

Abstract
Liquid chromatography–tandem mass spectrometry is an increasingly powerful tool for studying proteins in the context of disease. As technological advances in instrumentation and data analysis have enabled deeper profiling of proteomes and peptidomes, the need for a rigorous, standardized approach to validate individual peptide–spectrum matches (PSMs) has emerged. To address this need, we developed a novel and broadly applicable workflow: PSM validation with internal standards (P-VIS). In this approach, the fragmentation spectrum and chromatographic retention time of a peptide within a biological sample are compared with those of a synthetic version of the putative peptide sequence match. Similarity measurements obtained for a panel of internal standard peptides are then used to calculate a prediction interval for valid matches. If the observed degree of similarity between the biological and the synthetic peptide falls within this prediction interval, then the match is considered valid. P-VIS enables systematic and objective assessment of the validity of individual PSMs, providing a measurable degree of confidence when identifying peptides by mass spectrometry.
Keywords: peptide–spectrum match, validation, peptide splicing, hybrid insulin peptides, peptidomics, immunopeptidomics, post-translational modification, immunotherapy
Graphical Abstract
INTRODUCTION
Recently, there has been rapidly growing interest in using liquid chromatography–tandem mass spectrometry (LC–MS/MS) to identify naturally occurring peptides (peptidomics), particularly immunologically relevant peptides presented by human leukocyte antigen (HLA) molecules (immunopeptidomics). For example, the Human Immunopeptidome Project (HIPP) was established to identify the entire immunopeptidome,1,2 immunopeptidome-wide association studies (IWAS) have been proposed,3 and early efforts have demonstrated potential for using immunopeptidomics to identify neoantigen targets for personalized cancer immunotherapy.4–6 However, technical hurdles still exist before the full potential of peptidomics can be realized. The recent debate surrounding the identification of proteasomally spliced peptides showcases some of these challenges.7–12 Several publications have established that the proteasome can join noncontiguous regions from a single protein or two different proteins to generate spliced peptides.13 However, efforts to assess the extent to which these spliced peptides are presented on HLA class I molecules have produced vastly different estimates.7,8 Ultimately, these differences originate from disagreement over specific peptide–spectrum matches (PSMs). If peptidomics is to be used as a keystone technology for personalized medicine, then a rigorous and standardized method for validating individual PSMs is essential.
In routine bottom-up mass spectrometry approaches, complex protein samples are proteolytically digested, and the resultant peptide mixtures are analyzed by LC–MS/MS. Because the presence of a given protein in the sample is inferred from the identification of several peptides derived from that protein, less confidence is needed in any individual PSM. In contrast, for experiments in which the goal is to identify specific peptides or to determine if specific residues in a protein are modified, conclusions may need to be based on only a single PSM. Consequently, a much higher degree of confidence in a single match is needed.14 Furthermore, as mass spectra are often ambiguous—in many cases, a single spectrum can be matched to multiple peptide sequences—a high level of confidence may be difficult to attain for individual peptides.
This central problem can be exacerbated by other factors. For example, when proteins are digested with a protease during sample preparation, the search space can be limited to only those peptide sequences predicted by in silico digestion of protein sequences in a database. However, if native peptides are analyzed, as is the case in immunopeptidomics experiments then this restriction cannot be imposed. This leads to larger search spaces and an increased risk of false discoveries. Furthermore, while digestion with trypsin generates peptides with a basic residue on their C-termini, which often yield high-quality fragmentation spectra, native peptides may lack such basic C-terminal residues and yield spectra that are difficult to interpret confidently. Additionally, if post-translational modifications (PTMs) are considered, then the search space, and consequently the risk of false-positive identifications, can increase dramatically.
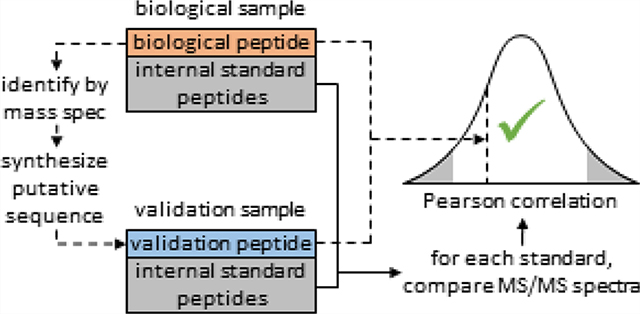
Given these challenges, routine methodologies alone do not always provide sufficient confidence when identifying native peptides or modified sequences; thus, further validation is required. A common approach for validating PSMs is to compare the spectrum of interest to the spectrum for a synthetic version of the putative sequence match using a similarity metric such as the Pearson correlation coefficient (PCC).15 These calculations can be cumbersome, and results can differ depending on which spectral peaks are considered in the calculation.16 Because two spectra for even the same peptide will not be completely identical, the overarching problem is in determining what degree of similarity is sufficient to indicate a correct match; currently, this decision is made subjectively. To address the need for a rigorous approach to directly validate PSMs, we developed a broadly applicable workflow—PSM validation with internal standards (P-VIS)—that reduces the subjectivity when evaluating the validity of PSMs. We created a computer program, PSM_validator, to automate and standardize the data analysis portion of the workflow. The P-VIS workflow is particularly useful for validating identifications based on a single PSM, making it a powerful tool in applications such as peptidomics, immunopeptidomics, and the discovery of post-translational modifications.
METHODS
P-VIS Workflow
A detailed overview of the P-VIS workflow is provided in Figure 1. The investigator first uses LC–MS/MS and traditional data analysis, such as a database search, to identify a PSM of interest for a peptide in a biological sample. The putative peptide sequence is considered the query sequence. A synthetic version of the query sequence is then obtained for validation purposes. This validation peptide and the biological sample are each spiked with a panel of internal standard peptides (ISPs). The spiked samples are then analyzed on the same LC–MS/MS system, using identical settings for both samples. PSM_validator is then used to compare the fragmentation spectrum of the biological peptide to the spectrum of the validation peptide by calculating the PCC. The panel of ISPs, which is present in both samples, provides several pairs of matching peptides. These matching peptides are used by PSM_validator to assess the similarity of fragmentation spectra for a given peptide between the two samples and calculate a prediction interval for the PCC of a valid match. If the degree of correlation between the spectrum for the biological peptide and the spectrum for the validation peptide falls within the prediction interval, then the biological peptide and the validation peptide are reported as a match (indicating that the PSM is valid). A similar analysis is conducted based on the chromatographic retention time (RT) of each peptide in the biological run and the validation run, thereby providing an additional measurement of validity.
Figure 1.

Schematic of the PSM validation with internal standards (P-VIS) workflow. (a) Complex mixture of biological peptides is analyzed by LC–MS/MS. (b) Peptide–spectrum matches (PSMs) are generated using a traditional data analysis workflow, and a PSM of interest for a biological peptide is identified. The putative sequence match is considered the query sequence. (c) Sample containing a synthetic version of the query sequence is prepared (validation peptide). (d) Set of internal standard peptides (ISPs) is added to the biological sample and the validation sample, and (e) both samples are analyzed by LC–MS/MS. Data is then evaluated using PSM_validator. (f) For each ISP and the query sequence, PSM_validator finds the best spectrum in the biological sample data and the validation sample data and (g) calculates the Pearson correlation coefficient (PCC) between the two spectra. (h) For each ISP, and for the biological peptide and the validation peptide, PSM_validator determines the chromatographic retention time (RT) in the biological sample run and the validation sample run and (i) calculates an adjusted delta RT (using a linear spline model to align the runs). (j) Using the ISP results, a normal distribution is modeled for the PCC and for the delta RT of true matches (comparison of the same ISP in two different runs). (k) Percentile is calculated for the PCC and for the delta RT for the comparison of the biological peptide and the validation peptide. If the percentile for both the PCC and for the delta RT surpasses a user-defined threshold, then the analysis is given a passing result, indicating that the biological peptide and the validation peptide have the same sequence (the PSM is correct).
