

Abstract
Complex karyotype identified by chromosome-banding analysis has been shown to have prognostic value in chronic lymphocytic leukemia (CLL). Genomic arrays offer high-resolution genomewide detection of copy-number alterations (CNA) and could therefore be well equipped to detect the presence of a complex karyotype. Current knowledge on genomic arrays in CLL is based on outcomes of single-center studies, in which different cutoffs for CNA calling were used. To further determine the clinical utility of genomic arrays for CNA assessment in CLL diagnostics, we retrospectively analyzed 2,293 arrays from 13 diagnostic laboratories according to established standards. CNA were found outside regions captured by CLL fluorescence in situ hybridization probes in 34% of patients, and several of them, including gains of 8q, deletions of 9p and 18p (P<0.01), were linked to poor outcome after correction for multiple testing. Patients (n=972) could be divided into three distinct prognostic subgroups based on the number of CNA. In multivariable analysis only high genomic complexity, defined as ≥5 CNA, emerged as an independent adverse prognosticator for time to first treatment (hazard ratio: 2.15; 95% confidence interval: 1.36-3.41; P=0.001) and overall survival (hazard ratio: 2.54, 95% confidence interval: 1.54-4.17; P<0.001; n=528). Lowering the size cutoff to 1 Mb in 647 patients did not significantly improve risk assessment. Genomic arrays detected more chromosomal abnormalities and, in terms of risk stratification, performed at least as well as simultaneous chromosome banding analysis as carried out in 122 patients. Our findings indicate that genomic array is an accurate tool for CLL risk stratification.
Introduction
Chronic lymphocytic leukemia (CLL) is both clinically and genetically highly heterogeneous. The advent of efficacious, albeit expensive, targeted treatment regimens for CLL has highlighted the need for more accurate risk stratification. Diverse CLL-specific biomarkers are used to predict clinical course and survival in CLL including Rai1 and Binet2 staging, immunoglobulin heavy variable (IGHV) gene somatic hypermutation status,3,4 TP53 mutation status and fluorescence in situ hybridization (FISH)-detected specific chromosomal alterations.5,6
Chromosome banding analysis (CBA) of in vitro mitogen- stimulated peripheral blood samples provides a lowresolution whole-genome view of the cytogenetic landscape of CLL, thereby overcoming the targeted nature of FISH which precludes a comprehensive assessment of the genomic landscape of CLL.7-12 Relatively small studies employing CBA in CLL identified the presence of a complex karyotype (CK; defined as ≥3 cytogenetically-visible structural and/or numerical aberrations) as a prognostic marker for refractoriness not only to chemoimmunotherapy, 9,12,13 but also to novel targeted agents such as ibrutinib14 and venetoclax.15 Within the European Research Initiative on CLL (ERIC), we recently performed a retrospective study of 5,290 patients with available CBA data and found that cytogenetic complexity with ≥5 but not ≥3 chromosomal aberrations is an independent prognostic factor with adverse outcome in CLL.12 These observations demonstrate that the genome-wide analysis of genetic aberrations provides valuable additional prognostic information compared to FISH analysis alone, hence refining risk-adapted stratification of patients.
Genomic array platforms such as array-based comparative genomic hybridization and single nucleotide polymorphism arrays allow the entire genome to be screened for copy-number alterations (CNA) in a single experiment. Current array platforms enable the identification of CNA of 10-100 kb, and have been implemented in the diagnostic work-up of hematologic malignancies, including CLL.16-22 In contrast to CBA analyses, input of array platforms only includes DNA and obviates the need for fresh isolated tumor cells and the need for in vitro mitogen stimulation. In order to focus on tumor-associated abnormalities and to avoid the detection of benign constitutional variants (present in the population), a conservative size cutoff of ≥5 megabases (Mb) has been recommended clinically for CNA other than those occurring in specific loci recurrently affected in CLL, i.e., del(13)(q14) (RB1/DLEU/MIR15A/MIR16-1) [del(13q)], del(11)(q22.3) (ATM) [del(11q)], and del(17)(p13.1) (TP53) [del(17p)].23,24 Consequently, it remains unclear whether lowering the cutoff might identify clinically relevant CNA, with potential to improve risk stratification, or reduce assay sensitivity through the inclusion of non-pathogenic somatic or normal germline variations. Array-based studies have been performed with lower size cutoffs than 5 or even 1 Mb, zooming in on (new) potentially relevant chromosome regions containing cancer-specific genes.19,21,23 These studies have been inconclusive with respect to the value of such higher resolution approaches for risk assessment.
A number of genomic array studies have observed an association between genomic complexity and adverse outcome in CLL.12,19,25-27 These studies were performed in single centers, used different cutoffs for CNA calling, and had relatively short follow-ups, precluding definitive conclusions regarding the applicability of genomic arrays for risk assessment in CLL. The emergence of ≥5 chromosomal abnormalities determined by CBA as an independent prognosticator and the discovery of heterogeneity within CK patients, urged for a reappraisal of the performance of genomic arrays. Realizing this, ERIC conducted the present multicenter analysis of genomic arrays in 2,293 CLL patients from 13 European CLL laboratories.
