

Abstract
Flavin-dependent ‘ene’-reductases (ERED) can generate stabilized alkyl radicals when irradiated with visible light, however, they are not known to form unstabilized radicals. Here, we report an enantioselective radical cyclization using alkyl iodides as precursors to unstabilized nucleophilic radicals. Evidence suggests this species is accessed by photoexcitation of a charge-transfer complex that forms between flavin and substrate within the protein active site. Stereoselective delivery of a hydrogen atom from the flavin semiquinone to the prochiral radical formed after cyclization provides high levels of enantioselectivity across a variety of substrates. Overall, this transformation demonstrates that photoenzymatic catalysis can address long-standing selectivity challenges in the radical literature.
Graphical Abstract

Radicals are versatile intermediates that enable retrosynthetic disconnections distinct from those available to other reactive species.1 While the advent of modern methods of radical formation has spurred the development of new chemical transformations, general catalytic strategies for controlling the stereochemical outcome of these reactions are underdeveloped.2 The lack of control provided by small-molecule catalysts across a range of radical elementary steps highlights the need for new approaches to controlling this family of reactive intermediates.
Enzymes are attractive catalysts for asymmetric synthesis because they can mediate reactions with unparalleled levels of stereochemical control.3 This feature has enabled them to address some of the most daunting selectivity challenges in organic synthesis.4 Despite significant recent progress, only a small number of synthetic challenges have been tackled with enzymatic catalysis due to a perceived inability to catalyze non-natural reactions.5,6 Inspired by the selectivity challenges inherent to non-natural radical reactions, our group has sought to develop strategies for controlling radical intermediates within enzyme active sites.7,8
In our previous studies, we found that flavin-dependent ‘ene’-reductases (EREDs) can catalyze radical reactions with high levels of stereoselectivity using an electron transfer mechanism that is distinct from the hydride transfer responsible for their native reactivity.9,10 Specifically, we found that electron-deficient α-chloroamides form charge-transfer (CT) complexes with the flavin hydroquinone (FMNhq) within the ERED active site. When this complex is irradiated with visible light, an electron is promoted from flavin to the substrate, generating an electrophilic radical. In general, the strength of association in CT complexes is dependent on the electron affinity of the acceptor, the degree of electron transfer in the ground state, and a high degree of planarity in both the donor and acceptor to maximize orbital interactions.11 Consequently, their use in organic synthesis has been restricted to substrates with low reduction potentials that often possess a large degree of π-conjugation, as these tend to favor the formation of CT complexes in solution.12 Our previous study suggested that enzymes can overcome this requirement by forcing molecular interactions between the electron donor (FMNhq) and acceptor (substrate) that do not occur in solution (Figure 1A). In theory, this feature should enable enzymes to use less activated substrates as radical precursors while also providing a mechanism for restricting radical formation to the protein active site. Based on this understanding, we questioned whether a similar mechanism could be employed to generate nucleophilic radicals from unactivated alkyl iodides (Figure 1B).
Figure 1.
