

Abstract
In proteogenomic studies, genomic and transcriptomic variants are incorporated into customized protein databases for the identification of proteoforms, especially proteoforms with sample-specific variants. Most proteogenomic research has been focused on combining genomic or transcriptomic data with bottom-up mass spectrometry data. In the last decade, top-down mass spectrometry has attracted increasing attention because of its capacity to identify various proteoforms with alterations. However, top-down proteogenomics, in which genomic or transcriptomic data are combined with top-down mass spectrometry data, has not been widely adopted, and there is still a lack of software tools for top-down proteogenomic data analysis. In this paper, we introduce TopPG, a proteogenomic tool for generating proteoform sequence databases with genetic alterations and alternative splicing events. Experiments on top-down proteogenomic data of DLD-1 colorectal cancer cells showed that TopPG coupled with database search confidently identified proteoforms with sample-specific alterations.
Keywords: proteogenomics, top-down mass spectrometry, RNA-seq
Graphical Abstract
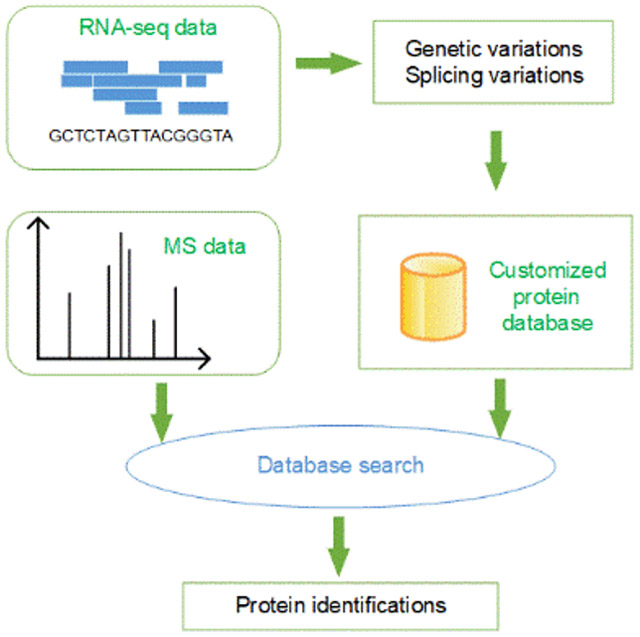
1. Introduction
Top-down mass spectrometry (MS) has been widely used in proteoform identification and characterization because of its ability to sequence whole proteoforms 1. In a top-down MS experiment 2, intact proteoforms are separated by a separation platform such as a liquid chromatography (LC) system and then analyzed by tandem mass spectrometry (MS/MS) to generate MS1 spectra of proteoforms and MS/MS spectra of proteoform fragments. Each of the mass spectra contains a list of peaks measuring the mass-to-charge ratios (m/z values) and abundances of proteoforms or their fragments 3, 4.
Database search is the dominant method for top-down MS-based proteoform identification and characterization 5, 6. In this method, a top-down MS/MS spectrum is searched against a proteoform sequence database to identify a proteoform that best explains the peaks in the spectrum. Proteoforms identified by top-down MS are often modified forms of sequences in the database. These proteoforms can be further characterized to localize alterations and find their combinatorial alteration patterns using probabilistic models 7, 8.
A key challenge in proteoform identification is that proteoforms contain various alterations, such as sequence mutations, splicing events, and post-translational modifications (PTMs) 9, 10. Intact proteoforms with hundreds of amino acids tend to have more alterations than short peptides analyzed in bottom-up MS. However, most proteoform databases used in proteomics studies, such as UniProt databases, contain only reference sequences, not proteoforms with various alterations 11.
There are three categories of computational methods that have been proposed for identifying proteoforms with alterations 12. In the first approach, extended databases containing proteoforms with alterations are built from protein sequence annotations, or genomic or transcriptomic data 13. A spectrum of a proteoform A with alterations can be easily identified if the extended database contains A. Even if the extended database does not contain A, but contains another proteoform B that is similar to A, it also facilitates spectral identification because proteoform B is a good reference sequence.
In the second approach, an open search method is used to identify proteoforms with unexpected alterations, which has been widely used in bottom-up 14 and top-down MS 13, 15 data analysis. It is capable of identifying proteoforms with one unexpected alteration using unmodified proteoform fragments. The main limitation of the method is that only one unexpected mass shift can be identified in proteoforms.
In the third approach, mass spectra are aligned against database sequences 16, 17, which are capable of identifying proteoforms with several unexpected alterations. Some alignment algorithms allow users to provide expected PTMs 7, 15, 18, 19, making it feasible to identify highly modified proteoforms, such as histone proteoforms.
Most existing tools adopt one or two of the three approaches. ProSightPC 13 uses the extended database and open database search methods; TopPIC 20, TopMG 21, SPECTRUM 22, and MS-PathFinder 15 use the open search and spectral alignment methods 23. Because of the complementary strengths of the approaches, combining extended databases and the other two methods can increase the sensitivity in the identification of complex proteoforms.
Proteogenomic methods build extended proteoform databases using DNA/RNA sequencing data 24, which contain information of sample-specific alterations. For example, based on RNA-Seq data, a customized proteoform database can be built to include sequences with sample-specific genetic alterations and alternative splicing events 25, 26. Using such a database in MS data analysis increases proteoform identifications with these alterations.
Proteogenomic methods also facilitate proteoform characterization in top-down spectral interpretation. Although top-down MS is capable of identifying complex proteoforms, many modified proteoforms are not characterized because top-down MS/MS spectra lack enough fragment ions. Genomic and transcriptomic data provide additional information for identifying and characterizing alterations in proteoforms. If a mutation in a proteoform sequence is supported by both MS and transcriptomic data, the confidence of the identification is significantly increased 25.