Methods
Patients and established biomarkers
Overall, 2,293 CLL patients, diagnosed according to International Workshop on CLL criteria,28 were included in the present multicenter, retrospective study. Thirteen different CLL diagnostic laboratories connected to ERIC were involved (Online Supplementary Figure S1). In total, 572 of the 2,293 patients have been included in previous publications.19,20,29 Cases included for survival analyses were mostly recruited within 2 years (69.1%) or 1 year (59.1%) after diagnosis. An overview of the patients’ characteristics (demographics and biological features) is provided in Online Supplementary Table S1.
TP53 mutation analysis (Online Supplementary Table S2) and determination of the somatic hypermutation status of the clonotypic rearranged IGHV genes were performed and interpreted according to ERIC guidelines (Online Supplementary Methods).30,31
In a subgroup of patients (260/2,293) interphase FISH analysis using probes for the regions 13q14 (RB1 and/or DLEU), 11q22.3 (ATM), 17p13.1 (TP53) and trisomy 12 was performed concurrently with genomic array analysis (i.e., same date of sampling). Simultaneous CBA (i.e., same date of sampling) was conducted in 122/2,293 patients. Metaphases for CBA were induced with stimulation protocols based on phorbol-12-myristate-13-acetate or immunostimulatory cytosine guanine dinucleotide-oligonucleotide DSP30 plus interleukin as previously described.12 The study was performed according to national and international ethical and legal recommendations with approval from the local Ethics Review Committees of the participating institutes
Genomic array analysis
Genomic arrays were performed using several commercially available high-density (array-based comparative genomic hybridization/single nucleotide polymorphism array) platforms applied for diagnostics in the participating centers (Online Supplementary Table S3 and Online Supplementary Methods). CNA routinely assessed in CLL by targeted FISH revealed by genomic arrays in this study were included in all analyses irrespective of size, while for other CNA a size cutoff of ≥5 Mb was used according to Schoumans et al.24 Copy-number neutral loss of heterozygosity events, detected by single nucleotide polymorphism arrays, were not included in the counting of CNA. CNA with a distance ≤5 Mb between the two CNA, and putative chromothripsis events were counted as one event.
Statistical analysis
Statistical analysis was performed using SPSS version 24 and R. A Pearson c2 test was used to evaluate the independence between categorical factors, and a Kruskal-Wallis test was applied to compare the distributions of continuous variables between groups of patients.
Time-dependent receiver operating characteristic (ROC) curve analysis was performed evaluating different time-points from the date of array analysis (Online Supplementary Methods).
Overall survival (OS) and time to first treatment (TTFT) were calculated from the date of sampling for array analysis to the last date of follow-up. Only patients untreated at the date of sampling (n=972) were included in the survival analyses unless otherwise stated. The median follow-up was 44 months with a total of 353 survival events. Kaplan-Meier survival curves were compared by log-rank testing. Univariable Cox regression analysis was used to evaluate the effect of CNA, for which there were at least ten events and established clinico-biological features, on OS. Factors found to be statistically significant in univariable analysis were included in multivariable analysis (for TTFT and OS). A P-value <0.05 signified a statistically significant result. Correction for multiple tests was applied when appropriate using the Bonferroni approach.
Data sharing statement
A list of curated array profiles is provided in the Online Supplementary Excel File. Please contact either c.h.mellink@amc.nl or a.p.kater@amc.nl for the original data.
Results
Genomic arrays detect aberrations not captured by fluorescence in situ hybridization in over one-third of patients with chronic lymphocytic leukemia
CNA including aneuploidy were detected in 78.9% of patients enrolled in this study. Reflecting the heterogeneous genetic landscape of CLL, the total number of CNA per patient varied considerably (median 1; range, 0-17). Increased numbers of CNA were identified in samples from previously treated patients (2.23 vs. 1.66; P<0.0001) (Online Supplementary Figure S2A) and in patients with aggressive disease (Binet B/C; 2.16 vs. 1.57, unmutated IGHV (U-CLL); 2.15 vs. 1.38, and TP53 mutation and/or del(17p) (TP53abn); 2.93 vs. 1.45; P<0.0001) (Online Supplementary Figure 2B-D). In 34.1% of patients, CNA were outside the established CLL regions captured by FISH (13q14, 11q22.3 [ATM], 17p13.1 [TP53] and chromosome 12) and included chromosomal deletions (n=1,064), chromosomal gains (n=597), monosomies (n=69), and trisomies (n=68) (Figure 1A-E). Complex patterns (n=32), indicative of chromothripsis, were also detected. Cases with putative chromothripsis were all del(11q) or TP53abn-positive, and chromosomes 2 (n=3), 3 (n=6), 6 (n=3), 8 (n=4) and 17 (n=3) were mainly targeted (Figure 1F). Putative chromothripsis was associated with adverse outcome (median OS: 3.7 years, 95% confidence interval [95% CI]: 0.6-6.8 years; P<0.001) (Online Supplementary Figure S3A) and a high total number of CNA (median 6). Surivival of cases with chromothripsis was also worse than that of cases with TP53abn or del(11q) without chromothripsis (P=0.03) (Online Supplementary Figure S3B).