PSM_validator Algorithm
The PSM_validator source code accompanies the manuscript as the Supporting Information. Both the PSM_validator source code and a standalone executable of PSM_validator are freely available at https://github.com/Delong-Lab/PSM_validator/releases under the Creative Commons Attribution 4.0 International Public License. The PSM_validator code is written in Python 3.7.4. The standalone executable was generated using PyInstaller 3.5. The executable may be the preferred method for running PSM_validator for most users. Dependencies for executing the source code are the Matplotlib17 and SciPy18 libraries, which can be separately installed or are part of the default installation of the Anaconda distribution of Python.
Input.
Prior to analyzing LC–MS/MS data with PSM_validator, each raw mass spectrometry data file must be converted to both a Mascot generic format (MGF) file and an MS1 file. The name of the MGF file and the MS1 file for a given sample must be the same. For file conversion, we used the open-source tool MSConvert.19 The time required for PSM_validator analysis can be reduced by including filters during conversion with MSConvert to remove low abundance peaks or peaks outside the m/z range of the ISPs and the peptides to be validated. The MGF files and the MS1 files for the biological sample and validation sample must be placed in a single directory. The PSM_validator.exe file (or the source code) is associated with a directory titled “parameters” that contains four CSV files that allow for user input: “amino_acids.csv”, “queries.csv”, “settings.csv”, and “standards.csv”. PSM_validator.exe (or the source code) and the “parameters” directory must be kept within the same directory for the program to run. The CSV files in “parameters” can be manipulated in a spreadsheet editor such as Microsoft Excel and must be saved after editing for the changes to take effect. “amino_acids.csv” contains a list of the 20 canonical amino acids (single-letter code) and their monoisotopic residue masses. The user can add custom amino acids to this list by providing a single-character name (case sensitive) and an exact mass, thus allowing PSM_validator to handle modified amino acids. In “queries.csv”, the user enters the analyses to be performed in batch format. For each analysis, the user indicates the name of the biological files and the name of the validation files, the directory containing the files, and the putative peptide sequence (the query sequence) being validated. If the sequence contains an N-terminal and/or a C-terminal modification, this can be indicated by entering an N-terminal and/or a C-terminal mass shift. Multiple analyses are performed automatically in the order listed. “settings.csv” provides access to all user-adjustable algorithm parameters, such as mass tolerances, along with a description of each and information on acceptable values. PSM_validator is capable of processing data generated by collision-induced dissociation (CID), which is usually dominated by b and y ions, or electron transfer dissociation (ETD), which is usually dominated by c and z ions. Within “settings.csv”, the user can indicate if the algorithm should consider either b and y ions or c and z ions. “standards.csv” contains the amino acid sequence for each of the ISPs. By default, the ProteomeTools Calibration Standard (PROCAL; JPT Peptide Technologies)20 sequences are listed. PROCAL is a mixture of 40 non-natural peptides spanning a broad range of hydrophobicities. After these files have been updated, saved, and closed, the user clicks on the “PSM_validator.exe” icon (or executes the source code using a Python interpreter) to begin execution of the analyses.
Output.
Execution of PSM_validator opens a console window in which real-time results are displayed, allowing the user to monitor progress and results. All analyses dictated in “queries.csv” are performed automatically in the order listed. Analysis time can vary widely, depending on various factors such as the size of the data files, the number of spectra with the correct precursor mass, and the computer being used to run the analysis. The analyses presented here were performed on a laptop computer with an Intel Core i7 processor, 32 GB of RAM, and a 64-bit operating system; validation of a single peptide sequence (which consists of comparing the fragmentation spectrum and RT between the biological sample run and the validation sample run for each ISP and the peptide sequence being validated) generally took 1–3 min. After all analyses are complete, a time-stamped, high-level batch result summary file containing the spectrum comparison result and the RT comparison result for each analysis is saved to the directory containing the MGF and MS1 data files used for the first analysis. For each individual analysis, a time-stamped folder named after the peptide sequence is saved in the directory containing the MGF and MS1 data files used for that analysis. This folder contains a detailed results summary file, which contains most of the information displayed in the real-time output, and two subfolders: “Figures” and “Tables”. “Figures” contains nine automatically generated plots, providing an intuitive representation of the results and allowing the user to manually check the veracity of the assessment and evaluate if appropriate settings were used. Selecting the “verbose” option for a given analysis in “queries.csv” will cause a subfolder named “ISPs” to be generated that contains figures detailing results for each ISP. “Tables” contains detailed scoring results and other details from the analysis in tabular form.
Scoring.
Validation of each individual peptide listed in “queries.csv” involves three major steps: scoring, comparison of fragmentation spectra, and retention time (RT) analysis. Each of these steps includes not only assessment of the peptide being validated but also each of the ISPs. PSM_validator works through the list of peptides in “queries.csv” in order, performing the entire validation process for a single peptide (including the necessary evaluation of all ISPs in the samples as well) then moving to the next peptide in the list of queries until all peptides have been assessed.
The PSM_validator algorithm first generates filtered MGF files that contain only spectra with precursor masses that match (within a user-defined window for mass error tolerance) the query peptide mass or one of the ISP masses. Next, the program identifies in the filtered MGF file for the biological sample the spectrum with the highest backbone coverage of the query amino acid sequence. Backbone coverage refers to the percentage of peptide bonds in the sequence that are represented by fragment ions in the observed spectrum. A backbone coverage of 100% indicates that the fragmentation spectrum provides direct evidence for the entire amino acid sequence of the peptide. If coverage of a region of the backbone is missing, then the spectrum provides less certainty of the precise sequence in that region. If multiple spectra provide the same backbone coverage, then the algorithm selects the spectrum in which the summed intensity of all the backbone coverage ions accounts for the highest percentage of the total ion signal in the spectrum. The precursor 12C peak is excluded from the calculation of the total ion signal. The program then finds the best-matching spectrum in the filtered MGF file for the validation sample. Selection of the bestmatching validation spectrum is limited to spectra with the same precursor charge state as the spectrum chosen for the biological sample. The best spectra are also identified for each of the ISPs.
Comparison of Fragmentation Spectra.
Once the best-matching spectrum in the biological file and the validation file are found for the peptide of interest and each ISP, PSM_validator calculates the PCC for each pair of spectra. Some previous studies have included in the PCC analysis only those peaks that correspond to ions predicted by the chosen sequence interpretation.7,9,15 This, however, introduces bias to the analysis. For example, consider the scenario in which a peptide in a biological sample is erroneously matched to sequence X in a database. A fragmentation spectrum is obtained for a synthetic version of sequence X (the validation peptide). Because the biological peptide and validation peptide have similar (but different) sequences, the fragmentation spectra are very similar. However, the spectrum for the biological peptide contains a dominant peak that does not correspond to a predicted fragment ion for sequence X and is not present in the validation peptide spectrum. Although this peak suggests that the biological peptide is not the same as the validation peptide, it would be omitted from the analysis since it would not be predicted based on sequence X. This would result in an artificially high PCC value.
