

Abstract
The treacherous nature of tuberculosis (TB) combined with the ubiquitous presence of the drug-resistant (DR) forms pose this disease as a growing public health menace. Therefore, it is imperative to develop new chemotherapeutic agents with a novel mechanism of action to circumvent the cross-resistance problems. The unique architecture of the Mycobacterium tuberculosis (M. tb) outer envelope plays a predominant role in its pathogenesis, contributing to its intrinsic resistance against available therapeutic agents. The mycobacterial membrane protein large 3 (MmpL3), which is a key player in forging the M. tb rigid cell wall, represents an emerging target for TB drug development. Several indole-2-carboxamides were previously identified in our group as potent anti-TB agents that act as inhibitor of MmpL3 transporter protein. Despite their highly potent in vitro activities, the lingering Achilles heel of these indoleamides can be ascribed to their high lipophilicity as well as low water solubility. In this study, we report our attempt to improve the aqueous solubility of these indole-2-carboxamides while maintaining an adequate lipophilicity to allow effective M. tb cell wall penetration. A more polar adamantanol moiety was incorporated into the framework of several indole-2-carboxamides, whereupon the corresponding analogues were tested for their anti-TB activity against drug-sensitive (DS) M. tb H37Rv strain. Three adamantanol derivatives 8i, 8j and 8l showed nearly 2- and 4-fold higher activity (MIC = 1.32 – 2.89 μM) than ethambutol (MIC = 4.89 μM). Remarkably, the most potent adamantanol analogue 8j demonstrated high selectivity towards DS and DR M. tb strains over mammalian cells [IC50 (Vero cells) ≥ 169 μM], evincing its lack of cytotoxicity. The top eight active compounds 8b, 8d, 8f, 8i, 8j, 8k, 8l and 10a retained their in vitro potency against DR M. tb strains and were docked into the MmpL3 active site. The most potent adamantanol/adamantane-based indoleamides 8j/8k displayed a two-fold surge in potency against extensively DR (XDR) M. tb strains with MIC values of 0.66 and 0.012 μM, respectively. The adamantanol-containing indole-2-carboxamides exhibited improved water solubility both in silico and experimentally, relative to the adamantane counterparts. Overall, the observed antimycobacterial and physicochemical profiles support the notion that adamantanol moiety is a suitable replacement to the adamantane scaffold within the series of indole-2-carboxamide-based MmpL3 inhibitors.
Keywords: Tuberculosis, MmpL3, Indoleamides, Adamantane, Adamantanol, MDR-TB, XDR-TB, Cytotoxicity, Water solubility
Graphical Abstract
1. Introduction
Tuberculosis (TB) is a life-threatening insidious disease that has afflicted humanity for millennia, taking a heavy toll on human life and health [1]. Mycobacterium tuberculosis (M. tb), the etiologic agent of TB, is a highly communicable airborne pathogen. It mainly affects the lungs and is capable of hijacking the host immune system, often persisting for years without causing any symptoms (termed latent TB) [2–6]. The status of bacteria switches from being dormant to active mode, when the host immune system becomes compromised, allowing the bacteria to invade the host and causing the disease to be symptomatic and contagious [7–9]. Globally, in 2018, TB was contracted by 10 million people; an infection rate that remained steady for the past few years, and claimed the lives of an estimated 1.5 million people, including around 0.25 million in the HIV-positive cohort [10]. The World Health Organisation (WHO) also declared that one quarter of the human population are latently infected with TB [10]. This relentless burden places TB as the number one infectious disease killer (outranking HIV/AIDS) and one of the top 10 causes of death worldwide [10]. Due to the complexity and heterogeneity of the M. tb infection, the treatment protocol is protracted and burdensome [11, 12]. The directly observed TB treatment, short course (TB-DOTS) comprises an initial 2-months intensive therapy with a cocktail of the first-line drugs: isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA) and ethambutol (EMB), followed by a 4-months continuation phase with INH and RIF [10, 13]. Poor patient compliance to this multi-drug regimen (due to side effects, high pill count and lengthy duration of therapy) together with HIV co-epidemic have fuelled the emergence of drug-resistant TB (DR-TB) [12]. Hence, new labels were assigned to the disease, namely multi-, extensively and totally drug-resistant TB (MDR-, XDR-, TDR-TB, respectively), perpetuating TB as a global health threat [14–20]. Besides the increasing prevalence of these refractory strains, the TB resistance crisis is further exacerbated by the limited drug options thereof which require administration for a longer duration (up to 2 years) and are less efficacious, more expensive and cytotoxic compared to the front-line regimen [10, 21]. Accordingly, there is an urgent need to identify novel anti-TB agents directed against new cellular targets, different from the ones targeted by the current anti-TB drugs, in order for them to be effective against drug-sensitive TB (DS-TB) and DR-TB.
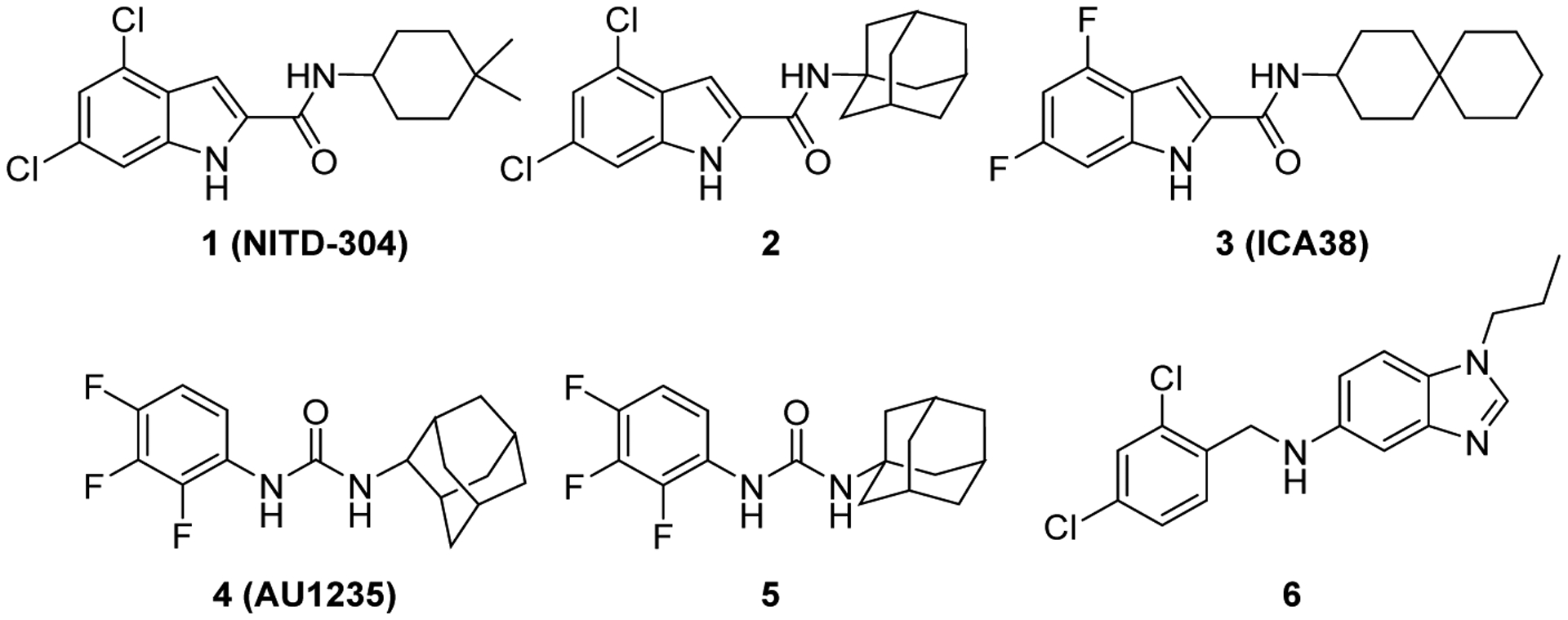
Genetic and lipid profiling studies identified a membrane transporter, the mycobacterial membrane protein large 3 (MmpL3) as a promising antimycobacterial target [22–25]. MmpL3 plays an important role in the heme uptake into the bacterial cell. More importantly, it is responsible for the translocation of mycolic acids (MAs) precursors, trehalose monomycolates (TMMs), across the plasma membrane [26–30]. MAs are the major lipid components of the mycobacterial outer membrane, which are responsible for its hydrophobicity and impermeability to a range of therapeutic agents [31–33]. We previously detailed the MAs biosynthetic machinery and their assembly into the mycobacterial cell envelope [34]. Not only is this intricate protective lipid coating impervious to exogenous substances, including many antibiotics, it also insulates the mycobacteria against the host’s immune system, accentuating its importance in the mycobacterial growth and survival in host [35–37]. Conditional depletion of MmpL3 in Mycobacterium smegmatis (M. smegmatis) led to the loss of cell wall mycolation concomitant with intracellular accumulation of TMM, verifying the role of MmpL3 in shuttling TMM across the cytoplasmic membrane [24, 26, 38]. A downregulation of mmpL3 expression was in fact followed by an abrogation in cell division and rapid cell death [39, 40]. Several MmpL3 inhibitors with varied chemical entities have been reported to date by various high throughput screening (HTS) campaigns, further validating MmpL3 as a viable target for anti-TB drugs. Five structurally distinct analogues, including the indole-2-carboxamides and adamantyl ureas, were recently found to directly inhibit MmpL3 [41–43]. NITD-304 (1, Figure 1) was previously identified as a preclinical agent for treating MDR-TB [23]. In our previous work, the lead indoleamide 2 and ICA38 3 were also highlighted as highly potent anti-TB agents [44, 45]. Unfortunately, both compounds were found to be inactive in vivo, likely due to their poor bioavailability [44, 45].
Figure 1.
Anti-TB derivatives: Indole-2-carboxamides 1, 2 and 3, adamantyl ureas 4 and 5, and benzimidazole 6.
Very recently, the crystal structure of MmpL3 in M. smegmatis was disclosed, serving as an excellent paradigm for the M. tb counterpart due to the high homology between these two mycobacteria [25]. The MmpL3 binding site therein is divided into five subsites (S1-S5) with S4 being the only hydrophilic one. Upon binding, both the indole-2-carboxamides and adamantyl ureas occupy the S3-S5 subsites in the proton-translocation channel, elaborating a common inhibitory mechanism (Figure 2) [25]. These findings were in harmony with the homology model previously constructed by our group [45]. These compounds were found to be likely disrupting the key components of the S4 subsite (the two Asp-Tyr pairs) implicated in the proton relay, thereby blocking the proton motive force (PMF) for substrate translocation. Indeed, this was further substantiated by another study that employed in vitro and whole-cell-based approaches where they found that the adamantyl urea AU1235 4 dissipates the PMF in M. tb from which the MmpL3 transporter derives its energy (indirect mechanism) [42]. Nevertheless, the same group provided evidence that the indole-2-carboxamide derivatives, NITD-304 1 and its analogous difluoro compound NITD-349, in addition to AU1235 4, are able to inhibit MmpL3 via a direct mechanism and that whether or not dissipation of PMF is observed, can be regarded as an additional quality that potentiates the overall activity [42].
Figure 2.
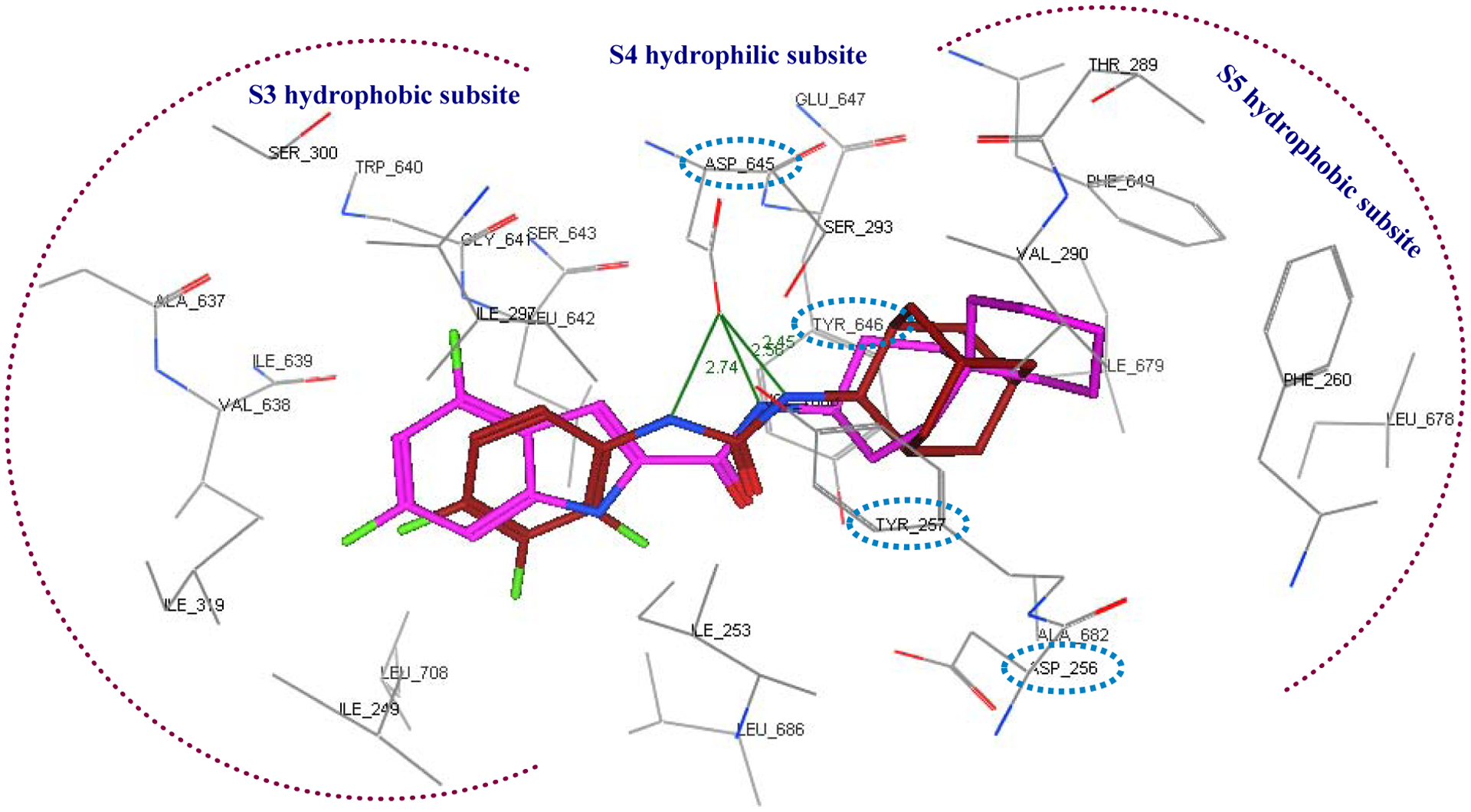
Close-up view of the MmpL3 binding site showing the S3-S5 subsites in which the indole-2-carboxamide ICA38 (magenta) and the adamantyl urea AU1235 (maroon) are stabilised by a set of hydrophobic interactions and hydrogen bonding. The two key Asp-Tyr pairs implicated in the proton relay in the S4 subpocket are marked in cyan hashed ovals.