We focused on the ten most frequent CNA other than del(13q), del(11q), trisomy 12 and del(17p) in our cohort (indicated by asterisks in Online Supplementary Figure S4) and investigated whether these CNA correlated with IGHV gene somatic hypermutation status and CNA captured by FISH probes applied in CLL. This resulted in several significant associations, such as (i) U-CLL with gain of 2p and 8q and loss of 6q (Online Supplementary Figure S5); (ii) del(11q) with gain of 2p and 8q and loss of 4p; (iii) trisomy 12 with loss of 14q; (iv) del(13q) with loss of 18p and 14q; and, (v) del(17p) with gain of 3q and 8q, and loss of 4p, 8p, 15q and 18p (Online Supplementary Figure S6AD). A summary of all significant correlations of CNA (corrected P<0.01) detected in the entire cohort with predefined CLL prognostic subgroups is presented in Table 1 and for all correlations in Online Supplementary Table S4.
In a subgroup of 260 patients with FISH and genomic array results available from the same sample, we observed 92.2% concordance (906/983 of regions analyzed by FISH) between the two approaches. Compared to genomic arrays, FISH detected statistically more cases of del(13q) (58.6% vs. 49.8%, P<0.001) and del(11q) (31.3% vs. 25.3%, P=0.003); while the detection rates of trisomy 12 (12.9% vs. 10.4%, P=0.07) and del(17p) (9.3 vs. 8.9%, P=1.00) were not statistically different (Online Supplementary Table S5). FISH-identified CNA not captured by genomic arrays (60/983) were mostly subclonal (median FISH clone size of 19%). Vice versa, 17/983 CNA were detected by genomic arrays only, including six cases of del(17p) (median size 19.11 Mb) and four of del(11q) (median size 17.67 Mb). To investigate the discriminatory power of FISH versus genomic arrays, a concordance index was calculated for genomic arrays versus FISH for del(11q) and del(17p) for both TTFT and OS. The higher number of del(11q) detected by FISH did result in a higher concordance index for TTFT (57% vs. 53%) while the concordance index for OS was 54% for both techniques. The concordance index for del(17p) was similar for genomic arrays and FISH for both TTFT (52% vs. 51%) and OS (57% vs. 55%). These data confirm that genomic arrays show high concordance with FISH with the advantage that (other) potentially clinically relevant abnormalities can be elucidated on a genome-wide scale.
Figure 1.

Overview of copy-number alterations detected by genomic arrays. (A) Percentages of patients with del(13)(q14), del(11)(q22.3) (ATM), trisomy 12 (+12) or del(17)(p13.1) (TP53) detected by genomic arrays irrespective of size. (B-E) Percentages of patients with different chromosomal losses (B), gains (C), monosomies (D) and trisomies (E). (F) Percentages of patients with putative chromothripsis events containing TP53abn in black and cases without TP53abn but del(11q)-positive in white. In four del(11q) cases, TP53 mutation status was not determined.
Table 1.
Correlations of copy-number alterations with predefined prognostic subgroups of chronic lymphocytic leukemia.

Recurrent copy-number alterations detected by genomic arrays are associated with adverse outcome
The effect of CNA detected by genomic arrays on OS was tested in untreated patients at the date of sampling (n=972), mostly within 2 years from diagnosis (69.1%; median time after diagnosis, 7 months). A significant impact of well-established risk factors for CLL, including Binet B/C, U-CLL, TP53abn and del(11q) (Figure 2 and Online Supplementary Figure S7) on OS was confirmed. Next, we analzed the effects of CNA outside the established CLL regions which were detected in at least ten cases (Figure 2). Minimal common regions of deletion or amplification are presented in Online Supplementary Table S6. Gains of 8q (P=0.01; encompassing MYC in >95% of cases), loss of 9p (P<0.001; involving SMARCA2 [46%] and CDKN2A [85%]) and 18p (P<0.001; including USP14 [100%]) were significantly associated with a shorter OS in univariable Cox regression analysis after correction for multiple testing.
