

Abstract
Glioblastomas are highly lethal cancers defined by resistance to conventional therapies and rapid recurrence. While new brain tumor cell-specific drugs are continuously becoming available, efficient drug delivery to brain tumors remains a limiting factor. We developed a multicomponent nanoparticle, consisting of an iron oxide core and a mesoporous silica shell that can effectively deliver drugs across the blood–brain barrier into glioma cells. When exposed to alternating low-power radiofrequency (RF) fields, the nanoparticle’s mechanical tumbling releases the entrapped drug molecules from the pores of the silica shell. After directing the nanoparticle to target the near-perivascular regions and altered endothelium of the brain tumor via fibronectin-targeting ligands, rapid drug release from the nanoparticles is triggered by RF facilitating wide distribution of drug delivery across the blood-brain tumor interface.
1. Introduction
Drug-loaded nanoparticles often release their drug cargo either slowly at the target site or prematurely while circulating in the bloodstream. We developed a multicomponent drug carrier based on a mesoporous silica nanoparticle (MSN) with a switchable drug release mechanism. Drug release from the nanoparticle can be delayed by hours, while it can be triggered by the physics of external radiofrequency (RF) fields, rather than the chemistry or in vivo breakdown of the particle. MSNs are all-purpose nanoparticles that can be fabricated in high quantities and conveniently accommodate a high cargo of various drugs.1,2 To demonstrate the capabilities of the nanoparticle, we used the nanoparticles to deliver a cytotoxic drug to glioblastoma (GBM), a hard-to-treat cancer.
GBMs remain highly lethal despite maximal surgical resection, concurrent radiation and chemotherapy, and adjuvant Temozolomide (TMZ).3,4 Despite advancements, the recurrence of gliomas is >90%. This stems from the fact that gliomas are characteristically diffuse with infiltrating edges, resistant to drugs and nearly inaccessible to systemic therapies due to the blood–brain barrier (BBB).5–7 Even though new potent agents are continuously becoming available, drug delivery to brain tumors is limited, which has become a liability to the success of newly developed therapeutics. While the BBB barrier is partially breached in regions with glioma cells, the ‘compromised’ BBB barrier still presents a major roadblock, resulting in the failure of most drugs to reach glioma cells. Notably, the BBB of invasive sites with dispersing glioma cells has a very high likelihood to remain completely intact.8 To tackle these issues of today’s systemic therapies, we used a multicomponent nanoparticle, termed Fe@MSN, which consists of an iron oxide core and a mesoporous silica shell. As shown in Fig. 1, our approach is based on the development of an effective systemic therapy by incorporating delivery of cytotoxic agents across the BBB, and minimal toxicity to normal brain.
Fig. 1.
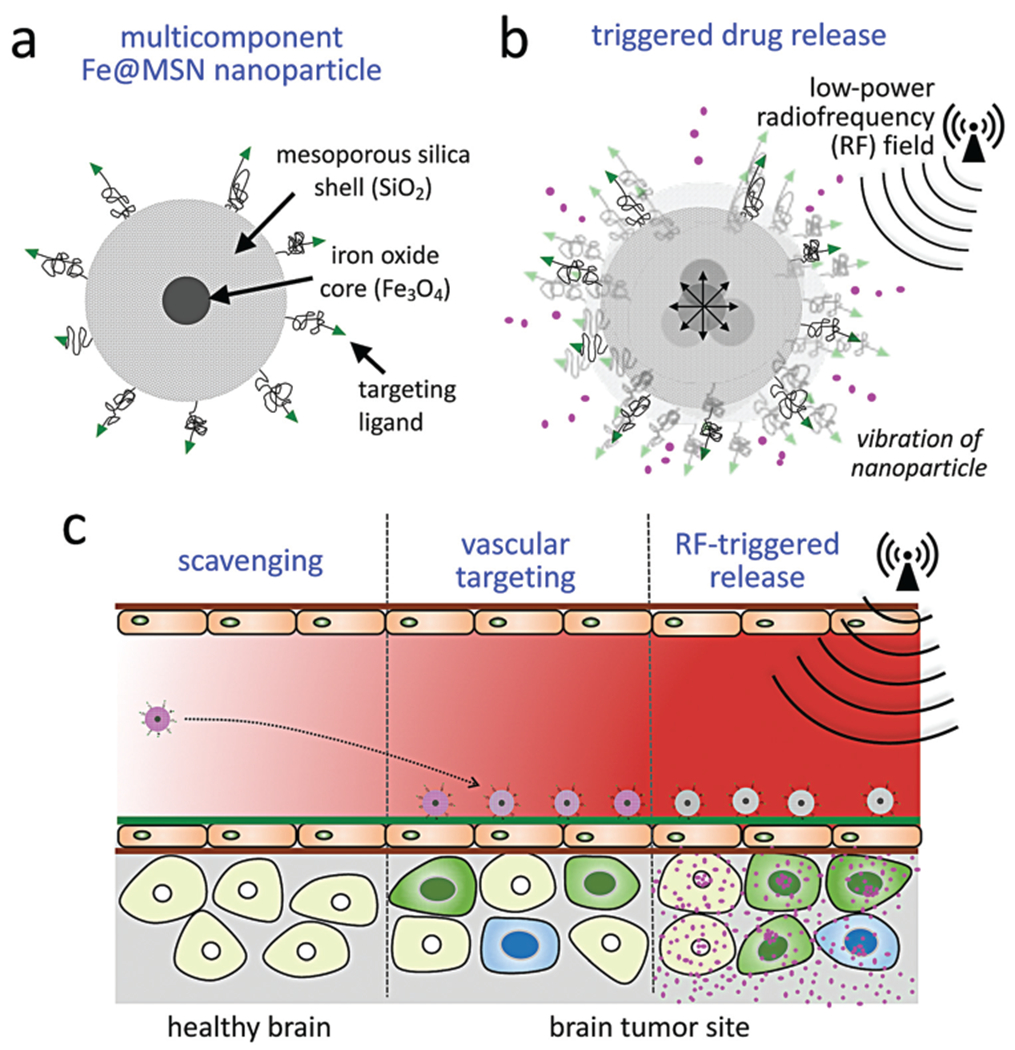
Illustration of therapeutic strategy. (a) The nanoparticle, termed (Fe@MSN), is comprised of an iron core surrounded by a drug-loaded a mesoporous silica shell. (b) Drug release from the Fe@MSN nanoparticles is triggered by an external low-power radiofrequency (RF) field at frequencies of about 50 kHz, which makes the nanoparticle to vibrate giving kinetic energy to the drug molecules to escape the pores of the silica shell. (c) The therapeutic strategy consists of vascular targeting of the nanoparticle to the endothelium of glioma sites, and RF-triggered release of drug cargo from Fe@MSN nanoparticles resulting in effective drug delivery across the BBB to glioma cells.
To circumvent the BBB, we utilized a two-step targeting approach. First, rather than targeting cancer cells deep in the tumor interstitium, we direct the Fe@MSN nanoparticles to the altered endothelium and near-perivascular regions of GBM by targeting fibronectin, which is an overexpressed perivascular biomarker in GBMs.9–13 Fibronectin is also associated with migrating, invasive glioma cells and hypoxic regions, while its expression is insignificant on the endothelium of healthy brain. While nanoparticles have shown some promise to passively deposit through compromised BBB and ‘smuggle’ drugs into intracranial tumors due to the enhanced permeation and retention (EPR) effect, the EPR effect is heterogeneous and significantly attenuated in GBM.5–7 On the other hand, the endothelium is the closest point-of-contact for circulating nanoparticles. Further, the size and multivalent avidity make nanoparticles ideal for targeting vascular biomarkers. Our studies show that the vascular targeting scheme leads to highly specific and rapid deposition of Fe@MSN nanoparticles on the endothelium of GBM throughout the brain tumor.