The PSM_validator algorithm addresses this concern by initially considering every peak in each of the two spectra being analyzed. The algorithm works through each peak in the validation spectrum one at a time. For each peak, the algorithm finds the peak in the biological spectrum with the closest m/z. If the difference between the m/z of the two peaks is within a user-defined window, then the peaks are paired. If no peak is found within this window, then the peak in the validation spectrum is paired with an intensity of zero for the biological spectrum. Any peaks in the biological spectrum that remain unmatched are paired with an intensity of zero for the validation spectrum. If a peak in one spectrum is matched to more than one peak in the other spectrum, then the algorithm pairs it with the peak that has the closest m/z value. Each spectrum began as a list of paired m/z and intensity values. Following pairing between the two spectra, a single list of pairs is generated, with one member of each pair being the intensity of the peak in the biological spectrum and the other member being the intensity in the validation spectrum. Peaks that correspond to an intact precursor are excluded. This list is then filtered to include only those intensity pairs in which at least one member is above both of two different abundance thresholds. The first is a basic noise threshold (abund_thresh) that can be adjusted by the user based on the level of noise typically seen in fragmentation spectra generated by the instrument being used. The second threshold, PCC_abund_thresh, which can also be adjusted by the user, requires that a peak have an abundance that is greater than the user-specified percentage of the abundance of the largest peak in the spectrum. (We recommend a PCC_abund_thresh of 10%.) This allows for exclusion of peaks that are not noise but are minor peaks in the spectrum. Including multiple relatively small peaks can “buffer” the PCC, leading to high PCC values that do not reflect large differences in more prominent peaks. The PCC is then calculated for the filtered list.
In complex biological samples, multiple precursors are sometimes co-isolated for fragmentation, yielding a composite fragmentation spectrum. Because the algorithm initially considers all peaks rather than just those that match the proposed sequence interpretation, it should be noted that co-isolation could result in a low PCC even when the same peptide is being compared between the biological sample and the validation sample. If this occurs for an ISP, then the prediction interval may become broader, increasing the risk of false negatives. To address this problem, a minimum PCC threshold can be set by the user. ISPs with a PCC below this threshold are excluded when calculating the prediction interval. We recommend setting the threshold at 0.7. It should also be noted that co-isolation of another precursor with the biological peptide of interest could result in a low PCC that causes a valid match to be reported as invalid.
As discussed above, a major challenge is determining what level of similarity between spectra is necessary in order to be confident that the spectra originated from fragmentation of the same peptide sequence. Since each ISP is present in both samples, the PCC values calculated for comparisons between the best spectrum in each sample for the ISPs are used to model the distribution of PCC values for correct matches. Prior to analysis, the user defines a percentile for a one-sided prediction interval. By selecting a 95% prediction interval, for example, the user asserts that the match will be considered correct if the PCC between the biological spectrum and the validation spectrum is greater than or equal to the lower bound of the estimated 95% prediction interval for PCC values of correct matches. (Because only a low PCC, and not a high PCC, would suggest an invalid match, a one-tailed prediction interval, rather than a two-tailed prediction interval, is used.) In this way, an objective decision about the validity of a match is made based on what level of similarity can be expected between the two spectra when the match is correct. Because PCC values are bound by 1 and –1, Fisher’s Z-transformation is used when calculating the prediction interval. The D’Agostino–Pearson omnibus normality test is used to determine if the transformed PCC values for the ISPs follow a normal distribution. The results of this test are reported in the “normality” subfolder in the “Figures” output. Note that for the normality test, the null hypothesis is that the population is normally distributed; the null hypothesis can be rejected if the p-value is less than the chosen significance level (we chose α = 0.05). Thus, a p-value greater than 0.05 would be considered a passing result.
Retention Time (RT) Analysis.
After completing the fragmentation spectrum-based analysis, PSM_validator next assesses the validity of the match based on chromatographic RT. The MS1 data files are used for RT analysis. An extracted ion chromatogram is generated for a given precursor, and the time at which the chromatogram is the highest is considered the RT for that precursor. To reduce analysis time, extraction is limited to a time window centered on the time when the best-scoring spectrum was acquired. The users define the size of the time window in “settings.csv” based on the typical chromatographic peak width observed for their system.
RT for the same peptide varies between runs in a nonlinear fashion. This relationship could be modeled using a polynomial. However, a low-order polynomial will sometimes provide a poor fit. While increasing the order of the polynomial can sometimes improve the fit to a nonlinear data set, it may decrease the accuracy of interpolation (Runge’s phenomenon). To avoid this problem, we used a linear spline approach as described previously.16 The algorithm determines the RT for the peptide of interest in the biological sample run and the RT for each of the two flanking ISPs. It then finds the RT for these two ISPs in the validation run and calculates a linear model of the RT relationship for that segment of the two runs. This model is used to predict what the RT of the biological peptide would have been in the validation run if it had been present in the validation sample. The difference between this predicted RT and the observed RT for the validation peptide is considered the delta RT.
Even if the biological peptide and the validation peptide are the same, delta RT may not be zero as the linear spline model is not perfect. The algorithm therefore performs the same modeling and prediction approach for each ISP, treating the ISP as the peptide of interest and using the two flanking ISPs to generate a prediction model. This calculation is not done for the first and last ISP to elute in the biological sample run as these are not flanked by an ISP on both sides. These calculations generate a list of delta RTs for the ISPs and the peptide of interest. As with the PCC values, a normal distribution is calculated based on the ISP results, and the percentile for the delta RT of the validation peptide is reported. The user-defined percentile threshold is used to classify the biological peptide and the validation peptide as a match or mismatch based on RT analysis. The results of the D’Agostino–Pearson omnibus normality test of the ISP delta RTs are provided in the “normality” subfolder of the “Figures” output. In our experience, run-to-run RT variability increases at the very beginning and end of runs. Furthermore, with the ISPs and gradients used in our experiments, fewer ISPs elute in these regions. These factors sometimes reduce the reliability of the linear spline model in these regions. To account for this, “settings.csv” contains an option for the user to manually define a time range for the RT analyses. This allows the user to inspect the data and exclude ranges early and late in the run where the linear spline model is likely to perform poorly.
Handling of Spliced and Hybrid Peptides.
PSM_validator handles spliced and hybrid peptides the same as any other peptides. Inclusion of a hyphen in the amino acid sequence at the site of fusion/splicing indicates to PSM_validator that the sequence is hybrid/spliced. This does not affect the mainstream analysis, such as the selection of the best matching spectra or calculation of the PCC, in any way. However, it will activate two features. For hybrid/spliced sequences, PSM_validator will report the backbone coverage for the left and right sides of the peptide in addition to the backbone coverage for the entire peptide sequence. In the mirror plot, ions will be labeled as left ions (L ions) and right ions (R ions) rather than b and y ions or c and z ions. L ions are those ions (whether b, y, c, or z ions) that result from fragmentation of peptide bonds that are to the left of the hybrid/spliced junction. R ions are those that correspond to fragmentation of bonds that are to the right of the junction. A b or c ion corresponding to fragmentation at the junction bond is labeled as an L ion, and a y or z ion corresponding to fragmentation at the junction bond is labeled as an R ion. Although these features do not affect the validation results, they allow the user to assess if the spectrum provides direct sequence evidence for both sides of the hybrid/spliced sequence.
Synthetic Peptides and ISPs
PROCAL20 (JPT Peptide Technologies, Germany) was used for ISPs. Synthetic 6.9HIP peptide was obtained from SynPeptide. Native and spliced benchmarking peptides were obtained from GenScript at >95% purity.