The valuable insights brought forth by this crystal structure prompted us to reinvigorate our efforts to develop new anti-TB agents that could be endowed with better lead-like properties than the current lead candidates. Therefore, herein, we describe the design, synthesis, and in vitro biological evaluation of adamantane and adamantanol analogues tethered to the framework of various indole-2-caboxamides, phenylureas and benzimidazoles, aspiring to improve water solubility while retaining potent antitubercular activity. All final compounds were screened in vitro against DS M. tb H37Rv strain. The top potent compounds in our study were further evaluated against two nontuberculous mycobacterial (NTM) strains as well as one DS and four DR M. tb strains. In parallel, we tested their cytotoxicity in Vero cells and assessed their drug-likeness in silico to predict their oral bioavailability. We also measured the kinetic water solubility for two pairs of analogues 8b/8c and 8k/8l, as representatives of the adamantane/adamantanol counterparts. Molecular docking analysis was also performed on the top active compounds using the MmpL3 crystal structure.
2. Design
Lipophilicity is an important parameter in the design of novel anti-TB analogues to ensure their permeation through the capsular lipids of M. tb. Unfortunately, high lipophilicity can also be detrimental especially when accompanied with low water solubility and subsequently poor bioavailability. For instance, the bioavailability of the highly lipophilic lead indole-2-carboxamide derivatives 2 and 3 (ClogP = 5.67 and 5.89, respectively) was associated with low water solubility [45]. Their limited aqueous solubility stems from the high lipophilicity of the indole and carbocyclic rings, fulfilling the requisite requirements for the hydrophobic binding pockets of MmpL3 (S3 and S5). Although compounds with high lipophilicity may have improved cellular permeability through the lipid rich mycobacterial cell wall, they need to be sufficiently water soluble at the site of absorption (the aqueous gastrointestinal fluids) in order to traverse through membranes. In other words, both water solubility and lipophilicity are intertwined limiting factors that are crucial for achieving the desired bioavailability and cellular diffusion. In light of these data, the general strategy for the chemical modifications in the present work is mainly aimed at improving the drug-like physicochemical properties of the indole-2-carboxamides by enhancing the water solubility while retaining sufficient lipophilicity to elicit high anti-TB activity. This was attained by integrating water solubilising groups as outlined below.
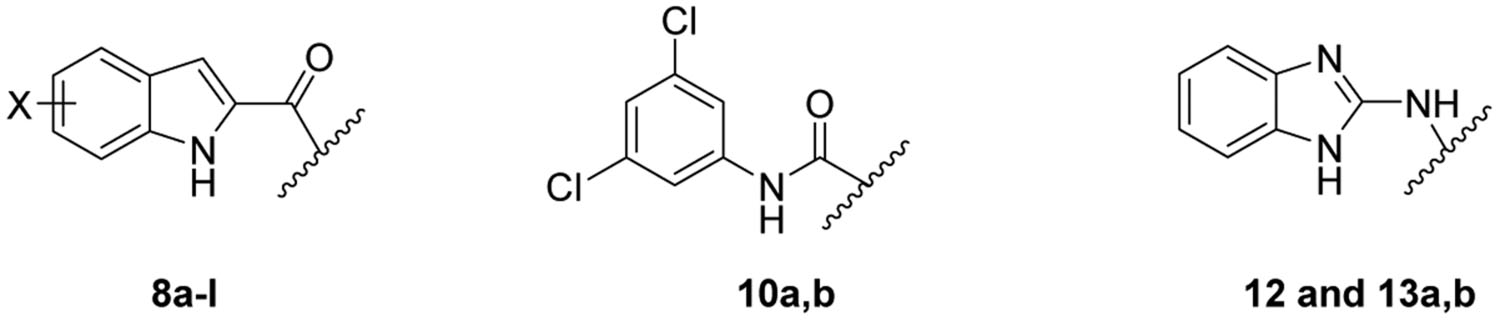
First, in an unprecedented approach, we appended a 3-adamantanol moiety to the indole-2-carboxamides. We reasoned that the insertion of a polar functional substituent (OH group) on the adamantane moiety could be tolerated in terms of steric hindrance while potentially providing the new compounds with improved water solubility. The anti-TB activities of the new 3-hydroxyadamantane analogues were compared to the previously reported and current study’s novel bare adamantane counterparts (8a-l). It is worth noting that adamantane is a privileged scaffold in medicinal chemistry that is considered a “lipophilic bullet”, functioning as an add-on used in the modification of known pharmacophores to provide the needed lipophilicity and enhance the stability of the drugs [46]. On the other hand, adamantyl ureas 4 and 5 were also identified as potent anti-TB compounds (Figure 1) [43]. As the adamantyl urea AU1235 4 was proven to target MmpL3 [25, 26, 42], we scrutinised the activity of two novel 3,5-dichlorophenyl urea analogues bearing an adamantane moiety 10a,b. Similar to the indolecarboxamides, we also assessed the influence of introducing a polar OH group on the adamantane moiety in the urea derivative 10b in comparison to the plain adamantane analogue counterpart 10a.
Additionally, benzimidazole urea analogue 12 was prepared to explore the activity of the benzimidazole scaffold (containing an extra ionisable nitrogen) as a replacement to the phenyl core in the adamantyl ureas. It is noteworthy that benzimidazole derivative 6 (Figure 1) was previously reported to inhibit MmpL3, exhibiting an IC90 value of 16 μM against M. tb H37Rv strain [47]. Besides lowering the lipophilicity, we explored whether the extra NH spacer in the urea analogue 12 would lead to an improved binding with the hydrophilic residues in the S4 subsite, similar to AU1235, thereby exhibiting an enhanced biological activity. We also investigated the activity of the benzimidazole nucleus as a bioisosteric replacement to the indole core to improve the water solubility. Towards this, two benzimidazole amides 13a,b were synthesised and evaluated for their anti-TB activity.
3. Chemistry
The synthetic routes for the preparation of target compounds 8a-l, 10a,b, 12 and 13a,b are delineated in Scheme 1 and 2. The synthesis of the indole-2-carboxamide derivatives 8a-l was accomplished in a one-step amide coupling reaction in which commercially available indole-2-carboxylic acids 7a-h were coupled with 1-adamantylamine or 3-amino-1-adamantanol (Scheme 1) following either amide coupling conditions A or B. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl), hydroxybenzotriazole hydrate (HOBt) and N,N-diisopropylethylamine (DIPEA) were the coupling reagents used to get most of our carboxamide analogues (Method A). Alternatively, we also treated carboxylic acid derivatives 7b and 7d with oxalyl chloride to generate the acid chloride which was subsequently reacted in situ with 1-adamantylamine in the presence of triethylamine to provide the final amides 8b and 8f (Method B). To afford the urea analogues 10a,b, a one pot reaction was carried out in which imidazocarbonylation of 3,5-dichloroaniline (9) was initially conducted under anhydrous conditions at an elevated temperature. The 1,1′-carbonyldiimidazole (CDI)-mediated amidation protocol is a modified approach from the one described by Padiya and co-workers [48]. The resulting carbonylimidazolides were subsequently subjected to a nucleophilic substitution reaction with the corresponding 1-adamantylamine or 3-amino-1-adamantanol in situ to provide the asymmetrical urea analogues 10a,b, respectively (Scheme 2). Padiya et al used water as a solvent and reported that under their conditions neither aromatic amines with a deactivated ring (due to their low nucleophilicity) nor adamantylamine reacted with CDI to yield any product. Hence, we employed more extreme conditions (i.e. heated at 90 °C) and anhydrous dimethylformamide (DMF) as a solvent to render the desired ureas with poor to moderate yields (20–48%).
Scheme 1.
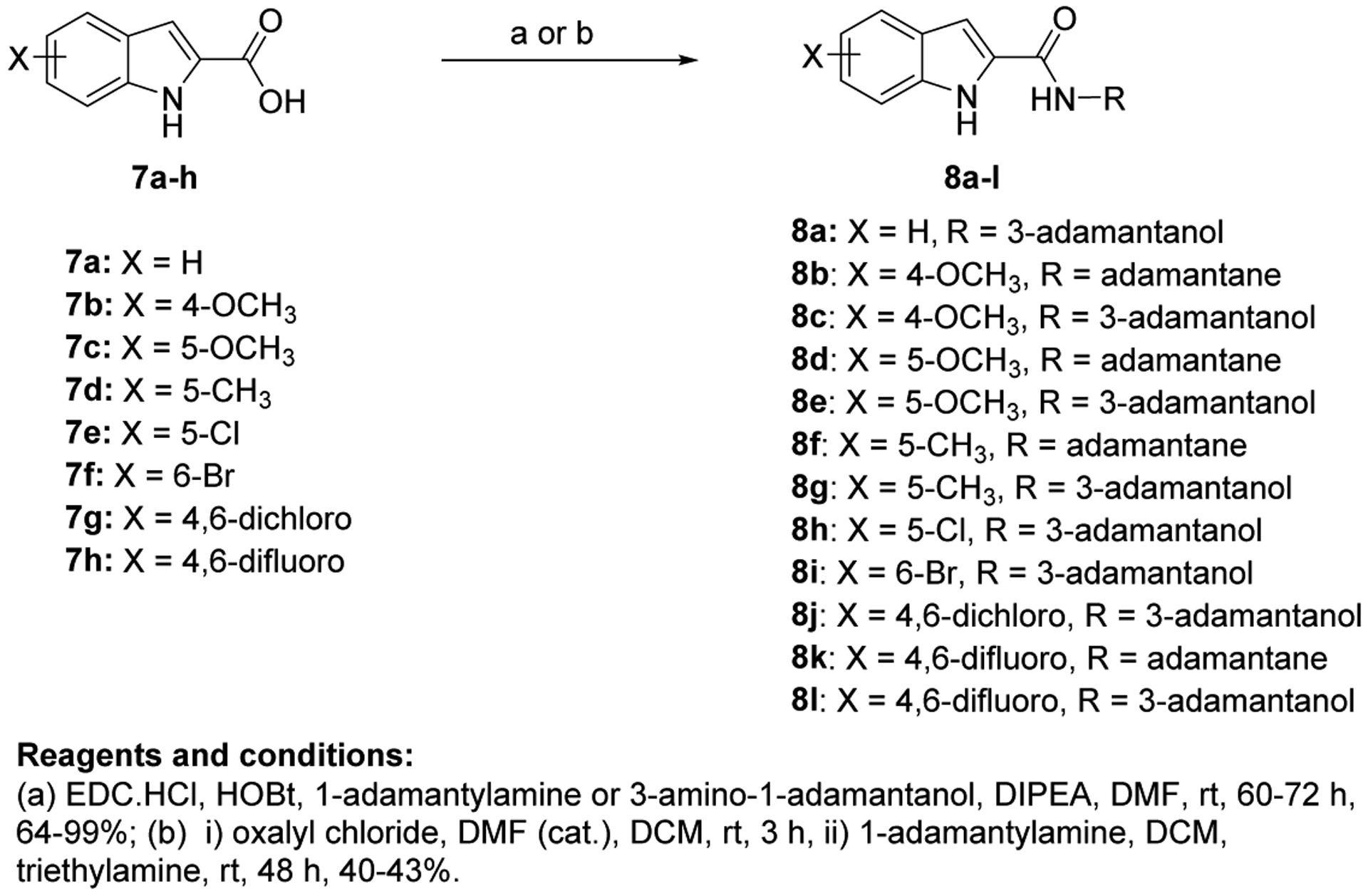
Synthetic conditions for compounds 8a-l.
Scheme 2.