Second, rapid drug release from the Fe@MSN nanoparticles is triggered by an external low-power radiofrequency (RF) field facilitating drug delivery across the BBB to glioma cells. Using the endothelium as a docking site, vascular targeting establishes well-distributed drug reservoirs throughout the brain tumor, which can subsequently spread free drug across the brain–tumor interface into the tumor interstitium using low-power magnetic fields at frequencies of 50 kHz as an external trigger. By overwhelming the BBB barrier and its efflux pumps with a rapidly elevated concentration of drug molecules, a surge of the liberated drug crosses the endothelium at the brain–tumor interface. Unlike nanoparticles, the liberated drug molecules, being low molecular weight compounds, are able to rapidly diffuse into the tumor interstitium, thereby reaching to distant and hard-to-reach brain tumor cell populations. The triggered release mechanism is unique to the Fe@MSN structure and relies on mechanical vibration (rather than heating) facilitating rapid drug release at any tissue depth. Because the wavelength of these fields is on the order of kilometers, they pass through the body as if it was transparent. When magnetic nanoparticles are subjected to an external magnetic field, they must overcome thermal and viscous forces to achieve magnetic reversal and align with the applied field. The Néel and Brownian relaxation mechanisms govern this behavior.14 Used for hyperthermia, Neel relaxation, a competition between magnetocrystalline energy and thermal energy, is dominant for nanoparticles smaller than 15 nm. In our case, the governing mechanism is the Brownian relaxation, which is the physical rotation of the entire magnetic core, which is larger than 15 nm. In its effort to align with the alternating RF field, the Fe@MSN particle tumbles, providing kinetic energy to the drug molecules and helping them to overcome the electrostatic interactions keeping them within the pores of the silica shell. This added kinetic energy enables drugs to be liberated from the Fe@MSN nanoparticle. Contrary to other triggered release mechanisms that depend on high-energy electromagnetic fields or significant changes in environmental factors (e.g. hyperthermia, pH), the mechanical forces required for our strategy are generated at low RF energies (5–10 W). Due to the ease of transmitting the required low-energy RF throughout the brain, drugs are rapidly released from very low concentrations of Fe@MSN particles and at any tissue depth. Using two orthotopic models of GBM in mice, we show that RF-triggered release from Fe@MSN nanoparticles facilitated widespread distribution of drug molecules in GBMs, resulting in remarkable anticancer outcomes and tumor shrinkage.
2. Experimental section
2.1. Synthesis of Fe@MSN nanoparticles
We synthesized the iron oxide cores using a coprecipitation method. Briefly, FeCl3·6H2O and FeCl2·4H2O were dissolved in deoxygenated water followed by the addition of the iron precursor solution at 80 °C under argon. After magnetic separation and washing steps, the iron oxide cores were coated with citric acid. The silica shell was synthesized using a base-catalyzed sol–gel process with modifications. Upon dispersing the iron oxide cores in a solution of CTAB, tetraethylorthosilicate (TEOS) was added followed by phosphonate functionalization and silane–PEG–NH2. By adjusting the pH according to the pKa of the drug, drugs were conveniently loaded into the nanoparticles via co-incubation for 12 h under mild mixing. 1400 W was loaded into Fe@MSN nanoparticles in PBS at a pH of 8. DOX was loaded at a pH of 7.4. Any unbound drug was removed from the particles by washing with PBS several times and repeated centrifugation. To evaluate drug loading capacity, the residual drug was measured after the loading procedure. The washing solutions were collected and the residual drug content was measured by UV-Vis absorption spectroscopy at λ = 480 nm for DOX or an HPLC assay for 1400 W. For the concentration of 1400 W, an HPLC assay was run in a C18 5μ reverse-phase column (isocratic; mobile phase: 50% water, 25% methanol and 25% acetonitrile; flow rate of 0.5 ml min−1; detection at 254 nm). Infrared analyses for DOX and 1400 W were obtained using a Thermo Nexus 870 FTIR spectrometer with an attenuated total reflection (ATR) accessory. Spectra over the 4000–500 cm−1 range were obtained by the co-addition of 64 scans with a resolution of 4 cm−1. The content of Fe and Si in the Fe@MSN nanoparticle was determined via ICP-OES (Optima 7000 DV; PerkinElmer). First, the sample was digested with hydrofluoric acid (HF) in a 50 mL polyethylene tubes at room temperature. The sample was held at room temperature for about 6 h. Then, 25% by mass fraction of tetramethyl ammonium hydroxide (TMAH) was added to neutralize the solution. Finally, an aliquot of 0.2 M HNO3 was added for iron digestion. The sample was then analyzed with ICP-OES.
The fibronectin-targeting peptide CREKA15,16 was conjugated on the surface of the Fe@MSN particle via its distal end of PEG–NH2 using standard conjugation chemistry. Briefly, the thiol of the cysteine residue on the peptide was conjugated to the amine of PEG–NH2 via the sulfo-SMCC crosslinker. Sulfo-SMCC contains an amine-reactive NHS ester and a sulfhydryl-reactive maleimide group to form stable amide and thioether bonds. To reassure complete conjugation of the available PEG-NH2, we used a 2-fold molar excess of the peptides over the PEG molecules followed by a cleaning step. A detailed description of the synthesis is provided in ESI.†
2.2. Tumor models
This study was performed in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals (8th Edition) and the United States Department of Agriculture and the Public Health Service policy and regulations. All animal procedures were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University. For the CNS-L rodent glioma tumor model, 5–8-week-old female athymic nude mice (~25 g) were housed in the Athymic Animal Core Facility at Case Western Reserve University according to institutional policies. CNS-L cells were infected with green fluorescent protein (GFP) encoding lentivirus, harvested for intracranial implantation by trypsinization, and concentrated in PBS. Mice were anesthetized by intraperitoneal administration of ketamine and xylazine and fitted into a stereotaxic rodent frame. Cells were implanted at AP = +0.5 and ML = −2.0 from bregma at a rate of 1 μL min−1 in the right striatum at a depth of −3 mm from dura. A total of 200 000 cells were implanted per mouse. Similar procedures were employed for the GL261 glioma cells. After tumor implantation, mice were randomized into groups for subsequent studies. All cell lines were authenticated using CellCheck including Short tandem repeat (STR) profile, interspecies contamination testing and Mycoplasma contamination.
2.3. Bioluminescence imaging
Using the IVIS Spectrum system, bioluminescence imaging (BLI) was performed 10 min after intraperitoneal administration of 200 μl of d-luciferin (10 mg ml−1). BLI was performed every 2 days until the terminal point of the study. At the terminal point, organs were extracted for ex vivo organ imaging or histological analysis.
2.4. Evaluation of targeting brain tumors
The deposition of CREKA-targeted Fe@MSN nanoparticles in gliomas was evaluated in mice bearing orthotopic CNS-1 brain tumors. Using a tail vein injection, free DOX or DOX-loaded nanoparticles were systemically injected at a dose of 5 mg per kg b.w. After 1, 3 or 8 hours from injection, animals were anesthetized and transcardially perfused with heparinized PBS. Organs and brain tumors were then retrieved, washed, blotted dry, weighed, and DOX was measured following an established protocol.17 Total doxorubicin content of each sample was analyzed (λex = 480, λem = 590) using a fluorescence spectrometer (Synergy HT, Biotek). Organs and brain tumors from animals treated with a saline injection were used to correct for background fluorescence.
