

Abstract
Ambient fine particulate matter (PM2.5) exposure correlates with adverse cardiometabolic effects. The underlying mechanisms have not yet been fully understood. Hypothalamic-pituitary-adrenal (HPA) axis, as the central stress response system, regulates cardiometabolic homeostasis and is implicated in the progression of various adverse health effects caused by inhalational airborne pollutant exposure. In this study, we investigated whether ambient PM2.5 exposure activates HPA axis and its effect mediating PM2.5-induced pulmonary inflammation. C57Bl/6J mice were intratracheally instilled with different concentrations of diesel exhaust PM2.5 (DEP), and plasma was harvested at different times. Assessments of plasma stress hormones revealed that DEP instillation dose- and time-dependently increased mouse circulating corticosterone and adrenocorticotropic hormone (ACTH) levels, strongly supporting that DEP instillation activates HPA axis. To determine which components of DEP activate HPA axis, C57Bl/6J mice were intratracheally instilled with water-soluble and -insoluble fractions of DEP. Plasma analyses showed that water-insoluble but not -soluble fraction of DEP increased circulating corticosterone and ACTH levels. Consistently, concentrated ambient PM2.5 (CAP) exposure significantly increased mouse urine and hair corticosterone levels, corroborating the activation of HPA axis by ambient PM2.5. Furthermore, deletion of stress hormones by total bilateral adrenalectomy alleviated PM2.5-induced pulmonary inflammation, providing insights into the contribution of central neurohormonal mechanisms in modulating adverse health effects caused by exposure to PM2.5.
Keywords: PM2.5, HPA axis, stress hormones, pulmonary inflammation
1. Introduction
Ambient fine particulate matter (with aerodynamic diameter ≤ 2.5 μm, also known as PM2.5) pollution is one of the leading challenges for global public health (Lepeule et al., 2012). It has been shown that most inhaled ambient particles deposit in the lower airway instead of entering the systemic circulation. However, in addition to pulmonary effects, exposure to PM2.5 also correlates to various systemic abnormalities such as endothelial dysfunction, systemic inflammation, increased blood pressure, insulin resistance, and accelerated atherosclerotic progression (Brook et al., 2010). Currently, how these PM2.5 in the lower airway induce systemic effects remains to be determined.
The nervous system exerts control over all activity in the body and is the ultimate control center for the maintenance of homeostasis. It has been well known that exposure to ambient PM2.5 results in a variety of neural outcomes (Calderon-Garciduenas and Torres-Jardon, 2015; Miller et al., 2016; Wilker et al., 2015). For example, a large number of works have shown that acute PM2.5 exposure upregulates heart rate variability, an indicative of imbalanced autonomic nervous activity (Buteau and Goldberg, 2016). We previously reported that exposure to CAP resulted in hypothalamic inflammation, and the latter subsequently led to the imbalanced activation of autonomic nervous system and development of hypertension (Ying et al., 2014). Notably, in addition to automatic nervous system, the hypothalamus also controls over endocrine system via the pituitary gland, which includes the central stress response system known as the hypothalamic pituitary adrenal (HPA) axis. Most recently, inhalation of ozone, another criteria airborne pollutant, has been shown to increases circulating stress hormones in humans, implicating the HPA axis in the pathogenesis of systemic adverse effects of air pollution (Miller et al., 2016). There are also several studies showing that ambient PM2.5 exposure increases circulating stress hormone of animal models in various contexts such as co-exposure to ozone and one day after CAP exposure (Sirivelu et al., 2006; Thomson et al., 2013). In this study, to determine whether the HPA axis is involved in the progression of various adverse systemic effects caused by ambient PM2.5 exposure, we investigated systemically the effects of PM2.5 exposure on the HPA axis activity in murine models. The present results revealed that exposures to PM2.5 from different sources activated HPA axis in mouse models, and that deletion of stress hormones using total bilateral adrenalectomy alleviated pulmonary inflammation in mice, strongly supporting that HPA axis is implicated in the pathophysiology due to exposure to PM2.5.
2. Methods
2.1. Animals
Female C57Bl/6J mice (8 weeks old) for intratracheal instillation of diesel exhaust PM2.5 (DEP) were purchased from Jackson Laboratories (Bar Harbor, Maine, USA). Female C57Bl/6J mice (3 weeks old) for inhalational exposure of concentrated ambient PM2.5 (CAP) and female C57Bl/6J mice (9 weeks old) for adrenalectomy surgery were purchased from the Animal Centre, Shanghai Medical School, Fudan University (Shanghai, China). All procedures were approved by the Institutional Animal Care and Use Committees of Fudan University or University of Maryland, Baltimore. All mice were offered with standard feed and water ad lib, and kept at room temperature (20–25°C) in a relative humidity of 30–70% with a 12-hour dark/12-hour light cycle.
2.2. Sources of DEP
DEP powders were purchased from the National Institute of Standards and Technology (SRM 2975; NIST, Gaithersburg, USA). They were kept away from the direct sunlight and stored at 4 °C before usage. DEP samples were suspended using normal sterile saline (4 mg/ml or diluted as needed), aliquoted and stored in −80 °C until using for instillation. On the day of instillation, all the DEP suspensions were sonicated (Clifton Ultrasonic Bath, Clifton, NJ, USA) for 20 minutes and vortexed for 30 seconds before each instillation to minimize aggregation.
2.3. Soluble and insoluble DEP fractions
DEP suspensions in normal saline were then separated to insoluble and soluble fractions. Briefly, suspension was vortexed for 30 seconds and then centrifuged for 30 minutes in the speed of 12850×g. The supernatant was transferred to a new vial as the soluble fraction, while the pellet was resuspended using the same volume of sterile normal saline as the initial suspension and used as the insoluble fraction. The volume of soluble fraction was also finally adjusted to the volume of the initial DEP suspension.