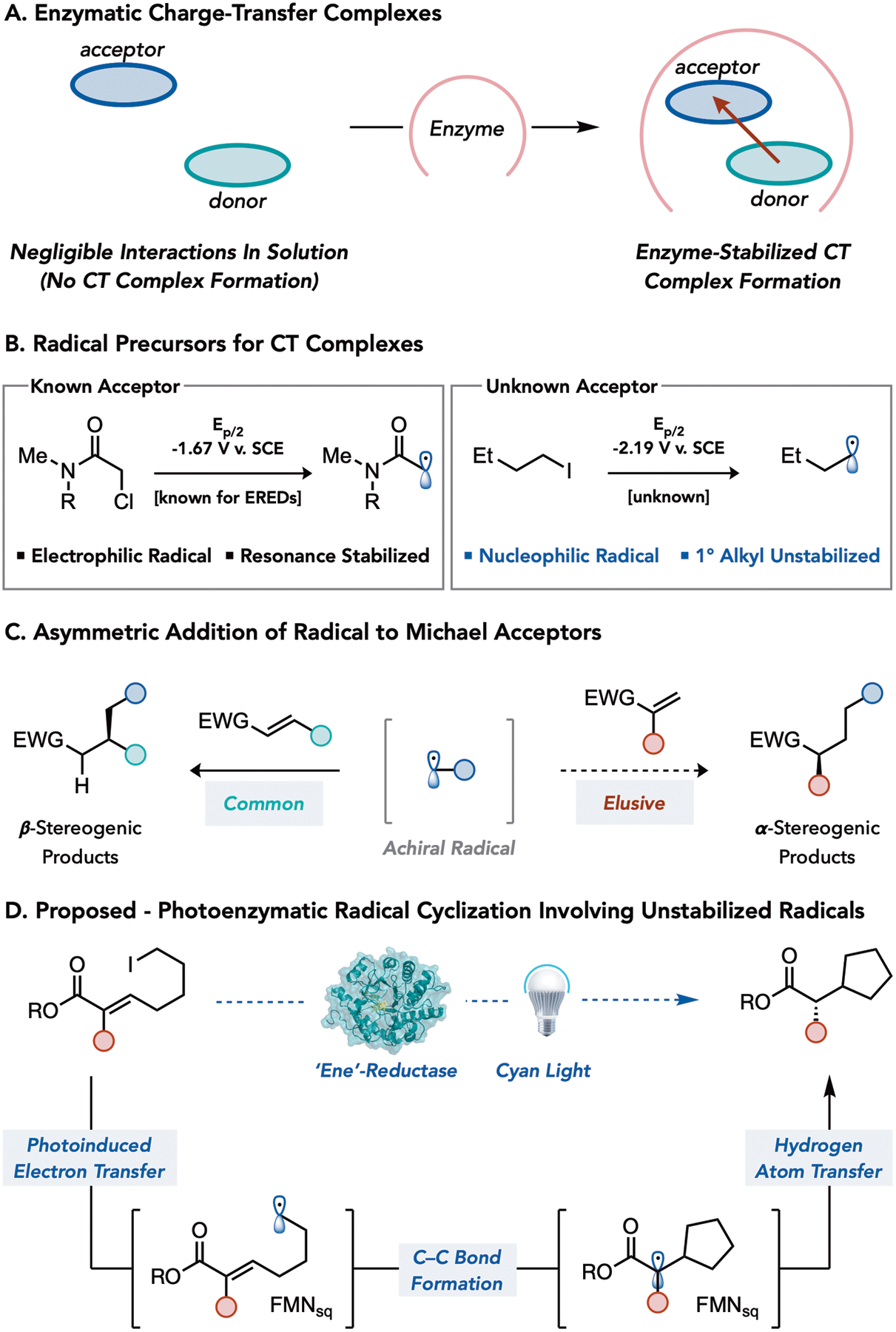
Principals of Charge-Transfer Complexes and Challenges Associated with Asymmetric Radical Conjugate Additions.
The reaction of nucleophilic radicals with electrophilic alkenes is one of the most widely utilized transformations in the radical literature.13 While chiral Lewis acid and amine catalysts have proven to be effective at setting the β-stereocenter through the controlled addition of achiral radicals to the prochiral alkenes,14 they are largely ineffective at setting the α-stereocenter via hydrogen atom transfer (HAT) (Figure 1C).15 In our previous reports, we found that EREDs can catalyze a radical hydrodehalogenation reaction with high levels of enantioselectivity. Central to this reactivity is the protein’s ability to control the stereoselective delivery of a hydrogen atom from flavin semiquinone (FMNsq) to prochiral α-acyl radicals.2 Inspired by the selectivity achieved in this step, we sought to develop a photoenzymatic cyclization involving the addition of nucleophilic radicals to Michael acceptors terminating in enantioselective HAT.
We targeted unactivated alkyl iodides as precursors to nucleophilic alkyl radicals.16 In contrast to α-halocarbonyl compounds that possess low reduction potentials and produce electrophilic radicals, unactivated alkyl iodides are more challenging to reduce and have lower electron affinities (Figure 1B).17 Consequently, they are not known to form stable CT complexes with common electron donors in solution.18 We reasoned that substrates with alkyl iodides primed for intramolecular Giese cyclizations would be capable of forming CT complexes with flavin (Figure 1D).