Patients with chronic lymphocytic leukemia can be grouped into three prognostic subgroups based on copy-number alteration profile
ROC curve analysis revealed that based on the total number of CNA above the 5 Mb cutoff, including aneuploidy events (see the Online Supplementary Methods for details of this analysis), the cohort could be subdivided into three prognostic subgroups: low-genomic complexity (GC) (0-2 CNA; n=793), intermediate-GC (3-4 CNA; n=122) and high-GC (≥5 CNA; n=57). The results of the ROC curve analysis were replicated by a different statistical approach, using maximally selected rank statistics (see the Online Supplementary Methods for details of this analysis).32
Results were supported with bootstrapping based on 100 random bootstrap samples from the original sample (with replacement) and showed relatively high percentages for the Youden index and the maximally selected estimated cut-point (Online Supplementary Methods). Low-GC cases were associated with indolent disease: Binet A, IGHV mutated-CLL, and low incidences of TP53abn and del(11q) (P<0.01). Intermediate-GC and high-GC cases were associated with a more advanced clinical stage compared to low-GC cases (P=0.003). High-GC cases were enriched for TP53abn and U-CLL (P<0.001) compared to intermediate-GC and low-GC cases (Table 2). Calculation of associations of established biomarkers and CNA with prognostic subgroups identified differences between the prognostic GC subgroups (Figure 3). Interestingly, loss of 9p and loss of 15q were strongly associated with high-GC (P<0.001), not significantly associated with intermediate- GC and negatively associated with low-GC (P<0.001). A positive correlation with high-GC was observed for all CNA except for trisomies 12,18 and 19.
Low-GC was associated with a more favorable clinical outcome (median OS: 10.17 years, 95% CI: 8.92-11.42 years) compared to the outcomes of cases with intermediate- GC (median OS: 7.02 years, 95% CI: 4.79-9.25 years, P=0.001) and high-GC, who had the shortest OS (median OS: 3.05 years, 95% CI: 1.14-4.96 years, P<0.001) (Figure 4A). Similar results were observed for TTFT (Figure 4B) between the three different prognostic subgroups (P=0.001).
High genomic complexity is an independent prognostic risk factor in chronic lymphocytic leukemia
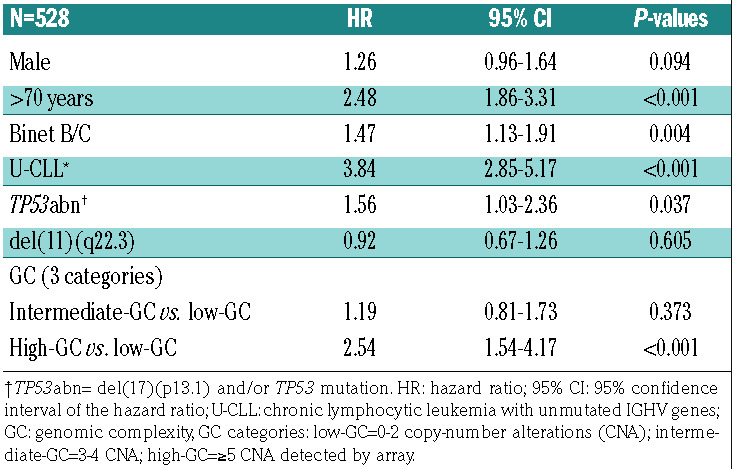
Univariable Cox regression analysis revealed that the GC subgroups intermediate-GC and high-GC (P<0.001) as well as established CLL prognostic factors – male sex, age >70, Binet stage B/C disease, U-CLL, TP53abn and del(11q) – were associated with adverse outcome (P<0.05). (Online Supplementary Tables S7 and S8). High- GC cases had significantly shorter TTFT and OS in the CLL subsets U-CLL (P<0.001), IGHV gene-mutated CLL (P=0.001) and TP53abn/del(11q) (P=0.01) compared to low-GC cases (Online Supplementary Figures S8-S10). Furthermore, including the established factors – male gender, age >70, Binet stage B/C, U-CLL, TP53abn and del(11q) – in the multivariable Cox regression model, high-GC retained statistical significance for TTFT (hazard ratio [HR]: 2.15; 95% CI: 1.36-3.41; P=0.001) (Table 3) and OS (HR: 2.54; 95% CI: 1.54-4.17; P<0.001) (Table 4). Similarly, high-GC retained statistical significance as a binary predictor (with the categories ≥5 CNA and <5 CNA) including the above-mentioned factors in the multivariable Cox regression model for TTFT and OS (P=0.002) (Online Supplementary Tables S9 and S10).
Figure 2.

Effect of copy-number alterations on clinical outcome in chronic lymphocytic leukemia. Forest plot representing the results of univariable Cox regression analysis of copy-number alterations detected by array (with at least 10 events) and the effect of chronic lymphocytic leukemia (CLL) prognostic factors. A hazard ratio of less than 1.00 indicates a lower risk of overall survival. The size of each square is proportional to the amount of data available. U-CLL denotes patients with an unmutated immunoglobulin heavy chain variable gene and TP53abn denotes patients with del(17p) and/or a TP53 mutation. Loss13q.other are patients with a del(13q) not containing the established 13q14 minimally deleted region detectable by the diagnostic fluorescence in situ hybridization probe, and TRIS denotes patients with a trisomy. Only patients untreated at the date of DNA sampling were included for survival analysis. 95% CI: 95% confidence interval.