2.4. Intratracheal administration of DEP
DEP instillation was carried out with minor modifications as previously described (Kyjovska et al., 2015) In brief, mice were first anesthetized using isoflurane (3%), and placed supine on an angled board. Then a Becton Dickinson 18 Gauge cannula was inserted into the mice trachea via the mouth. 50 μl of saline or DEP suspension (prepared as demonstrated before) was then intratracheally instilled with 150 μl of air bolus using a sterile syringe. Finally, mice were placed in a vertical position for 5 minutes with their heads up ensuring the maintenance of instilled DEP in the hung as well as avoiding the blockage of airways after the remove of intubation catheter. To minimize the effects of instillation on HPA axis activity, the animals were kept anesthetized with 1.5% isoflurane during the intratracheal administration. For the dose-dependency experiment, DEP suspensions at the concentrations of 0, 0.2, 1, 2 and 4 mg/ml were applied. And DEP suspension of 4 mg/ml were used for the time-dependency experiment. To check the effects of different DEP fractions, mice were instilled with vehicle (sterile PBS), total DEP (4 mg/ml), water-soluble and insoluble fractions derived from DEP suspension of 4 mg/ml. 24 hours after intratracheal administration, mice were sacrificed and plasma were collected for corticosterone and ACTH levels assessment.
2.5. Inhalation exposure to concentrated ambient PM2.5 (CAP)
After acclimation for 1 week, female C57BL/6J mice (4 weeks old) were subjected to CAP or filtered air (FA) exposure (n= 10/group) using a versatile aerosol concentration enrichment system (VACES) modified for long-term exposures as described previously in Fudan University (130 Dong’an Rd, Shanghai, China)(Wang et al., 2018). The exposure was carried out from May to August 2016, comprising exposures for 5 days/week (with no exposure in the weekend) and 6 hours/day. After 3 months of FA/CAP exposure, 24-hour urine samples were collected with metabolic cages following 1-week acclimation to housing in the metabolic cages. Approximately 100 mg of hair samples were collected from each mouse on the back. Mice were then sacrificed, and their lungs were weighted and harvested for proinflammatory cytokines expressions assessment. The animal experiments protocol was approved by the Animal Care and Use Committee of Fudan University, and all the animals were treated humanely with regard for suffering alleviation.
2.6. Adrenalectomy surgery
After acclimation for one week, female C57BL/6J mice (10 weeks old) were anesthetized using isoflurane (3%) followed by two types of surgeries: bilateral total adrenalectomy (ADREX) with both adrenal glands removed or control sham surgery (SHAM) with identical procedures except for the removal of adrenal glands (n=10/group). Mice were maintained on the same diet and provided with 0.9% saline water for 2 weeks. Intratracheal administration of DEP (4 mg/ml) was then performed, and 24 hours after that mice were sacrificed.
2.7. Mouse corticosterone and ACTH assessments
To collect the plasma for corticosterone and ACTH assessments, mice bloods were collected from the trunk after decapitation. Before the experiment, collected hair samples were weighed and cut into small pieces. After homogenization using Precellys24 (Bertin Instruments) in methanol (1 mL for hair from one mouse), samples were incubated at 52 °C overnight with shaking. Supernatants were then collected into new vials after centrifugation. Evaporation of the methanol in supernatant was conducted under the protection of nitrogen using a dry bath (Thermolyne® Dri-Bath), and sample was then resuspended using phosphate buffered saline (PBS, 200 μL, pH 8.0) and vortexed for 1 minute to ensure complete mix. The corticosterone levels in mouse plasma, urine and hair were measured with the Mouse and Rat Corticosterone ELISA (Alpco Diagnostics®, Windham, NH). The ACTH levels in mouse plasma were assessed using the Mouse/Rat Adrenocorticotropic Hormone (ACTH) ELISA (Sigma-Aldrich) according to the manufacturers’ instructions. To check the depletion of stress hormone by ADREX/SHAM surgery, plasma samples were collected from caudal vein for corticosterone assessment before and after fasting.
2.8. Bronchoalveolar lavage and real-time RT-PCR
After euthanasia by overdose of isoflurane, the mouse trachea was cannulated, and the left primary bronchus was closed off with a ligation. Sterile phosphate buffered saline (PBS) with 0.1mM ethylenediaminetetraacetic acid (EDTA) was instilled and withdrawn for 3 times (0.5mL each) to recover bronchoalveolar lavage fluid (BALF). The total cell number of BALF (around 1.5mL) was estimated using a hemocytometer. Following BALF collection, the other half lung tissue was harvested and snap frozen using liquid nitrogen and kept at −80°C. Total RNA was extracted and purified using the Trizol reagent (Invitrogen). The quality of RNA was assessed by determination of the ratio of absorbance at 260nm to that at 280nm by Nanodrop. RNA was then reverse transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems™). Real time RT-PCR was performed using SYBR qPCR Master Mix (Vazyme) with a 7500 Real-time PCR System (Applied Biosystems™). The specific sense and antisense primers for tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-1β and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (the reference housekeeping gene) were previously described(Chen et al., 2018b). 2ΔCt were calculated as the relative expression level of detected genes(Hu et al., 2017). The levels of pro-inflammatory cytokines and chemokines (IL-6; VEGF, vascular endothelial growth factor; MIP-1α, macrophage inflammatory protein-1 alpha) in BALF were measured using the MILLPLEX MAP Mouse Cytokine/Chemokine Panel 1 (Merk, Millipore) according to the manufacturers’ instruction.
2.9. Statistical analysis
All the data are demonstrated as mean ± SEM. All the collected data was used for statistical analysis otherwise noted. Statistical significances were evaluated by t-test, one-way or two-way ANOVA analyses using GraphPad Prism Software. The significance was set at p < 0.05.
3. Results
3.1. Exposure to DEP activates mouse HPA axis
To test if exposure to ambient PM2.5 activates HPA axis, C57Bl/6J mice were exposed to DEP, a prominent source of ambient fine particles in urban areas and widely-used model for studying PM2.5 toxicology, through intratracheal instillation and their plasma corticosterone levels were then determined. Figures 1A and C show that intratracheal instillation of DEP time- and dose-dependently increased mouse plasma corticosterone levels, strongly supporting that exposure to ambient PM2.5 activates HPA axis. Consistently, Figures 1B and D reveal that intratracheal instillation of DEP time- and dose-dependently increased the plasma levels of ACTH, another biomarker of HPA axis activity. To further investigate the effects of different constitutes on the progression of PM2.5 induced adverse health effects, we prepared water-soluble and -insoluble DEP fractions, and examined the effects of instillation of these fractions on the activity of HPA axis. Figures 1E and F demonstrate that water-insoluble but not water-soluble fraction of DEP increased mouse plasma corticosterone and ACTH.