2.5. Histological evaluation
Immunohistochemistry was performed to evaluate the distribution of nanoparticles and drugs and the topology of fibronectin expression with respect to glioma cells and blood vessels. The mice were anesthetized with an IP injection of ketamine/xylazine and transcardially perfused with heparinized PBS followed by 4% paraformaldehyde in PBS. Brains were explanted and post-fixed overnight in 4% paraformaldehyde in PBS. The tissues were soaked in 30% sucrose (w/v) in PBS at 4 °C for cryosectioning. Serial tissue sections of 12 μm in thickness were obtained. Direct fluorescence of GFP (green) imaging was performed for imaging the location of glioma cells. To visualize the tumor microvasculature and fibronectin, the tissue slices were immunohistochemically stained for the endothelial antigen CD31 or anti-fibronectin primary antibody (BD Biosciences, Pharmingen). Prussian blue stain was used to detect iron. Direct fluorescence (red) imaging of tumor sections was performed for imaging DOX. To label the hypoxic regions, we delivered the substrate pimonidazole hydrochloride to mice bearing orthotopic GL261 tumors before harvesting brains. As pimonidazole is reductively activated in hypoxic cells and forms stable adducts with thiol (sulfhydryl) groups in proteins, peptides, and amino acids, it can be recognized by a monoclonal antibody to mark the hypoxic regions in tumors. Tissue sections were imaged at 5, 10 or 40× on the Zeiss Axio Observer Z1 motorized FL inverted microscope. To obtain an image of large tissue sections, a montage of each section was made using the automated tiling function of the microscope.
2.6. Survival study
Mice bearing orthotopic CNS-1 or GL261 brain tumors were injected intravenously with free drugs or drug-loaded Fe@MSN at a dose of 5 mg kg−1 DOX. Under anesthesia, RF was applied as described previously. Treatments were given 4 times at days 2, 3, 6 and 7 after tumor inoculation. Mice were monitored daily for any abnormal symptoms. The well-being of the animals took priority in decisions regarding euthanasia or other interventions. When animals showed a 10% loss of body weight, they were euthanized in a CO2 chamber. The 10% weight loss was the primary endpoint criterion. Time of death was determined to be the following day.
2.7. Statistical analysis
Statistics were performed in Prism version 7 for Mac (GraphPad Software, La Jolla, CA, USA). Data are represented as mean ± s.d. Statistical significance between survival curves was determined using the log-rank (Mantel-Cox) test. In cases where data met the assumptions necessary for parametric statistics, analysis of differences between two groups was performed using two-tailed Student’s t-test assuming equal variance. Data from three or more groups were analyzed with a one-way analysis of variance (ANOVA) that was corrected for multiple comparisons using the Holm-Sidak method.
3. Results and discussion
3.1. Synthesis and characterization of drug-loaded Fe@MSN nanoparticles
We initially synthesized the iron oxide cores using the coprecipitation method. After magnetic separation and washing steps, the iron oxide cores were coated with citric acid. The silica shell was synthesized using a base-catalyzed sol–gel process. Upon dispersing the iron oxide cores in a CTAB solution, tetraethylorthosilicate (TEOS) was added followed by phosphonate functionalization and silane–PEG–NH2. Details on the synthesis and characterization of the nanoparticles are provided in ESI.† Fig. 2a shows TEM images of the nanoparticles showing the iron oxide core and porous structure of the silica shell. The final Fe@MSN nanoparticle exhibits an overall size of ~80 nm with the iron oxide core being ~20 nm. Prior to loading the particle with drugs, elemental analysis indicates that the Fe@MSN composition is 96% Si and 4% Fe (Fig. 2b). In 1 mg of sample, we measured 4.17 × 1011 particles using the Archimedes particle metrology system. Fig. 2c shows the FTIR spectrum of Fe@MSN nanoparticle. The bands at 1080, 900, and 800 cm−1 are due to the stretching vibration of Si–O–Si, Si–OH, and Si–O, respectively, which are characteristic vibration modes of SiO2. The broad band at 3400 cm−1 can be attributed to the O–H groups, whereas the band at 1600 cm−1 can be assigned to the bending vibration of O–H. Additionally, the bands at 2900 cm−1 correspond to the C–H stretching vibrations. Upon surface modification with silane–PEG–NH2, functionalization with the fibronectin-targeting peptide (CREKA) resulted in conjugation of ~3000 peptides on each Fe@MSN nanoparticle as measured by a Bio-Rad DC protein assay (Fig. 2d). Zeta potential measurements indicated that surface charge of these nanoparticles was −32.5 mV (Fig. 2e). Fig. 2f shows the hydrodynamic size of the starting iron oxide core and final Fe@MSN nanoparticle.
Fig. 2.
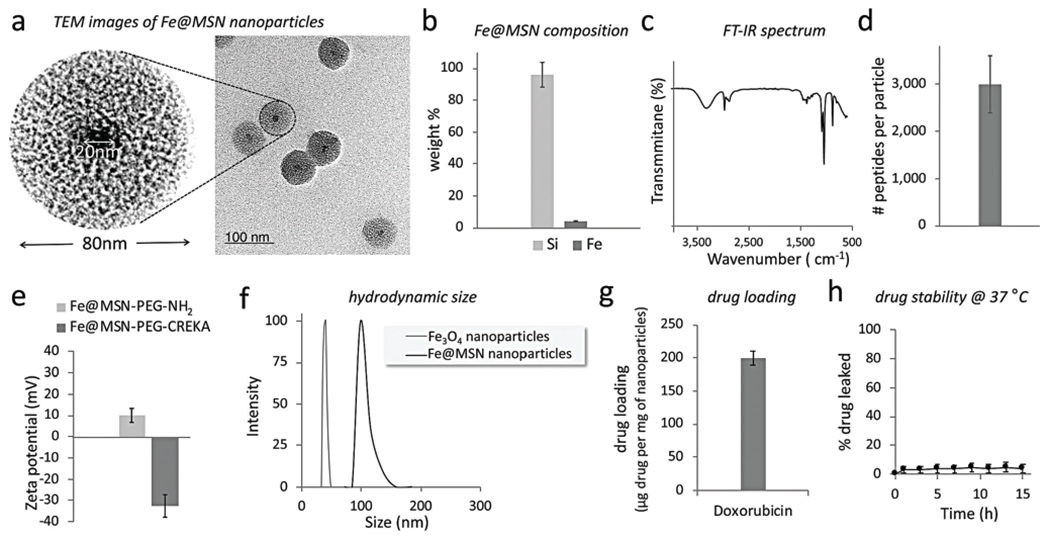
Characterization of the nanoparticles. (a) TEM image of the Fe@MSN nanoparticle. (b) Elemental analysis of the composition of the Fe@MSN nanoparticle was performed using ICP-OES. (c) FT-IR spectrum of the nanoparticle. (d) The number of peptides on each Fe@MSN nanoparticle was measured using a Bio-Rad DC protein assay. (e) The zeta potential of the DOX-loaded Fe@MSN nanoparticle was measured in 1 M KCl before and after conjugation with the targeting peptide CREKA using a Malvern Zeta Potential Analyzer. (f) Size distribution of the starting iron oxide core and the final Fe@MSN nanoparticles obtained by DLS. (g) The drug cargo of the Fe@MSN nanoparticles is shown. (h) Drug loading into the particles demonstrated good stability in PBS with 10% FBS at 37 °C.