Many proteogenomic pipelines and software tools have been proposed for combining genomic or transcriptomic data with bottom-up MS data 27, 28. Most tools like customProDB29, MutationDB, MSProGene30, PGA26, and JUMPg 31 generate sample-specific sequences with mutations and/or splice junctions from RNA-Seq data. SpliceDB 32 and Splicify 33 produce customized protein databases with only splicing variants. SpectroGene 34 builds protein sequence databases by six-frame translation from open reading frames in genomes. PGTools 35 utilizes annotations of Ensembl 36 to build various protein sequence databases with mutations and splicing events. Askenazi et al. developed a proteogenomic tool called PGx, which maps identified peptides onto their putative genomic coordinates 37.
Top-down proteogenomics has its unique advantages and challenges compared with bottom-up proteogenomics. A primary benefit of top-down MS is that its spectra cover whole proteoform sequences instead of short peptides, making it possible to identify sequence alterations on peptides unidentified by bottom-up MS and analyze splicing events not covered by single peptides. And it paves the way for studying combinatorial patterns of sequence alterations of proteoforms. One challenge of top-down proteogenomics is that its protein coverage is low in proteome-wide studies: only hundreds of high abundance proteins can be identified. Protein purification is usually needed to analyze low abundance proteins, increasing the complexity of MS experiments. Another limitation is that the fragment coverage of top-down MS/MS spectra tends to be lower than bottom-up spectra. As a result, we may fail to confidently characterize sequence alterations.
Only several studies have been carried out in top-down proteogenomics. If the genomic annotation of an organism is unavailable or incomplete, proteoform databases are generated from its genomic data using six-frame translation 34 or from transcriptomic data using de novo assembly 38. Otherwise, the annotation is often utilized to align RNA-Seq reads to the reference genome to identify genomic alterations and novel splicing junctions 38, 39. One top-down proteogenomic pilot study identified 41 single amino acid variations (SAAVs) and 11 novel splicing junctions from patient-derived mouse xenograft samples of breast cancer 39.
Here we present TopPG, a software tool for combining RNA-Seq and top-down MS data for proteoform identification. TopPG builds sample-specific proteoform sequence databases using genomic alterations and splicing junctions identified by aligning RNA-Seq reads to the reference genome. We assessed TopPG on a top-down MS data set of DLD-1 colorectal cancer cells and demonstrated that TopPG increased the number of proteoform identifications and identified many proteoforms with sample-specific mutations and splicing events compared with reference databases from Ensembl 36.
2. Methods
2.1. Data sets
An RNA-seq data set of DLD-1 colorectal cancer cells was downloaded from the sequence read archive (accession number: SRR6926366), which contains 48.8 million paired short reads (150 bp). The RNA sample was prepared using the Illumina TruSeq RNA sample preparation protocol and deeply sequenced with an Illumina HiSeq 3000 40. Phred quality scores (Q scores) reported by FastQC (version 0.11.8, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) showed that more than 90% of the short reads reached Q30 (99.9% base call accuracy).
A top-down MS data set of DLD-1 cells was downloaded from the MassIVE repository (ID: MSV000079978) 41. In the MS experiment, a protein sample of DLD-1 cells was first separated into 12 fractions by a gel-eluted liquid-fraction entrapment electrophoresis (GELFrEE) fractionation system. Then the first 8 fractions were analyzed by reverse-phase liquid chromatography (RPLC) coupled with a 21 T Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer. A total of 14588 MS/MS spectra were collected. Details of the experiment can be found in ref. 41.
A bottom-up MS data set of DLD-1 cells was also downloaded from the MassIVE repository (ID: MSV000080374) 42. In the bottom-up MS experiment, tryptic peptides of DLD-1 cells were first separated into 15 fractions by a basic RPLC system, and then each fraction was analyzed by a Thermo Orbitrap-Velos mass spectrometer coupled with a reversed-phase high-performance liquid chromatography (HPLC) system. A total of 53976 MS1 and 115987 MS/MS spectra were collected.
2.2. Building sample-specific proteoform sequence databases
We develop a top-down proteogenomic pipeline that uses RNA-Seq data to build customized proteoform sequence databases. In the pipeline, RNA short reads are aligned to a reference genome to identify genomic alterations and alternative splicing events, based on which proteoform sequence databases are generated.
2.2.1. RNA-seq data analysis
The GATK pipeline (version 4.1.0.0) 43 is used for the analysis of RNA-seq short reads. First, short reads are aligned to the hg38 reference genome (downloaded from the GATK resource bundle) using the two-pass mode of STAR (version 2.7.0.c) 44, in which splicing junctions identified in the first round guide the second round of alignment. Second, Picard (version 2.18.26, http://picard.sourceforge.net) is used to remove duplicates from short-read alignment BAM files, then sort and add indexes to the files. Finally, we use SplitNCigarReads to remove skipped regions in sequence alignments and apply the BQSR method to recalibrate base quality scores of short reads. The parameter settings and commands of the tools are given in the supplementary material.
The HaploTypeCaller tool in GATK is used to call variants from short-read alignment files. The minimum phred-scaled confidence threshold is set to 20 (the error rate is lower than 1%); soft-clipped bases are excluded to reduce false positives. To further optimize the sensitivity and specificity, VariantFiltration is employed to filter out single-nucleotide variant (SNV) clusters with three SNVs in a window of 35 bases 45. All SNVs, insertions, and deletions (indels) reported by HaploTyperCaller are annotated by ANNOVAR 46 (version April 16, 2018).