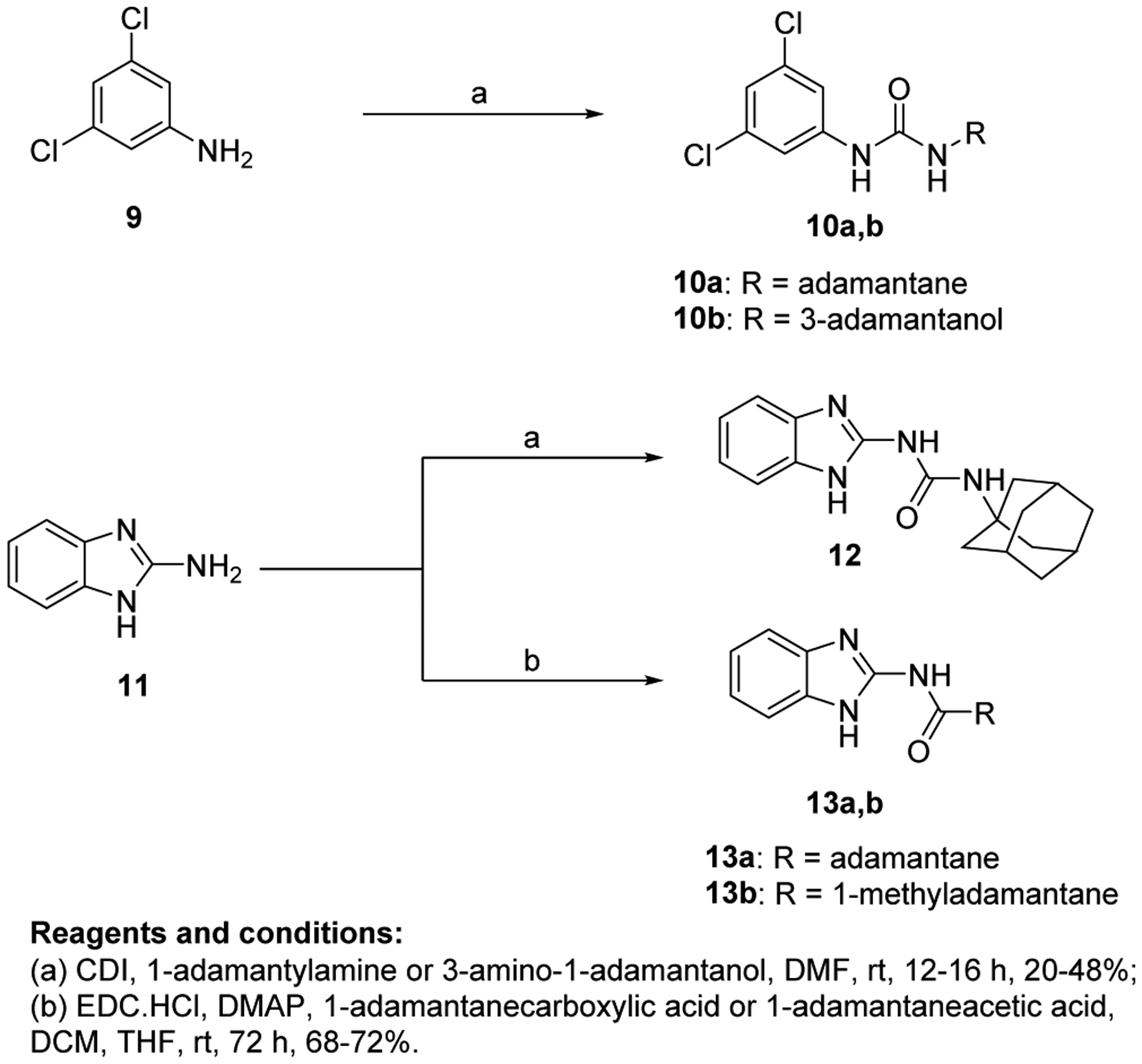
Synthetic conditions for compounds 10a,b, 12, and 13a,b.
On the other hand, analogous to 10a,b, the benzimidazole urea derivative 12 was obtained by treating 2-aminobenzimidazole 11 and 1-adamantylamine with CDI as a crosslinking agent (Scheme 2). Finally, 2-aminobenzimidazole was subjected to amide coupling conditions C [EDC.HCl and 4-dimethylaminopyridine (DMAP)] with 1-adamantanecarboxylic acid and 1-adamantaneacetic acid to deliver the requisite amides 13a,b, respectively.
4. Results and discussion
4.1. Biological Evaluation (DS M. tb H37Rv) and SAR Analysis
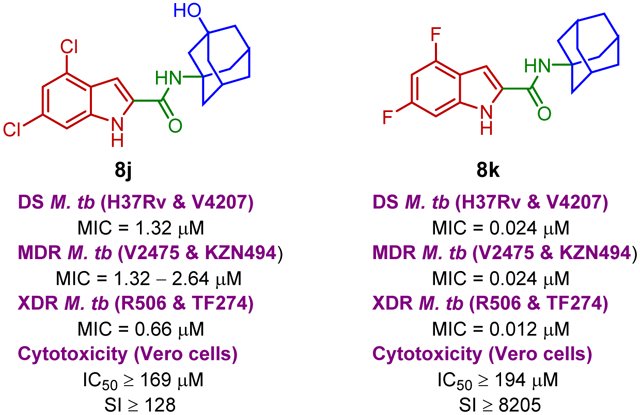
The synthesised seventeen compounds 8a-l, 10a,b, 12 and 13a,b were screened in vitro against M. tb H37Rv strain. The obtained activities, determined as the minimum inhibitory concentration of the drug (MIC) inhibiting the growth of M. tb by at least 90% using the microplate alamarBlue assay (MABA), are summarised in Table 1. The first round of investigation, intended for increasing the polarity and accordingly the water solubility, entailed tethering a 3-hydroxyadamantane moiety to several indole-2-carboxamides and examining their anti-TB activities between the adamantane/adamantanol analogues. The unsubstituted indole-2-carboxamide attached to 3-adamantanol moiety 8a showed a drastic drop in activity (MIC = 51.5 μM) compared to the previously reported adamantane analogue counterpart (MIC = 0.68 μM [49]). However, the 4-methoxyindole derivative 8b possessing an unsubstituted adamantane moiety exhibited an excellent anti-TB activity (MIC = 0.096 μM). Compound 8b is approximately 3-fold more active than the standard first line anti-TB drug INH (MIC = 0.29 μM). In contrast, its hydroxy adamantane analogue 8c showed a modest anti-TB activity (MIC = 11.7 μM). Shifting the methoxy group to position 5 of the indole ring 8d led to an approximately 8-fold loss of activity (MIC = 0.77 μM) compared to compound 8b (MIC = 0.096 μM). Again, the less lipophilic 3-adamantanol analogue 8e (MIC = 47.0 μM, ClogP = 2.73) was nearly 61-fold less active than the adamantane counterpart 8d (MIC = 0.77 μM, ClogP = 4.12). Similar to compounds pair 8d/8e, the 5-methylindole derivative containing an adamantanol moiety 8g displayed a dramatic attenuation of activity compared to compound 8f (MIC = 24.7 and 0.20 μM, respectively). Of particular note, compound 8f is approximately 1.5- and 4-fold more potent than INH and 8d, respectively. This signifies that methyl substituents are generally more favourable than the more polar methoxy groups due to their enhanced lipophilicity (8d: ClogP = 4.12, 8f: ClogP = 4.60). However, the nearly 8- and 2-fold increase in activity manifested in the 4-methoxyindole analogue 8b compared to the 5-methoxyindole 8d and 5-methylindole 8f counterparts, respectively, epitomises the consequential role of the substitution pattern in the indole ring on the activity. The 5-chloroindole derivative 8h bearing a 3-adamantanol moiety was bereft of anti-TB activity (MIC > 93 μM), as opposed to the previously observed high potency of the N-adamantane-5-chloroindole analogue (MIC = 0.38 μM [44]). This is a noteworthy result because the lipophilicity of compound 8h (ClogP = 3.53) is more than that of the above-mentioned adamantanols 8a, 8c, 8e and 8g, yet 8h displayed lower potency than the foregoing analogues. Intriguingly, compound 8i, a 6-bromoindole carrying a 3-adamantanol moiety, was approximately 2-fold more active than the first line anti-TB drug EMB (MIC = 2.57, 4.89 μM, respectively). Despite the gap in the activity between 8i and its previously reported adamantane counterpart (MIC = 0.042 μM [45, 50]), the satisfactory activity of 8i establishes it as a promising anti-TB compound. In this case, 8i still retained a decent anti-TB activity while being significantly less lipophilic than its adamantane analogue (ClogP = 3.68 and 5.07, respectively). Next, we explored the effect of disubstitution with chloro and fluoro groups on the indole ring. Indoleamides bearing the 4,6-dihalogen groups, for instance compounds 2 and 3, were previously deemed more advantageous than the 4,6-dimethyl counterpart as the methyl groups presented metabolic liability (prone to metabolic oxidation) [45]. In this regard, the dihalogen groups circumvent this metabolism issue while equipping the indole ring with similar lipophilicity. It was pleasing to find that the dichloro analogue 8j (MIC = 1.32 μM) bearing an adamantanol moiety is about 4- and 2-fold more active than EMB and compound 8i, respectively. Importantly, the dichloroindole 8j, which is the most lipophilic derivative in the N-(3-hydroxyadamantane)indole-2-carboxamide series (ClogP = 4.27), proved to be the most active compound in this set of adamantanol derivatives. These findings in turn clearly reflect the positive correlation between lipophilicity and anti-TB activity. A similar trend in activity was observed for compounds 8k and 8l with 4,6-difluoro substitution on the indole ring. The bare adamantane analogue of the 4,6-difluoroindole 8k (MIC = 0.024 μM) was the most active compound in our study, displaying more potency than the adamantanol counterpart 8l (MIC = 2.89 μM). It is important to note, however, that the activity of the adamantanol analogue 8l is still high, possessing nearly a 2-fold improvement in activity compared to EMB and about a 2-fold inferior activity compared to 8j. Interestingly, compound 8l is less lipophilic than 8i (ClogP = 3.13, 3.68, respectively) and yet both molecules displayed comparable potencies with MIC values of 2.89 and 2.57 μM, respectively. In addition, while compound 8k showed a 2-fold decrease in activity when compared to the previously reported dichloroindole counterpart (MIC = 0.011 μM [45]), its lipophilicity was more optimal than the 4,6-dichloroindole analogue (ClogP = 4.53 and 5.67, respectively). Thus far, these findings support the notion that the anti-TB activity is lipophilicity-driven in these indole-2-carboxamides. A polar hydroxy group placed at the adamantane moiety impacted the MIC values, but substituting the indole ring at position 4 and/or 6 appeared to salvage the anti-TB activity, in agreement with our previous findings on the indole-2-carboxamides [45].
Table 1.
In vitro anti-TB activity of target compounds 8a-l, 10a,b, 12, and 13a,b as well as reference standards INH, EMB, 1, 2, 4, 5, and 6.
| Comp | X | R | MICa μM | ClogPb | Comp | X | R | MICa μM | ClogPb |
|---|---|---|---|---|---|---|---|---|---|
| 8a | H | 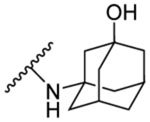 |
51.5 | 2.71 | 10a | - | 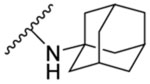 |
1.47 | 5.59 |
| 8b | 4-OCH3 | 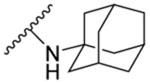 |
0.096 | 4.12 | 10b | - | 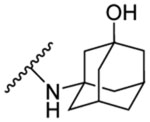 |
>90 | 4.15 |
| 8c | 4-OCH3 | 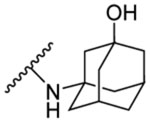 |
11.7 | 2.73 | 12 | - | 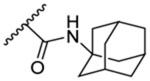 |
25.8 | 4.15 |
| 8d | 5-OCH3 | 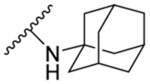 |
0.77 | 4.12 | 13a | - | 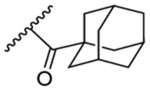 |
13.5 | 4.07 |
| 8e | 5-OCH3 | 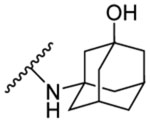 |
47.0 | 2.73 | 13b | - | 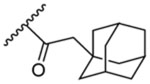 |
>103 | 5.73 |
| 8f | 5-CH3 | 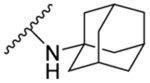 |
0.20 | 4.60 | INH | - | - | 0.29 [22] | −0.67 |
| 8g | 5-CH3 | 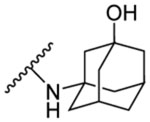 |
24.7 | 3.21 | EMB | - | - | 4.89 [22] | 0.12 |
| 8h | 5-Cl | 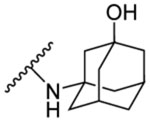 |
>93 | 3.53 | 1 | - | - | 0.015 [23] | 6.08 |
| 8i | 6-Br | 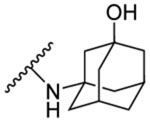 |
2.57 | 3.68 | 2 | - | - | 0.011 [45] | 5.67 |
| 8j | 4,6- dichloro | 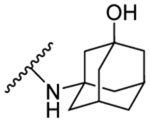 |
1.32 | 4.27 | 4 | - | - | 0.03–0.3 [26, 43] | 5.08 |
| 8k | 4,6- difluoro | 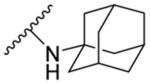 |
0.024 | 4.53 | 5 | - | - | 1.23 [43] | 4.04 |
| 8l | 4,6- difluoro | 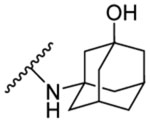 |
2.89 | 3.13 | 6 | - | - | 16 [47] | 5.76 |
The lowest concentration of drug diminishing at least 90% of bacterial growth by the microplate alamarBlue assay (MABA). The reported MIC values are an average of three individual measurements.