We initiated our studies by exploring the reductive cyclization of alkyl iodide 1a to chiral lactone 1b catalyzed by EREDs. Mutation of a conserved active site tyrosine is known to inhibit ERED’s native reactivity, so in order to avoid undesired reduction of the alkene of the starting material, this tyrosine was mutated to phenylalanine for a small panel of EREDs.19 When this panel of mutated enzymes was provided 1a and irradiated with a cyan LED (λmax = 497 nm), every enzyme afforded the desired cyclized product 1b, demonstrating promiscuous reactivity across a diverse set of EREDs (Supplemental Figure 1). The variant from Gluconobacter (GluER-Y177F) proved most effective, providing 1b in good yield and enantioselectivity, and with no observed hydrodehalogenated product (Table 1, entry 1). Alternatively, the opposite enantiomer could be obtained using OYE2-Y197F (Table 1, entry 2). No reaction occurred in the absence of light and enzyme (Table 1, entries 3–4).
TABLE 1.
Asymmetric Reductive Radical Cyclization
| entrya | variation | yield (%) | er |
|---|---|---|---|
| 1 | nonea | 56 | 92:8 |
| 2 | OYE2-Y197F | 41 | 14:86 |
| 2 | no GluER-Y177F | 0 | - |
| 3 | no light | 0 | - |
| 4 | FMN instead of GluER-Y177F | 0 | - |
| 5 | GluER instead of GluER-Y177F | 47 | 90:10 |
| 6 | freeze-pump-thaw degassing | 90 | 92:8 |
| 7 | degassing and 12 °C | 92 | 92:8 |
| 8 | preparative scale reaction | 82 | 92:8 |
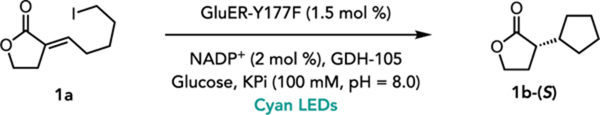
1a (10 μmol), ‘ene’-reductase (0.15 μmol), NADP+ (0.2 μmol) GDH-105 (1 mg/rxn), glucose (60 μmol), KPi (100 mM, pH = 8.0, 900 μL), iPrOH (100 μL), 48 h, rt, anaerobic.
In optimizing this reaction, several elements proved to be essential for achieving high and reproducible yields. Notable is the conserved tyrosine mutation as wild type GluER afforded product in lower yield and enantioselectivity (Table 1, entry 5), consistent with results from our previous hydrodehalogenation.8 Moreover, oxygen provided to be detrimental to the reaction, with degassed reactions providing product in improved yields (Table 1, entry 6).20 Finally, cooling the reaction from room temperature to 12 °C significantly improved reproducibility and provided a slight increase in yield (Table 1, entry 7).21 Under these conditions, the reaction could be carried out on preparative scale and isolated in nearly identical yields and enantioselectivity (Table 1, entry 8).
Next, we conducted experiments to elucidate the mechanism of this transformation. At the onset, we hypothesized that electron transfer was occurring via photoexcitation of a CT complex that forms between the alkyl iodide 1a and FMNhq. To probe this possibility, we analyzed our reaction mixture via UV-Vis. Under reaction conditions, FMN is fully reduced to FMNhq, and in the absence of substrate, there is negligible absorption around 500 nm. However, when substrate is added, a new absorption feature appears with a maximum absorption at 485 nm (Figure 2a), suggestive of the formation of a CT complex. Importantly, the same spectral feature is not observed in the absence of protein (Supplemental Figure 11), suggesting further that binding to the protein active site is required for CT complex formation.10,22 As no reaction is observed with a brominated analog of 1a, we hypothesize that the CT complex forms between flavin and the σ*C–I orbital rather than the π* of the α,-unsaturated ester. Mechanistically, excitation of this complex facilitates electron transfer from flavin to substrate, which results in loss of iodide and the formation of an unstabilized alkyl radical, which then cyclizes to form a resonance stabilized α-acyl radical.
Figure 2.