2.2.2. Building proteoform sequence databases with genomic variants
We use nonsynonymous SNVs (nsSNVs) and indels reported by ANNOVAR to generate three types of transcript/segments: homozygous transcripts, heterozygous transcript segments, and decoy transcript segments (Fig. 1). Given a reference transcript sequence database, we incorporate all homozygous SNVs and indels to generate a homozygous transcript database. If a reference transcript does not contain any homozygous SNVs or indels, the sequence itself is treated as a homozygous transcript. Otherwise, all homozygous variants on the reference transcript are added to produce a homozygous transcript. Consequently, reference and homozygous databases have the same size.
Figure 1.

Generation of a homozygous transcript sequence, heterozygous transcript segments (blue), and decoy transcript segments (green) from a reference transcript. Sequence A is a reference transcript with 3600 nucleotide bases, a homozygous SNV, and 3 heterozygous SNVs. The homozygous SNV is added to A to generate a homozygous transcript B, which is broken into three overlapping segments of length 1800: C1, C2, and C3. The first heterozygous SNV is added to C1 to generate a heterozygous segment D1, and a random SNV is added to C1 to generate a random segment E1. Three heterozygous segments are produced from C3: D3 and D4 each with one SNV and D5 with two SNVs. Three decoy segments E3, E4, and E5 are generated from C3 with random SNVs.
Next, we generate heterozygous transcript segments with heterozygous nsSNVs and/or indels. Let S be a homozygous transcript sequence with n bases and t heterozygous nsSNVs and/or indels. There are 2t proteoform sequences of S with different heterozygous variant combinations. To address the combinatorial explosion problem, we follow the method proposed by Kolmogorov et al. 34 to produce short transcript segments with alterations. Because most mass spectrometers identify only proteoforms with a molecular mass of less than 50 kDa, we choose 1800 as the length of short transcript segments, which corresponds to 600 amino acids. We split the transcript sequence S into overlapping segments S1, S2, …, Sk with a window of length L=1800, where . The overlapping region of two segments Si and Si+1 is for 1 ≤ i ≤ k − 1. If Si (1 ≤ i ≤ k) contains j > 2 heterozygous variants, we generate only transcript segments with one or two heterozygous variants: j segments each with one heterozygous variant and segments each with two heterozygous variants (Fig. 1). Note that more than one heterozygous nsSNV may occur at the same location and that the number of heterozygous nsSNVs may be larger than the number of SNV sites. If a segment Si (1 ≤ i ≤ k) does not contain any heterozygous nsSNVs and indels, no new transcript segments are generated.
In addition to heterozygous transcript segments, we generate decoy transcript segments for estimating false discovery rates (FDRs) of variant identifications. For each heterozygous segment obtained from a segment Si and a heterozygous nsSNV, we generate a decoy segment from Si by adding a random nsSNV. Similarly, for each heterozygous segment obtained from a segment Si and two heterozygous nsSNVs, we generate a decoy segment from Si by adding two random nsSNV (Fig. 1). Similarly, decoy segments with indels are produced. The number of decoy segments is the same as that of heterozygous segments.
Finally, the homozygous transcripts, heterozygous segments, and decoy segments are combined and translated into proteoform sequences and segments. When a transcript sequence has frameshift indels, we find the first downstream stop codon with the new frame and use the new frame and the stop codon to generate a proteoform sequence or segment. The FDR of identified heterozygous variants in database search is estimated by the ratio of the numbers of identified random variants in decoy segments and identified heterozygous variants in heterozygous segments.
2.2.3. Building proteoform databases with alternative splicing events
We use the Multivariate Analysis of Transcript Splicing (MATS) tool (version 4.0.2) 47 to identify alternative splicing events from RNA-seq data. MATS reports five different splicing events: exon skipping, mutually exclusive exons, alternative 3’ splice sites, alternative 5’ splice sites, and intron retention (Fig. S1), in which exon skipping events are the most common form (~30%) 48. We generate only proteoforms with exon-skipping events. An exon-splicing event involves three exons: the upstream exon, the downstream exon, and the cassette (skipped) exon, and results in two transcript forms: the inclusive form contains the cassette exon, and the exclusive form does not. If a transcript in genome annotation contains the upstream and downstream exons (with or without the cassette exon) of an exon splicing event, then the transcript is matched to the event.
We use the homozygous transcript database reported in Section 2.2.2 to generate transcripts with alternative splicing events (Fig. 2). We search each transcript in the database against the reported exon-skipping events to find matches, then generate an inclusive form for each matched event of the exclusive form and vice versa. When a transcript is matched to k exon-skipping events, k transcripts will be generated, which are referred to as alternative splicing transcripts. If an alternative splicing transcript is the same as another sequence in the homozygous transcript database, then we will not generate any new transcript and only update the description of the homozygous transcript to include the annotation of the splicing event. As a result, it is possible that a transcript is a database sequence for one splicing event and an alternative splicing one for another event.
Figure 2.

Generation of transcripts with exon skipping events. Sequence A is a reference transcript with three exons, a homozygous SNV, and a heterozygous SNV. The length of the cassette exon is l. A homozygous transcript B is generated from A by adding the homozygous SNV. An alternative splicing transcript C is generated from B by removing the cassette exon. A decoy transcript D is generated from B by remove a random RNA sequence of length l, and a decoy transcript E is generated from C by adding a random exon of length l. By adding the heterozygous SNV, four heterozygous transcripts B’, C’, D’, and E’ are generated from B, C, D, and E, respectively.
For a pair of a homozygous transcript B and an alternative splicing transcript C, we generate two decoy transcripts for estimating FDRs of identified alternative splicing events. Suppose that B is the inclusive form, C is the exclusive form, and the length of the cassette exon is l. We generate the first decoy transcript by removing a random RNA sequence of length l from B, and the second by adding a random RNA sequence of length l into C (Fig. 2).