Estimated using ChemDraw 16.0.
In line with the activity and framework of the dichloroindole derivative 8j, we were interested in evaluating the 3,5-dichlorophenyl urea derivative bearing either adamantane 10a or adamantanol 10b. Our attention was drawn to the adamantyl urea skeleton because they are well-known MmpL3 inhibitors with potent anti-TB activity, wherein the halogenated derivative AU1235 (4) stands out as an exemplar of this class [25, 42, 43, 51]. The 3,5-dichloro analogue 10a exhibited potent anti-TB activity (MIC = 1.47 μM) which is approximately 3-fold more than EMB and around 5-fold less than AU1235. It is worth mentioning that AU1235 was initially reported to have an MIC value of 0.03 μM and in the subsequent reports of the same group they identified its MIC to be 0.3 μM [26, 43, 52]. Indeed, in our previous evaluation of AU1235, the observed MIC was 0.096 – 0.19 μM [22]. Although compound 10a was less active than the N-(2-adamantyl)urea analogue AU1235, it exhibited similar activity to the more homologous N-(1-adamantyl)urea analogue 5 (MIC = 1.23 μM [43]). The discrepancies in the activities of compound 10a and 5 in comparison to AU1235 is ostensibly correlated to the preference for the 2-adamantyl urea scaffold over the 1-adamantyl counterpart. This was further supported by the fact that despite the higher lipophilicity of compound 10a compared to AU1235 (ClogP = 5.59 and 5.08, respectively), 10a was less potent than AU1235. Unfortunately, the adamantanol counterpart 10b was devoid of anti-TB activity (MIC > 90 μM), featuring once again the apparent impact of lipophilicity on activity (ClogP = 5.59 and 4.15 corresponding to compounds 10a and 10b, respectively).
On the other hand, replacing the phenyl core in the urea analogue 10a with a benzimidazole nucleus (entailing an ionisable nitrogen) yielded the urea derivative 12 (MIC = 25.8 μM) which showed more activity than 10b, despite their similar lipophilicity (ClogP = 4.15). Additionally, our group previously reported some benzimidazole-2-carboxamides as potent anti-TB compounds [44]. Therefore, in consonance with our endeavour to achieve more water-soluble compounds, we further examined the benzimidazole scaffold as a replacement to the indole core. We were particularly interested in probing the influence of reversing the amide linker in the benzimidazoleamide derivatives. The reversed amide compound 13a showed moderate activity with a MIC value of 13.5 μM which is 2-fold higher than that of compound 12. This activity was significantly lower than the evaluated N-adamantyl benzimidazole-2-carboxamide derivatives in our previous study (MIC = 0.39 and 1.5 μM [44]), suggesting that reversing the amide linker in this framework is unfavourable. Nevertheless, 13a was more potent than the most active benzimidazole derivative 6 in Stanley et al study (MIC = 16 μM [47]). Finally, the influence of extending the length between the benzimidazole nucleus and the adamantane ring was further scrutinised. In this respect, a methylene spacer was inserted between the carboxamide linker and the adamantane moiety, yielding the highly lipophilic compound 13b (ClogP = 5.73), which surprisingly showed a large loss of activity (MIC > 103 μM) compared to 13a. This finding is discrepant with our previous results on the indole-2-carboxamides, in which the addition of a methylene spacer next to the cycloaliphatic ring was tolerated [44]. These data suggest that lipophilicity seems to not be the main factor driving the anti-TB potency in these benzimidazoles.
4.2. In vitro Activity of the Most Potent Compounds against NTM Strains and Clinical Isolates of M. tb along with Their Cytotoxicity Evaluation
Based on their satisfactory results in vitro against the wild-type M. tb H37Rv strain, the most promising eight compounds, namely 8b, 8d, 8f, 8i, 8j, 8k, 8l and 10a were selected for further in vitro studies against two NTM strains and a panel of clinical isolates of M. tb (Table 2). Ciprofloxacin (CPF), one of the second-line anti-TB drugs (used in the treatment of MDR-TB), was simultaneously evaluated against the same mycobacterial strains, serving as a positive control. Interestingly, N-adamantyl-4-methoxyindole-2-carboxamide (8b) exhibited a decent activity against Mycobacterium abscessus (M. abs) which is 2-fold higher than that of CPF (MIC = 12.3 and 24.1, respectively). However, compound 8b was devoid of activity against Mycobacterium avium (M. avium) in comparison to CPF (MIC > 197 and 0.75 μM, respectively). Conversely, the adamantyl urea derivative 10a exhibited more potency against M. avium than M. abs (MIC = 47.2 and > 189 μM, respectively). Meanwhile, the rest of the compounds were bereft of activity in the M. abs and M. avium assays (MIC > 164 μM), suggesting the selective activity of these compounds against M. tb. On the other hand, pleasingly, all eight compounds retained their high activity against a panel of DS and DR M. tb strains, originally procured from pulmonary TB patients. The M. tb clinical isolates comprise one DS (V4207), two MDR (V2475, KZN494), and two XDR (R506, TF274) strains. Three derivatives 8b, 8f, and 8k were more potent than CPF when tested against DS M. tb H37Rv and V4207 strains (CPF MIC = 0.75 μM) and the preceding MDR M. tb isolates, whilst the activity of compound 8d against these strains was comparable to CPF. Notably, the most active adamantanol and adamantane-based indoleamides 8j and 8k fortuitously displayed a 2-fold increase in activity against the forenamed XDR M. tb strains (MIC = 0.66 and 0.012 μM, respectively). In fact, all eight compounds showed significantly higher activities against XDR-strains compared to CPF. The remarkable potencies of these compounds spotlight not only their potential to treat DS- and DR-TB, but also the prospective lack of cross resistance between these derivatives and the currently used medications. We also further evaluated the toxicity of the top potent compounds against Vero cells, expressed as IC50 values, and their selectivity index (SI) were subsequently calculated. All compounds, except for 10a, exhibited high SI values, demonstrating the potential lack of cytotoxicity of these analogues against mammalian cells (Table 2). In particular, compound 8j (IC50 ≥ 169 μM), the most active compound in the 3-adamantanol set of derivatives, displayed higher IC50 value than the other two analogous compounds 8i and 8l in the same series (IC50 = 82.2 and 92.4 μM, respectively). Similar to 8j, the tested N-(1-adamantyl)-indole-2-carboxamides exhibited high selectivity against DS and DR M. tb strains over mammalian cells (IC50 ≥ 194 μM, SI ≥ 256). Meanwhile, the urea analogue 10a showed some toxicity against Vero cells with IC50 of 5.9 μM (SI = 4.0). This result was in fact counterintuitive due to the previously reported limited cytotoxicity of the analogous adamantyl urea AU1235 4 and compound 5 (IC50 = 675 and 2238 μM, respectively [43]).
Table 2.
In vitro activities of the top eight potent compounds and CPF against M. abs, M. avium and selected clinical isolates of M. tb [MIC (μM)] as well as their cytotoxicity against Vero cells [IC50 (μM)].
| Comp | M. abs (μM) | M. avium (μM) | M. tb MIC (μM) | IC50 (μM) | |||||
|---|---|---|---|---|---|---|---|---|---|
| V4207/DS | V2475/MDRa | KZN494/MDRa | R506/XDRb | TF274/XDRb | Vero cells | SIc | |||
| 8b | 12.3 | >197 | 0.096 | 0.096 | 0.048 | 0.024–0.048 | 0.024–0.048 | ≥197 | ≥2048 |
| 8d | >197 | >197 | 0.39–0.77 | 0.39–0.77 | 0.39 | 0.39 | 0.39 | ≥197 | ≥256 |
| 8f | >207 | >207 | 0.20 | 0.20 | 0.20 | 0.10–0.20 | 0.10–0.20 | ≥207 | ≥1024 |
| 8i | >164 | >164 | 5.14 | 5.14 | 2.57–5.14 | 2.57 | 2.57 | 82.2 | 32 |
| 8j | >169 | >169 | 1.32 | 2.64 | 1.32 | 0.66 | 0.66 | ≥169 | ≥128 |
| 8k | >194 | >194 | 0.024 | 0.024 | 0.024 | 0.012 | 0.012 | ≥194 | ≥8205 |
| 8l | >185 | >185 | 2.89 | 5.77 | 2.89 | 2.89 | 1.44 | 92.4 | 32 |
| 10a | >189 | 47.2 | 1.47 | 1.47–2.95 | 1.47 | 0.74 | 0.74 | 5.9 | 4.0 |
| CPF | 24.1 | 0.75 | 0.75 | 0.75 | 0.75 | 6.04 | 6.04 | ≥193 | ≥256 |
Resistant to INH and rifampin (RIF)
Resistant to INH, RIF, levofloxacin, ofloxacin, and kanamycin
Selectivity index (SI) = IC50(Vero)/MIC(H37Rv)
4.3. Molecular Docking
Considering that MmpL3 is most likely the target of our compounds, a molecular modelling study was performed on the most active derivatives (MIC ≤ 3 μM) to gain some perspectives on their binding mechanism within the active site of this membrane transporter (PDB ID: 6AJJ). Initially, reference ligands ICA38 3 and AU1235 4 were redocked into the MmpL3 binding pocket to validate the docking protocol, using MOE 2008.10 modelling software (Molecular Operating Environment), as published previously [53, 54]. The bulky S3 and S5 hydrophobic subsites harboured the 4,6-dichloroindole/trifluorophenyl scaffolds and the spirocarbocyclic/adamantane groups, respectively, with regard to ligands 3/4, forming extensive set of hydrophobic contacts with the surrounding residues (Figure 2). The amide and urea linkers, meanwhile, were positioned in the S4 hydrophilic subsite, whereupon the amide NH of the spirocyclic ligand ICA38 formed only one hydrogen bond with Asp645, whereas two hydrogen bonds connected Asp645 and the NH groups of the urea motif in AU1235, in accordance with Zhang et al crystal structure report [25]. Notably, Asp645 is an integral piece of the two Asp-Tyr pairs, the key elements of the S4 subsite, implicated in proton relay. Upon binding, ICA38 and AU1235 were found to inhibit MmpL3 by occupying S3-S5 subsites in the proton translocation channel, disrupting the two Asp-Tyr pairs, wherefore they dissipate the PMF for substrate transport [25]. The binding mode of lead compound 2 in the MmpL3 receptor was similar to ICA38 and AU1235 [54].
Next, the most active compounds in our study 8b, 8d, 8f, 8i, 8j, 8k, 8l and 10a were docked into the MmpL3 active site. London dG scoring was used to rank the generated poses after the forcefield refinement stage, in which lower scores designate more favourable ligand configurations. As expected, all eight compounds were oriented inside the MmpL3 binding pocket in a manner resembling that of ICA38 and AU1235, whereby they all showed high binding affinities (docking score −12.5 – −13.6 -kcal/mol). The docking poses of the indole-2-carboxamides, showing the lowest binding energy (most favourable poses), retained the main interactions discerned in ICA38. In the S4 hydrophilic subsite, in addition to the hydrogen bond formed between the amide NH and Asp645 (distance = 2.37 – 2.43 Å), another one took place between Asp645 and the indole NH in the adamantane and adamantanol-based indoleamide analogues (distance = 2.64 – 2.77 Å), similar to lead compound 2. Meanwhile, the indole ring and the adamantane/adamantanol moiety were embedded in the S3 and S5 hydrophobic subsites, respectively. The binding fashion and overlapping of compounds 8j and 8k with ICA38 are illustrated in Figure 3, respectively, exemplifying the 3-adamantanol and adamantane series, respectively. Compound 10a adopted nearly a superimposed orientation with both ICA38 and AU1235 4 as depicted in Figure 4. Akin to AU1235 4, the dichlorophenyl group and adamantane moiety of 10a were lodged in the S3 and S5 hydrophobic subsites, respectively, whilst the urea linker occupied the S4 hydrophilic subsite forming two hydrogen bonds with Asp645 (distance = 2.61 and 2.36 Å). The binding mode and superimposed orientation between the most active compounds in the current study and MmpL3 inhibitors ICA38 and AU1235 suggest that our analogues presumably inhibit the same target, likely by disrupting the two Asp-Tyr pairs involved in the proton relay. The results from another study also favoured a direct mechanism of MmpL3 inhibition by the indole-2-carboxamides and adamantyl ureas [42].
Figure 3.