Mass Spectrometry Analysis
For nano-flow LC–MS/MS analyses, samples were analyzed on an Agilent 1200 series UHPLC system with a nano-flow adapter and an Agilent 6550 Q-TOF equipped with a nano-ESI source. Benchmarking samples were separated by online reversed-phase liquid chromatography using a Thermo Acclaim Pepmap 100 C18 trap column (75 μm × 2 cm; 3 μm particles; 100 Å pores) and Thermo Acclaim Pepmap RSLC C18 analytical column (75 μm × 15 cm; 2 μm particles; 100 Å pores) in a trap forward-elute configuration. The following 90 min linear gradient was used (buffer A = 0.1% formic acid/water; buffer B = 90% acetonitrile/0.1% formic acid/water; flowrate ≈ 300 nL/min; column temperature = 40 °C): 0–1 min = 3% B, 66 min = 30% B, 71–72 min = 100% B, 73–90 min = 3% B. Centroided mass spectrometry data were collected in positive ion mode with an MS scan range of 290–1700 m/z and an MS acquisition rate of 5 spectra/s. A maximum of 10 precursors per cycle was selected for fragmentation. Precursor isolation width was set at narrow. Fragmentation was performed by CID. The Agilent software determines collision energy using the following equation: (slope) × (m/z)/100 + (offset). A slope of 3.1 and an offset of 1 were used for precursors of all charge states. MS/MS scans were collected with a scan range of 50–1700 m/z and a minimum scan rate of 3 spectra/s. Abundance-dependent accumulation, which varies the MS/MS scan speed based on precursor abundance, was enabled with a target of 80,000 counts/spectrum. The theoretical mass-to-charge ratio (m/z) of doubly and triply charged forms of each of the spliced peptides, native peptides, and ISPs was added to a preferred ion list. Precursor selection was limited to this list to ensure the collection of high-quality spectra and fair comparison between biological and validation samples.
Micro-flow LC–MS/MS analysis was performed using a similar configuration and settings as described above. Peptides were separated by direct loading onto an XBridge Peptide BEH C18 column, 130 Å, 3.5 μm, 1 mm × 150 mm (Waters) at a flow rate of 55 μL/min. Peptides were introduced to the mass spectrometer using the Agilent dual Agilent jet stream (dual-AJS) ESI source.
Mouse Islet Analysis
The mouse islet experiment was performed as published previously.16 Briefly, pancreatic islets isolated from non-obese diabetic (NOD) mice were lysed in triflouroethanol, and proteins were separated by size exclusion chromatography, digested with the protease AspN, and analyzed by nano-LC–MS/MS.
Data Availability
Benchmarking data and mouse islet data, as well as all PSM_validator result files, have been deposited to the ProteomeXchange Consortium21 via the PRIDE22 partner repository with the dataset identifier PXD020901.
RESULTS AND DISCUSSION
To evaluate performance of the P-VIS workflow, we turned to the recent dispute regarding the frequency of proteasomally spliced peptides in the human immunopeptidome. In a publication by Liepe et al.,7 the investigators assessed the extent to which spliced peptides are presented on the surface of human cells. By re-analyzing a published dataset23 obtained from LC–MS/MS analysis of HLA class I-eluted peptides, Liepe et al. concluded that spliced peptides constitute approximately one-third of the HLA class I immunopeptidome.7 An independent group, Mylonas et al., reanalyzed the data using a different approach and contended that 2–6% was a more reasonable upper limit for the contribution of spliced peptides and that the actual proportion was likely even smaller.8 Many of the spectra that were matched by Liepe et al. to spliced peptide sequences were matched by Mylonas et al. to non-spliced, native peptide sequences in the UniProt human proteome database.
To use P-VIS to determine if the spliced or native PSM was validated for any given spectrum, we would need to spike the original samples with ISPs and reanalyze them by mass spectrometry. As this was not feasible, we designed an experiment to model this situation. First, a list of spectra that were matched by Liepe et al.7 to spliced peptides and by Mylonas et al.8 to native peptides was obtained from the supplementary data provided by Mylonas et al.8 This list was then sorted by RT, and the spectra were divided into 10 min retention time windows from 30 to 80 min. Within each time window, the two or three spectra with the highest-scoring matches in Liepe et al.’s search (spliced peptide interpretations) were chosen, resulting in a list of 11 disputed spectra. (In the 40–50 minute window, the first and third spectra were chosen. One of the peptide matches for the second spectrum—the native sequence VEGEGEEEGEEY—lacked any basic residues and was thus expected to ionize poorly.) Next, for each of the disputed spectra, we obtained a synthetic version of the spliced peptide sequence match (proposed by Liepe et al.) and the native peptide sequence match (proposed by Mylonas et al.), resulting in a set of 11 spliced peptides and 11 corresponding native peptides. We then pooled the 11 synthetic spliced peptides in one sample and the 11 synthetic native peptides in another sample. Additionally, we added a standardized HeLa cell protein digest to an aliquot of each pool for use as a mock-biological sample, creating a final total of four samples: native pool, native pool + HeLa, spliced pool, and spliced pool + HeLa (Figure 2). A set of 40 ISPs (PROCAL) was spiked into each of the four samples. Final samples contained spliced or native peptides at 250 fmol/μl/ peptide and PROCAL ISPs at 150 fmol/μl/peptide in 2.7% acetonitrile/0.1% formic acid/water. Mock-biological samples contained HeLa protein digest (Thermo Pierce) at a concentration of 0.15 μg/μl. The following spliced peptides were not observed by mass spectrometry when included at 250 fmol/μl/peptide and were therefore included at 2.5 pmol/μl/ peptide: KESTISVAQK, KRDGTAGILK, KPTTGKELALK, and KFSVEDMAELT. Manual inspection of the data confirmed that, following this adjustment, the observed precursor intensities for all 22 peptides (11 native and 11 spliced) were sufficient to enable the collection of fragmentation spectra.
Figure 2.

Experimental design for benchmarking the P-VIS workflow. Eleven spectra for which Liepe et al. proposed spliced peptide matches and Mylonas et al. proposed native (non-spliced) matches were chosen. Synthetic peptides were obtained for each of these proposed sequences. All 11 spliced peptides were pooled together into a single sample, and all 11 native peptides were pooled together into a single sample. For each of these two samples, two aliquots were made, and HeLa cell protein extract was added to one of the aliquots to mimic the complexity of a biological sample. A mixture of 40 internal standard peptides (ISPs) was added to each of the four samples. The four samples were then analyzed to be either nano-flow or micro-flow LC–MS/MS. Arrows indicate the four pairwise data comparisons that were performed using PSM_validator. The labels for these comparisons (roman numerals I–IV) are referenced throughout the remainder of the manuscript.
All four samples were next analyzed by nano-flow LC–MS/MS, injecting 2 μL of each sample per analysis. Samples were also analyzed by micro-flow LC–MS/MS, which is less sensitive but has been shown to have superior RT reproducibility.24 Approximately 10 times as much material was loaded for micro-flow analyses. Samples were run in the following order to prevent carryover and avoid biasing results: native + HeLa, blank, spliced, blank, native, blank, spliced + HeLa. Running the samples in an order such as [native + HeLa, native, spliced + HeLa, spliced] may have favored the desired outcome by running matched peptides closer together in time while running mismatched peptides farther apart in time. The same samples were run in the same order on three different days using the nano-flow LC–MS/MS system, providing three technical replicates for the analyses. Two technical replicates were obtained by micro-flow LC–MS/MS analysis. To improve mass accuracy and spectrum quality, raw Agilent .d files were preprocessed with the Agilent spectrum mill data extractor using the following settings: MS/MS merging based on precursor selection purity, spectral similarity, RT (tolerance = +/− 60 s), and m/z (tolerance = +/− 1.4 m/z); find precursor charge and 12C precursor m/z; minimum MS1 signal-to-noise ratio = 10; MS noise threshold = 100 counts. Using MSConvert,19 MS1 files were generated from the raw Agilent .d files, and MGF files were generated from the mzXML files produced by Spectrum Mill following preprocessing. When generating MS1 files using MSConvert, data were filtered to include only those peaks in the m/z range of 300–750 with an abundance >500; this filtering step greatly reduced the time required for PSM_validator analyses.