Heterozygous nsSNVs and indels are used to generate transcripts from homozygous transcripts, alterative splicing transcripts, and decoy transcripts. If a transcript contains j heterozygous variants, then j new transcripts will be generated, each containing a heterozygous variant. Finally, the transcripts with and without heterozygous variants are combined and translated into proteoform sequences. The FDR of identified alternative splicing events in database search is estimated by the ratio of the numbers of identified random events in decoy sequences and identified events in homozygous or alterative splicing sequences.
2.2.4. Proteoform identification by top-down MS
The raw files of the DLD-1 top-down MS data were centroided and converted into mzML files using msconvert in ProteoWizard 49, and further deconvoluted into msalign files containing monoisotopic masses of fragment and precursor ions using TopFD (version 1.3.4). The deconvoluted spectra were searched against proteoform databases using TopPIC (version 1.3.4) 20. A shuffled decoy database with the same size was concatenated with the target database. The error tolerances for precursor and fragment masses were set to 15 parts per million (ppm). A proteoform database reported by TopPG contains some sequences with decoy SNV sites and alternative splicing events. These sequences are similar to reference database protein sequences and are treated as a part of the target database in the estimation of FDRs of proteoform-spectrum-match (PrSM) identifications. The reason is that the spectrum is usually matched to the correct protein in a PrSM identification with such a sequence even though the identified alterations are incorrect. Identifications of PrSMs were filtered using a 1% spectrum-level FDR. Identified proteoforms were treated as the same if their spectra were generated from the same LC-MS feature reported by TopFD or they were from the same protein (gene) and had similar precursor masses (within an error tolerance of 2.2 Da). Using the method, we grouped identified PrSMs into clusters, each of which corresponds to a proteoform. Then the identifications of proteoforms and proteins were filtered using a 1% proteoform-level FDR. The parameter settings of TopPIC are given in Table S1.
2.2.5. Peptide identification by bottom-up MS
The raw files of the bottom-up MS data were centroided and converted to mzML files using msconvert 49. MS-GF+ (v2020.08.05)50 was used to search bottom-up mass spectra against protein sequence databases. In MS-GF+, the target-decoy method was employed to estimate spectrum-level FDRs, the mass tolerance was set to 20 ppm, cysteine carbamidomethylation as a fixed modification, oxidation as a variable modification, and other parameters were set to default values.
3. Results
3.1. Comparison of proteoform reference databases
Three human proteome databases were compared for proteoform identification by database search. The first two databases were generated using the basic and comprehensive annotations of GENCODE (version 28) 51 and were referred to as the BASIC (57089 entries) and COMP (97713 entries) databases, respectively. All sequences in BASIC are included in COMP. The third one, referred to as the SWISS database (19236 entries), is a subset of BASIC containing only proteoforms matched to entries in the Swiss-Prot human proteome database (20380 entries, March 2019).
We searched the spectra in the DLD-1 top-down MS data against the three proteoform reference databases separately using TopPIC (See Section 2). With a 1% spectrum-level FDR, TopPIC identified 3857, 3590, and 3535 PrSMs from the SWISS, BASIC, and COMP databases, respectively (Fig. 3a). We identified more proteoforms and proteins with the SWISS database than the other two. The reason may be that the increase of the database size introduced many decoy identifications in the target-decoy approach and increased the Q-values of identifications in BASIC and COMP. Because of this, some identifications in the BASIC and COMP database search were filtered out. In addition, the BASIC and COMP database searches identified some proteoforms not included in the SWISS database (Fig. 3b).
Figure 3.

Comparison of the three proteoform databases SWISS, BASIC, and COMP on proteoform identification using the DLD-1 MS data set. (a) Numbers of identified PrSMs, proteoforms, and proteins. (b) Overlaps of the proteoforms identified from the three databases.
3.2. Sample specific databases with genomic alterations
The RNA-Seq data of DLD-1 cells were analyzed by the GATK pipeline for short-read alignment and SNV calling (See Section 2). ANNOVAR 46 reported 18133 genomic variants (Fig. S2) with the basic annotation of GENCODE (version May-06-2018), including 9283 nsSNVs, 503 frameshift indels and 152 non-frameshift indels. The nsSNVs were mapped to 5420 genes and 14135 transcripts, most of which contained 1 or 2 nsSNVs (Fig. S3(a)). Of the 14135 transcripts, 36 were not located on protein-coding regions, and the others were matched to 5014 and 14099 proteoform sequences in the SWISS and BASIC databases, respectively. The 5014 proteoforms in the SWISS database covered 93% of the 5420 genes with nsSNVs. The 14099 matched proteoforms in the BASIC database had 1.5 mutations on average. The frameshift indels and non-frameshift indels were also used to generate proteoform sequences with alterations. Using the basic annotation of GENCODE, the indels were mapped to 605 genes and 1553 transcripts, of which 1391 transcripts contained only one indel (Fig. S3(b)). We used the matched SWISS proteoform sequences to generate a database SWISS-M with 36904 entries: 17417 without variants, 1819 homozygous sequences, 8834 heterozygous segments (including 2002 segments with both heterozygous and homozygous variants), and 8834 decoy segments. Combinations of two heterozygous variants were not included in the database. Similarly, based on BASIC proteoform sequences, we generated a database BASIC-M with 105559 entries: 52192 without variants, 4897 homozygous sequences, 24235 heterozygous segments (including 4926 segments with both heterozygous and homozygous variants), and 24235 decoy segments.