Mechanistic Experiments
We then conducted a series of experiments targeted at determining the mechanism of termination for the α-acyl radical. While we hypothesized that radical termination occurs via HAT, we recognized that single electron reduction to the corresponding enolate followed by protonation would provide the same product. To differentiate between these two pathways, we conducted isotopic labelling experiments. As a probe for the HAT mechanism, flavin was labelled in situ using d1-glucose and glucose dehydrogenase (GDH-105). When this reaction was examined, the lactone product 1b was observed in 90% yield with 92:8 er and 76% deuterium incorporation, supporting HAT from flavin as the primary mechanism of radical termination (Figure 2b). Alternatively, when the standard reaction is carried out in deuterated buffer, only 16% deuterium is observed in the product (Figure 2b). Collectively, these results suggest that α-stereocenter is set via HAT from flavin. This contrasts the native mechanism, where the α-stereocenter is set via protonation by the conserved tyrosine following hydride transfer from FMNhq. As the conserved tyrosine and FMN reside on opposite prochiral faces of the substrate, we hypothesized that reduction of a structurally related butenolide by wild-type enzyme should provide the opposite enantiomer to the one obtained in the radical cyclization. Consistent with this hypothesis, when the butenolide reduction was conducted with wild-type GluER, the reduced product was isolated as the (R)-enantiomer (Figure 2c).
With a working understanding of the mechanism and ideal conditions in hand, we explored the scope and limitations of this reaction (Figure 3). We were pleased to find that simple esters are effective substrates with GluER-Y177F, providing product in good yields and enantioselectivities (Figure 3, 2b-5b). In general, smaller substituents (Figure 3, 2b-3b) afford product with slightly attenuated enantiomeric ratios by comparison to larger substituents (Figure 3, 4b-5b). Additional functional handles such as an alkene and alkyl bromide are well tolerated off the ester (Figure 3, 6b-7b). Importantly, all these substrates lack secondary coordinating groups, providing a synthetic benefit by comparison to traditional approaches using chiral Lewis acids.2 The reaction also accommodates different substituents at the α-position. Alkyl substituents provide product with good levels of enantioselectivity (Figure 3, 8b-9b). An acetamide functional group is well tolerated by the enzyme but afford cyclized product with low levels of enantioselectivity (Figure 3, 10b). Other heteroatoms are well tolerated, providing product in good yields and modest levels of enantioselectivity (Figure 3, 11b-13b). Notably, valuable fluorinated 12b and chlorinated 13b stereocenters can be obtained.
Figure 3.

Substrate Scope
a Substrate (10 μmol), GluER-Y177F (0.15 μmol), NADP+ (0.2 μmol), GDH-105 (1 mg/rxn), glucose (60 μmol), KPi (100 mM, pH = 8.0, 900 μL), iPrOH (100 μL), 72 h, 12 °C, anaerobic. b OYE3-Y197F used instead of GluER-Y177F. c ‘ene’-reductase (0.3 μmol. 3 mol %), NADP+ (0.4 μmol).
Next, we examined other carbonyl compounds in this reaction. Ketone 14a is an effective substrate for the desired cyclization, but product 14b is formed with low levels of enantioselectivity due to competitive radical termination mechanisms (Supplemental Figure 8). Less electrophilic amide 15a was also effective but required another ERED from our panel, OYE3-Y197F, to achieve synthetically useful yield and enantioselectivity. Finally, we explored a 6-exo-trig radical cyclization and the impact that ring substituents have on the transformation. In general, the 6-exo-trig substrates were less reactive than the 5-exo-trig substrates, requiring increased enzyme loading to achieve comparable yields (Figure 3, 16b-18b).
Having identified that alkyl iodides form CT complexes with flavin, we questioned whether other alkyl radical precursors would be compatible in this reaction, and if selectivity would be maintained in the resulting cyclization. N-Hydroxyphthalimide (NHPI) esters have been demonstrated to produce alkyl radicals upon single electron reduction.23,24,25 Indeed, when the redox active ester precursor 19 was prepared and subjected to the reaction conditions, the expected ester product was formed in 93% yield with 92:8 er (Figure 4, 2b). Moreover, while alkyl iodides were only modestly effective for 6-exo-trig cyclizations, requiring high catalyst loadings (3 mol %) to achieve acceptable yields, the reaction can be accomplished using the corresponding NHPI ester at lower catalyst loadings with comparable yields and levels of selectivity (Figure 4, 16b).
Figure 4.