Loading of the drug into the mesoporous silica shell of the Fe@MSN nanoparticle was achieved via electrostatic interactions. To improve drug loading and loading stability, we functionalized the surface of the pores with the hydrophilic phosphonate group. The phosphonate functionalization increases the negative charge of the pores in the silica shell resulting in increased electrostatic interactions with the positively charged drug molecules.18 Because of the low molecular weight and size of the drug molecules, drug was loaded into the pores of the nanoparticles via co-incubation of drug and nanoparticles for 12 h under mild mixing. The pH of the suspension was adjusted to 7.4. Due to its cationic nature, DOX is mostly protonated at pH 7.4 (pKa ~8.2). Free drug molecules were removed from the drug-loaded nanoparticles by washing with PBS several times and repeated centrifugation. To evaluate drug loading capacity, the residual drug was measured after the loading procedure. The washing solutions were collected and the residual drug content was measured by UV-Vis absorption spectroscopy (λ = 480 nm) for DOX. To confirm that the drug was stably loaded in the nanoparticle, we performed repeated washes and centrifugation steps over the period of 1 week. Fig. 2g shows that high amounts of both drugs were loaded in the Fe@MSN particles. DOX loading into the Fe@MSN was not possible when the pH was decreased below 6. The importance of electrostatic interactions for drug loading was further verified by the fact DOX loading was very low when we used non-functionalized MSN (only silanol groups) at pH 7.4. Notably, negligible leakage of drugs from the Fe@MSN particles was observed using a dialysis membrane with a 100 000 Da MWCO at 37 °C against PBS with 10% fetal bovine serum (Fig. 2h).
Even though TMZ is the first-line chemotherapy for GBM, we selected DOX due to our prior experience with its antitumor efficacy against brain tumors.17,19 Further, clinical studies had shown that DOX-loaded nanoparticles produced significant therapeutic outcomes in patients with GBM.20 To confirm the anticancer effect of DOX against glioma cells, we performed cell cytotoxicity studies to evaluate the (ESI Fig. S1†). DOX was highly cytotoxic with an IC50 of 0.28 μM against glioma cells. In addition, the mild fluorescence properties of DOX molecules suited very well the nature of our studies including histological analyses to evaluate the spread of drug into brain tumor tissues upon application of the RF.
3.2. RF-triggered drug release in vitro
Contrary to other triggered release systems, the mechanism of drug release from Fe@MSN is concentration-independent and is based on mechanical vibrations of the nanoparticle upon application of a low-power RF field. The components of the RF system were “off-the-shelf” audio power amplifiers and inexpensive electromagnets (amplitude B = 5 mT, frequency f = 1–50 kHz, RF power = 2–30 Watts). The RF coil was a custom-made solenoid (N = 105 turns, solenoid’s resistance ~5 Ohms, inner diameter = 4 cm). In a typical experiment, a small vial containing 1 mL of the Fe@MSN nanoparticles was positioned in the middle of the RF coil. After exposure to the RF field at different energy outputs and periods of time, drug release was measured using Amicon ultra centrifugal filters with 100 000 Da MWCO followed by fluorescence spectroscopy for DOX (λex/em = 485/590 nm). The primary findings of a series of in vitro mechanistic studies are summarized here: (1) The drug release rate of DOX was triggered in a controlled manner under the RF field (Fig. 3a). The release rate could be modulated by adjusting the operating parameters of the RF field. The release rate increased by increasing the RF frequency from 1 to 10 and then 50 kHz. (2) Upon application of the RF field (5 mT, 50 kHz) for 30 min, about 75% of the Fe@MSN nanoparticle’s cargo was released (Fig. 3b). (3) No temperature increase occurred in the Fe@MSN suspension after a 60 min exposure to the RF field. Further, when the temperature of the Fe@MSN suspension was elevated to 60 °C for 60 min by external heating, negligible drug was released (Fig. 3c). These two findings demonstrate that the triggered release mechanism is not based on hyperthermic effects, thereby minimizing the role of Néel relaxation. (4) Under the same RF field, suspensions of Fe@MSN with dramatically different particle concentrations exhibited the same release rate per particle (Fig. 3d). (5) The drug release rate from Fe@MSN was similar at different positions in the RF field (Fig. 3e). The sample was positioned in the center of the RF coil or 1 cm from the center of the coil or 2 cm from the center of the coil (close to the edge of the coil).
Fig. 3.
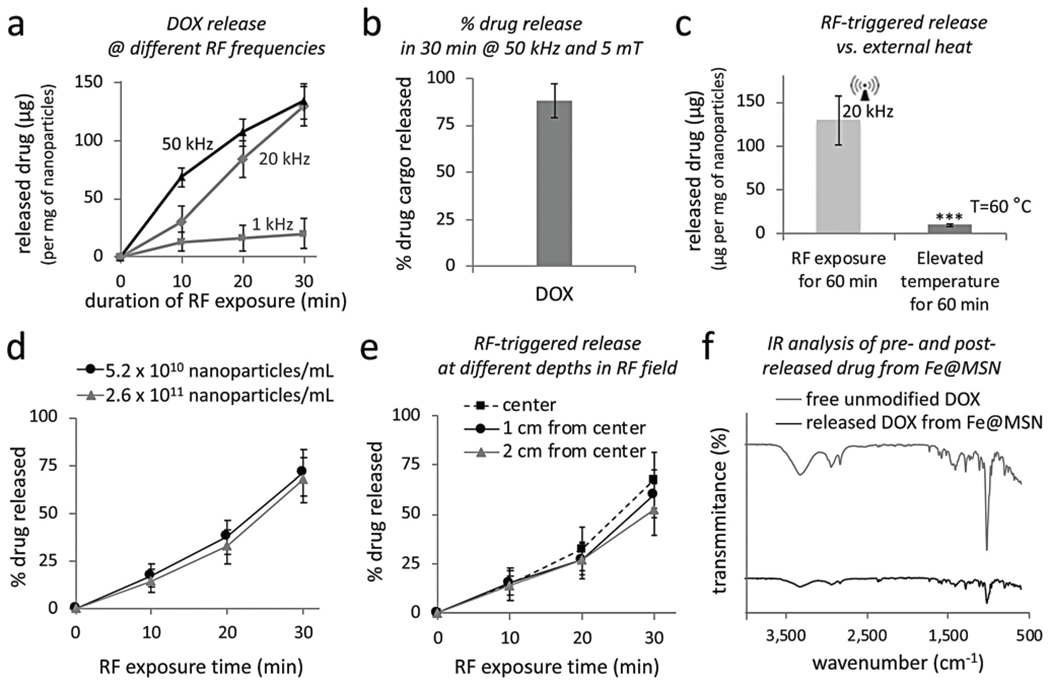
In vitro evaluation of radiofrequency (RF)-triggered drug release from Fe@MSN nanoparticles. (a) The release of DOX was triggered from Fe@MSN particles using an RF field at different frequencies (1, 20, 50 and 380 kHz; n = 4). (b) The percent released drug of the nanoparticle’s cargo is shown upon application of the RF field at 50 kHz for 30 min (c) Effect of elevated temperature on the drug release from Fe@MSN particles with an incubation time of 60 min (n = 4; unpaired t-test P < 0.0001). (d) Drug release from Fe@MSN at different particle concentration under an RF field at 50 kHz. (e) Drug release from Fe@MSN particles at different depths in the RF source (RF field: 50 kHz). (f) FT-IR spectra of free unmodified DOX and DOX released from Fe@MSN nanoparticle.
We then evaluated the integrity of the drugs by comparing the FT-IR spectra of free unmodified DOX and DOX being released from Fe@MSN (Fig. 3f). The absorption bands of DOX in its unmodified form and after being released from Fe@MSN do not exhibit any significant shift, suggesting that the structure of the molecule remained unaltered. The distinct peaks of DOX appear at 687, 1021, 1015, 1412, 1589, 1732, 2156, 2360, 2935, and 3445.65 cm−1, which correspond to alkyne C–H, aromatic C–H, aromatic C–H, carboxylic acid, alkenyl C═C stretch, C–O bond, NH3, charged amines C═NH+, C–H stretch, and O–H stretch, respectively.