ANNOVAR reported 18682 genomic variants with the comprehensive annotation of GENCODE (9576 nsSNVs, 526 frameshift indels, and 154 non-frameshift indels). The nsSNVs were mapped to 17983 transcripts of 5549 genes. Similarly, the nsSNVs, frameshift, and non-frameshift indels were used to generate a customized database COMP-M with 154108 entries: 91612 without variants, 6028 homozygous sequences, 28234 heterozygous segments (including 5256 segments with both heterozygous and homozygous variants), and 28234 decoy segments.
With a 1% spectrum-level FDR, TopPIC identified 1200, 1192, and 1168 proteoforms from the SWISS-M, BASIC-M, and COMP-M databases, respectively (Fig. 4a). The numbers of identifications were similar for the three databases. The identified proteoforms covered some SNV sites, but no indels reported by RNA-Seq data. We mapped the proteoforms to their corresponding RNA transcripts and checked whether the transcripts contain nsSNV sites. Of the 1200 proteoforms identified by SWISS-M, 112 proteoforms covered 43 target SNV sites and 13 decoy SNV sites (some SNV sites were covered by more than one proteoform), and 1088 did not cover any SNV sites. The FDR for identified SNV sites was about 23.2%. We further manually inspected the PrSMs and found that 37 of the 43 SNV sites were confidently identified (Fig. S4 and Table S2). The other seven SNV sites were not confident identifications because they were covered by proteoforms containing unexpected mass shifts near the sites (Fig. S5). We identified 43 SNV sites with manual validation from BASIC-M and the same number of sites from COMP-M (Table S2), and the FDRs of the identifications were 27.9% and 23.2% for BASIC-M and COMP-M, respectively.
Figure 4.

Comparison of the three proteoform databases SWISS-M, BASIC-M, and COMP-M on proteoform identification using the DLD-1 top-down MS data set: (a) identified proteoforms, (b) identified proteins, and (c) identified SNV sites with manual inspection.
The comparison of the three databases on the numbers of identified proteoforms, proteins, and SNV sites is summarized in Fig. 4. COMP-M and BASIC-M identified similar numbers of proteoforms and SNVs sites because they have similar database sizes. The reason that SWISS-M identified fewer SNV sites than the other two databases may be that SWISS-M included fewer proteoforms compared with the other two databases.
To identify proteoforms with two heterozygous SNVs, we extended the BASIC-M database by adding 14507 heterozygous segments and 14507 decoy segments, each of which contained 2 SNVs (Fig. 1). The resulting database is referred to as BASIC-C. With a 1% spectrum-level FDR, we identified 3323 PrSMs and 1198 proteoforms from BASIC-C, which covered 53 SNV sites. By manual inspection, we removed 6 SNV sites not confidently identified. Of the remaining 47 SNV sites, 43 were also identified by BASIC-M, and the other 4 sites were on two segments, each with 2 heterozygous SNVs (Fig. S6).
3.3. Proteoform identifications with splicing events
MATS 47 reported 22774 exon-skipping events in the DLD-1 RNA-Seq data. We generated a proteoform database using the BASIC annotation of GENCODE and the exon-skipping events (See Section 2). Of the 57089 transcripts in the BASIC annotation, 13014 were matched one or more reported exon-skipping events, and the remaining 44075 did not. From the 13014 transcripts, we generated 21101 alternative splicing transcripts, of which 2201 were included in the BASIC transcripts, and the other 18900 were novel transcripts. In addition, we generated 42202 decoy transcripts. The resulting BASIC-ES database contained 118191 entries, including 57089 BASIC transcripts, 18900 alternative splicing transcripts, and 42202 decoy transcripts (Fig. 2).
With a 1% spectrum-level FDR, TopPIC identified 3324 PrSMs and 1175 proteoforms from the BASIC-ES database, of which 128 proteoforms covered 139 exon-skipping events (132 inclusive forms and 9 exclusive forms) and 8 proteoforms covered 8 decoy splicing events (two inclusive forms and six exclusive forms). The FDR for identified splicing variations was 5.4%. After manual inspection, we kept 124 inclusive forms and 6 exclusive forms that were confidently identified, including seven inclusive and five exclusive novel forms (Table S3). We identified one exon-skipping event with both inclusive and exclusive forms, and three exon-skipping events covered by two or more proteoforms. In addition, some identified proteoforms covered more than one splicing event (Fig. S7).
For each of the inclusive or exclusive form identification, we computed the RNA expression level of the gene, and the percentage of the expressed transcript isoforms containing the exon, called the percent spliced in index (PSI) value, from the RNA-Seq data. The Reads Per Kilobase per Million mapped reads (RPKMs) of genes after logarithm transformation were used as RNA expression levels. Most of the inclusive form identifications have a high PSI value close to 1.0, and most of the exclusive form identifications have a low PSI value close to 0 (Fig. 5), showing that the identifications are consistent with the PSI values of the splicing events on the transcript level. In addition, most of the proteoform identifications have a high expression level at the transcript level, demonstrating that top-down MS tends to identify only highly expressed proteoforms.
Figure 5.

Gene expression levels and PSI values of the splicing events in the DLD-1 data set. Splicing events with proteoform identifications are labeled.
3.4. Comparison of bottom-up and top-down approaches
We generated a sample-specific protein sequence database BASIC-B using customProDBJ 52 with the default parameter setting. The input of customProDBJ was the BASIC database and the nsSNVs reported by ANNOVAR with the basic annotation of GENCODE in the previous subsection. BASIC-B contained 82253 entries, in which 25294 entries were sequences with nsSNVs and the others were sequences in BASIC without any variants. With a 0.1% spectrum-level FDR, MS-GF+ identified 11340 peptides from the BASIC-B database, including 186 peptides with SNV sites. A total of 175 SNV sites were identified in the 186 peptides (Table S4). Only seven SNV sites were identified by both top-down and bottom-up approaches (Fig. 6), showing that the two approaches are complementary for identifying SNV sites. Top-down MS is capable of identifying some SNV sites missed by bottom-up MS in highly abundant proteoforms.