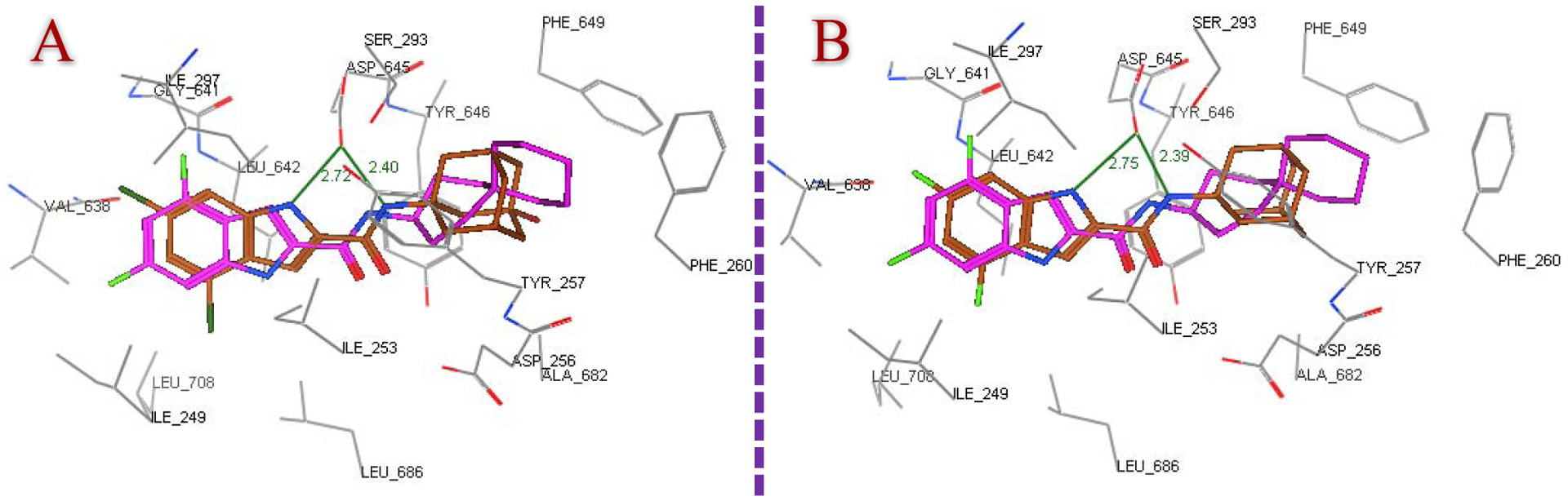
Superposition of the top ranked docking pose of 8j (A) and 8k (B) (brown) and co-crystallised ligand ICA38 (magenta) in the MmpL3 binding pocket, portraying both compounds having similar binding pattern as ICA38.
Figure 4.
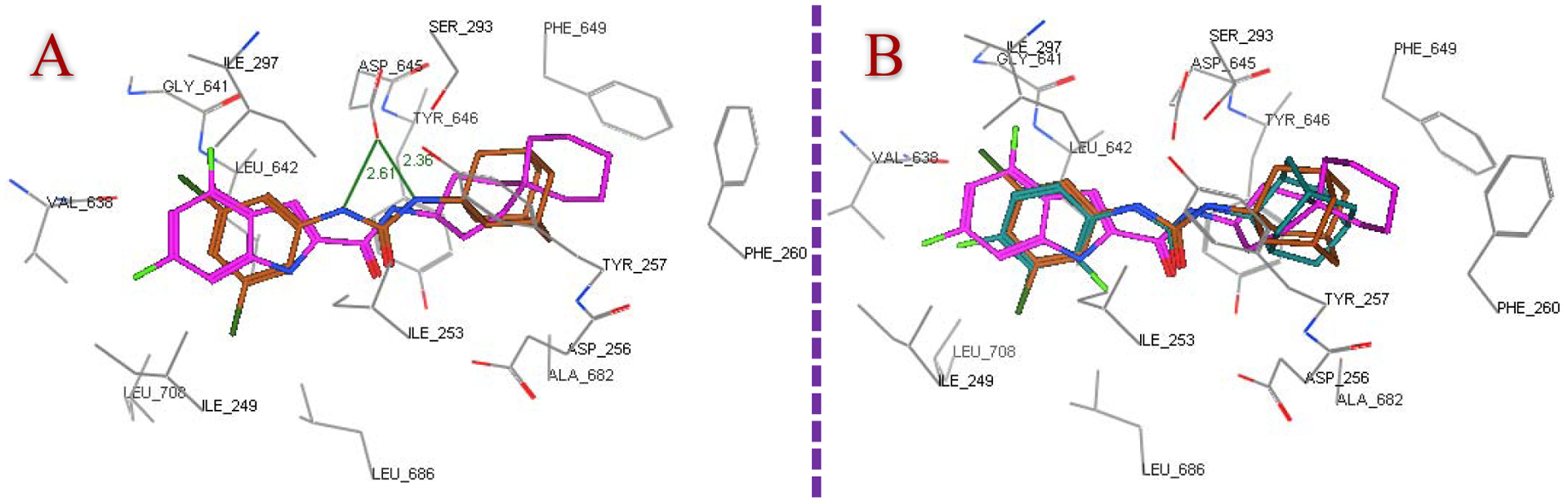
Putative binding mode of compound 10a (brown) superpositioned with ICA38 (magenta) (A) and their overlay with AU1235 (dark cyan) (B) in the MmpL3 active site.
4.4. In silico ADME Profiling
The physicochemical properties of adamantane/adamantanol-based derivatives 8a-l and 10a,b as well as lead compounds 2 and 4 were predicted using ACD/Labs Percepta 2016 Build 2911 (13 Jul 2016) (Table 3). We particularly assessed the compliance of these compounds with Lipinski’s rule of five (RO5) in order to evaluate their drug-likeness [55]. All compounds showed zero violation to the RO5, except for 10a and lead compound 2. The one violation observed in lead compound 2, related to its high lipophilicity, may also explain the lack of in vivo activity of this derivative. Importantly, the most active indoleamides 8b, 8d, 8f, 8i, 8j, 8k, and 8l conform to the RO5, suggesting the drug-like attributes of these compounds, including their prospective in vivo drug absorption and permeation. In fact, all the indoleamides derivatives evaluated in our study exhibited desirable lipophilicities [55] (logP = 2.94 – 4.57). In general, ClogP values obtained from Chemdraw resembled those from ACD/Labs Percepta. Indeed, all the adamantanol-derived compounds in our study were predicted to have higher hydrophilicity/lower lipophilicity than their bare adamantane counterparts. It is noteworthy that the lipophilicity of the most active compound in our study 8k (logP = 4.32) is significantly less than that of bedaquiline (logP > 7 [56]). In 2012, bedaquiline was approved by the United States Food and Drug Administration (FDA), representing the first anti-TB medication with a novel mechanism of action approved by the FDA since 1971 [57]. Several adverse effects correlated with bedaquiline are likely ascribed to its high lipophilicity, therefore it is provisionally recommended for people with pulmonary MDR-TB when no other effective treatment regimen can be designed [56, 57]. Importantly, compound 8k (MIC = 0.012 – 0.024 μM) displayed higher potency than bedaquiline (MIC = 0.11 μM [58]) against both DS- and DR-TB. On the other hand, the Caco-2 permeability values of 8a-l and 10a,b fluctuated between 94 and 214 × 10−6 cm/s, therefore they all are expected to traverse the cell membrane. The plasma protein binding (PPB) degree of the preceding compounds was also estimated to range from 87 to 98%, suggesting their long plasma half-life (T1/2), low volume of distribution and low clearance.
Table 3.
In silico physicochemical and pharmacokinetic parameters of the adamantane/adamantanol-based analogues 8a-l and 10a,b in addition to reference compounds 2 and 4 as per ACD/Labs Percepta 2016 Build 2911 (13 Jul 2016).
| Comp | MW | HBD | HBA | NRB | ClogP (Chem draw) | logP | TPSA | Solubility (mg/L) | Caco-2 (x 10−6 cm/s) | PPB (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 8a | 310.39 | 3 | 4 | 2 | 2.71 | 2.94 | 65.12 | 60 | 151 | 91 |
| 8b | 324.42 | 2 | 4 | 3 | 4.12 | 4.04 | 54.12 | 20 | 214 | 93 |
| 8c | 340.42 | 3 | 5 | 3 | 2.73 | 3.03 | 74.35 | 40 | 155 | 92 |
| 8d | 324.42 | 2 | 4 | 3 | 4.12 | 4.07 | 54.12 | 20 | 214 | 93 |
| 8e | 340.42 | 3 | 5 | 3 | 2.73 | 3.02 | 74.35 | 40 | 155 | 92 |
| 8f | 308.42 | 2 | 3 | 2 | 4.60 | 4.57 | 44.89 | 20 | 194 | 92 |
| 8g | 324.42 | 3 | 4 | 2 | 3.21 | 3.26 | 65.12 | 30 | 168 | 92 |
| 8h | 344.84 | 3 | 4 | 2 | 3.53 | 3.51 | 65.12 | 30 | 177 | 97 |
| 8i | 389.29 | 3 | 4 | 2 | 3.68 | 3.83 | 65.12 | 20 | 180 | 93 |
| 8j | 379.28 | 3 | 4 | 2 | 4.27 | 4.26 | 65.12 | 5 | 167 | 95 |
| 8k | 330.37 | 2 | 3 | 2 | 4.53 | 4.32 | 44.89 | 20 | 207 | 91 |
| 8l | 346.37 | 3 | 4 | 2 | 3.13 | 3.10 | 65.12 | 60 | 161 | 87 |
| 10a | 339.26 | 2 | 3 | 2 | 5.59 | 5.47 | 41.13 | 1 | 94 | 98 |
| 10b | 355.26 | 3 | 4 | 2 | 4.15 | 4.57 | 61.36 | 4 | 144 | 98 |
| 2 | 363.28 | 2 | 3 | 2 | 5.67 | 5.49 | 44.89 | 1 | 91 | 96 |
| 4 | 324.34 | 2 | 3 | 2 | 5.08 | 4.59 | 41.13 | 10 | 194 | 92 |
MW: molecular weight, HBD: H-bond donors, HBA: H-bond acceptors, NRB: number of rotatable bonds, logP: octanol–water partition coefficient, TPSA: topological polar surface area, PPB: plasma protein binding
4.5. Kinetic Water Solubility
Aqueous solubility is a critical parameter for the absorption and oral bioavailability of compounds, in which derivatives equipped with good water solubility are often better qualified for clinical advancement. MmpL3 inhibitors are generally lipophilic due to the hydrophobic nature of the binding site, thereby having an intrinsic poor aqueous solubility which can hamper their drug-like properties as seen in Table 3. This interplay between water solubility and lipophilicity govern the bioavailability of these inhibitors and their ability to penetrate the hydrophobic cell wall of M. tb to elicit anti-TB activity. This was further corroborated by the fact that despite the high activity of the previously evaluated N-(1-adamantyl)-4,6-dichloroindole-2-carboxamide 2 (MIC = 0.011 μM, logP > 5), it turned out to be inactive in vivo due to its poor bioavailability and was not pursued for further studies [45]. The kinetic water solubility assessment using high performance liquid chromatography (HPLC), entailing the solution-precipitation technique using DMSO stock solutions is well established in the literature [59]. Water solubility (mg/L) was measured using analytical HPLC for the N-adamantanol-derived indoles 8c and 8l, serving as representatives of this class, in addition to their bare adamantane counterparts 8b and 8k, respectively. The adamantanol derivatives 8c and 8l showed a conspicuous improvement in water solubility in comparison to their homologous more lipophilic adamantane analogues 8b and 8k. The N-adamantanol-4-methoxy indole 8c was 6-fold more soluble than the corresponding adamantane analogue 8b with aqueous solubility values of 30 and 5 mg/L, respectively. Likewise, the N-adamantanol-4,6-difluoro indole 8l possessed a moderate water solubility (19 mg/L) which is approximately 3-fold better than the respective adamantane derivative 8k (7 mg/L). It is important to note that the ACD/Lab Percepta solubility values of the adamantanol analogues 8c and 8l were predicted to be 40 and 60 mg/L, respectively, compared to 20 mg/L for both adamantane counterparts 8b and 8k, amounting to 2- and 3-fold increase in solubility in compounds pairs 8b/8c and 8k/8l, respectively. Hence, it seems that the computationally estimated water solubility data of our analogues are in harmony with the HPLC-based aqueous solubility experimental values.
5. Conclusions
A series of adamantane and adamantanol analogues were designed and synthesised geared toward identifying potent anti-TB compounds possessing optimal lipophilicity and improved water solubility. All the N-(1-adamantyl)indole-2-carboxamide analogues evaluated in this study were highly potent (MIC = 0.024 – 0.77 μM) when tested against the M. tb H37Rv strain. In case of the analogous 3-adamantanol counterparts, mixed results were obtained. In general, indoles bearing an adamantane moiety were more potent than their corresponding 3-adamantanol derivatives. However, three of these relatively polar adamantanol analogues 8i, 8j and 8l displayed promising activities (MIC = 1.32 – 2.89 μM), higher than the activity of EMB (MIC = 4.89 μM). Evidently, substitutions at position 4 and/or 6, especially with halogens, on the indole ring are optimal for activity. On the other hand, the adamantylurea derivative 10a (MIC = 1.47 μM) exhibited higher potency than EMB, whereas its adamantanol counterpart 10b was inactive. The benzimidazole derivatives 12 and 13a,b in which the extra ionisable nitrogen is expected to confer higher aqueous solubility, showed moderate or negligible activities. The excellent activities displayed by our eight most active compounds 8b, 8d, 8f, 8i, 8j, 8k, 8l, and 10a were not duplicated against M. abs and M. avium. However, compound 8b still managed to elicit better activity against M. avium than CPF. To our delight, all eight compounds maintained the same high activities against MDR and XDR M. tb strains. In particular, 8j and 8k stood out as the most potent compounds in the adamantanol and adamantane set of derivatives, respectively, in addition to exhibiting high selectivity against the tested M. tb strains over mammalian cells, denoting their potential lack of cytotoxicity. The top potent eight derivatives were also docked into the MmpL3 active site, in which they were accommodated in the S3-S5 subsites, recapitulating the binding modes and alignment of ICA38 and AU1235. Importantly, the enhanced kinetic aqueous solubility of 8c and 8l, typifying the 3-adamantanol class, compared to the bare more lipophilic adamantane counterparts 8b and 8k, respectively, is in agreement with the predicted figures obtained in silico. These findings foreground the N-(3-adamantanol)-indole-2-carboxamides as good anti-TB candidates with improved water solubility while maintaining potency against both DS and DR M. tb strains.