3.3. Targeting of Fe@MSN nanoparticles to brain tumors
The tumor deposition of fibronectin-targeting Fe@MSN nanoparticles was evaluated in the CNS-1 rodent glioma model at different time points after tail vein injection of the formulation at a dose of 5 mg kg−1 DOX (n = 5 mice per time point). Drug levels were directly measured in homogenized brain tumors using an established method.17 Compared to negligible amounts in healthy brains, targeting of Fe@MSN particles resulted in rapid deposition to glioma sites. Within 1 h after injection, 2% of the injected dose accumulated in brain tumors, whereas deposition in glioma sites plateaued within 3 h to 4.7% of the dose (Fig. 4a). In comparison to the standard unmodified DOX and non-targeted DOX-loaded Fe@MSN, Fig. 4b shows the intratumoral drug levels of targeted Fe@MSN were about 23 and 3-fold higher than free drug and non-targeted Fe@MSN, respectively. Considering that passive intratumoral deposition typically requires longer periods of time, it should be noted that the drug levels were measured 3 and 24 h after injection for targeted and non-targeted DOX-loaded Fe@MSN, respectively. As expected, the majority of the nanoparticles ended up being cleared by the liver and spleen (Fig. 4c), while the drug levels in the heart, lungs, and kidney were very low.
Fig. 4.
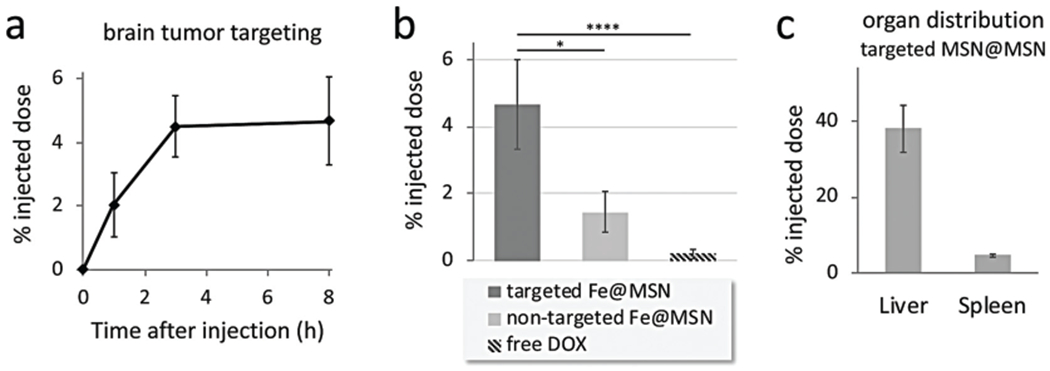
Targeting of CREKA-targeted Fe@MSN nanoparticles to brain tumors. The deposition of CREKA-targeted DOX-loaded Fe@MSN particles in GBM was evaluated in mice bearing orthotopic CNS-1 brain tumors. (a) The accumulation of targeted Fe@MSN particles in brain tumors is shown after 1, 3 and 8 h from i.v. administration at a dose of 5 mg DOX per kg b.w. (n = 5 mice in each group). (b) The accumulation of targeted Fe@MSN in brain tumors was compared to non-targeted Fe@MSN or free DOX using a dose of 5 mg kg−1 DOX for all formulations. Grouped analysis ANOVA; correct for multiple comparisons using the Holm–Sidak method. P values: * <0.01, **** <0.0001. (c) The accumulation of targeted Fe@MSN in liver and spleen is shown after 8 h from i.v. administration at a dose of 5 mg kg−1 DOX (n = 5 mice in each group).
3.4. Histological evaluation of RF-triggered drug release from Fe@MSN in vivo
The near-perivascular region and the endothelium associated with brain tumors offer a diverse set of targetable biomarkers, which differ from those in healthy brain. Such a biomarker is fibronectin.10–13 We histologically evaluated the expression of fibronectin in the orthotopic model CNS-1 in mice, which is a highly invasive rodent GBM. Fibronectin showed upregulation in brain tumors in the primary and invasive sites. The main findings are summarized here (Fig. 5a): (1) near-perivascular regions and the altered endothelium in GBM regions were exactly the locations with highly targetable fibronectin (Fig. 5a; top left and right). (2) Abundance and selective expression of fibronectin could be seen in the primary and invasive GBM sites. Fibronectin was not present on the endothelium of healthy brain tissues. (3) Mice with orthotopic CNS-1 tumors were euthanized 3 h after intravenous injection of targeted Fe@MSN particles loaded with DOX were intravenously injected via the tail vein. The predominant deposition of the targeted Fe@MSN nanoparticles was in the near-perivascular regions with overexpression of fibronectin (Fig. 5a; bottom). Similar results were observed in the GL261 glioma model in mice (ESI Fig. S2†).
Fig. 5.
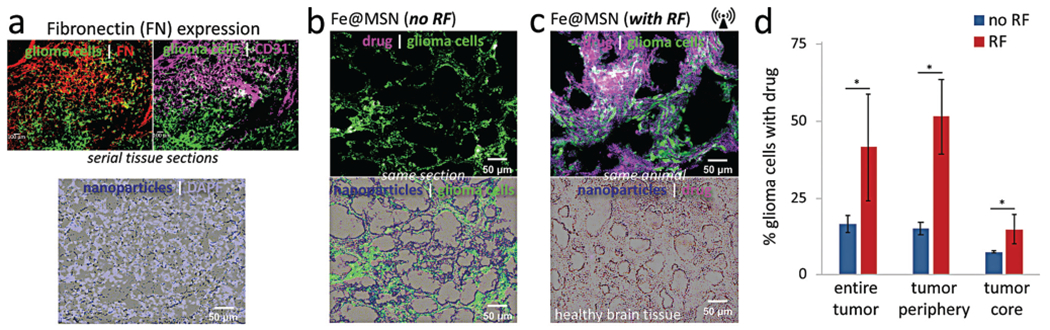
Histological evaluation of the anticancer effect of RF-triggered drug release from the Fe@MSN nanoparticles in vivo. Histological analysis was performed 3 h after the animals were treated with a single dose of DOX-loaded Fe@MSN particles. (a) The degree and topology of fibronectin was assessed in the orthotopic CNS-1 model in mice (20× magnification; green: glioma cells; red: fibronectin; purple: endothelial cells). Microdistribution of Fe@MSN particles was visualized by staining iron with Prussian blue. (b, c) Using the fluorescence properties of DOX, fluorescence microscopy shows the widespread distribution of DOX molecules (purple: DOX) after a 60 min application of RF (10× magnification). The distribution of DOX molecules is shown (b) without or (c) with RF. (d) The percent of DOX-stained cells relative to the total number of glioma cells was measured by counting the total number of glioma cells (GFP-GL261) and DOX-stained nuclei in two histological sections per tumor (n = 4 mice in each group). We also drew ROIs to distinguish the periphery from the core of the tumor (unpaired t-test; P values: *0.034, *0.011 and *0.044).
Today, systemic therapies are not the primary treatment of choice for GBMs due to the BBB. Due to the tight junctions between endothelial cells, the brain is inaccessible to any hydrophilic molecule via passive transport. Further, the brain vasculature contains various transmembrane transporter proteins (e.g., the P-glycoprotein efflux pump) that prohibit systemic therapies from reaching the brain parenchyma. Traditionally, drug delivery strategies have aimed directly for the interstitium of brain tumors. Targeting ligands have been employed to direct nanoparticles to upregulated receptors on glioma cells. Unfortunately, the microenvironment of brain tumors does not favor deep penetration of nanoparticles into the tumor interstitium with glioma cells. Precise targeting of glioma cells requires that the nanoparticles successfully extra-vasate across the brain tumor’s endothelium, navigate through the extracellular space and come in close proximity to glioma cells to interact meaningfully with the intended cell–surface receptors on the target cells. Here, we showed that the endothelium and near-perivascular regions associated with glioma cells may serve as the obvious targetable site of the disease to direct drug-loaded nanoparticles to brain tumors. By having direct access to the altered endothelium and the near-perivascular regions, circulating nanoparticles continuously scavenge the endothelium for biomarkers of disease. In the previous section, we showed that targeting of dug-loaded Fe@MSN nanoparticles resulted in a 23-fold higher intratumoral drug deposition in brain tumors in mice than free drug. However, our targeting scheme only facilitated delivery of the drug-loaded nanoparticle in the vascular and near-perivascular regions of GBM. Ideally, the drug-loaded nanoparticles should release their drug cargo at the target site to achieve cytotoxic drug levels in the tumor tissues.
