; Vascotto F; Hefesha H; Grunwitz C; Vormehr M; Husemann Y; Selmi A; Kuhn AN; Buck J; Derhovanessian E; Rae R; Attig S; Diekmann J, et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [DOI] [PubMed] [Google Scholar]
- 65.Kreiter S; Vormehr M; van de Roemer N; Diken M; Lower M; Diekmann J; Boegel S; Schrors B; Vascotto F; Castle JC; Tadmor AD; Schoenberger SP; Huber C; Tureci O; Sahin U Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520, 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hilf N; Kuttruff-Coqui S; Frenzel K; Bukur V; Stevanovic S; Gouttefangeas C; Platten M; Tabatabai G; Dutoit V; van der Burg SH; Thor Straten P; Martinez-Ricarte F; Ponsati B; Okada H; Lassen U; Admon A; Ottensmeier CH; Ulges A; Kreiter S; von Deimling A, et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [DOI] [PubMed] [Google Scholar]
- 67.Temizoz B; Kuroda E; Ohata K; Jounai N; Ozasa K; Kobiyama K; Aoshi T; Ishii KJ TLR9 and STING Agonists Synergistically Induce Innate and Adaptive Type II IFN. Eur J Immunol 2015, 45, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Atukorale PU; Raghunathan SP; Raguveer V; Moon TJ; Zheng C; Bielecki PA; Wiese ML; Goldberg AL; Covarrubias G; Hoimes CJ; Karathanasis E Nanoparticle Encapsulation of Synergistic Immune Agonists Enables Systemic Codelivery to Tumor Sites and IFNβ-Driven Antitumor Immunity. Cancer Res 2019, 79, 5394–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Collier MA; Junkins RD; Gallovic MD; Johnson BM; Johnson MM; Macintyre AN; Sempowski GD; Bachelder EM; Ting JP; Ainslie KM Acetalated Dextran Microparticles for Codelivery of STING and TLR7/8 Agonists. Mol Pharm 2018, 15, 4933–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaffney BL; Veliath E; Zhao J; Jones RA One-Flask Syntheses of C-Di-Gmp and the [Rp,Rp] and [Rp,Sp] Thiophosphate Analogues. Org Lett 2010, 12, 3269–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karttunen J; Sanderson S; Shastri N Detection of Rare Antigen-Presenting Cells by the Lacz T-Cell Activation Assay Suggests an Expression Cloning Strategy for T-Cell Antigens. Proc Natl Acad Sci U S A 1992, 89, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.