6. Experimental Section
6.1. Chemistry
General information
All indole-2-carboxylic acids, 1-adamantylamine, 3-amino-1-adamantanol, 1-adamantaneacetic acid were purchased from Fluorochem, while 1-adamantanecarboxylic acid was purchased from Sigma-Aldrich. 2-Aminobenzimidazole and 3,5-dichloroaniline were obtained from AlfaAesar. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III spectrometer at 400 and 100 MHz, respectively, with TMS as an internal standard. Standard abbreviations indicating multiplicity were as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, td = triplet of doublets, q = quadruplet, m = multiplet and br = broad. HRMS experiments were done on a Thermo Scientific Q-Exactive Orbitrap mass spectrometer. TLC was carried out on SiliCycle SiliaPlate TLC plates (200 μm, 20 × 20 cm). Flash chromatography was performed using a Teledyne Isco CombiFlash Rf system with RediSep columns or manually using SiliCycle SiliaFlash® P60 Silica Gels [40–63 μm (230–400 mesh)]. Final compounds were purified by preparative HPLC unless otherwise stated. The preparative HPLC (Shimadzu) employed a Phenomenex Luna® Omega 5 μm Polar C18 100A (150 × 21.2 mm) column, with detection at 254 and 280 nm on a Shimadzu SPD-20A detector, flow rate = 25.0 mL/min. Method 1: 40–100% acetonitrile/Milli-Q water (CH3CN/H2O) in 15 min; 100% CH3CN in 10 min; 100–40% CH3CN/H2O in 10 min. Method 2: 60–100% CH3CN/H2O in 10 min; 100% CH3CN in 15 min; 100–60% CH3CN/H2O in 10 min. Both solvents contained 0.05% of trifluoroacetic acid (TFA). Purities of final compounds were established by analytical HPLC, which was conducted using Waters HPLC system (1525 binary pump, 2487 dual wavelength absorbance detector, and 717 plus autosampler) with a Phenomenex Luna® 5μ C18(2) 100 Å (150 × 4.6 mm) column. Analytical HPLC method: flow rate = 1 mL/min; gradient elution over 30 min. Gradient: 20–100% CH3CN/H2O in 10 min; 100% CH3CN in 10 min; 100–20% CH3CN/H2O in 10 min. 0.05% of TFA was incorporated in both solvents. The purity of all tested compounds was >95% as determined by the method described above.
6.1.1. General procedure for amide coupling (Method A)
To a solution of the appropriate carboxylic acid (1 mmol) in anhydrous dimethylformamide (DMF, 10 mL), 1-ethyl-3-(3-(dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 1.2 mmol), hydroxybenzotriazole hydrate (HOBt, 1.2 mmol) and the corresponding amine (1.2 mmol) were added at room temperature (rt). The reaction was then basified with N,N-Diisopropylethylamine (DIPEA, 1.5 equiv) and the mixture was stirred at room temperature (rt) until the disappearance of the starting material (usually 60–72 h). After this time water (50 mL) was added, and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with water (5 × 25 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was initially purified by flash chromatography using dichloromethane/methanol (DCM/MeOH) gradient prior to further preparative HPLC purification unless otherwise stated.
6.1.2. General procedure for amide coupling (Method B)
To a solution of the indole-2-carboxylic acid derivative (1 mmol) in anhydrous dichloromethane (DCM, 10 mL), DMF (0.1 mL) and oxalyl chloride (2 mmol) were added. After stirring for 3 h at rt, the mixture was concentrated under vacuum and the residue was dissolved in anhydrous DCM. Thereafter, 1-adamantylamine (1.5 mmol) and triethylamine (2 mmol) were added and the mixture was stirred at rt for 48 h and filtered off. The precipitate was washed with DCM (2 × 25 mL) and the collected 50 mL DCM combined with the filtrate were evaporated in vacuo and the crude residue was purified by flash chromatography using DCM/MeOH gradient. The obtained compounds were already >95% pure after flash chromatography.
6.1.3. General procedure for amide coupling (Method C)
To a stirred solution of 2-aminobenzimidazole (1 mmol) in a 1:1 mixture of tetrahydrofuran (THF) and DCM, EDC.HCl (1.2 mmol), the corresponding carboxylic acid (1.2 mmol), and 4-dimethylaminopyridine (DMAP, 1.2 mmol) were added and the reaction mixture was stirred at room temperature for 72 h. The solvent was then removed under vacuum and the residue was purified by flash or manual chromatography using DCM/MeOH gradient prior to further HPLC purification unless otherwise stated.
6.1.4. General procedure for urea formation (Method D)
A mixture of the aromatic amine (1 mmol) and 1,1′-carbonyldiimidazole (CDI) (1.2 mmol) in anhydrous DMF (10 mL) was stirred at 90 °C for two hours, followed by the addition of 1-adamantylamine or 3-amino-1-adamantanol (1.1 mmol) and stirring was continued for 48 h at 90 °C. Water (50 mL) was then added to the reaction mixture, followed by extraction with EtOAc (3 × 50 mL). The organic layers were separated, washed with water (5 × 25 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude was then purified by flash chromatography using DCM/MeOH gradient prior to HPLC purification.
N-(3-hydroxyadamantan-1-yl)-1H-indole-2-carboxamide (8a)
The title compound was synthesised from indole-2-carboxylic acid (7a) and 3-amino-1-adamantanol following general procedure A. Off white solid, yield: 97%.1H NMR (DMSO-d6) δ 11.41 (s, 1H), 7.63 (s, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.42 (dd, J = 8.2, 0.8 Hz, 1H), 7.20 – 7.13 (m, 2H), 7.02 (ddd, J = 8.0, 7.1, 0.9 Hz, 1H), 4.56 (s, 1H), 2.19 (s, 2H), 2.06 – 1.90 (m, 6H), 1.64 – 1.43 (m, 6H); 13C NMR (DMSO-d6) δ 161.0, 136.7, 133.0, 127.6, 123.6, 121.8, 120.0, 112.6, 103.4, 67.8, 54.5, 49.6, 44.7, 35.4, 30.6; HRMS (ESI) m/z calcd for C19H22N2O2 ([M + H]+) m/z 311.1754; found 311.1748.
N-(1-Adamantyl)-4-methoxy-1H-indole-2-carboxamide (8b)
The title compound was obtained from 4-methoxyindole-2-carboxylic acid (7b) and 1-adamantylamine employing method B. Yellow solid, yield: 40%.1H NMR (DMSO-d6) δ 11.40 (s, 1H), 7.52 (s, 1H), 7.25 (dd, J = 2.2, 0.7 Hz, 1H), 7.07 (t, J = 7.9 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.49 (dd, J = 7.6, 0.5 Hz, 1H), 3.87 (s, 3H), 2.09 (s, 6H), 2.06 (s, 3H), 1.67 (s, 6H); 13C NMR (DMSO-d6) δ 160.9, 154.0, 138.1, 131.7, 124.5, 118.5, 105.8, 100.9, 99.6, 55.4, 52.0, 41.6, 36.6, 29.4; HRMS (ESI) m/z calcd for C20H24N2O2 ([M + H]+) m/z 325.1911; found 325.1903.
N-(3-hydroxyadamantan-1-yl)-4-methoxy-1H-indole-2-carboxamide (8c)
This compound was synthesised from 4-methoxyindole-2-carboxylic acid (7b) and 3-amino-1-adamantanol according to method A. White solid, yield: 90%.1H NMR (DMSO-d6) δ 11.39 (d, J = 1.4 Hz, 1H), 7.61 (s, 1H), 7.26 (d, J = 1.7 Hz, 1H), 7.08 (t, J = 7.9 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.49 (d, J = 7.5 Hz, 1H), 4.55 (s, 1H), 3.87 (s, 3H), 2.18 (s, 2H), 2.03 – 1.90 (m, 6H), 1.65 – 1.41 (m, 6H); 13C NMR (DMSO-d6) δ 160.9, 154.0, 138.1, 131.6, 124.6, 118.5, 105.8, 100.9, 99.6, 67.9, 55.4, 54.5, 49.5, 44.7, 35.4, 30.6; HRMS (ESI) m/z calcd for C20H24N2O3 ([M + H]+) m/z 341.1860; found 341.1858.
N-(1-Adamantyl)-5-methoxy-1H-indole-2-carboxamide (8d)
The title compound was obtained from 5-methoxyindole-2-carboxylic acid (7c) and 1-adamantylamine employing method A. It was >95% pure after flash chromatography. White solid, yield: 98%.1H NMR (DMSO-d6) δ 11.26 (s, 1H), 7.49 (s, 1H), 7.30 (d, J = 8.9 Hz, 1H), 7.04 (d, J = 1.4 Hz, 1H), 7.03 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 8.9, 2.5 Hz, 1H), 3.75 (s, 3H), 2.09 (s, 6H), 2.06 (s, 3H), 1.67 (s, 6H); 13C NMR (DMSO-d6) δ 160.9, 154.1, 133.5, 132.0, 127.9, 114.6, 113.4, 103.1, 102.4, 55.7, 52.0, 41.5, 36.5, 29.4; HRMS (ESI) m/z calcd for C20H24N2O2 ([M + H]+) m/z 325.1911; found 325.1908.
N-(3-hydroxyadamantan-1-yl)-5-methoxy-1H-indole-2-carboxamide (8e)
This compound was synthesised from 5-methoxyindole-2-carboxylic acid (7c) and 3-amino-1-adamantanol following method A. Dusty pink solid, yield: 96%.1H NMR (DMSO-d6) δ 11.28 (s, 1H), 7.58 (s, 1H), 7.32 (d, J = 8.9 Hz, 1H), 7.07 (d, J = 1.5 Hz, 1H), 7.04 (d, J = 2.3 Hz, 1H), 6.83 (dd, J = 8.9, 2.4 Hz, 1H), 4.55 (s, 1H), 3.76 (s, 3H), 2.19 (s, 2H), 2.06 – 1.91 (m, 6H), 1.67 – 1.43 (m, 6H); 13C NMR (DMSO-d6) δ 160.9, 154.1, 133.4, 132.0, 127.8, 114.6, 113.4, 103.1, 102.5, 67.8, 55.7, 54.5, 49.6, 44.7, 35.4, 30.6; HRMS (ESI) m/z calcd for C20H24N2O3 ([M + H]+) m/z 341.1860; found 341.1852.
N-(1-Adamantyl)-5-methyl-1H-indole-2-carboxamide (8f)
This compound was obtained from 5-methylindole-2-carboxylic acid (7d) and 1-adamantylamine according to method B. White solid, yield: 43%.1H NMR (DMSO-d6) δ 11.28 (s, 1H), 7.49 (s, 1H), 7.34 (s, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 1.8 Hz, 1H), 6.98 (dd, J = 8.4, 1.2 Hz, 1H), 2.36 (s, 3H), 2.10 (s, 6H), 2.07 (s, 3H), 1.67 (s, 6H); 13C NMR (DMSO-d6) δ 161.0, 135.1, 133.1, 128.5, 127.8, 125.4, 121.1, 112.3, 102.8, 52.0, 41.6, 36.5, 29.4, 21.6; HRMS (ESI) m/z calcd for C20H24N2O ([M + H]+) m/z 309.1961; found 309.1953.
N-(3-hydroxyadamantan-1-yl)-5-methyl-1H-indole-2-carboxamide (8g)
This compound was synthesised from 5-methylindole-2-carboxylic acid (7d) and 3-amino-1-adamantanol following method A. White solid, yield: 65%.1H NMR (DMSO-d6) δ 11.27 (s, 1H), 7.58 (s, 1H), 7.34 (s, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 1.4 Hz, 1H), 6.98 (dd, J = 8.4, 1.1 Hz, 1H), 4.53 (s, 1H), 2.35 (s, 3H), 2.18 (s, 2H), 2.05 – 1.88 (m, 6H), 1.67 – 1.41 (m, 6H); 13C NMR (DMSO-d6) δ 161.0, 135.1, 133.0, 128.5, 127.8, 125.4, 121.1, 112.3, 102.9, 67.8, 54.5, 49.6, 44.7, 35.4, 30.6, 21.6; HRMS (ESI) m/z calcd for C20H24N2O2 ([M + H]+) m/z 325.1911; found 325.1914.
5-chloro-N-(3-hydroxyadamantan-1-yl)-1H-indole-2-carboxamide (8h)
The title compound was obtained from 5-chloroindole-2-carboxylic acid (7e) and 3-amino-1-adamantanol employing method A. White solid, yield: 64%.1H NMR (DMSO-d6) δ 11.63 (s, 1H), 7.74 (s, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.42 (d, J = 8.7 Hz, 1H), 7.20 – 7.13 (m, 2H), 4.56 (s, 1H), 2.18 (s, 2H), 2.03 – 1.90 (m, 6H), 1.64 – 1.43 (m, 6H); 13C NMR (DMSO-d6) δ 160.6, 135.1, 134.5, 128.6, 124.5, 123.6, 120.9, 114.2, 102.9, 67.8, 54.6, 49.5, 44.7, 35.3, 30.6; HRMS (ESI) m/z calcd for C19H21ClN2O2 ([M + H]+) m/z 345.1364; found 345.1364.