Thus, we histologically determined the effect of RF on the degree and topology of drug delivery from targeted Fe@MSN localized at the brain-tumor interface in GBM (Fig. 5b and c). Since the deposition of the targeted Fe@MSN nanoparticle reaches a plateau in 3 hours after systemic injection, the RF field was applied at that time point. The RF field (50 kHz, 5 mT) was applied for 60 min with the head of the animal being positioned in the center of the RF coil in such manner that the magnetic field was directed towards the brain. The animals were euthanized after the application of RF. The glioma cells were stably transfected with GFP, while the mild red fluorescence of DOX allowed visualisation of drug delivery and spread in histology. Specifically, fluorescence quenching occurred when DOX remained loaded in the Fe@MSN particles, which resulted in negligible red fluorescence being generated without RF. More specifically, very little penetration of DOX molecules was observed into the tumor interstitium in the case of Fe@MSN-treated animals that were not exposed to RF (Fig. 5b). In the absence of RF application, the histological analysis indicated that the actual drug remained in the Fe@MSN nanoparticles and did not gain access into glioma cells. However, upon RF-triggered release, red fluorescence allowed visualization of DOX in its released free form. While no widespread delivery of DOX was observed in absence of RF, application of the RF field facilitated the release of drug from nanoparticles and transport across the BBB barrier with extensive distribution throughout the entire brain tumor volume (Fig. 5c; top panel). No nanoparticles or drug were found in healthy brain tissues of the same animal (Fig. 5c; bottom panel).
Various triggered release mechanisms have been applied in the design of nanoparticle systems, including temperature, ultrasound, light or pH sensitive systems. The release mechanism of the Fe@MSN nanoparticle is fundamentally different from thermosensitive or echogenic particles. These systems cannot consistently achieve triggered release at different locations due to either attenuation of the required high-energy waves at increased depth in tissues, or heat dissipation or insufficient particle concentration for heat generation. For example, magnetic nanoparticles of size smaller than 15 nm convert high-energy alternating magnetic fields into heat energy (Néel relaxation). In the case of Fe@MSN, the release mechanism is based on low-power RF fields and mechanical tumbling of the 20 nm iron oxide core (Brownian relaxation) rather than heating. The low-power RF fields required for drug release from Fe@MSN are very safe, penetrate deep tissues with ease, and can easily be generated throughout the entire brain. Given these relatively low RF frequencies (50 kHz) compared to high field MRI (>300 MHz), the magnetic fields are well understood. Thus, the design, cost and clinical deployment of the RF system present a low degree of translational challenge.
To obtain a quantitative evaluation, the total number of glioma cells (GFP-expressing) was counted in multiple histological sections (minimum 2 slices) per tumor (n = 4 mice per condition), whereas successful drug delivery was quantified based on DOX-stained nuclei. The percent of DOX-stained cells relative to the total number of cancer cells was used as a measure of the anticancer efficacy. We also drew ROIs to distinguish the periphery from the core of the tumor. Fig. 5d shows that 16.5% of the glioma cells had DOX-stained nuclei in the case of animals treated with the targeted Fe@MSN without RF. On the other hand, RF application resulted in 41.5% of the glioma cells having DOX-stained nucleus, which was a significant increase compared to the no RF condition (P < 0.05). Importantly, the DOX-stained nuclei were equally elevated in the core as well the periphery of the tumors. Application of RF on Fe@MSN-treated tumors exhibited a 3.4 and 2-fold increase of DOX-stained nuclei in the core of tumors compared to the no RF condition. Since DOX is a weak fluorophore, it is possible that fluorescence microscopy underestimated the number of glioma cells that contained DOX.
In additional to visualization of the drug spread in histology, we also evaluated the effect of the Fe@MSN treatment and application of RF in a macroscopic manner. Mice bearing orthotopic brain tumors were euthanized 48 h after treatment with targeted Fe@MSN loaded with DOX. Fig. 6a shows ex vivo photographs of these brains highlighting the anticancer effect of the RF-triggered release of DOX in animals treated with Fe@MSN. RF-applied animals show a dramatic tumor shrinkage compared to animals without RF application (n = 4 mice per condition). After the brains were explanted, they were precisely sliced in 500 μm sections using a mouse brain slicer and the tumor size was measured (Fig. 6b). Being ~2.4 mm at the beginning of treatment, the tumor size of the mice treated with Fe@MSN followed by RF reduced to 1.16 mm (sd = ±0.19) 48 h after treatment. In the case of the Fe@MSN treatment without RF, the tumor size grew to 4.6 mm (sd = ±0.83).
Fig. 6.
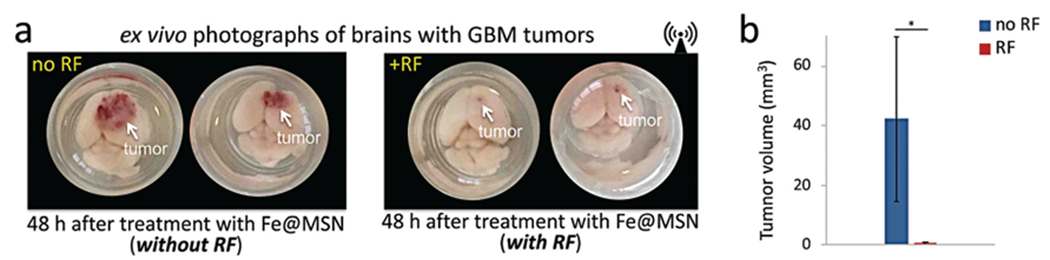
Macroscopic ex vivo evaluation of the therapeutic efficacy of the Fe@MSN treatment. (a) Using the orthotopic CNS-1 model in mice, photographs of brains show the treatment response of the RF-triggered release of DOX in animals treated with Fe@MSN. All animals were euthanized 48 h after treatment with DOX-loaded Fe@MSN particles at a dose of 5 mg kg−1 DOX. Animals were perfused and whole brains were excised and photographed from the top. (b) After euthanasia, tumors were excised and their size was measured (n = 4 mice in each group; unpaired t-test; P value *0.011).
3.5. Evaluation of therapeutic efficacy in vivo
We tested the in vivo therapeutic efficacy of DOX-loaded Fe@MSN in mice with intracranial brain tumors. The glioma cells stably expressed firefly luciferase, which allowed in vivo bioluminescence Imaging (BLI). Using BLI signal as a measure of short-term tumor response to various treatments, we compared free unmodified DOX, TMZ, and the DOX-loaded Fe@MSN. Considering the short lifespan of GBM models in mice, animals were treated three times with free unmodified drugs or drug-loaded Fe@MSN formulations (with or without RF) on day 6, 7 and 9 after tumor inoculation. The average tumor size was 1.1 mm (sd = ±0.14) at the beginning of treatment. After the last treatment on day 9, we monitored the response of the tumor to various drugs and formulations. As a metric of the response to various treatments, quantification of BLI signal was used (Fig. 7). Animals treated with free unmodified TMZ with or without the application of RF showed no therapeutic benefits. The treatment with the free unmodified DOX also had negligible therapeutic benefits. Previous preclinical and clinical studies have shown that topoisomerase I or II inhibitors (e.g., DOX) are effective against brain tumors.21–25 In our studies, DOX had similar therapeutic efficacy against GBM as TMZ. While the targeted Fe@MSN loaded with DOX treatment (without RF) exhibited an insignificant effect, the application of RF on animals treated with DOX-loaded Fe@MSN exhibited a significant therapeutic outcome. On the other hand, treatment with non-targeted Fe@MSN loaded with DOX did not exhibit any therapeutic benefits even with the application of RF.