Figure 6.

Comparison of the numbers of SNV sites identified from DLD-1 cells by bottom-up MS and top-down MS.
4. Discussion and conclusions
In this study, we present a new proteogenomics tool TopPG, which is capable of identifying proteoforms with genomic alterations and alternative splicing events. TopPG builds customized proteoform sequence databases from RNA-Seq data using nsSNVs, indels, and exon-skipping events. The experiments on the DLD-1 data set demonstrated that databases generated by TopPG facilitated the identification and characterization of sample-specific genomic variants and exon splicing events. In addition, the analysis of exon-skipping events showed that their percent spliced in levels were consistent in the transcript and proteoform levels.
Top-down proteogenomics still has many limitations in practice. One limitation is the low proteoform coverage of top-down MS in proteome-level studies. A single shot top-down MS experiment usually identifies only hundreds of proteins, which is a small fraction of genes identified in the transcript level. Consequently, only a small number of SNVs and other alterations can be identified by top-down MS.
Another limitation is that some proteoforms with genomic and/or transcriptomic alterations cannot be fully characterized. Manual inspection of the proteoforms identified from the DLD-1 top-down MS data showed that many SNV sites are close to other unknown alterations in identified proteoforms and that the SNV sites cannot be confidently identified because the spectra lack enough fragment ions to characterize them. Similarly, many exon-skipping events cannot be confidently identified because the spectra lack enough fragment ions to distinguish between the inclusive and the exclusive forms. Increasing proteoform sequence coverage is essential to identifying sample-specific genomics and transcriptomic alterations.
Supplementary Material
Parameters and commands for software tools used in RNA-seq analysis.
Figure S1: The diagram of five types of alternative splicing events.
Figure S2: The distributions of the types of variants reported by the ANNOVAR.
Figure S3: The distributions of the numbers of nonsynonymous substitutions and indels per transcript reported by ANNOVAR.
Figure S4: An example PrSM of the confident identifications from SWISS-M database.
Figure S5: An example PrSM of the unconfident identifications from SWISS-M database.
Figure S6: The PrSMs containing two heterozygous variants per sequence.
Figure S7: An example PrSM of the identifications from BASIC-ES database.
Table S1: Parameter setting for TopPIC.
Table S2: Proteoforms covering SNV sites identified from SWISS-M, BASIC-M and COMP-M.
Table S3: Proteoforms covering exon skipping events identified from BASIC-ES.
Table S4: Peptides covering SNV sites identified in bottom-up MS.
5. Acknowledgment
The research was supported by the National Institute of General Medical Sciences, National Institutes of Health (NIH) through Grants R01GM118470, R01GM125991, and R01AI141625.
Footnotes
Availability: TopPG is available at http://proteomics.informatics.iupui.edu/software/toppg/.
REFERENCES
- 1.Domon B; Aebersold R, Mass spectrometry and protein analysis. Science 2006, 312 (5771), 212–217. [DOI] [PubMed] [Google Scholar]
- 2.Catherman AD; Skinner OS; Kelleher NL, Top down proteomics: facts and perspectives. Biochemical and biophysical research communications 2014, 445 (4), 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chait BT, Mass spectrometry: bottom-up or top-down? Science 2006, 314 (5796), 65–66. [DOI] [PubMed] [Google Scholar]
- 4.Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M; Wu C; Sweet SMM; Early BP; Siuti N; LeDuc RD; Compton PD; Thomas PM; Kelleher NL, Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480 (7376), 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kertész-Farkas A; Reiz B; P Myers M; Pongor S, Database searching in mass spectrometry based proteomics. Current Bioinformatics 2012, 7 (2), 221–230. [Google Scholar]
- 6.Sadygov RG; Cociorva D; Yates JR III, Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nature methods 2004, 1 (3), 195. [DOI] [PubMed] [Google Scholar]
- 7.Liu X; Hengel S; Wu S; Tolić N; Pasa-Tolic L; Pevzner PA, Identification of ultramodified proteins using top-down tandem mass spectra. Journal of proteome research 2013, 12 (12), 5830–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durbin KR; Fornelli L; Fellers RT; Doubleday PF; Narita M; Kelleher NL, Quantitation and identification of thousands of human proteoforms below 30 kDa. Journal of proteome research 2016, 15 (3), 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith LM; Kelleher NL; Linial M; Goodlett D; Langridge-Smith P; Goo YA; Safford G; Bonilla L; Kruppa G; Zubarev R; Rontree J; Chamot-Rooke J; Garavelli J; Heck A; Loo J; Penque D; Hornshaw M; Hendrickson C; Pasa-Tolic L; Borchers C; Chan D; Young N; Agar J; Masselon C; Gross M; McLafferty F; Tsybin Y; Ge Y; Sanders I; Langridge J; Whitelegge J; Marshall A, Proteoform: a single term describing protein complexity. Nature methods 2013, 10 (3), 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kou Q; Wu S; Liu X, Systematic Evaluation of Protein Sequence Filtering Algorithms for Proteoform Identification Using Top-Down Mass Spectrometry. Proteomics 2018, 18 (3-4), 1700306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.UniProt: the universal protein knowledgebase. Nucleic acids research 2016, 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kou Q; Zhu B; Wu S; Ansong C; Tolic N; Paša-Tolić L; Liu X, Characterization of