6-bromo-N-(3-hydroxyadamantan-1-yl)-1H-indole-2-carboxamide (8i)
This compound was synthesised from 6-bromoindole-2-carboxylic acid (7f) and 3-amino-1-adamantanol according to method A. White solid, yield: 93%.1H NMR (DMSO-d6) δ 11.57 (s, 1H), 7.72 (s, 1H), 7.57 (s, 1H), 7.55 (s, 1H), 7.20 (d, J = 0.9 Hz, 1H), 7.15 (dd, J = 8.5, 1.8 Hz, 1H), 4.54 (s, 1H), 2.18 (s, 2H), 2.04 – 1.88 (m, 6H), 1.64 – 1.43 (m, 6H); 13C NMR (DMSO-d6) δ 160.6, 137.5, 133.9, 126.6, 123.7, 123.1, 116.3, 115.1, 103.4, 67.8, 54.6, 49.5, 44.7, 35.3, 30.6; HRMS (ESI) m/z calcd for C19H21BrN2O2 ([M + H]+) m/z 389.0859; found 389.0866.
4,6-dichloro-N-(3-hydroxyadamantan-1-yl)-1H-indole-2-carboxamide (8j)
This compound was obtained from 4,6-dichloroindole-2-carboxylic acid (7g) and 3-amino-1-adamantanol employing method A. It was >95% pure after flash chromatography. Buff solid, yield: 99%.1H NMR (DMSO-d6) δ 11.93 (d, J = 1.5 Hz, 1H), 7.92 (s, 1H), 7.41 (dd, J = 1.6, 0.8 Hz, 1H), 7.35 (dd, J = 2.2, 0.8 Hz, 1H), 7.20 (d, J = 1.7 Hz, 1H), 4.55 (s, 1H), 2.18 (s, 2H), 2.05 – 1.90 (m, 6H), 1.64 – 1.42 (m, 6H); 13C NMR (DMSO-d6) δ 160.1, 137.1, 134.7, 128.0, 126.7, 125.3, 119.7, 111.5, 101.5, 67.8, 54.8, 49.4, 44.6, 35.3, 30.6; HRMS (ESI) m/z calcd for C19H20Cl2N2O2 ([M + H]+) m/z 379.0975; found 379.0977.
N-(1-Adamantyl)-4,6-difluoro-1H-indole-2-carboxamide (8k)
This compound was obtained from 4,6-difluoroindole-2-carboxylic acid (7h) and 1-adamantylamine following method A. It was >95% pure after flash chromatography. Off white solid, yield: 94%.1H NMR (DMSO-d6) δ 11.83 (s, 1H), 7.67 (s, 1H), 7.30 (d, J = 0.5 Hz, 1H), 7.02 (dd, J = 9.5, 1.4 Hz, 1H), 6.85 (td, J = 10.4, 2.1 Hz, 1H), 2.08 (s, 6H), 2.07 (s, 3H), 1.67 (s, 6H); 13C NMR (DMSO-d6) δ 160.1, 159.5 (dd, J = 238.1, 12.1 Hz), 156.1 (dd, J = 248.4, 15.6 Hz), 137.9 (dd, J = 15.2, 13.4 Hz), 134.0 (d, J = 3.3 Hz), 113.5 (dd, J = 21.9, 0.6 Hz), 99.0, 95.4 (dd, J = 29.6, 23.3 Hz), 95.0 (dd, J = 25.9, 4.4 Hz), 52.2, 41.4, 36.5, 29.4; HRMS (ESI) m/z calcd for C19H20F2N2O ([M + H]+) m/z 331.1610; found 331.1610.
4,6-difluoro-N-(3-hydroxyadamantan-1-yl)-1H-indole-2-carboxamide (8l)
This compound was synthesised from 4,6-difluoroindole-2-carboxylic acid (7h) and 3-amino-1-adamantanol employing method A. It was >95% pure after flash chromatography. Light buff solid, yield: 87%.1H NMR (DMSO-d6) δ 11.83 (s, 1H), 7.75 (s, 1H), 7.30 (d, J = 1.5 Hz, 1H), 7.02 (dd, J = 9.5, 1.5 Hz, 1H), 6.85 (td, J = 10.4, 2.0 Hz, 1H), 4.54 (s, 1H), 2.18 (s, 2H), 2.06 – 1.87 (m, 6H), 1.66 – 1.41 (m, 6H); 13C NMR (DMSO-d6) δ 160.2, 159.5 (dd, J = 238.1, 12.1 Hz), 156.1 (dd, J = 248.4, 15.6 Hz), 137.9 (dd, J = 15.2, 13.4 Hz), 134.0 (d, J = 3.3 Hz), 113.5 (d, J = 21.8 Hz), 99.1, 95.5 (dd, J = 29.6, 23.3 Hz), 95.0 (dd, J = 25.8, 4.4 Hz), 67.8, 54.7, 49.5, 44.7, 35.3, 30.6; HRMS (ESI) m/z calcd for C19H20F2N2O2 ([M + H]+) m/z 347.1566; found 347.1559.
1-(1-Adamantyl)-3-(3,5-dichlorophenyl)urea (10a)
The title compound was obtained from 3,5-dichloroaniline (9) and 1-adamantylamine following method D. White solid, yield: 48%.1H NMR (DMSO-d6) δ 8.67 (s, 1H), 7.39 (d, J = 1.8 Hz, 2H), 7.02 (t, J = 1.8 Hz, 1H), 6.04 (s, 1H), 2.02 (s, 3H), 1.92 (s, 6H), 1.62 (s, 6H); 13C NMR (DMSO-d6) δ 153.8, 143.5, 134.4, 120.2, 115.8, 50.6, 41.9, 36.4, 29.3; HRMS (ESI) m/z calcd for C17H20Cl2N2O ([M + H]+) m/z 339.1025; found 339.1030.
1-(3,5-dichlorophenyl)-3-(3-hydroxyadamantan-1-yl)urea (10b)
This compound was obtained from 3,5-dichloroaniline (9) and 3-amino-1-adamantanol employing method D. White solid, yield: 20%.1H NMR (DMSO-d6) δ 8.64 (s, 1H), 7.40 (d, J = 1.9 Hz, 2H), 7.04 (t, J = 1.9 Hz, 1H), 6.14 (s, 1H), 4.51 (s, 1H), 2.14 (s, 2H), 1.92 – 1.71 (m, 6H), 1.61 – 1.35 (m, 6H); 13C NMR (DMSO-d6) δ 153.9, 143.5, 134.4, 120.3, 115.8, 67.8, 53.2, 49.9, 44.6, 35.2, 30.6; HRMS (ESI) m/z calcd for C17H20Cl2N2O2 ([M + H]+) m/z 355.0975; found 355.0980.
1-(1-adamantyl)-3-(1H-benzo[d]imidazol-2-yl)urea (12)
This compound was obtained from 2-aminobenzimidazole (11) and 1-adamantylamine employing method D. White solid, yield: 36%.1H NMR (DMSO-d6) δ 7.64 – 7.57 (m, 2H), 7.40 (s, 1H), 7.39 – 7.34 (m, 2H), 2.14 (s, 3H), 2.08 (s, 6H), 1.73 (s, 6H); 13C NMR (DMSO-d6) δ 151.4, 145.6, 130.4, 124.0, 113.3, 51.5, 41.5, 36.3, 29.3; HRMS (ESI) m/z calcd for C18H22N4O ([M + H]+) m/z 311.1866; found 311.1865.
N-(1H-benzo[d]imidazol-2-yl)adamantane-1-carboxamide (13a)
The title compound was synthesised from 2-aminobenzimidazole (11) and 1-adamantanecarboxylic acid according to method C. White solid, yield: 68%.1H NMR (DMSO-d6) δ 7.61 (s, 2H), 7.32 (s, 2H), 2.05 (s, 3H), 1.97 (s, 6H), 1.72 (s, 6H); 13C NMR (DMSO-d6) δ 177.6, 145.3, 131.1, 124.2, 114.0, 41.9, 37.9, 36.1, 27.8; HRMS (ESI) m/z calcd for C18H21N3O ([M + H]+) m/z 296.1756; found 296.1755.
2-(1-Adamantyl)-N-(1H-benzo[d]imidazol-2-yl)acetamide (13b)
This compound was obtained from 2-aminobenzimidazole (11) and 1-adamantaneacetic acid following method C. It was >95% pure after flash chromatography. White solid, yield: 72%.1H NMR (DMSO-d6) δ 12.05 (s, 1H), 11.41 (s, 1H), 7.43 (s, 2H), 7.15 – 6.95 (m, 2H), 2.19 (s, 2H), 1.91 (s, 3H), 1.74 – 1.49 (m, 12H);13C NMR (DMSO-d6) δ 170.9, 147.0, 140.8, 133.0, 121.5, 121.4, 117.3, 112.0, 50.0, 42.4, 36.8, 33.3, 28.5; HRMS (ESI) m/z calcd for C19H23N3O ([M + H]+) m/z 310.1914; found 310.1911.
6.2. Biology
Microplate alamarBlue assay (MABA) was used in the MIC evaluation against the tested mycobacteria as previously reported [60, 61]. MABA format was also used in the cytotoxicity assessment on Vero cells and the corresponding IC50 values were subsequently determined [22].
6.3. Molecular Modelling
Computational docking analysis was performed using the Molecular Operating Environment MOE software version 2008.10. The MmpL3 crystal structure in complex with ICA38 (6AJJ) was downloaded from the protein data bank (PDB), implementing the same docking protocol in our previous report [54]. In brief, the most potent compounds were docked into the binding pocket of ICA38 using MOE-DOCK function. Triangle Matcher placement method was applied, and the generated poses were rescored according to London dG methodology. The same scoring function was also used to then rank the poses output after being relaxed (energy minimised) through the conventional molecular mechanics forcefield refinement setup. The top 30 poses were retained and the highest-ranked pose with the best docking score (i.e. the lowest binding energy) pertaining to each ligand was selected. It is noteworthy that when Pharmacophore placement method, in lieu of Triangle Matcher, was used to filter the ligand poses while docking, identical results were produced. In this context, final poses that do not satisfy the pharmacophore model were automatically eliminated.
6.4. Water Solubility Measurement
Kinetic water solubility was experimentally determined utilising analytical HPLC [Waters HPLC system (1525 Binary pump, 2487 dual wavelength absorbance detector, and 717 plus Autosampler) with a Phenomenex Luna® 5μ C18(2) 100 Å (150 × 4.6 mm) column]. The following gradient was operated: 20–100% CH3CN/water over 15 min, maintained at 100% CH3CN for 10 min, and finally returned to 20% CH3CN in water over 5 min. Both CH3CN and Milli-Q water solvents used were containing 0.05% TFA and the flow rate was set at 1.0 mL/min. First, 4000 mg/L stock solutions in DMSO were created for compounds 8b, 8c, 8k, and 8l, followed by serial dilutions using CH3CN/H2O (9:1) to create a calibration curve for each compound by plotting absorbance versus concentrations of 200 mg/L, 400 mg/L, 600 mg/L, 800 mg/L and 1000 mg/L. The absorbance values of each concentration are an average of two individual measurements. Next, from the 4000 mg/L stock solution in DMSO, three samples of each compound were diluted 1:20 (200 mg/L) in deionised water and centrifuged. A 100 μL sample from the supernatant was diluted 1:1 with 100 μL neat CH3CN, mixed, filtered using a 0.45 μM filter, and 100 μL sample was taken for analytical HPLC testing. Two absorbance values of each sample were measured and averaged, followed by appropriate calculation and extrapolation to the established calibration curve.
Supplementary Material
Highlights:
The most potent adamantanol/adamantane-based indoleamides 8j/8k displayed a two-fold surge in potency against extensively DR (XDR) M. tb strains with MIC values of 0.66 and 0.012 μM, respectively.
The adamantanol-containing indole-2-carboxamides generally exhibited improved water solubility both in silico and experimentally, relative to the adamantane counterparts. Overall, the observed antimycobacterial and physicochemical profiles support the notion that adamantanol moiety is a suitable replacement to the adamantane scaffold within the series of indole-2-carboxamide-based MmpL3 inhibitors.