We next tested the accuracy of P-VIS at determining when two peptides are the same or different by conducting four different pairwise comparisons (Figure 2). We first compared the 11 peptides in the “native + HeLa” (mock-biological) sample with the 11 peptides in the “native” (validation) sample. This modeled the scenario in which an investigator analyzes a biological sample containing the 11 native peptides, correctly identifies the native sequences, and analyzes synthetic versions of the native peptides to validate the matches. Alternatively, an investigator could have erroneously identified 11 spliced peptides in the mock-biological sample. In this case, synthetic spliced peptides would be analyzed for validation purposes and, if performing ideally, P-VIS would identify all PSMs as incorrect. To model this scenario, we compared the peptides in the “native + HeLa” (mock-biological) sample and the peptides in the “spliced” (validation) sample. Similarly, we performed a comparison of the peptides in the “spliced + HeLa” sample with the peptides in the “spliced” sample and a comparison of the peptides in the “spliced + HeLa sample” with the peptides in the “native” sample (Figure 2).
For analysis with PSM_validator, the following settings were used: pre_mz_tol = 10 ppm, pro_mz_tol = 20 ppm, abund_thresh = 500, PCC_abund_thresh = 10%, min_score = 15%, min_weighted_score = 10%, min_pairs_PCC = 8, min_PCC = 0.7, RTtol = 0.6 min, manual_RTdev_thresh = 0 min, min_intstd = 15, percentile_thresh = 5%, ion_type = b/y. These parameters are described in “settings.csv”. For microflow data, RT analysis was limited to the 12–70 min time range (min_RT = 12 min, max_RT = 70 min) to exclude the beginning and end of the run where the RT model was expected to be less accurate. For nano-flow experiments, many of the peptides eluted very early in the runs, resulting in several incomplete analyses when limiting the time range; thus, the entire time range was included (min_RT = 0 min, max_RT = 90 min). Note that in the results summary file automatically generated for each analysis, PSM_validator records the settings used. PSM_validator performs a D’Agostino–Pearson omnibus normality test on the PCCs and delta RTs for the ISPs. Results of these tests are shown in Figure 3. Using a threshold of 0.05, the data passed the normality test in most cases. However, the ISP delta RTs failed the normality test in five out of 12 cases for the nano-flow data likely because the entire time range was included in the analysis.
Figure 3.

Results of normality tests for PCCs and delta RTs obtained from ISPs. (a) Representative examples of plots showing the distribution of PCCs and delta RTs for ISPs along with a normal fit and p-value for the normality test (taken from comparison IV of micro-flow replicate 1). These plots are generated automatically by PSM_validator and are saved in the “normality” subfolder of the “Figures” folder for each analysis. (b) Results of all normality tests. For analyses involving micro-flow data, RT analysis was limited to the 12–70 min time range. For the nano-flow runs, the entire time range had to be included to avoid a large number of incomplete analyses. Gray boxes indicate analyses where the full time range was included. Bolded p-values were less than or equal to the significance threshold (α = 0.05), indicating that the distribution did not pass the test for normality.
For benchmarking purposes, if both the spectrum and RT outcome for a particular comparison satisfied the percentile threshold, then the result was reported as “PASS”. If one or both of the tests returned a result of “FAIL”, then the analysis outcome was reported as “FAIL”. If the outcome of the spectrum comparison was “FAIL” but the RT test was not completed, then the outcome was still reported as “FAIL”. If the outcome of the spectrum comparison was “PASS” but the RT comparison was not completed, or if neither test could be completed, then the analysis was considered incomplete. Outcomes of the benchmarking analyses are reported in Figure 4a as true positives (TPs), true negatives (TNs), false positives (FPs), or false negatives (FNs). If the same peptide was present in both samples (mock-biological and validation) and PSM_validator returned a result of “PASS” (indicating that the match was valid), then the benchmarking outcome was a TP; if the returned result was “FAIL” (indicating that the match was invalid), then the outcome was an FN. If one sample contained the spliced peptide, the other contained the native peptide, and PSM_validator returned a result of “FAIL”, then the benchmarking outcome was a TN; if the returned result was “PASS”, then the outcome was a FP. In some instances, PSM_validator could not complete the analysis, as indicated in Figure 4a. This can occur, for example, when the spectrum acquired for the peptide of interest was not of sufficient quality to pass the preliminary scoring step of the algorithm or the peptide of interest eluted outside the range of the ISPs.
Figure 4.

Summary of nano-flow and micro-flow LC–MS/MS P-VIS benchmarking results using a statistically-determined PCC threshold and a statistically-determined delta RT threshold. (a) Accuracy of P-VIS at identifying matched and mismatched peptides was evaluated using the data from each technical replicate. For each replicate, the numbers reported are totals across all four pairwise comparisons performed for that dataset. Three different iterations of the data analysis were performed. First, performance was evaluated when the results of both spectrum analysis and retention time (RT) analysis were used to determine the validity of matches. Second, performance was evaluated when spectrum analysis alone was used to validate matches. Third, performance was evaluated when RT analysis alone was used. The same PSM_validator settings were used in all three iterations of the data analysis. Both the PCC threshold and delta RT threshold were statistically determined based on the results for the ISPs. RT analysis for micro-flow data was limited to the 12–70 min time range; RT analysis for nano-flow data included the entire time range of the experiment. The PSM_validator percentile threshold was set at 5% for all analyses. Pos = positive; neg = negative; incomplete = analyses not completed. (b) Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated for each iteration of the data analysis (spectrum and RT, spectrum analysis only, and RT analysis only). Results from nano-flow experiments are shown with open shapes (three replicates), and results from micro-flow experiments are shown with filled shapes (two replicates). The equation for calculating each metric is indicated.
The sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) are performance metrics commonly used to describe the accuracy of a test.25 In the context of P-VIS, sensitivity provides the probability that P-VIS will correctly identify a valid match as valid (identify two peptides as being the same when they are the same), and specificity provides the probability that P-VIS will correctly identify an invalid match as invalid (identify two different peptides as being different). When both spectrum analysis and RT analysis were used, sensitivity of P-VIS ranged from 0.38 to 0.81 for the five technical replicates, and the specificity was always 1 (Figure 4b). A specificity of 1 reflects the fact that PVIS did not generate any FPs. An FP would occur if the peptide in the mock-biological sample and the peptide in the validation sample were different, but PSM_validator determined that the match was valid. Since P-VIS was designed as a rigorous method for ensuring the validity of reported matches, this lack of FPs was desired. Specificity was likewise 1 when RT analysis alone was used. Using spectrum analysis only provided greater sensitivity but resulted in a small number of FPs. PPV provides the probability that a match is indeed valid if P-VIS determines it is valid, and NPV provides the probability that a match is invalid when P-VIS determines it is invalid. When both spectrum analysis and RT analysis were used, PPV was always 1 for the five technical replicates, and NPV ranged from 0.59 to 0.83 (Figure 4b). No clear difference was seen between the performance of nano-flow and micro-flow data in the analyses.
Although high specificity was seen when using both spectrum and RT analysis, sensitivity was variable. For one of the nano-flow replicates, sensitivity was 0.38, while the sensitivity for the other four replicates (two nano-flow replicates and two micro-flow replicates) was much better, ranging from 0.63 to 0.81. Manual inspection of the detailed PSM_validator results suggested that the statistical approach used to distinguish between valid and invalid matches based on RT may have been too stringent for this dataset. In most cases, valid and invalid matches could easily be distinguished manually based on the delta RT generated by the linear spline model. We therefore added a user-specified manual delta RT threshold to the PSM_validator algorithm. To fail the RT analysis, the delta RT for the peptide of interest must fall outside the prediction interval (which is statistically determined based on the delta RTs of the ISPs) and must also exceed the manual threshold. We reanalyzed the data with this new approach using a manual threshold of +/− 0.5 min (manual_RTdev_thresh = 0.5 min). Results are shown in Figure 5. Implementation of the manual delta RT threshold reduced the number of FNs (increased sensitivity) without compromising specificity (no FPs were observed when RT analysis was used) and reduced the variability between replicates. For the five replicates, sensitivity ranged from 0.84 to 0.95 and specificity was always 1. The sensitivity, specificity, PPV, and NPV were high when spectrum analysis alone was used to determine validity. However, FPs were completely eliminated only when RT comparison was used. Although RT analysis is often omitted from validation efforts, our results demonstrate that RT analysis in the P-VIS workflow can increase the confidence in a match without the need for costly isotopically labeled peptides or extra effort. It is our intention and recommendation that both spectrum comparison and RT comparison be used when determining the validity of PSMs.