Lower copy-number alteration size cutoff does not significantly improve risk stratification
In routine clinical practice, a 5 Mb threshold for reporting CNA other than del(13q), del(11q) and del(17p) is used, as described in the publication of Schoumans et al.24 In order to identify whether the recommended 5 Mb threshold or a 1 Mb threshold may be a better discriminator for prognostication, a subgroup of genomic arrays (n=647; with both 1 Mb and 5 Mb cutoff results available) was analyzed applying both cutoffs. Lowering the size cutoff to 1 Mb resulted in the detection of 290 additional aberrations in these 647 patients and revealed an increased percentage of high-GC (≥5 CNA) cases (10.4 vs. 7.3; P<0.001). Next, the discriminatory power was determined by calculating a concordance index to study the effects of lowering the array cutoff to 1 Mb (Online Supplementary Methods) on TTFT and OS. The concordance index was identical for TTFT (77%) and slightly decreased for OS (74% vs. 75%), indicating no further improvement when lowering the cutoff to 1Mb.
Table 2.
Correlations of the three genomic complexity subgroups (based on the complexity of the array profile) with other chronic lymphocytic leukemia risk factors

Genomic arrays do not underperform compared with chromosome-banding analysis
CLL patients with ≥5 chromosomal abnormalities detected by CBA were recently shown to have a dismal prognosis and CBA is considered the gold standard for defining a CK.12 Direct comparison of 122 patients with simultaneous CBA and genomic array results available, showed chromosomal abnormalities in 86.1% of patients by genomic arrays and in 73.8% by CBA (P=0.015) (Online Supplementary Figure S11). Analysis of a subset of patients (n=96) for whom prognostic factors gender, age, Binet stage, del(11q), del(17p) and IGHV mutation status were known, revealed a higher concordance index for OS (77% vs. 72%) and similar concordance index for TTFT (68% vs. 67%) for genomic arrays versus CBA.
Discussion
In our study CLL patients with ≥5 CNA emerged as a separate subgroup with aggressive disease and an adverse outcome, which was independent of several well-established factors in CLL, such as IGHV and TP53 status. A disadvantage of a collection of retrospective multicenter data is the risk of variation introduced by combining results from different genomic array platforms. Nevertheless, marked variation in this study is less likely since the data were methodically reviewed and uniformly interpreted using commonly accepted guidelines which provide tools for filtering and calling CNA and single nucleotide polymorphism data.17,23,24,33 Another clear disadvantage of such real-world retrospective data is that clinical correlations, although indicative, must be considered with great caution as treatments were not uniform and we were limited to the date of sample collection (instead of diagnosis) for the TTFT analysis. In current International Working Group CLL guidelines,34 targeted FISH is recommended for the detection of genomic aberrations. In our multicenter study overall concordance between arraybased comparative genomic hybridization data and (targeted) FISH results (subgroup, n=260) was high (>90%). Nevertheless, genomic arrays resulted in the detection of fewer CNA for regions captured by FISH, which could be explained by the superior detection limit of FISH.17,25,28,35-38 However, the increased detection of CNA by FISH was not directly associated with a better risk assessment, in agreement with the findings in other genomic array studies,17,18,22 providing a rationale that genomic arrays can be used, besides FISH, in CLL diagnostics.17 Direct comparison of genomic arrays with FISH in prospective clinical trials is necessary before genomic arrays can be accepted to replace FISH in routine clinical practice for CLL prognostics.
Figure 3.

Heatmap of correlations for different categories of genomic complexity based on total number of copy-number alterations. Genomic complexity (GC) was subdivided into three prognostic subgroups: low-GC (0-2 copy-number alterations [CNA]), intermediate-GC (3-4 CNA) and high-GC (≥5 CNA). Colors are based on the results of Kendall’s tau_b which assumes values between -1 and 1. Correlation levels are depicted according to the color scale with increased correlation ranging from green to red. Only statistically significant correlations with a P<0.01 are colored. Non-significant correlations are shown as uncolored crossed squares. U-CLL: chronic lymphocytic leukemia with an unmutated immunoglobulin heavy chain variable gene; TP53abn: cases with del(17p) and/or a TP53 mutation; Loss13q.other: cases with a del(13q) not containing the established 13q14 minimally deleted region detectable by the diagnostic fluorescence in situ hybridization probe; TRIS trisomy.
Since our data were collected retrospectively from patients during follow-up after various chemoimmunotherapy regimens, we could not correlate the effects of CNA on OS with specific treatments. CNA such as del(4p), del(9p) and del(18p) co-occurred with other CNA and were associated with other CLL biomarkers. For this reason, the prognostic impact of these individual CNA must be considered with caution. Furthermore, the applicability of our results needs to be established for novel agents, as the prognostic value following chemoimmunotherapy might be different. This is exemplified by a recent update of the RESONATE-2 study in which del(11q) was prognostically favorable in ibrutinib-treated patients.39 Additionally, del(8p) was recently correlated with resistance to ibrutinib.40 The inferior outcome of patients with loss of 9p is in line with the findings of a recent study on the effect of loss of 9p24.3 containing SMARCA2.41 Loss of 9p24.3 and/or mutations in the SWISNF chromatin remodeling complex were identified in all nonresponders to ibrutinib and venetoclax. Deletion of SMARCA2 results in upregulation of the prosurvival protein BCL-XL which could protect cells from apoptosis.41
Figure 4.