Acknowledgments
SSRA is thankful for the support of Curtin International Postgraduate Research Scholarship (CIPRS). WRB acknowledges the support of NIH grants AI 37856 and HL 133190. HG is the recipient of an Australian Research Council Discovery Early Career Researcher Award (DE160100482) funded by the Australian Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Barberis I, Bragazzi NL, Galluzzo L, Martini M, The history of tuberculosis: from the first historical records to the isolation of Koch’s bacillus, J. Prev. Med. Hyg 58(1) (2017) E9–E12. [PMC free article] [PubMed] [Google Scholar]
- [2].Blomgran R, Desvignes L, Briken V, Ernst JD, Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells, Cell Host Microbe 11(1) (2012) 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cambier CJ, Falkow S, Ramakrishnan L, Host evasion and exploitation schemes of Mycobacterium tuberculosis, Cell 159(7) (2014) 1497–509. [DOI] [PubMed] [Google Scholar]
- [4].Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM, Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity, Infect. Immun 70(8) (2002) 4501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, Desauvage FJ, Lund FE, Cooper AM, Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection, J. Exp. Med 203(7) (2006) 1805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wayne LG, Dormancy of Mycobacterium tuberculosis and latency of disease, Eur. J. Clin. Microbiol. Infect. Dis 13(11) (1994) 908–14. [DOI] [PubMed] [Google Scholar]
- [7].Blaser MJ, Kirschner D, The equilibria that allow bacterial persistence in human hosts, Nature 449(7164) (2007) 843–9. [DOI] [PubMed] [Google Scholar]
- [8].Kaufmann SH, McMichael AJ, Annulling a dangerous liaison: vaccination strategies against AIDS and tuberculosis, Nat. Med 11(4 Suppl) (2005) S33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pawlowski A, Jansson M, Skold M, Rottenberg ME, Kallenius G, Tuberculosis and HIV co-infection, PLoS Pathog 8(2) (2012) e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].World Health Organization Global Tuberculosis Report, Geneva, 2019. [Google Scholar]
- [11].Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, Fujiwara P, Grzemska M, Hopewell PC, Iseman MD, Jasmer RM, Koppaka V, Menzies RI, O’Brien RJ, Reves RR, Reichman LB, Simone PM, Starke JR, Vernon AA, C.f.D.C. American Thoracic Society, Prevention, S. the Infectious Diseases, American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis, Am. J. Respir. Crit. Care Med 167(4) (2003) 603–62. [DOI] [PubMed] [Google Scholar]
- [12].Keshavjee S, Farmer PE, Tuberculosis, drug resistance, and the history of modern medicine, N. Engl. J. Med 367(10) (2012) 931–6. [DOI] [PubMed] [Google Scholar]
- [13].Dheda K, Barry CE 3rd, Maartens G, Tuberculosis, Lancet 387(10024) (2016) 1211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Caminero JA, World Health O, American Thoracic S, British Thoracic S, Treatment of multidrug-resistant tuberculosis: evidence and controversies, Int. J. Tuberc. Lung Dis 10(8) (2006) 829–37. [PubMed] [Google Scholar]
- [15].Chan ED, Laurel V, Strand MJ, Chan JF, Huynh ML, Goble M, Iseman MD, Treatment and outcome analysis of 205 patients with multidrug-resistant tuberculosis, Am. J. Respir. Crit. Care Med 169(10) (2004) 1103–9. [DOI] [PubMed] [Google Scholar]
- [16].Eker B, Ortmann J, Migliori GB, Sotgiu G, Muetterlein R, Centis R, Hoffmann H, Kirsten D, Schaberg T, Ruesch-Gerdes S, Lange C, German TG, Multidrug- and extensively drug-resistant tuberculosis, Germany, Emerg. Infect. Dis 14(11) (2008) 1700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Migliori GB, Loddenkemper R, Blasi F, Raviglione MC, 125 years after Robert Koch’s discovery of the tubercle bacillus: the new XDR-TB threat. Is “science” enough to tackle the epidemic?, Eur. Respir. J 29(3) (2007) 423–7. [DOI] [PubMed] [Google Scholar]
- [18].Mitnick CD, Shin SS, Seung KJ, Rich ML, Atwood SS, Furin JJ, Fitzmaurice GM, Alcantara Viru FA, Appleton SC, Bayona JN, Bonilla CA, Chalco K, Choi S, Franke MF, Fraser HS, Guerra D, Hurtado RM, Jazayeri D, Joseph K, Llaro K, Mestanza L, Mukherjee JS, Munoz M, Palacios E, Sanchez E, Sloutsky A, Becerra MC, Comprehensive treatment of extensively drug-resistant tuberculosis, N. Engl. J. Med 359(6) (2008) 563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Velayati AA, Farnia P, Masjedi MR, The totally drug resistant tuberculosis (TDR-TB), Int. J. Clin. Exp. Med 6(4) (2013) 307–9. [PMC free article] [PubMed] [Google Scholar]
- [20].Parida SK, Axelsson-Robertson R, Rao MV, Singh N, Master I, Lutckii A, Keshavjee S, Andersson J, Zumla A, Maeurer M, Totally drug-resistant tuberculosis and adjunct therapies, J. Intern. Med 277(4) (2015) 388–405. [DOI] [PubMed] [Google Scholar]
- [21].Caminero JA, Sotgiu G, Zumla A, Migliori GB, Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis, Lancet Infect. Dis 10(9) (2010) 621–9. [DOI] [PubMed] [Google Scholar]
- [22].Lun S, Guo H, Onajole OK, Pieroni M, Gunosewoyo H, Chen G, Tipparaju SK, Ammerman NC, Kozikowski AP, Bishai WR, Indoleamides are active against drug-resistant Mycobacterium tuberculosis, Nat. Commun 4 (2013) 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rao SP, Lakshminarayana SB, Kondreddi RR, Herve M, Camacho LR, Bifani P, Kalapala SK, Jiricek J, Ma NL, Tan BH, Ng SH, Nanjundappa M, Ravindran S, Seah PG, Thayalan P, Lim SH, Lee BH, Goh A, Barnes WS, Chen Z, Gagaring K, Chatterjee AK, Pethe K, Kuhen K, Walker J, Feng G, Babu S, Zhang L, Blasco F, Beer D, Weaver M, Dartois V, Glynne R, Dick T, Smith PW, Diagana TT, Manjunatha UH, Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis, Sci. Transl. Med 5(214) (2013) 214ra168. [DOI] [PubMed] [Google Scholar]
- [24].Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE 3rd, Boshoff HI, SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis, Antimicrob. Agents Chemother 56(4) (2012) 1797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang B, Li J, Yang X, Wu L, Zhang J, Yang Y, Zhao Y, Zhang L, Yang X, Yang X, Cheng X, Liu Z, Jiang B, Jiang H, Guddat LW, Yang H, Rao Z, Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target, Cell 176(3) (2019) 636–648 e13. [DOI] [PubMed] [Google Scholar]
- [26].Grzegorzewicz AE, Pham H, Gundi VA, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SE, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M, Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane, Nat. Chem. Biol 8(4) (2012) 334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Owens CP, Chim N, Goulding CW, Insights on how the Mycobacterium tuberculosis heme uptake pathway can be used as a drug target, Future Med. Chem 5(12) (2013) 1391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Owens CP, Chim N, Graves AB, Harmston CA, Iniguez A, Contreras H, Liptak MD, Goulding CW, The Mycobacterium tuberculosis secreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11, J. Biol. Chem 288(30) (2013) 21714–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, Iniguez A, Kimmey JM, Sawaya MR, Whitelegge JP, Horwitz MA, Goulding CW, Discovery and characterization of a unique mycobacterial heme acquisition system, Proc. Natl. Acad. Sci. U S A 108(12) (2011) 5051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xu Z, Meshcheryakov VA, Poce G, Chng SS, MmpL3 is the flippase for mycolic acids in mycobacteria, Proc. Natl. Acad. Sci. U S A 114(30) (2017) 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brennan PJ, Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis, Tuberculosis (Edinb) 83(1–3) (2003) 91–7. [DOI] [PubMed] [Google Scholar]
- [32].Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H, Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure, Proc. Natl. Acad. Sci. U S A 105(10) (2008) 3963–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu J, Barry CE 3rd, Besra GS, Nikaido H, Mycolic acid structure determines the fluidity of the mycobacterial cell wall, J. Biol. Chem 271(47) (1996) 29545–51. [DOI] [PubMed] [Google Scholar]
- [34].Alsayed SSR, Beh CC, Foster NR, Payne AD, Yu Y, Gunosewoyo H, Kinase Targets for Mycolic Acid Biosynthesis in Mycobacterium tuberculosis, Curr. Mol. Pharmacol 12(1) (2019) 27–49. [DOI] [PubMed] [Google Scholar]
- [35].Brennan PJ, Nikaido H, The envelope of mycobacteria, Annu. Rev. Biochem 64 (1995) 29–63. [DOI] [PubMed] [Google Scholar]
- [36].Nataraj V, Varela C, Javid A, Singh A, Besra GS, Bhatt A, Mycolic acids: deciphering and targeting the Achilles’ heel of the tubercle bacillus, Mol. Microbiol 98(1) (2015) 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Takayama K, Wang C, Besra GS, Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis, Clin. Microbiol. Rev 18(1) (2005) 81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Varela C, Rittmann D, Singh A, Krumbach K, Bhatt K, Eggeling L, Besra GS, Bhatt A, MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria, Chem. Biol 19(4) (2012) 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Degiacomi G, Benjak A, Madacki J, Boldrin F, Provvedi R, Palu G, Kordulakova J, Cole ST, Manganelli R, Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression, Sci. Rep 7 (2017) 43495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li W, Obregon-Henao A, Wallach JB, North EJ, Lee RE, Gonzalez-Juarrero M, Schnappinger D, Jackson M, Therapeutic Potential of the Mycobacterium tuberculosis Mycolic Acid Transporter, MmpL3, Antimicrob. Agents Chemother 60(9) (2016) 5198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Campanico A, Moreira R, Lopes F, Drug discovery in tuberculosis. New drug targets and antimycobacterial agents, Eur. J. Med. Chem 150 (2018) 525–545. [DOI] [PubMed] [Google Scholar]
- [42].Li W, Stevens CM, Pandya AN, Darzynkiewicz Z, Bhattarai P, Tong W, Gonzalez-Juarrero M, North EJ, Zgurskaya HI, Jackson M, Direct Inhibition of MmpL3 by Novel Antitubercular Compounds, ACS Infect. Dis 5(6) (2019) 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Brown JR, North EJ, Hurdle JG, Morisseau C, Scarborough JS, Sun D, Kordulakova J, Scherman MS, Jones V, Grzegorzewicz A, Crew RM, Jackson M, McNeil MR, Lee RE, The structure-activity relationship of urea derivatives as anti-tuberculosis agents, Bioorg. Med. Chem 19(18) (2011) 5585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Onajole OK, Pieroni M, Tipparaju SK, Lun S, Stec J, Chen G, Gunosewoyo H, Guo H, Ammerman NC, Bishai WR, Kozikowski AP, Preliminary structure-activity relationships and biological evaluation of novel antitubercular indolecarboxamide derivatives against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains, J. Med. Chem 56(10) (2013) 4093–103. [DOI] [PubMed] [Google Scholar]
- [45].Stec J, Onajole OK, Lun S, Guo H, Merenbloom B, Vistoli G, Bishai WR, Kozikowski AP, Indole-2-carboxamide-based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection, J. Med. Chem 59(13) (2016) 6232–47. [DOI] [PubMed] [Google Scholar]
- [46].Wanka L, Iqbal K, Schreiner PR, The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives, Chem. Rev 113(5) (2013) 3516–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stanley SA, Grant SS, Kawate T, Iwase N, Shimizu M, Wivagg C, Silvis M, Kazyanskaya E, Aquadro J, Golas A, Fitzgerald M, Dai H, Zhang L, Hung DT, Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening, ACS Chem. Biol 7(8) (2012) 1377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Padiya KJ, Gavade S, Kardile B, Tiwari M, Bajare S, Mane M, Gaware V, Varghese S, Harel D, Kurhade S, Unprecedented “In Water” imidazole carbonylation: paradigm shift for preparation of urea and carbamate, Org. Lett 14(11) (2012) 2814–7. [DOI] [PubMed] [Google Scholar]
- [49].Franz ND, Belardinelli JM, Kaminski MA, Dunn LC, Calado V Nogueira de Moura, M.A. Blaha, D.D. Truong, W. Li, M. Jackson, E.J. North, Design, synthesis and evaluation of indole-2-carboxamides with pan anti-mycobacterial activity, Bioorg. Med. Chem 25(14) (2017) 3746–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kozikowski AP, Onajole OK, Stec J, Dupont C, Viljoen A, Richard M, Chaira T, Lun S, Bishai W, Raj VS, Ordway D, Kremer L, Targeting Mycolic Acid Transport by Indole-2-carboxamides for the Treatment of Mycobacterium abscessus Infections, J. Med. Chem 60(13) (2017) 5876–5888. [DOI] [PubMed] [Google Scholar]
- [51].Scherman MS, North EJ, Jones V, Hess TN, Grzegorzewicz AE, Kasagami T, Kim IH, Merzlikin O, Lenaerts AJ, Lee RE, Jackson M, Morisseau C, McNeil MR, Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability, Bioorg. Med. Chem 20(10) (2012) 3255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li W, Upadhyay A, Fontes FL, North EJ, Wang Y, Crans DC, Grzegorzewicz AE, Jones V, Franzblau SG, Lee RE, Crick DC, Jackson M, Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis, Antimicrob. Agents Chemother 58(11) (2014) 6413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].MOE Chemical Computing Group, lnc., Montreal, http://www.chemcomp.com. [Google Scholar]
- [54].Alsayed SSR, Lun S, Luna G, Beh CC, Payne AD, Foster N, Bishai WR, Gunosewoyo H, Design, synthesis, and biological evaluation of novel arylcarboxamide derivatives as anti-tubercular agents, RSC Advances 10(13) (2020) 7523–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev 46(1–3) (2001) 3–26. [DOI] [PubMed] [Google Scholar]
- [56].Sutherland HS, Tong AST, Choi PJ, Conole D, Blaser A, Franzblau SG, Cooper CB, Upton AM, Lotlikar MU, Denny WA, Palmer BD, Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles, Bioorg. Med. Chem 26(8) (2018) 1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Worley MV, Estrada SJ, Bedaquiline: a novel antitubercular agent for the treatment of multidrug-resistant tuberculosis, Pharmacotherapy 34(11) (2014) 1187–97. [DOI] [PubMed] [Google Scholar]
- [58].Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V, A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis, Science 307(5707) (2005) 223–7. [DOI] [PubMed] [Google Scholar]
- [59].Yamamoto K, Ikeda Y, Kinetic solubility and lipophilicity evaluation connecting formulation technology strategy perspective, J. Drug Deliv. Sci. Technol 33 (2016) 13–18. [Google Scholar]
- [60].Collins L, Franzblau SG, Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium, Antimicrob. Agents Chemother 41(5) (1997) 1004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pieroni M, Tipparaju SK, Lun S, Song Y, Sturm AW, Bishai WR, Kozikowski AP, Pyrido[1,2-a]benzimidazole-based agents active against tuberculosis (TB), multidrug-resistant (MDR) TB and extensively drug-resistant (XDR) TB, ChemMedChem 6(2) (2011) 334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.