Fig. 7.
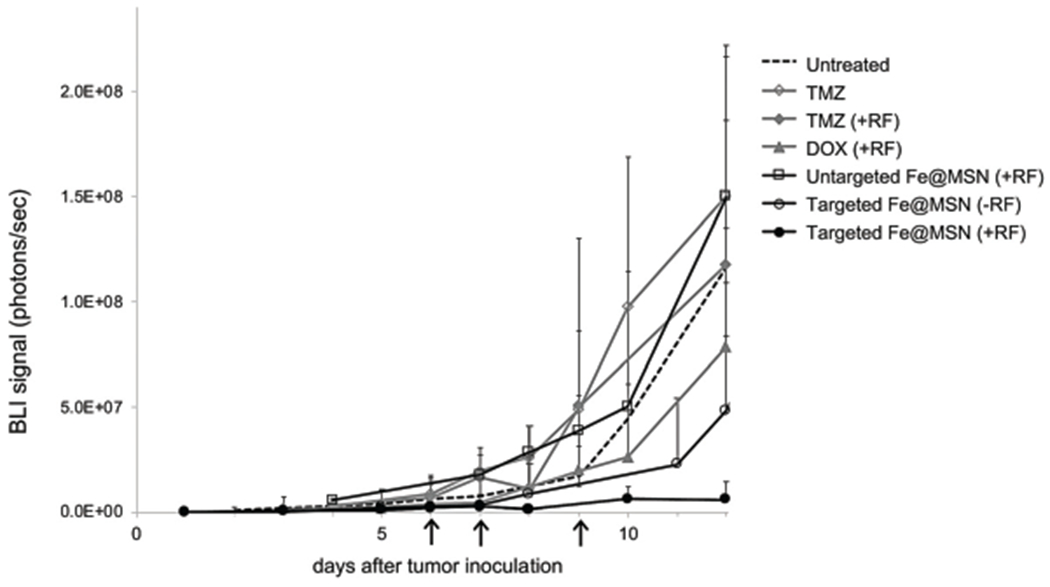
Evaluation of therapeutic efficacy of Fe@MSN treatments in vivo. (a) Various formulations were i.v. injected in mice bearing orthotopic GL261 brain tumor on day 6, 7 and 9 after tumor inoculation. Treatments included free TMZ, DOX, and DOX-loaded Fe@MSN (5 mg kg−1). In the case of treatments combined with the RF field, animals were exposed for 60 min to the RF field (5 mT, 50 kHz). The response to treatment was monitored using longitudinal bioluminescence imaging (BLI). Quantification of the whole head BLI light emission is shown (n = 7 mice in each group).
The animal studies were performed using an administered dose of ~6 mg of silica per kg of body weight. This corresponds to a relatively low dose of systemically administered silica compared to the typical dose of silica mesoporous nanoparticles in preclinical studies. The animals tolerated well the dose of the nanoparticles as indicated by negligible loss of weight and immediate recovery after IV injection as well as long-term survival. However, we have not yet completed detailed toxicity studies to evaluate short-term acute toxicity (e.g., LD50) and long-term tissue residence times. The safety–toxicity of the nanoparticle is subject of ongoing investigation. However, many reports on the toxicology of silica show that silica nanoparticles are generally well tolerated with a large maximum tolerated dose.2,26,27 For example, a long-term toxicity study of mesoporous silica nanoparticles showed no negative consequences in behavior, histology or hematology, when mice were injected twice per week for two months with 50 mg kg−1 of silica.26
Overall, effective delivery of a potent chemotherapeutic into brain tumors was able to significantly decrease tumor growth. On the contrary, this data also show that ineffective delivery of drugs resulted in continuous rapid growth of this highly aggressive animal model. As our data show in the next section, GBMs grow back rapidly once treatment with chemotherapy is discontinued. GBMs display remarkable cellular heterogeneity, which contributes to their high rates of therapeutic resistance and rapid recurrence.28–30 Previous studies identified the presence of a self-renewing, tumor-initiating subset of cells within GBMs, often called glioma stem cells or brain tumor initiating cells (BTICs).28–30 This small subpopulation of cells exhibits significant chemoresistance. As a result, chemotherapy alone is not capable to cure the disease. In future studies, we will exploit the incorporation of targeted therapeutic molecules selective towards BTICs into the Fe@MSN nanoparticle to eliminate the fraction of glioma cells that are resistant and can cause tumor recurrence.31–34
To assess the therapeutic efficacy of the targeted Fe@MSN treatment, we also measured survival times (Fig. 8). Control treatments included free unmodified DOX, and Fe@MSN without RF. Treatments were administered twice. The median survival of the untreated control group was 9 days. In agreement to the BLI studies, the tumor response to free unmodified DOX and Fe@MSN (without RF) was modest and did not prolong survival. On the other hand, RF application on Fe@MSN-treated animals resulted in a 2-fold increase in survival.
Fig. 8.
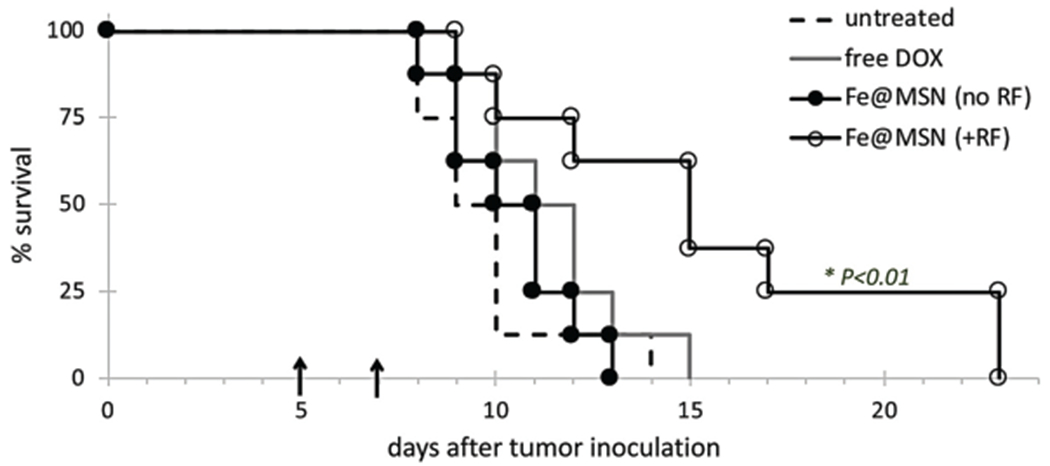
Survival curves in the orthotopic CNS-1 glioma model in mice. The survival time of animals treated with DOX-loaded nanoparticles (+RF) was compared to that of animals treated with free unmodified DOX and the untreated group (n = 8 mice in each group). Each formulation was administered at a dose of 5 mg DOX per kg of body weight. Treatments were systemically administered via a tail vein injection twice at days 5 and 7 after tumor inoculation (arrows). Statistical significance was determined using the log-rank (Mantel-Cox) test.