Effect of genomic complexity on clinical outcome in chronic lymphocytic leukemia. (A, B) Kaplan-Meier plots presenting the overall survival (A) and time to first treatment (B) of patients with chronic lymphocytic leukemia divided into three categories of genomic complexity (GC) based on the total number of copy-number alterations (CNA): low-GC (0-2 CNA), intermediate-GC (3-4 CNA) and high-GC (≥5 CNA) and defined by receiver operating characteristic curve analysis. 95% CI: 95% confidence interval.
Table 3.
Multivariable analysis for time to first treatment.
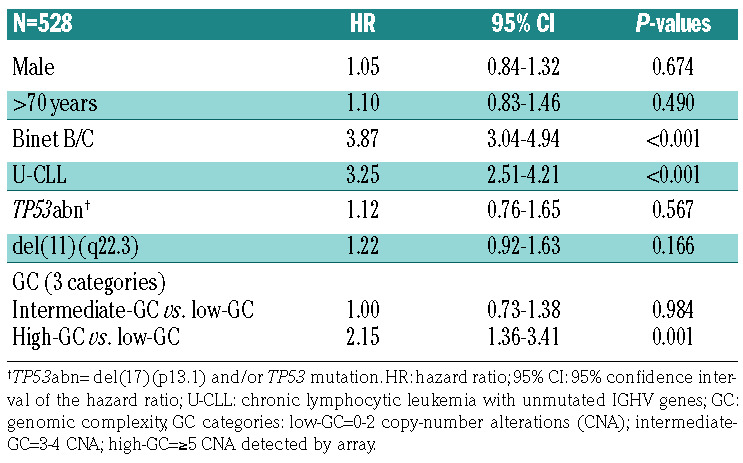
Table 4.
Multivariable analysis for overall survival.
The differences in OS and TTFT in the distinct GC subcategories based on ROC analysis in this study are in agreement with recent findings regarding CBA-defined CK and the independent effect of high-CK (with ≥5 numerical and/or structural abnormalities),12 which were characterized by a distinct distribution of aberrations affecting a broad spectrum of chromosomes.12 Similarly, high-GC detected by genomic arrays was negatively associated with trisomy 12 and, in contrast to intermediate- GC, not positively associated with trisomy 18 and 19 (Figure 3). High-CK cases detected by CBA and lacking del(17p) or a Sanger-detected (‘clonal’) TP53 mutation were all negative for subclonal TP53 mutations when sequenced by next-generation sequencing.12 It is conceivable that recently identified potential drivers for high-GC in CLL, such as SAMHD1 and SETD2, attributed to high- GC in a subset of patients.9,42-44 The resolutions of the genomic array platforms used in this study are higher than the resolution that can be achieved by CBA. Differences in the detection of chromosomal aberrations between genomic arrays and CBA in our study could be related to the lower resolution of CBA and/or the use of phorbol-12- myristate-13-acetate in most CBA cases instead of the more sensitive method involving cytosine guanine dinucleotides plus interleukin 2.12 In contrast to genomic arrays, CBA selects for cells with proliferative potential as a prognostic marker and is able to detect balanced translocations. Although balanced rearrangements (such as balanced translocations) cannot be detected by genomic arrays, chromosome regions involved in unbalanced translocations can be recognized more precisely by arrays, possibly revealing new recurrent abnormalities. Our finding that, in terms of risk stratification, genomic arrays do not underperform compared with CBA implies that the inability of genomic arrays to detect balanced rearrangements does not impede adequate risk assessment. This could be explained by the fact that, in contrast to other mature B-cell tumors, CLL is defined by the presence of CNA with a paucity of balanced rearrangements.11,45
In CLL diagnostics genomic arrays are cost-effective as a stand-alone method when compared to FISH plus CBA. Only recently, the integration of genomic arrays has been recommended by the Cancer Genomics Consortium working group as having evidence-based clinical utility for a broad spectrum of hematologic malignancies.33,46-49
In conclusion, our study highlights the strength of genome-wide approaches for risk stratification in CLL and we identified CLL patients with high-GC, defined as ≥5 CNA detected by genomic arrays, as an independent subgroup with dismal clinical outcome. We, therefore, recommend implementation of comprehensive genomic profiling in future clinical trials.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge all patients who contributed to this study. This study was partly funded by an unrestricted contribution from Janssen Pharmaceuticals and from GILEAD Sciences SA. The funding sources had no role in identifying statements, abstracting data, synthesizing results, grading evidence or preparing the manuscript, or in the decision to submit the manuscript for publication. ACL is supported by the Peters van der Laan foundation. JCS was funded by Bloodwise (11052, 12036), the Kay Kendall Leukaemia Fund (873), Cancer Research UK (C34999/A18087, ECMC C24563/A15581), Wessex Medical Research and the Bournemouth Leukaemia Fund. KP, MJ, and SP are supported by the project MHCR DRO n. 65269705, the research infrastructures NCMG LM2015091, and EATRIS-CZ LM2015064, and the project CEITEC2020 LQ1601, funded by MEYS CR. RR is supported by the Swedish Cancer Society, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, Karolinska Institutet, Karolinska University Hospital, and Radiumhemmets Forskningsfonder, Stockholm.