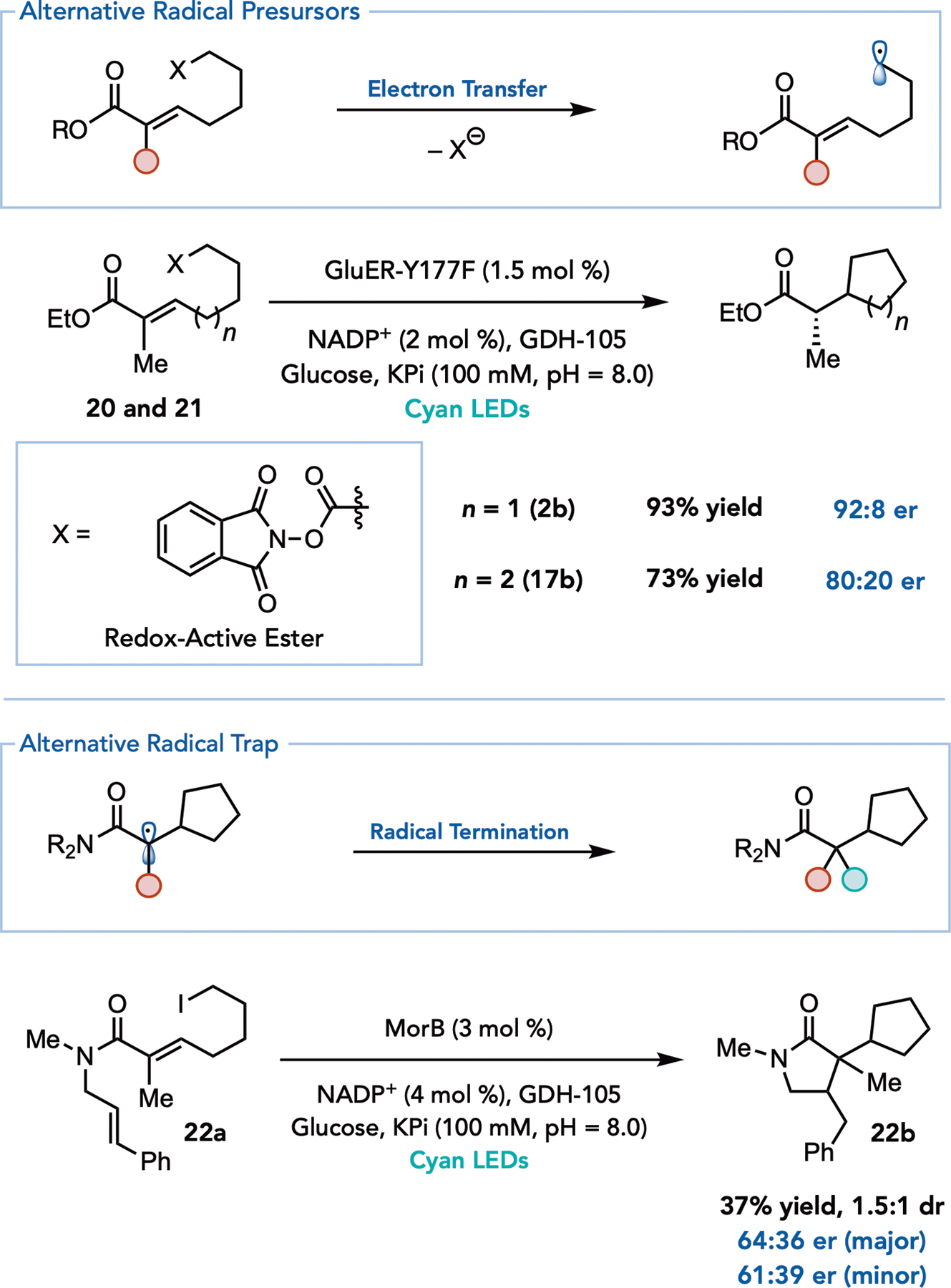
Redox Active Esters and Polycyclization
Lastly, we questioned whether the α-acyl radical intermediate formed during this reaction could be trapped with alternative radical coupling partners to build further molecular complexity. We recently demonstrated that EREDs can catalyze reductive radical cyclizations using α-acyl radical intermediates. We reasoned that an enzyme with a large active site could accommodate a substrate primed for a polycyclization (Figure 4, 21a). Indeed, morphinone reductase (MorB), can catalyze the polycyclization of a substrate bearing a cinnamyl amide in promising yields (Figure 4, 21b). This proof of concept demonstrates the potential of EREDs to be applied to complex polyene-cyclizations.