Figure 1. Instillation of DEP activates mouse HPA axis.
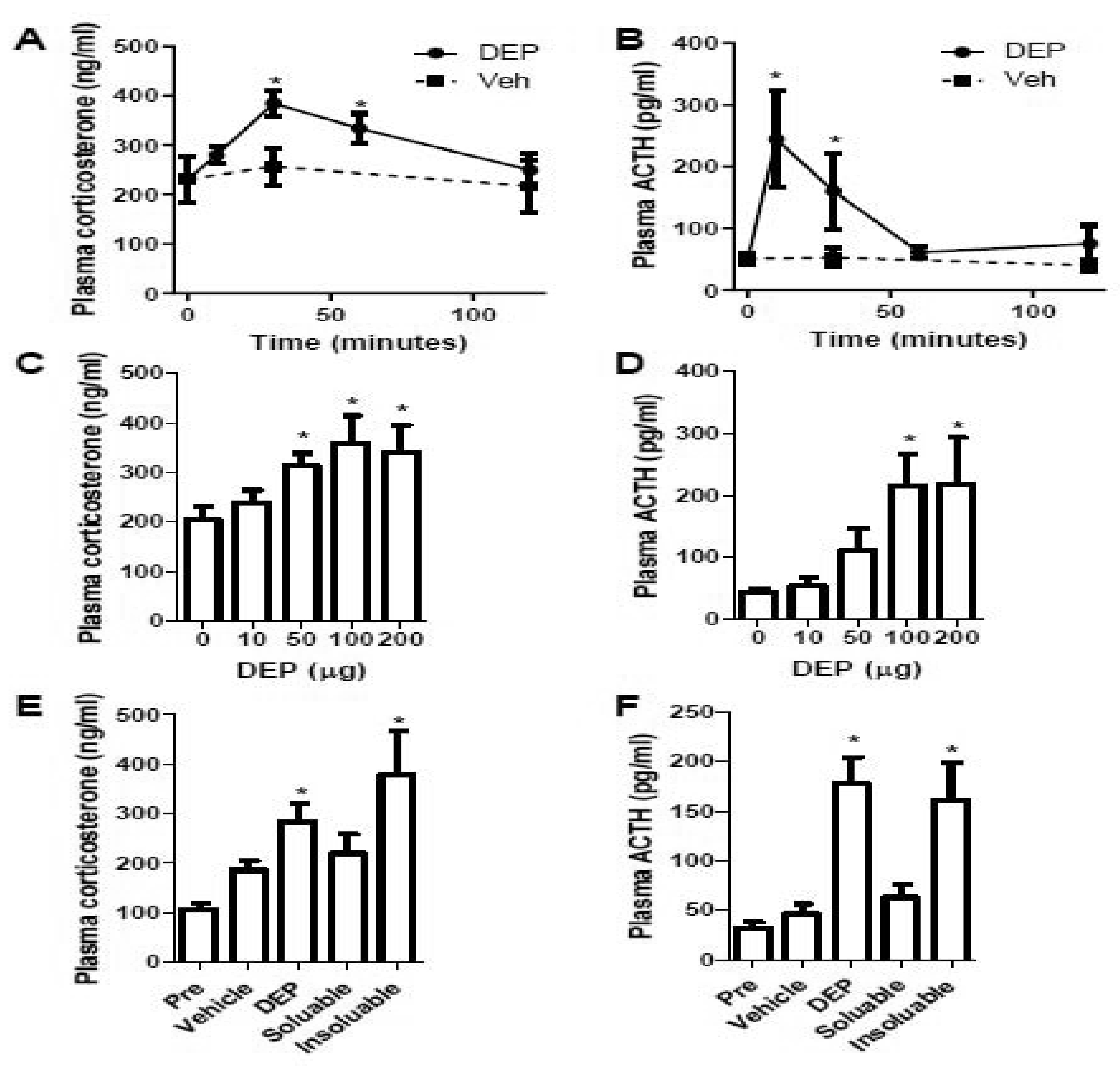
A and B, female C57Bl/6J mice were instilled with DEP (200 μg in 50 μl/mouse) and euthanized at the indicated time-points. Plasma corticosterone (A) and ACTH (B) were assessed and presented. n = 6–8/group. *p<0.05 versus 0 (minutes), one-way ANOVA. C and D, female C57Bl/6J mice were instilled with the indicated concentration of DEP and euthanized after 30 minutes. Plasma corticosterone (A) and ACTH (B) were assessed and presented. n = 6/group. *p<0.05 versus 0 (μg), one-way ANOVA. E and F, female C57Bl/6J mice were instilled with vehicle (50 μl sterile PBS), total DEP (DEP, 200 μg in 50 μl/mouse), soluble DEP (soluble, 50 μl), and insoluble DEP (insoluble, 50 μl), and euthanized after 30 minutes. Plasma corticosterone (E) and ACTH (F) were assessed and presented. n = 6/group. *p<0.05 versus Vehicle, one-way ANOVA.
3.2. Exposure to ambient PM2.5 induces pulmonary inflammation and activates HPA axis
As intratracheal instillation may cause stress which could confound the observation, we next examined the influence of inhalational CAP exposure on the activity of mouse HPA axis. Due to the big variation in mouse plasma corticosterone levels, we did not observe a significant increase in mouse plasma corticosterone levels after 6-hour exposure to CAP (Data not shown). As many adverse effects of ambient PM2.5 exposure appear to be cumulative, we next assessed the effect of chronic CAP exposure on the activity of HPA axis. CAP exposure was performed from May to August 2016. The average concentration of ambient PM2.5 during this period was 42.1 ± 23.5 μg/m3, and the average PM2.5 concentrations in CAP and FA chambers were 276.2 ± 170.1 and 12.3 ± 5.8 μg/m3, respectively.