References
- 1.Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood. 1975;46(2):219-234. [DOI] [PubMed] [Google Scholar]
- 2.Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer. 1981;48(1):198-206. [DOI] [PubMed] [Google Scholar]
- 3.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854. [PubMed] [Google Scholar]
- 4.Fais F, Ghiotto F, Hashimoto S, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J Clin Invest. 1998;102(8):1515-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910-1916. [DOI] [PubMed] [Google Scholar]
- 6.Krober A, Bloehdorn J, Hafner S, et al. Additional genetic high-risk features such as 11q deletion, 17p deletion, and V3-21 usage characterize discordance of ZAP-70 and VH mutation status in chronic lymphocytic leukemia. J Clin Oncol. 2006;24(6): 969-975. [DOI] [PubMed] [Google Scholar]
- 7.Blanco G, Puiggros A, Baliakas P, et al. Karyotypic complexity rather than chromosome 8 abnormalities aggravates the outcome of chronic lymphocytic leukemia patients with TP53 aberrations. Oncotarget. 2016;7(49):80916-80924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haferlach C, Dicker F, Schnittger S, Kern W, Haferlach T. Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia. 2007;21(12):2442-2451. [DOI] [PubMed] [Google Scholar]
- 9.Herling CD, Klaumunzer M, Rocha CK, et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood. 2016;128(3): 395-404. [DOI] [PubMed] [Google Scholar]
- 10.Rigolin GM, Saccenti E, Guardalben E, et al. In chronic lymphocytic leukaemia with complex karyotype, major structural abnormalities identify a subset of patients with inferior outcome and distinct biological characteristics. Br J Haematol. 2018;181(2): 229-233. [DOI] [PubMed] [Google Scholar]
- 11.Baliakas P, Iskas M, Gardiner A, et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: a systematic reappraisal of classic cytogenetic data. Am J Hematol. 2014;89(3):249-255. [DOI] [PubMed] [Google Scholar]
- 12.Baliakas P, Jeromin S, Iskas M, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations and clinical impact. Blood. 2019;133(11): 1205-1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Bris Y, Struski S, Guieze R, et al. Major prognostic value of complex karyotype in addition to TP53 and IGHV mutational status in first-line chronic lymphocytic leukemia. Hematol Oncol. 2017;35(4):664-670. [DOI] [PubMed] [Google Scholar]
- 14.Thompson PA, O'Brien SM, Wierda WG, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinibbased regimens. Cancer. 2015;121(20): 3612-3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson MA, Tam C, Lew TE, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood. 2017;129(25): 3362-3370. [DOI] [PubMed] [Google Scholar]
- 16.Sargent R, Jones D, Abruzzo LV, et al. Customized oligonucleotide array-based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia. J Mol Diagn. 2009;11(1):25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunn SR, Mohammed MS, Gorre ME, et al. Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia. J Mol Diagn. 2008;10(5): 442-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Malley DP, Giudice C, Chang AS, et al. Comparison of array comparative genomic hybridization (aCGH) to FISH and cytogenetics in prognostic evaluation of chronic lymphocytic leukemia. Int J Lab Hematol. 2011;33(3):238-244. [DOI] [PubMed] [Google Scholar]
- 19.Gunnarsson R, Mansouri L, Isaksson A, et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica. 2011;96(8):1161-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puiggros A, Puigdecanet E, Salido M, et al. Genomic arrays in chronic lymphocytic leukemia routine clinical practice: are we ready to substitute conventional cytogenetics and fluorescence in situ hybridization techniques? Leuk Lymphoma. 2013;54(5): 986-995. [DOI] [PubMed] [Google Scholar]
- 21.Edelmann J, Holzmann K, Miller F, et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood. 2012;120(24):4783-4794. [DOI] [PubMed] [Google Scholar]
- 22.Stevens-Kroef MJ, van den Berg E, Olde Weghuis D, et al. Identification of prognostic relevant chromosomal abnormalities in chronic lymphocytic leukemia using microarray-based genomic profiling. Mol Cytogenet. 2014;7(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simons A, Sikkema-Raddatz B, de Leeuw N, Konrad NC, Hastings RJ, Schoumans J. Genome-wide arrays in routine diagnostics of hematological malignancies. Hum Mutat. 2012;33(6):941-948. [DOI] [PubMed] [Google Scholar]
- 24.Schoumans J, Suela J, Hastings R, et al. Guidelines for genomic array analysis in acquired haematological neoplastic disorders. Genes Chromosomes Cancer. 2016;55(5):480-491. [DOI] [PubMed] [Google Scholar]
- 25.Ouillette P, Collins R, Shakhan S, et al. Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood. 2011;118(11):3051-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu L, Kim HT, Kasar S, et al. Survival of Del17p CLL depends on genomic complexity and somatic mutation. Clin Cancer Res. 2017;23(3):735-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight SJ, Yau C, Clifford R, et al. Quantification of subclonal distributions of recurrent genomic aberrations in paired pre-treatment and relapse samples from patients with B-cell chronic lymphocytic leukemia. Leukemia. 2012;26(7):1564-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745-2760. [DOI] [PubMed] [Google Scholar]