In conclusion, we have developed a reductive radical cyclization of unactivated alkyl iodides to afford chiral esters, amides, and lactones. Radical formation is achieved via photoexcitation of a CT complex that forms between flavin and the substrate. This report expands the types of reductive radical cyclizations reactions available to biocatalysts, enhancing their utility in solving long-standing challenges in asymmetric chemical synthesis.
Supplementary Material
ACKNOWLEDGMENT
Financial support provided by the NIH (R01 GM127703), the Searle Scholars Foundation, and the Sloan Research Fellowship.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3296. [DOI] [PubMed] [Google Scholar]
- (3).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed 2018, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Liang J; Mundorff E; Voladri R; Jenne S; Gilson L; Conway A; Krebber A; Wong J; Huisman G; Truesdell S; Lalonde J Highly Enantioselective Reduction of a Small Heterocycle Ketone: Biocatalytic Reduction of Tetrahedyrothiophene-3-one to the Corresponding (R)-Alcohol. Org. Process Res. Dev 2010, 14, 188–192. [Google Scholar]; (b) Chen K; Arnold FH Engineering New Catalytic Activities in Enzymes. Nat. Catal 2020, 3, 203–213. [Google Scholar]; (c) Huffman MA et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366, 1255–1259. [DOI] [PubMed] [Google Scholar]; (d) Schober M et al. Chiral Synthesis of LSD1 inhibitor GSK2879552 enabled by direction evolution of an imine reductase. Nat. Catal 2019, 2, 909–915. [Google Scholar]
- (5).Bornscheuer UT; Huisman GW; Kazlauskas RJ; Lutz S; Moore JC; Robins K Engineering the Third Wave of Biocatalysis. Nature 2012, 48, 185. [DOI] [PubMed] [Google Scholar]
- (6).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (7).Hyster TK Radical Biocatalysis: Using Non-Natural Single Electron Transfer Mechanisms to Access New Enzymatic Functions. Synlett 2020, 31, 248–254. [Google Scholar]
- (8).Nakano Y; Biegasiewicz KF; Hyster TK Biocatalytic Hydrogen Atom Transfer: An Invigorating Approach to Free-Radical Reactions. Curr. Opin. Chem. Biol 2019, 49, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sandoval BA; Meichan AJ; Hyster TK Enantioselective Hydrogen Atom Transfer: Discovery of Catalytic Promiscuity in Flavin-Dependent ‘Ene’-Reductases. J. Am. Chem. Soc 2017, 139, 11313–11316. [DOI] [PubMed] [Google Scholar]
- (10).Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Goetz KP’ Vermeulen D; Payne ME; Kloc C; McNeil LE; Jurchescu OD Charge-transfer complexes: new perspectives on an old class of compounds. J. Mat. Chem. C 2014, 2, 3065. [Google Scholar]
- (12).(a) Crisenza GEM; Mazzarella D; Melchiorre P Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu M-C; Shang R; Zhao B; Wang B; Fu Y Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide. Science 2019, 363, 1429–1434. [DOI] [PubMed] [Google Scholar]; (c) Bahamonde A; Melchiorre P Mechanism of Stereoselective α-alkylation of aldehyde driven by the photochemical activity of enamines. J. Am. Chem. Soc 2016, 138, 8019–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Woźniak Ł; Murphy JJ; Melchiorre P Photo-organocatalytic Enantioselective Perfluorialkylation of β-ketoesters. J. Am. Chem. Soc 2015, 137, 5678–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Srikanth GSC; Castle SL Advances in Radical Conjugate Additions. Tetrahedron 2005, 6, 10377–10441. [Google Scholar]