Figure 2A shows that compared to FA-exposed controls, urine corticosterone levels of CAP-exposed animals were significantly increased, strongly indicating that inhalational ambient PM2.5 exposure activates mouse HPA axis. Hair samples of those FA/CAP-exposed animals, another reliable biomarker of long-term activity of HPA axis, were also collected and their corticosterone levels were assessed. Figure 2B demonstrates that the hair corticosterone levels of CAP-exposed animals were significantly increased, corroborating that inhalation exposure to ambient PM2.5 is sufficient to activate HPA axis. In addition, no change was found on the weights of body and lung of FA/CAP-exposed mice (Figures 2C–D). As indicated in Figures 2E–G, proinflammatory cytokines expression assessment by real time RT-PCR demonstrated that CAP exposure significantly upregulated levels of TNFα and IL-1β but not IL-6, indicating that chronic exposure to ambient PM2.5 induced pulmonary inflammation.
Figure 2. Exposure to ambient PM2.5 activates HPA axis.
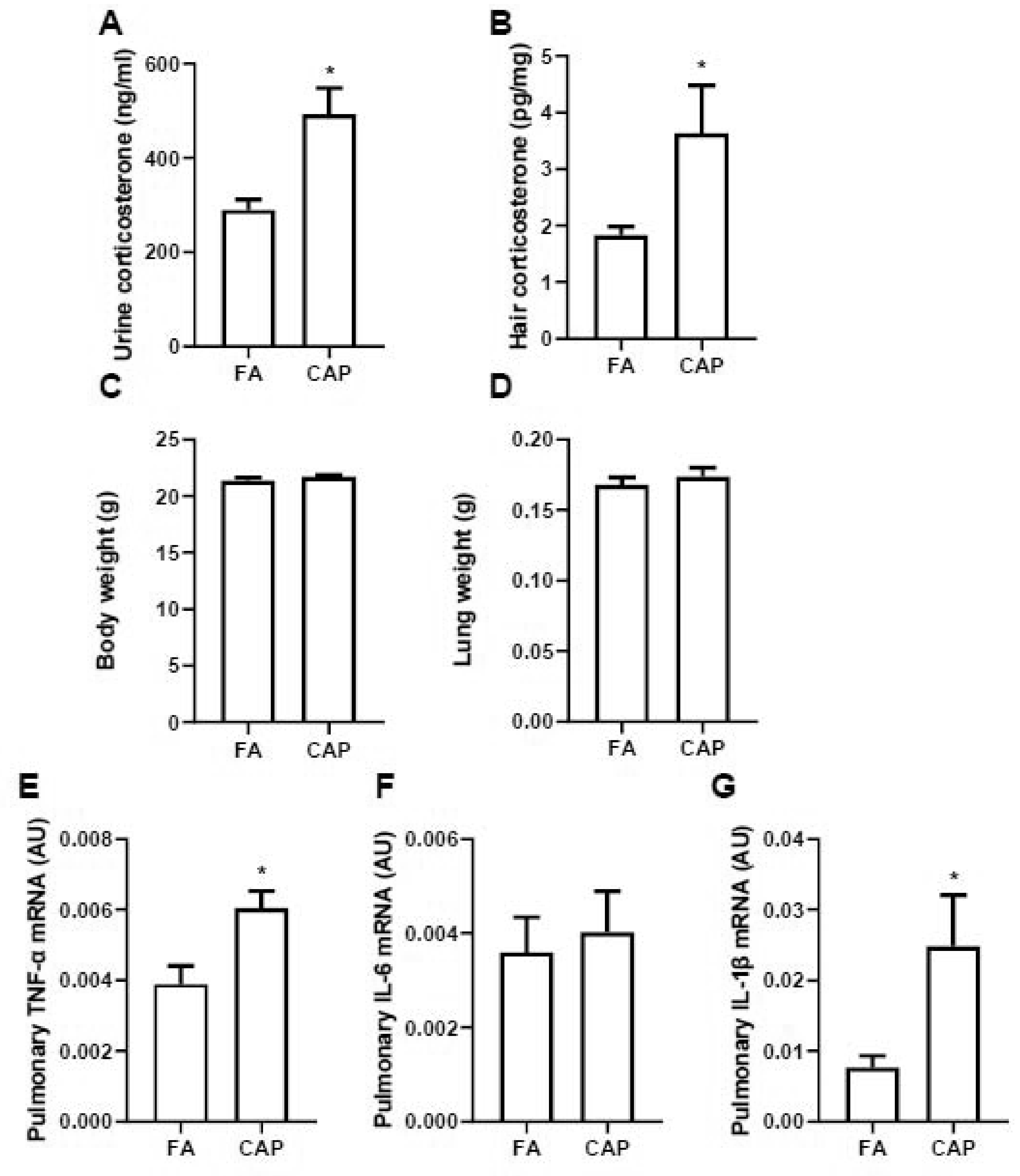
A and B, female C57Bl/6J mice were exposed to FA or CAP for 3 months and then the 24-hour urine and hair samples were collected for corticosterone level assessment. Corticosterone levels in urine (A) and hairs (B) were assessed and presented. Their body weight (C) and lung weight (D) were collected and pulmonary inflammatory gene expressions (E, TNF-α; F, IL-6; G, IL-1β) were assessed using real time RT-PCR. n = 10/group. *p<0.05 versus FA, student t test.
3.3. HPA axis mediates PM2.5-induced pulmonary inflammation
To determine the role of HPA axis on the PM2.5-induced adverse health effects, ADREX surgery was performed on female mice (10 weeks old), and depletion of stress hormone corticosterone was successfully introduced by the surgery as shown in Figure 3A. 2 weeks after recovery by 0.9% saline water, mice was intratracheally instilled with DEP and pulmonary inflammation level was assessed 24 hours after PBS/DEP treatment. Figures 3B reveals that intratracheal instillation of DEP significantly increased BALF total cells in SHAM group. While for ADREX mice, BALF cell numbers were not significantly different between DEP- vs. PBS-instilled mice, which suggests that HPA axis may mediate PM2.5-induced pulmonary inflammation. In addition, real time RT-PCR results (Figures 3C–E) show that pulmonary TNFα, IL-6 and IL-1β expressions are higher after intratracheal instillation of DEP compared with PBS-treated mice, and the differences were only significant in SHAM group but not ADREX group. Consistent with gene expression results, the BALF pro-inflammatory cytokine/chemokine analysis results (Figures 4A–C) also show that DEP instillation induced the pulmonary inflammation in SHAM group with levels of IL-6, VEGF and MIP-1α significantly higher for DEP- versus PBS-instilled mice, and the dramatic differences were attenuated by ADREX surgery for IL-6 and VEGF, but not for MIP-1α.