Figure 5.

Summary of nano-flow and micro-flow LC–MS/MS P-VIS benchmarking results using a statistically-determined PCC threshold and a manually-determined delta RT threshold. (a) Accuracy of P-VIS at identifying matched and mismatched peptides was evaluated using the data from each technical replicate. For each replicate, the numbers reported are totals across all four pairwise comparisons performed for that dataset. Three different iterations of the data analysis were performed. First, performance was evaluated when the results of both spectrum analysis and retention time (RT) analysis were used to determine the validity of matches. Second, performance was evaluated when spectrum analysis alone was used to validate matches. Third, performance was evaluated when RT analysis alone was used. The same PSM_validator settings were used in all three iterations of the data analysis. The PCC threshold was statistically determined based on the results for the ISPs. A manual delta RT threshold of +/− 0.5 min was used for RT analysis. RT analysis for micro-flow data was limited to the 12–70 min time range; RT analysis for nano-flow data included the entire time range of the experiment. The PSM_validator percentile threshold was set at 5% for all analyses. Pos = positive; neg = negative; incomplete = analyses not completed. (b) Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated for each iteration of the data analysis (spectrum and RT, spectrum analysis only, and RT analysis only). Results from nano-flow experiments are shown with open shapes (three replicates), and results from micro-flow experiments are shown with filled shapes (two replicates). The equation for calculating each metric is indicated.
Figure 6 provides the validation results for each individual comparison in the three nano-flow replicates using the manual delta RT approach and illustrates the accuracy of P-VIS at distinguishing valid and invalid matches. PSM_validator was even capable of distinguishing very similar peptides. For example, in all three technical replicates, it successfully distinguished the peptides TKVGPNATY and KTVGPNATY, which differ by the transposition of only two amino acids, while always returning “PASS” when one of these peptides was present in both samples (Figure 6). However, it should be noted that validation with synthetic peptides has limitations. Transposition of threonine (T) and lysine (K), which have very distinct side chains, could be expected to result in noticeable differences in the fragmentation spectrum. In contrast, transposition of more similar amino acids (e.g., threonine and serine) might result in more subtle differences that elude detection by the P-VIS approach. Even with the rigor and objectivity provided by implementing the P-VIS workflow, the researcher must be aware that two different but similar peptides can potentially be indistinguishable in terms of their fragmentation spectra and retention times. This is particularly a concern when putative spliced peptides are being identified using large spliced peptide databases as spliced sequences often differ only slightly from native sequences. In instances where such ambiguity is suspected, it is advisable to synthesize both potential peptide matches and test each for validity using P-VIS. If the correct match cannot be determined, then other strategies for identifying the peptide of interest need to be employed. For example, using different proteases during sample preparation can sometimes produce distinct peptides that cover the same sequence region and can be matched unambiguously.
Figure 6.

Nano-flow LC–MS/MS benchmarking of the P-VIS workflow using native and spliced peptides (spectrum comparison plus RT comparison; manual delta RT threshold = 0.5 min). Results are based on the combination of spectrum comparison and retention time comparison results. For RT comparison, the entire RT range was included, and a manual delta RT threshold of 0.5 min was used. Blue shading indicates that PSM_validator correctly identified the two peptides as either a match or mismatch. Red shading indicates that the program’s assessment was incorrect. Regions of the validation peptide that differ from the mock-biological peptide are underlined. Experiments are technical replicates (i.e., the same samples were run on three consecutive days). Rep = technical replicate. TP = true positive. TN = true negative. FP = false positive. FN = false negative. In cases where the analysis could not be completed, the reason is indicated as follows: a = in the mock-biological run, no spectrum satisfying minimum scoring requirements; b = in the validation run, no spectrum satisfying minimum scoring requirements (for this dataset, there were no instances where both runs lacked a satisfactory spectrum); c = too few ion pairs between the two spectra to complete Pearson analysis.
It is unclear why the statistical approach to RT analysis did not perform better. When the statistical approach was used for RT comparison, a high number of FNs was observed in replicate 2 of the nano-flow experiment compared to the other replicates. This could reflect a high level of variability inherent to nano-flow liquid chromatography. However, we anticipated that an approach that uses internal standards would accommodate such variability. Future experiments could explore if micro-flow LC–MS/MS might exhibit better reproducibility than nano-flow LC–MS/MS in the P-VIS workflow, but the data presented here are not sufficient for making such a determination. Matrix effects have been shown to cause surprising behaviors in liquid chromatography analysis, such as shifting of retention times or even the generation of two peaks for a single compound.26 Considering that the matrix of the mock-biological samples was quite different from the matrix of the validation samples due to the addition of HeLa protein extract, it is possible that the FNs observed were caused by matrix effects that influenced some of the spliced or native peptides differently than the ISPs. This possibility deserves further investigation.
Implementation of a manual delta RT threshold introduces an element of subjectivity that we intended to avoid in the P-VIS approach. However, the statistical approach to spectrum comparison, which performed very well in the benchmarking experiments, provides an objective metric. Since a match is considered invalid if it fails either the spectrum analysis or the RT analysis, the use of even a very liberal manual delta RT threshold will either improve specificity or, at worst, have no effect at all on specificity. As a protection against FNs (a loss of sensitivity) that might otherwise occur if the manual delta RT threshold was set too low, the RT analysis only returns a result of “FAIL” if the observed delta RT falls outside of both the statistically determined prediction interval and the window set by the manual threshold. Although ultimately a more objective approach is desirable, the reasons discussed here and the results of the benchmarking experiments suggest that the judicious use of a manual delta RT threshold is a viable approach.
Having confirmed the reliability of P-VIS, we next used the approach to validate the presence of a hybrid insulin peptide (HIP) in mouse islets. HIPs consist of fragments of insulin post-translationally fused to other peptides via a peptide bond, forming amino acid sequences that are not encoded in the genome.27 We have demonstrated that HIPs are targets of the autoimmune response in type 1 diabetes (T1D).27–31 The discovery of HIPs has afforded new insight into the pathogenesis of T1D, providing potential for identifying novel biomarkers30,31 and therapeutic targets.32 However, confident identification of HIPs by mass spectrometry poses many of the same challenges as identifying proteasomally-spliced peptides. Consequently, we recently established guidelines to ensure rigor when making HIP identifications.16 Following those guidelines, we identified HIPs in the pancreatic islets of mice and humans by mass spectrometry.16 As P-VIS provides a means of assessing validity that builds upon these guidelines, we analyzed mouse islets by nano-flow LC–MS/MS and used P-VIS to further validate the identification of 6.9HIP, a previously identified HIP consisting of a peptide fragment of insulin fused to a peptide fragment of an islet amyloid polypeptide (IAPP).16,27,29,33 PSM_validator analysis supported the conclusion that the 6.9HIP sequence DLQTLAL-NAAR (hyphen indicates the hybrid junction) is present in mouse islets (PCC percentile = 45.3%; RT percentile = 59.3%; percentile threshold = 5%). Figure 7 shows some of the key graphical outputs generated by PSM_validator. The original statistical approach to RT analysis was used in this experiment; the 6.9HIP would have also been determined valid had the manual delta RT threshold of +/− 0.5 min been used (6.9HIP delta RT was –0.09 min). Figures and detailed tabular results for this analysis and each of the benchmarking analyses are available as compressed folders in the ProteomeXchange Consortium21 via the PRIDE22 partner repository (dataset identifier PXD020901).