4. Conclusions
Using an external RF field, we designed a spatiotemporal method for release of drug from targeted nanoparticles. Since the drug-carrying component of Fe@MSN is a mesoporous silica shell, it should be mentioned that mesoporous silica nanoparticles are all-purpose, versatile drug carriers that have demonstrated loading of a variety of drug types. Mesoporous silica nanoparticles with an iron oxide core can be fabricated in high quantities with a high degree of consistency and conveniently accommodate high cargos of drugs, due to the high surface and pore volume, and available charges via various silane chemistries.2 Overall, the combination of vascular targeting with the multicomponent silica nanoparticle and low-power RF facilitated effective delivery of a potent drug across the BBB into deep interstitial regions of GMB, which are otherwise inaccessible. These results illustrate that more effective delivery and wider distribution of anticancer drugs in brain tumors can improve the survival of GBM patients.
Supplementary Material
Acknowledgements
This work was partially supported by grants from the National Cancer Institute (R01CA177716, U01CA198892), the Prayers from Maria Children’s Glioma Foundation, the Alex’s Lemonade Stand Foundation and the Angie Fowler AYA Cancer Research Initiative of the Case Comprehensive Cancer Center (E. K.). P. A. B. was supported by a graduate research fellowship from NSF. G. C. was supported by a fellowship from the NIH Interdisciplinary Biomedical Imaging Training Program (T32EB007509) administered by the Department of Biomedical Engineering, Case Western Reserve University. We also acknowledge the Case Center for Imaging Research and the core facilities at Case Comprehensive Cancer Center.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr02876e
Conflicts of interest
There are no conflicts to declare.
References
- 1.Liu X, Situ A, Kang Y, Villabroza KR, Liao Y, Chang CH, Donahue T, Nel AE and Meng H, ACS Nano, 2016, 10, 2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liberman A, Mendez N, Trogler WC and Kummel AC, Surf. Sci. Rep, 2014, 69, 132–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO and R. European Organisation for, T. Treatment, of Cancer Brain, G. Radiation Oncology and G. National Cancer Institute of Canada Clinical Trials, Lancet Oncol., 2009, 10, 459–466. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO and R. European Organisation for, T. Treatment, of Cancer Brain, G. Radiotherapy and G. National Cancer Institute of Canada Clinical Trials, N. Engl. J. Med, 2005, 352, 987–996. [DOI] [PubMed] [Google Scholar]
- 5.Juratli TA, Schackert G and Krex D, Pharmacol. Ther, 2013, 139, 341–358. [DOI] [PubMed] [Google Scholar]
- 6.Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK and Bigner DD, Expert Opin. Invest. Drugs, 2009, 18, 1061–1083. [DOI] [PubMed] [Google Scholar]
- 7.Black KL and Pikul BK, Clin. Neurosurg, 1999, 45, 160–163. [PubMed] [Google Scholar]
- 8.Iacob G and Dinca EB, J. Med. Life, 2009, 2, 386–393. [PMC free article] [PubMed] [Google Scholar]
- 9.Castellani P, Borsi L, Carnemolla B, Biro A, Dorcaratto A, Viale GL, Neri D and Zardi L, Am. J. Pathol, 2002, 161, 1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serres E, Debarbieux F, Stanchi F, Maggiorella L, Grall D, Turchi L, Burel-Vandenbos F, Figarella-Branger D, Virolle T, Rougon G and Van Obberghen-Schilling E, Oncogene, 2014, 33, 3451–3462. [DOI] [PubMed] [Google Scholar]
- 11.Ohnishi T, Hiraga S, Izumoto S, Matsumura H, Kanemura Y, Arita N and Hayakawa T, Clin. Exp. Metastasis, 1998, 16, 729–741. [DOI] [PubMed] [Google Scholar]
- 12.Neri D and Bicknell R, Nat. Rev. Cancer, 2005, 5, 436–446. [DOI] [PubMed] [Google Scholar]
- 13.Borsi L, Balza E, Bestagno M, Castellani P, Carnemolla B, Biro A, Leprini A, Sepulveda J, Burrone O, Neri D and Zardi L, Int. J. Cancer, 2002, 102, 75–85. [DOI] [PubMed] [Google Scholar]
- 14.Rauwerdink AM and Weaver JB, Med. Phys, 2011, 38, 1136–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, Qutaish M, Han Z, Schur RM, Liu Y, Wilson DL and Lu ZR, Nat. Commun, 2015, 6, 7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perera VS, Covarrubias G, Lorkowski M, Atukorale P, Rao A, Raghunathan S, Gopalakrishnan R, Erokwu BO, Liu Y, Dixit D, Brady-Kalnay SM, Wilson D, Flask C, Rich J, Peiris PM and Karathanasis E, Nanoscale, 2017, 9, 9659–9667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peiris PM, Abramowski A, Mcginnity J, Doolittle E, Toy R, Gopalakrishnan R, Shah S, Bauer L, Ghaghada KB, Hoimes C, Brady-Kalnay SM, Basilion JP, Griswold MA and Karathanasis E, Cancer Res, 2015, 75, 1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI and Nel AE, ACS Nano, 2010, 4, 4539–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McNeeley KM, Karathanasis E, Annapragada AV and Bellamkonda RV, Biomaterials, 2009, 30, 3986–3995. [DOI] [PubMed] [Google Scholar]
- 20.Koukourakis MI, Koukouraki S, Fezoulidis I, Kelekis N, Kyrias G, Archimandritis S and Karkavitsas N, Br. J. Cancer, 2000, 83, 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saito R, Krauze MT, Noble CO, Drummond DC, Kirpotin DB, Berger MS, Park JW and Bankiewicz KS, Neuro-Oncology, 2006, 8, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noble CO, Krauze MT, Drummond DC, Yamashita Y, Saito R, Berger MS, Kirpotin DB, Bankiewicz KS and Park JW, Cancer Res, 2006, 66, 2801–2806. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita Y, Krauze MT, Kawaguchi T, Noble CO, Drummond DC, Park JW and Bankiewicz KS, Neuro-Oncology, 2007, 9, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammond LA, Eckardt JR, Ganapathi R, Burris HA, Rodriguez GA, Eckhardt SG, Rothenberg ML, Weiss GR, Kuhn JG, Hodges S, Von Hoff DD and Rowinsky EK, Clin. Cancer Res, 1998, 4, 1459–1467. [PubMed] [Google Scholar]
- 25.Krauze MT, Noble CO, Kawaguchi T, Drummond D, Kirpotin DB, Yamashita Y, Kullberg E, Forsayeth J, Park JW and Bankiewicz KS, Neuro-Oncology, 2007, 9, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Liong M, Li Z, Zink JI and Tamanoi F, Small, 2010, 6, 1794–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, Li L, Teng X, Huang X, Liu H, Chen D, Ren J, He J and Tang F, Biomaterials, 2011, 32, 1657–1668. [DOI] [PubMed] [Google Scholar]
- 28.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J and Dirks PB, Cancer Res, 2003, 63, 5821–5828. [PubMed] [Google Scholar]
- 29.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB, Nature, 2004, 432, 396–401. [DOI] [PubMed] [Google Scholar]
- 30.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN, Nature, 2006, 444, 756–760. [DOI] [PubMed] [Google Scholar]
- 31.Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca KL, Lathia JD, Forrester MT, Lee J, Stamler JS, Goldman SA, Bredel M, McLendon RE, Sloan AE, Hjelmeland AB and Rich JN, Cell, 2011, 146, 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raspollini MR, Amunni G, Villanucci A, Boddi V, Baroni G, Taddei A and Taddei GL, Gynecol. Oncol, 2004, 92, 806–812. [DOI] [PubMed] [Google Scholar]
- 33.Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC and Grimm EA, Clin. Cancer Res, 2000, 6, 4768–4775. [PubMed] [Google Scholar]
- 34.Loibl S, Buck A, Strank C, von Minckwitz G, Roller M, Sinn HP, Schini-Kerth V, Solbach C, Strebhardt K and Kaufmann M, Eur. J. Cancer, 2005, 41, 265–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.