Figure 3. HPA axis mediates PM2.5-induced pulmonary inflammation responses.
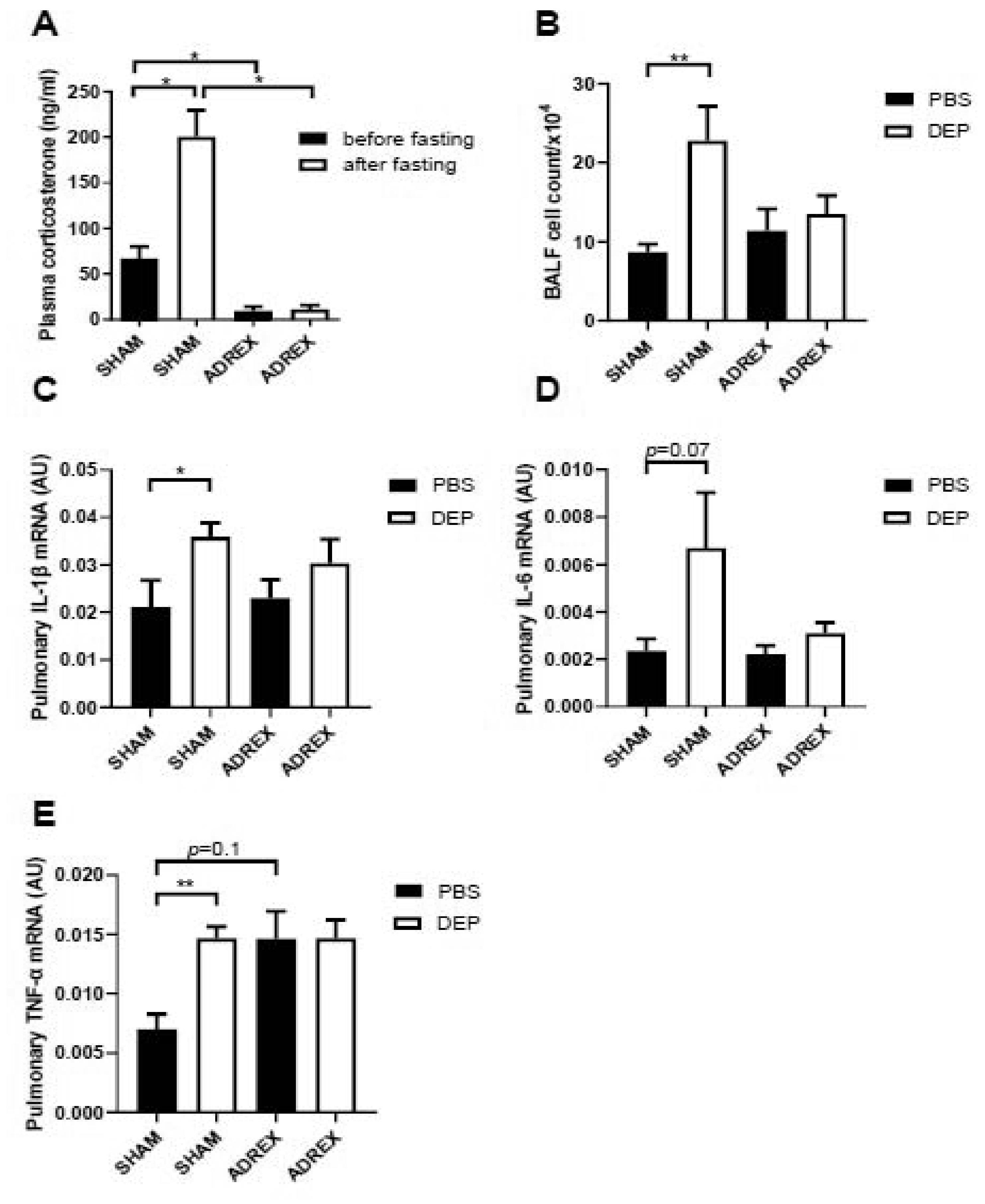
A, female C57Bl/6J mice were performed with control sham surgery or bilateral total adrenalectomy, and their plasma corticosterone levels were assessed before and after fasting for 24 hours. n=10/group. *p<0.05 versus sham or versus before fasting, two-way ANOVA. B-E, 2 weeks after recovery from sham or bilateral total adrenalectomy surgery, mice were instilled with DEP suspension (200 μg in 50 μl/mouse), and BALF cell number (B), pulmonary inflammatory gene expressions (C, IL-1β; D, IL-6; E, TNF-α) were assessed using real time RT-PCR. n=10/group. *p<0.05, **p<0.01 versus sham or versus PBS, two-way ANOVA.
Figure 4. Pro-inflammatory cytokines and chemokines levels in BALF after PBS/DEP instillation.