- 29.Parker H, Rose-Zerilli MJ, Parker A, et al. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia. 2011;25(3):489-497. [DOI] [PubMed] [Google Scholar]
- 30.Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemiaupdate on methodological approaches and results interpretation. Leukemia. 2018;32(5):1070-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenquist R, Ghia P, Hadzidimitriou A, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia. 2017; 31(7):1477-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hothorn T, Zeileis A. Generalized maximally selected statistics. Biometrics. 2008; 64(4):1263-1269. [DOI] [PubMed] [Google Scholar]
- 33.Chun K, Wenger GD, Chaubey A, et al. Assessing copy number aberrations and copy-neutral loss-of-heterozygosity across the genome as best practice: an evidencebased review from the Cancer Genomics Consortium (CGC) working group for chronic lymphocytic leukemia. Cancer Genet. 2018;228-229:236-250. [DOI] [PubMed] [Google Scholar]
- 34.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for diagnosis, indications for treatment, response assessment and supportive management of chronic lymphocytic leukemia. Blood. 2018;131(25):2745-2760. [DOI] [PubMed] [Google Scholar]
- 35.Hagenkord JM, Monzon FA, Kash SF, Lilleberg S, Xie Q, Kant JA. Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of Affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays. J Mol Diagn. 2010;12(2):184-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehmann S, Ogawa S, Raynaud SD, et al. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia. Cancer. 2008;112(6):1296-1305. [DOI] [PubMed] [Google Scholar]
- 37.Tyybakinoja A, Vilpo J, Knuutila S. Highresolution oligonucleotide array-CGH pinpoints genes involved in cryptic losses in chronic lymphocytic leukemia. Cytogenet Genome Res. 2007;118(1):8-12. [DOI] [PubMed] [Google Scholar]
- 38.Kolquist KA, Schultz RA, Slovak ML, et al. Evaluation of chronic lymphocytic leukemia by oligonucleotide-based microarray analysis uncovers novel aberrations not detected by FISH or cytogenetic analysis. Mol Cytogenet. 2011;16;4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barr PM, Robak T, Owen C, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica. 2018;103(9):1502-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;20;7:11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agarwal R, Chan YC, Tam CS, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119-129. [DOI] [PubMed] [Google Scholar]
- 42.Clifford R, Louis T, Robbe P, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123(7):1021-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker H, Rose-Zerilli MJ, Larrayoz M, et al. Genomic disruption of the histone methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia. 2016;30(11):2179-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mar BG, Chu SH, Kahn JD, et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood. 2017;130(24):2631-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leeksma AC, Taylor J, Wu B, et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia. 2018;33(2):390-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanagal-Shamanna R, Hodge JC, Tucker T, et al. Assessing copy number aberrations and copy neutral loss of heterozygosity across the genome as best practice: an evidence based review of clinical utility from the Cancer Genomics Consortium (CGC) working group for myelodysplastic syndrome, myelodysplastic/myeloproliferative and myeloproliferative neoplasms. Cancer Genet. 2018;228-229:197-217. [DOI] [PubMed] [Google Scholar]
- 47.Pugh TJ, Fink JM, Lu X, et al. Assessing genome-wide copy number aberrations and copy-neutral loss-of-heterozygosity as best practice: an evidence-based review from the Cancer Genomics Consortium working group for plasma cell disorders. Cancer Genet. 2018;228-229:184-196. [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Bryke C, Sukhanova M, et al. Assessing copy number abnormalities and copy-neutral loss-of-heterozygosity across the genome as best practice in diagnostic evaluation of acute myeloid leukemia: an evidence-based review from the Cancer Genomics Consortium (CGC) myeloid neoplasms working group. Cancer Genet. 2018;228-229:218-235. [DOI] [PubMed] [Google Scholar]
- 49.Mikhail FM, Biegel JA, Cooley LD, et al. Technical laboratory standards for interpretation and reporting of acquired copy-number abnormalities and copy-neutral loss of heterozygosity in neoplastic disorders: a joint consensus recommendation from the American College of Medical Genetics and Genomics (ACMG) and the Cancer Genomics Consortium (CGC). Genet Med. 2019;21(9):1903-1915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.