proteoforms with unknown post-translational modifications using the MIScore. Journal of proteome research 2016, 15 (8), 2422–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamdborg L; LeDuc RD; Glowacz KJ; Kim Y-B; Viswanathan V; Spaulding IT; Early BP; Bluhm EJ; Babai S; Kelleher NL, ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic acids research 2007, 35 (suppl_2), W701–W706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chick JM; Kolippakkam D; Nusinow DP; Zhai B; Rad R; Huttlin EL; Gygi SP, A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nature biotechnology 2015, 33 (7), 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park J; Piehowski PD; Wilkins C; Zhou M; Mendoza J; Fujimoto GM; Gibbons BC; Shaw JB; Shen Y; Shukla AK, Informed-Proteomics: open-source software package for top-down proteomics. Nature methods 2017, 14 (9), 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank AM; Pesavento JJ; Mizzen CA; Kelleher NL; Pevzner PA, Interpreting top-down mass spectra using spectral alignment. Analytical chemistry 2008, 80 (7), 2499–2505. [DOI] [PubMed] [Google Scholar]
- 17.Tsur D; Tanner S; Zandi E; Bafna V; Pevzner PA In Identification of post-translational modifications via blind search of mass-spectra, 2005 IEEE Computational Systems Bioinformatics Conference (CSB'05), IEEE: 2005; pp 157–166. [DOI] [PubMed] [Google Scholar]
- 18.Sun R-X; Luo L; Wu L; Wang R-M; Zeng W-F; Chi H; Liu C; He S-M, pTop 1.0: a high-accuracy and high-efficiency search engine for intact protein identification. Analytical chemistry 2016, 88 (6), 3082–3090. [DOI] [PubMed] [Google Scholar]
- 19.Kou Q; Wu S; Tolić N; Pasa-Tolić L; Liu X, Mass graphs and their applications in top-down proteomics. bioRxiv 2015, 031997. [Google Scholar]
- 20.Kou Q; Xun L; Liu X, TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32 (22), 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kou Q; Wu S; Tolić N; Paša-Tolić L; Liu Y; Liu X, A mass graph-based approach for the identification of modified proteoforms using top-down tandem mass spectra. Bioinformatics 2017, 33 (9), 1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basharat AR; Iman K; Khalid MF; Anwar Z; Hussain R; Kabir HG; Tahreem M; Shahid A; Humayun M; Hayat HA; Mustafa M; Shoaib MA; Ullah Z; Zarina S; Ahmed S; Uddin E; Sadia H; Ahmad F; Chaudhary SU, SPECTRUM–A MATLAB Toolbox for Proteoform Identification from Top-Down Proteomics Data. Scientific reports 2019, 9 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X; Sirotkin Y; Shen Y; Anderson G; Tsai YS; Ting YS; Goodlett DR; Smith RD; Bafna V; Pevzner PA, Protein identification using top-down spectra. Molecular & cellular proteomics 2012, 11 (6), M111. 008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nesvizhskii AI, Proteogenomics: concepts, applications and computational strategies. Nature methods 2014, 11 (11), 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X; Slebos RJ; Wang D; Halvey PJ; Tabb DL; Liebler DC; Zhang B, Protein identification using customized protein sequence databases derived from RNA-Seq data. Journal of proteome research 2011, 11 (2), 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen B; Xu S; Zhou R; Zhang B; Wang X; Liu X; Xu X; Liu S, PGA: an R/Bioconductor package for identification of novel peptides using a customized database derived from RNA-Seq. BMC bioinformatics 2016, 17 (1), 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo S; Cha SW; Merrihew G; He Y; Castellana N; Guest C; MacCoss M; Bafna V, Proteogenomic database construction driven from large scale RNA-seq data. Journal of proteome research 2013, 13 (1), 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimitrakopoulos L; Prassas I; Diamandis EP; Nesvizhskii A; Kislinger T; Jaffe J; Drabovich A, Proteogenomics: opportunities and caveats. Clinical chemistry 2016, 62 (4), 551–557. [DOI] [PubMed] [Google Scholar]
- 29.Wang X; Zhang B, customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics 2013, 29 (24), 3235–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zickmann F; Renard BY, MSProGene: integrative proteogenomics beyond six-frames and single nucleotide polymorphisms. Bioinformatics 2015, 31 (12), i106–i115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y; Wang X; Cho J-H; Shaw TI; Wu Z; Bai B; Wang H; Zhou S; Beach TG; Wu G; Zhang J; Peng J, JUMPg: an integrative proteogenomics pipeline identifying unannotated proteins in human brain and cancer cells. Journal of proteome research 2016, 15 (7), 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burset M; Seledtsov IA; Solovyev VV, SpliceDB: database of canonical and non-canonical mammalian splice sites. Nucleic acids research 2001, 29 (1), 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komor MA; Pham TV; Hiemstra AC; Piersma SR; Bolijn AS; Schelfhorst T; Delis-van Diemen PM; Tijssen M; Sebra RP; Ashby M; Meijer GA; Jimenez CR; Fijneman RJA, Identification of differentially expressed splice variants by the proteogenomic pipeline Splicify. Molecular & Cellular Proteomics 2017, 16 (10), 1850–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolmogorov M; Liu X; Pevzner PA, SpectroGene: a tool for proteogenomic annotations using top-down spectra. Journal of proteome research 2015, 15 (1), 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagaraj SH; Waddell N; Madugundu AK; Wood S; Jones A; Mandyam RA; Nones K; Pearson JV; Grimmond SM, PGTools: a software suite for proteogenomic data analysis and visualization. Journal of proteome research 2015, 14 (5), 2255–2266. [DOI] [PubMed] [Google Scholar]