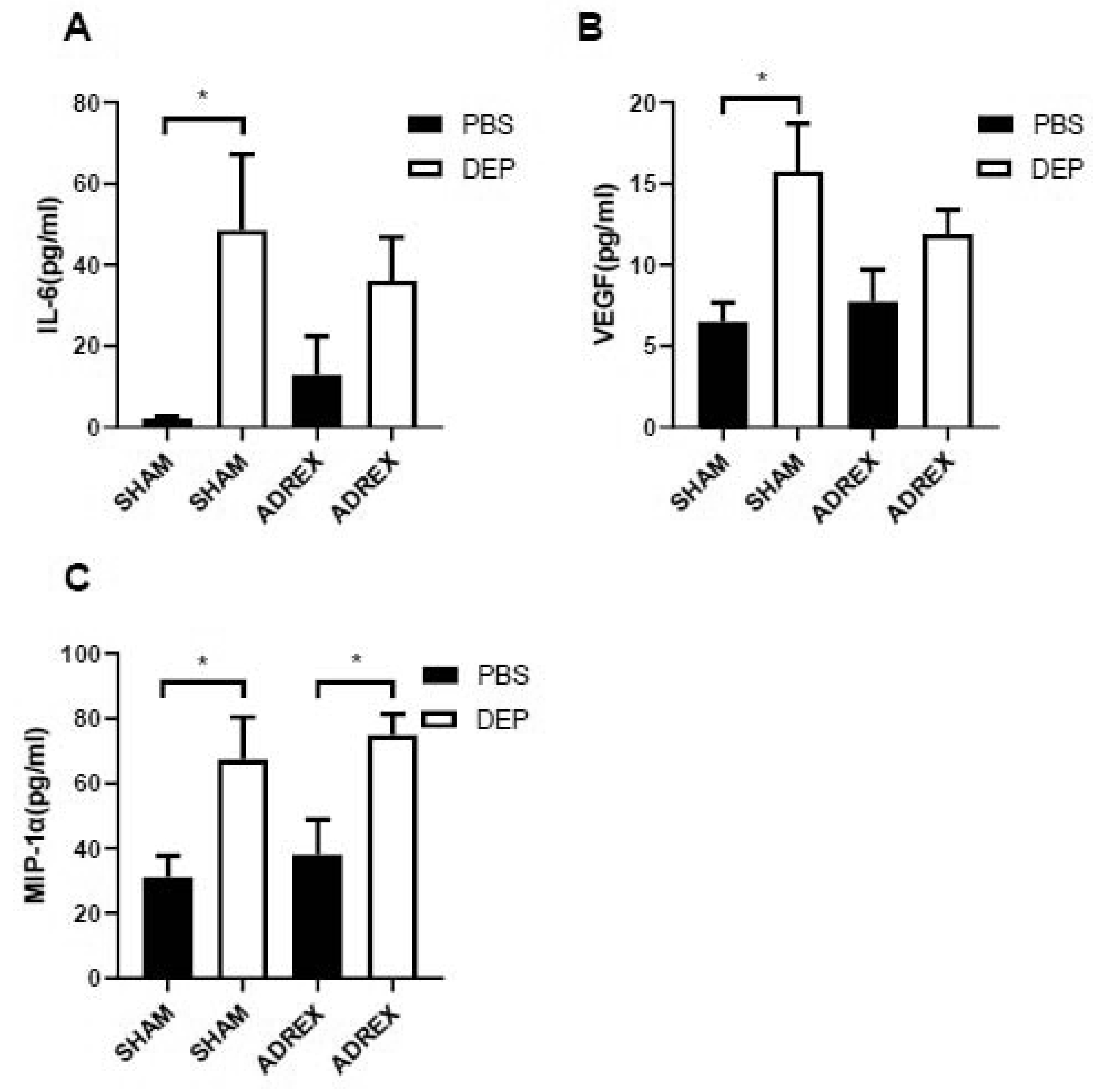
A-C, female C57Bl/6J mice were performed with control sham surgery or bilateral total adrenalectomy, instilled with PBS/DEP after recovery, and their BALF pro-inflammatory cytokines and chemokines (A, IL-6; B, VEGF; C, MIP-1α) levels were assessed. n=10/group. *p<0.05 versus sham or versus PBS, two-way ANOVA.
4. Discussion
Ambient PM2.5 exposure is associated with various cardiometabolic abnormalities that may lead to premature mortality, but the underlying biological mechanisms remain elusive (Brook et al., 2010). As the central stress response system, HPA axis plays a crucial role in the maintenance of cardiometabolic homeostasis. In this study, we revealed that PM2.5 exposure increases stress hormones in mouse models. Our results are consistent with previous studies demonstrating that airborne particulate matter exposure results in an increase in circulating stress hormone in rats (Sirivelu et al., 2006; Thomson et al., 2013). Most recently, our metabolomics analysis showed that the ambient PM2.5 level was associated with circulating stress hormones in apparently healthy college students (Li et al., 2017). Given the central role of the HPA axis in the regulation of cardiometabolic homeostasis, our data and others’ collectively implicate the HPA axis in the progression of adverse cardiometabolic effects caused by PM2.5 exposure.
The time-dependency analysis in this study reveals that PM2.5 instillation rapidly and transiently increased plasma corticosterone and ACTH levels in mice. This time course is reminiscent of the rapid, transient increase in blood pressure following exposure to concentrated ambient PM2.5 (CAP) shown in several controlled human exposure studies (Brook and Rajagopalan, 2009). Along with the well-known hypertensive effect of HPA axis activation (Vanitallie, 2002), this similarity in time course suggests that HPA axis activation may be more than a component of acute response to PM2.5 exposure and may also participate in the mediation of acute cardiometabolic effects of PM2.5 exposure.
Due to the well-known habituation process of neural responses, the neural mechanism is generally believed to mediate acute response to ambient PM2.5 exposure only (Brook et al., 2010). However, chronic psychosocial stress has been long known to correlate to various cardiometabolic diseases (Black and Garbutt, 2002). Furthermore, there is increasing evidence that neural mechanisms may contribute to the progression of various adverse health effects caused by chronic ambient PM2.5 exposure (Ying et al., 2014). Consistent with these, this study shows that chronic PM2.5 exposure significantly increased urine and hair stress hormone (Figure 2), suggesting that this HPA axis-mediated stress response may play a role in the pathophysiology due to chronic exposure to ambient PM2.5. Notably, in this study, we failed to observe a significant upregulation in plasma stress hormone following a 6-hour exposure to CAP, which appeared to be due to the failure of control of other stressors accompanying the exposure procedure as evidenced by the big variance of individual mice. This is consistent with one previous work demonstrating that a 4-hour exposure to particulate matter non-significantly increased plasma ACTH in rats (Thomson et al., 2013). Furthermore, CAP-exposed rats were shown to have increased circulating corticosterone one day after 8-hour exposure to CAP (Sirivelu et al., 2006). Together, our results demonstrated that both acute and chronic exposure to ambient PM2.5 activated HPA axis. These data suggest that the effect of PM2.5 exposure on HPA axis activity may be cumulative. This is consistent with the mode of cardiometabolic actions of PM2.5 exposure (EPA, 2009).