Figure 7.

P-VIS workflow confirms that the insulin-IAPP hybrid peptide 6.9HIP is present in mouse islets. Proteins extracted from mouse islets were fractionated by size exclusion chromatography (SEC), digested with the protease AspN, and analyzed by nano-flow LC–MS/MS. A database search identified the predicted 6.9HIP AspN cleavage product DLQTLAL-NAAR. To validate this identification, we applied the P-VIS workflow. Plots automatically generated by PSM_validator are shown. (a) Mirror plot displaying the spectrum from the biological sample (positive y-axis) and the spectrum from the validation sample (negative y-axis) that best matched the query sequence. Peak intensities are normalized to the most intense peak in each spectrum. The intensity of the tallest peak in each spectrum is indicated. In the sequence map, “\”, “/”, and “|” indicate that the spectrum from the biological sample contained a b ion, y ion, or both, respectively, corresponding to fragmentation at a given bond. The hyphen in the sequence indicates the location of the hybrid peptide junction. Horizontal dotted lines indicate the user-defined threshold for consideration of peaks in the PCC calculation. Ions corresponding to fragmentation of a bond to the left (L ions) or right (R ions) of the hybrid junction are labeled in red and cyan, respectively. (b) Raw intensities in the biological spectrum and the validation spectrum for all peaks included in calculation of the PCC. (c) Distribution of PCCs for internal standard peptides (ISPs). The distribution passed the test for normality (p = 0.146). Green shading indicates the 95% prediction interval based on one-tailed analysis. The PCC comparing the biological spectrum and the validation spectrum is also shown, and the percentile (%tile) is reported. (d) Extracted ion chromatogram showing the raw retention time for the biological peptide and the validation peptide. (e) Difference in RT for each of the ISPs between the two sample runs and the RT difference between the biological peptide and the validation peptide. Vertical lines indicate the time range considered in RT analysis (9–70 min). (f) Linear spline model based on the ISP data was used to model the relationship between RT in the two sample runs. The model was used to predict the RT of each peptide in the validation sample run based on the RT in the biological sample run. The difference between the expected and observed RT in the validation sample run (expected RT – syn RT) is reported. The distribution passed the test for normality (p = 0.393). Green shading indicates the 95% prediction interval based on two-tailed analysis. The difference between the biological peptide and the validation peptide is also shown, and the percentile (% tile) is reported.
CONCLUSIONS
Our results establish P-VIS as a systematic approach to determining the validity of individual PSMs. P-VIS allows for simultaneous validation of multiple peptides while requiring very little effort and time. Once validation peptides are synthesized (whether in-house or commercially), the biological and validation peptide samples then only need to be spiked with an internal peptide mixture such as PROCAL (JPT Peptide Technologies), which can be easily obtained commercially, and analyzed by LC–MS/MS. Afterward, the data can be converted to the appropriate formats using MSConvert or another freely available tool and analyzed using PSM_validator, which requires only limited user input, quickly performs all needed calculations, and returns detailed results with publication-ready figures.
Improved computational approaches for predicting peptide fragmentation and chromatographic RT are rapidly emerging (e.g., DeepRT,34 Prosit,35 and pValid36). In these approaches, algorithms are often trained using large datasets specific to a given set of conditions such as instrument configuration, instrument settings, and the protease used for digestion. The training dataset is used to develop a model for predicting fragmentation and RT based on the peptide sequence. The validity of a match generated from a standard database search can then be evaluated by comparison of the observed data to these predictions. P-VIS does not require a separate training dataset and assesses validity of a match based on the actual observed fragmentation and RT of a synthetic validation peptide rather than predictions. We encourage the use of prediction-based approaches for initial validation of data when feasible. Specific peptides of interest can then be selected from the curated data for final, direct validation using the P-VIS approach.
We demonstrated that P-VIS could have been a useful tool in determining the validity of individual UniProt (native) and spliced peptide identifications in the analyses performed by Liepe et al.7 and Mylonas et al.8 The data presented here should not be interpreted as supporting or refuting the claims by either group; rather, we established that, if the original biological samples had been analyzed using P-VIS, interpretations could have been assigned more objectively and with greater confidence. We also showed how P-VIS can be used to provide an objective assessment of the validity of HIP matches. Although we have demonstrated the utility of P-VIS in the confident identification of spliced peptides and HIPs, the workflow is broadly applicable. For example, P-VIS can be used to validate proteotypic peptides (bolstering confidence in the identification of proteins) or naturally-occurring peptides that are unmodified or contain conventional post-translational modifications. Recent studies have demonstrated the potential of immunopeptidomics in clinical applications, such as the design of personalized cancer immunotherapy.4–6 Integration of the P-VIS workflow could help in the selection of valid peptide candidates in such applications, preventing the waste of valuable resources pursuing peptides that do not exist in vivo. P-VIS can ensure high confidence in proteomic experiments, advancing LC–MS/MS as a powerful technology for answering critical biological questions.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Roger Powell and Zach McGrath for helpful suggestions and discussion, Katie Haskins for providing mouse islets, K. Scott Beard for isolating mouse islets, and Mylinh Dang for preparing mouse islets for LC–MS/MS analysis. The following funding sources supported this research: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R01 DK119529 (T.D.) and JDRF postdoctoral fellowship 3-PDF-2019-746-A-N (T.A.W.).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00355.
Table of contents (PDF)
PSM_validator source code (ZIP)
Contributor Information
Timothy Aaron Wiles, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, Colorado 80045-0508, United States States.
Laura M. Saba, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, Colorado 80045-0508, United States States.
Thomas Delong, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, Colorado 80045-0508, United States States.