- 36.Yates A; Akanni W; Amode MR; Barrell D; Billis K; Carvalho-Silva D; Cummins C; Clapham P; Fitzgerald S; Gil L; Gordon L; Hourlier T; Hunt SE; Janacek SH; Johnson N; ón C. G. ı. G.; Juettemann T; Keenan S; Lavidas I; Martin FJ; Maurel T; McLaren W; Murphy DN; Nag R; Nuhn M; Parker A; Patricio M; Pignatelli M; Rahtz M; Riat HS; Sheppard D; Taylor K; Thormann A; Vullo A; Wilder SP; Zadissa A; EwanBirney; Harrow J; Muffato M; Perry E; Ruffier M; Spudich G; Trevanion SJ; Cunningham F; Aken BL; Zerbino DR; Flicek P, Ensembl 2016. Nucleic acids research 2015, 44 (D1), D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Askenazi M; Ruggles KV; Fenyö D, PGx: putting peptides to BED. Journal of proteome research 2016, 15 (3), 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z; He B; Feng W, Evaluation of bottom-up and top-down mass spectrum identifications with different customized protein sequences databases. Bioinformatics 2020, 36 (4), 1030–1036. [DOI] [PubMed] [Google Scholar]
- 39.Ntai I; LeDuc RD; Fellers RT; Erdmann-Gilmore P; Davies SR; Rumsey J; Early BP; Thomas PM; Li S; Compton PD; Ellis MJC; Ruggles KV; Fenyo D; Boja ES; Rodriguez H; Townsend RR; Kelleher NL, Integrated bottom-up and top-down proteomics of patient-derived breast tumor xenografts. Molecular & Cellular Proteomics 2016, 15 (1), 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan Y; Hu Y; Xiao Q; Tang Y; Chen H; He J; Chen L; Jiang K; Wang Z; Yuan Y; Ding K, Silencing of brain-expressed X-linked 2 (BEX2) promotes colorectal cancer metastasis through the Hedgehog signaling pathway. International Journal of Biological Sciences 2020, 16 (2), 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson LC; DeHart CJ; Kaiser NK; Fellers RT; Smith DF; Greer JB; LeDuc RD; Blakney GT; Thomas PM; Kelleher NL, Identification and characterization of human proteoforms by top-down LC-21 tesla FT-ICR mass spectrometry. Journal of proteome research 2017, 16 (2), 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J; Mouradov D; Wang X; Jorissen RN; Chambers MC; Zimmerman LJ; Vasaikar S; Love CG; Li S; Lowes K, Colorectal cancer cell line proteomes are representative of primary tumors and predict drug sensitivity. Gastroenterology 2017, 153 (4), 1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenna A; Hanna M; Banks E; Sivachenko A; Cibulskis K; Kernytsky A; Garimella K; Altshuler D; Gabriel S; Daly M; DePristo MA, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010, 20 (9), 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A; Davis CA; Schlesinger F; Drenkow J; Zaleski C; Jha S; Batut P; Chaisson M; Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29 (1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adetunji MO; Lamont SJ; Abasht B; Schmidt CJ, Variant analysis pipeline for accurate detection of genomic variants from transcriptome sequencing data. PloS one 2019, 14 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K; Li M; Hakonarson H, ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 2010, 38 (16), e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen S; Park JW; Lu Z.-x.; Lin L; Henry MD; Wu YN; Zhou Q; Xing Y, rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proceedings of the National Academy of Sciences 2014, 111 (51), E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y; Liu J; Huang B; Xu YM; Li J; Huang LF; Lin J; Zhang J; Min QH; Yang WM; Wang X, Mechanism of alternative splicing and its regulation. Biomedical reports 2015, 3 (2), 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kessner D; Chambers M; Burke R; Agus D; Mallick P, ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24 (21), 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S; Mischerikow N; Bandeira N; Navarro JD; Wich L; Mohammed S; Heck AJ; Pevzner PA, The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Molecular & Cellular Proteomics 2010, 9 (12), 2840–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrow J; Frankish A; Gonzalez JM; Tapanari E; Diekhans M; Kokocinski F; Aken BL; Barrell D; Zadissa A; Searle S; Barnes I; Bignell A; Boychenko V; Hunt T; Kay M; Mukherjee G; Rajan J; Despacio-Reyes G; Saunders G; Steward C; Harte R; Lin M; Howald C; Tanzer A; Derrien T; Chrast J; Walters N; Balasubramanian S; Pei B; Tress M; Rodriguez JM; Ezkurdia I; Baren J. v. ; Brent M; Haussler D; Kellis M; Valencia A; Reymond A; Gerstein M; Guigó R; Hubbard TJ, GENCODE: the reference human genome annotation for The ENCODE Project. Genome research 2012, 22 (9), 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen B; Li K; Zhang Y; Zhang B, Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nature communications 2020, 11 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Parameters and commands for software tools used in RNA-seq analysis.
Figure S1: The diagram of five types of alternative splicing events.
Figure S2: The distributions of the types of variants reported by the ANNOVAR.
Figure S3: The distributions of the numbers of nonsynonymous substitutions and indels per transcript reported by ANNOVAR.
Figure S4: An example PrSM of the confident identifications from SWISS-M database.
Figure S5: An example PrSM of the unconfident identifications from SWISS-M database.
Figure S6: The PrSMs containing two heterozygous variants per sequence.
Figure S7: An example PrSM of the identifications from BASIC-ES database.
Table S1: Parameter setting for TopPIC.
Table S2: Proteoforms covering SNV sites identified from SWISS-M, BASIC-M and COMP-M.
Table S3: Proteoforms covering exon skipping events identified from BASIC-ES.
Table S4: Peptides covering SNV sites identified in bottom-up MS.