Emission of PM2.5 from road vehicles is found to be a prominent source of ambient PM2.5 in urban areas like Shanghai and New York. Diesel exhaust PM2.5 (DEP) with defined composition is commercially available and thus frequently used as a model for PM2.5 toxicological studies (Chen et al., 2018a; Chen et al., 2017; Heal et al., 2012; Weldy et al., 2013; Zou et al., 2018). Another crucial finding in this study is that HPA axis is activated by water-insoluble but not -soluble constituents of diesel exhaust PM2.5. This is consistent with the majority of literature showing that compared to the water-soluble constituents of ambient PM2.5 or PM10, their water-insoluble constituents are more relevant to their toxicity (Soukup and Becker, 2001; Walters et al., 2001). However, it should be noted that there is also a literature showing that the soluble metallic components of ambient PM2.5 may contribute to the progression of airway inflammation caused by ambient PM2.5 exposure (Pardo et al., 2015a). Therefore, further study may still be needed to determine if there is a synergy between the water-insoluble and -soluble constituents of ambient PM2.5 in the activation of the HPA axis. Researchers have found that other than the compositions, water-soluble and -insoluble fractions of PM2.5 may have different capacities to regulate cell function, induce oxidative stress and lipid peroxidation (Pardo et al., 2015b; Soukup and Becker, 2001). In addition, due to their difference in the accessibility to the systemic circulation, water-soluble and insoluble constituents of PM2.5 exert extra-pulmonary effects through different mechanisms (EPA, 2009). While water-soluble constituents directly cause various extra-pulmonary effects through the direct entrance to circulation, water-insoluble constituents impact extra-pulmonary organs by various mediators. The latter has not yet been fully understood. Therefore, the demonstration of HPA axis activation by water-insoluble PM2.5 component markedly furthers our understanding of mechanisms whereby PM2.5 inhalation leads to various extra-pulmonary effects.
By performing the bilateral total adrenalectomy surgery, stress hormones were depleted in mice and PM2.5-induced pulmonary inflammation level was alleviated, providing strong evidence that HPA axis may play a crucial role mediating PM2.5-induced adverse health effects. As shown in Figures 3 and 4, compared with SHAM surgery group, the pulmonary inflammation level in mice with ADREX was slightly higher, which may due to the anti-inflammatory effect of glucocorticoid (Vandewalle et al., 2018). Generally, in response to HPA activation, the adrenal cortex secretes glucocorticoids, while the adrenal medulla can be directly stimulated by the sympathetic efferent nerves and secrete epinephrine and/or norepinephrine(Goldstein, 2010). In addition, some studies have found that acute stressors-mediated increases of corticosteroids and epinephrine can simulate leukocyte trafficking and extravasation at the injury sites in an immune-cell specific manner(Dhabhar, 2009; Dhabhar et al., 2012). Altogether, the marked suppression of pulmonary inflammation in ADREX mice provides insights into the contribution of central neurohormonal mechanisms in modulating pulmonary inflammatory responses induced by PM2.5.
Although our work offers compelling evidence that inhalational PM2.5 exposure activates HPA axis which may help mediate PM2.5 exposure-induced pulmonary inflammation effect, the mechanism linking PM2.5 in the airway to HPA axis has to be determined. Notably, inhalation of ozone was shown to activate the nucleus tractus solitarius (NTS) and stress responsive regions of the hypothalamus through stimulation of pulmonary vagal C-fibers (Gackiere et al., 2011). Furthermore, PM2.5 has been shown to activate TRPA1, and TRPA1 agonists can activate vagal bronchopulmonary C-fibers (Birrell et al., 2009; Wang et al., 1996). Therefore, it is likely that inhalation exposure to PM2.5 may activate central stress response system through activation of vagal C-fibers.
5. Conclusions
In this study, we demonstrate compelling evidence that inhalational PM2.5 exposure activates HPA axis and HPA axis may help mediate PM2.5-induced pulmonary inflammation, offering a deep mechanistic insight into the development of adverse health effects due to exposure to PM2.5.
Highlights.
DEP exposure dose and time-dependently affects corticosterone and ACTH levels
Water-insoluble but not soluble component of DEP contributes to HP A axis activation
Chronic CAP exposure significantly increases mouse urine and hair corticosterone levels
Deletion of stress hormones alleviated PM2.5-induced pulmonary inflammation
Funding
This work was supported by the National Institutes of Health (R01ES024516 to ZY), the American Heart Association (13SDG17070131 to ZY), Shanghai Committee of Science and Technology (19DZ1204603 to YX) and the National Natural Science Foundation of China (Grant No. 81770805 to YX, 81500216 to MC, 91643205 to HK and 81302452 to LQ).
List of abbreviations
- FA
filtered air
- PM2.5
ambient fine particles
- CAP
concentrated ambient PM2.5
- HPA axis
hypothalamic-pituitary-adrenal axis
- DEP
diesel exhaust PM2.5
- ACTH
adrenocorticotropic hormone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics approval and consent to participate
UMB and Fudan University are AAALAC accredited institutions. All procedures of this study were approved by the Institutional Animal Care and Use Committee (IACUC) at UMB or Fudan University, and all the animals were treated humanely and with regard for alleviation of suffering.