- (14).(a) Sibi MP; Zimmerman J; Rheault T Enantioselective Conjugate Radical Addition to β-Acyloxy Acrylate Acceptors: An Approach to Acetate Aldol-Type Products. Angew. Chem. Int. Ed 2003, 42, 4521–4523. [DOI] [PubMed] [Google Scholar]; (b) Ruiz Espelt L; McPherson IS; Wiensch EM; Yoon TP Enantioselective Conjugate Additions of α-Amino Radicals via Cooperative Photoredox and Lewis Acid Catalysis. J. Am. Chem. Soc 2015, 137, 2452–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huo H; Harms K; Meggers E Catalytic, Enantioselective Addition of Alkyl Radicals to Alkenes via Visible-Light-Activated Photoredox Catalysis with a Chiral Rhodium Complex. J. Am. Chem. Soc 2016, 138, 6936–6939. [DOI] [PubMed] [Google Scholar]; (d) Murphy JJ; Bastida D; Paria S; Fagnoni M; Melchiorre P Asymmetric Catalytic Formation of Quaternary Carbons by Iminium Ion Trapping of Radicals. Nature 2016, 532, 218–222. [DOI] [PubMed] [Google Scholar]
- (15).(a) Sibi MP; Patil K Enantioselective H-Atom Transfer Reaction: A Strategy to Synthesize Formaldehyde Aldol Products. Org. Lett 2005, 7, 1453–1456. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP; Sausker JB The Role of the Achiral Template in Enantioselective Transformations. Radical Conjugate Additions to α-Methacrylates Followed by Hydrogen Atom Transfer. J. Am. Chem. Soc 2002, 124, 984–991. [DOI] [PubMed] [Google Scholar]; (c) Aechtner T; Dressel M; Bach T Hydrogen Bond Mediated Enantioselectivity of Radical Reactions. Angew. Chem. Int. Ed 2004, 43, 5849–5851. [DOI] [PubMed] [Google Scholar]
- (16).Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CRJ Engaging Unactivated Alkyl, Alkenyl and Aryl Iodides in Visible-Light-Mediated Free Radical Reactions. Nat. Chem 2012, 4, 854. [DOI] [PubMed] [Google Scholar]
- (17).Rondini S; Mussini PR; Muttini P; Sello G Silver as a powerful electrocatalyst for organic halide reduction: the critical role of molecular structure. Electrochimica Acta 2001, 46, 3245–3258. [Google Scholar]
- (18).Lima CGS; Lima TM; Duarte C; Jurberg ID; Paixão MW Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. [Google Scholar]
- (19).Kohli RM; Massey V The Oxidative Half-Reaction of Old Yellow Enzyme THE ROLE OF TYROSINE 196. J. Biol. Chem 1998, 273, 32763–32770. [DOI] [PubMed] [Google Scholar]
- (20).Baier J; Maisch T; Maier M; Engle E Landthaler M; Bäumler. Singlet Oxygen Geneation by UVA Light Exposure of Endogenous Photosensitizer. BioPhys J. 2006, 91, 1452–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). Using tryptic digestions coupled with LC-MS/MS, we found that the substrate alkylates the protein throughout the reaction. See SI for details.
- (22).Emmanuel MA; Greenberg NR; Oblinsky DG; Hyster TK Accessing Non-Natural Reactivity by Irradiating Nicotinamide-Dependent Enzymes with Light. Nature 2016, 540, 414. [DOI] [PubMed] [Google Scholar]
- (23).Okada Keiji.; Okamoto Kazushige.; Oda Masaji. A New and Practical Method of Decarboxylation: Photosensitized Decarboxylation of N-Acyloxyphthalimides via Electron-Transfer Mechanism. J. Am. Chem. Soc 1988, 110, 8736–8738. [Google Scholar]
- (24).(a) Murarka S N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]; (b) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A General Alkyl-Alkyl Cross-Coupling Enabled by Redox-Active Esters and Alkylzinc Reagents. Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Fawcett A; Pradeilles J; Wang Y; Matsuga T; Myers EL; Aggarwal VK Photoinduced decarboxylative borylation of carboxylic acids. Science 2017, 357, 283–286. [DOI] [PubMed] [Google Scholar]; (b) Zhang J; Li Y; Xu R; Chen Y Donor-Acceptor Comeplex Enables Alkoxyl Radical Generation form Metal-Free C(sp3)-C(sp3) Cleavage and Allylation/Alkenylation. Angew. Chem. Int. Ed 2017, 56, 12619–12623. [DOI] [PubMed] [Google Scholar]; (c) Bosque I; Bach T 3-Acetoxyquinuclidine as Catalyst in Electron Donor-Acceptor Complex-Mediated Reactions Triggered by Visible Light. ACS Catal. 2019, 9, 9103–9109. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.