REFERENCES
- (1).Admon A; Bassani-Sternberg M The Human Immunopeptidome Project, a suggestion for yet another postgenome next big thing. Mol. Cell. Proteomics 2011, 10, O111011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Caron E; Aebersold R; Banaei-Esfahani A; Chong C; Bassani-Sternberg M A Case for a Human Immuno-Peptidome Project Consortium. Immunity 2017, 47, 203–208. [DOI] [PubMed] [Google Scholar]
- (3).Vizcaíno JA; Kubiniok P; Kovalchik KA; Ma Q; Duquette JD; Mongrain I; Deutsch EW; Peters B; Sette A; Sirois I; Caron E The Human Immunopeptidome Project: A Roadmap to Predict and Treat Immune Diseases. Mol. Cell. Proteomics 2020, 19, 31–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chen R; Fulton KM; Twine SM; Li J IDENTIFICATION OF MHC PEPTIDES USING MASS SPECTROMETRY FOR NEOANTIGEN DISCOVERY AND CANCER VACCINE DEVELOPMENT. Mass Spectrom. Rev. 2019, DOI: 10.1002/mas.21616. Online ahead of print [DOI] [PubMed] [Google Scholar]
- (5).Ghosh M; Gauger M; Marcu A; Nelde A; Denk M; Schuster H; Rammensee H-G; Stevanović S. Guidance Document: Validation of a High-Performance Liquid Chromatography-Tandem Mass Spectrometry Immunopeptidomics Assay for the Identification of HLA Class I Ligands Suitable for Pharmaceutical Therapies. Mol. Cell. Proteomics 2020, 19, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chong C; Müller M; Pak H; Harnett D; Huber F; Grun D; Leleu M; Auger A; Arnaud M; Stevenson BJ; Michaux J; Bilic I; Hirsekorn A; Calviello L; Simó-Riudalbas L; Planet E; Lubiński J; Bryśkiewicz M; Wiznerowicz M; Xenarios I; Zhang L; Trono D; Harari A; Ohler U; Coukos G; Bassani-Sternberg M Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11, 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Liepe J; Marino F; Sidney J; Jeko A; Bunting DE; Sette A; Kloetzel PM; Stumpf MPH; Heck AJR; Mishto M A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [DOI] [PubMed] [Google Scholar]
- (8).Mylonas R; Beer I; Iseli C; Chong C; Pak H-S; Gfeller D; Coukos G; Xenarios I; Müller M; Bassani-Sternberg M Estimating the Contribution of Proteasomal Spliced Peptides to the HLA-I Ligandome. Mol. Cell. Proteomics 2018, 17, 2347–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Faridi P; Li C; Ramarathinam SH; Vivian JP; Illing PT; Mifsud NA; Ayala R; Song J; Gearing LJ; Hertzog PJ; Ternette N; Rossjohn J; Croft NP; Purcell AW A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands. Sci. immunol. 2018, 3, eaar3947. [DOI] [PubMed] [Google Scholar]
- (10).Rolfs Z; Müller M; Shortreed MR; Smith LM; Bassani-Sternberg M Comment on ″A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands″. Sci. immunol. 2019, 4, eaaw1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Faridi P; Li C; Ramarathinam SH; Illing PT; Mifsud NA; Ayala R; Song J; Gearing LJ; Croft NP; Purcell AW Response to Comment on ″A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands″. Sci. immunol. 2019, 4, eaaw8457. [DOI] [PubMed] [Google Scholar]
- (12).Rolfs Z; Solntsev SK; Shortreed MR; Frey BL; Smith LM Global Identification of Post-Translationally Spliced Peptides with Neo-Fusion. J. Proteome Res. 2019, 18, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vigneron N; Ferrari V; Stroobant V; Abi Habib J; Van den Eynde BJ Peptide splicing by the proteasome. J. Biol. Chem. 2017, 292, 21170–21179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Faridi P; Purcell AW; Croft NP In Immunopeptidomics We Need a Sniper Instead of a Shotgun. Proteomics 2018, 18, No. e1700464. [DOI] [PubMed] [Google Scholar]
- (15).Fälth M; Svensson M; Nilsson A; Sköld K; Fenyö D; Andren PE Validation of endogenous peptide identifications using a database of tandem mass spectra. J. Proteome Res. 2008, 7, 3049–3053. [DOI] [PubMed] [Google Scholar]
- (16).Wiles TA; Powell R; Michel CR; Beard KS; Hohenstein A; Bradley B; Reisdorph N; Haskins K; Delong T Identification of Hybrid Insulin Peptides (HIPs) in Mouse and Human Islets by Mass Spectrometry. J. Proteome Res. 2019, 18, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hunter JD Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar]
- (18).Virtanen P; Gommers R; Oliphant TE; Haberland M; Reddy T; Cournapeau D; Burovski E; Peterson P; Weckesser W; Bright J; van der Walt SJ; Brett M; Wilson J; Millman KJ; Mayorov N; Nelson ARJ; Jones E; Kern R; Larson E; Carey CJ; Polat I; Feng Y; Moore EW; VanderPlas J; Laxalde D; Perktold J; Cimrman R; Henriksen I; Quintero EA; Harris CR; Archibald AM; Ribeiro AH; Pedregosa F; van Mulbregt P SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kessner D; Chambers M; Burke R; Agus D; Mallick P ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics (Oxford, England) 2008, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zolg DP; Wilhelm M; Yu P; Knaute T; Zerweck J; Wenschuh H; Reimer U; Schnatbaum K; Kuster B PROCAL: A Set of 40 Peptide Standards for Retention Time Indexing, Column Performance Monitoring, and Collision Energy Calibration. Proteomics 2017, 17, 1700263. [DOI] [PubMed] [Google Scholar]
- (21).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang M; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–d1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vizcaíno JA; Csordas A; Del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; Xu Q-W; Wang R; Hermjakob H 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bassani-Sternberg M; Pletscher-Frankild S; Jensen LJ; Mann M Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 2015, 14, 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yin X; Baig F; Haudebourg E; Blankley RT; Gandhi T; Müller S; Reiter L; Hinterwirth H; Pechlaner R; Tsimikas S; Santer P; Willeit J; Kiechl S; Witztum JL; Sullivan A; Mayr M Plasma Proteomics for Epidemiology: Increasing Throughput With Standard-Flow Rates. Circ.: Cardiovasc. Genet. 2017, 10, e001808. [DOI] [PubMed] [Google Scholar]
- (25).Parikh R; Mathai A; Parikh S; Chandra Sekhar G; Thomas R Understanding and using sensitivity, specificity and predictive values. Indian J. Ophthalmol. 2008, 56, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fang N; Yu SY; Ronis MJJ; Badger TM Matrix effects break the LC behavior rule for analytes in LC-MS/MS analysis of biological samples. Exp. Biol. Med. 2015, 240, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Delong T; Wiles TA; Baker RL; Bradley B; Barbour G; Reisdorph R; Armstrong M; Powell RL; Reisdorph N; Kumar N; Elso CM; DeNicola M; Bottino R; Powers AC; Harlan DM; Kent SC; Mannering SI; Haskins K Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Babon JAB; DeNicola ME; Blodgett DM; Crèvecoeur I; Buttrick TS; Maehr R; Bottino R; Naji A; Kaddis J; Elyaman W; James EA; Haliyur R; Brissova M; Overbergh L; Mathieu C; Delong T; Haskins K; Pugliese A; Campbell-Thompson M; Mathews C; Atkinson MA; Powers AC; Harlan DM; Kent SC Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat. Med. 2016, 22, 1482–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wiles TA; Delong T; Baker RL; Bradley B; Barbour G; Powell RL; Reisdorph N; Haskins K An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J. Autoimmun. 2017, 78, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Baker RL; Jamison BL; Wiles TA; Lindsay RS; Barbour G; Bradley B; Delong T; Friedman RS; Nakayama M; Haskins K CD4 T Cells Reactive to Hybrid Insulin Peptides Are Indicators of Disease Activity in the NOD Mouse. Diabetes 2018, 67, 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Baker RL; Rihanek M; Hohenstein AC; Nakayama M; Michels A; Gottlieb PA; Haskins K; Delong T Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes 2019, 68, 1830–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jamison BL; Neef T; Goodspeed A; Bradley B; Baker RL; Miller SD; Haskins K Nanoparticles Containing an Insulin-ChgA Hybrid Peptide Protect from Transfer of Autoimmune Diabetes by Shifting the Balance between Effector T Cells and Regulatory T Cells. J. Immunol. 2019, 203, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wan X; Vomund AN; Peterson OJ; Chervonsky AV; Lichti CF; Unanue ER The MHC-II peptidome of pancreatic islets identifies key features of autoimmune peptides. Nat. Immunol. 2020, 21, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ma C; Ren Y; Yang J; Ren Z; Yang H; Liu S Improved Peptide Retention Time Prediction in Liquid Chromatography through Deep Learning. Anal. Chem. 2018, 90, 10881–10888. [DOI] [PubMed] [Google Scholar]
- (35).Gessulat S; Schmidt T; Zolg DP; Samaras P; Schnatbaum K; Zerweck J; Knaute T; Rechenberger J; Delanghe B; Huhmer A; Reimer U; Ehrlich HC; Aiche S; Kuster B; Wilhelm M Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [DOI] [PubMed] [Google Scholar]
- (36).Zhou WJ; Yang H; Zeng WF; Zhang K; Chi H; He SM pValid: Validation Beyond the Target-Decoy Approach for Peptide Identification in Shotgun Proteomics. J. Proteome Res. 2019, 18, 2747–2758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Benchmarking data and mouse islet data, as well as all PSM_validator result files, have been deposited to the ProteomeXchange Consortium21 via the PRIDE22 partner repository with the dataset identifier PXD020901.