Consent for publication
Not applicable
Availability of data and material
The datasets during and/or analysed during the current study available from the corresponding author on reasonable request.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Birrell MA, et al. , 2009. TRPA1 agonists evoke coughing in guinea pig and human volunteers. Am J Respir Crit Care Med. 180, 1042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black PH, Garbutt LD, 2002. Stress, inflammation and cardiovascular disease. J Psychosom Res. 52, 1–23. [DOI] [PubMed] [Google Scholar]
- Brook RD, Rajagopalan S, 2009. Particulate matter, air pollution, and blood pressure. J Am Soc Hypertens. 3, 332–50. [DOI] [PubMed] [Google Scholar]
- Brook RD, et al. , 2010. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 121, 2331–78. [DOI] [PubMed] [Google Scholar]
- Buteau S, Goldberg MS, 2016. A structured review of panel studies used to investigate associations between ambient air pollution and heart rate variability. Environ Res. 148, 207–47. [DOI] [PubMed] [Google Scholar]
- Calderon-Garciduenas L, Torres-Jardon R, 2015. The impact of air pollutants on the brain. JAMA Psychiatry. 72, 529–30. [DOI] [PubMed] [Google Scholar]
- Chen M, et al. , 2018a. Prenatal exposure to diesel exhaust PM2.5 causes offspring beta cell dysfunction in adulthood. Am J Physiol Endocrinol Metab. 315, E72–E80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, et al. , 2017. Prenatal and postnatal mothering by diesel exhaust PM2.5-exposed dams differentially program mouse energy metabolism. Part Fibre Toxicol. 14, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, et al. , 2018b. Concentrated Ambient PM2.5-Induced Inflammation and Endothelial Dysfunction in a Murine Model of Neural IKK2 Deficiency. Environ Health Perspect. 126, 027003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, 2009. A hassle a day may keep the pathogens away: The fight-or-flight stress response and the augmentation of immune function. Integrative and Comparative Biology. 49, 215–236. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, et al. , 2012. Stress-induced redistribution of immune cells-From barracks to boulevards to battlefields: A tale of three hormones - Curt Richter Award Winner. Psychoneuroendocrinology. 37, 1345–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA, U. S. A., Integrated Science Assessment (ISA) for Particulate Matter. Vol. 2017 U.S. Environmental Protection Agency, 2009. [PubMed] [Google Scholar]
- Gackiere F, et al. , 2011. Ozone inhalation activates stress-responsive regions of the CNS. J Neurochem. 117, 961–72. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, 2010. Adrenal Responses to Stress. Cellular and Molecular Neurobiology. 30, 1433–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal MR, et al. , 2012. Particles, air quality, policy and health. Chem Soc Rev. 41, 6606–30. [DOI] [PubMed] [Google Scholar]
- Hu Z, et al. , 2017. Inactivation of TNF/LT locus alters mouse metabolic response to concentrated ambient PM2.5. Toxicology. 390, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyjovska ZO, et al. , 2015. DNA strand breaks, acute phase response and inflammation following pulmonary exposure by instillation to the diesel exhaust particle NIST1650b in mice. Mutagenesis. 30, 499–507. [DOI] [PubMed] [Google Scholar]
- Lepeule J, et al. , 2012. Chronic exposure to fine particles and mortality: an extended follow-up of the Harvard Six Cities study from 1974 to 2009. Environ Health Perspect. 120, 965–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. , 2017. Particulate Matter Exposure and Stress Hormone Levels: A Randomized, Double-Blind, Crossover Trial of Air Purification. Circulation. 136, 618–627. [DOI] [PubMed] [Google Scholar]
- Miller DB, et al. , 2016. Ozone Exposure Increases Circulating Stress Hormones and Lipid Metabolites in Humans. Am J Respir Crit Care Med. 193, 1382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo M, et al. , 2015a. Single Exposure to near Roadway Particulate Matter Leads to Confined Inflammatory and Defense Responses: Possible Role of Metals. Environ Sci Technol. 49, 8777–85. [DOI] [PubMed] [Google Scholar]
- Pardo M, et al. , 2015b. Single Exposure to near Roadway Particulate Matter Leads to Confined Inflammatory and Defense Responses: Possible Role of Metals. Environmental Science & Technology. 49, 8777–8785. [DOI] [PubMed] [Google Scholar]
- Sirivelu MP, et al. , 2006. Activation of the stress axis and neurochemical alterations in specific brain areas by concentrated ambient particle exposure with concomitant allergic airway disease. Environ Health Perspect. 114, 870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup JM, Becker S, 2001. Human alveolar macrophage responses to air pollution particulates are associated with insoluble components of coarse material, including particulate endotoxin. Toxicol Appl Pharmacol. 171, 20–6. [DOI] [PubMed] [Google Scholar]
- Thomson EM, et al. , 2013. Mapping acute systemic effects of inhaled particulate matter and ozone: multiorgan gene expression and glucocorticoid activity. Toxicol Sci. 135, 169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle J, et al. , 2018. Therapeutic Mechanisms of Glucocorticoids. Trends in Endocrinology and Metabolism. 29, 42–54. [DOI] [PubMed] [Google Scholar]
- Vanitallie TB, 2002. Stress: a risk factor for serious illness. Metabolism. 51, 40–5. [DOI] [PubMed] [Google Scholar]
- Walters DM, et al. , 2001. Ambient urban Baltimore particulate-induced airway hyperresponsiveness and inflammation in mice. Am J Respir Crit Care Med. 164, 1438–43. [DOI] [PubMed] [Google Scholar]
- Wang AL, et al. , 1996. Vagal bronchopulmonary C-fibers and acute ventilatory response to inhaled irritants. Respir Physiol. 104, 231–9. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. , 2018. Exposure to concentrated ambient PM2.5 alters the composition of gut microbiota in a murine model. Part Fibre Toxicol. 15, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldy CS, et al. , 2013. In utero and early life exposure to diesel exhaust air pollution increases adult susceptibility to heart failure in mice. Part Fibre Toxicol. 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilker EH, et al. , 2015. Long-term exposure to fine particulate matter, residential proximity to major roads and measures of brain structure. Stroke. 46, 1161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, et al. , 2014. Long-term exposure to concentrated ambient PM2.5 increases mouse blood pressure through abnormal activation of the sympathetic nervous system: a role for hypothalamic inflammation. Environ Health Perspect. 122, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, et al. , 2018. PM2.5 Induced the Expression of Fibrogenic Mediators via HMGB1-RAGE Signaling in Human Airway Epithelial Cells. Can Respir J. 2018, 1817398. [DOI] [PMC free article] [PubMed] [Google Scholar]